FORM
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
(Mark One)
For the fiscal year ended
OR
For the transition period from _________ to __________.
Commission file number
(Exact name of registrant as specified in its charter) |
| ||
(State or other jurisdiction of incorporation or organization) |
| (I.R.S. Employer Identification No.) |
|
|
|
| ||
(Address of principal executive offices) |
| (Zip Code) |
Registrant's telephone number, including area code: (
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
| Trading Symbol |
| Name of each exchange on which registered |
|
|
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. ☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an “emerging growth company”. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | ☐ | Accelerated filer | ☐ |
☒ | Smaller reporting company | ||
|
| Emerging Growth Company |
If an emerging growth company, indicate by checkmark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act): Yes
The aggregate market value of the voting stock held by non-affiliates of the Registrant, based upon the closing sale price of the registrant’s common stock on March 31, 2022, as quoted on the NYSE American, was $
As of December 14, 2022, the Registrant had
Documents Incorporated by Reference: None
FORWARD-LOOKING STATEMENTS
This report contains “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995, Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. You can generally identify these forward-looking statements by forward-looking words such as “anticipates,” “believes,” “expects,” “intends,” “future,” “could,” “estimates,” “plans,” “would,” “should,” “potential,” “continues” and similar words or expressions (as well as other words or expressions referencing future events, conditions or circumstances). These forward-looking statements involve risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by such forward-looking statements, including, but not limited to:
| · | the progress and timing of, and the amount of expenses associated with, our research, development and commercialization activities for our product candidates, including Multikine; |
| · | our expectations regarding the timing, costs and outcome of any pending or future litigation matters, lawsuits or arbitration proceeding; |
| · | the success of our clinical studies for our product candidates; |
| · | our ability to obtain U.S. and foreign regulatory approval for our product candidates and the ability of our product candidates to meet existing or future regulatory standards; |
| · | our expectations regarding federal, state and foreign regulatory requirements; |
| · | the therapeutic benefits and effectiveness of our product candidates; |
| · | the safety profile and related adverse events of our product candidates; |
| · | our ability to manufacture sufficient amounts of Multikine or our other product candidates for use in our clinical studies or, if approved, for commercialization activities following such regulatory approvals; |
| · | our plans with respect to collaborations and licenses related to the development, manufacture or sale of our product candidates; |
| · | business disruption and related risks resulting from the recent pandemic of the novel coronavirus 2019 (COVID-19); |
| · | our expectations as to future financial performance, expense levels and liquidity sources; |
| · | our ability to compete with other companies that are or may be developing or selling products that are competitive with our product candidates; |
| · | anticipated trends and challenges in our potential markets; |
| · | our ability to attract, retain and motivate key personnel; |
| · | our ability to continue as a going concern; and |
| · | our liquidity. |
All forward-looking statements are expressly qualified in their entirety by this cautionary statement. The forward-looking statements contained in this report speak only as of their respective dates. Except to the extent required by applicable laws and regulations, we undertake no obligation to update these forward-looking statements to reflect new information, events or circumstances or to reflect the occurrence of unanticipated events. In light of these risks and uncertainties, the forward-looking events and circumstances described in this report may not occur and actual results could differ materially from those anticipated or implied in such forward-looking statements. Accordingly, you are cautioned not to place undue reliance on these forward-looking statements.
| 2 |
PART I
ITEM 1. BUSINESS
CEL-SCI Corporation (CEL-SCI) is a clinical-stage biotechnology company focused on finding the best way to activate the immune system to fight cancer and infectious diseases. Its lead investigational therapy Multikine® (Leukocyte Interleukin, Injection) completed a pivotal Phase 3 clinical trial for patients who are newly diagnosed with locally advanced (stage III and IV) primary (not yet treated) squamous cell carcinoma of the head and neck (SCCHN). Multikine has received Orphan Drug Status from the U.S. Food and Drug Administration(FDA) for this indication. The study is believed to be the biggest Phase 3 head and neck cancer study ever with 928 patients and lasted almost 10 years. This trial was under the management of two clinical research organizations(CROs): ICON plc.(ICON) and Ergomed Clinical Research Limited (Ergomed).
On June 28, 2021, the Company announced top line results from its pivotal Phase 3 study for Multikine. The Phase 3 results showed a long-term 5-year overall survival (OS) benefit in the treatment arm that received Multikine treatment followed by surgery and radiation (the lower risk to recurrence treatment arm). This survival benefit was robust and durable and added no toxicity to the overall treatment, something not commonly seen with cancer drugs. In fact, the survival benefit increased over time and at 5-years the overall survival benefit reached an absolute 14.1% advantage for the Multikine treated arm over control (n=380, total study patients treated with surgery plus radiation), control arm 48.6%, Multikine arm 62.7% survival.
The study used the standard of care treatment for advanced primary head and neck cancer patients as a comparison. The patients received surgery followed by either radiation or chemoradiation (chemotherapy and radiation at the same time), as determined by the physician. This means that there were 2 treatment arms, 1) surgery plus radiation or 2) surgery plus chemoradiation. The arm that received Multikine treatment followed by surgery and radiation showed great survival benefit, but when chemotherapy was added in the second treatment arm, the immunological effect of Multikine was negated. Therefore, when the two treatment arms were combined the study did not achieve its primary endpoint of a 10% improvement in overall survival.
However, the analysis of the separate treatment arms was prespecified in the protocol and carried out prior to the Company becoming unblinded. The OS benefit of 14.1% at 5 years for this treatment arm exceeded the 10% OS benefit set out for the study population as a whole. The OS results for this treatment arm are significant (two-sided p=0.0236, HR=0.68) and the effect is robust, durable and increasing over time. In addition, the study had a significant number of patients who had partial and even complete tumor responses following the 3-week treatment with Multikine. Most of those were among the patients who did not receive chemotherapy. It was also discovered that patients who have tumor burden reductions (early tumor responders) have significantly improved survival. The results from the Phase 3 cancer study proved that Multikine met all of the protocol required benefits stated in the study protocol in patients in the treatment arm receiving surgery and radiation as their standard therapies. The Company will be filing for and seeking FDA approval for the use of Multikine in the treatment of advanced primary head and neck cancer in this patient population of about 210,000 patients annually worldwide.
CEL-SCI’s investigational immunotherapy Multikine is being used in a different way than cancer immunotherapy is usually used. It is given before any other therapy has been administered because that is when the immune system is thought to be strongest (as a neoadjuvant). It is also administered locally around the tumors and near the draining lymph node. In the Phase 3 clinical trial, Multikine was given locally for three weeks, five days per week as a first line treatment before surgery, radiation or radiochemotherapy. The goal is to help the intact immune system “see” the cancer and kill the micro metastases that usually cause recurrence of the cancer. In short, CEL-SCI believes that local administration and administration of Multikine before weakening of the immune system by surgery and radiation will result in improved outcomes and better overall survival rates for patients suffering from head and neck cancer.
CEL-SCI is also investigating a peptide-based immunotherapy (CEL-4000) as a vaccine for rheumatoid arthritis using its LEAPS technology platform.
CEL-SCI was formed as a Colorado corporation in 1983. CEL-SCI’s principal office is located at 8229 Boone Boulevard, Suite 802, Vienna, VA 22182. CEL-SCI’s telephone number is 703-506-9460 and its website is www.cel-sci.com. CEL-SCI does not incorporate the information on its website into this report, and you should not consider it part of this report.
CEL-SCI makes its electronic filings with the Securities and Exchange Commission (SEC), including its annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to these reports. These filings are available on its website free of charge as soon as practicable after they are filed or furnished to the SEC.
| 3 |
CEL-SCI’S PRODUCTS
CEL-SCI is a clinical-stage biotechnology company dedicated to research and development directed at improving the treatment of cancer and other diseases by using the immune system, the body’s natural defense system. CEL-SCI is currently focused on the development of the following product candidates and technologies:
| 1) | Multikine, an investigational immunotherapy under development for the potential treatment of certain head and neck cancers; |
|
|
|
| 2) | L.E.A.P.S. (Ligand Epitope Antigen Presentation System) technology, or LEAPS, with a product candidate CEL-4000, under development for the potential treatment of rheumatoid arthritis. |
None of CEL-SCI’s product candidates have been approved for sale, barter or exchange by the FDA or any other regulatory agency for any use to treat disease in humans nor has the safety or efficacy of these products been established for any use. There can be no assurance that obtaining marketing approval from the FDA in the United States and by comparable agencies in most foreign countries will be granted.
MULTIKINE
CEL-SCI’s lead investigational therapy, Multikine, is currently being developed as a potential therapeutic agent directed at using the immune system to produce an anti-tumor immune response. Data from Phase 1 and Phase 2 clinical trials suggest that Multikine may help the immune system “see” the tumor and then attack it, enabling the body’s own anti-tumor immune response to fight the tumor. Multikine is the trademark that CEL-SCI has registered for this investigational therapy, and this proprietary name is subject to review by the FDA, in connection with CEL-SCI’s future anticipated regulatory submission for approval in the United States. Multikine has not been licensed or approved for sale, barter or exchange by the FDA or any other regulatory agency, such as the European Medicine Agency(EMA), and neither its safety nor its efficacy been established.
Multikine is an immunotherapy product candidate comprised of a patented defined mixture of 14 human natural cytokines. If commercial approval is obtained, CEL-SCI intends to manufacture Multikine in a proprietary manner in CEL-SCI’s manufacturing facility near Baltimore, Maryland, USA. CEL-SCI spent over 10 years and more than $100 million developing and validating the manufacturing process for Multikine. The pro-inflammatory cytokine mixture includes interleukins, interferons, chemokines and colony-stimulating factors, which contain elements of the body’s natural mix of defenses against cancer.
Multikine is designed to be used in a different way than cancer immunotherapy is generally being used. Generally, cancer immunotherapy is given to patients who have already failed other treatments such as surgery, radiation and/or chemotherapy and most of the time it is administered systemically. Multikine on the other hand is administered locally to treat tumors and their microenvironment before any other therapy has been administered because it is believed that this is the time when the immune system would be strongest and most amenable to activation against the tumor. For example, in the Phase 3 clinical trial, Multikine was injected locally around the tumor and near the adjacent draining lymph nodes for three weeks, five days a week as a first treatment before surgery, radiation and/or chemotherapy. The goal is to help the intact immune system recognize and kill the tumor micro metastases that usually cause recurrence of the cancer. In short, CEL-SCI believes that the local administration of Multikine before weakening of the immune system by surgery, chemotherapy and radiation will result in better anti-tumor response than if Multikine were administered after surgery and radiation. In clinical studies of Multikine, administration of the investigational therapy to head and neck cancer patients has demonstrated the potential for lesser or no appreciable toxicity.
| 4 |
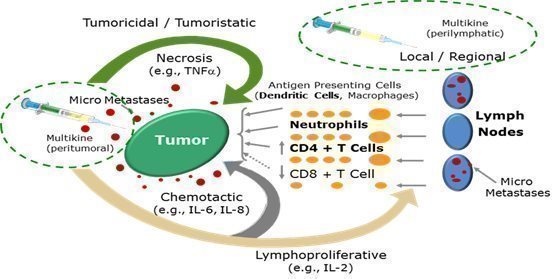
Source: Adapted from Timar et al., Journal of Clinical Oncology 23(15) May 20, 2005
The first indication CEL-SCI is pursuing for its investigational drug product candidate Multikine is an indication for the neoadjuvant therapy in patients with squamous cell carcinoma of the head and neck, or SCCHN (hereafter also referred to as advanced primary head and neck cancer).
On May 27, 2022, CEL-SCI announced the American Society of Clinical Oncology (ASCO) published two abstracts related to its pivotal Phase 3 Multikine head and neck cancer clinical trial. The poster was presented by CEL-SCI’s Chief Scientific Officer, Eyal Talor, Ph.D. at the 2022 ASCO Annual Meeting on June 6, 2022 in Chicago, Illinois. The abstract titles and corresponding links are as follows:
| · | “Leukocyte interleukin injection (LI) immunotherapy extends overall survival (OS) in treatment-naive low-risk (LR) locally advanced primary squamous cell carcinoma of the head and neck: The IT-MATTERS study.” |
| o | Link to abstract: https://meetings.asco.org/abstracts-presentations/207201 |
| o | Link to poster: https://cel-sci.com/wp-content/uploads/2022/06/CEL-SCI-ASCO-2022-Poster-6032-June-6-Head-and-Neck-Cancer-1.pdf |
| · | “Novel algorithm for assigning risk/disease-directed treatment (DDT) choice in locally advanced primary squamous cell carcinoma of the head and neck (SCCHN): Using pretreatment data only.” |
| o | Link to abstract: https://meetings.asco.org/abstracts-presentations/207202/ |
At ASCO 2022, CEL-SCI presented data that showed the following:
| · | 14.1% absolute advantage in overall survival (OS) at 5-years in the lower-risk-for-recurrence treatment arm (62.7% vs 48.6%) of patients with previously untreated locally advanced primary squamous cell carcinoma of the head and neck (Multikine+CIZ) versus the standard of care (SOC) control patients. Patients in the “lower-risk-for-recurrence” treatment arm are those with no adverse features discovered during surgery and who are therefore supposed to receive radiotherapy only after surgery. However, “lower-risk” does not mean low risk, as this treatment arm without CEL-SCI’s investigational Multikine still saw less than 50% survival 5-years post standard of care treatment alone (control group). |
|
|
|
| · | Nearly 4-year increase in median survival in this treatment arm (101.7 months for Multikine+CIZ versus 55.2 months for the SOC alone). |
| 5 |
| · | Objective response before surgery (partial and complete tumor responses): |
| o | In 8.5% (45/529) of Multikine-treated patients in the intent-to-treat (ITT) population (n=923) versus zero in the SOC alone (control). |
| o | In 16.0% (34/212) of Multikine-treated patients in this treatment arm (n=380) versus zero in the SOC alone (control). |
| · | Complete tumor response before surgery in five of the early responders, all five of which were in the Multikine+CIZ treatment arm. |
| · | Objective responses before surgery were prognostic for improved survival and significant for reduced death rate: |
| o | In the overall ITT population, 22.2% death rate (n=45) among objective responders before surgery versus 54.1% death rate for the Multikine non-responders (n=484). |
| o | In the Proposed Indication, 17.6% death rate (n=34) among objective responders before surgery versus 42.7% death rate for the Multikine non-responders (n=178). |
| · | Histopathological analysis confirmed the effect of Multikine, as 61 markers, ratios, and combinations showed a statistically significant effect (two-sided p<0.05) favoring the Multikine+CIZ treatment arm versus the SOC alone (control) for OS, Progression Free Survival (PFS), and Locoregional Control (LRC) outcomes. |
| · | Additional (confirmatory) progression-free survival (PFS) benefit in the Proposed Indication was observed for Multikine+CIZ versus the SOC alone. |
| · | Pre-specified analysis of the Proposed Indication was noted and discussed in the original study protocol and pre-specified in the statistical analysis plan. The Proposed Indication comprised about 40% of all study participants. |
| · | The overall incidence of adverse events and serious adverse events in the Multikine arms was not substantially different versus the SOC alone. |
CEL-SCI also presented a selection process (algorithm) that allows physicians to select before surgery those patients who are intended to receive only radiotherapy after surgery.
On September 12, 2022, CEL-SCI announced two poster presentations were delivered at the European Society for Medical Oncology (EMSO) annual Congress. Data presented were from CEL-SCI’s pivotal Phase 3 study and summarized below:
Poster Presentation: Early response to Neoadjuvant Leukocyte Interleukin Injection (LI) immunotherapy extends overall survival (OS) in locally advanced primary squamous cell carcinoma (SCC) of the head & neck (HN): the IT-MATTERS Study
| · | Early tumor response (early response) to neoadjuvant Multikine-Treatment is noted before surgery (occurring at median 5 weeks post-randomization) adding credibility to the isolated impact of early treatment |
| · | Early response provides a positive signal to both patients and care providers (early in the treatment course) |
| · | Early response was noted only in the Multikine* (Leukocyte Interleukin Injection) treatment groups and not in the control group |
| · | Early response occurs in both the Lower Risk and Higher Risk groups for recurrence (Risk as defined per NCCN Guidelines) |
| · | Early response is prognostic and predictive for overall survival in: |
| · | The overall population; and |
| · | The Lower Risk population |
| · | Benefit was also seen in Multikine-treated Lower Risk non-responders |
| · | Link to poster: https://cel-sci.com/wp-content/uploads/2022/09/CEL-SCI-ESMO-690P-Early-Responders-Poster-FINAL.pdf |
| · | Link to video presentation: https://youtu.be/zMoFtweVGzs |
| 6 |
Poster Presentation: Histopathology (HP) biomarkers confirm Leukocyte Interleukin Injection (LI) treatment (Tx) outcome in naïve locally advanced primary head & neck squamous cell carcinoma (SCCHN) the IT-MATTERS Study
| · | Pre-defined markers, ratios, and combinations derived from Multikine treated tumor samples at surgery contribute to Multikine efficacy for all three efficacy endpoints (OS), progression free survival (PFS), and local regional control (LRC) |
| · | Broad representation of markers, ratios, and combinations overall and for Lower Risk (LR) for the OS, PFS, LRC efficacy study endpoints |
| · | There were 61 (21.9%) favorable overall and 54 (19.4%) favorable Lower Risk treatment group outcomes (much beyond 2.5% chance) and only a total of five instances (1.9%) [all High Risk] having unfavorable treatment group outcome (within the realm of chance) |
| · | These biomarkers were prognostic for superior efficacy of the post-surgery adjuvant radiotherapy as compared to adjuvant chemoradiotherapy |
| · | The results support the Lower Risk treatment advantage (0.68 HR, Wald p<0.05) significantly favoring Multikine+CIZ+ SOC vs SOC alone |
| · | Link to poster: https://cel-sci.com/wp-content/uploads/2022/09/CEL-SCI-ESMO-128P-HP-Poster-FINAL.pdf |
The study used the standard of care treatment for advanced primary head and neck cancer patients as a comparison. The patients received surgery followed by either radiation or chemoradiation (chemotherapy and radiation at the same time), as determined by the physician based on pathology from surgery. This means that there were 2 distinctly different treatment arms, 1) surgery plus radiation or 2) surgery plus chemoradiation. The arm that received Multikine treatment followed by surgery and radiation showed great survival benefit, but when chemotherapy was added in the second treatment arm, the immunological effect of Multikine was negated. Therefore, when the two treatment arms were combined the study did not achieve its primary endpoint of a 10% improvement in overall survival.
However, the analysis of the separate treatment arms was prespecified in the protocol and carried out prior to the Company becoming unblinded. The OS benefit of 14.1% at 5 years for this treatment arm exceeded the 10% OS benefit set out for the study population as a whole. The OS results for this treatment arm are significant (two-sided p=0.0236, HR=0.68) and the effect is robust, durable and increasing over time. The results from the Phase 3 cancer study proved that Multikine met all of the protocol required benefits stated in the study protocol in patients in the treatment arm receiving surgery and radiation as their standard therapies.
CEL-SCI also presented data at the ESMO cancer meeting in September 2022. Particularly important is the early tumor response (ER) seen as a direct result of the 3-week Multikine treatment. LR means lower risk for recurrence which is scheduled to be treated with surgery and radiotherapy, HR means higher risk for recurrence which is scheduled to be treated with surgery followed by radiotherapy and concurrent chemotherapy. MK means Multikine. OS means Overall Survival.
There were 45 objective Early Tumor Responses (5 complete and 40 partial responders) and 462 deaths (50.1%); the 5 complete responders [CRs] were all confirmed by pathology. ERs were only observed in the two Multikine-treated groups (8.5% combined MK; 16% LR combined MK vs 3.7% HR MK; 15.2% LR MK+CIZ+SOC). ERs were more commonly seen in the LR group (16.0%) vs the HR group (3.7%). No responders were seen in the SOC patients. In the MK-treated groups, death rates fell significantly for ERs vs non-responders (54.1% vs 22.2% combined MK; 42.5% vs 17.6% LR combined MK groups; 40.7% vs 12.5% LR (MK+CIZ+SOC); the corresponding hazard ratios (HZR) were 0.301 (Wald p<0.0001) overall, 0.348 (p=0.0067) for LR MK, and 0.246 (p=0.01) for LR MK=CIZ=SO in support of ER being supportive.
| 7 |
Assuming no OS prolongation for the remaining 84.8%, this equates to a 46.5% OS gain corresponding to the observed 0.68 HZR (1/1.465) for the ITT Lower Risk LI (MK)+CIZ+SOC group; this was consistent with the observed 46.5-month median OS advantage for LR LI+CIZ+SOC (101.7 months) vs LR SOC (55.2 months). ER was also predictive; among the 45 responders; there were only 10 deaths (22.2%) in contrast to 452 (51.5%) for the overall ITT population. Thus, ER was predictive from both a modeling and outcome perspective. The conclusions were as follows: Objective ER was only observed for the MK treatments. Multiple ERs were observed across LR (n=34), HR (n=10), and UC (n=1). ER from MK treatment is not only prognostic but also predicts a most favorable OS outcome.
Since CEL-SCI launched its Phase 3 clinical trial for Multikine, CEL-SCI has incurred expenses of approximately $64.1 million as of September 30, 2022 on direct costs for the Phase 3 clinical trial. CEL-SCI estimates it will incur additional expenses of approximately $0.6 million for the remainder of the Phase 3 clinical trial and the filing of the clinical study report to the FDA. It should be noted that this estimate is based only on the information currently available from the CROs responsible for managing the Phase 3 clinical trial and does not include other related costs, e.g., preparations for the potential commercial manufacture of the drug.
Ultimately, the decision as to whether CEL-SCI’s drug product candidate is safe and effective can only be made by the FDA and/or by other regulatory authorities based upon an assessment of all of the data from an entire drug development program submitted as part of an application for marketing approval. As detailed in the Risk Factors section of this report, the current Phase 3 clinical study for CEL-SCI’s investigational drug may or may not be able to be used as the pivotal study supporting a marketing application in the United States, and, if not, at least one entirely new Phase 3 pivotal study would need to be conducted to support a marketing application in the United States. However, CEL-SCI does not believe that this would be ethical or supported by the survival data for this unmet medical need.
Development Agreements for Multikine
In August 2008, CEL-SCI signed an agreement with Teva Pharmaceutical Industries Ltd., or Teva, that gives Teva the exclusive right and license to market, distribute and sell Multikine, if approved, in Israel and Turkey for treatment of head and neck cancer. The agreement terminates on a country-by-country basis 10 years after the product launch in each country or upon a material breach or upon bankruptcy of either party. The agreement will automatically extend for additional two-year terms unless either party gives notice of its intent not to extend the agreement. If CEL-SCI develops Multikine for other oncology indications and Teva indicates a desire to participate, the parties have agreed to negotiate in good faith with respect to Teva’s participation and contribution in future clinical trials.
Teva has agreed to use all reasonable efforts to obtain regulatory approval to market and sell Multikine in its territory at its own cost and expense. Pursuant to the agreement, it is CEL-SCI’s responsibility to supply Multikine and Teva’s responsibility to sell Multikine, if approved by regulatory authorities in the relevant countries. Net sales will be divided 50/50 between the two parties. Teva also initially agreed to fund certain activities relating to the conduct of a clinical trial in Israel as part of the global Phase 3 trial for Multikine. In January 2012, pursuant to an assignment and assumption agreement between CEL-SCI, Teva and GCP Clinical Studies Ltd., or GCP, Teva transferred all of its rights and obligations concerning the Phase 3 trial in Israel to GCP.
In July 2011, Serbia and Croatia were added to Teva’s territory, pursuant to a joinder agreement between CEL-SCI and PLIVA Hrvatska d.o.o., or PLIVA, an affiliate of Teva’s, subject to similar terms as described above.
| 8 |
In consideration for the rights granted by CEL-SCI to PLIVA under the joinder agreement, CEL-SCI will be paid by PLIVA (in U.S. dollars):
| · | $100,000 upon EMA grant of Marketing Authorization for Multikine; |
|
|
|
| · | $50,000 upon Croatia’s grant of reimbursement status for Multikine in Croatia; and |
|
|
|
| · | $50,000 upon Serbia’s grant of reimbursement status for Multikine in Serbia. |
In November 2000, CEL-SCI signed an agreement with Orient Europharma Co., Ltd., or Orient Europharma, of Taiwan, which was amended in October 2008 and again in June 2010. Pursuant to this agreement, as amended, Orient Europharma has the exclusive marketing and distribution rights to Multikine, if approved by regulatory authorities, for head and neck cancer, naso-pharyngeal cancer and potentially cervical cancer indications in Taiwan, Singapore, Malaysia, Hong Kong, the Philippines, South Korea, Australia and New Zealand. CEL-SCI has granted Orient Europharma the first right of negotiation with respect to Thailand and China.
The agreement requires Orient Europharma to fund 10% of the cost of the clinical trials needed to obtain marketing approvals in these countries for head and neck cancer, naso-pharyngeal cancer and potentially cervical cancer.
If Multikine is approved for sale, Orient Europharma will purchase Multikine from CEL-SCI for 35% of the gross selling price in each country.Orient Europharma is obligated to use the same diligent efforts to develop, register, market, sell and distribute Multikine in its territory as with its own products or other licensed products.
The agreement will terminate on a country-by-country basis 15 years after the product approval for Multikine in each country, at which point the agreement will be automatically extended for successive two year periods, unless either party gives notice of its intent not to extend the agreement. The agreement may also be terminated upon the bankruptcy of either party or material misrepresentations that are not cured within 60 days. If the agreement ends before the 15-year term through no fault of either party, CEL-SCI will reimburse Orient Europharma for a prorated part of Orient Europhorma’s costs towards the clinical trials of Multikine. If Orient Europharma fails to make certain minimum purchases of Multikine during the term of the agreement, Orient Europhorma’s rights to the territory will become non-exclusive.
CEL-SCI has a licensing agreement with Byron Biopharma LLC (Byron) under which CEL-SCI granted Byron an exclusive license to market and distribute Multikine in the Republic of South Africa, if approved. This license will terminate 20 years after marketing approval in South Africa or after the bankruptcy or uncured material breach by either party. After the 20-year period has expired, the agreement will be automatically extended for successive two-year periods, unless either party gives notice of its intent not to extend the agreement.
Pursuant to the agreement, Byron will be responsible for registering Multikine in South Africa. If Multikine is approved for sale in South Africa, CEL-SCI will be responsible for manufacturing the product, while Byron will be responsible for sales in South Africa. Sales revenues will be divided equally between CEL-SCI and Byron.
LEAPS
CEL-SCI’s patented T-cell Modulation Process, referred to as LEAPS (Ligand Epitope Antigen Presentation System), uses “heteroconjugates” to direct the body to choose a specific immune response. LEAPS is designed to stimulate the human immune system to more effectively fight bacterial, viral and parasitic infections as well as autoimmune conditions, allergies, transplantation rejection and cancer, when it cannot do so on its own. LEAPS combines T-cell binding ligands with small, disease associated, peptide antigens and may provide a new method to treat and prevent certain diseases.
The ability to generate a specific immune response is important because many diseases are often not combated effectively due to the body’s selection of the “inappropriate” immune response. The capability to specifically reprogram an immune response may offer a more effective approach than existing vaccines and drugs in attacking an underlying disease.
| 9 |
LEAPS Candidates: CEL-2000, CEL-4000 and DerG-PG275(Cit)
On September 19, 2017, CEL-SCI announced that it had been awarded a Phase 2 Small Business Innovation Research (SBIR) grant in the amount of $1.5 million from the National Institute of Arthritis and Musculoskeletal and Skin Diseases(NIAMS), which is part of the U.S. National Institutes of Health (NIH). This grant provided funding to allow CEL-SCI to advance its first LEAPS product candidate, CEL-4000, towards an Investigational New Drug (IND) application for a Phase 1 safety study, by funding IND enabling studies and additional mechanism of action studies, among other preclinical development activities. Work on CEL-4000 was conducted at CEL-SCI’s research laboratory and Rush University Medical Center in Chicago, Illinois in the laboratories of Tibor Glant, MD, Ph.D., Jorge O. Galante Professor of Orthopedic Surgery and Katalin Mikecz, MD, Ph.D. Professor of Orthopedic Surgery & Biochemistry. The SBIR grant was awarded based on published data described below by Dr. Glant's team in collaboration with CEL-SCI showing that the administration of a proprietary peptide using CEL-SCI's LEAPS technology prevented the development, and lessened the severity, including inflammation, of experimental proteoglycan induced arthritis (PGIA or GIA) when it was administered after the disease was induced in animals. This grant has been fully expended.
In May 2019, CEL-SCI announced that a newly discovered LEAPS conjugate acts alone and can complement CEL-4000 therapeutically when administered in combination to an animal model of Rheumatoid Arthritis (RA). This new LEAPS conjugate appears to act on T cell pathways by a new mechanism that is different from the pathways used by the CEL-4000 vaccine. The data was presented at the American Association of Immunologists 103rd Annual Meeting (Immunology 2019) by Daniel Zimmerman, Ph.D., CEL-SCI’s Senior Vice President of Research, Cellular Immunology. The work was performed in conjunction with researchers at Rush University Medical Center, Chicago, Illinois and was funded by the SBIR Phase 2 Grant.
In July 2019, one of CEL-SCI’s collaborators from Rush, Dr. Adrienn Markovics, presented new LEAPS data at i-Chem2019, International Conference on Immunity and Immunochemistry. Data presented was for a new second RA conjugate discovered which acts alone and can complement the existing CEL-4000 RA vaccine in an animal model of RA. The combination of the two RA conjugates provided not only broader epitope coverage, but also a greater therapeutic effect than either conjugate alone. The LEAPS work was performed in conjunction with researchers at CEL-SCI on CEL-4000 and a newly discovered LEAPS conjugate, DerG-PG275Cit. Both conjugates were evaluated alone and in combination in the model of proteoglycan (PG) induced arthritis (PGIA) called recombinant PG G1 domain-induced arthritis (GIA), an autoimmune mouse model of RA.
In February 2017 and November 2016, CEL-SCI announced preclinical data that demonstrate its investigational new drug candidate CEL-4000 has the potential to treat rheumatoid arthritis. This study was supported in part by the SBIR Phase I Grant and was conducted in collaboration with Drs. Katalin Mikecz and Tibor Glant, and their research team at Rush University Medical Center in Chicago, IL. This work was published in an article entitled “An epitope-specific DerG-PG70 LEAPS vaccine modulates T cell responses and suppresses arthritis progression in two related murine models of rheumatoid arthritis” and can be found online at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5568759/.
Prior to the SBIR Phase 2 grant in 2014, CEL-SCI was awarded a Phase 1 SBIR grant in the amount of $225,000 from NIAMS. This grant funded the development of CEL-SCI’s LEAPS technology as a potential treatment for rheumatoid arthritis, an autoimmune disease of the joints. The work was conducted at Rush University Medical Center in Chicago, Illinois in the laboratories of Tibor Glant, MD, Ph.D., Katalin Mikecz, MD, Ph.D., and Allison Finnegan, Ph.D. Professor of Medicine.
| 10 |
With the support of these SBIR grants, CEL-SCI is developing several new drug candidates, CEL-2000 and CEL-4000, as potential rheumatoid arthritis therapeutic treatments. The data from animal studies using the CEL-2000 treatment suggests that it could be used against rheumatoid arthritis with fewer administrations than those required by other anti-rheumatoid arthritis treatments currently on the market for arthritic conditions associated with the Th17 signature cytokine TNF-a. The preclinical data indicates these peptides could be used against rheumatoid arthritis where a Th1 signature cytokine (IFN-γ) is dominant. CEL-2000 and CEL-4000 each have the potential to become a personalized, disease-specific therapy that acts at an earlier step in the disease process than current therapies, and which may be useful in patients not responding to existing rheumatoid arthritis therapies. CEL-SCI believes this represents a large unmet medical need in the rheumatoid arthritis market.
In March 2015, CEL-SCI and its collaborators published a review article on vaccine therapies for rheumatoid arthritis based in part on work supported by the SBIR Phase 1 grant. The article is entitled “Rheumatoid arthritis vaccine therapies: perspectives and lessons from therapeutic Ligand Epitope Antigen Presentation System vaccines for models of rheumatoid arthritis” and was published in Expert Review of Vaccines 1 - 18 and can be found online at http://www.ncbi.nlm.nih.gov/pubmed/25787143.
Accordingly, even though the various LEAPS candidates have not yet been given to humans, they have been tested in vitro with human cells. They have induced similar cytokine responses that were seen in these animal models, which may indicate that the LEAPS technology might translate to humans. The LEAPS candidates have demonstrated protection against lethal herpes simplex virus (HSV1) and H1N1 influenza infection as a prophylactic or therapeutic agent in animals. They have also shown some level of activity in animals in two autoimmune conditions, curtailing and sometimes preventing disease progression in arthritis and myocarditis animal models.
None of the LEAPS investigational products have been approved for sale, barter or exchange by the FDA or any other regulatory agency for any use to treat disease in animals or humans. The safety or efficacy of these products has not been established for any use. Lastly, no definitive conclusions can be drawn from the early-phase, preclinical-trials data involving these investigational products. Before obtaining marketing approval from the FDA in the United States, and by comparable agencies in most foreign countries, these product candidates must undergo rigorous preclinical and clinical testing which is costly and time consuming and subject to unanticipated delays. There can be no assurance that these approvals will be granted.
INTELLECTUAL PROPERTY
Patents and other proprietary rights are essential to CEL-SCI’s business. CEL-SCI files patent applications to protect its technologies, inventions and improvements that CEL-SCI considers important to the development of its business. CEL-SCI’S intellectual property portfolio covers its proprietary technologies, including Multikine and LEAPS, by multiple issued patents and pending patent applications in the United States and in key foreign markets.
Multikine is protected by a U.S. patent, which is a composition-of-matter patent issued in May 2005 that, in its current format, expires in 2023. Additional composition-of-matter patents for Multikine have been issued in Germany (issued in June 2011 and currently set to expire in 2025), China (issued in May 2011 and currently set to expire in 2024), Japan (issued in November 2012 and currently set to expire in 2025), and three in Europe (issued in September 2015, May 2016 and October 2017, currently set to expire in 2025 and 2026). The most recent patent issued in October 2017, patent # EP 1 879 618 B1, titled “A Method for Modulating HLA Class II Tumor Cell Surface Expression With A Cytokine Mixture,” addresses Multikine’s mechanism of action to make tumors more visible to the immune system. This new patent is important because, along with the other Multikine issued patents, it addresses how Multikine enables the immune system to recognize and attack the tumor. One way tumor cells evade the immune system is by expressing human leukocyte antigens (HLA) on the tumor cell surface, thus appearing as ‘self’ to the immune cells and therefore the tumor cells are not attacked. It is important to note that the tumors of the Multikine-treated best responders in CEL-SCI’s prior Phase 2 studies had no HLA Class II expressed on the cell surface following Multikine treatment as compared to controls. This points to Multikine’s ability to modulate HLA expression on the tumor cell surface, thereby allowing the immune system to recognize and attack the tumor.
| 11 |
In addition to the patents that offer certain protections for Multikine, the method of manufacture for Multikine, a complex biological product, is held by CEL-SCI as a trade secret. CEL-SCI considers this to be its best protection from competitors.
LEAPS is protected by patents in the United States issued between January 2019 and June 2021. The LEAPS patents, which expire between 2027 and 2032, include overlapping claims, with composition of both matter (new chemical entity), process and methods-of-use, to maximize and extend the coverage in their current format. One issued U.S. application is a joint application with Northeast Ohio Medical University (“Neoucom”) and CEL-SCI will share the ability to use the patent, unless CEL-SCI licenses the rights to the patent from Neoucom. In October 2017 and October 2020, patents were issued in Europe for LEAPS, which expire in 2029 and 2034, respectively.
CEL-SCI has five patent applications pending in the United States for LEAPS, which, if issued, would extend protection through 2040, subject to any potential patent term extensions.
As of December 27, 2022, there were no contested proceedings and/or third-party claims with respect to CEL-SCI’s patents or patent applications.
MANUFACTURING FACILITY
Before starting the Phase 3 clinical trial, for reasons related to regulatory considerations, CEL-SCI built a dedicated manufacturing facility to produce its investigational biological product candidate Multikine. This facility produced multiple clinical lots for the Phase 3 clinical trial and has also passed quality systems review by a European Union Qualified Person on several occasions. CEL-SCI expanded the manufacturing facility so CEL-SCI will be able to meet the expected demand for Multikine, if FDA approval is granted. This expansion was completed at the end of 2021, allowing CEL-SCI employees to return to work inside the manufacturing facility. The facility is currently undergoing validation.
CEL-SCI’s lease on the manufacturing facility expires on October 31, 2028. At that time CEL-SCI can either purchase the facility or extend its lease. See Item 2 of this report for more information concerning the terms of this lease.
GOVERNMENT REGULATION
The FDA and other regulatory authorities at federal, state and local levels and in foreign countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing and post-approval monitoring and reporting of biologics such as those CEL-SCI is developing. CEL-SCI, along with third party contractors, will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries in which it wishes to conduct studies or seek approval or licensure of its product candidates. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations requires the expenditure of substantial time and financial resources.
| 12 |
U.S. Food and Drug Administration Regulation of Biological Products
In the United States, the FDA regulates biological products under the Federal Food, Drug, and Cosmetic Act, or FDCA, and the Public Health Service Act, or PHSA, and their implementing regulations. The process required by the FDA before biological product candidates may be marketed in the United States generally involves the following:
| · | completion of preclinical laboratory tests and animal studies performed in accordance with the FDA’s Good Laboratory Practice (GLP) regulations; |
|
|
|
| · | submission to the FDA of an investigational new drug application, or IND, which must become effective before clinical trials may begin and must be updated annually; |
|
|
|
| · | approval by an independent Institutional Review Board, or IRB, or ethics committee at each clinical site before the trial is initiated; |
|
|
|
| · | performance of adequate and well-controlled human clinical trials in compliance with Good Clinical Practice (GCP) regulations to establish the safety, purity and potency of the proposed biologic product candidate for its intended purpose; |
|
|
|
| · | preparation of and submission to the FDA of a Biologics License Application (BLA) after completion of clinical trials; |
|
|
|
| · | satisfactory completion of an FDA Advisory Committee review, if applicable; |
|
|
|
| · | a determination by the FDA within 60 days of its receipt of a BLA to file the application for review; |
|
|
|
| · | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the proposed product is produced to assess compliance with current Good Manufacturing Practice (cGMP) requirements and to assure that the facilities, methods and controls are adequate to preserve the biological product’s continued safety, purity and potency, and of selected clinical investigations to assess compliance with GCPs; and |
|
|
|
| · | FDA review and approval of the BLA to permit commercial marketing of the product for particular indications for use in the United States. |
Prior to commencing the first clinical trial with a product candidate in the U.S., CEL-SCI must submit an IND to the FDA. An IND is a request for authorization from the FDA to administer an investigational product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for human studies. The IND also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises safety concerns or questions about the proposed clinical trial. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before the clinical trial can begin. Submission of an IND therefore may or may not result in FDA authorization to commence a clinical trial.
| 13 |
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical study. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments. Furthermore, an independent IRB for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial commences at that site, and must monitor the study until completed. Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk. Some studies also include oversight by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board (DSMB) or independent data monitoring committee (IDMC), which provides recommendations for whether or not a study should move forward at designated check points based on access to certain data from the study and may suggest halting the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing clinical studies and clinical study results to public registries.
For purposes of approval of a Biologics License Application, or BLA, human clinical trials are typically conducted in three or four sequential phases that may overlap.
● Phase 1 — The investigational product is initially introduced into healthy human subjects or patients with the target disease or condition. These studies are designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans and the side effects associated with increasing doses.
● Phase 2 — The investigational product is administered to a limited patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials.
● Phase 3 — The investigational product is administered to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the investigational product and to provide an adequate basis for product approval.
● Phase 4 — In some cases, the FDA may require, or companies may voluntarily pursue, additional clinical trials after a product is approved to gain more information about the product. The FDA may also make these so-called Phase 4 or post-marketing studies a condition to approval of the BLA.
Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within a specified period, if at all, and there can be no assurance that the data collected will support FDA approval or licensure of the product. Concurrent with clinical trials, companies may complete additional animal studies and develop additional information about the biological characteristics of the product candidate, and must finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
| 14 |
BLA Submission and Review by the FDA
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development, nonclinical studies and clinical trials are submitted to the FDA as part of a BLA requesting approval to market the product for one or more indications. The BLA must include all relevant data available from pertinent preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other things. Data can come from company-sponsored clinical studies intended to test the safety and effectiveness of a use of the product, or from a number of alternative sources, including studies initiated by investigators.
In most cases, the submission of a BLA is subject to a substantial application user fee. Under the goals and policies agreed to by the FDA under the Prescription Drug User Fee Act, or PDUFA, for original BLAs, the FDA’s goal is to review the BLA within ten months after it accepts the application for filing, or, if the product relates to an unmet medical need in a serious or life-threatening indication and has received a priority review designation, six months after the FDA accepts the application for filing.
After filing the marketing application, the FDA reviews a BLA to determine, among other things, whether a product is safe, pure and potent and the facility in which it is manufactured, processed, packed, or held meets standards designed to assure the product’s continued safety, purity and potency. Before approving a BLA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve a biological product for marketing unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCPs. If the FDA determines that the data provided in the application, or the manufacturing process or manufacturing facilities for the product are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. The FDA also may refer applications for novel biologic candidates which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions, if any. The FDA is not bound by recommendations of an advisory committee, but it considers such recommendations when making decisions on approval.
After the FDA evaluates a BLA and conducts inspections of manufacturing facilities where the biological product and/or its drug substance will be produced, the FDA may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete but the application is not ready for approval. A Complete Response Letter may request additional information or clarification, including new clinical studies. The FDA may delay or refuse approval of a BLA if applicable regulatory criteria are not satisfied, require additional testing or information and/or require post-marketing testing and surveillance to monitor safety or efficacy of a product. If a Complete Response Letter is issued, the applicant may either resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application. Even if such data and information are submitted, the FDA may decide that the re-submitted BLA does not satisfy the criteria for approval.
| 15 |
If a product receives regulatory approval, such approval is limited to the conditions of use (e.g., patient population, indication) described in the application. Further, depending on the specific risk(s) to be addressed, the FDA may require that contraindications, warnings or precautions be included in the product labeling, require that post-approval trials, including Phase 4 clinical trials, be conducted to further assess a product’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a Risk Evaluation and Mitigation Strategy, or REMS, plan if it determines that a REMS is necessary to ensure that the benefits of the product outweigh its risks and to assure the safe use of the biological product, which can materially affect the potential market and profitability of the product. The REMS plan could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling or the development of adequate controls and specifications. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing regulatory standards is not maintained or if problems occur after the product reaches the marketplace. The FDA may prevent or limit further marketing of a product based on the results of post-marketing trials or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Expedited Review and Approval
A sponsor may seek approval of its product candidate under programs designed to accelerate the FDA’s review and approval of new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for fast track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. For a fast track product, the FDA may consider sections of the BLA for review on a rolling basis before the complete application is submitted if relevant criteria are met. A fast track designated product candidate may also qualify for priority review.
Under the accelerated approval program, the FDA may approve a BLA on the basis of either a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Post-marketing studies or completion of ongoing studies after marketing approval are generally required to verify the biologic’s clinical benefit in relationship to the surrogate endpoint or ultimate outcome in relationship to the clinical benefit. In addition, the Food and Drug Administration Safety and Innovation Act, or FDASIA, which was enacted and signed into law in 2012, established the new Breakthrough Therapy designation. A sponsor may seek FDA designation of its product candidate as a breakthrough therapy if the product candidate is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the therapy may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Sponsors may request the FDA to designate a breakthrough therapy at the time of or any time after the submission of an IND, but ideally before an end-of-phase 2 meeting with the FDA. If the FDA designates a breakthrough therapy, it may take actions appropriate to expedite the development and review of the application, which may include holding meetings with the sponsor and the review team throughout the development of the therapy; providing timely advice to, and interactive communication with, the sponsor regarding the development of the drug to ensure that the development program to gather the nonclinical and clinical data necessary for approval is as efficient as practicable; involving senior managers and experienced review staff, as appropriate, in a collaborative, cross-disciplinary review; assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the review team and the sponsor; and considering alternative clinical trial designs when scientifically appropriate, which may result in smaller trials or more efficient trials that require less time to complete and may minimize the number of patients exposed to a potentially less efficacious treatment.
Fast Track designation, priority review and breakthrough therapy designation do not change the standards for approval but may expedite the development or approval process.
| 16 |
Post-Approval Requirements
All therapeutic products manufactured or distributed pursuant to FDA approval or licensure are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to record-keeping, reporting of adverse experiences, periodic reporting, product sampling and distribution, and advertising and promotion of the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual user fee requirements under the PDUFA for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications containing clinical data. Biologic manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP requirements, which impose significant procedural and documentation requirements. Changes to the manufacturing process are strictly regulated and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting requirements on manufacturers. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance. CEL-SCI cannot be certain that it, or CEL-SCI’s present or future suppliers, will be able to comply with the cGMP regulations and other FDA regulatory requirements. If CEL-SCI is not able to comply with these requirements, the FDA may, among other things, take enforcement action or seek sanctions against use, impose restrictions on a product or its manufacturer, require CEL-SCI to recall a product from distribution, or withdraw approval of the BLA.
The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
| · | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
|
|
|
| · | fines, warning letters or holds on post-approval clinical studies; |
|
|
|
| · | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
|
|
|
| · | product seizure or detention, or refusal to permit the import or export of products; |
|
|
|
| · | injunctions or the imposition of civil or criminal penalties; and |
|
|
|
| · | consent decrees, corporate integrity agreements, debarment, or exclusion from federal healthcare programs; or mandated modification of promotional materials and labeling and the issuance of corrective information. |
| 17 |
The FDA closely regulates the marketing, labeling, advertising and promotion of drugs and biologics. A company can make only those claims relating to safety and efficacy, purity and potency that are approved by the FDA and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of unapproved, or “off-label,” uses. Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available products for uses that are not described in the product’s labeling and that differ from those tested and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturer’s communications on the subject of off-label use of their products.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs or biologics intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the United States, or if it affects more than 200,000 individuals in the United States and there is no reasonable expectation that the cost of developing and making the drug for this type of disease or condition will be recovered from sales in the United States.
In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. In addition, if a product receives the first FDA approval for the indication for which it has orphan designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other application to market a drug for the same indication for a period of 7 years, except in limited circumstances, such as a showing of clinical superiority over the product with orphan exclusivity. Orphan drug exclusivity also could block the approval of one of CEL-SCI’s products for seven years if a competitor obtains approval of a product before CEL-SCI does, as defined by the FDA, for the same indication CEL-SCI is seeking, or if CEL-SCI’s product candidate is determined to be contained within the scope of the competitor’s product for the same indication or disease. If one of CEL-SCI’s products designated as an orphan drug receives marketing approval for an indication broader than that which is designated, it may not be entitled to orphan drug exclusivity. Orphan drug status in the European Union has similar, but not identical, requirements and benefits.
Orphan drug designation must be requested before submitting a BLA to the FDA for review and approval. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
Multikine has received Orphan Drug Status from the FDA.
Other U.S. Health Care Laws
CEL-SCI’s sales, promotion, medical education and other activities following product approval will be subject to regulation by numerous regulatory and law enforcement authorities in the United States in addition to the FDA, including potentially the Federal Trade Commission, the Department of Justice, the Centers for Medicare and Medicaid Services, other divisions of the Department of Health and Human Services and state and local governments. CEL-SCI’s promotional and scientific/educational programs must comply with the anti-kickback provisions of the Social Security Act, the Foreign Corrupt Practices Act, the False Claims Act, the Physician Payments Sunshine Act, the Veterans Health Care Act and similar state laws.
| 18 |
Depending on the circumstances, failure to meet these applicable regulatory requirements can result in criminal prosecution, fines or other penalties, exclusion from government health care programs, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre-marketing product approvals, private “qui tam” actions brought by individual whistleblowers under the False Claims Act in the name of the government or refusal to allow CEL-SCI to enter into supply contracts, including government contracts.
Coverage, Pricing and Reimbursement in the U.S.
Sales of pharmaceutical products depend significantly on the availability of third-party coverage and reimbursement. Third-party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. These third-party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products and new drug classes, including biological products such as CEL-SCI’s product candidates. CEL-SCI may need to conduct expensive clinical studies to demonstrate the comparative cost-effectiveness of its products. The product candidates that CEL-SCI develops may not be considered cost-effective. It is time consuming and expensive for CEL-SCI to seek reimbursement from third- party payors. Reimbursement may not be available or sufficient to allow CEL-SCI to sell its products on a competitive and profitable basis.
The United States and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to change the healthcare system in ways that could affect CEL-SCI’s ability to sell its products profitably. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
Foreign Regulation
In addition to regulations in the United States, CEL-SCI will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of its products to the extent CEL-SCI chooses to develop or sell any products outside of the United States. The approval process varies from country to country and the time may be longer or shorter than that required to obtain FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
ITEM 1B. RISK FACTORS
The risks described below could adversely affect the price of CEL-SCI’s common stock.
Risk Factor Summary
CEL-SCI has incurred significant losses since its inception and anticipates that it will continue to incur significant losses for the foreseeable future and may never achieve or maintain profitability.
CEL-SCI will require substantial additional capital to remain in operation. A failure to obtain this necessary capital when needed could force CEL-SCI to delay, limit, reduce or terminate the product candidates’ development or commercialization efforts.
The costs of the product candidates’ development and clinical trials are difficult to estimate and will be very high for many years, preventing CEL-SCI from making a profit for the foreseeable future, if ever.
| 19 |
CEL-SCI has not established a definite plan for the marketing of Multikine, if approved.
CEL-SCI depends heavily on the success of Multikine, for which top line Phase 3 data has been announced, while its other candidates are still in preclinical phases. CEL-SCI’s product candidates must undergo rigorous preclinical and clinical testing and regulatory approvals, which could be costly and time-consuming and subject CEL-SCI to unanticipated delays or prevent CEL-SCI from marketing any products. If CEL-SCI is unable to advance its product candidates in clinical development, obtain regulatory approval and ultimately commercialize its product candidates, or experiences significant delays in doing so, CEL-SCI’s business will be materially harmed.
Even if CEL-SCI obtains regulatory approval for its investigational products, CEL-SCI will be subject to stringent, ongoing government regulation.
CEL-SCI’s product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial utility of an approved prescribing label, or result in significant negative consequences following marketing approval, if any.
Biologics carry unique risks and uncertainties, which could have a negative impact on future results of CEL-SCI’s operations.
The current and future relationships with healthcare professionals, principal investigators, consultants, potential customers and third-party payors in the United States and elsewhere may be subject, directly or indirectly, to applicable healthcare laws and regulations.
CEL-SCI’s commercial success depends, in part, upon attaining significant market acceptance of its product candidates, if approved, among physicians, patients, healthcare payors and major operators of cancer clinics.
CEL-SCI’s patents might not protect its technology from competitors, in which case CEL-SCI may not have any advantage over competitors in selling any products that CEL-SCI may develop.
Much of CEL-SCI’s intellectual property is protected as trade secrets or confidential know-how, not by patents.
You may experience future dilution as a result of future equity offerings or other equity issuances by CEL-SCI.
The price of CEL-SCI’s common stock has been volatile and is likely to continue to be volatile, which could result in substantial losses for CEL-SCI’s shareholders.
CEL-SCI faces business disruption and related risks resulting from the recent pandemic of COVID-19 which could have a material adverse effect on CEL-SCI's business plan.
| 20 |
Risks Related to CEL-SCI
CEL-SCI faces business disruption and related risks resulting from the recent pandemic of COVID-19 which could have a material adverse effect on our business plan.
The development of CEL-SCI’s product candidates could be disrupted and materially adversely affected by the ongoing COVID-19 pandemic. CEL-SCI knows that COVID delayed the time to reach data lock for its Phase 3 clinical trial, delayed the expansion of CEL-SCI’s manufacturing facility for Multikine and could potentially affect the regulatory pathway for Multikine through the FDA and/or other regulatory agencies throughout the world. CEL-SCI is continually assessing its business plans and the impact COVID-19 may have on its ability to seek regulatory approval for Multikine and conduct the preclinical studies and clinical trials other than CEL-SCI’s Phase 3 trial, but there can be no assurance that this analysis will enable CEL-SCI to avoid part or all of any impact from the spread of COVID-19 or its consequences, including downturns in business sentiment generally. The extent to which the COVID-19 pandemic and global efforts to contain its spread will impact CEL-SCI’s operations will depend on future developments, which are highly uncertain and cannot be predicted at this time, and include the duration, severity and scope of the pandemic and the actions taken to contain or treat the COVID-19 pandemic.
CEL-SCI has incurred significant losses since its inception and anticipates that it will continue to incur significant losses for the foreseeable future and may never achieve or maintain profitability.
CEL-SCI has a history of net losses, expects to incur substantial losses and have negative operating cash flow for the foreseeable future, and may never achieve or maintain profitability. Since the date of its formation and through September 30, 2022, CEL-SCI incurred net losses of approximately $456 million. CEL-SCI has relied principally upon the proceeds from the public and private sales of its securities to finance its activities to date. To date, CEL-SCI has not commercialized any products or generated any revenue from the sale of products, and CEL-SCI does not expect to generate any product revenue for the foreseeable future. CEL-SCI does not know whether or when it will generate product revenue or become profitable.
CEL-SCI is heavily dependent on the success of Multikine for which Phase 3 data has been announced and presented at ASCO 2022 and ESMO 2022. CEL-SCI cannot be certain that Multikine will receive regulatory approval or be successfully commercialized even if CEL-SCI receives regulatory approval. Multikine is the only product candidate in late-stage clinical development and CEL-SCI’s business currently depends heavily on its successful development, regulatory approval and commercialization. CEL-SCI has no drug products for sale currently and may never be able to develop approved and marketable drug products.
Even if CEL-SCI succeeds in developing and commercializing one or more of its product candidates, CEL-SCI expects to continue to incur significant operating and capital expenditures as CEL-SCI:
| · | continues to undertake preclinical development and clinical trials for product candidates; |
|
|
|
| · | seeks regulatory approvals for product candidates; and |
|
|
|
| · | implements additional internal systems and infrastructure. |
To become and remain profitable, CEL-SCI must succeed in developing and commercializing product candidates which must generate significant revenue. This will require CEL-SCI to be successful in a range of challenging activities, including completing preclinical testing and clinical trials of its product candidates, discovering or acquiring additional product candidates, obtaining regulatory approval for these product candidates and manufacturing, marketing and selling any products for which CEL-SCI may obtain regulatory approval. CEL-SCI is only in the preliminary stages of most of these activities. CEL-SCI may never succeed in these activities and, even if CEL-SCI does, may never generate revenue that is significant enough to achieve profitability.
Even if CEL-SCI does achieve profitability, it may not be able to sustain or increase profitability on a quarterly or annual basis. The failure to become and remain profitable could depress the value of CEL-SCI’s common stock and could impair its ability to raise capital, expand its business, maintain research and development efforts, diversify product offerings or even continue in operation. A decline in the value of CEL-SCI’s common stock could cause its stockholders to lose all or part of their investment.
| 21 |
CEL-SCI has identified conditions and events that raise substantial doubt about its ability to continue as a going concern for more than twelve months from the date of these financial statements.
Primarily as a result of CEL-SCI’s losses incurred to date, CEL-SCI’s expected continued future losses, and the uncertainties associated with obtaining regulatory approval and ultimately commercializing its products, management has identified conditions and events that raise substantial doubt about CEL-SCI’s ability to continue as a going concern. CEL-SCI’s ability to continue as a going concern is contingent upon, among other factors, the sale of its securities or obtaining alternate financing.
CEL-SCI will require substantial additional capital to remain in operation. A failure to obtain this necessary capital when needed could force CEL-SCI to delay, limit, reduce or terminate the product candidates’ development or commercialization efforts.
As of September 30, 2022, CEL-SCI had cash and cash equivalents of approximately $22.7 million. CEL-SCI believes that it will continue to expend substantial resources for the foreseeable future developing Multikine, LEAPS and any other product candidates or technologies that it may develop or acquire. These expenditures will include costs associated with research and development, obtaining regulatory approvals and having the products manufactured, as well as marketing and selling products approved for sale, if any. In addition, other unanticipated costs may arise. CEL-SCI cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of the product candidates.
CEL-SCI’s future capital requirements depend on many factors, including:
| · | the cost of completing Phase 3 clinical development of Multikine for the treatment of certain head and neck cancers; |
|
|
|
| · | the results of the applications to and meetings with the FDA, the EMA and other regulatory authorities and the consequential effect on operating costs; |
|
|
|
| · | the cost, timing and outcome of the efforts to obtain marketing approval for Multikine in the United States, Europe and in other jurisdictions, including the preparation and filing of regulatory submissions for Multikine with the FDA, the EMA and other regulatory authorities; |
|
|
|
| · | the scope, progress, results and costs of additional preclinical, clinical, or other studies for additional indications for Multikine, LEAPS and other product candidates and technologies that CEL-SCI may develop or acquire; |
|
|
|
| · | the timing of, and the costs involved in, obtaining regulatory approvals for LEAPS if clinical studies are successful; |
|
|
|
| · | the cost and timing of future commercialization activities for the products, if any of the product candidates are approved for marketing, including product manufacturing, marketing, sales and distribution costs; |
|
|
|
| · | the revenue, if any, received from commercial sales of the product candidates for which CEL-SCI receives marketing approval; |
|
|
|
| · | the cost of having the product candidates manufactured for clinical trials and in preparation for commercialization; |
|
|
|
| · | the ability to establish and maintain strategic collaborations, licensing or other arrangements and the financial terms of such agreements; |
|
|
|
| · | the costs involved in preparing, filing and prosecuting patent applications and maintaining, defending and enforcing its intellectual property rights, including litigation costs, and the outcome of such litigation; and |
|
|
|
| · | the extent to which CEL-SCI acquires or in-licenses other products or technologies. |
| 22 |
CEL-SCI will need to raise additional funds in order to continue its operations and additional funds may not be available when CEL-SCI needs them on terms that are acceptable to CEL-SCI, or at all. If adequate funds are not available to CEL-SCI on a timely basis, CEL-SCI may be required to delay, limit, reduce or terminate preclinical studies, clinical trials or other development activities for Multikine, LEAPS, or any other product candidates or technologies that CEL-SCI develops or acquires, or delay, limit, reduce or terminate its sales and marketing activities that may be necessary to commercialize its product candidates. Due to recurring losses from operations and future liquidity needs, there is substantial doubt about CEL-SCI’s ability to continue as a going concern without additional capital becoming available. The substantial doubt about CEL-SCI’s ability to continue as a going concern could have an adverse impact on CEL-SCI’s ability to execute its business plan, result in the reluctance on the part of certain suppliers to do business with CEL-SCI, or adversely affect CEL-SCI’s ability to raise additional debt or equity capital.
The costs of the product candidates’ development and clinical trials are difficult to estimate and will be very high for many years, preventing CEL-SCI from making a profit for the foreseeable future, if ever.
Clinical and other studies necessary to obtain approval of a new drug or biologic can be time consuming and costly, especially in the United States, but also in foreign countries. The estimates of the costs associated with future clinical trials and research may be substantially lower than what CEL-SCI actually experiences. It is impossible to predict what CEL-SCI will face in the development of a product candidate such as Multikine. The purpose of clinical trials is to provide both CEL-SCI and regulatory authorities with safety and efficacy data in humans. The difficult and often complex steps necessary to obtain regulatory approval, especially that of the FDA and the EMA, involve significant costs and may require several years to complete. CEL-SCI expects that it will need substantial additional financing over an extended period of time in order to fund the costs of future clinical trials, related research, and general and administrative expenses.
CEL-SCI’s LEAPS technology may not result in a viable drug candidate
Using CEL-SCI’s LEAPS technology, CEL-SCI is also developing a LEAPS immunotherapy for the potential treatment of Rheumatoid Arthritis. However, the development of this technology is in a preliminary stage. There can be no assurance that the LEAPS technology will be successful in treating any disease. CEL-SCI’s primary focus at present is seeking the approval of Multikine for the treatment of head and neck cancer.
An adverse determination in any future legal proceedings could have a material adverse effect on CEL-SCI.
CEL-SCI may be the target of claims asserting violations of securities fraud and derivative actions, or other litigation or arbitration proceedings in the future. Any future litigation could result in substantial costs and divert management’s attention and resources. These legal proceedings may result in large judgments or settlements against CEL-SCI, any of which could have a material adverse effect on its business, operating results, financial condition and liquidity.
| 23 |
Compliance with changing regulations concerning corporate governance and public disclosure may result in additional expenses.
Changing laws, regulations and standards relating to corporate governance and public disclosure may create uncertainty regarding compliance matters. New or changed laws, regulations and standards are subject to varying interpretations in many cases. As a result, their application in practice may evolve over time. CEL-SCI is committed to maintaining high standards of corporate governance and public disclosure. Complying with evolving interpretations of new or changing legal requirements may cause CEL-SCI to incur higher costs as CEL-SCI revises current practices, policies and procedures, and may divert management’s time and attention from potential revenue-generating activities to compliance matters. If the efforts to comply with new or changed laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, CEL-SCI’s reputation may also be harmed. Further, CEL-SCI’s board members, chief executive officer, and other executive officers could face an increased risk of personal liability in connection with the performance of their duties. As a result, CEL-SCI may have difficulty attracting and retaining qualified board members and executive officers, which could harm its business.
CEL-SCI has not established a definite plan for the marketing of Multikine, if approved.
CEL-SCI has not established a definitive plan for marketing Multikine nor has CEL-SCI established a price structure for any of its product candidates, if approved. However, CEL-SCI intends, if it is in a position to do so, to sell Multikine itself in certain markets where it is approved and/or to enter into written marketing agreements with various third parties with established sales forces in such markets. The sales forces in turn would, CEL-SCI believes, focus on selling Multikine to targeted cancer centers, physicians and clinics involved in the treatment of head and neck cancer. CEL-SCI has already licensed future sales of Multikine, if approved, to three companies: Teva Pharmaceutical Industries Ltd. in Israel, Turkey, Serbia and Croatia; Orient Europharma in Taiwan, Singapore, Hong Kong, Malaysia, South Korea, the Philippines, Australia and New Zealand; and Byron BioPharma, LLC in South Africa. CEL-SCI believes that these companies will have the resources to market Multikine appropriately in their respective territories, if approved, but there is no guarantee that they will. There is no assurance that CEL-SCI will be able to find qualified third-party partners to market its products in other areas, on terms that are favorable to CEL-SCI, or at all.
CEL-SCI may encounter problems, delays and additional expenses in developing marketing plans with third parties. In addition, even if Multikine, if approved, is cost-effective and demonstrated to increase overall patient survival, CEL-SCI may experience other limitations involving the proposed sale of Multikine, such as uncertainty of third-party coverage and reimbursement. There is no assurance that CEL-SCI can successfully market Multikine, if approved, or any other product candidates it may develop.
CEL-SCI hopes to expand its clinical development capabilities in the future, and any difficulties hiring or retaining key personnel or managing this growth could disrupt its operations.
CEL-SCI is highly dependent on the principal members of its management and development staff. CEL-SCI expects to expand its clinical development and manufacturing capabilities, which will involve hiring additional employees. Future growth will require CEL-SCI to continue to implement and improve its managerial, operational and financial systems and continue to retain, recruit and train additional qualified personnel, which may impose a strain on its administrative and its operational infrastructure. The competition for qualified personnel in the biopharmaceutical field is intense. CEL-SCI is highly dependent on its ability to attract, retain and motivate highly qualified management and specialized personnel required for clinical development. Due to limited resources, CEL-SCI may not be able to effectively manage the expansion of its operations or recruit and train additional qualified personnel. If CEL-SCI is unable to retain key personnel or manage its future growth effectively, CEL-SCI may not be able to implement its business plan.
| 24 |
If product liability or patient injury lawsuits are brought against CEL-SCI, CEL-SCI may incur substantial liabilities and may be required to limit clinical testing or future commercialization of Multikine or its other product candidates.
CEL-SCI faces an inherent risk of product liability as a result of the clinical testing of Multikine and other product candidates and will face an even greater risk if CEL-SCI is able to commercialize any of its product candidates. For example, CEL-SCI may be sued if its Multikine or LEAPS product candidates, or any other future product candidates, allegedly cause injury or are found to be otherwise unsuitable during clinical testing, manufacturing or, if approved, marketing, sale or during administration to patients. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product candidate, negligence, strict liability and a breach of warranties. Claims could also be asserted under state consumer protection acts.
Furthermore, Multikine is made, in part, from components of human blood. There are inherent risks associated with products that involve human blood such as possible contamination with viruses, including hepatitis or HIV. Any possible contamination could cause injuries to patients who receive contaminated Multikine, or could require CEL-SCI to destroy batches of Multikine, thereby subjecting CEL-SCI to possible financial losses, lawsuits and harm to its business.
If CEL-SCI cannot successfully defend itself against product liability claims, CEL-SCI may incur substantial liabilities or be required to limit or cease the clinical testing or commercialization of its product candidates, if approved. Even a successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| · | decreased demand for Multikine or other product candidates, if approved and commercialized, or clinical holds or suspension of the IND while the candidates are still in clinical development; |
|
|
|
| · | injury to CEL-SCI’s reputation; |
|
|
|
| · | withdrawal of existing, or failure to enroll additional, clinical trial participants; |
|
|
|
| · | costs to defend any related litigation; |
|
|
|
| · | a diversion of management’s time and resources; |
|
|
|
| · | substantial monetary awards to trial participants or patients; |
|
|
|
| · | recalls of approved products, withdrawal of BLA approvals or new labeling, marketing or promotional restrictions; |
|
|
|
| · | loss of revenue; |
|
|
|
| · | inability to commercialize Multikine or other product candidates; and |
|
|
|
| · | a decline in the price of CEL-SCI’s common stock. |
Although CEL-SCI has product liability insurance for Multikine in the amount of $10 million, the successful prosecution of a product liability case against CEL-SCI could have a materially adverse effect upon its business if the amount of any judgment exceeds the insurance coverage. Any claim that may be brought against CEL-SCI could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by CEL-SCI’s insurance or that is in excess of the limits of the insurance coverage. CEL-SCI’s insurance policies also have various exclusions, and CEL-SCI may be subject to a claim for which CEL-SCI has no coverage. CEL-SCI may have to pay any amounts awarded by a court or negotiated in a settlement that exceed the coverage limitations or that are not covered by its insurance, and CEL-SCI may not have, or be able to obtain, sufficient capital to pay such amounts. CEL-SCI commenced the Phase 3 clinical trial for Multikine in December 2010. Although no claims have been brought to date, participants in the clinical trials could bring civil actions against CEL-SCI for any unanticipated harmful effects allegedly arising from the use of Multikine or any other product candidate that CEL-SCI may attempt to develop.
| 25 |
CEL-SCI’s commercial success depends, in part, upon attaining significant market acceptance of its product candidates, if approved, among physicians, patients, healthcare payors and major operators of cancer clinics.
Even if CEL-SCI obtains regulatory approval for its product candidates, any resulting product may not gain market acceptance among physicians, healthcare payors, patients and the medical community, which are critical to commercial success. Market acceptance of any product candidate for which CEL-SCI receives approval depends on a number of factors, including:
| · | the efficacy and safety of the products as demonstrated in clinical trials; |
|
|
|
| · | the timing of market introduction of such product as well as competitive products; |
|
|
|
| · | the clinical indications for which the biological product is approved; |
|
|
|
| · | the approval, availability, market acceptance and reimbursement for the companion diagnostic, if appropriate or necessary; |
|
|
|
| · | acceptance by physicians, major operators of cancer clinics and patients of the biologic as a safe and effective treatment; |
|
|
|
| · | the potential and perceived advantages of such product candidate over alternative treatments, especially with respect to patient subsets that are targeted with such product; |
|
|
|
| · | the safety of such product seen in a broader patient group, including its use outside the approved indications; |
|
|
|
| · | the cost of treatment in relation to alternative treatments; |
|
|
|
| · | the availability of adequate reimbursement and pricing by third-party payors and government authorities; |
|
|
|
| · | relative convenience and ease of administration; |
|
|
|
| · | the prevalence and severity of adverse side effects; and |
|
|
|
| · | the effectiveness of sales and marketing efforts. |
If CEL-SCI’s product candidates are approved but fail to achieve an adequate level of acceptance by physicians, healthcare payors and patients, CEL-SCI will not be able to generate significant revenues, and CEL-SCI may not become or remain profitable.
Under CEL-SCI’s amended bylaws, stockholders that initiate certain proceedings may be obligated to reimburse CEL-SCI and its officers and directors for all fees, costs and expenses incurred in connection with such proceedings if the claim proves unsuccessful.
On February 18, 2015, CEL-SCI adopted new bylaws which include a fee-shifting provision in Article X for stockholder claims. Article X provides that in the event any stockholder initiates or asserts a claim against CEL-SCI, or any of its officers or directors, including any derivative claim or claim purportedly filed on CEL-SCI’s behalf, and the stockholder does not obtain a judgment on the merits that substantially achieves, in substance and amount, the full remedy sought, then the stockholder will be obligated to reimburse CEL-SCI and any of its officers or directors named in the action, for all fees, costs and expenses of every kind and description that CEL-SCI or its officers or directors may incur in connection with the claim. In adopting Article X, it is the intent that:
| 26 |
| · | all actions, including federal securities law claims, would be subject to Article X; |
|
|
|
| · | the phrase “a judgment on the merits” means the determination by a court of competent jurisdiction on the matters submitted to the court; |
|
|
|
| · | the phrase “substantially achieves, in both substance and amount” means the plaintiffs in the action would be awarded at least 90% of the relief sought; |
|
|
|
| · | only persons who were stockholders at the time an action was brought would be subject to Article X; and |
|
|
|
| · | in addition to CEL-SCI, only the directors or officers named in the action would be allowed to recover. |
The fee-shifting provision contained in Article X of the bylaws is not limited to specific types of actions, but is rather potentially applicable to the fullest extent permitted by law. Fee-shifting bylaws are relatively new and untested. The case law and potential legislative action on fee-shifting bylaws are evolving and there exists considerable uncertainty regarding the validity of, and potential judicial and legislative responses to, such bylaws. For example, it is unclear whether the ability to invoke the fee-shifting bylaw in connection with claims under the federal securities laws would be pre-empted by federal law. Similarly, it is unclear how courts might apply the standard that a claiming stockholder must obtain a judgment that substantially achieves, in substance and amount, the full remedy sought. The application of the fee-shifting bylaw in connection with such claims, if any, will depend in part on future developments of the law. CEL-SCI cannot assure its shareholders that CEL-SCI will or will not invoke the fee-shifting bylaw in any particular dispute. In addition, given the unsettled state of the law related to fee-shifting bylaws, such as CEL-SCI’s, CEL-SCI may incur significant additional costs associated with resolving disputes with respect to such bylaw, which could adversely affect CEL-SCI’s business and financial condition.
If a stockholder that brings any such claim, suit, action or proceeding is unable to obtain the required judgment, the attorneys’ fees and other litigation expenses that might be shifted to a claiming stockholder are potentially significant. This fee-shifting bylaw may therefore dissuade or discourage stockholders and their attorneys from initiating lawsuits or claims against CEL-SCI or its directors and officers. In addition, it may impact the fees, contingency or otherwise, required by potential plaintiffs’ attorneys to represent the stockholders or otherwise discourage plaintiffs’ attorneys from representing the stockholders at all. As a result, this bylaw may limit the ability of stockholders to alter CEL-SCI’s management and direction, particularly through litigation or the threat of litigation.
The provision of the amended bylaws requiring exclusive venue in the U.S. District Court for Delaware for certain types of lawsuits may have the effect of discouraging lawsuits against CEL-SCI and its directors and officers.
Article X of CEL-SCI’s amended bylaws provides that stockholder claims brought against CEL-SCI, or its officers or directors, including any derivative claim or claim purportedly filed on its behalf, must be brought in the U.S. District Court for the district of Delaware and that with respect to any such claim, the laws of Delaware will apply.
The exclusive forum provision may limit a stockholder’s ability to bring a claim in a judicial forum the stockholder finds favorable for disputes with CEL-SCI or its directors or officers and may have the effect of discouraging lawsuits with respect to claims that may benefit CEL-SCI or its stockholders.
| 27 |
A provision in our Bylaws regarding shareholder claims may not be enforceable.
Article X of our bylaws provides that stockholder claims brought against us, or our officers or directors, including any derivative claim or claim purportedly filed on our behalf, must be brought in the U.S. District Court for the district of Delaware.
Although it is our intent that this provision applies to actions arising under the Securities Act of 1933 and the Securities Exchange Act of 1934 there is uncertainty as to whether a court would enforce this provision since Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations under the Securities Act.
In addition, since this provision in our bylaws applies to state law claims there is uncertainty as to whether any court would enforce this provision.
Risks Related to Clinical Development, Government Approvals and the Marketing of Biopharmaceutical Products
CEL-SCI depends heavily on the success of Multikine, for which Phase 3 data has been presented, while its other candidates are still in preclinical phases. CEL-SCI’s product candidates must undergo rigorous preclinical and clinical testing and regulatory approvals, which could be costly and time-consuming and subject CEL-SCI to unanticipated delays or prevent CEL-SCI from marketing any products. If CEL-SCI is unable to advance its product candidates in clinical development, obtain regulatory approval and ultimately commercialize its product candidates, or experiences significant delays in doing so, CEL-SCI’s business will be materially harmed.
CEL-SCI currently has no products approved for sale and CEL-SCI cannot guarantee that it will ever have marketable products. CEL-SCI’s product candidates are subject to premarket approval from the FDA in the United States, the EMA in the European Union, and by comparable agencies in most foreign countries before they can be sold. Before obtaining marketing approval, these product candidates must undergo costly and time consuming preclinical and clinical testing which could subject CEL-SCI to unanticipated delays and may prevent CEL-SCI from marketing the product candidates in the future. There can be no assurance that such approvals will be granted on a timely basis, if at all.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of preclinical studies and early clinical trials of the product candidates may not be predictive of the results of later-stage clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials. CEL-SCI’s current and future clinical trials may not be successful.
CEL-SCI is in the early development stages for the candidates designed using its LEAPS technology and has not yet initiated any clinical studies for any of those product candidates. Clinical trials can be delayed for a variety of reasons, including delays related to:
| · | the availability of financial resources needed to commence and complete the planned trials; |
|
|
|
| · | obtaining regulatory approval to commence a trial; |
|
|
|
| · | reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
|
|
|
| · | obtaining Institutional Review Board, or IRB, approval at each clinical trial site; |
|
|
|
| · | recruiting suitable patients to participate in a trial; |
|
|
|
| · | having patients complete a trial or return for post-treatment follow-up; |
|
|
|
| · | clinical trial sites deviating from trial protocol or dropping out of a trial; |
|
|
|
| · | adding new clinical trial sites; or |
|
|
|
| · | manufacturing sufficient quantities of the product candidate for use in clinical trials. |
| 28 |
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors including the competence of the CRO running the study, size and nature of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, competing clinical trials and clinicians' and patients’ perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications CEL-SCI is investigating. Furthermore, CEL-SCI relies on CROs and clinical trial sites to ensure the proper and timely conduct of the clinical trials and while CEL-SCI has agreements governing their committed activities, CEL-SCI has limited influence over their actual performance.
CEL-SCI could also encounter significant delays and/or need to terminate a development program for a product candidate if physicians encounter unresolved ethical issues associated with enrolling patients in clinical trials of the product candidates while existing treatments have established safety and efficacy profiles. Further, a clinical trial may be suspended or terminated by CEL-SCI, one or more of the IRBs for the institutions in which such trials are being conducted, by CEL-SCI upon a final recommendation by the Independent Data Monitoring Committee, or IDMC, with which CEL-SCI agrees for such trial, or by FDA or other regulatory authorities due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or the clinical protocols, as a result of inspection of the clinical trial operations or trial site(s) by FDA or other regulatory authorities, the imposition of a clinical hold or partial clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a product candidate, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. The occurrence of any one or more of these events would have significant and severe material consequences for CEL-SCI and could impact CEL-SCI’s ability to continue as a going concern.
If CEL-SCI experiences termination of, or delays in the completion of, any clinical trial of its product candidates, the commercial prospects for the product candidates will be harmed, and the ability to generate product revenues will be delayed. In addition, any delays in completing the clinical trials will increase the costs, slow the product development and approval process and jeopardize the ability to commence product sales and generate revenues. Any of these occurrences may harm CEL-SCI’s business, prospects, financial condition and results of operations significantly. Many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to a delay or the denial of regulatory approval for the product candidates.
CEL-SCI cannot be certain when or under what conditions it will undertake future clinical trials. Early trials for the other product candidates, or the plans for later trials, may not satisfy the requirements of regulatory authorities, such as the FDA. CEL-SCI may fail to find subjects willing to enroll in the trials. Accordingly, the clinical trials relating to the product candidates may not be completed on schedule, the FDA or foreign regulatory agencies may order CEL-SCI to stop or modify research, or these agencies may not ultimately approve any of the product candidates for commercial sale. Varying interpretations of the data obtained from pre-clinical and clinical testing could delay, limit or prevent regulatory approval of the product candidates. The data collected from the clinical trials may not be sufficient to support regulatory approval of the various product candidates, including Multikine. The failure to adequately demonstrate the safety and efficacy of any of the product candidates would delay or prevent regulatory approval of the product candidates in the United States, which could prevent CEL-SCI from achieving profitability.
| 29 |
The development and testing of product candidates and the process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. FDA sanctions could include, among other actions, refusal to approve pending applications, withdrawal of an approval, a clinical hold, termination of the Phase 3 study, warning letters, product recalls or withdrawals from the market, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, and payment of civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on CEL-SCI.
The requirements governing the conduct of clinical trials, manufacturing and marketing of the product candidates, including Multikine, outside the United States vary from country to country. Foreign approvals may take longer to obtain than FDA approvals and can require, among other things, additional testing and different trial designs. Foreign regulatory approval processes include all of the risks associated with the FDA approval process. Some of those agencies also must approve prices for products approved for marketing. Approval of a product by the FDA or the EMA does not ensure approval of the same product by the health authorities of other countries. In addition, changes in regulatory requirements for product approval in any country during the clinical trial process and regulatory agency review of each submitted new application may cause delays or rejections.
CEL-SCI has only limited experience in filing and pursuing applications necessary to gain regulatory approvals. The lack of experience may impede its ability to obtain timely approvals from regulatory agencies, if at all. CEL-SCI will not be able to commercialize Multikine and other product candidates until CEL-SCI has obtained regulatory approval. In addition, regulatory authorities may also limit the types of patients to which CEL-SCI or its third-party partners may market Multikine (if approved) or the other product candidates. Any failure to obtain or any delay in obtaining required regulatory approvals may adversely affect CEL-SCI’s or its third-party partners’ ability to successfully market the product candidates after they are approved.
Even if CEL-SCI obtains regulatory approval for its investigational products, CEL-SCI will be subject to stringent, ongoing government regulation.
If CEL-SCI’s investigational products receive regulatory approval, either in the United States or internationally, those products will be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval and may contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance of the safety and efficacy of the investigational products. CEL-SCI will continue to be subject to extensive regulatory requirements. These regulations are wide-ranging and govern, among other things:
| · | product design, development and manufacture; |
|
|
|
| · | product application and use |
|
|
|
| · | adverse drug experience monitoring reporting; |
|
|
|
| · | product advertising and promotion; |
|
|
|
| · | product manufacturing, including compliance with good manufacturing practices |
|
|
|
| · | record keeping requirements; |
|
|
|
| · | registration and listing of products with the FDA, EMA and other state and national agencies; |
|
|
|
| · | product storage and shipping; |
|
|
|
| · | drug sampling and distribution requirements; |
|
|
|
| · | electronic record and signature requirements; and |
|
|
|
| · | labeling changes or modifications. |
| 30 |
CEL-SCI and any of its third-party manufacturers or suppliers must continually adhere to federal regulations setting forth human drug and biologic manufacturing requirements, known as current Good Manufacturing Practices, or cGMPs, and their foreign equivalents, which are enforced by the FDA, the EMA and other national regulatory bodies through their facilities inspection programs. If the facilities, or the facilities of the contract manufacturers or suppliers, cannot pass a pre-approval inspection by regulators or fail such inspections in the future, the FDA, EMA or other national regulators will not approve the marketing applications for the product candidates, or may withdraw any prior approval. In complying with cGMP and foreign regulatory requirements, CEL-SCI and any of its potential third-party manufacturers or suppliers will be obligated to expend time, money and effort in production, record-keeping and quality control to ensure that the product candidates meet applicable specifications and other requirements.
If CEL-SCI does not comply with regulatory requirements at any stage, whether before or after marketing approval is obtained, CEL-SCI may be subject to, among other things, license suspension or revocation, criminal prosecution, seizure, injunction, fines, be forced to remove a product from the market or experience other adverse consequences, including restrictions or delays in obtaining regulatory marketing approval for such products or for other product candidates for which CEL-SCI seeks approval. This could materially harm CEL-SCI’s financial results, reputation and stock price. Additionally, CEL-SCI may not be able to obtain the labeling claims necessary or desirable for product promotion. If CEL-SCI or other parties identify adverse effects after any of the products are on the market, or if manufacturing problems occur, regulatory approval may be suspended or withdrawn. CEL-SCI may be required to reformulate products, conduct additional clinical trials, make changes in product labeling or indications of use, or submit additional marketing applications to support any changes. If CEL-SCI encounters any of the foregoing problems, its business and results of operations will be harmed and the market price of its common stock may decline.
The FDA and other governmental authorities’ policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of CEL-SCI’s product candidates. If CEL-SCI is slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if CEL-SCI is not able to maintain regulatory compliance, CEL-SCI may lose any marketing approval that it may have obtained, which would adversely affect its business, prospects and ability to achieve or sustain profitability. CEL-SCI cannot predict the extent of adverse government regulations which might arise from future legislative or administrative action. Without government approval, CEL-SCI will be unable to sell any of its product candidates.
CEL-SCI’s product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial utility of an approved prescribing label, or result in significant negative consequences following marketing approval, if any.
Undesirable side effects caused by its product candidates could cause CEL-SCI or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign authorities. Results of the clinical trials could reveal a high and unacceptable severity and/or prevalence of these or other side effects. In such an event, the trials could be suspended or terminated and the FDA or comparable foreign regulatory authorities could order CEL-SCI to cease further development of, or deny approval of, the product candidates for any or all targeted indications. The drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. Any of these occurrences may harm CEL-SCI’s business, financial condition and prospects significantly.
| 31 |
Additionally, if one or more of the product candidates receives marketing approval, and CEL-SCI or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including but not limited to the following:
| · | regulatory authorities may withdraw approvals of such product or require product recalls; |
|
|
|
| · | regulatory authorities may require additional warnings on the label or impose restrictions on product distribution or use; |
|
|
|
| · | regulatory authorities may require CEL-SCI to conduct new post-marketing studies or clinical trials; |
|
|
|
| · | CEL-SCI could receive warning or untitled letters from the FDA or comparable notices of violations from foreign regulatory authorities; |
|
|
|
| · | CEL-SCI may be required to create a medication guide outlining the risks of such side effects for distribution to patients; |
|
|
|
| · | CEL-SCI could be sued and held liable for harm caused to patients; and |
|
|
|
| · | CEL-SCI’s reputation may suffer. |
Any of these events could prevent CEL-SCI from achieving or maintaining market acceptance of a particular product candidate, if approved, and could significantly harm its business, results of operations and prospects.
CEL-SCI relies on third parties to conduct its preclinical and clinical trials. If these third parties do not successfully carry out their contractual duties and meet regulatory requirements, or meet expected deadlines, CEL-SCI may not be able to obtain regulatory approval for or commercialize the product candidates and its business could be substantially harmed.
CEL-SCI does not have the ability to independently conduct large clinical trials. CEL-SCI has relied upon and plans to continue to rely upon third-party CROs to prepare for, conduct, monitor and manage data for its ongoing preclinical and clinical programs, including the global Phase 3 trial for Multikine. CEL-SCI relies on these parties for all aspects of the execution of its clinical trials and although CEL-SCI diligently oversees and carefully manages the CROs, CEL-SCI directly controls only certain aspects of their activities and relies upon them to provide timely, complete, and accurate reports on the conduct of the studies. Although such third parties provide support and represent CEL-SCI for regulatory purposes in the context of the clinical trials, ultimately CEL-SCI is responsible for ensuring that each of the studies is conducted in accordance with the applicable protocol, legal, regulatory, and scientific standards, and the reliance on the CROs does not relieve CEL-SCI of its regulatory responsibilities. CEL-SCI and the CROs acting on CEL-SCI’s behalf, as well as principal investigators and trial sites, are required to comply with Good Clinical Practice, or GCP, and other applicable requirements, which are implemented through regulations and guidelines enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area, or EEA, and comparable foreign regulatory authorities for all of the products in clinical development. Regulatory authorities enforce these GCPs through periodic inspections of trial sponsors, principal investigators, and trial sites. If CEL-SCI or any of the CROs fail to comply with applicable GCPs or other applicable regulations, the clinical data generated in the clinical trials may be determined to be unreliable and CEL-SCI may therefore need to enroll additional subjects in the clinical trials, or the FDA, EMA or comparable foreign regulatory authorities may require CEL-SCI to perform an additional clinical trial or trials before approving the marketing applications. Moreover, if CEL-SCI or any of the CROs, principal investigators, or trial sites, fail to comply with applicable regulatory and GCP requirements, CEL-SCI, the CROs, principal investigators, or trial sites may be subject to enforcement actions, such as fines, warning letters, untitled letters, clinical holds, civil or criminal penalties, and/or injunctions. CEL-SCI cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of the clinical trials comply with cGCP regulations. In addition, the clinical trials must be conducted with product produced under cGMP regulations. The failure to comply with these regulations may require CEL-SCI to delay or repeat clinical trials, which would delay the regulatory approval process.
| 32 |
If any of the relationships with the third-party CROs terminate, CEL-SCI may not be able to enter into arrangements with alternative CROs or to do so on commercially reasonable terms. In addition, the CROs are not CEL-SCI’s employees, and except for remedies available to CEL-SCI under the agreements with such CROs, CEL-SCI cannot control whether or not they devote sufficient time and resources to the on-going clinical, nonclinical and preclinical programs. If CROs do not successfully fulfill their regulatory obligations, carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to the clinical protocols, regulatory requirements or for other reasons, the clinical trials may be extended, delayed or terminated, and CEL-SCI may not be able to obtain regulatory approval for, or successfully commercialize, the product candidates. As a result, CEL-SCI’s results of operations and the commercial prospects for the product candidates would be harmed, the costs could increase and the ability to generate revenues could be delayed.
Switching or adding additional CROs involves additional cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays may occur, which can materially impact CEL-SCI’s ability to meet the desired clinical development timelines. Though CEL-SCI diligently oversees and carefully manages its relationships with the CROs, there can be no assurance that CEL-SCI will not encounter similar challenges or delays in clinical development in the future or that these delays or challenges will not have a material adverse impact on CEL-SCI’s business, financial condition and prospects.
CEL-SCI has obtained orphan drug designation from the FDA for Multikine for neoadjuvant, or primary, therapy in patients with squamous cell carcinoma of the head and neck, but CEL-SCI may be unable to maintain the benefits associated with orphan drug designation, including the potential for market exclusivity.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug or biologic intended to treat a rare disease or condition, which is defined as one occurring in a patient population of fewer than 200,000 in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug or biologic will be recovered from sales in the United States. In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. In addition, if a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications, including a full BLA, to market the same biologic for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity or where the manufacturer is unable to assure sufficient product quantity.
Even though CEL-SCI has received orphan drug designation for Multikine for the treatment of squamous cell carcinoma of the head and neck, CEL-SCI may not be the first to obtain marketing approval of a product for the orphan-designated indication due to the uncertainties associated with developing pharmaceutical products. CEL-SCI believes this to be unlikely since it does not know of any other company with a similar technology in Phase 3 studies. In addition, exclusive marketing rights in the United States may be limited if CEL-SCI seeks approval for an indication broader than the orphan-designated indication, or may be lost if the FDA later determines that the request for designation was materially defective or if CEL-SCI is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition. Further, even if CEL-SCI obtains orphan drug exclusivity for a product candidate, that exclusivity may not effectively protect the product candidate from competition because different drugs with different active moieties can be approved for the same condition. Even after an orphan product is approved, the FDA can subsequently approve another drug with the same active moiety for the same condition if the FDA concludes that the later drug is safer, more effective, or makes a major contribution to patient care. Orphan drug designation neither shortens the development time or regulatory review time of a drug nor gives the drug any advantage in the regulatory review or approval process.
| 33 |
Biologics carry unique risks and uncertainties, which could have a negative impact on future results of CEL-SCI’s operations.
The successful discovery, development, manufacturing and sale of biological products like CEL-SCI’s candidates is a long, expensive and uncertain process. There are unique risks and uncertainties with biologics. For example, access to and supply of necessary biological materials, such as cell lines, may be limited and governmental regulations restrict access to and regulate the transport and use of such materials. In addition, the development, manufacturing and sale of biologics is subject to regulations that are often more complex and extensive than the regulations applicable to other pharmaceutical products. Manufacturing biologics, especially in large quantities, is often complex and may require the use of innovative technologies. Such manufacturing also requires facilities specifically designed and validated for this purpose and sophisticated quality assurance and quality control procedures. Biologics are also frequently costly to manufacture because production inputs are derived from living animal or plant material, and some biologics cannot be made synthetically. Failure to successfully discover, develop, manufacture and sell its biological product candidates would adversely impact CEL-SCI’s business and future results of operations.
CEL-SCI may face substantial competition, which may result in others discovering, developing or commercializing competing products more quickly or more successfully than CEL-SCI.
The development and commercialization of new drug and biological products is highly competitive. CEL-SCI expects to face competition with respect to any product candidates that CEL-SCI may seek to develop or commercialize in the future, from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
Many of the companies which CEL-SCI may compete with in the future have significantly greater financial resources and expertise in research and development, manufacturing, nonclinical studies, conducting clinical trials, obtaining marketing approvals and marketing approved products than CEL-SCI. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of CEL-SCI’s potential competitors. Smaller and early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties may compete with CEL-SCI in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, CEL-SCI’s programs.
| 34 |
CEL-SCI may be unable to successfully scale-up manufacturing of its lead product candidate Multikine in sufficient quality and quantity, which would delay or prevent CEL-SCI from commercializing this product, if approved for marketing by the FDA or other regulatory agencies.
In order to commercialize its product candidates, CEL-SCI will need to manufacture them in large quantities. At the present time, CEL-SCI is not manufacturing Multikine while the manufacturing plant for Multikine is being validated to produce commercial quantities of Multikine, if approved. CEL-SCI may be unable to successfully increase the manufacturing capacity for its lead product candidate Multikine due to issues that may arise during scale-up activities.
Further, in order to release product and demonstrate stability of product candidates for future commercial use, CEL-SCI’s analytical methods must be validated in accordance with regulatory guidelines. CEL-SCI may not be able to successfully validate or maintain validation of its analytical methods during scale-up or demonstrate adequate purity, stability or comparability of its biological product candidates in a timely or cost-effective manner, or at all. Even if CEL-SCI believes its manufacturing processes meets all of the regulatory manufacturing requirements, the FDA will review those processes and the manufacturing facility as part of the review of the future BLA for Multikine when submitted. If CEL-SCI is unable to successfully scale up the manufacture of Multikine in sufficient quality and quantity, or if CEL-SCI encounters validation issues, the development, testing, and clinical trials of future product candidates, may be delayed or infeasible, and regulatory approval or commercial launch of any resulting product, including Multikine, may be delayed or may not be successfully achieved.
The current and future relationships with healthcare professionals, principal investigators, consultants, potential customers and third-party payors in the United States and elsewhere may be subject, directly or indirectly, to applicable healthcare laws and regulations.
Although CEL-SCI does not currently have any products on the market, if CEL-SCI begins commercializing its product candidates, CEL-SCI will be subject to additional healthcare statutory and regulatory requirements and oversight by federal and state governments as well as foreign governments in the jurisdictions in which CEL-SCI conducts its business. Healthcare providers, physicians and third-party payors in the United States and elsewhere will play a primary role in the recommendation and prescription of any drug candidates for which CEL-SCI obtains marketing approval. The current and future arrangements with healthcare professionals, principal investigators, consultants, potential customers and third-party payors may expose CEL-SCI to broadly applicable healthcare laws, including, without limitation:
| · | the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, lease, order or recommendation of, any good, facility, item or service, for which payment may be made, in whole or in part, under federal and state healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation. In addition, the Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; |
|
|
|
| · | federal civil and criminal false claims laws, including the federal False Claims Act, which impose criminal and civil penalties, including civil whistleblower actions, against individuals or entities for, among other things, knowingly presenting, or causing to be presented, to the federal government, including the Medicare and Medicaid programs, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
|
|
|
| · | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private), knowingly and willfully embezzling or stealing from a health care benefit program, willfully obstructing a criminal investigation of a healthcare offense and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters; |
| 35 |
| · | HIPAA, which also imposes obligations on covered entities, including healthcare providers, health plans, and healthcare clearinghouses, as well as their respective business associates that create, receive, maintain or transmit individually identifiable health information for or on behalf of a covered entity, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
|
|
|
| · | the U.S. federal physicians payment transparency requirements, sometimes called the “Sunshine Act” and its implementing regulations, which requires certain manufacturers of drugs, devices, biologicals and medical supplies that are reimbursable under Medicare, Medicaid, or the Children’s Health Insurance Program to report to the Centers for Medicare & Medicaid Services, or CMS, information related to physician payments and “other transfers of value” to physicians and teaching hospitals (and, beginning in 2021, for transfers of value to other healthcare providers), as well as the ownership and investment interests held by physicians and their immediate family members; |
|
|
|
| · | analogous state and foreign laws, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government which otherwise restrict payments that may be made to healthcare providers; state and foreign laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts; |
|
|
|
| · | the U.S. federal laws that require pharmaceutical manufacturers to report certain calculated product prices to the government or provide certain discounts or rebates to government authorities or private entities, often as a condition of reimbursement under federal healthcare programs; and |
|
|
|
| · | state and foreign laws that govern the privacy and security of health information in certain circumstances, including state security breach notification laws, state health information privacy laws and federal and state consumer protection laws, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
Efforts to ensure that the future business arrangements with third parties will comply with applicable healthcare laws and regulations may involve substantial costs. It is possible that governmental authorities will conclude that the business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws. If CEL-SCI’s operations are found to be in violation of any of these laws or any other governmental regulations, CEL-SCI may be subject to significant civil, criminal and administrative penalties, including, without limitation, damages, fines, imprisonment, exclusion from participation in government healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of the operations, all of which could significantly harm CEL-SCI’s business. If any of the physicians or other healthcare providers or entities with whom CEL-SCI expects to do business, including current and future collaborators, are found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from participation in government healthcare programs, which could also adversely affect CEL-SCI’s business.
| 36 |
Failure to obtain or maintain adequate coverage and reimbursement for the product candidates, if approved, could limit the ability to market those products and decrease CEL-SCI’s ability to generate revenue.
Sales of CEL-SCI’s product candidates will depend substantially, both domestically and abroad, on the extent to which the costs of the approved products will be paid by health maintenance, managed care, pharmacy benefit, and similar healthcare management organizations, or reimbursed by government authorities, private health insurers and other third-party payors. CEL-SCI anticipates that government authorities and other third-party payors will continue efforts to contain healthcare costs by limiting the coverage and reimbursement levels for new drugs and biologics. If coverage and reimbursement are not available, or are available only to limited levels, CEL-SCI may not be able to successfully commercialize its product candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow CEL-SCI to establish or maintain pricing sufficient to realize a return on its investment. It is difficult to predict at this time what third-party payors will decide with respect to the coverage and reimbursement for CEL-SCI’s product candidates.
Moreover, in some countries, particularly the countries of the European Union, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, CEL-SCI may be required to conduct a clinical trial that compares the cost-effectiveness of its product candidate to other available therapies. If reimbursement of its products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, CEL-SCI’s business could be harmed, possibly materially.
Even if any product candidate receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success.
If CEL-SCI’s product candidates do not achieve an adequate level of acceptance, CEL-SCI may not generate significant product revenues and CEL-SCI may not become profitable. The degree of market acceptance of its product candidates, including Multikine if approved for commercial sale, will depend on a number of factors, including:
| · | the timing of CEL-SCI’s receipt of any marketing approvals; |
|
|
|
| · | the terms of any approvals and the countries in which approvals are obtained; |
|
|
|
| · | the efficacy and safety and potential advantages and disadvantages compared to alternative treatments, including future alternative treatments; |
|
|
|
| · | the prevalence and severity of any side effects associated with CEL-SCI’s product candidates; |
|
|
|
| · | the indications for which its products are approved and the scope of risk information required to be included in the product labeling; |
|
|
|
| · | adverse publicity about CEL-SCI’s products or favorable publicity about competing products; |
|
|
|
| · | the approval of other products for the same indications as CEL-SCI’s products; |
|
|
|
| · | CEL-SCI’s ability to offer its products for sale at competitive prices; |
|
|
|
| · | the convenience and ease of administration compared to alternative treatments; |
| 37 |
| · | the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; |
|
|
|
| · | the success of CEL-SCI’s physician education programs; |
|
|
|
| · | the strength of CEL-SCI’s marketing and distribution support; and |
|
|
|
| · | the availability of third-party coverage and adequate reimbursement. |
If any product candidate CEL-SCI commercializes fails to achieve market acceptance, it could have a material and adverse effect on CEL-SCI’s business, financial condition, results of operation and prospects.
CEL-SCI currently has no marketing and sales force. If CEL-SCI is unable to establish effective sales or marketing capabilities or enter into agreements with third parties to sell or market its product candidates, CEL-SCI may not be able to effectively sell or market its product candidates, if approved, or generate product revenues.
CEL-SCI currently has no sales and marketing infrastructure due to the fact that all of its product candidates are still in clinical development. To achieve commercial success for any approved product candidate for which CEL-SCI retains sales and marketing responsibilities, CEL-SCI must build its sales, marketing, managerial, and other non-technical capabilities or make arrangements with third parties to perform these services. For example, CEL-SCI has entered into agreements with certain foreign distributors to commercialize Multikine, if approved, within their respective territories. However, CEL-SCI may determine that there is a need to build its own sales force in the United States for the future marketing of Multikine, if approved, rather than seeking a U.S. co-marketing partner or relying on a contracted sales force. There are risks involved with either establishing its own sales and marketing capabilities or entering into arrangements with third parties to perform these services. For example, recruiting and training a sales force is expensive and time consuming and could delay any product launch. If the commercial launch of a product candidate for which CEL-SCI recruits a sales force and establishes marketing capabilities is delayed or does not occur for any reason, CEL-SCI would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and its investment would be lost if CEL-SCI cannot retain or reposition its sales and marketing personnel.
Factors that may inhibit CEL-SCI’s efforts to commercialize its product candidates on its own include:
| · | CEL-SCI’s inability to recruit, hire, retain and incentivize adequate numbers of effective sales and marketing personnel; |
|
|
|
| · | the inability of sales personnel to obtain access to physicians or persuade adequate numbers of physicians to use and administer CEL-SCI’s future products; |
|
|
|
| · | the lack of complementary products to be offered by sales personnel, which may put CEL-SCI at a competitive disadvantage relative to companies with more extensive product lines; and |
|
|
|
| · | unforeseen costs and expenses associated with establishing an independent sales and marketing organization. |
If CEL-SCI does not establish sales and marketing capabilities successfully, either on its own or in collaboration with third parties, CEL-SCI will not be successful in commercializing Multikine, if approved, or any of its other product candidates that receive marketing approval or any such commercialization may experience delays or limitations.
| 38 |
CEL-SCI’s business activities may be subject to the Foreign Corrupt Practices Act and similar anti-bribery and anti-corruption laws of other countries in which CEL-SCI operates or will operate in the future.
CEL-SCI has conducted and has ongoing studies in international locations and may in the future initiate additional studies in countries other than the United States. Moreover, CEL-SCI has entered into agreements with foreign distributors to commercialize Multikine, if approved, in various territories outside of the United States. As a result, CEL-SCI’s business activities may be subject to the Foreign Corrupt Practices Act, or FCPA, and similar anti-bribery or anti-corruption laws, regulations or rules of other countries in which CEL-SCI operates. The FCPA generally prohibits offering, promising, giving or authorizing others to give anything of value, either directly or indirectly, to a non-U.S. government official in order to influence official action or otherwise obtain or retain business. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect the transactions of the corporation and to devise and maintain an adequate system of internal accounting controls.
CEL-SCI’s business is heavily regulated and therefore involves significant interaction with public officials, including officials of non-U.S. governments. Additionally, in many other countries, the healthcare providers who prescribe biopharmaceuticals are employed by their government, and the purchasers of biopharmaceuticals are government entities; therefore, CEL-SCI’s dealings with these prescribers and purchasers are subject to regulation under the FCPA. Recently the SEC and Department of Justice have increased their FCPA enforcement activities with respect to biotechnology and pharmaceutical companies. There is no certainty that all of CEL-SCI’s employees, agents or contractors conducting business abroad will comply with all applicable laws and regulations, particularly given the high level of complexity of these laws. Violations of these laws and regulations could result in fines, criminal sanctions against CEL-SCI, its officers or employees, the closing of CEL-SCI’s facilities, requirements to obtain export licenses, cessation of business activities in sanctioned countries, implementation of compliance programs and prohibitions on the conduct of CEL-SCI’s business. Any such violations could include prohibitions on CEL-SCI’s ability to offer its product candidates, if approved, in one or more countries and could materially damage its reputation, its brand, its future international marketing efforts, its ability to attract and retain employees and its business, prospects, operating results and financial condition.
Healthcare legislative reform measures may have a material adverse effect on CEL-SCI’s business and results of operations.
Existing regulatory policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of CEL-SCI’s product candidates. CEL-SCI cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If CEL-SCI is slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if CEL-SCI is not able to maintain regulatory compliance, CEL-SCI may lose any marketing approval that it may have obtained and it may not achieve or sustain profitability.
In the United States, there have been and continue to be a number of legislative initiatives to contain healthcare costs that may result in more limited coverage or downward pressure on the price CEL-SCI may otherwise receive for its product candidates. For example, in March 2010, Congress passed the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Affordable Care Act, which expanded healthcare coverage through Medicaid expansion and the implementation of the individual mandate for health insurance coverage and which included changes to the coverage and reimbursement of drug products under federal healthcare programs. The ACA contains a number of provisions that affect coverage and reimbursement of drug and biological products and/or that could potentially reduce the demand for pharmaceutical products such as increasing drug rebates under state Medicaid programs for brand name prescription drugs and extending those rebates to Medicaid managed care and assessing a fee on manufacturers and importers of brand name prescription drugs reimbursed under certain government programs, including Medicare and Medicaid. Tax reform legislation was enacted at the end of 2017 that eliminates the tax penalty established under the ACA for individuals who do not maintain mandated health insurance coverage beginning in 2019. In a May 2018 report, the Congressional Budget Office estimated that, compared to 2018, the number of uninsured will increase by 3 million in 2019 and 6 million in 2028, in part due to the elimination of the individual mandate. The ACA has also been subject to judicial challenge. In December 2018, a federal district court, in a challenge brought by a number of state attorneys general, found the ACA unconstitutional in its entirety because, once Congress repealed the individual mandate provision, there was no longer a basis to rely on Congressional taxing authority to support enactment of the law. Pending appeals, which could take some time, the ACA is still operational in all respects.
| 39 |
CEL-SCI’s industry continues to face potential changes in the legal and regulatory landscape on the federal, state and international levels. Additional legislative actions to control U.S. healthcare or other costs have passed. The Budget Control Act, as amended, resulted in the imposition of 2% reductions in Medicare (but not Medicaid) payments to providers in 2013 and will remain in effect through 2027 unless additional Congressional action is taken. There has also been increasing and considerable public and government interest in the United States with respect to specialty drug pricing practices, including proposed federal and state legislation designed to bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, put in place limits and caps on pharmaceutical prices, request rebates for certain pharmaceutical products, and reform government program reimbursement methodologies for drugs. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what biopharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. In markets outside of the United States, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and control the prices of medicinal products for human use.
CEL-SCI expects that current or future healthcare reform measures may result in more rigorous coverage criteria and in additional downward pressure on the price that it receives for any approved product, including Multikine if it is approved for commercialization. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent CEL-SCI from being able to generate revenue, attain profitability or commercialize its product candidates.
Legislative and regulatory proposals also have been made to expand post-approval requirements and restrict sales and promotional activities for biotechnology products. CEL-SCI cannot be sure whether additional legislative changes will be enacted, or whether FDA regulations, guidance or interpretations for biological products will be changed, or what the impact of such changes on the marketing approvals of its product candidates, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval and decision-making processes may significantly delay or prevent marketing approval, as well as subject CEL-SCI to more stringent product labeling and post-marketing testing and other requirements.
Foreign governments often impose strict price controls, which may adversely affect CEL-SCI’s future profitability.
CEL-SCI intends to seek approval to market its lead investigational product, Multikine, in both the United States and foreign jurisdictions. If CEL-SCI obtains approval in one or more foreign jurisdictions, CEL-SCI will be subject to rules and regulations in those jurisdictions relating to Multikine. In some foreign countries, particularly in the European Union, prescription drug pricing is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a drug candidate. Coverage and reimbursement decisions in one foreign jurisdiction may impact decisions in other countries. To obtain reimbursement or pricing approval in some countries, CEL-SCI may be required to conduct clinical trials that demonstrate the product candidate is more effective than current treatments and that compare the cost-effectiveness of Multikine to other available therapies. If reimbursement of Multikine is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, CEL-SCI may be unable to achieve or sustain profitability.
| 40 |
If CEL-SCI fails to comply with environmental, health and safety laws and regulations, CEL-SCI could become subject to fines or penalties or incur costs that could harm its business.
CEL-SCI is subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes generated in its biologic manufacturing facility. CEL-SCI cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from its use of hazardous materials, including radioactive materials used in its research laboratory, CEL-SCI could be held liable for any resulting damages, and the amount of the liability could exceed its resources. CEL-SCI also could incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
Risks Related to Intellectual Property
CEL-SCI may not be able to achieve or maintain a competitive position, and other technological developments may result in its proprietary technologies becoming uneconomical or obsolete.
CEL-SCI is involved in a biomedical field that is undergoing rapid and significant technological change. The pace of change continues to accelerate. The successful development of product candidates from the compounds, compositions and processes, through research financed by CEL-SCI, or as a result of possible third-party licensing arrangements with pharmaceutical or other companies, is not assured. CEL-SCI may fail to apply for patents on important technologies or product candidates in a timely fashion, or at all.
Many companies are working on drugs designed to cure or treat cancer. Many of these companies have financial, research and development, and marketing resources which are much greater than CEL-SCI’s and are capable of providing significant long-term competition either by establishing in-house research groups or by forming collaborative ventures with other entities. In addition, smaller companies and non-profit institutions are active in research relating to cancer and infectious diseases. The future market share of Multikine or the other product candidates, if approved, will be reduced or eliminated if the competitors develop and obtain approval for products that are safer or more effective than CEL-SCI’S product candidates. Moreover, the patent positions of pharmaceutical companies are highly uncertain and involve complex legal and factual questions for which important legal principles are often evolving and remain unresolved. As a result, the validity and enforceability of patents cannot be predicted with certainty. In addition, CEL-SCI does not know whether:
| · | CEL-SCI was the first to make the inventions covered by each of its issued patents and pending patent applications; |
|
|
|
| · | CEL-SCI was the first to file patent applications for these inventions; |
|
|
|
| · | others will independently develop similar or alternative technologies or duplicate any of CEL-SCI’s technologies; |
|
|
|
| · | any of the pending patent applications will result in issued patents; |
|
|
|
| · | any of the patents will be valid or enforceable; |
| 41 |
| · | any patents issued to CEL-SCI or its collaboration partners will provide CEL-SCI with any competitive advantages or will be challenged by third parties; |
|
|
|
| · | CEL-SCI will be able to develop additional proprietary technologies that are patentable; |
|
|
|
| · | the U.S. government will exercise any of its statutory rights to CEL-SCI’s intellectual property that was developed with government funding; or |
|
|
|
| · | its business may infringe the patents or other proprietary rights of others. |
CEL-SCI’s patents might not protect its technology from competitors, in which case CEL-SCI may not have any advantage over competitors in selling any products that CEL-SCI may develop.
CEL-SCI’s commercial success will depend in part on its ability to obtain additional patents and protect its existing patent position, as well as its ability to maintain adequate intellectual property protection for the technologies, product candidates, and any future products in the United States and other countries. If CEL-SCI does not adequately protect its technology, product candidates and future products, competitors may be able to use or practice them and erode or negate any competitive advantage CEL-SCI may have, which could harm CEL-SCI’s business and its ability to achieve profitability. The laws of some foreign countries do not protect the proprietary rights to the same extent or in the same manner as U.S. laws, and CEL-SCI may encounter significant problems in protecting and defending its proprietary rights in these countries. CEL-SCI will be able to protect its proprietary rights from unauthorized use by third parties only to the extent that its proprietary technologies, product candidates and any future products are covered by valid and enforceable patents or are effectively maintained as trade secrets.
Certain aspects of CEL-SCI’s technologies are covered by U.S. and foreign patents. In addition, CEL-SCI has a number of new patent applications pending. There is no assurance that the applications which are pending or which may be filed in the future will result in the issuance of any patents. Furthermore, there is no assurance as to the breadth and degree of protection any issued patents might afford CEL-SCI. Disputes may arise between CEL-SCI and others as to the scope and validity of these or other patents. Any defense of the patents could prove costly and time consuming and there can be no assurance that CEL-SCI will be in a position, or will deem it advisable, to carry on such a defense. A suit for patent infringement could result in increasing costs, delaying or halting development, or even forcing CEL-SCI to abandon a product candidate. Other private and public concerns, including universities, may have filed applications for, may have been issued, or may obtain additional patents and other proprietary rights to technology potentially useful or necessary to CEL-SCI. CEL-SCI is not currently aware of any such patents, but the scope and validity of such patents, if any, and the cost and availability of such rights are impossible to predict.
Much of CEL-SCI’s intellectual property is protected as trade secrets or confidential know-how, not as a patent.
CEL-SCI considers proprietary trade secrets and/or confidential and unpatented know-how to be important to its business. Much of the intellectual property pertains to CEL-SCI’s manufacturing methodologies, certain aspects of which may not be suitable for patent filings and must be protected as trade secrets and/or confidential know-how. This type of information must be protected diligently by CEL-SCI to protect its disclosure to competitors, since legal protections after disclosure may be minimal or non-existent. Accordingly, much of the value of this intellectual property is dependent upon the ability of CEL-SCI to keep its trade secrets and know-how confidential.
To protect this type of information against disclosure or appropriation by competitors, CEL-SCI’s policy is to require its employees, consultants, contractors and advisors to enter into confidentiality agreements with CEL-SCI. However, current or former employees, consultants, contractors and advisers may unintentionally or willfully disclose the confidential information to competitors, and confidentiality agreements may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. Enforcing a claim that a third party obtained illegally, and is using, trade secrets and/or confidential know-how is expensive, time consuming and unpredictable. The enforceability of confidentiality agreements may vary from jurisdiction to jurisdiction.
| 42 |
In addition, in some cases a regulator considering the application for product candidate approval may require the disclosure of some or all of the proprietary information. In such a case, CEL-SCI must decide whether to disclose the information or forego approval in a particular country. If CEL-SCI is unable to market its product candidates in key countries, CEL-SCI’s opportunities and value may suffer.
Failure to obtain or maintain trade secrets and/or confidential know-how trade protection could adversely affect CEL-SCI’S competitive position. Moreover, competitors may independently develop substantially equivalent proprietary information and may even apply for patent protection in respect of the same. If successful in obtaining such patent protection, competitors could limit the use of such trade secrets and/or confidential know-how.
CEL-SCI may be subject to claims challenging the inventorship or ownership of its patents and other intellectual property.
CEL-SCI may also be subject to claims that former employees, collaborators or other third parties have an ownership interest in its patents or other intellectual property. CEL-SCI may be subject to ownership disputes in the future arising, for example, from conflicting obligations of consultants or others who are involved in developing the product candidates. Litigation may be necessary to defend against these and other claims challenging inventorship or ownership. If CEL-SCI fails in defending any such claims, in addition to paying monetary damages, CEL-SCI may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on its business. Even if CEL-SCI is successful in defending against such claims, litigation could result in substantial costs and be a distraction to CEL-SCI’s management and employees.
Risks Related to CEL-SCI’s common stock
Future equity offerings or other equity issuances may result in dilution to existing shareholders.
CEL-SCI expects that significant additional capital will be needed in the future to continue its planned operations. To raise additional capital, CEL-SCI may in the future offer additional shares of its common stock or other securities convertible into or exchangeable for its common stock. To the extent CEL-SCI raises additional capital by issuing equity securities, CEL-SCI’s stockholders may experience substantial dilution and new investors could gain rights superior to existing stockholders.
The exercise of outstanding warrants and options will cause dilution.
As of September 30, 2022, there were outstanding warrants which allow the holders to purchase 1,310,822 shares of common stock, with a weighted average exercise price of $2.97 per share, and outstanding options which allow the holders to purchase up to 12,964,014 shares of common stock, with a weighted average exercise price of $9.06 per share. The exercise of these outstanding warrants and options will cause dilution to holders of our common stock.
| 43 |
Since CEL-SCI does not intend to pay dividends on its common stock, any potential return to investors will result only from any increases in the price of its common stock.
At the present time, CEL-SCI intends to use available funds to finance its operations. Accordingly, while payment of dividends rests within the discretion of its board of directors, no common stock dividends have been declared or paid by CEL-SCI and CEL-SCI has no intention of paying any common stock dividends in the foreseeable future. Additionally, any future debt financing arrangement may contain terms prohibiting or limiting the amount of dividends that may be declared or paid on CEL-SCI’s common stock. Any return to CEL-SCI’s shareholders will therefore be limited to appreciation in the price of its common stock, which may never occur. If CEL-SCI’s stock price does not increase, CEL-SCI’s shareholders are unlikely to receive any return on their investments in CEL-SCI’s common stock.
The price of CEL-SCI’s common stock has been volatile and is likely to continue to be volatile, which could result in substantial losses for CEL-SCI’s shareholders.
CEL-SCI’s stock price has been, and is likely to continue to be, volatile. As a result of this volatility, CEL-SCI’s shareholders may not be able to sell their shares at or above the price they paid for their shares. The market price for CEL-SCI’s common stock may be influenced by many factors, including:
| · | actual or anticipated fluctuations in CEL-SCI’s financial condition and operating results; |
|
|
|
| · | actual or anticipated changes in CEL-SCI’s growth rate relative to competitors; |
|
|
|
| · | competition from existing products or new products or product candidates that may emerge; |
|
|
|
| · | development of new technologies that may make CEL-SCI’s technology less attractive; |
|
|
|
| · | changes in physician, hospital or healthcare provider practices that may make CEL-SCI’s product candidates less useful; |
|
|
|
| · | announcements by CEL-SCI, its partners or competitors of significant acquisitions, strategic partnerships, joint ventures, collaborations or capital commitments; |
|
|
|
| · | developments or disputes concerning patent applications, issued patents or other proprietary rights; |
|
|
|
| · | the recruitment or departure of key personnel; |
|
|
|
| · | failure to meet or exceed financial estimates and projections of the investment community; |
|
|
|
| · | actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts; |
|
|
|
| · | variations in its financial results or those of companies that are perceived to be similar to CEL-SCI; |
|
|
|
| · | changes to coverage and reimbursement levels by commercial third-party payors and government payors, including Medicare, and any announcements relating to reimbursement levels; |
|
|
|
| · | general economic, industry and market conditions; and |
|
|
|
| · | the other factors described in this “Risk Factors” section. |
CEL-SCI has identified material weaknesses in its internal control over financial reporting. Failure to achieve and maintain effective internal controls over financial reporting could adversely affect CEL-SCI’s ability to report its results of operations and financial condition accurately and in a timely manner, which could have an adverse impact on CEL-SCI’s business.
CEL-SCI’s management is responsible for establishing and maintaining adequate internal control over financial reporting designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with GAAP. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement in an entity’s annual or interim consolidated financial statements might not be prevented or detected on a timely basis.
As described in Item 9A of this Annual Report, management noted material weaknesses in its internal control over financial reporting as of September 30, 2022. CEL-SCI’s management concluded that material weaknesses existed relating to ineffective information technology general controls in the areas of user access and segregation of duties related to certain information technology systems that support CEL-SCI’s financial reporting process and ineffective design of certain management review controls across a significant portion of CEL-SCI’s financial statement areas, particularly with regard to the precision of the review and evidence of review procedures performed. Although these control weaknesses did not result in any material misstatement of CEL-SCI’s financial statements for the periods presented, they could lead to a material misstatement of account balances or disclosures. As a result, CEL-SCI’s management concluded that its internal controls over financial reporting were not effective as of September 30, 2022.
In order to remediate these material weaknesses, CEL-SCI will change certain control activities over financial reporting to include, but are not limited to, the following: (i) evaluating and implementing enhanced process controls around user access management and segregation of duties and (ii) expanding the documentation over user access and system controls and enhancing the level of evidence maintained in management’s review controls.
If CEL-SCI is unable to remediate the material weaknesses timely and sufficiently or if CEL-SCI identifies future material weaknesses in its internal control over financial reporting or is unable to comply with the requirements of Section 404 in a timely manner or assert that its internal control over financial reporting is effective, CEL-SCI may experience a loss of investor confidence in the accuracy and completeness of its financial statements, incur material misstatements in its financial statements, incur difficulty accessing capital on favorable terms, or at all, be subject to fines, penalties or judgments, incur reputational harm, and the market price of its common stock may be adversely affected.
ITEM 1B. UNRESOLVED SEC COMMENTS
None
ITEM 2. PROPERTIES
CEL-SCI leases office space at 8229 Boone Blvd., Suite 802, Vienna, Virginia at a monthly rental of approximately $8,000. The lease on the office space expires on November 30, 2025. CEL-SCI believes this arrangement is adequate for the conduct of its present business.
| 44 |
CEL-SCI has a 17,900 square foot laboratory located in Baltimore, Maryland. The laboratory is leased by CEL-SCI at a cost of approximately $22,000 per month. During the year ended September 30, 2021, the laboratory lease was extended ten years and expires on February 29, 2032.
In August 2007, CEL-SCI leased a building near Baltimore, Maryland (the San Tomas lease). The building, which consists of approximately 73,000 square feet, has been remodeled in accordance with CEL-SCI’s specifications so that it can be used by CEL-SCI to manufacture Multikine for CEL-SCI’s Phase 3 clinical trial and sales of the drug, if approved by the FDA. The lease expires on October 31, 2028 and requires annual base rent payments of approximately $2.5 million during the twelve months ended September 30, 2022. The annual base rent escalates each year at 3% beginning on November 1st. CEL-SCI is also required to pay all real estate and personal property taxes, insurance premiums, maintenance expenses, repair costs and utilities, which were approximately $65,000 per month as of September 30, 2022. The lease allows CEL-SCI, at its election, to extend the lease for two ten-year periods or to purchase the building at the end of the 20-year lease.
In August 2020, the Company entered into an amendment to the San Tomas lease agreement under which the landlord agreed to allow the Company to substantially upgrade the manufacturing facility in preparation for the potential commercial production of Multikine. The total cost of the upgrades was $11.1 million. The landlord agreed to finance the final $2.4 million of the costs incurred. The Company started repaying these costs on March 1, 2021 through increased lease payments which will continue over the remaining lease term. The repayment includes a base rent which escalates at 3% each year plus interest that accrues at 13.75% per year on the $2.4 million financed.
ITEM 3. LEGAL PROCEEDINGS
Not applicable.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
ITEM 5. MARKET FOR CEL-SCI'S COMMON EQUITY AND RELATED STOCKHOLDER MATTERS.
As of September 30, 2022, there were approximately 525 holders of record of CEL-SCI’s common stock. CEL-SCI’s common stock is traded on the NYSE American under the symbol “CVM”.
Shown below are the range of high and low quotations for CEL-SCI’s common stock for the periods indicated as reported by the NYSE American.
Quarter Ending |
| High |
|
| Low |
| ||
12/31/2020 |
| $ | 16.70 |
|
| $ | 10.76 |
|
3/31/2021 |
| $ | 40.91 |
|
| $ | 11.88 |
|
6/30/2021 |
| $ | 27.86 |
|
| $ | 8.20 |
|
9/30/2021 |
| $ | 12.90 |
|
| $ | 7.08 |
|
|
|
|
|
|
|
|
|
|
12/31/2021 |
| $ | 12.82 |
|
| $ | 7.06 |
|
3/31/2022 |
| $ | 7.73 |
|
| $ | 3.80 |
|
6/30/2022 |
| $ | 6.14 |
|
| $ | 2.49 |
|
9/30/2022 |
| $ | 5.42 |
|
| $ | 3.09 |
|
| 45 |
Holders of common stock are entitled to receive dividends as may be declared by CEL-SCI’s Board of Directors out of legally available funds and, in the event of liquidation, to share pro rata in any distribution of CEL-SCI’s assets after payment of liabilities. CEL-SCI’s Board of Directors is not obligated to declare a dividend. CEL-SCI has not paid any dividends on its common stock and CEL-SCI does not have any current plans to pay any common stock dividends.
The provisions in CEL-SCI’s Articles of Incorporation relating to CEL-SCI’s preferred stock allow CEL-SCI’s directors to issue preferred stock with rights to multiple votes per share and dividend rights which would have priority over any dividends paid with respect to CEL-SCI’s common stock. The issuance of preferred stock with such rights may make more difficult the removal of management even if such removal would be considered beneficial to shareholders generally, and will have the effect of limiting shareholder participation in certain transactions such as mergers or tender offers if such transactions are not favored by incumbent management.
The market price of CEL-SCI’s common stock, as well as the securities of other biopharmaceutical and biotechnology companies, have historically been highly volatile, and the market has from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. Factors such as fluctuations in CEL-SCI’s operating results, announcements of technological innovations or new therapeutic products by CEL-SCI or its competitors, governmental regulation, developments in patent or other proprietary rights, public concern as to the safety of products which may be developed by CEL-SCI or other biotechnology and pharmaceutical companies, and general market conditions may have a significant effect on the market price of CEL-SCI’s common stock.
ITEM 6. SELECTED FINANCIAL DATA
CEL-SCI is a smaller reporting company as defined by Rule 12b-2 of the Securities and Exchange Commission and is not required to provide the information required under this item.
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion should be read in conjunction with the financial statements and the related notes thereto appearing elsewhere in this report.
Multikine (Leukocyte Interleukin, Injection) which, for simplicity, is referred to in this report as Multikine, is the trademark that the Company has registered for this investigational therapy, and this proprietary name is subject to FDA review under the Company’s future anticipated regulatory submission for approval. Multikine has not been licensed or approved by the FDA or any other regulatory agency. Neither has its safety or efficacy been established for any use.
CEL-SCI also owns and is developing a pre-clinical technology called LEAPS (Ligand Epitope Antigen Presentation System). CEL-SCI is using its LEAPS technology platform to investigate its lead peptide-based immunotherapy (CEL-4000) as a vaccine treatment for rheumatoid arthritis.
All of CEL-SCI’s projects are under development. As a result, CEL-SCI cannot predict when it will be able to generate any revenue from the sale of any of its products.
Since inception, CEL-SCI has financed its operations through the issuance of equity securities, convertible notes, loans and certain research grants. CEL-SCI will likely continue to generate net operating losses as it continues the development of Multikine and brings other drug candidates into clinical trials. Until such time as CEL-SCI becomes profitable, any or all of these financing vehicles or others may be utilized to assist CEL-SCI’s capital requirements.
| 46 |
Results of Operations
The Company incurred a net operating loss of approximately $36.1 million for the twelve months ended September 30, 2022. This net operating loss consists of significant non-cash expenses accounting for approximately 42% of the operating loss. The non-cash operating expenses include approximately $11.4 million in stock-based employee compensation and approximately $3.8 million in depreciation and amortization expense.
During the year ended September 30, 2022, research and development expenses increased by approximately $2.2 million, or 10%, compared to the year ended September 30, 2021. Major components of this increase include approximately $0.5 million increase in employee stock compensation expense, approximately $1.6 million increase in depreciation, primarily related to leasehold improvements to the manufacturing facility that were placed in service in October 2021, an increase of approximately $1.2 million of costs incurred to prepare for the potential commercial sale of Multikine, and an increase of approximately $0.2 million in other miscellaneous research and development expenses. These increases were offset by a decrease of approximately $1.3 million in costs related to the Phase 3 clinical study.
During the year ended September 30, 2022, general and administrative expenses decreased by approximately $2.4 million, or 18%, compared to the year ended September 30, 2021. This decrease is primarily due to a decrease in employee stock compensation expense of approximately $2.8 million offset by a $0.4 million increase in other net general and administrative expenses.
During the years ended September 30, 2022 and 2021, CEL-SCI recorded a derivative gain of approximately $0.4 million and a loss of approximately $0.7 million, respectively. This variation was the result of the change in fair value of the derivative liabilities during the period which was caused by fluctuations in the share price of CEL-SCI’s common stock.
During the year ended September 30, 2022, other non-operating gain decreased by approximately $1.7 million compared to the year ended September 30, 2021 resulting in a loss of approximately $31,000 for the year ended September 30, 2022. This gain for the year ended September 30, 2021 relates to the Securities Purchase Agreement (SPA) described in Note 12 to the financial statements included as part of this report. Under the SPA, the Company issued Ergomed shares of common stock and the net proceeds from the sales of those shares reduces outstanding amounts due Ergomed. Upon issuance, the Company expensed the full value of the shares as other non-operating gain/loss and subsequently offset the gain or loss as amounts were realized through the sale by Ergomed and reduced accounts payable to Ergomed. The amount of the gain or loss is a result of the timing of shares issued to Ergomed and the subsequent re-sale of those shares. There was no activity under the agreement during the year ended September 30, 2022. During the year ended September 30, 2021, the Company realized approximately $1.7 million in value upon the resale of shares. Ergomed resold the final balance of shares issued in the quarter ended September 30, 2021.
| 47 |
Research and Development Expenses
CEL-SCI’s research and development efforts involved Multikine and LEAPS. The table below shows the research and development expenses associated with each project during the reporting periods.
|
| Year ended September 30, |
| |||||
|
| 2022 |
|
| 2021 |
| ||
Multikine |
| $ | 24,251,629 |
|
| $ | 21,508,026 |
|
LEAPS |
|
| 1,103,717 |
|
|
| 1,600,871 |
|
Total research and development |
| $ | 25,355,346 |
|
| $ | 23,108,897 |
|
CEL-SCI’s Phase 3 clinical trial began in December 2010 after the completion and validation of CEL-SCI’s dedicated manufacturing facility. The Phase 3 clinical trial was fully enrolled in September 2016, reached its primary endpoint in April 2020 and achieved database lock in December 2020. The data was unblinded in June 2021, the primary endpoint results were announced in June 2021, additional data was presented at ASCO 2022 and ESMO 2022 in June 2022 and September 2022, respectively. CEL-SCI is currently preparing the BLA to submit to the FDA.
As explained in Item 1 of this report, clinical and other studies necessary to obtain regulatory approval of a new drug involve significant costs and require several years to complete. The extent of the Company’s clinical trials and research programs are primarily based upon the amount of capital available to the Company and the extent to which the Company has received regulatory approvals for clinical trials. The inability of the Company to conduct clinical trials or research, whether due to a lack of capital or regulatory approval, will prevent the Company from completing the studies and research required to obtain regulatory approval for any products which the Company is developing. Without regulatory approval, the Company will be unable to sell any of its products. Since all of the Company’s projects are under development, the Company cannot predict when it will be able to generate any revenue from the sale of any of its products.
Liquidity and Capital Resources
CEL-SCI has had only limited revenues from operations since its inception in March 1983. CEL-SCI has relied primarily upon capital generated from the public and private offerings of its common stock and convertible notes. In addition, CEL-SCI has utilized short-term loans to meet its capital requirements. Capital raised by CEL-SCI has been used to acquire an exclusive worldwide license to use, and later purchase, certain patented and unpatented proprietary technology and know-how relating to the human immunological defense system and for clinical trials. Capital has also been used for patent applications, debt repayment, research and development, administrative costs, and for CEL-SCI’s laboratory and manufacturing facilities. CEL-SCI does not anticipate realizing significant revenues until it enters into licensing arrangements regarding its technology and know-how or until it receives regulatory approval to sell its products (which could take a number of years). As a result, CEL-SCI has been dependent primarily upon the proceeds from the sale of its securities to meet all of its liquidity and capital requirements and anticipates having to do so in the future. During fiscal years 2022 and 2021, CEL-SCI raised net proceeds of approximately $0.1 million and $54.0 million, respectively, through a combination of the sale of common stock and the exercise of warrants and options.
In August 2007, CEL-SCI leased a building near Baltimore, Maryland. The building, which consists of approximately 73,000 square feet, has been remodeled in accordance with CEL-SCI’s specifications so that it can be used by CEL-SCI to manufacture Multikine for CEL-SCI’s Phase III clinical trials and sales of the drug if approved by the FDA. The lease expires on October 31, 2028, and required annual base rent payments of approximately $2.5 million during the twelve months ended September 30, 2022. See Item 2 of this report for more information concerning the terms of this lease.
In June 2021, the Company sold 1,400,000 shares of common stock at a public offering price of $22.62 per share and received aggregate net proceeds of approximately $29.4 million. Under the terms of the Underwriting Agreement the Company granted the Underwriters a 30-day option to purchase up to an additional 210,000 shares of common stock solely to cover over-allotments. The underwriter fully exercised this option in June 2021 resulting in additional net proceeds to the Company of approximately $4.4 million.
| 48 |
In December 2020, the Company sold 1,000,000 shares of common stock at a public offering price of $14.65 per share and received aggregate net proceeds of approximately $13.6 million.
The following charts list the warrants that were exercised and the proceeds received during the years ended September 30, 2022 and 2021.
Fiscal Year 2022
Warrants |
| Warrants Exercised |
|
| Exercise Price |
|
| Proceeds |
| |||
Series CC |
|
| 15,205 |
|
| $ | 5.00 |
|
| $ | 76,025 |
|
Series NN |
|
| 10,000 |
|
| $ | 2.52 |
|
|
| 25,200 |
|
|
|
| 25,205 |
|
|
|
|
|
| $ | 101,225 |
|
Fiscal Year 2021
Warrants |
| Warrants Exercised |
|
| Exercise Price |
|
| Proceeds |
| |||
Series Z |
|
| 79,200 |
|
| $ | 13.75 |
|
| $ | 1,089,000 |
|
Series ZZ |
|
| 20,000 |
|
| $ | 13.75 |
|
|
| 275,000 |
|
Series AA |
|
| 100,000 |
|
| $ | 13.75 |
|
|
| 1,375,000 |
|
Series CC |
|
| 132,798 |
|
| $ | 5.00 |
|
|
| 663,990 |
|
Series MM |
|
| 464,201 |
|
| $ | 1.86 |
|
|
| 863,414 |
|
Series NN |
|
| 138,755 |
|
| $ | 2.52 |
|
|
| 349,663 |
|
Series RR |
|
| 165,888 |
|
| $ | 1.65 |
|
|
| 273,715 |
|
Series SS |
|
| 126,064 |
|
| $ | 2.09 |
|
|
| 263,474 |
|
Series TT |
|
| 370,964 |
|
| $ | 2.24 |
|
|
| 830,959 |
|
|
|
| 1,597,870 |
|
|
|
|
|
| $ | 5,984,215 |
|
CEL-SCI entered into Securities Purchase Agreements (SPAs) with Ergomed plc, one of CEL-SCI’s Clinical Research Organizations responsible for managing the Phase 3 clinical trial, to facilitate payment of amounts due Ergomed. Under the Agreements, CEL-SCI issued Ergomed shares of common stock and the net proceeds from the sales of those shares reduces outstanding amounts due Ergomed. Upon issuance, CEL-SCI expensed the full value of the shares as other non-operating gain/loss and subsequently offsets the expense as amounts were realized through the sale by Ergomed and reduced accounts payable to Ergomed.
During the years ended September 30, 2022 and 2021, CEL-SCI did not issue Ergomed any shares of common stock. Additionally, no shares were sold during the year ended September 30, 2022. During the year ended September 30, 2021, the company recorded other non-operating gains of approximately $1.7 million upon the resale of shares from the SPA.
As of September 30, 2022 and 2021, Ergomed had no shares remaining for resale.
During the year ended September 30, 2022, the Company used approximately $19.6 million, or approximately $1.6 million per month, in cash, after considering the maturity and transfer to cash of the remaining $6.2 million in U.S. Treasury bills (T-bills). Significant components of this decrease include cash used to fund the Company’s regular operations of approximately $18.2 million, the purchase of property and equipment for approximately $0.6 million, approximately $0.2 million in share issuance costs and approximately $1.4 million in lease payments. These outflows are offset by approximately $0.8 million in lease incentives received from the landlord to partially offset costs of the manufacturing facility upgrade.
| 49 |
During the year ended September 30, 2022, 25,205 warrants were exercised at a weighted average exercise price of $4.02 for total proceeds of approximately $0.1 million.
Prepaid expenses decreased by approximately $0.2 million during the year ended September 30, 2022 as compared to September 30, 2021 primarily due to the timing of payments and recognition of related expenses relating to the Company’s Phase 3 clinical trial status.
Supplies are purchased for use in the Company’s manufacturing and R&D efforts. During the year ended September 30, 2022, the supplies increased by approximately $0.2 million in support of the work to validate and prepare the manufacturing facility to produce Multikine for commercial purposes and before the Company’s Biologics License Application (BLA) can be submitted to the FDA.
Primarily as a result of CEL-SCI’s losses incurred to date, its expected continued future losses, and limited cash balances, CEL-SCI has included a disclosure in its financial statements expressing substantial doubt about its ability to continue as a going concern. CEL-SCI has included this disclosure on numerous occasions in the preceding years.
Future Capital Requirements
CEL-SCI’s material capital commitments include funding operating losses, funding its research and development program and making required lease payments. Additionally, the Company recently completed upgrading the manufacturing facility to prepare for the potential commercial production of Multikine. Total costs of this upgrade were approximately $11.1 million. The landlord of the property agreed to finance the final $2.4 million of costs and allow for the repayment through increased lease payments which began on March 1, 2021. As of September 30, 2022, the landlord had provided approximately all $2.4 million of the agreed funding. Because of the change in lease payments, the new financing arrangement was considered a lease modification.
For information on employment contracts, see Item 11 of this report.
Further, CEL-SCI has contingent obligations with vendors for work that will be completed in relation to the Phase 3 trial. The timing of these obligations cannot be determined at this time. CEL-SCI estimates it will incur additional expenses of approximately $0.6 million for the filing of the clinical study report with the FDA. It should be noted that this estimate is based only on the information currently available from the CROs responsible for managing the Phase 3 clinical trial and does not include other related costs, e.g., the manufacturing of the drug.
CEL-SCI will need to raise additional funds, either through the exercise of outstanding warrants/options, through debt or equity financings or a partnering arrangement, to bring Multikine to market. The ability of CEL-SCI to complete the necessary clinical trials and obtain FDA approval for the sale of products to be developed on a commercial basis is uncertain. However, it is possible that CEL-SCI will not be able to generate enough cash to continue operations at its current level. CEL-SCI’s management has engaged in fundraising for over 25 years and believes that the manner in which it is proceeding will produce the best possible outcome for the shareholders. There can be no assurances that CEL-SCI will be successful in raising additional funds.
| 50 |
Clinical and other studies necessary to obtain regulatory approval of a new drug involve significant costs and require several years to complete. The extent of CEL-SCI’s clinical trials and research programs are primarily based upon the amount of capital available to CEL-SCI and the extent to which CEL-SCI has received regulatory approvals for clinical trials. The inability of CEL-SCI to conduct clinical trials or research, whether due to a lack of capital or regulatory approval, will prevent CEL-SCI from completing the studies and research required to obtain regulatory approval for any products which CEL-SCI is developing. Without regulatory approval, CEL-SCI will be unable to sell any of its products.
In the absence of revenues, CEL-SCI will be required to raise additional funds through the sale of securities, debt financings or other arrangements in order to continue with its research efforts. However, there can be no assurance that such financing will be available or be available on favorable terms. Ultimately, CEL-SCI must complete the development of its products, obtain appropriate regulatory approvals and obtain sufficient revenues to support its cost structure.
Since all of CEL-SCI’s projects are under development, CEL-SCI cannot predict with any certainty the funds required for future research and clinical trials, the timing of future research and development projects, or when it will be able to generate any revenue from the sale of any of its products.
CEL-SCI's cash flow and earnings are subject to fluctuations due to changes in interest rates on its bank accounts, and, to an immaterial extent, foreign currency exchange rates.
Critical Accounting Policies
CEL-SCI's significant accounting policies are more fully described in Note 3 to the financial statements included as part of this report. However, certain accounting policies are particularly important to the portrayal of CEL-SCI’s financial position and results of operations and require the application of significant judgments by management. As a result, the financial statements are subject to an inherent degree of uncertainty. In applying those policies, management uses its judgment to determine the appropriate assumptions to be used in the determination of certain estimates. These estimates are based on CEL-SCI's historical experience, terms of existing contracts, observance of trends in the industry and information available from outside sources, as appropriate.
Management believes that the following critical accounting policies require the most significant judgments and estimates with respect to the preparation of CEL-SCI’s financial statements.
Lease Accounting –The measurement of the finance and operating lease right-of-use asset and lease liabilities requires the determination of an estimated lease term and an incremental borrowing rate, which involves complex judgment by management. The determination of the incremental borrowing rates for new and modified lease contracts is a critical accounting policy. Significant judgment is required by management to develop inputs and assumptions used to determine the incremental borrowing rate for lease contracts.
Share-based Compensation– Share-based compensation cost to employees is measured at fair value as of the grant date in accordance with the provisions of ASC 718. The fair value of the stock options is calculated using the Black-Scholes option pricing model. The Black-Scholes model requires various judgmental assumptions including volatility and expected option life. The compensation cost is recognized as expense over the requisite service or vesting period. Performance-based options are valued using a Monte-Carlo simulation model, which requires inputs based on estimates, including the likelihood of the occurrence of performance and market conditions, volatility and expected option life.
| 51 |
Derivative Instruments– CEL-SCI enters into financing arrangements that consist of freestanding derivative instruments or hybrid instruments that contain embedded derivative features. CEL-SCI accounts for these arrangements in accordance with ASC 815, Accounting for Derivative Instruments and Hedging Activities, as well as related interpretations of these standards. In accordance with accounting principles generally accepted in the United States (“GAAP”), derivative instruments and hybrid instruments are recognized as either assets or liabilities in the statement of financial position and are measured at fair value with gains or losses recognized in earnings or other comprehensive income depending on the nature of the derivative or hybrid instruments. Fair value is generally calculated using a Black-Scholes valuation model. Embedded derivatives that are not clearly and closely related to the host contract are bifurcated and recognized at fair value with changes in fair value recognized as either a gain or loss in earnings if they can be reliably measured. When the fair value of embedded derivative features cannot be reliably measured, CEL-SCI measures and reports the entire hybrid instrument at fair value with changes in fair value recognized as either a gain or loss in earnings.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURE ABOUT MARKET RISKS
Not applicable.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
See the financial statements included with this report.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Not applicable
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Under the direction and with the participation of CEL-SCI’s management, including CEL-SCI’s Chief Executive Officer and Chief Financial Officer, CEL-SCI carried out an evaluation of the effectiveness of the design and operation of its disclosure controls and procedures as of September 30, 2022. CEL-SCI maintains disclosure controls and procedures that are designed to ensure that information required to be disclosed in its periodic reports with the Securities and Exchange Commission is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and regulations, and that such information is accumulated and communicated to CEL-SCI’s management, including its principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure. CEL-SCI’s disclosure controls and procedures are designed to provide a reasonable level of assurance of reaching its desired disclosure control objectives.
CEL-SCI’s Chief Executive and Principal Financial Officer has concluded that, due to the material weaknesses described below, CEL-SCI’s disclosure controls and procedures were not effective as of September 30, 2022.
Management’s Report on Internal Control over Financial Reporting
CEL-SCI’s management is responsible for establishing and maintaining adequate internal control over financial reporting and for the assessment of the effectiveness of internal control over financial reporting. As defined by the Securities and Exchange Commission, internal control over financial reporting is a process designed by, or under the supervision of CEL-SCI’s principal executive officer and principal financial officer and implemented by CEL-SCI’s Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of CEL-SCI’s financial statements in accordance with U.S. generally accepted accounting principles.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Under the supervision and with the participation of management, including CEL-SCI’s Chief Executive and Principal Financial Officer, CEL-SCI evaluated the effectiveness of its internal control over financial reporting as of September 30, 2022 based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission, or the COSO Framework. Management’s assessment included an evaluation of the design of CEL-SCI’s internal control over financial reporting and testing of the operational effectiveness of those controls. CEL-SCI's Chief Executive and Financial Officer concluded that as of such date, CEL-SCI's internal controls over financial reporting were not effective.
Material Weaknesses in Internal Control Over Financial Reporting
A material weakness (as defined in Rule 12b-2 under the Exchange Act) is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of CEL-SCI’s annual or interim financial statements will not be prevented or detected on a timely basis. CEL-SCI’s management concluded that a material weakness existed as of September 30, 2022 relating to ineffective information technology general controls in the areas of user access and segregation of duties related to certain information technology systems that support our financial reporting process. As a result, certain activity level controls were also deemed to be ineffective that are dependent on information derived from these information technology systems.
In addition, we identified a material weakness related to the ineffective design of certain management review controls across a significant portion of the Company’s financial statement areas, particularly with regard to the precision of the review and evidence of review procedures performed.
| 52 |
Remediation of Material Weaknesses
In order to remediate these material weaknesses, CEL-SCI will change certain control activities over financial reporting to include, but are not limited to, the following: (i) evaluating and implementing enhanced process controls around user access management and segregation of duties and (ii) expanding the documentation over user access and system controls and enhancing the level of evidence maintained in management review controls.
CEL-SCI is committed to maintaining a strong internal control environment and implementing measures designed to help ensure that control deficiencies contributing to the material weaknesses are remediated as soon as possible.
Notwithstanding the material weaknesses described above, CEL-SCI’s management has concluded that the consolidated financial statements included in this Annual Report on Form 10-K present fairly, in all material respects, our financial position, results of operations and cash flows in conformity with GAAP.
Changes in Internal Control over Financial Reporting
During the fiscal year ended September 30, 2022, CEL-SCI implemented additional controls over financial reporting which include a thorough review of all leases and relevant accounting guidance such that the accounting for any leases complies with U.S. GAAP.
Other than as described above, there have been no other changes in CEL-SCI’s internal control over financial reporting occurred during the fiscal year ended September 30, 2022 that has materially affected, or is reasonably likely to materially affect, CEL-SCI’s internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
None.
ITEM 10. DIRECTORS AND EXECUTIVE OFFICERS OF THE REGISTRANT
Officers and Directors
Name |
| Age |
| Position |
Geert R. Kersten, Esq. |
| 62 |
| Director, Chief Executive and Financial Officer and Treasurer |
Patricia B. Prichep |
| 71 |
| Senior Vice President of Operations and Corporate Secretary |
Dr. Eyal Talor |
| 66 |
| Chief Scientific Officer |
Dr. Daniel H. Zimmerman |
| 81 |
| Senior Vice President of Research, Cellular Immunology |
John Cipriano |
| 80 |
| Senior Vice President of Regulatory Affairs |
Dr. Peter R. Young |
| 77 |
| Director |
Bruno Baillavoine |
| 70 |
| Director |
Robert Watson |
| 65 |
| Director |
Dr. Gail Naughton |
| 66 |
| Director |
The directors of CEL-SCI serve in such capacity until the next annual meeting of CEL-SCI's shareholders or until their successors have been duly elected and qualified. The officers of CEL-SCI serve at the discretion of CEL-SCI's directors.
| 53 |
The principal occupations of CEL-SCI's officers and directors, during the past several years, are as follows:
Geert Kersten has served in his current leadership role at CEL-SCI since 1995. Mr. Kersten has been with CEL-SCI since 1987. He has been involved in the pioneering field of cancer immunotherapy for over three decades and has successfully steered CEL-SCI through many challenging cycles in the biotechnology industry. Mr. Kersten also provides CEL-SCI with significant expertise in the fields of finance and law and has a unique vision of how CEL-SCI's Multikine product could potentially change the way cancer is treated. Prior to joining CEL-SCI, Mr. Kersten worked at the law firm of Finley & Kumble and worked at Source Capital, an investment banking firm located in McLean, VA. He is a native of Germany, graduated from high school in England, and completed his studies in the US. Mr. Kersten received his Undergraduate Degree in Accounting and an M.B.A. from George Washington University, and a law degree (J.D.) from American University in Washington, DC.
Patricia B. Prichep joined CEL-SCI in 1992 and has been CEL-SCI's Senior Vice President of Operations since March 1994. Between December 1992 and March 1994, Ms. Prichep was CEL-SCI's Director of Operations. Ms. Prichep became CEL-SCI's Corporate Secretary in May 2000. She is responsible for all day-to-day operations of CEL-SCI, including human resources and is the liaison with CEL-SCI’s independent registered public accounting firm for financial reporting. From June 1990 to December 1992, Ms. Prichep was the Manager of Quality and Productivity for the NASD’s Management, Systems and Support Department and was responsible for the internal auditing and work flow analysis of operations. Between 1982 and 1990, Ms. Prichep was Vice President and Operations Manager for Source Capital, Ltd. She handled all operations and compliance for Source Capital and was licensed as a securities broker. Ms. Prichep received her B.A. from the University of Bridgeport in Connecticut.
Eyal Talor, Ph.D. joined CEL-SCI in October 1993. In October 2009, Dr. Talor was promoted to Chief Scientific Officer. Prior to this promotion, Dr. Talor was the Senior Vice President of Research and Manufacturing. He is a clinical immunologist with over 27 years of hands-on management of clinical research and drug development for immunotherapy application; pre-clinical to Phase III clinical trials, in the biopharmaceutical industry. His expertise includes biopharmaceutical R&D and Biologics product development, GMP (Good Manufacturing Practices) manufacture, Quality Control testing, and the design and building of GMP manufacturing and testing facilities. He served as Director of Clinical Laboratories (certified by the State of Maryland) and has experience in the design of pre-clinical and clinical trials (Phase I – III) and GCP (Good Clinical Practices) requirements. He also has broad experience in the different aspects of biological assay development, analytical methods validation, raw material specifications, and QC (Quality Control) tests development under FDA/GMP, USP, and ICH guidelines. He has extensive experience in the preparation of documentation for IND and other regulatory submissions. His scientific area of expertise encompasses immune response assessment. He is the author of over 25 publications and has published a number of reviews on immune regulations in relation to clinical immunology. Before coming to CEL-SCI, he was Director of R&D and Clinical Development at CBL, Inc., Principal Scientist - Project Director, and Clinical Laboratory Director at SRA Technologies, Inc. Prior to that he was a full-time faculty member at The Johns Hopkins University, Medical Institutions; School of Public Health. He has invented technologies which are covered by ten issued patents on Multikine’s composition of matter and method of use in cancer and two platform Peptide technologies, Antigen Directed Apoptosis of T-cells (‘Adapt’) and LEAPS, for the treatment of autoimmune diseases, asthma, allergy, transplantation rejection and infectious diseases. He also is responsible for numerous product and process inventions as well as a number of pending US and PCT patent applications. He received his Ph.D. in Microbiology and Immunology from the University of Ottawa, Ottawa, Ontario, Canada, and had post-doctoral training in clinical and cellular immunology at The Johns Hopkins University, Baltimore, Maryland, USA. He holds an Associate teaching position at the Johns Hopkins University Medical Institutions.
| 54 |
Daniel H. Zimmerman, Ph.D. was CEL-SCI's Senior Vice President of Research, Cellular Immunology between 1996 and December 2008 and again since November 2009. He joined CEL-SCI in January 1996 as the Vice President of Research, Cellular Immunology. Dr. Zimmerman founded CELL-MED, Inc. and was its president from 1987-1995. From 1973-1987, Dr. Zimmerman served in various positions at Electronucleonics, Inc. His positions included: Scientist, Senior Scientist, Technical Director and Program Manager. Dr. Zimmerman held various teaching positions at Montgomery College between 1987 and 1995. Dr. Zimmerman has invented technologies which are covered by over a dozen US patents as well as many foreign equivalent patents. He is the author of over 50 scientific publications in the area of immunology and infectious diseases. He has been awarded numerous grants from National Institutes of Health, or NIH, and the Department of Defense, or DOD. From 1969-1973, Dr. Zimmerman was a Senior Staff Fellow at NIH. For the following 25 years, he continued on at NIH as a guest worker. Dr. Zimmerman received a Ph.D. in Biochemistry in 1969, and a Masters in Zoology in 1966 from the University of Florida as well as a B.S. in Biology from Emory and Henry College in 1963.
John Cipriano was CEL-SCI’s Senior Vice President of Regulatory Affairs between March 2004 and December 2008 and again since October 2009. Mr. Cipriano brings to CEL-SCI over 30 years of experience with both biotech and pharmaceutical companies. In addition, he held positions at the United States Food and Drug Administration (FDA) as Deputy Director, Division of Biologics Investigational New Drugs, Office of Biologics Research and Review and was the Deputy Director, IND Branch, Division of Biologics Evaluation, Office of Biologics. Mr. Cipriano completed his B.S. in Pharmacy from the Massachusetts College of Pharmacy in Boston, Massachusetts and his M.S. in Pharmaceutical Chemistry from Purdue University in West Lafayette, Indiana.
Peter R. Young, Ph.D. has been a Director of CEL-SCI since August 2002. Dr. Young has been a senior executive within the pharmaceutical industry in the United States and Canada for most of his career, originally in organizations that are now part of Sanofi S.A. Over the last 20 years he has primarily held positions of Chief Executive Officer or Chief Financial Officer and has extensive experience with acquisitions and equity financing. Since November 2001, Dr. Young has been the President of Agnus Dei, LLC, which has acted as a partner in an organization managing immune system clinics which treat patients with diseases such as cancer, multiple sclerosis and hepatitis. Dr. Young was also the President and Chief Executive Officer of SRL Technology, Inc., a company involved in the development of pharmaceutical drug delivery systems. Between 1998 and 2001, Dr. Young was the Chief Financial Officer of Adams Laboratories, Inc., the developer of Mucinex®. Dr. Young received his Ph.D. in Organic Chemistry from the University of Bristol, England after obtaining his Bachelor's degree in Honors Chemistry, Mathematics and Economics. Subsequently, he qualified as a Fellow of the Chartered Institute of Management Accountants.
Bruno Baillavoine joined CEL-SCI’s board of directors in June 2015. Since 2017, Mr. Baillavoine has been the Director, Head of Pericles Group UK the subsidiary of the Paris-based leading French consulting firm Pericles Consulting Holdings SAS. Pericles is an expert in the fields of Banking, Finance, Asset Management and Insurance with over 350 institutional clients. He has also been an advisor to the Board of CSL Inc, Combatives Sports League, a US Mixed Martial Arts company since 2017 and was appointed to the Board in 2019. Between 2010-2016, Mr. Baillavoine was a partner of Globomass Holdings Limited, a London, England based developer of renewable energy projects from concept through final operations. From 2012 and 2016 Mr. Baillavoine was the Executive Chairman of Globomass Holdings. Globomass was acquired by CleanBay Inc. in 2016. Mr. Baillavoine remains a significant shareholder in CleanBay Inc. Between 1978 and 1982 he was the marketing manager of Ravenhead Ltd., a manufacturer of glass tableware, and part of United Distillers Group (later acquired by Grand Metropolitan). During this time Mr. Baillavoine became the UK Business Manager where he restored market share and profit for United Distillers. From 1982 to 1986 Mr. Baillavoine was Group Corporate Planning and Group Marketing Director for Prontaprint where he expanded the number of shops to 500 locations in four years. Mr. Baillavoine was with Grand Metropolitan Plc between 1986-1988 (now Diageo Plc), a FTSE 100 beverage, food, hotel and leisure company, as director in the Special Operations division. In this capacity, he developed plans for Grand Met’s trouble-shooting division for over 20,000 Grand Met retail outlets. From 1988-1991 he was the Managing Director of Nutri Systems (UK) Ltd., a subsidiary of the US based provider of professionally supervised weight loss programs. Between 1991 and 1995, Mr. Baillavoine was Director of BET Group plc, a multinational business support services group, and in 1992, was promoted to the Managing Director for the manufacturing businesses. The £2.3 billion turnaround of BET during his tenure is one of the most successful turnarounds of a top 100 FTSE company. Since 1995, Mr. Baillavoine has held a number of CEO positions across a wide range of industries and geographical locations. Mr. Baillavoine has European and American educations (US high school and University of Wisconsin Eau Claire 1972-1976).
| 55 |
Robert Watson has been a director of CEL-SCI since December 2017. Mr. Watson has been the President and CEO of Juvare, LLC (formerly Intermedix, Inc.) since July 2017. Immediately prior to joining Intermedix, now Juvare, he was the President and Chief Growth Officer of NantHealth, Inc. (Nasdaq: NH) from January 2015 to May 2017. Prior to NantHealth, he was President and CEO of Streamline Health, Inc. (Nasdaq: STRM) from January 2011 to January 2015. Mr. Watson has over 35 years of experience in the healthcare information technology industry as a CEO, board member and advisor to multiple healthcare information technology companies. He has participated in over 75 acquisitions, raised nearly $750 million in capital, completed three public offerings and successfully sold four companies. Mr. Watson holds a MBA from the Wharton School of Business at the University of Pennsylvania and a BA degree from Syracuse University.
Gail K. Naughton, Ph.D. has been a Director of CEL-SCI since August 2022. Dr. Naughton is a pioneer in the field of regenerative medicine for over 35 years. She was the founder of Advanced Tissue Sciences where she oversaw the design and development of the world’s first up-scaled manufacturing facility for cell-based products, established corporate development and marketing partnerships with companies including Smith & Nephew, Ltd., Medtronic and Inamed Corporation, was pivotal in raising over $350M from the public market and corporate partnerships, and brought four human cell-based products from concept through FDA approval and market launch. Dr. Naughton founded Histogen Inc. in 2007 and holds more than 125 U.S. and foreign patents and has been extensively published in the field. Dr. Naughton served as Dean of the College of Business Administration at San Diego State University from 2002 until 2011, where she helped to make SDSU the first US campus to establish a Ph.D./MBA in life sciences. In 2000, Dr. Naughton received the 27th Annual National Inventor of the Year award by the Intellectual Property Owners Association in honor of her pioneering work in the field of tissue engineering and regenerative medicine. Dr. Naughton received her Ph.D. and M.S. from NYU Medical Center, and an MBA from UCLA. She currently sits on the Board of directors of Therapeutics MD and is the Chair of the Board of the La Jolla Institute for Immunology.
CEL-SCI believes its directors are qualified to serve as directors for the following reasons:
Name | Reason | |
Geert Kersten, Esq. | Long standing relationship with CEL-SCI | |
Dr. Peter Young | Long standing relationship with CEL-SCI | |
Bruno Baillavoine | Long standing relationship with CEL-SCI | |
Robert Watson | Long standing relationship with CEL-SCI | |
Dr. Gail Naughton | Thirty-five years' experience in regenerative medicine |
All of CEL-SCI's officers devote substantially all of their time to CEL-SCI's business.
CEL-SCI’s Board of Directors does not have a “leadership structure”, as such, since each director is entitled to introduce resolutions to be considered by the Board and each director is entitled to one vote on any resolution considered by the Board. CEL-SCI’s Chief Executive Officer is not the Chairman of CEL-SCI’s Board of Directors.
CEL-SCI’s Board of Directors has the ultimate responsibility to evaluate and respond to risks facing CEL-SCI. CEL-SCI’s Board of Directors fulfills its obligations in this regard by meeting on a regular basis and communicating, when necessary, with CEL-SCI’s officers.
| 56 |
Dr. Peter R. Young, Bruno Baillavoine, Robert Watson, and Dr. Gail Naughton are independent directors as that term is defined in section 803 of the listing standards of the NYSE American.
CEL-SCI has adopted a Code of Ethics which is applicable to CEL-SCI’S principal executive, financial, and accounting officers and persons performing similar functions. The Code of Ethics is available on CEL-SCI’s website, located at www.cel-sci.com.
If a violation of this code of ethics act is discovered or suspected, any person (anonymously, if desired) may send a detailed note, with relevant documents, to CEL-SCI’s Audit Committee, c/o Dr. Peter Young, 208 Hewitt Drive, Suite 103-143, Waco, TX 76712.
For purposes of electing directors at its annual meeting, CEL-SCI has a nominating committee that is made up of CEL-SCI’s independent directors. The nominating committee selects the nominees to the Board of Directors which are subject to election by CEL-SCI’s shareholders.
CEL-SCI does not have any policy regarding the consideration of director candidates recommended by shareholders and under Colorado law, any shareholder can nominate a person for election as a director at the annual shareholders’ meeting. However, CEL-SCI’s Board of Directors will consider candidates recommended by shareholders. To submit a candidate for the Board of Directors the shareholder should send the name, address and telephone number of the candidate, together with any relevant background or biographical information, to CEL-SCI’s Chief Executive Officer, at the address shown on the cover page of this report. The Board has not established any specific qualifications or skills a nominee must meet to serve as a director. Although the Board does not have any process for identifying and evaluating director nominees, the Board does not believe there would be any differences in the manner in which the Board evaluates nominees submitted by shareholders as opposed to nominees submitted by any other person.
CEL-SCI does not have a policy with regard to Board member’s attendance at annual meetings. All Board members attended the last annual shareholder’s meeting held on June 13, 2022.
Holders of CEL-SCI’s common stock can send written communications to CEL-SCI’s entire Board of Directors, or to one or more Board members, by addressing the communication to “the Board of Directors” or to one or more directors, specifying the director or directors by name, and sending the communication to CEL-SCI’s offices in Vienna, Virginia. Communications addressed to the Board of Directors as whole will be delivered to each Board member. Communications addressed to a specific director (or directors) will be delivered to the director (or directors) specified.
A security holder communication not sent to the Board of Directors as a whole is not relayed to Board members which did not receive the communication.
ITEM 11.EXECUTIVE COMPENSATION
Components of Compensation — Executive Officers
CEL-SCI’s executive officers are compensated through the following three components:
| · | Base Salary |
| · | Long-Term Incentives (“LTIs”) (stock options and/or grants of stock) |
| · | Benefits |
These components provide a balanced mix of base compensation and compensation that is contingent upon each executive officer’s individual performance. A goal of the compensation program is to provide executive officers with a reasonable level of security through base salary and benefits. CEL-SCI wants to ensure that the compensation programs are appropriately designed to encourage executive officer retention and motivation to create shareholder value. The Compensation Committee believes that CEL-SCI’s stockholders are best served when CEL-SCI can attract and retain talented executives by providing compensation packages that are competitive but fair.
| 57 |
Base Salaries
Base salaries generally have been targeted to be competitive when compared to the salary levels of persons holding similar positions in other pharmaceutical companies and other publicly traded companies of comparable size. Each executive officer’s respective responsibilities, experience, expertise and individual performance are considered.
A further consideration in establishing compensation for the senior employees is their long term history with CEL-SCI. Taken into consideration are factors that have helped CEL-SCI survive in times when it was financially weak, such as: willingness to accept salary cuts, willingness not to be paid at all for extended time periods, and in general an attitude that helped CEL-SCI survive during financially difficult times.
Long-Term Incentives
Stock grants and option grants help to align the interests of CEL-SCI’s employees with those of its shareholders. Options and stock grants are made under CEL-SCI’s Stock Option, Incentive Stock Bonus, Stock Bonus and Stock Compensation Plans. Options are granted with exercise prices equal to the closing price of CEL-SCI’s common stock on the day immediately preceding the date of grant, with pro rata vesting at the end of each of the following three years.
CEL-SCI believes that grants of equity-based compensation:
| · | Enhance the link between the creation of shareholder value and long-term executive incentive compensation; |
| · | Provide focus, motivation and retention incentive; and |
| · | Provide competitive levels of total compensation. |
Benefits
In addition to cash and equity compensation programs, executive officers participate in the health and welfare benefit programs available to other employees. In a few limited circumstances, CEL-SCI provides other benefits to certain executive officers, such as car allowances.
All executive officers are eligible to participate in CEL-SCI’s 401(k) plan on the same basis as its other employees. CEL-SCI matches 100% of each employee’s contribution up to 6% of his or her salary.
| 58 |
The following table sets forth in summary form the compensation received by (i) the Chief Executive and Financial Officer of CEL-SCI and (ii) by each other executive officer of CEL-SCI who received in excess of $100,000 during the two fiscal years ended September 30, 2022.
Name and Principal Position |
| Fiscal Year |
| Salary (1) |
|
| Bonus (2) |
|
| Stock Awards (3) |
|
| Option Awards (4) |
|
| All Other Compensation (5) |
|
| Total |
| ||||||
|
|
|
| $ |
|
| $ |
|
| $ |
|
| $ |
|
| $ |
|
| $ |
| ||||||
Geert R. Kersten, |
| 2022 |
|
| 616,355 |
|
|
| -- |
|
|
| 17,400 |
|
|
| 748,458 |
|
|
| 65,631 |
|
|
| 1,447,844 |
|
Chief Executive and Financial Officer and Treasurer |
| 2021 |
|
| 580,017 |
|
|
| -- |
|
|
| 17,325 |
|
|
| 201,280 |
|
|
| 55,631 |
|
|
| 854,253 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Patricia B. Prichep, |
| 2022 |
|
| 276,999 |
|
|
| -- |
|
|
| 17,160 |
|
|
| 499,970 |
|
|
| 9,031 |
|
|
| 803,160 |
|
Senior Vice President of Operations and Secretary |
| 2021 |
|
| 261,021 |
|
|
| -- |
|
|
| 16,201 |
|
|
| 97,920 |
|
|
| 9,031 |
|
|
| 384,173 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Eyal Talor, Ph.D., |
| 2022 |
|
| 334,557 |
|
|
| -- |
|
|
| 9,600 |
|
|
| 544,877 |
|
|
| 6,031 |
|
|
| 895,065 |
|
Chief Scientific Officer |
| 2021 |
|
| 314,833 |
|
|
| -- |
|
|
| 9,600 |
|
|
| 97,920 |
|
|
| 6,031 |
|
|
| 428,384 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Daniel Zimmerman, Ph.D., |
| 2022 |
|
| 256,825 |
|
|
| -- |
|
|
| 15,769 |
|
|
| 239,506 |
|
|
| 6,031 |
|
|
| 518,131 |
|
Senior Vice President of Research, Cellular Immunology |
| 2021 |
|
| 242,978 |
|
|
| -- |
|
|
| 14,939 |
|
|
| 54,400 |
|
|
| 6,031 |
|
|
| 318,348 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
John Cipriano, |
| 2022 |
|
| 239,074 |
|
|
| -- |
|
|
| -- |
|
|
| 194,599 |
|
|
| 31 |
|
|
| 433,704 |
|
Senior Vice President of Regulatory Affairs |
| 2021 |
|
| 225,333 |
|
|
| -- |
|
|
| -- |
|
|
| 54,400 |
|
|
| 31 |
|
|
| 279,764 |
|
(1) | The dollar value of base salary (cash and non-cash) earned. |
(2) | The dollar value of bonus (cash and non-cash) earned. |
(3) | For all persons listed in the table, the shares issued as CEL-SCI's contribution on behalf of the named officer who participates in CEL-SCI's 401(k) retirement plan. The value of all stock awarded during the periods covered by the table is calculated according to ASC 718-10-30-3 which represented the grant date fair value. |
(4) | The fair value of all stock options granted during the periods covered by the table are calculated on the grant date in accordance with ASC 718-10-30-3 which represented the grant date fair value. |
(5) | All other compensation received that CEL-SCI could not properly report in any other column of the table including the dollar value of any insurance premiums paid by, or on behalf of, CEL-SCI with respect to term life insurance for the benefit of the named executive officer and car allowances paid by CEL-SCI. Includes board of directors fees for Mr. Kersten. |
Employee Pension, Profit Sharing or Other Retirement Plans
CEL-SCI has a defined contribution retirement plan, qualifying under Section 401(k) of the Internal Revenue Code and covering substantially all CEL-SCI’s employees. CEL-SCI’s contribution to the plan is made in shares of CEL-SCI's common stock. Each participant's contribution is matched by CEL-SCI with shares of common stock which have a value equal to 100% of the participant's contribution, not to exceed the lesser of $10,000 or 6% of the participant's total compensation. CEL-SCI's contribution of common stock is valued each quarter based upon the closing price of its common stock. The fiscal 2022 expenses for this plan were approximately $6,000. Other than the 401(k) Plan, CEL-SCI does not have a defined benefit, pension plan, profit sharing or other retirement plan.
Compensation of Directors During Year Ended September 30, 2022
Name |
| Fees |
|
| Stock Awards (1) |
|
| Option Awards (2) |
|
| Total |
| ||||
Geert Kersten |
| $ | 50,000 |
|
|
| - |
|
|
| 748,458 |
|
| $ | 798,458 |
|
Peter R. Young |
|
| 57,500 |
|
|
| - |
|
|
| 191,605 |
|
|
| 249,105 |
|
Bruno Baillavoine |
|
| 55,000 |
|
|
| - |
|
|
| 191,605 |
|
|
| 246,605 |
|
Robert Watson |
|
| 57,500 |
|
|
| - |
|
|
| 191,605 |
|
|
| 249,105 |
|
Gail K. Naughton |
|
| 8,152 |
|
|
| - |
|
|
| 206,084 |
|
|
| 214,236 |
|
(1) | The fair value of stock issued for services. |
(2) | The fair value of options granted computed in accordance with ASC 718-10-30-3 on the date of grant which represents their grant date fair value. |
Directors’ fees paid to Geert Kersten are included in the Executive Compensation table.
| 59 |
Employment Contracts
Geert Kersten
On August 31, 2019, CEL-SCI entered into a four-year employment agreement with Geert Kersten, CEL-SCI’s Chief Executive Officer. The employment agreement with Mr. Kersten, which is essentially the same as Mr. Kersten’s prior employment agreement, as amended on August 30, 2016, provides that, during the term of the agreement, CEL-SCI would pay Mr. Kersten an annual salary of $559,052, plus any increases in proportion to salary increases granted to other senior executive officers of CEL-SCI, as well any increases approved by the Board of Directors during the term of the employment agreement.
During the employment term, Mr. Kersten will be entitled to receive any other benefits which are provided to CEL-SCI's executive officers or other full time employees in accordance with CEL-SCI's policies and practices and subject to Mr. Kersten’s satisfaction of any applicable conditions of eligibility.
If Mr. Kersten resigns after the occurrence of any of the following events:(i) a reduction in Mr. Kersten’s salary(ii) a relocation (or demand for relocation) of Mr. Kersten’s place of employment to a location more than ten(10) miles from his current place of employment, (iii) a significant and material reduction in Mr. Kersten’s authority, job duties or level of responsibility or the imposition of significant and material limitations on Mr. Kersten’s autonomy in his position, or (iv) a Change in Control, then the employment agreement will be terminated and Mr. Kersten will be entitled to receive a lump-sum payment from CEL-SCI equal to 24 months of salary ($1,232,710) and the unvested portion of any stock options would vest immediately. For purposes of the employment agreement a change in the control of CEL-SCI means: (1) the merger of CEL-SCI with another entity if after such merger the shareholders of CEL-SCI do not own at least 50% of voting capital stock of the surviving corporation; (2) the sale of substantially all of the assets of CEL-SCI; (3) the acquisition by any person of more than 50% of CEL-SCI's common stock; or (4) a change in a majority of CEL-SCI's directors which has not been approved by the incumbent directors.
The employment agreement will also terminate upon the death of Mr. Kersten or Mr. Kersten’s physical or mental disability. If the employment agreement is terminated for any of these reasons, Mr. Kersten, or his legal representatives, as the case may be, will be paid the salary provided by the employment agreement through the date of termination, any options or bonus shares of CEL-SCI then held by Mr. Kersten will become fully vested and the expiration date of any options which would expire during the four year period following his termination of employment will be extended to the date which is four years after his termination of employment. The employment agreement will also terminate upon the willful misconduct, an act of fraud against CEL-SCI, or a breach of the employment agreement by Mr. Kersten, in which case Mr. Kersten will be paid the salary provided by the employment agreement through the date of termination.
Patricia B. Prichep / Eyal Talor, Ph.D.
On August 31, 2019, CEL-SCI entered into a four-year employment agreement with Patricia B. Prichep, CEL-SCI’s Senior Vice President of Operations. The employment agreement with Ms. Prichep, which is essentially the same as Ms. Prichep’s prior employment agreement entered into on August 30, 2016 provides that, during the term of the agreement, CEL-SCI would pay Ms. Prichep an annual salary of $245,804 plus any increases approved by the Board of Directors during the period of the employment agreement.
| 60 |
On August 31, 2019, CEL-SCI entered into a four-year employment agreement with Eyal Talor, Ph.D., CEL-SCI’s Chief Scientific Officer. The employment agreement with Dr. Talor, which is essentially the same as Dr. Talor’s prior employment agreement entered into on August 30, 2016, provides that, during the term of the agreement, CEL-SCI would pay Dr. Talor an annual salary of $303,453 plus any increases approved by the Board of Directors during the period of the employment agreement.
If Ms. Prichep or Dr. Talor resigns after the occurrence of any of the following events: (i) a relocation (or demand for relocation) of employee’s place of employment to a location more than ten (10) miles from the employee’s current place of employment, (ii) a significant and material reduction in the employee’s authority, job duties or level of responsibility, (iii) the imposition of significant and material limitations on the employee’s autonomy in her or his position, or (iv) there is a change in the control of CEL-SCI, the employment agreements with Ms. Prichep and Dr. Talor allow Ms. Prichep and/or Dr. Talor (as the case may be) to resign from her or his position at CEL-SCI and receive a lump-sum payment from CEL-SCI equal to 18 months of salary ($406,498 and $501,836 respectively). In addition, the unvested portion of any stock options held by the employee will vest immediately. For purposes of the employment agreements, a change in the control of CEL-SCI means: (1) the merger of CEL-SCI with another entity if after such merger the shareholders of CEL-SCI do not own at least 50% of voting capital stock of the surviving corporation; (2) the sale of substantially all of the assets of CEL-SCI; (3) the acquisition by any person of more than 50% of CEL-SCI's common stock; or (4) a change in a majority of CEL-SCI's directors which has not been approved by the incumbent directors.
The employment agreements with Ms. Prichep and Dr. Talor will also terminate upon the death of the employee, the employee’s physical or mental disability, willful misconduct, an act of fraud against CEL-SCI, or a breach of the employment agreement by the employee. If the employment agreement is terminated for any of these reasons the employee, or her or his legal representatives, as the case may be, will be paid the salary provided by the employment agreement through the date of termination.
Compensation Committee Interlocks and Insider Participation
CEL-SCI has a compensation committee comprised of Mr. Bruno Baillavoine, Dr. Peter Young and Mr. Robert Watson, all of whom are independent directors.
During the year ended September 30, 2022, no director of CEL-SCI was also an executive officer of another entity which had an executive officer of CEL-SCI serving as a director of such entity or as a member of the compensation committee of such entity.
Stock Option, Bonus and Compensation Plans
CEL-SCI has Incentive Stock Option Plans, Non-Qualified Stock Option Plans, Stock Bonus Plans, Stock Compensation Plans and an Incentive Stock Bonus Plan. All Stock Option, Bonus and Compensation Plans have been approved by the stockholders. A summary description of these Plans follows. In some cases these Plans are collectively referred to as the "Plans".
Incentive Stock Option Plans. The Incentive Stock Option Plans authorize the issuance of shares of CEL-SCI's common stock to persons who exercise options granted pursuant to the Plans. Only CEL-SCI’s employees may be granted options pursuant to the Incentive Stock Option Plans.
Options may not be exercised until one year following the date of grant. Options granted to an employee then owning more than 10% of the common stock of CEL-SCI may not be exercisable by its terms after five years from the date of grant. Any other option granted pursuant to the Plans may not be exercisable by its terms after ten years from the date of grant.
| 61 |
The option exercise price is determined by CEL-SCI’s Compensation Committee but cannot be less than the fair market value of the common stock on the date of the grant of the option (or 110% of the fair market value in the case of a person owning more than 10% of CEL-SCI's outstanding shares).
Non-Qualified Stock Option Plans. The Non-Qualified Stock Option Plans authorize the issuance of shares of CEL-SCI's common stock to persons that exercise options granted pursuant to the Plans. CEL-SCI's employees, directors, officers, consultants and advisors are eligible to be granted options pursuant to the Plans, provided however that bona fide services must be rendered by such consultants or advisors and such services must not be in connection with a capital-raising transaction or promoting CEL-SCI’s common stock. The option exercise price is determined by CEL-SCI’s Compensation Committee.
Stock Bonus Plans. Under the Stock Bonus Plans, shares of CEL-SCI’s common stock may be issued to CEL-SCI's employees, directors, officers, consultants and advisors, provided however that bona fide services must be rendered by consultants or advisors and such services must not be in connection with a capital-raising transaction or promoting CEL-SCI’s common stock.
Stock Compensation Plans. Under the Stock Compensation Plans, shares of CEL-SCI’s common stock may be issued to CEL-SCI’s employees, directors, officers, consultants and advisors in payment of salaries, fees and other compensation owed to these persons. However, bona fide services must be rendered by consultants or advisors and such services must not be in connection with a capital-raising transaction or promoting CEL-SCI’s common stock.
Incentive Stock Bonus Plan. Under the 2014 Incentive Stock Bonus Plan, shares of CEL-SCI’s common stock may be issued to executive officers and other employees who contribute significantly to the success of CEL-SCI. The purpose of the Plan is to provide long term incentive for outstanding service to CEL-SCI and its shareholders, to assist in recruiting and retaining people of outstanding ability and initiative in executive and management positions to allow officers and employees to participate in CEL-SCI's future prosperity and growth, and to align the interests of CEL-SCI's officers and employees with those of its shareholders.
Other Information Regarding the Plans. The Plans are administered by CEL-SCI's Compensation Committee (“the Committee”), each member of which is a director of CEL-SCI. The members of the Committee were selected by CEL-SCI's Board of Directors and serve for a one-year tenure and until their successors are elected. A member of the Committee may be removed at any time by action of the Board of Directors. Any vacancies which may occur on the Committee will be filled by the Board of Directors. The Committee is vested with the authority to interpret the provisions of the Plans and supervise the administration of the Plans. In addition, the Committee is empowered to select those persons to whom shares or options are to be granted, to determine the number of shares subject to each grant of a stock bonus or an option and to determine when, and upon what conditions, shares or options granted under the Plans will vest or otherwise be subject to forfeiture and cancellation.
In the discretion of the Committee, any option granted pursuant to the Plans may include installment exercise terms such that the option becomes fully exercisable in a series of cumulating portions. The Committee may also accelerate the date upon which any option (or any part of any options) is first exercisable. Any shares issued pursuant to the Stock Bonus Plans or Stock Compensation Plans and any options granted pursuant to the Incentive Stock Option Plans or the Non-Qualified Stock Option Plans will be forfeited if the "vesting" schedule established by the Committee administering the Plans at the time of the grant is not met. For this purpose, vesting means the period during which the employee must remain an employee of CEL-SCI or the period of time a non-employee must provide services to CEL-SCI. At the time an employee ceases working for CEL-SCI (or at the time a non-employee ceases to perform services for CEL-SCI), any shares or options not fully vested will be forfeited and cancelled. At the discretion of the Committee payment for the shares of common stock underlying options may be paid through the delivery of shares of CEL-SCI's common stock having an aggregate fair market value equal to the option price, provided such shares have been owned by the option holder for at least one year prior to such exercise. A combination of cash and shares of common stock may also be permitted at the discretion of the Committee.
| 62 |
Options are generally non-transferable except upon the death of the option holder. Shares issued pursuant to the Stock Bonus Plans will generally not be transferable until the person receiving the shares satisfies the vesting requirements imposed by the Committee when the shares were issued.
The Board of Directors of CEL-SCI may at any time, and from time to time, amend, terminate, or suspend one or more of the Plans in any manner it deems appropriate, provided that such amendment, termination or suspension may not adversely affect rights or obligations with respect to shares or options previously granted.
During fiscal year ended September 30, 2022, CEL-SCI granted 250,000 performance-based stock options from the 2020 Non-Qualified Stock Option Plan to officers. Each option entitles the holder to purchase one share of CEL-SCI’s common stock at a price of $10.48 per share, the fair value on the date of issuance. The stock options will vest 100% upon approval of the first marketing application for any pharmaceutical based upon CEL-SCI’s Multikine technology in any of the USA, Canada, UK, Germany, France, Italy, Spain, Japan, or Australia. None of the options will be exercisable before November 19, 2022. All options which have not vested as of November 18, 2031 will be canceled.
Stock Options
The following table shows information concerning the options granted during the fiscal year ended September 30, 2022 to the persons named below:
Options Granted
Name |
| Grant Date |
| Options Granted |
|
| Price Per Share |
|
| Expiration Date | |||
Geert Kersten |
| 11/19/2021 |
|
| 50,000 |
|
| $ | 10.48 |
|
| 11/18/2031 | |
|
| 6/13/2022 |
|
| 250,000 |
|
| $ | 3.35 |
|
| 6/12/2032 | |
|
|
|
|
|
|
|
|
|
|
|
|
| |
Patricia Prichep |
| 11/19/2021 |
|
| 50,000 |
|
| $ | 10.48 |
|
| 11/18/2031 | |
|
| 6/13/2022 |
|
| 167,000 |
|
| $ | 3.35 |
|
| 6/12/2032 | |
|
|
|
|
|
|
|
|
|
|
|
|
| |
Eyal Talor |
| 11/19/2021 |
|
| 50,000 |
|
| $ | 10.48 |
|
| 11/18/2031 | |
|
| 6/13/2022 |
|
| 182,000 |
|
| $ | 3.35 |
|
| 6/12/2032 | |
|
|
|
|
|
|
|
|
|
|
|
|
| |
Dan Zimmerman |
| 11/19/2021 |
|
| 50,000 |
|
| $ | 10.48 |
|
| 11/18/2031 | |
|
| 6/13/2022 |
|
| 80,000 |
|
| $ | 3.35 |
|
| 6/12/2032 | |
|
|
|
|
|
|
|
|
|
|
|
|
| |
John Cipriano |
| 11/19/2021 |
|
| 50,000 |
|
| $ | 10.48 |
|
| 11/18/2031 | |
|
| 6/13/2022 |
|
| 65,000 |
|
| $ | 3.35 |
|
| 6/12/2032 | |
|
|
|
|
|
|
|
|
|
|
|
|
| |
Bruno Baillavoine |
| 6/13/2022 |
|
| 64,000 |
|
| $ | 3.35 |
|
| 6/12/2032 | |
|
|
|
|
|
|
|
|
|
|
|
|
| |
Peter Young |
| 6/13/2022 |
|
| 64,000 |
|
| $ | 3.35 |
|
| 6/12/2032 | |
|
|
|
|
|
|
|
|
|
|
|
|
| |
Robert Watson |
| 6/13/2022 |
|
| 64,000 |
|
| $ | 3.35 |
|
| 6/12/2032 | |
|
|
|
|
|
|
|
|
|
|
|
|
| |
Gail Naughton |
| 8/2/2022 |
|
| 64,000 |
|
| $ | 3.61 |
|
| 8/1/2032 | |
| 63 |
The following table shows the outstanding options held by the persons named below on September 30, 2022:
|
| Shares underlying unexercised |
|
|
|
|
|
| |||||||
|
| Option which are: |
|
| Exercise |
|
| Expiration | |||||||
Name |
| Exercisable |
|
| Unexercisable |
|
| Price |
|
| Date | ||||
Geert R. Kersten |
|
| 20,000 |
|
|
|
|
|
| 70.00 |
|
| 12/17/22 | ||
|
|
| 1,800 |
|
|
|
|
|
| 52.50 |
|
| 06/30/23 | ||
|
|
| 3,600 |
|
|
|
|
|
| 27.25 |
|
| 02/25/24 | ||
|
|
| 180,000 |
|
|
|
|
|
| 2.18 |
|
| 07/27/27 | ||
|
|
| 530,121 |
|
|
|
|
|
| 2.45 |
|
| 04/30/28 | ||
|
|
| 813,180 |
|
|
|
|
|
| 5.65 |
|
| 04/10/29 | ||
|
|
| 148,000 |
|
|
| 444,000 |
|
|
| 10.93 |
|
| 04/19/30 | |
|
|
| - |
|
|
| 592,000 |
|
|
| 20.61 |
|
| 05/13/31 | |
|
|
| - |
|
|
| 50,000 |
|
|
| 10.48 |
|
| 11/18/31 | |
|
|
| - |
|
|
| 250,000 |
|
|
| 3.35 |
|
| 06/12/32 | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
Patricia B. Prichep |
|
| 6,000 |
|
|
|
|
|
|
| 70.00 |
|
| 12/17/22 | |
|
|
| 1,200 |
|
|
|
|
|
|
| 52.50 |
|
| 06/30/23 | |
|
|
| 2,400 |
|
|
|
|
|
|
| 27.25 |
|
| 02/25/24 | |
|
|
| 120,000 |
|
|
|
|
|
|
| 2.18 |
|
| 07/27/27 | |
|
|
| 222,607 |
|
|
|
|
|
|
| 2.45 |
|
| 04/30/28 | |
|
|
| 405,631 |
|
|
|
|
|
|
| 5.65 |
|
| 04/10/29 | |
|
|
| 72,000 |
|
|
| 216,000 |
|
|
| 10.93 |
|
| 04/19/30 | |
|
|
| - |
|
|
| 288,000 |
|
|
| 20.61 |
|
| 05/13/31 | |
|
|
| - |
|
|
| 50,000 |
|
|
| 10.48 |
|
| 11/18/31 | |
|
|
| - |
|
|
| 167,000 |
|
|
| 3.35 |
|
| 06/12/32 | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
Eyal Talor, Ph.D |
|
| 6,000 |
|
|
|
|
|
|
| 70.00 |
|
| 12/17/22 | |
|
|
| 1,200 |
|
|
|
|
|
|
| 52.50 |
|
| 06/30/23 | |
|
|
| 2,400 |
|
|
|
|
|
|
| 27.25 |
|
| 02/25/24 | |
|
|
| 120,000 |
|
|
|
|
|
|
| 2.18 |
|
| 07/27/27 | |
|
|
| 193,934 |
|
|
|
|
|
|
| 2.45 |
|
| 04/30/28 | |
|
|
| 100,000 |
|
|
|
|
|
|
| 3.55 |
|
| 09/20/28 | |
|
|
| 382,712 |
|
|
|
|
|
|
| 5.65 |
|
| 04/10/29 | |
|
|
| 72,000 |
|
|
| 216,000 |
|
|
| 10.93 |
|
| 04/19/30 | |
|
|
| - |
|
|
| 318,000 |
|
|
| 20.61 |
|
| 05/13/31 | |
|
|
| - |
|
|
| 50,000 |
|
|
| 10.48 |
|
| 11/18/31 | |
|
|
| - |
|
|
| 182,000 |
|
|
| 3.35 |
|
| 06/12/32 | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
Dan Zimmerman, Ph.D |
|
| 900 |
|
|
|
|
|
|
| 52.50 |
|
| 06/30/23 | |
|
|
| 1,800 |
|
|
|
|
|
|
| 27.25 |
|
| 02/25/24 | |
|
|
| 8,000 |
|
|
|
|
|
|
| 27.50 |
|
| 08/05/24 | |
|
|
| 4,000 |
|
|
|
|
|
|
| 15.50 |
|
| 06/25/25 | |
|
|
| 4,000 |
|
|
|
|
|
|
| 11.75 |
|
| 07/21/26 | |
|
|
| 6,000 |
|
|
|
|
|
|
| 1.87 |
|
| 06/28/27 | |
|
|
| 20,000 |
|
|
|
|
|
|
| 1.59 |
|
| 09/17/27 | |
|
|
| 126,194 |
|
|
|
|
|
|
| 2.45 |
|
| 04/30/28 | |
|
|
| 211,459 |
|
|
|
|
|
|
| 5.65 |
|
| 04/10/29 | |
|
|
| 40,000 |
|
|
| 120,000 |
|
|
| 10.93 |
|
| 04/19/30 | |
|
|
| - |
|
|
| 160,000 |
|
|
| 20.61 |
|
| 05/13/31 | |
|
|
| - |
|
|
| 50,000 |
|
|
| 10.48 |
|
| 11/18/31 | |
|
|
| - |
|
|
| 80,000 |
|
|
| 3.35 |
|
| 06/12/32 | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
John Cipriano |
|
| 900 |
|
|
|
|
|
|
| 57.50 |
|
| 06/30/23 | |
|
|
| 1,800 |
|
|
|
|
|
|
| 27.25 |
|
| 02/25/24 | |
|
|
| 90,000 |
|
|
|
|
|
|
| 2.18 |
|
| 07/27/27 | |
|
|
| 148,347 |
|
|
|
|
|
|
| 2.45 |
|
| 04/30/28 | |
|
|
| 206,880 |
|
|
|
|
|
|
| 5.65 |
|
| 04/10/29 | |
|
|
| 40,000 |
|
|
| 120,000 |
|
|
| 10.93 |
|
| 04/19/30 | |
|
|
| - |
|
|
| 130,000 |
|
|
| 20.61 |
|
| 05/13/31 | |
|
|
| - |
|
|
| 50,000 |
|
|
| 10.48 |
|
| 11/18/31 | |
|
|
| - |
|
|
| 65,000 |
|
|
| 3.35 |
|
| 06/12/32 | |
| 64 |
Summary. The following shows certain information as of September 30, 2022 concerning the stock options and stock bonuses granted by CEL-SCI. Each option represents the right to purchase one share of CEL-SCI's common stock.
Name of Plan |
| Total Shares Reserved Under Plans |
|
| Shares Reserved for Outstanding Options |
|
| Shares Issued |
|
| Remaining Options/Shares Under Plans |
| ||||
Incentive Stock Option Plans |
|
| 138,400 |
|
|
| 75,329 |
|
|
| N/A |
|
|
| 213 |
|
Non-Qualified Stock Option Plans |
|
| 13,787,200 |
|
|
| 12,888,685 |
|
|
| N/A |
|
|
| 476,173 |
|
Stock Bonus Plans |
|
| 783,760 |
|
|
| N/A |
|
|
| 415,968 |
|
|
| 367,759 |
|
Stock Compensation Plans |
|
| 634,000 |
|
|
| N/A |
|
|
| 153,195 |
|
|
| 462,395 |
|
Incentive Stock Bonus Plan |
|
| 640,000 |
|
|
| N/A |
|
|
| 614,500 |
|
|
| 25,500 |
|
Of the shares issued pursuant to CEL-SCI's Stock Bonus Plans, 333,646 shares were issued as part of CEL-SCI's contribution to its 401(k) plan.
The following table shows the weighted average exercise price of the outstanding options granted pursuant to CEL-SCI’s Incentive and Non-Qualified Stock Option Plans as of September 30, 2022, CEL-SCI’s most recent fiscal year end. CEL-SCI's Incentive and Non-Qualified Stock Option Plans have been approved by CEL-SCI's shareholders.
Plan category |
| Number of Securities to be Issued Upon Exercise of Outstanding Options (a) |
|
| Weighted-Average Exercise Price of Outstanding Options |
|
| Number of Securities Remaining Available For Future Issuance Under Equity Compensation Plans, Excluding Securities Reflected in Column (a) |
| |||
Incentive Stock Option Plans |
|
| 75,329 |
|
| $ | 29.27 |
|
|
| 213 |
|
Non-Qualified Stock Option Plans |
|
| 12,888,685 |
|
| $ | 8.94 |
|
|
| 476,173 |
|
| 65 |
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table shows, as of December 1, 2022, information with respect to the only persons owning beneficially 5% or more of CEL-SCI’s outstanding common stock and the number and percentage of outstanding shares owned by each director and officer of CEL-SCI and by the officers and directors as a group. Unless otherwise indicated, each owner has sole voting and investment powers over his shares of common stock.
Name and Address |
| Number of Shares (1) |
|
| Percent of Class (2) |
| ||
Geert R. Kersten 8229 Boone Blvd., Suite 802 Vienna, VA 22182 |
|
| 3,844,893 | (3) |
|
| 8.3 | % |
|
|
|
|
|
|
|
|
|
Patricia B. Prichep 8229 Boone Blvd., Suite 802 Vienna, VA 22182 |
|
| 1,060,014 |
|
|
| 2.4 | % |
|
|
|
|
|
|
|
|
|
Eyal Talor, Ph.D. 8229 Boone Blvd., Suite 802 Vienna, VA 22182 |
|
| 985,402 |
|
|
| 2.2 | % |
|
|
|
|
|
|
|
|
|
Daniel H. Zimmerman, Ph.D. 8229 Boone Blvd., Suite 802 Vienna, VA 22182 |
|
| 542,579 |
|
|
| 1.2 | % |
|
|
|
|
|
|
|
|
|
John Cipriano 8229 Boone Blvd., Suite 802 Vienna, VA 22182 |
|
| 551,035 |
|
|
| 1.2 | % |
|
|
|
|
|
|
|
|
|
Peter R. Young, Ph.D. 208 Hewitt Drive, Suite 103-143 Waco, TX 76712 |
|
| 347,114 |
|
| * |
| |
|
|
|
|
|
|
|
|
|
Bruno Baillavoine 8229 Boone Blvd., Suite 802 Vienna, VA 22182 |
|
| 283,473 |
|
| * |
| |
|
|
|
|
|
|
|
|
|
Robert Watson 245 N. Highland Ave. NE Suite 230-296 Atlanta, GA 30307 |
|
| 254,431 |
|
| * |
| |
|
|
|
|
|
|
|
|
|
Gail Naughton, Ph.D. 8229 Boone Blvd., Suite 802 Vienna, VA 22182 |
|
| - |
|
| * |
| |
|
|
|
|
|
|
|
|
|
All Officers and Directors as a Group (9 persons) |
|
| 7,868,941 |
|
|
| 15.78 | % |
| * | Less than 1% |
(1) | Includes shares issuable prior to February 28, 2023 upon the exercise of options or warrants granted to the following persons: |
| 66 |
Name |
| Options or Warrants Exercisable Prior to February 28, 2023 |
| |
Geert R. Kersten, Esq. |
|
| 2,694,109 | (3) |
Patricia B. Prichep |
|
| 848,397 |
|
Eyal Talor, Ph.D. |
|
| 878,246 |
|
Daniel Zimmerman, Ph.D. |
|
| 422,353 |
|
John Cipriano |
|
| 487,927 |
|
Peter R. Young, Ph.D. |
|
| 318,200 |
|
Bruno Baillavoine |
|
| 277,500 |
|
Robert Watson |
|
| 250,000 |
|
Gail Naughton, Ph.D. |
|
| - |
|
(2) | Amount includes shares referred to in (1) above but excludes shares which may be issued upon the exercise of options and warrants previously issued by CEL-SCI. |
(3) | Amount includes 346,421 shares and 390,928 warrants held in the de Clara Trust, of which Mr. Kersten is a trustee and beneficiary. |
ITEM 13.CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
During the year ended September 30, 2022, no shares of the Company’s common stock were purchased by officers or directors. During the year ended September 30, 2021, officers and a director of the Company purchased 27,500 restricted shares of the Company’s common stock at an aggregate market value of approximately $220,000. The shares are subject to the conditions of Rule 144 under the Securities Act of 1933.
See Note 11 to the financial statements included as part of this report for additional information concerning related party transactions.
ITEM 14.PRINCIPAL ACCOUNTING FEES AND SERVICES
BDO USA, LLP served as CEL-SCI’s independent registered public accountant for the two years ended September 30, 2022. The following table shows the aggregate fees billed to CEL-SCI for these years by BDO USA, LLP:
|
| Year Ended September 30, |
| |||||
|
| 2022 |
|
| 2021 |
| ||
|
|
|
|
|
|
| ||
Audit Fees |
| $ | 358,899 |
|
| $ | 391,240 |
|
Audit Related Fees |
|
| - |
|
|
| - |
|
Tax Fees |
|
| - |
|
|
| - |
|
All Other Fees |
|
| - |
|
|
| - |
|
Audit fees represent amounts billed for professional services rendered for the audit of CEL-SCI’s annual financial statements and the reviews of the interim financial statements included in CEL-SCI’s 10-Q reports for the fiscal year and all regulatory filings.
Before BDO USA, LLP was engaged by CEL-SCI to render audit or non-audit services, the engagement was approved by CEL-SCI’s audit committee. CEL-SCI’s Board of Directors is of the opinion that the audit fees charged by BDO USA, LLP are consistent with BDO USA, LLP maintaining its independence from CEL-SCI.
| 67 |
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
Exhibits |
|
|
|
|
3(a) |
| Articles of Incorporation |
| Incorporated by reference to Exhibit 3(a) of CEL-SCI's combined Registration Statement on Form S-1 and Post-Effective Amendment ("Registration Statement"), Registration Nos. 2-85547-D and 33-7531. |
|
|
|
|
|
3(b) |
| Amended Articles |
| Incorporated by reference to Exhibit 3(a) of CEL-SCI's Registration Statement on Form S-1, Registration Nos. 2-85547-D and 33-7531. |
|
|
|
|
|
3(c) |
| Amended Articles (Name change only) |
| Incorporated by reference to Exhibit 3(c) of CEL-SCI's Registration Statement on Form S-1 Registration Statement (No. 33-34878). |
|
|
|
|
|
3(d) |
| Bylaws (as amended) |
| Incorporated by reference to Exhibit 3(d) of CEL-SCI's Post-Effective Amendment No. 3 to Registration Statement on Form S-1 (No. 333-229295). |
|
|
|
|
|
|
| Incorporated by reference to Exhibit 4 filed with CEL-SCI’s 8-K report dated October 30, 2020. | ||
|
|
|
|
|
|
| Incorporated by reference to Exhibit 4 (b) filed on October 7, 2016 with CEL-SCI’s registration statement on Form S¬8 (File number 333-214031). | ||
|
|
|
|
|
|
| Incorporated by reference to Exhibit 4 (b) filed on July 1, 2022 with CEL-SCI’s registration statement on Form S¬8 (File number 333-265994). | ||
|
|
|
|
|
|
| Incorporated by reference to Exhibit 4 (c) filed on February 9, 2018 with CEL-SCI’s registration statement on Form S¬8 (File number 333-222969). | ||
|
|
|
|
|
|
| Incorporated by reference to Exhibit 4 (c) filed on January 24, 2020 with CEL-SCI’s registration statement on Form S¬8 (File number 333-236063). | ||
|
|
|
|
|
|
| Filed with Amendment No. 2 to CEL-SCI’s annual report on Form 10-K for the year ended September 30, 2014. | ||
|
|
|
|
|
| First Amendment to Development Supply and Distribution Agreement with Orient Europharma. |
| Incorporated by reference to Exhibit 10(m) filed with CEL-SCI’s 10-K report for the year ended September 30, 2010. | |
|
|
|
|
|
| Exclusive License and Distribution Agreement with Teva Pharmaceutical Industries Ltd. |
| Incorporated by reference to Exhibit 10(n) filed with CEL-SCI’s 10-K report for the year ended September 30, 2010. | |
|
|
|
|
|
|
| Incorporated by reference to Exhibit 10(o) filed with CEL-SCI’s 10-K report for the year ended September 30, 2010. | ||
|
|
|
|
|
10(p) |
| Licensing Agreement with Byron Biopharma |
| Incorporated by reference to Exhibit 10(i) of CEL-SCI’s report on Form 8-K dated March 27, 2009 |
|
|
|
|
|
| Development, Supply and Distribution Agreement with Orient Europharma |
| Incorporated by reference to Exhibit 10(z) filed with CEL-SCI’s report on Form 10-K for the year ended September 30, 2003. | |
|
|
| ||
|
| Incorporated by reference to Exhibit 10(rr) of CEL-SCI’s report on Form 10-K/A report for the year ended September 30, 2014 dated April 17, 2015. |
| 68 |
| Service Agreement with GCP Clinical Studies, Ltd., together with Amendment 1 thereto* |
| Incorporated by reference to Exhibit 10(ss) of CEL-SCI’s first amendment to its Form 10-K report for the year ended September 30, 2014 dated April 17, 2015. | |
|
|
|
|
|
|
| Incorporated by reference to Exhibit 10(tt) of CEL-SCI’s first amendment to its Form 10-K report for the year ended September 30, 2014 dated April 17, 2015. | ||
|
|
|
|
|
| Master Service Agreement with Ergomed Clinical Research, Ltd., and Clinical Trial Orders thereunder |
| Incorporated by reference to Exhibit 10(uu) of CEL-SCI’s first amendment to its Form 10-K report for the year ended September 30, 2014 dated April 17, 2015. | |
|
|
|
|
|
|
| Incorporated by reference to Exhibit 10(vv) of CEL-SCI’s first amendment to its Form 10-K report for the year ended September 30, 2014 dated April 17, 2015. | ||
|
|
|
|
|
|
| Incorporated by reference to Exhibit 10(ww) of CEL-SCI’s first amendment to its Form 10-K report for the year ended September 30, 2014 dated April 17, 2015. | ||
|
|
|
|
|
|
| Incorporated by reference to Exhibit 10(xx) of CEL-SCI’s first amendment to its Form 10-K report for the year ended September 30, 2014 dated April 17, 2015. | ||
|
|
|
|
|
|
| Incorporated by reference to Exhibit 10(yy) of CEL-SCI’s first amendment to its Form 10-K report for the year ended September 30, 2014 dated April 17, 2015. | ||
|
|
|
|
|
| Project Agreement Number 1 with Aptiv Solutions, Inc. together with Amendments 1 and 2 thereto* |
| Incorporated by reference to Exhibit 10(zz) of CEL-SCI’s first amendment to its Form 10-K report for the year ended September 30, 2014 dated April 17, 2015. | |
|
|
|
|
|
| Second Amendment to Development Supply and Distribution Agreement with Orient Europharma |
| Incorporated by reference to Exhibit 10(aaa) of CEL-SCI’s first amendment to its Form 10-K report for the year ended September 30, 2014 dated April 17, 2015. | |
|
|
|
|
|
|
| Incorporated by reference to Exhibit 10 (iii) filed with CEL-SCI’s 10-K report for the year ended September 30, 2015. | ||
|
|
|
|
|
|
| Incorporated by reference to Exhibit 10(10.7) of CEL-SCI’s report on Form 8-K dated August 31, 2019. | ||
|
|
|
|
|
|
| Incorporated by reference to Exhibit 10(10.8) of CEL-SCI’s report on Form 8-K dated August 31, 2019. | ||
|
|
|
|
|
|
| Incorporated by reference to Exhibit 10(10.9) of CEL-SCI’s report on Form 8-K dated August 31, 2019. | ||
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
* Portions of this exhibit have been omitted pursuant to a request for confidential treatment filed with the Commission under Rule 24b-2 of the Securities Exchange Act of 1934. The omitted confidential material has been filed separately with the Commission. The location of the omitted confidential information is indicated in the exhibit with asterisks (*)
| 69 |
| CEL-SCI CORPORATION
Financial Statements for the Years Ended September 30, 2022 and 2021, and Report of Independent Registered Public Accounting Firm |
70 |
CEL-SCI CORPORATION
TABLE OF CONTENTS |
Page | ||||
F-2 | ||||
FINANCIAL STATEMENTS FOR THE YEARS ENDED SEPTEMBER 30, 2022 and 2021: | ||||
F-4 | ||||
F-5 | ||||
F-6 | ||||
F-7 | ||||
F-9 | ||||
| F-1 |
| Table of Contents |
Report of Independent Registered Public Accounting Firm
Stockholders and Board of Directors
CEL-SCI Corporation
Vienna, Virginia
Opinion on the Financial Statements
We have audited the accompanying balance sheets of CEL-SCI Corporation (the “Company”) as of September 30, 2022 and 2021, the related statements of operations, stockholders’ equity, and cash flows for each of the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company at September 30, 2022 and 2021, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern Uncertainty
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has suffered recurring losses from operations and has future liquidity needs that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
| F-2 |
| Table of Contents |
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Evaluation of Liquidity
As described in Note 2 to the financial statements, the Company has experienced recurring losses. The Company expects to continue incurring losses for the foreseeable future. Further, the Company has spent, and expects to continue to spend, a substantial amount of funds in connection with implementing its business strategy, including planned product development efforts, clinical trials and research and discovery efforts. The Company is dependent on its ability to raise additional funding from the capital markets in order to continue to fund its operations.
We identified management’s evaluation of the Company’s liquidity as a critical audit matter due to the significant judgments and assumptions used by management in (i) preparing its forecast of cash expenditures to support the Company’s drug development and clinical trials, and (ii) providing complete and accurate disclosures related to the Company’s liquidity. Auditing these judgments and assumptions involved especially challenging auditor judgment due to the nature and extent of audit effort required to address these matters.
The primary procedures we performed to address this critical audit matter included:
| · | Assessing the reasonableness of management’s key assumptions in forecasting cash expenditures by comparing the key assumptions to (i) historical forecasts and (ii) the Company’s ongoing drug development pipeline. |
|
|
|
| · | Testing the completeness and accuracy of underlying data used in the forecasted cash expenditures by (i) inspecting contractual arrangements with third-party clinical research organizations and suppliers, and (ii) considering current and past expenditures in evaluating the forecasted fixed and variable costs. |
|
|
|
| · | Evaluating the adequacy of management’s disclosure in the financial statements regarding the Company’s liquidity by comparing the disclosure to other audit evidence obtained to determine whether such evidence is consistent with or contradictory to the Company’s liquidity disclosure. |
/s/
We have served as the Company's auditor since 2005.
December 27, 2022
| F-3 |
| Table of Contents |
CEL-SCI CORPORATION
BALANCE SHEETS
SEPTEMBER 30, 2022 AND 2021
ASSETS |
| 2022 |
|
| 2021 |
| ||
Current assets: |
|
|
|
|
|
| ||
Cash and cash equivalents |
| $ |
|
| $ |
| ||
U.S. Treasury Bills |
|
|
|
|
|
| ||
Receivables |
|
|
|
|
|
| ||
Prepaid expenses |
|
|
|
|
|
| ||
Supplies used for R&D and manufacturing |
|
|
|
|
|
| ||
Total current assets |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
Finance lease right of use assets |
|
|
|
|
|
| ||
Operating lease right of use assets |
|
|
|
|
|
| ||
Property and equipment, net |
|
|
|
|
|
| ||
Patent costs, net |
|
|
|
|
|
| ||
Deposits |
|
|
|
|
|
| ||
Supplies used for R&D and manufacturing |
|
|
|
|
|
| ||
Total assets |
| $ |
|
| $ |
| ||
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS' EQUITY |
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
Accounts payable |
| $ |
|
| $ |
| ||
Accrued expenses |
|
|
|
|
|
| ||
Due to employees |
|
|
|
|
|
| ||
Derivative instruments, current portion |
|
|
|
|
|
| ||
Lease liabilities, current portion |
|
|
|
|
|
| ||
Total current liabilities |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
Finance lease liabilities, net of current portion |
|
|
|
|
|
| ||
Operating lease liabilities, net of current portion |
|
|
|
|
|
| ||
Other liabilities |
|
|
|
|
|
| ||
Total liabilities |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
STOCKHOLDERS' EQUITY |
|
|
|
|
|
|
|
|
Common stock, $ |
|
|
|
|
|
| ||
Additional paid-in capital |
|
|
|
|
|
| ||
Accumulated deficit |
|
| ( | ) |
|
| ( | ) |
Total stockholders' equity |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
TOTAL LIABILITIES AND STOCKHOLDERS' EQUITY |
| $ |
|
| $ |
| ||
| F-4 |
| Table of Contents |
CEL-SCI CORPORATION
STATEMENTS OF OPERATIONS
YEARS ENDED SEPTEMBER 30, 2022 AND 2021
|
| 2022 |
|
| 2021 |
| ||
|
|
|
|
|
|
|
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
Research and development |
| $ |
|
| $ |
| ||
General & administrative |
|
|
|
|
|
| ||
Total operating expenses |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
Operating loss |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
Other income (expense) |
|
|
|
|
| ( | ) | |
Gain (loss) on derivative instruments |
|
|
|
|
| ( | ) | |
Other non-operating (loss) gain |
|
| ( | ) |
|
|
| |
Interest expense, net |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
Net loss |
| $ | ( | ) |
| $ | ( | ) |
Modification of warrants |
|
| ( | ) |
|
| ( | ) |
Net loss available to common shareholders |
| $ | ( | ) |
| $ | ( | ) |
|
|
|
|
|
|
|
|
|
Net loss per common share - basic |
| $ | ( | ) |
| $ | ( | ) |
Weighted average common shares outstanding - basic |
| $ |
|
| $ |
| ||
|
|
|
|
|
|
|
|
|
Net loss per common share - diluted |
| $ | ( | ) |
| $ | ( | ) |
Weighted average common shares outstanding - diluted |
| $ |
|
| $ |
| ||
| F-5 |
| Table of Contents |
CEL-SCI CORPORATION
STATEMENT OF STOCKHOLDERS' EQUITY
YEAR ENDED SEPTEMBER 30, 2022
|
|
|
|
|
|
|
| Additional |
|
|
|
|
|
|
| |||||
|
| Common |
|
| Stock |
|
| Paid-In |
|
| Accumulated |
|
|
|
| |||||
|
| Shares |
|
| Amount |
|
| Capital |
|
| Deficit |
|
| Total |
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
BALANCE, SEPTEMBER 30, 2020 |
|
|
|
| $ |
|
| $ |
|
| $ | ( | ) |
| $ |
| ||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from the sale of common stock |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Warrant exercises |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Modification of warrants |
|
| - |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
401(k) contributions paid in common stock |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Stock issued to nonemployees for service |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Equity-based compensation - employees |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
| |||
Option exercises |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Purchase of stock by officers and directors |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Share issuance costs |
|
| - |
|
|
|
|
|
| ( | ) |
|
|
|
|
| ( | ) | ||
Net loss |
|
| - |
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BALANCE, SEPTEMBER 30, 2021 |
|
|
|
| $ |
|
| $ |
|
| $ | ( | ) |
| $ |
| ||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Warrant exercises |
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
| ||||
401(k) contributions paid in common stock |
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
| ||||
Stock issued to nonemployees for service |
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
| ||||
Equity-based compensation - employees |
|
| - |
|
|
| - |
|
|
|
|
|
| |
|
|
|
| ||
Option exercises |
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
| ||||
Share issuance costs |
|
| - |
|
|
| |
|
|
| ( | ) |
|
| |
|
|
| ( | ) |
Net loss |
|
| - |
|
|
| |
|
|
| |
|
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BALANCE, SEPTEMBER 30, 2022 |
|
|
|
| $ |
|
| $ |
|
| $ | ( | ) |
| $ |
| ||||
| F-6 |
| Table of Contents |
CEL-SCI CORPORATION
STATEMENTS OF CASH FLOWS
YEARS ENDED SEPTEMBER 30, 2022 AND 2021
|
|
|
|
|
|
| ||
|
| 2022 |
|
| 2021 |
| ||
CASH FLOWS FROM OPERATING ACTIVITIES: |
|
|
|
|
|
| ||
Net loss |
| $ | ( | ) |
| $ | ( | ) |
Adjustments to reconcile net loss to |
|
|
|
|
|
|
|
|
net cash used in operating activities: |
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
|
|
|
| ||
Non-cash lease expense |
|
| |
|
|
| |
|
Share-based payments for services |
|
|
|
|
|
| ||
Equity-based compensation |
|
|
|
|
|
| ||
Common stock contributed to 401(k) plan |
|
|
|
|
|
| ||
Gain on short term investments |
|
| ( | ) |
|
| ( | ) |
(Gain) loss on derivative instruments |
|
| ( | ) |
|
|
| |
Modification of warrants |
|
|
|
|
|
| ||
Impairment loss on abandonment of patents |
|
|
|
|
|
| ||
(Increase)/decrease in assets: |
|
|
|
|
|
|
|
|
Receivables |
|
|
|
|
|
| ||
Prepaid expenses |
|
|
|
|
|
| ||
Supplies used for R&D and manufacturing |
|
| ( | ) |
|
| ( | ) |
Deposits |
|
|
|
|
| ( | ) | |
Increase/(decrease) in liabilities: |
|
|
|
|
|
|
|
|
Accounts payable |
|
|
|
|
|
| ||
Accrued expenses |
|
| ( | ) |
|
|
| |
Due to employees |
|
|
|
|
| ( | ) | |
|
|
|
|
|
|
|
|
|
Net cash used in operating activities |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
CASH FLOWS FROM INVESTING ACTIVITIES: |
|
|
|
|
|
|
|
|
Net sales (purchases) of US Treasury bills |
|
|
|
|
| ( | ) | |
Purchases of property and equipment |
|
| ( | ) |
|
| ( | ) |
Expenditures for patent costs |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) investing activities |
|
|
|
|
| ( | ) | |
|
|
|
|
|
|
|
|
|
CASH FLOWS FROM FINANCING ACTIVITIES: |
|
|
|
|
|
|
|
|
Proceeds from issuance of common stock and warrants |
|
|
|
|
|
| ||
Payment of share issuance costs |
|
| ( | ) |
|
| ( | ) |
Proceeds from the exercise of warrants |
|
|
|
|
|
| ||
Proceeds from the exercise of options |
|
|
|
|
|
| ||
Proceeds from the purchase of stock by officers and directors |
|
|
|
|
|
| ||
Proceeds from landlord funding of lease improvements |
|
|
|
|
|
| ||
Payments on obligations under finance leases |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
Net cash (used in) provided by financing activities |
|
| ( | ) |
|
|
| |
|
|
|
|
|
|
|
|
|
NET (DECREASE) INCREASE IN CASH AND CASH EQUIVALENTS |
|
| ( | ) |
|
|
| |
|
|
|
|
|
|
|
|
|
CASH AND CASH EQUIVALENTS, BEGINNING OF YEAR |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
CASH AND CASH EQUIVALENTS, END OF YEAR |
| $ |
|
| $ |
| ||
| F-7 |
| Table of Contents |
CEL-SCI CORPORATION
STATEMENTS OF CASH FLOWS
YEARS ENDED SEPTEMBER 30, 2022 AND 2021
SUPPLEMENTAL DISCLOSURES OF NON-CASH INVESTING AND FINANCING ACTIVITIES: |
| 2022 |
|
| 2021 |
| ||
|
|
|
|
|
|
| ||
Property and equipment included in accrued expenses and accounts payable |
| $ |
|
| $ |
| ||
Capitalizable patent costs included in accrued expenses and accounts payable |
| $ |
|
| $ |
| ||
Changes to right of use assets and liabilities |
| $ |
|
| $ |
| ||
Finance lease obligation included in accounts payable |
| $ |
|
| $ |
| ||
Prepaid consulting services paid with issuance of common stock |
| $ |
|
| $ |
| ||
Fair value of warrant liabilities on date of exercise |
| $ |
|
| $ |
| ||
Assets purchased under finance leases |
| $ |
|
| $ |
| ||
Financing costs included in current liabilities |
| $ |
|
| $ |
| ||
Accrued consulting services to be paid with common stock |
| $ |
|
| $ |
| ||
|
|
|
|
|
|
|
|
|
SUPPLEMENTAL DISCLOSURE OF CASH FLOWS: |
|
|
|
|
|
|
|
|
Cash paid for interest |
| $ |
|
| $ |
| ||
| F-8 |
| Table of Contents |
CEL-SCI CORPORATION
NOTES TO FINANCIAL STATEMENTS
1. | ORGANIZATION |
CEL-SCI Corporation (the Company) was incorporated on March 22, 1983 in the state of Colorado to finance research and development in biomedical science and ultimately to engage in marketing and selling products.
The Company is focused on finding the best way to activate the immune system to fight cancer and infectious diseases. The Company has announced results from its Phase 3 study for its lead investigational therapy, Multikine® (Leukocyte Interleukin, Injection), involving head and neck cancer, for which the Company has received Orphan Drug Status from the United States Food and Drug Administration (FDA). Unlike other immune therapies, Multikine is administered locally at the site of the tumor as a first line treatment right after diagnosis, before surgery and radiation. The goal is to help the intact immune system kill the micro metastases that usually cause recurrence of the cancer to improve outcomes and better overall survival rates for patients suffering from head and neck cancer.
CEL-SCI is also investigating a peptide-based immunotherapy (CEL-4000) as a vaccine for rheumatoid arthritis using its LEAPS technology platform. CEL-SCI is in the process of completing pre-IND studies for CEL-4000.
2. | LIQUIDITY |
The Company has incurred significant costs since its inception for the acquisition of certain proprietary technology and scientific knowledge relating to the human immunological defense system, patent applications, research and development, administrative costs, construction and expansion of manufacturing and laboratory facilities and participation in clinical trials. The Company has funded such costs primarily with proceeds from loans and the public and private sale of its securities. The Company will be required to raise additional capital or find additional long-term financing to continue with its efforts to bring Multikine to market. The ability to raise capital may be dependent upon market conditions that are outside the control of the Company. The ability of the Company to obtain approval from the U.S. Food and Drug Administration (FDA) for the sale of products to be developed on a commercial basis is uncertain. Ultimately, the Company must complete the development of its products, obtain the appropriate regulatory approvals and obtain sufficient revenues to support its cost structure. The Company believes there is a high likelihood that it will continue to receive funds from private and public offerings and warrant exercises similar to the way it has funded operations in the past. However, there can be no assurance that the Company will be able to raise sufficient capital to support its operations.
To finance the Company through marketing approval, the Company plans to raise additional capital in the form of warrant exercises, corporate partnerships, and debt and/or equity financings. The Company believes that it will be able to obtain additional financing because it has done so consistently in the past and because it showed great survival benefit in the Phase 3 study in one of the two treatment arms for advanced primary head and neck cancer. However, there can be no assurance that the Company will be successful in raising additional funds on a timely basis or that the funds will be available to the Company on acceptable terms or at all. If the Company does not raise the necessary amounts of money, it may have to curtail its operations until such time as it is able to raise the required funding.
Due to the Company’s recurring losses from operations and future liquidity needs, there is substantial doubt about the Company’s ability to continue as a going concern. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Impact of the COVID-19 Pandemic
In response to the global outbreak of COVID-19 and the World Health Organization’s classification of the outbreak as a pandemic, the Company continues to take the necessary precautions to ensure the safety of its employees and to minimize interruptions to its operations. Management follows the Centers for Disease Control and Prevention’s (“CDC”) guidance and the recommendations and restrictions provided by state and local authorities. The full impact of the COVID-19 outbreak continues to evolve as of the date of this report. As such, it is uncertain as to the full impact the pandemic will have on the Company’s future financial condition, liquidity and results of operations. Management is actively monitoring the risks to public health and the impact of overall global business activity on the Company’s financial condition, liquidity, operations, suppliers, industry, and workforce.
| F-9 |
| Table of Contents |
3. | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
Cash and Cash Equivalents – Cash and cash equivalents consist principally of unrestricted cash on deposit and short-term money market funds. The Company considers all highly liquid investments with a maturity when purchased of less than three months as cash and cash equivalents.
U.S. Treasury Bills – U.S. Treasury Bills (“T-bills”) are highly liquid short-term investments with maturity dates of greater than 3 months, but less than one year. These investments are recorded at fair value.
Supplies used for R&D and manufacturing – Supplies are consumable items kept on hand to support the Company’s R&D and manufacturing operations. Supplies are recorded at cost and are charged to expense as they are used in operations.
Property and Equipment – Property and equipment are recorded at cost and depreciated using the straight-line method over estimated useful lives of five to seven years. Leasehold improvements are depreciated over the shorter of the estimated useful life of the asset or the term of the lease. Repairs and maintenance which do not extend the life of the asset are expensed when incurred. Property and equipment are reviewed on a quarterly basis to determine if any of the assets are impaired.
Patents – Patent expenditures are capitalized and amortized using the straight-line method over the shorter of the expected useful life or the legal life of the patent (
Leases – The Company accounts for contracts that convey the right to control the use of identified property, plant or equipment over a period of time in exchange for consideration, as leases upon inception. The Company leases certain real estate, machinery, laboratory equipment and office equipment over varying periods. Many of these leases include an option to either renew or terminate the lease. For purposes of calculating lease liabilities, these options are included in the lease term when it is reasonably certain that the Company will exercise such options. The incremental borrowing rate utilized to calculate the lease liabilities is based on the information available at the commencement date, as most of the leases do not provide an implicit borrowing rate. Short-term leases, defined as leases with initial terms of 12 months or less, are not reflected on the balance sheet. Lease expense for such short-term leases is not material.For purposes of calculating the finance and operating lease liabilities, lease and non-lease components are combined into a single element.
Derivative Instruments – The Company has financing arrangements that consist of freestanding derivative instruments that contain embedded derivative features. The Company accounts for these arrangements in accordance with Accounting Standards Codification (ASC) 815, Accounting for Derivative Instruments and Hedging Activities. In accordance with ASC 815, derivative instruments and hybrid instruments are recognized as either assets or liabilities on the balance sheet and are measured at fair value with gains or losses recognized in earnings or other comprehensive income depending on the nature of the derivative or hybrid instruments. The Company determines the fair value of derivative instruments and hybrid instruments based on available market data using appropriate valuation models considering all the rights and obligations of each instrument. The derivative liabilities are re-measured at fair value at the end of each interim period.
| F-10 |
| Table of Contents |
The Company adopted Accounting Standards Update (ASU) 2020-06, Debt—Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in an Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity effective October 1, 2021. The amendments in this ASU simplify and clarify the guidance in Subtopic 815-40. There was no financial impact upon adoption.
The Company adopted ASU 2021-04, Earnings Per Share (Topic 260), Debt—Modifications and Extinguishments (Subtopic 470-50), Compensation—Stock Compensation (Topic 718), and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40): Issuer’s Accounting for Certain Modifications or Exchanges of Freestanding Equity-Classified Written Call Options. This standard was issued to clarify and reduce diversity in an issuer’s accounting for modifications or exchanges of freestanding equity-classified written call options (for example, warrants) that remain equity classified after modification or exchange. ASU 2021-04 is effective for fiscal years beginning after December 15, 2021, however, as permitted, the Company has elected to prospectively adopt the standard effective as of October 1, 2021.
Stock-Based Compensation – Compensation cost for all stock-based awards is measured at fair value as of the grant date in accordance with the provisions of ASC 718, Compensation – Stock Compensation. The fair value of stock options is calculated using the Black-Scholes option pricing model. The Black-Scholes model requires various judgmental assumptions including volatility and expected option life. The stock-based compensation cost is recognized using the straight-line allocation method as expense over the requisite service or vesting period.
The Company has Incentive Stock Option Plans, Non-Qualified Stock Option Plans, Stock Compensation Plans, Stock Bonus Plans and an Incentive Stock Bonus Plan. These Plans are collectively referred to as the "Plans". All Plans have been approved by the Company’s stockholders.
The Company’s stock options are not transferable, and the actual value of the stock options that an employee may realize, if any, will depend on the excess of the market price on the date of exercise over the exercise price. For options issued with service conditions only, the assumption for stock price volatility is based on the variance of daily closing prices of the Company’s stock. The risk-free interest rate assumption is based on the U.S. Treasury rate at the date of grant with the term equal to the expected life of the option. Forfeitures are accounted for when they occur. The expected term of options represents the period that options granted are expected to be outstanding and has been determined based on an analysis of historical exercise behavior. If any of the assumptions used in the Black-Scholes model change significantly, stock-based compensation expense for new awards may differ materially in the future from that recorded in the current period.
Restricted stock granted under the Incentive Stock Bonus Plan and options granted under the 2021 and 2020 Non-Qualified Stock Option Plans are subject to service, performance and market conditions and meet the classification of equity awards. These awards were measured at fair value on the grant dates using a Monte Carlo simulation for issuances where the attainment of performance criteria is uncertain. The total compensation cost will be expensed over the estimated requisite service period.
Research and Development Costs - Research and development costs are expensed as incurred. Management accrues Clinical Research Organization (CRO) expenses and clinical trial study expenses based on services performed and relies on the CROs to provide estimates of those costs applicable to the completion stage of a study. Estimated accrued CRO costs are subject to revisions as such studies progress to completion. The Company records revisions to estimated expense in the period in which the facts that give rise to the revision become known.
| F-11 |
| Table of Contents |
Net Loss Per Common Share – The Company calculates net loss per common share in accordance with ASC 260, Earnings Per Share (ASC 260). Basic and diluted net loss per common share was determined by dividing net loss applicable to common shareholders by the weighted average number of common shares outstanding during the period. The Company’s potentially dilutive shares, which include outstanding common stock options, unvested restricted stock and common stock warrants, have not been included in the computation of diluted net loss per share for all periods as the result would be anti-dilutive.
Concentration of Credit Risk – Financial instruments, which potentially subject the Company to concentrations of credit risk, consist of cash and cash equivalents. The Company maintains its cash and cash equivalents with high quality financial institutions. At times, these accounts may exceed federally insured limits. The Company has not experienced any losses in such bank accounts. The Company believes it is not exposed to significant credit risk related to cash and cash equivalents. All non-interest bearing cash balances were fully insured up to $
Income Taxes – The Company uses the asset and liability method of accounting for income taxes. Under the asset and liability method, deferred tax assets and liabilities are recognized for future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating and tax loss carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. The Company records a valuation allowance to reduce the deferred tax assets to the amount that is more likely than not to be recognized. A full valuation allowance was recorded against the deferred tax assets as of September 30, 2022 and 2021.
The Company adopted ASU No. 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes, effective October 1, 2021. The new standard includes several provisions that simplify accounting for income taxes by removing certain exceptions to the general principles in Topic 740 and increasing consistency and clarity for the users of financial statements. The adoption of ASU 2019-12 had no impact on the Company’s financial statements.
On August 16, 2022, the Inflation Reduction Act of 2022 (IR Act) was signed into federal law. The IR Act provides for, among other things, a new U.S. federal 1% excise tax on certain repurchases of stock by publicly traded U.S. domestic corporations and certain U.S. domestic subsidiaries of publicly traded foreign corporations occurring on or after January 1, 2023. The excise tax is imposed on the repurchasing corporation itself, not its shareholders from which shares are repurchased. The amount of the excise tax is generally 1% of the fair market value of the shares repurchased at the time of the repurchase. However, for purposes of calculating the excise tax, repurchasing corporations are permitted to net the fair market value of certain new stock issuances against the fair market value of stock repurchases during the same taxable year. In addition, certain exceptions apply to the excise tax. The U.S. Department of the Treasury (Treasury) has been given authority to provide regulations and other guidance to carry out and prevent the abuse or avoidance of the excise tax.
Use of Estimates – The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and the accompanying disclosures. These estimates are based on management’s best knowledge of current events and actions the Company may undertake in the future. Estimates are used in accounting for, among other items, obsolescence of supplies used for R&D and manufacturing, accruals, stock options, useful lives for depreciation and amortization of long-lived assets, deferred tax assets and the related valuation allowance, and the valuation of derivative liabilities. Actual results could differ from estimates, although management does not believe such differences would materially affect the financial statements in any given year. However, regarding the valuation of derivative liabilities determined using the Black-Scholes pricing model, significant fluctuations may materially affect the financial statements in a given year. Additionally, in calculating the right of use assets and lease liabilities, estimates and assumptions were used to determine the incremental borrowing rates and the expected lease terms. The Company considers the estimates used in valuing the derivative liabilities and the lease assets and liabilities to be significant.
| F-12 |
| Table of Contents |
New Accounting Pronouncements
The Company has considered all recently issued accounting pronouncements and does not believe the adoption of such pronouncements will have a material impact on its financial statements.
4. | WARRANTS AND NON-EMPLOYEE OPTIONS |
The following warrants and non-employee options are outstanding at September 30, 2022:
Warrant/Options |
| Issue Date |
| Shares Issuable upon Exercise of Warrants/ Options |
|
| Exercise Price |
|
| Expiration Date | |||
Series N |
|
|
|
|
| $ |
|
| |||||
Series UU |
|
|
|
|
| $ |
|
| |||||
Series X |
|
|
|
|
| $ |
|
| |||||
Series Y |
|
|
|
|
| $ |
|
| |||||
Series MM |
|
|
|
|
| $ |
|
| |||||
Series NN |
|
|
|
|
| $ |
|
| |||||
Series RR |
|
|
|
|
| $ |
|
| |||||
Series SS |
|
|
|
|
| $ |
|
| |||||
Series TT |
|
|
|
|
| $ |
|
| |||||
Consultant Options |
|
|
|
|
| $ |
|
| |||||
The following warrants and non-employee options are outstanding at September 30, 2021:
Warrant/Options |
| Issue Date |
| Shares Issuable upon Exercise of Warrants/ Options |
|
| Exercise Price |
|
| Expiration Date | |||
Series N |
|
|
|
|
| $ |
|
| |||||
Series UU |
|
|
|
|
| $ |
|
| |||||
Series X |
|
|
|
|
| $ |
|
| |||||
Series Y |
|
|
|
|
| $ |
|
| |||||
Series Z |
|
|
|
|
| $ |
|
| |||||
Series CC |
|
|
|
|
| $ |
|
| |||||
Series HH |
|
|
|
|
| $ |
|
| |||||
Series AA |
|
|
|
|
| $ |
|
| |||||
Series MM |
|
|
|
|
| $ |
|
| |||||
Series NN |
|
|
|
|
| $ |
|
| |||||
Series RR |
|
|
|
|
| $ |
|
| |||||
Series SS |
|
|
|
|
| $ |
|
| |||||
Series TT |
|
|
|
|
| $ |
|
| |||||
Consultant Options |
|
|
|
|
| $ |
|
| |||||
| F-13 |
| Table of Contents |
A. | Warrant Liabilities |
Warrant liabilities outstanding at September 30 are as follows:
|
| 2022 |
|
| 2021 |
| ||
Series Z warrants |
| $ |
|
| $ |
| ||
Series AA warrants |
|
|
|
|
|
| ||
Series CC warrants |
|
|
|
|
|
| ||
Series HH warrants |
|
|
|
|
|
| ||
Total warrant liabilities |
| $ |
|
| $ |
| ||
The gains/(losses) on the warrant liabilities for the years ended September 30 are as follows:
|
| 2022 |
|
| 2021 |
| ||
Series W warrants |
| $ |
|
| $ |
| ||
Series Z warrants |
|
|
|
|
|
| ||
Series ZZ warrants |
|
|
|
|
| ( | ) | |
Series AA warrants |
|
|
|
|
| ( | ) | |
Series BB warrants |
|
|
|
|
|
| ||
Series CC warrants |
|
|
|
|
| ( | ) | |
Series HH warrants |
|
|
|
|
|
| ||
Net loss on warrant liabilities |
| $ |
|
| $ | ( | ) | |
The Company reviews all outstanding warrants in accordance with the requirements of ASC 815. This topic provides that an entity should use a two-step approach to evaluate whether an equity-linked financial instrument (or embedded feature) is indexed to its own stock, including evaluating the instrument’s contingent exercise and settlement provisions. The warrant agreements provide for adjustments to the exercise price for certain dilutive events. Under the provisions of ASC 815, the warrants are not considered indexed to the Company’s stock because future equity offerings or sales of the Company’s stock are not an input to the fair value of a “fixed-for-fixed” option on equity shares, and equity classification is therefore precluded.
In accordance with ASC 815, derivative liabilities must be measured at fair value upon issuance and re-valued at the end of each reporting period through expiration. Any change in fair value between the respective reporting periods is recognized as a gain or loss in the statement of operations.
Changes in Warrant Liabilities
In February 2022,
In December 2021,
In November 2021,
On August 22, 2021,
On October 28, 2020,
Exercise of Warrant Liabilities
The following warrants recorded as liabilities were exercised during the year ended September 30, 2022:
Warrants |
| Warrants Exercised |
|
| Exercise Price |
|
| Proceeds |
| |||
Series CC |
|
|
|
| $ |
|
| $ |
| |||
| F-14 |
| Table of Contents |
The following warrants recorded as liabilities were exercised during the year ended September 30, 2021:
Warrants |
| Warrants Exercised |
|
| Exercise Price |
|
| Proceeds |
| |||
Series Z |
|
|
|
| $ |
|
| $ |
| |||
Series ZZ |
|
|
|
| $ |
|
|
|
| |||
Series AA |
|
|
|
| $ |
|
|
|
| |||
Series CC |
|
|
|
| $ |
|
|
|
| |||
|
|
|
|
|
|
|
|
| $ |
| ||
B. | Equity Warrants |
Changes in Equity Warrants
On June 13, 2022, the expiration dates of the Series N, Series X, Series Y, Series UU, Series MM and Series NN warrants were extended two years. The incremental costs of the Series N, Series X and Series Y warrant extensions were recorded as a deemed dividend and totaled approximately $
On June 28, 2021, the expiration dates of the Series N, Series X, Series Y and Series UU warrants were extended one year. On December 7, 2020, the expiration dates of the Series N, Series X, Series Y and Series UU warrants were extended six months. The incremental costs of the warrant extensions were recorded consistent with the accounting for the initial warrant issuances. The incremental costs of the Series N and Series X warrant extensions were recorded as a deemed dividend and totaled approximately $
| F-15 |
| Table of Contents |
Exercise of Equity Warrants
The following equity warrants were exercised during the year ended September 30, 2022.
Warrants |
| Warrants Exercised |
|
| Exercise Price |
|
| Proceeds |
| |||
Series NN |
|
|
|
| $ |
|
| $ |
| |||
The following equity warrants were exercised during the year ended September 30, 2021.
Warrants |
| Warrants Exercised |
|
| Exercise Price |
|
| Proceeds |
| |||
Series MM |
|
|
|
| $ |
|
| $ |
| |||
Series NN |
|
|
|
| $ |
|
|
|
| |||
Series RR |
|
|
|
| $ |
|
|
|
| |||
Series SS |
|
|
|
| $ |
|
|
|
| |||
Series TT |
|
|
|
| $ |
|
|
|
| |||
|
|
|
|
|
|
|
|
| $ |
| ||
C. | Options and Shares Issued to Consultants |
The Company typically enters into consulting arrangements in exchange for common stock or stock options. During the years ended September 30, 2022 and 2021, the Company issued
During the years ended September 30, 2022 and 2021, the Company recorded total expense of approximately $
No options were issued to consultants during the year ended September 30, 2022. During the year ended September 30, 2021, the Company issued 5,000 options to a consultant to purchase common stock with an exercise price of $11.61 and an expiration of November 17, 2022. As of September 30, 2022, 15,000 options issued to consultants as payment for services remained outstanding, all of which were issued from the Non-Qualified Stock Option plan and are fully vested.
5. | PROPERTY AND EQUIPMENT |
Property and equipment consisted of the following at September 30:
|
| 2022 |
|
| 2021 |
| ||
Research equipment |
| $ |
|
| $ |
| ||
Furniture and equipment |
|
|
|
|
|
| ||
Leasehold improvements |
|
|
|
|
|
| ||
|
|
|
|
|
|
| ||
Accumulated depreciation and amortization |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
Net property and equipment |
| $ |
|
| $ |
| ||
Depreciation expense for the years ended September 30, 2022 and 2021 totaled approximately $
| F-16 |
| Table of Contents |
6. | PATENTS |
Patents consist of the following at September 30:
|
| 2022 |
|
| 2021 |
| ||
Patents |
| $ |
|
| $ |
| ||
Accumulated amortization |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
Patents, net |
| $ |
|
| $ |
| ||
During the years ended September 30, 2022 and 2021, there was no impairment of patent costs. Amortization expense for the years ended September 30, 2022 and 2021 totaled approximately $
Years ending September 30, |
| |||
2023 |
| $ |
| |
2024 |
|
|
| |
2025 |
|
|
| |
2026 |
|
|
| |
2027 |
|
|
| |
Thereafter |
|
|
| |
|
| $ |
| |
7. | INCOME TAXES |
At September 30, 2022 and 2021, the Company had net deferred tax assets of $
Pursuant to Section 382 of the Internal Revenue Code, or IRC, annual use of the Company’s
| F-17 |
| Table of Contents |
The Company had federal NOL carryforwards of approximately $
Significant components of the Company’s deferred tax assets and liabilities as of September 30, 2022 and 2021 are listed below:
|
| 2022 |
|
| 2021 |
| ||
|
|
|
|
|
|
| ||
NOL carryforwards |
| $ |
|
| $ |
| ||
Capitalized R&D |
|
|
|
|
|
| ||
Stock-based compensation |
|
|
|
|
|
| ||
Lease liabilities |
|
|
|
|
|
| ||
R&D credit |
|
|
|
|
|
| ||
Vacation and other |
|
|
|
|
|
| ||
Total deferred tax assets |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
Right of use assets |
|
| ( | ) |
|
| ( | ) |
Fixed assets and intangibles |
|
| ( | ) |
|
| ( | ) |
Total deferred tax liability |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
Net deferred tax asset |
|
|
|
|
|
| ||
Valuation allowance |
|
| ( | ) |
|
| ( | ) |
Ending balance |
| $ |
|
| $ |
| ||
The Company has no federal or state current or deferred tax expense or benefit. The Company’s effective tax rate differs from the applicable federal statutory tax rate. The reconciliation of these rates is as follows for the years ended September 30:
|
| 2022 |
|
| 2021 |
| ||
|
|
|
|
|
|
| ||
Federal rate |
|
| % |
|
| % | ||
State tax rate change |
|
| ( | ) |
|
| ( | ) |
State tax rate, net of federal benefit |
|
|
|
|
|
| ||
Other adjustments |
|
| ( | ) |
|
| ( | ) |
Permanent differences |
|
|
|
|
|
| ||
Change in valuation allowance |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
Effective tax rate |
|
| % |
|
| % | ||
The Company applies the provisions of ASC 740, “Accounting for Uncertainty in Income Taxes,” which requires financial statement benefits to be recognized for positions taken for tax return purposes when it is more likely than not that the position will be sustained. The Company has elected to reflect any tax penalties or interest resulting from tax assessments on uncertain tax positions as a component of income tax expense. The Company has generated federal net operating losses during the tax years ended September 30, 1999 through 2022. The Company files income tax returns in the U.S. federal jurisdiction and various states. With few exceptions, the Company is no longer subject to U.S. federal and state income tax examinations by tax authorities for years September 30, 2018 and prior.
| F-18 |
| Table of Contents |
8. | STOCK COMPENSATION |
The Company recognized the following expenses for options issued or vested and restricted stock awarded during the year:
|
| Year Ended September 30, |
| |||||
|
| 2022 |
|
| 2021 |
| ||
Employees |
| $ |
|
| $ |
| ||
Non-employees |
| $ |
|
| $ |
| ||
During the years ended September 30, 2022 and 2021 the fair value of each option grant was estimated on the date of grant using the Black-Scholes option pricing model using the following assumptions.
|
| 2022 |
|
| 2021 |
| ||
Expected stock price volatility |
|
|
|
| ||||
Risk-free interest rate |
|
|
|
| ||||
Expected life of options |
|
|
|
| ||||
Expected dividend yield |
|
| - |
|
|
| - |
|
Non-Qualified Stock Option Plans –
During the year ended September 30, 2022, the Company adopted the 2022 Non-Qualified Stock Option Plan, which provides for the issuance of up to
| F-19 |
| Table of Contents |
During the year ended September 30, 2021, the Company adopted the 2021 Non-Qualified Stock Option Plan, which provides for the issuance of up to
At September 30, 2022, the Company has collectively authorized the issuance of
Incentive Stock Option Plans – At September 30, 2022, the Company had collectively authorized the issuance of
Activity in the Company’s Non-Qualified and Incentive Stock Option Plans for the two years ended September 30, 2022 is summarized as follows:
Non-Qualified and Incentive Stock Option Plans
|
| Outstanding |
|
| Exercisable |
| ||||||||||||||||||||||||||
|
| Number of Shares |
|
| Weighted Average Exercise Price |
|
| Weighted Average Remaining Contractual Term (Years) |
|
| Aggregate Intrinsic Value |
|
| Number of Shares |
|
| Weighted Average Exercise Price |
|
| Weighted Average Remaining Contractual Term (Years) |
|
| Aggregate Intrinsic Value |
| ||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||
Outstanding at September 30, 2020 |
|
|
|
| $ |
|
|
|
|
| $ |
|
|
|
|
| $ |
|
|
|
|
| $ |
| ||||||||
Vested |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| $ |
|
|
|
|
|
|
|
|
| ||
Granted (a) |
|
|
|
| $ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
Exercised |
|
| ( | ) |
| $ |
|
|
|
|
|
|
|
|
|
|
| ( | ) |
| $ |
|
|
|
|
|
|
|
|
| ||
Forfeited |
|
| ( | ) |
| $ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
Expired |
|
| ( | ) |
| $ |
|
|
|
|
|
|
|
|
|
|
| ( | ) |
| $ |
|
|
|
|
|
|
|
|
| ||
Outstanding at September 30, 2021 |
|
|
|
| $ |
|
|
|
|
| $ |
|
|
|
|
| $ |
|
|
|
|
| $ |
| ||||||||
Vested |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| $ |
|
|
|
|
|
|
|
|
| ||
Granted |
|
|
|
| $ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
Exercised |
|
| ( | ) |
| $ |
|
|
|
|
|
|
|
|
|
|
| ( | ) |
| $ |
|
|
|
|
|
|
|
|
| ||
Forfeited |
|
| ( | ) |
| $ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
Expired |
|
| ) |
| $ |
|
|
|
|
|
|
|
|
|
|
| ( | ) |
| $ |
|
|
|
|
|
|
|
|
| |||
Outstanding at September 30, 2022 |
|
|
|
| $ |
|
|
|
|
| $ |
|
|
|
|
| $ |
|
|
|
|
| $ |
| ||||||||
(a) | Includes 1,872,000 performance based options issued to officers and directors, 728,000 options issued to employees and 5,000 options issued to consultants. |
| F-20 |
| Table of Contents |
A summary of the status of the Company’s non-vested options for the two years ended September 30, 2022 is presented below:
|
| Number of Options |
|
| Weighted Average Grant Date Fair Value |
| ||
Unvested at September 30, 2020 |
|
|
|
| $ |
| ||
Vested |
|
| ( | ) |
|
|
|
|
Granted |
|
|
|
|
|
|
| |
Forfeited |
|
| ( | ) |
|
|
|
|
Unvested at September 30, 2021 |
|
|
|
| $ |
| ||
Vested |
|
| ( | ) |
|
|
|
|
Granted |
|
|
|
|
|
|
| |
Forfeited |
|
| ( | ) |
|
|
|
|
Unvested at September 30, 2022 |
|
|
|
| $ |
| ||
Incentive Stock Bonus Plan – On September 30, 2022,
A summary of the status of the Company’s restricted common stock issued from the Incentive Stock Bonus Plan for the two years ended September 30, 2022 is presented below:
|
| Number of Shares |
|
| Weighted Average Grant Date Fair Value |
| ||
|
|
|
|
|
|
| ||
Unvested at September 30, 2020 |
|
|
|
| $ |
| ||
Forfeited |
|
| ( | ) |
|
|
|
|
Vested |
|
| ( | ) |
|
|
|
|
Unvested at September 30, 2021 |
|
| 151,250 |
|
| $ |
| |
Forfeited |
|
| - |
|
|
|
|
|
Vested |
|
| - |
|
|
|
|
|
Unvested at September 30, 2022 |
|
|
|
| $ |
| ||
| F-21 |
| Table of Contents |
Stock Bonus Plans – As of September 30, 2022, the Company authorized to issue up to
Stock Compensation Plans – On September 30, 2022,
9. | EMPLOYEE BENEFIT PLAN |
The Company maintains a defined contribution retirement plan, qualifying under Section 401(k) of the Internal Revenue Code, subject to the Employee Retirement Income Security Act of 1974, as amended, and covering substantially all Company employees. Each participant’s contribution is matched by the Company with shares of common stock that have a value equal to 100% of the participant’s contribution, not to exceed the lesser of $
10. | COMMITMENTS AND CONTINGENCIES |
Clinical Research Agreements
In April 2013, the Company entered into a co-development and revenue sharing agreement with Ergomed. Under the agreement, Ergomed will contribute up to $
| F-22 |
| Table of Contents |
Lease Agreements
The Company leases a manufacturing facility near Baltimore, Maryland (the San Tomas lease). The building was remodeled in accordance with the Company’s specifications so that it can be used by the Company to manufacture Multikine for the Company’s Phase 3 clinical trial and sales of the drug if approved by the FDA. The lease is for a term of twenty years and requires annual base rent to escalate each year at 3%. The Company is required to pay all real estate and personal property taxes, insurance premiums, maintenance expenses, repair costs and utilities. The lease allows the Company, at its election, to extend the lease for two ten-year periods or to purchase the building at the end of the 20-year lease, which expires in October 2028. The renewal options are not included in the calculation of the right of use asset and lease liability because exercise of those options is not reasonably certain.
Upon adoption of ASC 842 on October 1, 2019, the Company recorded a finance lease right of use asset of approximately $
In August 2020, the Company entered into an amendment to the San Tomas lease agreement under which the landlord agreed to allow the Company to substantially upgrade the manufacturing facility in preparation for the potential commercial production of Multikine. The upgrades were completed and the improvements were placed in service in October 2021. The total cost of the upgrades was $
During June 2021, the Company determined that it used an incorrect discount rate to calculate the opening ROU asset and lease liability balances upon adoption of ASC 842. Management engaged an outside valuation specialist to perform a synthetic credit rating analysis which resulted
The Company was required to deposit the equivalent of one year of base rent in accordance with the original lease. Under the landlord’s $
| F-23 |
| Table of Contents |
Approximate future minimum lease payments under finance leases as of September 30, 2022 are as follows:
Year ending September 30, |
|
|
| |
2023 |
| $ |
| |
2024 |
|
|
| |
2025 |
|
|
| |
2026 |
|
|
| |
2027 |
|
|
| |
Thereafter |
|
|
| |
Total future minimum lease obligations |
|
|
| |
Less imputed interest on finance lease obligations |
|
| ( | ) |
Net present value of finance lease obligations |
| $ |
| |
The Company leases two facilities under operating leases. The lease for the Company’s office headquarters will expire on November 30, 2025. The lease for its research and development laboratory was renewed in September 2021 for an additional ten years and will expire on February 29, 2032. The renewal was considered a modification for accounting purposes and the right of use asset and liability were remeasured as of the date of the renewal. This resulted in an increase of approximately $
Upon adoption of ASC 842 on October 1, 2019, the Company recorded an operating lease right of use asset and an operating lease liability of approximately $
As of September 30, 2022, future minimum lease payments under operating leases are as follows:
Year ending September 30, |
|
|
| |
2023 |
| $ |
| |
2024 |
|
| |
|
2025 |
|
|
| |
2026 |
|
|
| |
2027 |
|
|
| |
Thereafter |
|
|
| |
Total future minimum lease obligations |
|
|
| |
Less imputed interest on operating lease obligation |
|
| ( | ) |
Net present value of operating lease obligation |
| $ |
| |
| F-24 |
| Table of Contents |
Vendor Obligations
The Company has contingent obligations with vendors for work that will be completed in relation to the Phase 3 clinical trial. The timing of these obligations cannot be determined at this time. The Company estimates it will incur additional expenses of approximately $
11. | RELATED PARTY TRANSACTIONS |
During the year ended September 30, 2022, no restricted shares of the Company’s common stock were purchased by related parties. During the year ended September 30, 2021, the de Clara Trust, of which the Company’s CEO, Geert Kersten, is a trustee and beneficiary, and two directors purchased a total of
On June 13, 2022, the expiration dates of the Series N, Series X, Series Y, Series UU, Series MM and Series NN warrants were extended two years (Note 4). The incremental costs of the Series N, Series X and Series Y warrant extensions were recorded as a deemed dividend and totaled approximately $
In June 2021, the expiration dates of the Series N, Series X, and Series UU warrants were extended one year. In December 2020, the expiration dates of the Series N, Series X, Series UU warrants were extended six months. The incremental costs of the warrant extensions were recorded consistent with the accounting for the initial warrant issuances. The incremental costs of the Series N and Series X warrant extensions were recorded as a deemed dividend and totaled approximately $
| F-25 |
| Table of Contents |
12. | STOCKHOLDERS’ EQUITY |
Exercise of Warrants
During the years ended September 30, 2022 and 2021, the Company received proceeds of approximately $
Sales of Securities
There were no sales of securities during the fiscal year ended September 30, 2022.
In June 2021, the Company sold
In December 2020, the Company sold
Other Equity Transactions
The Company entered into Securities Purchase Agreements (SPA) with Ergomed plc, one of the Company’s Clinical Research Organizations responsible for managing the Company’s Phase 3 clinical trial, to facilitate payment of amounts due Ergomed. Under the Agreements, the Company issued Ergomed shares of common stock and the net proceeds from the sales of those shares reduces outstanding amounts due Ergomed. Upon issuance, the Company expenses the full value of the shares as Other non-operating gain/loss and subsequently offsets the expense as amounts are realized through the sale by Ergomed and reduces accounts payable to Ergomed.
During the years ended September 30, 2022 and 2021, CEL-SCI did not issue Ergomed any shares of common stock. Additionally, no shares were sold during the year ended September 30, 2022. During the year ended September 30, 2021, the company recorded Other Non-operating gains of approximately $
13. | FAIR VALUE MEASUREMENTS |
In accordance with the provisions of ASC 820, “Fair Value Measurements,” the Company determines fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The Company generally applies the income approach to determine fair value. This method uses valuation techniques to convert future amounts to a single present amount. The measurement is based on the value indicated by current market expectations with respect to the future amounts.
| F-26 |
| Table of Contents |
ASC 820 establishes a fair value hierarchy that prioritizes the inputs used to measure fair value. The hierarchy gives the highest priority to active markets for identical assets and liabilities (Level 1 measurement) and the lowest priority to unobservable inputs (Level 3 measurement). The Company classifies fair value balances based on the observability of those inputs. The three levels of the fair value hierarchy are as follows:
| o | Level 1 – Observable inputs such as quoted prices in active markets for identical assets or liabilities |
|
|
|
| o | Level 2 – Inputs other than quoted prices that are observable for the asset or liability, either directly or indirectly. These include quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active and amounts derived from valuation models where all significant inputs other than quoted prices are observable for the asset or liability. |
|
|
|
| o | Level 3 – Unobservable inputs that reflect management’s assumptions |
For disclosure purposes, assets and liabilities are classified in their entirety in the fair value hierarchy level based on the lowest level of input that is significant to the overall fair value measurement. The Company’s assessment of the significance of a particular input to the fair value measurement requires judgment and may affect the placement within the fair value hierarchy levels.
The Company purchased short-term T-bills during the year ended September 30, 2021 that are classified as trading securities. Quoted market prices were applied to determine the fair value of short-term investments, therefore they were categorized as Level 1 on the fair value hierarchy. The T-bills were recorded at fair market value, which includes an unrealized gain of approximately $
As of September 30, 2022 and 2021, all of the Company’s derivative liabilities are classified as Level 3.
The following sets forth the reconciliation of beginning and ending balances related to fair value measurements using significant unobservable inputs (Level 3), as of September 30:
|
| 2022 |
|
| 2021 |
| ||
Beginning balance |
| $ |
|
| $ |
| ||
Issuances |
|
|
|
|
|
| ||
Exercises |
|
| ( | ) |
|
| ( | ) |
N Net realized and unrealized derivative loss |
|
| ( | ) |
|
|
| |
|
|
|
|
|
|
|
|
|
Ending balance |
| $ |
|
| $ |
| ||
The fair values of the Company’s derivative instruments disclosed above under Level 3 are primarily derived from valuation models where significant inputs such as historical price and volatility of the Company’s stock were used. At September 30, 2022, the Company does not have any Level 3 derivative instruments. At September 30, 2021, the Company’s Level 3 derivative instruments have a weighted average fair value of $
| F-27 |
| Table of Contents |
14. | NET LOSS PER COMMON SHARE |
Basic loss per share is computed by dividing net loss available to common shareholders by the weighted average number of common shares outstanding during the period. The Company’s potentially dilutive shares, which include outstanding common stock options, common stock warrants and restricted stock are not included in the computation of diluted net loss per share if their effect would be anti-dilutive.
The following table provides a reconciliation of the numerators and denominators of the basic and diluted per-share computations:
|
| Year ended September 30, |
| |||||
|
| 2022 |
|
| 2021 |
| ||
Loss per share – basic |
|
|
|
|
|
| ||
Net loss available to common shareholders - basic |
| $ | ( | ) |
| $ | ( | ) |
Weighted average shares outstanding - basic |
|
|
|
|
|
| ||
Basic loss per common share |
| $ | ( | ) |
| $ | ( | ) |
|
|
|
|
|
|
|
|
|
Loss per share – diluted |
|
|
|
|
|
|
|
|
Net loss available to common shareholders - basic |
| $ | ( | ) |
| $ | ( | ) |
Unrealized gain on derivatives (1) |
|
|
|
|
| ( | ) | |
Net loss available to common shareholders - diluted |
| $ | ( | ) |
| $ | ( | ) |
|
|
|
|
|
|
|
|
|
Weighted average shares outstanding – basic |
|
|
|
|
|
| ||
Incremental shares underlying dilutive warrants (1) |
|
| - |
|
|
|
| |
Weighted average shares outstanding - diluted |
|
|
|
|
|
| ||
Diluted loss per common share |
| $ | ( | ) |
| $ | ( | ) |
(1) Includes Series Z, AA, CC and HH warrants for the year ended September 30, 2021.
In accordance with the contingently issuable shares guidance of FASB ASC Topic 260, Earnings Per Share, the calculation of diluted net loss per share excludes the following dilutive securities because their inclusion would have been anti-dilutive as of September 30:
|
| 2022 |
|
| 2021 |
| ||
Options and Warrants |
|
|
|
|
|
| ||
Unvested Restricted Stock |
|
|
|
|
|
| ||
Total |
|
|
|
|
|
| ||
15. | SUBSEQUENT EVENTS |
On October 28, 2022, the expiration date of the Series RR warrants were extended two years. The incremental cost of extending the Series RR warrants will be recorded as a deemed dividend, and totaled approximately $
Between October 1, 2022 and December 27, 2022 the company received approximately $
| F-28 |
In accordance with Section 13 or 15(a) of the Securities Exchange Act of 1934, the Registrant has caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized on the 27 day of December 2022.
CEL-SCI CORPORATION | |||
| By: | /s/ Geert Kersten | ||
|
| Geert Kersten, Chief Executive Officer | |
Pursuant to the requirements of the Securities Act of l934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
Signature |
| Title |
| Date |
|
|
|
|
|
/s/ Geert Kersten |
| Chief Executive, Principal Accounting, |
| December 27, 2022 |
Geert R. Kersten |
| Principal Financial Officer and a Director |
|
|
|
|
|
|
|
/s/Peter Young |
| Director |
| December 27, 2022 |
Dr. Peter R. Young |
|
|
|
|
|
|
|
|
|
/s/ Bruno Baillavoine |
| Director |
| December 27, 2022 |
Bruno Baillavoine |
|
|
|
|
|
|
|
|
|
/s/Robert Watson |
| Director |
| December 27, 2022 |
Robert Watson |
|
|
|
|
|
|
|
|
|
/s/Gail Naughton |
| Director |
| December 27, 2022 |
Gail Naughton |
|
|
|
|