UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
|
x ANNUAL REPORT PURSUANT TO SECTION 13 or 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
For the fiscal year ended December 31, 2010
|
|
OR
|
|
o TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
for the transition period from to
|
Commission File No. 0-14710
XOMA Ltd.
(Exact name of registrant as specified in its charter)
|
Bermuda
|
52-2154066
|
|
(State or other jurisdiction of incorporation or organization)
|
(I.R.S. Employer Identification No.)
|
|
2910 Seventh Street, Berkeley, California 94710
|
(510) 204-7200
|
|
(Address of principal executive offices, including zip code)
|
(Telephone Number)
|
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class
|
Name of each exchange on which registered
|
|
Common Shares, U.S. $0.0075 par value
|
The NASDAQ Global Market
|
|
Preference Share Purchase Rights
|
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes o No x
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act.
Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large Accelerated Filer o Accelerated Filer x Non-Accelerated filer o Smaller reporting company o
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act of 1934). Yes ¨ No x
The aggregate market value of voting shares held by non-affiliates of the registrant is $104,378,157 as of June 30, 2010
Number of Common Shares outstanding as of March 8, 2011: 29,510,963
DOCUMENTS INCORPORATED BY REFERENCE:
Portions of the Company’s Proxy Statement for the Company’s 2010 Annual General Meeting of Shareholders are incorporated by reference into Part III of this Report.
2010 FORM 10-K ANNUAL REPORT
TABLE OF CONTENTS
|
Item 1.
|
1
|
|
|
Item 1A.
|
17
|
|
|
Item 1B.
|
33
|
|
|
Item 2.
|
33
|
|
|
Item 3.
|
33
|
|
|
Item 4.
|
34
|
|
|
34
|
||
|
PART II
|
||
|
Item 5.
|
35
|
|
|
Item 6.
|
36
|
|
|
Item 7.
|
38
|
|
|
Item 7A.
|
53
|
|
|
Item 8.
|
53
|
|
|
Item 9.
|
53
|
|
|
Item 9A.
|
54
|
|
|
Item 9B.
|
55
|
|
|
PART III
|
||
|
Item 10.
|
56
|
|
|
Item 11.
|
56
|
|
|
Item 12.
|
56
|
|
|
Item 13.
|
56
|
|
|
Item 14.
|
56
|
|
|
PART IV
|
||
|
Item 15.
|
57
|
|
|
58
|
||
|
F-1
|
||
|
i
|
||
PART I
|
Item 1.
|
Overview
XOMA Ltd. (“XOMA”), a Bermuda company, is a biopharmaceutical company focused on the discovery, development and manufacture of therapeutic antibodies designed to treat autoimmune, infectious, inflammatory and oncological diseases. Our proprietary development pipeline includes XOMA 052, an antibody that inhibits interleukin-1 beta (“IL-1 beta”), which is expected to advance into Phase 3 development for the treatment of Behcet’s uveitis and is in Phase 2 clinical development for Type 2 diabetes with cardiovascular biomarkers; XOMA 3AB, a biodefense anti-botulism product candidate comprised of a combination, or cocktail, of antibodies; and preclinical antibody discovery programs in several indications, including autoimmune, cardio-metabolic, inflammatory, and oncological diseases. We have a fully integrated product development platform, extending from preclinical science and clinical development to scale-up development and manufacturing.
We have entered into a license and collaboration agreement with Les Laboratoires Servier (“Servier”), to jointly develop and commercialize XOMA 052 in multiple indications. XOMA 052 is designed to inhibit the pro-inflammatory cytokine IL-1 beta that is believed to be a primary trigger of pathologic inflammation in multiple diseases. Under the terms of the agreement, Servier has worldwide rights to diabetes and cardiovascular disease indications and rights outside the U.S. and Japan to Behcet’s uveitis and other inflammatory disease and oncology indications. XOMA retains development and commercialization rights for Behcet's uveitis and other inflammatory disease and oncology indications in the U.S. and Japan, and has an option to reacquire rights to diabetes and cardiovascular disease indications from Servier in these territories. Should we exercise our option to reacquire rights to the diabetes and cardiovascular disease indications in the U.S. and Japan, we will be required to pay Servier an option fee and partially reimburse their incurred development expenses.
Our biodefense initiatives currently include a $65 million multiple-year contract funded by the National Institute of Allergy and Infectious Diseases (“NIAID”), a part of the National Institutes of Health (“NIH”), to support our ongoing development of XOMA 3AB toward clinical trials in the treatment of botulism poisoning. XOMA also develops products with premier pharmaceutical companies including Novartis AG (“Novartis”) and Takeda Pharmaceutical Company Limited (“Takeda”).
We have a premier antibody discovery and development platform that incorporates a collection of antibody phage display libraries and proprietary Human Engineering™ (“HE™”), affinity maturation, Bacterial Cell Expression (“BCE”) and manufacturing technologies that enhance our ability and that of our collaboration partners to discover and develop new therapeutic antibodies. BCE is a key biotechnology for the discovery and manufacturing of antibodies and other proteins. To date, more than 50 pharmaceutical and biotechnology companies have signed BCE licenses, and a number of licensed product candidates are in clinical development. We continue to develop and commercialize additional antibody-related technologies including proprietary display technologies to enable antibody discovery and optimization. Our technologies have contributed to the success of the marketed antibody products LUCENTIS® (ranibizumab injection), for wet age-related macular degeneration and macular edema following retinal vein occlusion, and CIMZIA® (certolizumab pegol), for rheumatoid arthritis and Crohn’s disease.
Strategy
We are advancing a pipeline of biologic products using our proven expertise, technologies and capabilities from antibody discovery through product development. We seek to expand our pipeline by developing proprietary products and technologies, providing contract services to government agencies responsible for biodefense and entering into licensing and collaborative arrangements with pharmaceutical and biotechnology companies. The principal elements of our strategy are to:
|
|
·
|
Focus on advancing XOMA 052, our lead product candidate. Using our proprietary antibody technologies, capabilities and expertise, we discovered XOMA 052, an antibody that inhibits IL-1 beta. XOMA 052 has the potential to address the underlying inflammatory causes of a wide range of unmet medical needs by targeting IL-1 beta, a cytokine that triggers inflammatory pathways in the body. In 2010, we completed a successful Phase 2 proof-of-concept trial of XOMA 052 in Behcet’s uveitis and initiated two Phase 2 clinical trials in Type 2 diabetes patients and one Phase 2 trial in Type 1 diabetes patients.
|
|
|
In January of 2011, we announced interim results from three months’ treatment with XOMA 052 or placebo in the 74 patient Phase 2a Type 2 diabetes trial, showing that XOMA 052 was well-tolerated and demonstrated evidence of biological activity. We expect to report top line, six month results from the Phase 2b Type 2 diabetes trial, in which 420 patients were enrolled, in the first quarter of 2011, and results from the full six months’ treatment in the Phase 2a trial in the second quarter of 2011.
|
|
|
In 2010, we also completed and announced positive results from an open-label pilot study of XOMA 052 in patients with uveitis of Behcet’s disease who were suffering from vision-threatening exacerbations despite maximal doses of immunosuppressive medicines. XOMA 052 has been designated as an orphan drug for the treatment of Behcet’s disease by the U.S. Food and Drug Administration (“FDA”) and the European Medicines Agency (“EMA”).
|
|
|
In December of 2010, we entered into an agreement with Servier to jointly develop and commercialize XOMA 052. This collaboration agreement substantially increases our cash resources while reducing future cash requirements, provides the funding to move XOMA 052 into Phase 3 development in 2011 in Behcet’s uveitis, and supports further development in diabetes and cardiovascular diseases.
|
|
|
·
|
Continue building our biodefense business. To date, we have been awarded three contracts, totaling nearly $100 million, from NIAID, to support our ongoing development of XOMA 3AB and additional product candidates toward clinical trials in the treatment of botulism poisoning. In addition, our biodefense programs include two subcontracts with SRI International totaling $4.3 million, funded through NIAID, for the development of antibodies to neutralize H1N1 and H5N1 influenza viruses and the virus that causes severe acute respiratory syndrome (“SARS”). We will continue to seek further opportunities to work with government and other institutions.
|
|
|
·
|
Advancing our proprietary preclinical pipeline candidates. We will continue to develop our proprietary preclinical pipeline, which includes candidates in development for autoimmune, cardio-metabolic, inflammatory, and oncological diseases.
|
|
|
·
|
Generate collaboration and licensing revenue. We have generated significant revenue from collaborations and licensing related to our proprietary technologies, including our phage display libraries, BCE, HE™, and Targeted Affinity Enhancement (“TAE™”) technologies. Historically, we have established technology collaborations with several companies to provide access to multiple proprietary antibody discovery and optimization technologies. In addition, we have licensed our BCE technology to more than 50 companies in exchange for license, milestone and other fees, royalties and complementary technologies, and a number of licensed product candidates are in clinical development. We believe we can continue to generate significant revenue from our proprietary technologies in the future.
|
Proprietary Products
As part of our strategy, we are focusing our technology and resources on advancing our emerging proprietary pipeline. Below is a summary of our proprietary products:
|
|
·
|
XOMA 052 is a potent monoclonal antibody with the potential to improve the treatment of patients with a wide variety of inflammatory diseases. XOMA 052 binds strongly to IL-1 beta, a pro-inflammatory cytokine involved in the development of Behcet’s uveitis, Type 2 diabetes, cardiovascular disease, rheumatoid arthritis, gout and other diseases. By binding to IL-1 beta, XOMA 052 inhibits the activation of the IL-1 receptor, thereby preventing the cellular signaling events that produce inflammation. XOMA 052 is a humanized IgG2 antibody. Based on its binding properties, specificity for IL-1 beta and half-life in the body, XOMA 052 may provide convenient dosing of once per month or less frequently.
|
During 2010, we completed patient enrollment in the Phase 2a and Phase 2b clinical trials of XOMA 052 in Type 2 diabetes patients. The primary goal of the 74 patient Phase 2a trial was to gain additional XOMA 052 safety information in Type 2 diabetes patients on a background of stable metformin monotherapy. The patients were randomized at approximately a 3:1 ratio to receive three months of treatment with either XOMA 052 at a single dose level or placebo, respectively, after which patients in the XOMA 052 group received an additional three months of treatment at the same, a higher or a lower dose of XOMA 052. In January of 2011, we announced an interim review of 3-month data from the Phase 2a trial where XOMA 052 was shown to be well-tolerated with no significant differences in adverse events, lab abnormalities or vital signs between the XOMA 052 and placebo groups and no drug-related serious adverse events. At the time of this 3-month review, evidence of biological activity was observed including a reduction in C-reactive protein levels and a modest reduction in hemoglobin A1c (“HbA1c”) levels. C-reactive protein is a biomarker of cardiovascular risk, and HbA1c is a measure indirectly reflecting blood glucose levels as averaged over a period estimated to be 90 to 120 days. Separately, we anticipate reporting top line, six month results from the Phase 2b trial by the end of the first quarter of 2011. The primary goal of the 420 patient Phase 2b trial was to further evaluate the use of multiple dose regimens on the safety, pharmacodynamics and efficacy of XOMA 052 in cardiometabolic and other diseases, and based on positive results to select doses for pivotal Phase 3 studies.
Also during 2010, XOMA announced positive results from a Phase 2 proof-of-concept clinical trial evaluating XOMA 052 in Behcet’s uveitis, a vision-threatening complication of Behcet’s disease, demonstrating rapid improvement in vision-threatening disease exacerbations in all seven treated patients despite discontinuation of immunosuppressive drugs such as cyclosporine and/or azathioprine. Follow-up results demonstrated that each of the five patients re-treated with XOMA 052 after they experienced a new uveitis exacerbation responded again to XOMA 052 treatment and maintained their response for several months. The drug was well-tolerated in this trial, and no drug-related adverse events were reported.
In August of 2010, we obtained FDA orphan drug status for XOMA 052 for the treatment of Behcet’s disease. The designation offers a number of potential incentives, which may include, among others, a seven-year period of U.S. marketing exclusivity from the date of marketing authorization, written guidance on the non-clinical and clinical studies needed to obtain marketing approval, and tax credits for certain clinical research. In October of 2010, XOMA 052 was granted orphan drug status for the treatment of Behcet’s disease by the EMA. The designation generally provides EU market exclusivity for up to ten years following approval for the given indication. Other potential benefits include protocol assistance, direct access to centralized marketing authorization procedures and financial incentives.
|
|
·
|
XOMA 3AB is a multi-antibody product designed to neutralize the most potent of the botulinum toxins, Type A, which causes paralysis and is a bioterrorism threat. Our anti-botulism program was recently expanded to include additional product candidates and is the first of its kind to combine multiple human antibodies to target a broad spectrum of the most toxic botulinum toxins, including the three most toxic serotypes of botulism, Types A, B and E. The antibodies are designed to bind to each toxin and enhance the clearance of the toxin from the body. The use of multiple antibodies increases the likelihood of clearing the harmful toxins by providing specific protection against each toxin type. In contrast to existing agents that treat botulism, XOMA uses advanced human monoclonal antibody technologies in an effort to achieve superior safety, potency and efficacy, and avoid life-threatening immune reactions associated with animal-derived products.
|
XOMA 3AB is currently in preclinical studies to assess safety through funding provided by NIAID. We have a history of successfully providing contract services to the U.S. government for the development of anti-botulinum neurotoxin antibodies.
|
|
·
|
Preclinical Product Pipeline: We are pursuing additional opportunities to further broaden our preclinical product pipeline. These include internal discovery programs, product development collaborations with other pharmaceutical and biotechnology companies and evaluation of product in-licensing, in-kind product trades and acquisition opportunities.
|
Partnership Products
Historically, XOMA has provided contract research and development services for world-class organizations, such as Novartis, Takeda, and Schering Plough Research Institute, a division of Schering Corporation, now a subsidiary of Merck & Co. (referred to herein as “Merck/Schering-Plough”), in pursuit of new antibody products. In more recent years, we have been evolving our business focus from a service provider model to a proprietary product development model. However, we will continue to capitalize on collaborative partnership arrangements as opportunities arise. Below is a list of activities in 2010 through such collaborations:
|
|
·
|
Therapeutic Antibodies with Takeda: Since 2006, Takeda has been a collaboration partner for therapeutic monoclonal antibody discovery and development against multiple targets selected by them. In February of 2009, we expanded our existing collaboration to provide Takeda with access to multiple antibody technologies, including a suite of research and development technologies and integrated information and data management systems. In the first quarter of 2010, we received a $1.0 million payment from Takeda for achieving a pre-established, pre-clinical milestone under our collaboration agreement and may receive potential milestones and royalties on sales of antibody products in the future.
|
|
|
·
|
Therapeutic Antibodies with Novartis: In November of 2008, we restructured our product development collaboration with Novartis. Under the restructured agreement, Novartis received control over the two ongoing programs under the original product development collaboration entered into in 2004 with Novartis (then Chiron Corporation). In exchange, we recognized $13.7 million in revenue in 2008 and may, in the future, receive milestones and double-digit royalty rates for the programs and options to develop or receive royalties from four additional programs.
|
|
|
·
|
Therapeutic Antibodies with Merck/Schering-Plough: Merck/Schering-Plough has been a collaboration partner since 2006 for therapeutic monoclonal antibody discovery and development against multiple targets selected by them. In January of 2011, we successfully completed the services to Merck/Schering-Plough and the collaboration agreement in now complete.
|
Technology Licenses and Royalties
Technology Licenses
Below is a summary of certain proprietary technologies owned by us and available for licensing to other companies:
|
|
·
|
Antibody discovery technologies: XOMA uses human antibody phage display libraries in its discovery of therapeutic candidates, and we offer access to multiple libraries, including novel libraries developed internally, as part of our collaboration business. We believe that access to multiple libraries offers a number of benefits to XOMA and its collaboration partners, because it enables use of libraries best suited to the needs of a particular discovery project to increase the probability of technical and business success in finding rare and unique functional antibodies directed to targets of interest.
|
|
|
·
|
Bacterial Cell Expression: The production or expression of antibodies using bacteria is an enabling technology for the discovery and selection, as well as the development and manufacture, of recombinant protein pharmaceuticals, including diagnostic and therapeutic antibodies for commercial purposes. Genetically engineered bacteria are used in the recombinant expression of target proteins for biopharmaceutical research and development. Reasons include the relative simplicity of gene expression in bacteria as well as many years of experience culturing such species as E. coli in laboratories and manufacturing facilities. XOMA scientists have developed bacterial expression technologies for producing antibodies and other recombinant protein products.
|
We have granted more than 50 licenses to biotechnology and pharmaceutical companies to use our patented and proprietary technologies relating to bacterial expression of recombinant pharmaceutical products. Bacterial antibody expression is also a key technology used in multiple systems for high-throughput screening of antibody domains. Expression of antibodies by phage display technology, for example, depends upon the expression and secretion of antibody domains from bacteria as properly folded, functional proteins.
Many licensees of our bacterial cell expression technology have developed, or are in the process of developing, antibodies for which we may be entitled to future milestone payments and royalties on product sales. Under the terms of our license agreement with Pfizer, Inc. (“Pfizer”), signed in 2007, we received an up-front cash payment of $30 million and from 2008 through 2010 we received milestone payments relating to five undisclosed product candidates, including a payment of $0.5 million for the initiation of a Phase 3 clinical trial. We may also be eligible for additional milestone payments aggregating up to $6.4 million relating to these five product candidates and low single-digit royalties on future sales of all products subject to this license. In addition, we may receive potential milestone payments aggregating up to $1.7 million for each additional qualifying product candidate. Our right to milestone payments expires on the later of the expiration of the last-to-expire licensed patent or the tenth anniversary of the effective date. Our right to royalties expires upon the expiration of the last-to-expire licensed patent.
Current licensees include but are not limited to the following companies:
|
Active Biotech AB
|
Centocor Ortho Biotech (now a member of Johnson & Johnson)
|
MorphoSys AG
|
|
Affimed Therapeutics AG
|
Crucell Holland B.V. (now a member of Johnson & Johnson)
|
Novartis AG
|
|
Affitech AS
|
Dompe, s.p.a.
|
Pfizer Inc.
|
|
Alexion Pharmaceuticals, Inc.
|
Dyax Corp.
|
Takeda Pharmaceutical Company Ltd.
|
|
Applied Molecular Evolution, Inc. (now a subsidiary of Eli Lilly and Company)
|
Eli Lilly and Company
|
The Medical Research Council
|
|
Avecia Limited
|
Genentech, Inc. (now a member of the Roche Group)
|
UCB S.A.
|
|
Aventis Pharma Deutschland GmbH (Hoechst) (now Sanofi-Aventis)
|
Invitrogen Corporation
|
Verenium Corporation
|
|
Bayer Healthcare AG
|
Merck & Co., Inc.
|
Wyeth Pharmaceuticals Division (now a member of Pfizer Inc.)
|
|
BioInvent International AB
|
Mitsubishi Tanabe Pharma Corporation
|
ZymoGenetics, Inc. (now a member of Bristol-Myers Squibb Company)
|
These licenses are sometimes associated with broader agreements which may include expanded license rights, cell line development and process development.
|
|
·
|
Human Engineering™: HE™ is a proprietary technology that allows modification of non-human monoclonal antibodies to reduce or eliminate detectable immunogenicity and make them suitable for medical purposes in humans. The technology uses a unique method developed by us, based on analysis of the conserved structure-function relationships among antibodies. The method defines which residues in a non-human variable region are candidates to be modified. The result is a HE™ antibody with preserved antigen binding, structure and function, and with eliminated or greatly reduced immunogenicity. Human Engineering™ technology is used in development of XOMA 052 and certain other antibody products.
|
|
|
·
|
Targeted Affinity Enhancement™: TAE™ is a proprietary technology involving the assessment and guided substitution of amino acids in antibody variable regions, enabling efficient optimization of antibody binding affinity and selectivity modulation. TAE™ generates a comprehensive map of the effects of amino acid mutations likely to impact binding. The technology is utilized by XOMA scientists and has been licensed to a number of our collaborators.
|
We also have access to certain intellectual property rights and services that augment our existing integrated antibody technology platform and development capabilities and further compress product development timelines. This broad antibody technology platform and expertise is available for building our antibody product pipeline as well as those of our collaborators.
Royalties
In August of 2010, XOMA sold its royalty interest in CIMZIA® (certolizumab pegol) to an undisclosed buyer for gross proceeds of $4.0 million. Prior to the sale, XOMA earned low single digit royalties on sales of CIMZIA® in the U.S. and Canada from UCB Celltech, a branch of UCB S.A. (“UCB”). Royalties earned from these sales were $0.5 million in 2010, $0.5 million in 2009 and $0.1 million in 2008. CIMZIA®, an anti-tumor necrosis factor product, was approved by the FDA in April of 2008 for the treatment of moderate-to-severe Crohn’s disease in adults who have not responded to conventional therapies. In addition, CIMZIA® was approved for the treatment of moderate-to-severe rheumatoid arthritis in adults by the FDA in May of 2009 and in Canada in September of 2009. UCB is responsible for the marketing and sales effort in support of this product. We will no longer receive royalties on sales of CIMZIA®.
Proprietary Product Summary:
The following table describes important information related to the proprietary products we are currently developing:
|
Program
|
Description
|
Indication
|
Status
|
Developer
|
|
XOMA 052
|
HE™ antibody to IL-1 beta
|
Behcet’s uveitis, Type 2 diabetes, Type 1 diabetes and cardiovascular disease
|
Phase 2 for Behcet’s uveitis, Type 2 diabetes, Type 1 diabetes and cardiovascular disease
|
Proprietary (in collaboration with Servier)
|
|
XOMA 3AB
|
Therapeutic antibodies to multiple botulinum neurotoxins
|
Botulism poisoning
|
Preclinical
|
Proprietary (NIAID-funded)
|
|
Multiple preclinical programs
|
Fully human monoclonal antibodies to undisclosed disease targets
|
Inflammatory, autoimmune, infectious and oncological diseases
|
Preclinical
|
Proprietary
|
Partnership Product Summary:
The following table describes important information related to certain products that we are currently developing or have developed in the past, for which we may earn royalties on product sales in the future:
|
Program
|
Description
|
Indication
|
Status
|
Developer
|
|
HCD 122 and LFA 102
|
Fully human antibody to CD40 and other monoclonal antibodies to undisclosed disease targets
|
Hematologic tumors and other undisclosed diseases
|
Phase 1 and 2 and Phase 1
|
Novartis
|
|
Therapeutic antibodies
|
Fully human monoclonal antibodies to undisclosed disease targets
|
Undisclosed
|
Preclinical
|
Takeda (fully-funded)
|
|
Therapeutic antibodies
|
Fully human monoclonal antibodies to undisclosed disease targets
|
Non-small cell lung cancer
|
Phase 2
|
AVEO (fully-funded)
|
Licensed Product Summary:
The following table describes important information related to certain products developed under licenses with us, for which we earn or may earn royalties on product sales in the future:
|
Program
|
Description
|
Indication
|
Status
|
Developer
|
|
Various products in development by Pfizer
|
Various monoclonal antibodies to undisclosed disease targets
|
Undisclosed diseases
|
Various phases of clinical and preclinical development
|
Pfizer
|
|
Various products in development by other licensees
|
Various monoclonal antibodies to undisclosed disease targets
|
Undisclosed diseases
|
Various phases of clinical and preclinical development
|
Various licensees
|
Financial and Legal Arrangements of Product Collaborations, Licensing and Other Arrangements
Collaboration and Licensing Agreements
Servier
We have entered into a license and collaboration agreement with Servier, to jointly develop and commercialize XOMA 052 in multiple indications, which provides for a non-refundable upfront payment of $15 million that was received by us in January of 2011. Under the terms of the agreement, Servier has worldwide rights to diabetes and cardiovascular disease indications and rights outside the U.S. and Japan to Behcet’s uveitis and other inflammatory and oncology indications. XOMA retains development and commercialization rights for Behcet's uveitis and other inflammatory disease and oncology indications in the U.S. and Japan, and has an option to reacquire rights to diabetes and cardiovascular disease indications from Servier in these territories (the “Cardiometabolic Indications Option”). Should we exercise the Cardiometabolic Indications Option, we will be required to pay Servier an option fee and partially reimburse their incurred development expenses.
Under this agreement, Servier will fully fund activities to advance the global clinical development and future commercialization of XOMA 052 in diabetes and cardiovascular related diseases. Also, Servier will fund $50 million of future XOMA 052 global clinical development and chemistry and manufacturing controls (“CMC”) expenses and 50% of further expenses for the Behcet’s uveitis indication. We will also be responsible for manufacturing XOMA 052 throughout clinical development and launch.
In addition, under the agreement, we are eligible to receive a combination of Euro- and US Dollar (“USD”)-denominated, development and sales milestones for multiple indications aggregating to a potential maximum of approximately $470 million when converted using the December 31, 2010 Euro to USD exchange rate (the “12/31/10 Exchange Rate”), if XOMA reacquires diabetes and cardiovascular rights in the U.S. and Japan. If XOMA does not reacquire these rights, then the milestone payments aggregate to a potential maximum of approximately $770 million converted using the 12/31/10 Exchange Rate. Milestone payments for which XOMA will be eligible under the agreement include $20 million upon initiation of the first Phase 3 clinical trial for XOMA 052 by Servier in its licensed territory in Type 2 diabetes. Servier’s obligation to pay development and commercialization milestones will continue for so long as Servier is developing or selling products under the agreement.
We are also eligible to receive royalties on XOMA 052 sales, which are tiered based on sales levels and range from a mid-single digit to up to a mid-teens percentage rate. Our right to royalties with respect to a particular product and country will continue for so long as such product is sold in such country.
The collaboration will be carried out and managed by committees mutually established by the parties. In general, in the event of any disputes, each party will have decision-making authority over matters relating to its areas of responsibility and territory, but neither party will have unilateral decision-making rights if the decision would have a material adverse impact on the other party’s rights in its territory. The agreement contains customary termination rights relating to matters such as material breach by either party, safety issues and patents. Servier also has a unilateral right to terminate the agreement on a country-by-country basis or in its entirety on 6 months’ notice.
We have also entered into a loan agreement with Servier, which provides for an advance of up to €15 million, which converts to approximately $20 million using the 12/31/10 Exchange Rate. The loan was fully funded in January of 2011. This loan is secured by an interest in our intellectual property rights to all XOMA 052 indications worldwide, excluding the U.S. and Japan territories. The loan has a final maturity date in 2016; however, after a specified period prior to final maturity, the loan is required to be repaid (i) at Servier's option, by applying up to a significant percentage of any milestone or royalty payments owed by Servier under our collaboration agreement and (ii) using a significant percentage of any upfront, milestone or royalty payments we receive from any third party collaboration or development partner for rights to XOMA 052 in the U.S. and/or Japan. In addition, the loan becomes immediately due and payable upon certain customary events of default. Refer to Item 7: Management’s Discussion and Analysis of Financial Condition and Results of Operations: Subsequent Events for further information regarding our loan agreement with Servier.
NIAID
In March of 2005, we were awarded a $15 million competitive bid contract from NIAID to develop three anti-botulinum neurotoxin monoclonal antibodies. Under this contract, we created production cell lines using our proprietary antibody expression systems, built Master and Manufacturer’s Working Cell Banks, developed production processes and produced initial quantities of the three antibodies. The contract was performed over an 18-month period and was fully funded with federal funds from NIAID under Contract No. HHSN266200500004C (“NIAID 1”). Final acceptance of the project was received in October of 2006.
In July of 2006, we were awarded a $16.3 million NIAID contract under Contract No. HHSN266200600008C/N01-Al-60008 (“NIAID 2”) to produce monoclonal antibodies for the treatment of botulism to protect United States citizens against the harmful effects of botulinum neurotoxins used in bioterrorism. Under this contract, we created and produced XOMA 3A, an innovative injectable product comprised of three anti-type A botulinum neurotoxin monoclonal antibodies. This work was complete in the third quarter of 2010.
In September of 2008, we were awarded a third NIAID contract for $65 million under Contract No. HHSN272200800028C (“NIAID 3”) to continue development of our anti-botulinum antibody product candidates, including XOMA 3AB and additional product candidates. As part of the contract, we are developing, evaluating and producing the clinical supplies to support an IND filing with the FDA and conduct preclinical studies required to support human clinical trials.
SRI International
In the third quarter of 2009, we began work on two biodefense subcontract awards from SRI International, including a $2.1 million award to develop novel antibody drugs against the virus that causes SARS and a $2.2 million award to develop a novel antibody, known as F10, that has been shown to neutralize group 1 influenza A viruses, including the H1N1 and H5N1 strains. The subcontract awards are funded through NIAID.
Takeda
In November of 2006, we entered into a fully funded collaboration agreement with Takeda for therapeutic monoclonal antibody discovery and development under which we agreed to discover and optimize therapeutic antibodies against multiple targets selected by Takeda. Takeda agreed to make up-front, annual maintenance and milestone payments to us, fund our research and development and manufacturing activities for preclinical and early clinical studies and pay royalties on sales of products resulting from the collaboration. Takeda is responsible for clinical trials and commercialization of drugs after an IND submission and is granted the right to manufacture once a product enters into Phase 2 clinical trials. In the first quarter of 2010, a discovery and development program with Takeda under this collaboration was discontinued following the analysis of research data. The termination resulted in the recognition of the remaining unamortized balance in deferred revenue of $1.1 million in the first quarter of 2010, as no continuing performance obligations exist. Separately, we received a $1.0 million payment from Takeda for achieving a pre-established, preclinical milestone under the only currently active discovery and development program with Takeda. We recognized this milestone payment in revenue in the first quarter of 2010. We have completed a technology transfer and do not expect to perform any further contract research and development services under this program.
Under the terms of this agreement, we may receive milestone payments aggregating up to $20.75 million relating to one undisclosed product candidate and low single-digit royalties on future sales of all products subject to this license. In addition, in the event Takeda were to develop additional future qualifying product candidates under the terms of our agreement, we would be eligible for milestone payments aggregating up to $20.75 million for each such qualifying product candidate. Our right to milestone payments expires on the later of the receipt of payment from Takeda of the last amount to be paid under the agreement or the cessation of all research and development activities with respect to all program antibodies, collaboration targets and/or collaboration products. Our right to royalties expires on the later of 13.5 years from the first commercial sale of each royalty-bearing discovery product or the expiration of the last-to-expire licensed patent.
In February of 2009 we expanded our existing collaboration to provide Takeda with access to multiple antibody technologies, including a suite of research and development technologies and integrated information and data management systems. We may receive milestones of up to $3.25 million per discovery product candidate and low single-digit royalties on future sales of all antibody products subject to this license. Our right to milestone payments expires on the later of the receipt of payment from Takeda of the last amount to be paid under the agreement or the cessation of all research and development activities with respect to all program antibodies, collaboration targets and/or collaboration products. Our right to royalties expires on the later of 10 years from the first commercial sale of such royalty-bearing discovery product, or the expiration of the last-to-expire licensed patent.
Novartis
In November of 2008, we restructured our product development collaboration with Novartis, which involves six development programs including the HCD122 program. HCD122, which is a fully human anti-CD40 antagonist antibody, intended as a treatment for B-cell mediated diseases, including malignancies and autoimmune diseases, is currently recruiting patients for a Phase 1/2 lymphoma trial. The antibody has a dual mechanism of action that involves inhibition of CD40-ligand mediated growth and survival while recruiting immune effector cells to kill CD40-expressing tumor cells through a process known as antibody-dependent cellular cytotoxicity (ADCC). CD40, a member of the tumor necrosis factor, or TNF, family of antigens, is a cell surface antigen expressed in B-cell malignancies and involved in a broad variety of immune and inflammatory responses.
Under the restructured agreement, Novartis made a payment to us of $6.2 million in cash; reduced our existing debt by $7.5 million; will fully fund all future research and development expenses; may pay potential milestones of up to $14 million and royalty rates ranging from 10% to 20% for two ongoing product programs, HCD122 and LFA 102; and has provided us with options to develop or receive royalties on four additional programs. In exchange, Novartis has control over the HCD122 and LFA 102 programs, as well as the right to expand the development of these programs into additional indications outside of oncology. As part of the agreement, Novartis paid us for all project costs incurred after July 1, 2008. Our right to milestone payments expires at such time as no collaboration product or former collaboration product is being developed or commercialized anywhere in the world and no royalty-style payments on these products are due. Our right to royalty-style payments expires on the later of the expiration of any licensed patent covering each product or 20 years from the launch of each product that is produced from a cell line provided to Novartis by XOMA.
The collaboration between XOMA and Novartis (then Chiron Corporation) began in 2004 with the signing of an exclusive, worldwide, multi-product agreement to develop and commercialize multiple antibody products for the treatment of cancer. We shared expenses and revenue, generally on a 70-30 basis, with our share being 30 percent. Financial terms included initial payments to us in 2004 totaling $10 million and a note agreement, secured by our interest in the collaboration, to fund up to 75 percent of our share of expenses beginning in 2005. The secured note agreement with Novartis, which was executed in May of 2005, is due and payable in full in June of 2015. At December 31, 2010, the outstanding principal balance under this note agreement totaled $13.7 million and, pursuant to the terms of the arrangement as restructured in November of 2008, we will not make any additional borrowings on the Novartis note. In the first quarter of 2007, the mutual obligations of XOMA and Novartis to work together on an exclusive basis in oncology expired, except with respect to existing collaborative product development projects.
In December of 2008, we entered into a Manufacturing and Technology Transfer Agreement with Novartis, effective July 1, 2008. Under this agreement, XOMA was engaged by Novartis to perform research and development, process development, manufacturing and technology transfer activities with respect to the ongoing product programs now controlled by Novartis under the restructured product development collaboration. The work performed by XOMA under this agreement, which was fully funded by Novartis, was completed in the third quarter of 2009.
Arana
In September of 2009, we entered into an antibody discovery collaboration with Arana Therapeutics Limited (“Arana”), a wholly-owned subsidiary of Cephalon, Inc., involving multiple proprietary XOMA antibody research and development technologies, including a new antibody phage display library and a suite of integrated information and data management systems. Arana agreed to pay us a fee of $6.0 million, of which we received $4.0 million in the third quarter of 2009 and $2.0 million in the third quarter of 2010. Also, we may be entitled to future milestone payments, aggregating up to $3.0 million per product, and low single-digit royalties on product sales. Our right to milestone payments expires on the later of the receipt of payment from Arana of the last amount to be paid under the agreement, the cessation by Arana of the use of all research and development technologies or the cessation by Arana of the exercise of the patent rights granted to them. Our right to royalties expires five years from the first commercial sale of each royalty-bearing product.
Kaketsuken
In October of 2009, we entered into an antibody discovery collaboration with The Chemo-Sero-Therapeutic Research Institute, a Japanese research foundation known as Kaketsuken, involving multiple proprietary XOMA antibody research and development technologies, including a new antibody phage display library and a suite of integrated information and data management systems. Kaketsuken agreed to pay us a fee of $8.0 million, of which we received $6.0 million in the fourth quarter of 2009 and $2.0 million in the fourth quarter of 2010. Also, we may be entitled to future milestone payments, aggregating up to $0.2 million per product, and low single-digit royalties on product sales. Our right to milestone payments expires upon the receipt of payment from Kaketsuken of the last amount to be paid pursuant to the agreement. Our right to royalties expires 15 years from the first commercial sale of each royalty-bearing product.
Merck/Schering-Plough/AVEO Pharmaceuticals, Inc. (“AVEO”)
In April of 2006, we entered into an agreement with AVEO to utilize our HE™ technology to humanize AV-299, AVEO’s novel anti-HGF antibody, under which AVEO paid us an up-front license fee and development milestones. In addition, we will receive royalties on sales of products resulting from the agreement. Under this agreement we created four Human Engineering™ versions of the original AV-299, all of which met design goals and from which AVEO selected one as its lead development candidate. In September of 2006, as a result of the successful humanization of AV-299, we entered into a second agreement with AVEO to manufacture and supply AV-299 in support of early clinical trials. Under the agreement, we created AV-299 production cell lines, conducted process and assay development, and performed Good Manufacturing Practices (“cGMP”) manufacturing activities. AVEO retains all development and commercialization rights to AV-299 and may be required to pay XOMA annual maintenance fees, additional development milestone payments aggregating up to $6.3 million and low single-digit royalties on product sales in the future. Our right to milestone payments expires upon full satisfaction of all financial obligations of AVEO pursuant to the agreement. Our right to royalties expires on the later of 15 years from the first commercial sale of each royalty-bearing product or the expiration of the last-to-expire licensed patent. In the third quarter of 2010, the Company received a $0.8 million milestone payment related to AVEO’s initiation of a Phase 2 clinical trial to evaluate AV-299 for the treatment of non-small cell lung cancer. The Company recognized this milestone payment as revenue in the third quarter of 2010.
In April of 2007, Merck/Schering-Plough entered into a research, development and license agreement with AVEO concerning AV-299 and other anti-HGF molecules. In connection with the aforementioned license agreement, AVEO assigned its entire right, title and interest in, to and under its manufacturing agreement with XOMA to Merck/Schering-Plough. In the third quarter of 2010, AVEO regained its worldwide rights from Merck/Schering-Plough to develop and commercialize AV-299 and other anti-HGF molecules.
Merck/Schering-Plough
In May of 2006, we entered into a fully funded collaboration agreement with Merck/Schering-Plough for therapeutic monoclonal antibody discovery and development. Under the agreement, Merck/Schering-Plough made up-front, annual maintenance and milestone payments to us, funded our research and development activities related to the agreement and would have paid royalties on sales of products resulting from the collaboration. During the collaboration, we discovered therapeutic antibodies against multiple targets selected by Merck/Schering-Plough using multiple human antibody phage display libraries, optimized antibodies through affinity maturation or other protein engineering, used our proprietary HE™ technology to humanize antibody candidates generated by hybridoma techniques, performed preclinical studies to support regulatory filings, developed cell lines and production processes and produced antibodies for initial clinical trials. Merck/Schering-Plough selected the first target at the inception of the agreement and, in December of 2006, exercised its right to initiate the additional discovery and development programs. In January of 2011, we successfully completed the services to Merck/Schering-Plough and the collaboration agreement in now complete.
UCB
Celltech Therapeutics Ltd., now UCB Celltech, a branch of UCB, utilized our bacterial cell expression technology under license in the development of CIMZIA® for the treatment of moderate-to-severe Crohn’s disease in adults who have not responded to conventional therapies and for the treatment of moderate-to-severe rheumatoid arthritis in adults. The license provides for a low-single digit royalty on sales of CIMZIA® in countries where our bacterial cell expression technology is patented, which includes the U.S. and Canada, until the expiration of the last-to-expire licensed patent. In August of 2010, we sold our royalty interest in CIMZIA® to an undisclosed buyer for gross proceeds of $4.0 million. We will no longer receive royalties on sales of CIMZIA®.
Genentech
In April of 1996, we entered into a collaboration agreement with Genentech, Inc., a wholly-owned member of the Roche Group (referred to herein as “Genentech”) for the development of RAPTIVA®. In March of 2003, we entered into amended agreements which called for us to share in the development costs and called for Genentech to finance our share of development costs via a convertible subordinated loan. Under the loan agreement, upon FDA approval of the product, which occurred in October of 2003, we elected to pay $29.6 million of the development loan in convertible preference shares, which are convertible into approximately 0.3 million common shares at a price of $116.25 per common share.
In January of 2005, we restructured our arrangement with Genentech on RAPTIVA® under which we were entitled to receive mid-single digit royalties on worldwide sales of RAPTIVA® in all indications. The previous cost and profit sharing arrangement for RAPTIVA® in the U.S. was discontinued and Genentech was responsible for all operating and development costs associated with the product. In the first half of 2009, RAPTIVA® was withdrawn from the commercial drug markets and royalties ceased.
Genentech utilized our bacterial cell expression technology under license in the development of LUCENTIS® for the treatment of neovascular wet age-related macular degeneration. LUCENTIS® was approved by the FDA in June of 2006 and in the European Union in January of 2007. We were entitled to receive a low-single digit royalty on worldwide sales of LUCENTIS®. In the third quarter of 2009, we sold our LUCENTIS® royalty interest to Genentech for $25 million, including royalty revenue from the second quarter of 2009. We will not receive any further royalties from sales of LUCENTIS®.
Financing Agreements
Underwritten Offering
In February of 2010, we completed an underwritten offering of 2.8 million units, with each unit consisting of one of our common shares and a warrant to purchase 0.45 of a common share, for gross proceeds of approximately $21 million. As of December 31, 2010 all of these warrants were outstanding.
Registered Direct Offerings
In May of 2009, we entered into a definitive agreement with an institutional investor to sell 784,313 units, with each unit consisting of one of our common shares and a warrant to purchase 0.50 of a common share, for gross proceeds of approximately $10 million, before deducting placement agent fees and estimated offering expenses of $0.8 million, in a registered direct offering. The investor purchased the units at a price of $12.75 per unit. The warrants, which represent the right to acquire an aggregate of up to 392,157 common shares, were exercisable at any time on or after May 15, 2009 and prior to May 20, 2014 at an exercise price of $15.30 per share. In February of 2010, the holders of these warrants agreed to amend the terms of their warrants to remove the provisions that would have required a reduction of the warrant exercise price and an increase in the number of shares issuable on exercise of the warrants each time we sold common shares at a price less than the exercise price of such warrants (the “Eliminated Adjustment Provisions”) and the exercise price of these warrants was reduced from $15.30 per share to $0.015 per share. In the first quarter of 2010, the holders of these warrants exercised all warrants, acquiring 392,157 common shares for an aggregate exercise price of $5,882.
In June of 2009, we entered into a definitive agreement with certain institutional investors to sell 695,652 units, with each unit consisting of one of our common shares and a warrant to purchase 0.50 of a common share, for gross proceeds of approximately $12 million, before deducting placement agent fees and estimated offering expenses of $0.8 million, in a second registered direct offering. The investor purchased the units at a price of $17.25 per unit. The warrants, which represent the right to acquire an aggregate of up to 347,826 common shares, are exercisable at any time on or prior to December 10, 2014 at an exercise price of $19.50 per share. In February of 2010, the holders of these warrants agreed to amend the terms of their warrants to remove the Eliminated Adjustment Provisions and we made a cash payment of $4.5 million to these warrant holders, which was recorded in other income (expense). The exercise price of these warrants remained unchanged at $19.50 per share. As of December 31, 2010 all of these warrants were outstanding.
ATM Agreements
In the third quarter of 2009, we entered into an At Market Issuance Sales Agreement (the “2009 ATM Agreement”), under which we could sell up to 1.7 million of our common shares from time to time through Wm Smith & Co. (“Wm Smith”), as our agent for the offer and sale of the common shares. Wm Smith could sell these common shares by any method permitted by law deemed to be an “at the market” offering as defined in Rule 415 of the Securities Act of 1933, including but not limited to sales made directly on The NASDAQ Global Market, on any other existing trading market for the common shares or to or through a market maker. Wm Smith could also sell the common shares in privately negotiated transactions, subject to our approval. We paid Wm Smith a commission equal to 3% of the gross proceeds of all common shares sold through it as sales agent under the 2009 ATM Agreement but in no event less than $0.02 per share. Shares sold under the 2009 ATM Agreement were sold pursuant to a prospectus which formed a part of our registration statement on Form S-3 (File No. 333-148342) (the “Existing Registration Statement”) filed with the U.S. Securities and Exchange Commission (the “SEC”) on December 26, 2007 and declared effective by the SEC on May 29, 2008. From the inception of the 2009 ATM Agreement through October of 2010, the Company sold a total of 1.7 million common shares through Wm Smith, constituting all of the shares available for sale under the agreement, for aggregate gross proceeds of $12.2 million, including 1.4 million common shares sold in 2010 for aggregate gross proceeds of $9.3 million. Total offering expenses related to these sales were $0.4 million.
In the third quarter of 2010, we entered into an At Market Issuance Sales Agreement (the “2010 ATM Agreement”), with Wm Smith and McNicoll, Lewis & Vlak LLC (the “Agents”), under which we may sell common shares from time to time through the Agents, as our agents for the offer and sale of the common shares, in an aggregate amount not to exceed the amount that can be sold under the Existing Registration Statement. The Agents may sell the common shares by any method permitted by law deemed to be an “at the market” offering as defined in Rule 415 of the Securities Act, including without limitation sales made directly on The NASDAQ Global Market, on any other existing trading market for the common shares or to or through a market maker. The Agents may also sell the common shares in privately negotiated transactions, subject to our prior approval. We will pay the Agents, collectively, a commission equal to 3% of the gross proceeds of the sales price of all common shares sold through them as sales agents under the 2010 ATM Agreement. From the inception of the 2010 ATM Agreement through December 31, 2010, we sold a total of 6.7 million common shares under this agreement for aggregate gross proceeds of $29.7 million. Total offering expenses related to these sales were $0.9 million. Subsequent to December 31, 2010 through March 8, 2011, we have sold an additional 796,898 common shares through the Agents pursuant to the 2010 ATM Agreement for aggregate gross proceeds of $4.3 million. Total offering expenses related to these sales were $0.1 million.
Equity Line of Credit
In July of 2010, we entered into a common share purchase agreement (the “Purchase Agreement”) with Azimuth Opportunity Ltd. (“Azimuth”), pursuant to which we obtained a committed equity line of credit facility (the “Facility”) under which we could sell up to $30 million of our registered common shares to Azimuth over a 12-month period, subject to certain conditions and limitations. The Purchase Agreement provided that we could determine, in our sole discretion, the timing, dollar amount and floor price per share of each draw down under the Facility, subject to certain conditions and limitations and that the number and price of shares sold in each draw down were generally to be determined by a contractual formula designed to approximate fair market value, less a discount. The Purchase Agreement also provided that from time to time and in our sole discretion, we could grant Azimuth the right to exercise one or more options to purchase additional common shares during each draw down pricing period for the amount of shares based upon the maximum option dollar amount and the option threshold price specified by us. We also agreed to issue 111,111 common shares to Azimuth upon execution of the agreement relating to the Facility, in consideration of Azimuth’s execution and delivery of that agreement. Shares under the Facility and the shares we agreed to issue to Azimuth upon execution of the agreement relating to the Facility were sold pursuant to a prospectus which forms a part of a registration statement declared effective by the SEC on May 29, 2008. In August of 2010, we sold a total of 3,421,407 common shares under the Facility for aggregate gross proceeds of $14.2 million, representing the maximum number of shares that could be sold under the Facility. As a result, the Facility is no longer in effect, and no additional shares can be issued thereunder.
Research and Development
Our research and development expenses currently include costs of personnel, supplies, facilities and equipment, consultants, third party costs and other expenses related to preclinical and clinical testing. In 2010, our research and development expenses were $77.4 million compared with $58.1 million in 2009 and $82.6 million in 2008.
Our research and development activities can be divided into those related to our internal projects and those related to collaborative and contract arrangements, which are reimbursed by our customers. In 2010, research and development expenses related to internal projects were $58.1 million compared with $42.2 million in 2009 and $58.5 million in 2008. In 2010, research and development expenses related to collaborative and contract arrangements were $19.3 million compared with $15.9 million in 2009 and $24.1 million in 2008. Refer to Item 7: Management’s Discussion and Analysis of Financial Condition and Results of Operations- Research and Development Expenses for further information regarding our research and development expenses.
Competition
The biotechnology and pharmaceutical industries are subject to continuous and substantial technological change. Competition in the areas of recombinant DNA-based and antibody-based technologies is intense and expected to increase as new technologies emerge and established biotechnology firms and large chemical and pharmaceutical companies continue to advance in the field. A number of these large pharmaceutical and chemical companies have enhanced their capabilities by entering into arrangements with or acquiring biotechnology companies or entering into business combinations with other large pharmaceutical companies. Many of these companies have significantly greater financial resources, larger research and development and marketing staffs and larger production facilities than ours. Moreover, certain of these companies have extensive experience in undertaking preclinical testing and human clinical trials. These factors may enable other companies to develop products and processes competitive with or superior to ours. In addition, a significant amount of research in biotechnology is being carried out in universities and other non-profit research organizations. These entities are becoming increasingly interested in the commercial value of their work and may become more aggressive in seeking patent protection and licensing arrangements. Furthermore, many companies and universities tend not to announce or disclose important discoveries or development programs until their patent position is secure or, for other reasons, later. As a result, we may not be able to track development of competitive products, particularly at the early stages. There can be no assurance that developments by others will not render our products or technologies obsolete or uncompetitive.
Without limiting the foregoing, we are aware of the following competitors for the products and candidates shown in the table below. This table is not intended to be representative of all existing competitors in the market:
|
Product/Candidate
|
Competitors
|
|
XOMA 052
|
Biovitrum AB
Eli Lilly and Company
MedImmune
Novartis AG
Regeneron Pharmaceuticals, Inc.
|
|
XOMA 3AB
|
Cangene Corporation
Emergent BioSolutions, Inc.
|
Regulatory
Our products are subject to comprehensive preclinical and clinical testing requirements and to approval processes by the FDA and by similar authorities in other countries. Our products are primarily regulated on a product-by-product basis under the United States Food, Drug and Cosmetic Act and Section 351(a) of the Public Health Service Act. Most of our human therapeutic products are or will be classified as biologic products. Approval of a biologic for commercialization requires licensure of the product and the manufacturing facilities. The review of therapeutic biologic products is carried out by the FDA’s Center for Drug Evaluation and Research, the body that also reviews drug products.
The FDA regulatory process is carried out in several phases. Prior to beginning human clinical testing of a proposed new biologic product, an IND is filed with the FDA. This document contains scientific information on the proposed product, including results of testing of the product in animal and laboratory models. Also included is information on manufacturing the product and studies on toxicity in animals and a clinical protocol outlining the initial investigation in humans.
The initial stage of clinical testing, Phase 1, ordinarily encompasses safety, pharmacokinetic and pharmacodynamic evaluations. Phase 2 testing encompasses investigation in specific disease states designed to provide preliminary efficacy data and additional information on safety. Phase 3 studies are designed to further establish clinical safety and efficacy and to provide information allowing proper labeling of the product following approval. Phase 3 studies are most commonly multi-center, randomized, placebo-controlled trials in which rigorous statistical methodology is applied to clinical results. Other designs may also be appropriate in specific circumstances.
Following completion of clinical trials, a BLA is submitted to the FDA to request marketing approval. Internal FDA committees are formed that evaluate the application, including scientific background information, animal and laboratory efficacy studies, toxicology, manufacturing facility and clinical data. During the review process, a dialogue between the FDA and the applicant is established in which FDA questions are raised and additional information is submitted. During the final stages of the approval process, the FDA generally requests presentation of clinical or other data before an FDA advisory committee, at which point, some or all of such data may become available. Also, during the later stages of review, the FDA conducts an inspection of the manufacturing facility to establish that the product is made in conformity with good manufacturing practice. If all outstanding issues are satisfactorily resolved and labeling established, the FDA issues a license for the product and for the manufacturing facility, thereby authorizing commercial distribution.
The FDA has substantial discretion in both the product approval process and the manufacturing approval process. It is not possible to predict at what point, or whether, the FDA will be satisfied with our submissions or whether the FDA will raise questions which may delay or preclude product approval or manufacturing facility approval. As additional clinical data is accumulated, it will be submitted to the FDA and may have a material impact on the FDA product approval process. Given that regulatory review is an interactive and continuous process, we have adopted a policy of limiting announcements and comments upon the specific details of the ongoing regulatory review of our products, subject to our obligations under the securities laws, until definitive action is taken. There can be no assurance any of the products we have under development will be developed successfully, obtain the requisite regulatory approval or be successfully manufactured or marketed.
In Europe, most of our human therapeutic products are or will be classified as biological medicinal products which are assessed through a centralized procedure by the EMA. The EMA coordinates the evaluation and supervision of medicinal products throughout the European Union and the European Economic Area. The assessment of the Marketing Authorization Application (“MA”) is carried out by a Rapporteur and a Co-Rapporteur appointed by the Committee for Medicinal Products for Human Use (“CHMP”), which is the expert scientific committee of the EMEA.
The Rapporteur and Co-Rapporteur are drawn from the CHMP membership representing member states of the European Union. In addition to their responsibility for undertaking scientific assessments of an application for a MA, the Rapporteur and the Co-Rapporteur liaise with the applicant on behalf of the CHMP in an effort to provide answers to queries raised by the CHMP. Their assessment report(s) is circulated to and considered by the full CHMP membership, leading to the production ultimately of a CHMP opinion which is transmitted to the applicant and the European Commission. The final decision on the grant of a MA is made by the European Commission as the licensing authority of the European Community (“Community”). Under Community law, a positive decision issued by the European Commission represents the grant of a MA. Such an authorization allows a medicinal product to be placed on the European market. Upon the grant of an MA in the European Union, certain member states require pricing approval before the product can be placed into commercial distribution.
Under Community law, the applicant may request grant of a MA under exceptional circumstances if comprehensive data on the efficacy and safety of the drug, under normal conditions of use cannot be provided because its intended indications are encountered so rarely (such as in the case of a medicinal product intended for treating an orphan disease) that comprehensive evidence cannot reasonably be collected, the present state of scientific knowledge will not allow comprehensive information to be collected, or it would be against generally accepted medical ethics to collect comprehensive information. The Rapporteur, Co-Rapporteur and the other CHMP members will assess the justification/data submitted for exceptional circumstances as part of the overall assessment of the benefit/risk of the application. It is up to the CHMP, during the review, to ultimately decide on whether grant of a MA under exceptional circumstances is justified on the evidence before them. Approval under exceptional circumstances is subject to a requirement for specific procedures related to safety and results of its use and is reviewed annually to reassess the risk-benefit balance of the product. Once approval is granted, the product can be marketed under the single European MA in all member states of the European Union and the European Economic Area. Consistent with the single MA, the labeling for Europe is identical throughout all member states except that all labeling must be translated into the local language of the country of intended importation and in relation to the content of the so called “blue box” on the outer packaging in which locally required information may be inserted.
Orphan drugs are those intended for use in rare diseases or conditions. As a result of the high cost of development and the low return on investment for rare diseases, governments provide regulatory and commercial incentives for the development of drugs for small disease populations. In the United States, the term ‘‘rare disease or condition’’ means any disease or condition which affects less than 200,000 persons in the United States. Applications for United States orphan drug status are evaluated and granted by the Office of Orphan Products Development (“OOPD”) of the FDA. In the United States, orphan drugs are subject to the standard regulatory process for marketing approval but are exempt from the payment of user fees for licensure, receive market exclusivity for a period of seven years and some tax benefits, and are eligible for OOPD grants. In Europe, orphan medicinal products are those intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting not more than five in 10,000 persons in the Community. The EMA’s Committee for Orphan Medicinal Products (“COMP”) reviews applications seeking orphan designation. If the European Commission agrees with a positive assessment made by COMP, then the product will receive a positive designation through adoption of a decision by the European Commission. Orphan medicinal products are exempt from fees for protocol assistance and scientific advice from the Scientific Advice Working Party during development, reduction or exemption of MA and other fees, and ten-year market exclusivity upon granting of a MA in respect of the approved clinical indication. Moreover, manufacturers may be eligible for grants or other financial incentives from the Community and Member States programs.
Patents and Trade Secrets
Patent and trade secret protection is important to our business and our future will depend in part on our ability to obtain patents, maintain trade secret protection and operate without infringing on the proprietary rights of others. As a result of our ongoing activities, we hold and have filed applications for a number of patents in the United States and internationally to protect our products and important processes. We also have obtained or have the right to obtain exclusive licenses to certain patents and applications filed by others. However, the patent position of biotechnology companies generally is highly uncertain and no consistent policy regarding the breadth of allowed claims has emerged from the actions of the U.S. Patent and Trademark Office ("Patent Office") with respect to biotechnology patents. Accordingly, no assurance can be given that our patents will afford protection against competitors with similar technologies, or others will not obtain patents claiming aspects similar to those covered by our patent applications.
We have issued patents in the U.S. and Europe for XOMA 052. U.S. Patent Nos. 7,531,166 and 7,582,742 cover XOMA 052 and other antibodies and antibody fragments with similar binding properties for IL-1 beta, as well as nucleic acids, expression vectors and production cell lines for the manufacture of such antibodies and antibody fragments. The patents provide exclusivity in the U.S. into 2027 and 2026, respectively. In addition, in April of 2010, the U.S. Patent and Trademark Office issued U.S. Patent No. 7,695,718 covering methods of treating Type 2 diabetes with high affinity antibodies and antibody fragments that bind to IL-1 beta, including XOMA 052, and Patent No. 7,695,717 covering methods of treating IL-1 related inflammatory diseases, including rheumatoid arthritis and osteoarthritis, with XOMA 052 and other antibodies and antibody fragments with similar binding properties for human interleukin-1 beta (IL-1 beta). These patents provide coverage into 2027 and 2026, respectively. Further, in November of 2010, the U.S. Patent and Trademark Office issued U.S. Patent No. 7,829,093 relating to methods of treating diabetes mellitus Type 1 with XOMA 052 or other IL-1 beta antibodies having similar binding properties, and U.S. Patent No. 7,829,094 relating to methods of treating a cancer with XOMA 052 or other IL-1beta antibodies having similar binding properties, with the cancer being selected from multiple myeloma, acute myelogenous leukemia and chronic myelogenous leukemia. These patents provide coverage to 2026. Additionally, U.S. Patents Nos. 7,744,865 and 7,744,866 were issued June 29, 2010, covering additional IL-1 beta antibodies and antibody fragments into 2026. Also, the European Patent Office granted a patent for XOMA 052, as well as nucleic acids, expression vectors and production cell lines for the manufacture of XOMA 052. The patent provides exclusivity in Europe into 2026.
We have established a portfolio of patents related to our bacterial expression technology, including claims to novel promoter sequences, secretion signal sequences, compositions, methods for expression and secretion of recombinant proteins from bacteria, including immunoglobulin gene products, and improved methods and cells for expression of recombinant protein products. U.S. Patent Nos. 5,576,195 and 5,846,818 are related to DNA encoding a pectate lyase signal sequence, recombinant vectors, host cells and methods for production and externalization of recombinant proteins. U.S. Patent Nos. 5,595,898, 5,698,435 and 5,618,920 address secretable immunoglobulin chains, DNA encoding the chains and methods for their recombinant production. U.S. Patent Nos. 5,693,493, 5,698,417 and 6,204,023 relate to methods for recombinant production/secretion of functional immunoglobulin molecules. U.S. Patent Nos. 7,094,579 and 7,396,661 relate to eukaryotic signal sequences and their use in methods for prokaryotic expression of recombinant proteins. U.S. Patent No. 6,803,210 relates to improved bacterial host cells that are deficient in one or more of the active transport systems for an inducer of an inducible promoter, such as arabinose for an araB promoter, and methods for the use of such cells for the production of recombinant proteins. Most of the more important European patents in this portfolio expired in July of 2008 or earlier.
We have also established a portfolio of patent applications related to our mammalian expression technology, including U.S. Patent No. 7,192,737, related to methods for increasing the expression of recombinant polypeptides using expression vectors containing multiple copies of a transcription unit encoding a polypeptide of interest.
We have established a portfolio of patents and applications related to our Human Engineering™ technology, including U.S. Patent No. 5,766,886, directed to methods of modifying antibody variable domains to reduce immunogenicity. Related patents and applications are directed to antibodies engineered according to our patented methods. We believe that our patented Human Engineering™ technology provides an attractive alternative to other humanization technologies.
If certain patents issued to others are upheld or if certain patent applications filed by others issue and are upheld, we may require certain licenses from others in order to develop and commercialize certain potential products incorporating our technology. There can be no assurance that such licenses, if required, will be available on acceptable terms.
Where appropriate, we also rely on trade secrets to protect aspects of our technology. However, trade secrets are difficult to protect. We protect our proprietary technology and processes, in part, by confidentiality agreements with our employees, consultants and collaborators. These parties may breach these agreements, and we may not have adequate remedies for any breach. Our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that we or our consultants or collaborators use intellectual property owned by others, we may have disputes with our collaborators or consultants or other third parties as to the rights in related or resulting know-how and inventions.
International Operations
We believe that, because the pharmaceutical industry is global in nature, international activities will be a significant part of our future business activities and that, when and if we are able to generate income, a substantial portion of that income will be derived from product sales and other activities outside the United States.
A number of risks are inherent in international operations. Foreign regulatory agencies often establish standards different from those in the United States. An inability to obtain foreign regulatory approvals on a timely basis could have an adverse effect on our international business, financial condition and results of operations. International operations may be limited or disrupted by the imposition of government controls, export license requirements, political or economic instability, trade restrictions, changes in tariffs, restrictions on repatriating profits, taxation or difficulties in staffing and managing international operations. In addition, our business, financial condition and results of operations may be adversely affected by fluctuations in currency exchange rates. There can be no assurance that we will be able to successfully operate in any foreign market.
Financial information regarding the geographic areas in which we operate is included in Note 13 to the Financial Statements: Concentration of Risk, Segment and Geographic Information.
Concentration of Risk
In 2010, NIAID, UCB, and Takeda each accounted for more than 10% of our total revenue, none of which represents a related party to XOMA. These key customers accounted for 87% of our total revenue in 2010 and NIAID was responsible for 23% of the accounts receivable balance at December 31, 2010. Servier accounted for an additional 72% of the accounts receivable balance at December 31, 2010. The loss of one or more of these customers could have a material adverse effect on our business and financial condition.
In 2009, Takeda and Genentech each accounted for more than 10% of our total revenue, none of which represents a related party to XOMA. These key customers accounted for 65% of our total revenue in 2009, but were not responsible for any of the accounts receivable balance at December 31, 2009. NIAID, Arana, and Kaketsuken accounted for 90% of the accounts receivable balance at December 31, 2009. In 2008, Genentech, Novartis, and Merck/Schering-Plough each provided more than 10% of our total revenue, none of which represent a related party to XOMA.
Organization
We were incorporated in Delaware in 1981 and became a Bermuda company effective December 31, 1998, when we completed a shareholder-approved corporate reorganization, changing our legal domicile from Delaware to Bermuda and our name to XOMA Ltd. When referring to a time or period before December 31, 1998, or when the context so requires, the terms “Company” and “XOMA” refer to XOMA Corporation, a Delaware corporation and the predecessor of XOMA Ltd.
Employees
As of March 8, 2011, we employed approximately 230 full-time employees, none of which are unionized, at our facilities, principally in Berkeley, California. Our employees are primarily engaged in clinical, process development, research and product development, and in executive, business development, finance and administrative positions. We consider our employee relations to be excellent.
Available Information
For information on XOMA’s investment prospects and risks, please contact Investor Relations and Corporate Communications at (510) 204-7200 or by sending an e-mail message to investorrelations@xoma.com. Our principal executive offices are located at 2910 Seventh Street, Berkeley, California 94710, U.S.A. Our telephone number is (510) 204-7200.
The following information can be found on our website at http://www.xoma.com or can be obtained free of charge by contacting our Investor Relations Department:
|
|
·
|
Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and any amendments to those reports will be available as soon as reasonably practicable after such material is electronically filed with the United States Securities and Exchange Commission (“SEC”). All reports we file with the SEC can also be obtained free of charge via EDGAR through the SEC’s website at http://www.sec.gov.
|
|
|
·
|
Our policies related to corporate governance, including our Code of Ethics applying to our directors, officers and employees (including our principal executive officer and principal financial and accounting officer) that we have adopted to meet the requirements set forth in the rules and regulations of the SEC and its corporate governance principles are available.
|
|
|
·
|
The charters of the Audit, Compensation and Nominating & Governance Committees of our Board of Directors are available.
|
We intend to satisfy the applicable disclosure requirements regarding amendments to, or waivers from, provisions of our Code of Ethics by posting such information on our website.
|
Item 1A.
|
The following risk factors and other information included in this annual report should be carefully considered. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us also may impair our business operations. If any of the following risks occur, our business, financial condition, operating results and cash flows could be materially adversely affected.
Because our product candidates are still being developed, we will require substantial funds to continue; we cannot be certain that funds will be available and, if they are not available, we may have to take actions that could adversely affect your investment and may not be able to continue operations.
We will need to commit substantial funds to continue development of our product candidates and we may not be able to obtain sufficient funds on acceptable terms, or at all. If adequate funds are not available, we may have to raise additional funds in a manner that may dilute or otherwise adversely affect the rights of existing shareholders, curtail or cease operations, or file for bankruptcy protection in extreme circumstances. We have spent, and we expect to continue to spend, substantial funds in connection with:
|
|
•
|
research and development relating to our product candidates and production technologies,
|
|
|
•
|
various human clinical trials, and
|
|
|
•
|
protection of our intellectual property.
|
We finance our operations primarily through our multiple revenue streams resulting from the licensing of our antibody technologies, discovery and development collaborations, product royalties and biodefense contracts, and sales of our common shares. In September of 2009, we sold our royalty interest in LUCENTIS® to Genentech, Inc., a wholly-owned member of the Roche Group (referred to herein as “Genentech”) for gross proceeds of $25 million, including royalty revenue from the second quarter of 2009. These proceeds, along with other funds, were used to fully repay our loan from Goldman Sachs Specialty Lending Holdings, Inc. (“Goldman Sachs”). As a result, we no longer have a royalty interest in LUCENTIS®. In 2008, we received $8.8 million of revenue from this royalty interest. In August of 2010, we sold our royalty interest in CIMZIA® to an undisclosed buyer for gross proceeds of $4.0 million, including royalty revenue from the second quarter of 2010. As a result, we no longer have a royalty interest in CIMZIA®. We received revenue from this royalty interest of $0.5 million in 2010, $0.5 million in 2009 and $0.1 million in 2008.
Based on our cash reserves and anticipated spending levels, revenue from collaborations including our XOMA 052 collaboration agreement with Les Laboratoires Servier ("Servier"), biodefense contracts and licensing transactions and other sources of funding we believe to be available, we believe that we have sufficient cash resources to meet our anticipated net cash needs through the next twelve months. Any significant revenue shortfalls, increases in planned spending on development programs or more rapid progress of development programs than anticipated, as well as the unavailability of anticipated sources of funding, could shorten this period. If adequate funds are not available, we will be required to delay, reduce the scope of, or eliminate one or more of our product development programs and further reduce personnel-related costs. Progress or setbacks by potentially competing products may also affect our ability to raise new funding on acceptable terms. As a result, we do not know when or whether:
|
|
•
|
operations will generate meaningful funds,
|
|
|
•
|
additional agreements for product development funding can be reached,
|
|
|
•
|
strategic alliances can be negotiated, or
|
|
|
•
|
adequate additional financing will be available for us to finance our own development on acceptable terms, or at all.
|
Cash balances and operating cash flow are influenced primarily by the timing and level of payments by our licensees and development partners, as well as by our operating costs.
Global credit and financial market conditions may reduce our ability to access capital and cash and could negatively impact the value of our current portfolio of cash equivalents and our ability to meet our financing objectives.
Traditionally, we have funded a large portion of our research and development expenditures through raising capital in the equity markets. Recent events, including failures and bankruptcies among large commercial and investment banks, have led to considerable declines and uncertainties in these and other capital markets and may lead to new regulatory and other restrictions that may broadly affect the nature of these markets. These circumstances could severely restrict the raising of new capital by companies such as us in the future.
Volatility in the financial markets has also created liquidity problems in investments previously thought to bear a minimal risk. For example, money market fund investors, including us, have in the past been unable to retrieve the full amount of funds, even in highly-rated liquid money market accounts, upon maturity. Although as of December 31, 2010, we have received the full amount of proceeds from money market fund investments, an inability to retrieve funds from money market fund investments as they mature in the future could have a material and adverse impact on our business, results of operations and cash flows.
Our cash and cash equivalents are maintained in highly liquid investments with remaining maturities of 90 days or less at the time of purchase. While as of the date of this filing, we are not aware of any downgrades, material losses, or other significant deterioration in the fair value of our cash equivalents since December 31, 2010, no assurance can be given that further deterioration in conditions of the global credit and financial markets would not negatively impact our current portfolio of cash equivalents or our ability to meet our financing objectives.
Because all of our product candidates are still being developed, we have sustained losses in the past and we expect to sustain losses in the future.
We have experienced significant losses and, as of December 31, 2010, we had an accumulated deficit of $853.3 million.
For the year ended December 31, 2010, we had a net loss of approximately $68.8 million or $3.69 per common share (basic and diluted). For the year ended December 31, 2009, we had net income of approximately $0.6 million or $0.05 per common share (basic and diluted).
Our ability to achieve profitability is dependent in large part on the success of our development programs, obtaining regulatory approval for our product candidates and entering into new agreements for product development, manufacturing and commercialization, all of which are uncertain. Our ability to fund our ongoing operations is dependent on the foregoing factors and on our ability to secure additional funds. Because our product candidates are still being developed, we do not know whether we will ever achieve sustained profitability or whether cash flow from future operations will be sufficient to meet our needs.
We may issue additional equity securities and thereby materially and adversely affect the price of our common shares.
We are authorized to issue, without shareholder approval, 1,000,000 preference shares, of which 2,959 were issued and outstanding as of March 8, 2011, which may give other shareholders dividend, conversion, voting, and liquidation rights, among other rights, which may be superior to the rights of holders of our common shares. In addition, we are authorized to issue, generally without shareholder approval, up to 46,666,666 common shares, of which 28,491,318 were issued and outstanding as of December 31, 2010 and 29,510,963 were issued and outstanding as of March 8, 2011. If we issue additional equity securities, the price of our common shares may be materially and adversely affected.
In the third quarter of 2009, we had entered into an At Market Issuance Sales Agreement (the “2009 ATM Agreement”), with Wm Smith & Co. (“Wm Smith”), under which we could sell up to 1.7 million of our common shares from time to time through Wm Smith, as the agent for the offer and sale of the common shares. Wm Smith could sell these common shares by any method permitted by law deemed to be an “at the market” offering as defined in Rule 415 of the Securities Act of 1933, including but not limited to sales made directly on The NASDAQ Global Market, on any other existing trading market for the common shares or to or through a market maker. Wm Smith could also sell the common shares in privately negotiated transactions, subject to our approval. From the inception of this agreement through October 27, 2010, we sold a total of 1,666,666 common shares through Wm Smith, constituting all of the shares available for sale under the agreement, for aggregate gross proceeds of $12.2 million.
On February 5, 2010, we completed an underwritten offering of 2.8 million units, with each unit consisting of one of our common shares and a warrant to purchase 0.45 of a common share, for gross proceeds of approximately $21 million, before deducting underwriting discounts and commissions and estimated offering expenses of $1.7 million. The investors purchased the units at a price of $7.50 per unit. The warrants, which represent the right to acquire an aggregate of up to 1.26 million common shares, are exercisable beginning six months and one day after issuance and have a five-year term and an exercise price of $10.50 per share.
On July 23, 2010, we entered into a common share purchase agreement with Azimuth Opportunity, Ltd. (“Azimuth”), pursuant to which we obtained a committed equity line of credit facility under which we could sell up to $30 million of our registered common shares to Azimuth over a 12-month period, subject to certain conditions and limitations. In August of 2010, we sold a total of 3,421,407 common shares under this facility for aggregate proceeds of $14.2 million, representing the maximum number of shares that could be sold under this facility.
On October 26, 2010, we entered into an At Market Issuance Sales Agreement (the “2010 ATM Agreement”), with Wm Smith and McNicoll, Lewis & Vlak LLC (the “Agents”), under which we may sell common shares from time to time through the Agents, as our agents for the offer and sale of the common shares, in an aggregate amount not to exceed the amount that can be sold under our registration statement on Form S-3 (File No. 333-148342) filed with the U.S. Securities and Exchange Commission (the “SEC”) on December 26, 2007 and declared effective by the SEC on May 29, 2008. The Agents may sell the common shares by any method permitted by law deemed to be an “at the market” offering as defined in Rule 415 of the Securities Act, including without limitation sales made directly on The NASDAQ Global Market, on any other existing trading market for the common shares or to or through a market maker. The Agents may also sell the common shares in privately negotiated transactions, subject to our prior approval. From the inception of the 2010 ATM Agreement through December 31, 2010, we sold a total of 6.7 million common shares under this agreement for aggregate gross proceeds of $29.7 million. Subsequent to December 31, 2010 through March 8, 2011, we have sold an additional 796,898 common shares through Wm Smith for aggregate gross proceeds of $4.3 million.
On February 4, 2011, we entered into an At Market Issuance Sales Agreement (the “2011 ATM Agreement”), with McNicoll, Lewis & Vlak LLC (“MLV”), under which we may sell common shares from time to time through the MLV, as our agent for the offer and sale of the common shares, in an aggregate amount not to exceed the amount that can be sold under our registration statement on Form S-3 (File No. 333-172197) filed with the SEC on February 11, 2011, once such registration statement has been declared effective by the SEC. MLV may sell the common shares by any method permitted by law deemed to be an “at the market” offering as defined in Rule 415 of the Securities Act, including without limitation sales made directly on The NASDAQ Global Market, on any other existing trading market for the common shares or to or through a market maker. MLV may also sell the common shares in privately negotiated transactions, subject to our prior approval.
The financial terms of future collaborative or licensing arrangements could result in dilution of our share value.
Funding from collaboration partners and others has in the past and may in the future involve issuance by us of our shares. Because we do not currently have any such arrangements, we cannot be certain how the purchase price of such shares, the relevant market price or premium, if any, will be determined or when such determinations will be made. Any such issuance could result in dilution in the value of our issued and outstanding shares.
Our share price may be volatile and there may not be an active trading market for our common shares.
There can be no assurance that the market price of our common shares will not decline below its present market price or that there will be an active trading market for our common shares. The market prices of biotechnology companies have been and are likely to continue to be highly volatile. Fluctuations in our operating results and general market conditions for biotechnology stocks could have a significant impact on the volatility of our common share price. We have experienced significant volatility in the price of our common shares. From January 1, 2010 through March 8, 2011, our share price has ranged from a high of $12.60 to a low of $2.24. Factors contributing to such volatility include, but are not limited to:
|
|
·
|
sales and estimated or forecasted sales of products for which we receive royalties,
|
|
|
·
|
results of preclinical studies and clinical trials,
|
|
|
·
|
information relating to the safety or efficacy of products or product candidates,
|
|
|
·
|
developments regarding regulatory filings,
|
|
|
·
|
announcements of new collaborations,
|
|
|
·
|
failure to enter into collaborations,
|
|
|
·
|
developments in existing collaborations,
|
|
|
·
|
our funding requirements and the terms of our financing arrangements,
|
|
|
·
|
technological innovations or new indications for our therapeutic products and product candidates,
|
|
|
·
|
introduction of new products or technologies by us or our competitors,
|
|
|
·
|
government regulations,
|
|
|
·
|
developments in patent or other proprietary rights,
|
|
|
·
|
the number of shares issued and outstanding,
|
|
|
·
|
the number of shares trading on an average trading day,
|
|
|
·
|
announcements regarding other participants in the biotechnology and pharmaceutical industries, and
|
|
|
·
|
market speculation regarding any of the foregoing.
|
If we are unable to continue meet the requirements for continued listing on The NASDAQ Global Market, then we may be de-listed. In March of 2010, we received a Staff Determination letter from The NASDAQ Stock Market LLC (“NASDAQ”) indicating that we had not regained compliance with the minimum $1.00 per share requirement for continued inclusion on The NASDAQ Global Market, pursuant to NASDAQ Listing Rule 5450(a)(1). On August 18, 2010, we effected a reverse split of our common shares in order to regain compliance.
We face uncertain results of clinical trials of our potential products.
Our potential products, including XOMA 052 and XOMA 3AB, will require significant additional research and development, extensive preclinical studies and clinical trials and regulatory approval prior to any commercial sales. This process is lengthy and expensive, often taking a number of years. As clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals, the length of time necessary to complete clinical trials and to submit an application for marketing approval for a final decision by a regulatory authority varies significantly. As a result, it is uncertain whether:
|
|
·
|
our future filings will be delayed,
|
|
|
·
|
our preclinical and clinical studies will be successful,
|
|
|
·
|
we will be successful in generating viable product candidates to targets,
|
|
|
·
|
we will be able to provide necessary additional data,
|
|
|
·
|
results of future clinical trials will justify further development, or
|
|
|
·
|
we will ultimately achieve regulatory approval for any of these product candidates.
|
For example, in 2003, we completed two Phase 1 trials of XOMA 629, a BPI-derived topical peptide compound targeting acne, evaluating the safety, skin irritation and pharmacokinetics. In January of 2004, we announced the initiation of Phase 2 clinical testing in patients with mild-to-moderate acne. In August of 2004, we announced the results of a Phase 2 trial with XOMA 629 gel. The results were inconclusive in terms of clinical benefit of XOMA 629 compared with vehicle gel. In 2007, after completing an internal evaluation of this program, we chose to reformulate and focus development efforts on the use of this reformulated product candidate in superficial skin infections, including impetigo and the eradication of staphylococcus aureus. In the fourth quarter of 2008, we decided to curtail all spending on XOMA 629 in response to current economic conditions and to focus our financial resources on XOMA 052.
The timing of the commencement, continuation and completion of clinical trials may be subject to significant delays relating to various causes, including scheduling conflicts with participating clinicians and clinical institutions, difficulties in identifying and enrolling patients who meet trial eligibility criteria, and shortages of available drug supply. Patient enrollment is a function of many factors, including the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the existence of competing clinical trials and the availability of alternative or new treatments. In addition, we will conduct clinical trials in foreign countries in the future which may subject us to further delays and expenses as a result of increased drug shipment costs, additional regulatory requirements and the engagement of foreign clinical research organizations, as well as expose us to risks associated with foreign currency transactions insofar as we might desire to use U.S. dollars to make contract payments denominated in the foreign currency where the trial is being conducted.
All of our product candidates are prone to the risks of failure inherent in drug development. Preclinical studies may not yield results that would satisfactorily support the filing of an IND (or a foreign equivalent) with respect to our product candidates. Even if these applications would be or have been filed with respect to our product candidates, the results of preclinical studies do not necessarily predict the results of clinical trials. Similarly, early-stage clinical trials in healthy volunteers do not predict the results of later-stage clinical trials, including the safety and efficacy profiles of any particular product candidates. In addition, there can be no assurance that the design of our clinical trials is focused on appropriate indications, patient populations, dosing regimens or other variables which will result in obtaining the desired efficacy data to support regulatory approval to commercialize the drug. Preclinical and clinical data can be interpreted in different ways. Accordingly, Food and Drug Administration (“FDA”) officials or officials from foreign regulatory authorities could interpret the data in different ways than we or our development partners do which could delay, limit or prevent regulatory approval.
Administering any of our products or potential products may produce undesirable side effects, also known as adverse effects. Toxicities and adverse effects that we have observed in preclinical studies for some compounds in a particular research and development program may occur in preclinical studies or clinical trials of other compounds from the same program. Such toxicities or adverse effects could delay or prevent the filing of an IND (or a foreign equivalent) with respect to such products or potential products or cause us to cease clinical trials with respect to any drug candidate. In clinical trials, administering any of our products or product candidates to humans may produce adverse effects. These adverse effects could interrupt, delay or halt clinical trials of our products and product candidates and could result in the FDA or other regulatory authorities denying approval of our products or product candidates for any or all targeted indications. The FDA, other regulatory authorities, our development partners or we may suspend or terminate clinical trials at any time. Even if one or more of our product candidates were approved for sale, the occurrence of even a limited number of toxicities or adverse effects when used in large populations may cause the FDA to impose restrictions on, or stop, the further marketing of such drugs. Indications of potential adverse effects or toxicities which may occur in clinical trials and which we believe are not significant during the course of such clinical trials may later turn out to actually constitute serious adverse effects or toxicities when a drug has been used in large populations or for extended periods of time. Any failure or significant delay in completing preclinical studies or clinical trials for our product candidates, or in receiving and maintaining regulatory approval for the sale of any drugs resulting from our product candidates, may severely harm our reputation and business.
Given that regulatory review is an interactive and continuous process, we maintain a policy of limiting announcements and comments upon the specific details of regulatory review of our product candidates, subject to our obligations under the securities laws, until definitive action is taken.
Our therapeutic product candidates have not received regulatory approval. If these product candidates do not receive regulatory approval, neither our third party collaborators nor we will be able to manufacture and market them.
Our product candidates, including XOMA 052 and XOMA 3AB, cannot be manufactured and marketed in the United States and other countries without required regulatory approvals. The United States government and governments of other countries extensively regulate many aspects of our product candidates, including:
|
|
·
|
testing,
|
|
|
·
|
manufacturing,
|
|
|
·
|
promotion and marketing, and
|
|
|
·
|
exporting.
|
In the United States, the FDA regulates pharmaceutical products under the Federal Food, Drug, and Cosmetic Act and other laws, including, in the case of biologics, the Public Health Service Act. At the present time, we believe that most of our product candidates will be regulated by the FDA as biologics. Initiation of clinical trials requires approval by health authorities. Clinical trials involve the administration of the investigational new drug to healthy volunteers or to patients under the supervision of a qualified principal investigator. Clinical trials must be conducted in accordance with FDA and International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practices and the European Clinical Trials Directive under protocols that detail the objectives of the study, the parameters to be used to monitor safety and the efficacy criteria to be evaluated. Other national, foreign and local regulations may also apply. The developer of the drug must provide information relating to the characterization and controls of the product before administration to the patients participating in the clinical trials. This requires developing approved assays of the product to test before administration to the patient and during the conduct of the trial. In addition, developers of pharmaceutical products must provide periodic data regarding clinical trials to the FDA and other health authorities, and these health authorities may issue a clinical hold upon a trial if they do not believe, or cannot confirm, that the trial can be conducted without unreasonable risk to the trial participants. We cannot assure you that U.S. and foreign health authorities will not issue a clinical hold with respect to any of our clinical trials in the future.
The results of the preclinical studies and clinical testing, together with chemistry, manufacturing and controls information, are submitted to the FDA and other health authorities in the form of a new drug application for a pharmaceutical product, and in the form of a BLA for a biological product, requesting approval to commence commercial sales. In responding to a new drug application or an antibody license application, the FDA or foreign health authorities may grant marketing approvals, request additional information or further research, or deny the application if it determines that the application does not satisfy its regulatory approval criteria. Regulatory approval of a new drug application, BLA, or supplement is never guaranteed, and the approval process can take several years and is extremely expensive. The FDA and foreign health authorities have substantial discretion in the drug and biologics approval processes. Despite the time and expense incurred, failure can occur at any stage, and we could encounter problems that cause us to abandon clinical trials or to repeat or perform additional preclinical, clinical or manufacturing-related studies.
Changes in the regulatory approval policy during the development period, changes in, or the enactment of additional regulations or statutes, or changes in regulatory review for each submitted product application may cause delays in the approval or rejection of an application. State regulations may also affect our proposed products. The FDA has substantial discretion in both the product approval process and manufacturing facility approval process and, as a result of this discretion and uncertainties about outcomes of testing, we cannot predict at what point, or whether, the FDA will be satisfied with our or our collaborators’ submissions or whether the FDA will raise questions which may be material and delay or preclude product approval or manufacturing facility approval. As we accumulate additional clinical data, we will submit it to the FDA, and such data may have a material impact on the FDA product approval process.
Even once approved, a product may be subject to additional testing or significant marketing restrictions, its approval may be withdrawn or it may be voluntarily taken off the market.
Even if the FDA, the European Commission or another regulatory agency approves a product candidate for marketing, the approval may impose ongoing requirements for post-approval studies, including additional research and development and clinical trials, and the FDA, European Commission or other regulatory agency may subsequently withdraw approval based on these additional trials.
Even for approved products, the FDA, European Commission or other regulatory agency may impose significant restrictions on the indicated uses, conditions for use, labeling, advertising, promotion, marketing and/or production of such product.
Furthermore, a marketing approval of a product may be withdrawn by the FDA, the European Commission or another regulatory agency or such a product may be voluntarily withdrawn by the company marketing it based, for example, on subsequently-arising safety concerns. In February of 2009, the European Medicines Agency (“EMA”) announced that it had recommended suspension of the marketing authorization of RAPTIVA® in the European Union and that its Committee for Medicinal Products for Human Use (“CHMP”) had concluded that the benefits of RAPTIVA® no longer outweigh its risks because of safety concerns, including the occurrence of progressive multifocal leukoencephalopathy (“PML”) in patients taking the medicine. In the second quarter of 2009, Genentech announced and carried out a phased voluntary withdrawal of RAPTIVA® from the U.S. market, based on the association of RAPTIVA® with an increased risk of PML.
The FDA, European Commission and other agencies also may impose various civil or criminal sanctions for failure to comply with regulatory requirements, including withdrawal of product approval.
Certain of our technologies are relatively new and are in-licensed from third parties, so our capabilities using them are unproven and subject to additional risks.
We license technologies from third parties. These technologies include but are not limited to phage display technologies licensed to us in connection with our bacterial cell expression technology licensing program. However, our experience with some of these technologies remains relatively limited and, to varying degrees, we are still dependent on the licensing parties for training and technical support for these technologies. In addition, our use of these technologies is limited by certain contractual provisions in the licenses relating to them and, although we have obtained numerous licenses, intellectual property rights in the area of phage display are particularly complex. If the owners of the patent rights underlying the technologies we license do not properly maintain or enforce those patents, our competitive position and business prospects could be harmed. Our success will depend in part on the ability of our licensors to obtain, maintain and enforce our licensed intellectual property. Our licensors may not successfully prosecute the patent applications to which we have licenses, or our licensors may fail to maintain existing patents. They may determine not to pursue litigation against other companies that are infringing these patents, or they may pursue such litigation less aggressively than we would. Our licensors may also seek to terminate our license, which could cause us to lose the right to use the licensed intellectual property and adversely affect our ability to commercialize our technologies, products or services.
We do not know whether there will be, or will continue to be, a viable market for the products in which we have an ownership or royalty interest.
Even if other products in which we have an interest receive approval in the future, they may not be accepted in the marketplace. In addition, we or our collaborators or licensees may experience difficulties in launching new products, many of which are novel and based on technologies that are unfamiliar to the healthcare community. We have no assurance that healthcare providers and patients will accept such products, if developed. For example, physicians and/or patients may not accept a product for a particular indication because it has been biologically derived (and not discovered and developed by more traditional means) or if no biologically derived products are currently in widespread use in that indication. Similarly, physicians may not accept a product if they believe other products to be more effective or are more comfortable prescribing other products.
Safety concerns may also arise in the course of on-going clinical trials or patient treatment as a result of adverse events or reactions. For example, in February of 2009, the EMA announced that it had recommended suspension of the marketing authorization of RAPTIVA® in the European Union and EMD Serono Inc., the company that marketed RAPTIVA® in Canada (“EMD Serono”) announced that, in consultation with Health Canada, the Canadian health authority (“Health Canada”), it would suspend marketing of RAPTIVA® in Canada. In March of 2009, Merck Serono Australia Pty Ltd, the company that marketed RAPTIVA® in Australia (“Merck Serono Australia”), following a recommendation from the Therapeutic Goods Administration, the Australian health authority (“TGA”), announced that it was withdrawing RAPTIVA® from the Australian market. In the second quarter of 2009, Genentech announced and carried out a phased voluntary withdrawal of RAPTIVA® from the U.S. market, based on the association of RAPTIVA® with an increased risk of PML. As a result, sales of RAPTIVA® ceased in the second quarter of 2009.
Furthermore, government agencies, as well as private organizations involved in healthcare, from time to time publish guidelines or recommendations to healthcare providers and patients. Such guidelines or recommendations can be very influential and may adversely affect the usage of any products we may develop directly (for example, by recommending a decreased dosage of a product in conjunction with a concomitant therapy or a government entity withdrawing its recommendation to screen blood donations for certain viruses) or indirectly (for example, by recommending a competitive product over our product). Consequently, we do not know if physicians or patients will adopt or use our products for their approved indications.
We or our third party collaborators or licensees may not have adequate manufacturing capacity sufficient to meet market demand.
If any of our product candidates are approved, because we have never commercially introduced any pharmaceutical products, we do not know whether the capacity of our existing manufacturing facilities can be increased to produce sufficient quantities of our products to meet market demand. Also, if we or our third party collaborators or licensees need additional manufacturing facilities to meet market demand, we cannot predict that we will successfully obtain those facilities because we do not know whether they will be available on acceptable terms. In addition, any manufacturing facilities acquired or used to meet market demand must meet the FDA’s quality assurance guidelines.
Our agreements with third parties, many of which are significant to our business, expose us to numerous risks.
Our financial resources and our marketing experience and expertise are limited. Consequently, our ability to successfully develop products depends, to a large extent, upon securing the financial resources and/or marketing capabilities of third parties.
|
|
·
|
In April of 1996, we entered into an agreement with Genentech whereby we agreed to co-develop Genentech’s humanized monoclonal antibody product RAPTIVA®. In April of 1999, March of 2003, and January of 2005, the companies amended the agreement. In October of 2003, RAPTIVA® was approved by the FDA for the treatment of adults with chronic moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy and, in September of 2004, Merck Serono announced the product’s approval in the European Union. In January of 2005, we entered into a restructuring of our collaboration agreement with Genentech which ended our existing cost and profit sharing arrangement related to RAPTIVA® in the United States and entitled us to a royalty interest on worldwide net sales. In February of 2009, the EMEA announced that it had recommended suspension of the marketing authorization of RAPTIVA® in the European Union and EMD Serono announced that, in consultation with Health Canada, it would suspend marketing of RAPTIVA® in Canada. In March of 2009, Merck Serono Australia, following a recommendation from the TGA, announced that it was withdrawing RAPTIVA® from the Australian market. In the second quarter of 2009, Genentech announced and carried out a phased voluntary withdrawal of RAPTIVA® from the U.S. market, based on the association of RAPTIVA® with an increased risk of PML. As a result, sales of RAPTIVA® ceased in the second quarter of 2009.
|
|
|
·
|
In March of 2004, we announced we had agreed to collaborate with Chiron Corporation (now Novartis) for the development and commercialization of antibody products for the treatment of cancer. In April of 2005, we announced the initiation of clinical testing of the first product candidate out of the collaboration, HCD122, an anti-CD40 antibody, in patients with advanced chronic lymphocytic leukemia. In October of 2005, we announced the initiation of the second clinical trial of HCD122 in patients with multiple myeloma. In November of 2008, we announced the restructuring of this product development collaboration, which involves six development programs including the ongoing HCD122 and LFA 102 programs. In exchange for cash and debt reduction on our existing loan facility with Novartis, Novartis has control over the HCD122 and LFA 102 programs, as well as the right to expand the development of these programs into additional indications outside of oncology.
|
|
|
·
|
In March of 2005, we entered into a contract with the National Institute of Allergy and Infectious Diseases (“NIAID”) to produce three monoclonal antibodies designed to protect United States citizens against the harmful effects of botulinum neurotoxin used in bioterrorism. In July of 2006, we entered into an additional contract with NIAID for the development of an appropriate formulation for human administration of these three antibodies in a single injection. In September of 2008, we announced that we were awarded an additional contract with NIAID to support our on-going development of drug candidates toward clinical trials in the treatment of botulism poisoning.
|
|
|
·
|
In December of 2010, we entered into a license and collaboration agreement with Servier, to jointly develop and commercialize XOMA 052 in multiple indications. Under the terms of the agreement, Servier has worldwide rights to diabetes and cardiovascular disease indications and rights outside the U.S. and Japan to Behcet’s uveitis and other inflammatory and oncology indications. XOMA retains development and commercialization rights for Behcet’s uveitis and other inflammatory disease and oncology indications in the U.S. and Japan, and has an option to reacquire rights to diabetes and cardiovascular disease indications from Servier in these territories. Should we exercise this option, we will be required to pay Servier an option fee and partially reimburse their incurred development expenses. The agreement contains customary termination rights relating to matters such as material breach by either party, safety issues and patents. Servier also has a unilateral right to terminate the agreement on a country-by-country basis or in its entirety on 6 months’ notice.
|
|
|
·
|
In December of 2010, we also entered into a loan agreement with Servier, which provides for an advance of up to €15 million and was fully funded in January of 2011. This loan is secured by an interest in our intellectual property rights to all XOMA 052 indications worldwide, excluding the U.S. and Japan territories. The loan has a final maturity date in 2016; however, after a specified period prior to final maturity, the loan is required to be repaid (i) at Servier’s option, by applying up to a significant percentage of any milestone or royalty payments owed by Servier under our collaboration agreement and (ii) using a significant percentage of any upfront, milestone or royalty payments we receive from any third party collaboration or development partner for rights to XOMA 052 in the U.S. and/or Japan. In addition, the loan becomes immediately due and payable upon certain customary events of default.
|
|
|
·
|
We have licensed our bacterial cell expression technology, an enabling technology used to discover and screen, as well as develop and manufacture, recombinant antibodies and other proteins for commercial purposes, to over 50 companies. As of December 31, 2010, we were aware of two antibody products manufactured using this technology that have received FDA approval, Genentech’s LUCENTIS® (ranibizumab injection) for treatment of neovascular wet age-related macular degeneration and UCB’s CIMZIA® (certolizumab pegol) for treatment of Crohn’s disease and rheumatoid arthritis. In the third quarter of 2009, we sold our LUCENTIS® royalty interest to Genentech. In the third quarter of 2010, we sold our CIMZIA® royalty interest to an undisclosed buyer.
|
Because our collaborators and licensees are independent third parties, they may be subject to different risks than we are and have significant discretion in, and different criteria for, determining the efforts and resources they will apply related to their agreements with us. If these collaborators and licensees do not successfully develop and market these products, we may not have the capabilities, resources or rights to do so on our own. We do not know whether we, our collaborators or licensees will successfully develop and market any of the products that are or may become the subject of any of our collaboration or licensing arrangements. In some cases these arrangements provide for funding solely by our collaborators or licensees, and in other cases, such as our arrangement with Servier, all of the funding for certain projects and a significant portion of the funding for other projects is to be provided by our collaborator or licensee. In addition, third party arrangements such as ours also increase uncertainties in the related decision-making processes and resulting progress under the arrangements, as we and our collaborators or licensees may reach different conclusions, or support different paths forward, based on the same information, particularly when large amounts of technical data are involved. Furthermore, our contracts with NIAID contain numerous standard terms and conditions provided for in the applicable federal acquisition regulations and customary in many government contracts. Uncertainty exists as to whether we will be able to comply with these terms and conditions in a timely manner, if at all. In addition, we are uncertain as to the extent of NIAID’s demands and the flexibility that will be granted to us in meeting those demands.
Even when we have a collaborative relationship, other circumstances may prevent it from resulting in successful development of marketable products.
|
|
·
|
In September of 2004, we entered into a collaboration arrangement with Aphton Corporation (“Aphton”) for the treatment of gastrointestinal and other gastrin-sensitive cancers using anti-gastrin monoclonal antibodies. In January of 2006, Aphton announced that its common stock had been delisted from NASDAQ. In May of 2006, Aphton filed for bankruptcy protection under Chapter 11, Title 11 of the United States Bankruptcy Code.
|
|
|
·
|
In September of 2006, we entered into an agreement with Taligen Therapeutics, Inc. (“Taligen”) which formalized an earlier letter agreement, which was signed in May of 2006, for the development and cGMP manufacture of a novel antibody fragment for the potential treatment of inflammatory diseases. In May of 2007, we and Taligen entered into a letter agreement which provided that we would not produce a cGMP batch at clinical scale pursuant to the terms of the agreement entered into in September of 2006. In addition, the letter agreement provided that we would conduct and complete the technical transfer of the process to Avecia Biologics Limited or its designated affiliate (“Avecia”). The letter agreement also provided that, subject to payment by Taligen of approximately $1.7 million, we would grant to Avecia a non-exclusive, worldwide, paid-up, non-transferable, non-sublicensable, perpetual license under our owned project innovations. We received $0.6 million as the first installment under the payment terms of the letter agreement but not the two additional payments totaling approximately $1.1 million to which we were entitled upon fulfillment of certain obligations. In May of 2009, the matter was resolved by agreement of the parties in a manner that had no further impact on our financial position.
|
Although we continue to evaluate additional strategic alliances and potential partnerships, we do not know whether or when any such alliances or partnerships will be entered into.
Products and technologies of other companies may render some or all of our products and product candidates noncompetitive or obsolete.
Developments by others may render our products, product candidates, or technologies obsolete or uncompetitive. Technologies developed and utilized by the biotechnology and pharmaceutical industries are continuously and substantially changing. Competition in the areas of genetically engineered DNA-based and antibody-based technologies is intense and expected to increase in the future as a number of established biotechnology firms and large chemical and pharmaceutical companies advance in these fields. Many of these competitors may be able to develop products and processes competitive with or superior to our own for many reasons, including that they may have:
|
|
·
|
significantly greater financial resources,
|
|
|
·
|
larger research and development and marketing staffs,
|
|
|
·
|
larger production facilities,
|
|
|
·
|
entered into arrangements with, or acquired, biotechnology companies to enhance their capabilities, or
|
|
|
·
|
extensive experience in preclinical testing and human clinical trials.
|
These factors may enable others to develop products and processes competitive with or superior to our own or those of our collaborators. In addition, a significant amount of research in biotechnology is being carried out in universities and other non-profit research organizations. These entities are becoming increasingly interested in the commercial value of their work and may become more aggressive in seeking patent protection and licensing arrangements. Furthermore, many companies and universities tend not to announce or disclose important discoveries or development programs until their patent position is secure or, for other reasons, later; as a result, we may not be able to track development of competitive products, particularly at the early stages. Positive or negative developments in connection with a potentially competing product may have an adverse impact on our ability to raise additional funding on acceptable terms. For example, if another product is perceived to have a competitive advantage, or another product’s failure is perceived to increase the likelihood that our product will fail, then investors may choose not to invest in us on terms we would accept or at all.
The examples below pertain to competitive events in the market which we review quarterly and are not intended to be representative of all existing competitive events.
XOMA 052
We, in collaboration with Servier, are conducting clinical testing of XOMA 052, a potent anti-inflammatory monoclonal antibody targeting IL-1 beta, in Behcet’s uveitis patients, Type 2 diabetes patients and cardiovascular disease patients. Other companies are developing other products based on the same or similar therapeutic targets as XOMA 052 and these products may prove more effective than XOMA 052. We are aware that:
|
|
·
|
In June of 2009, Novartis announced it had received U.S. marketing approval for Ilaris® (canakinumab), a fully-human monoclonal antibody targeting IL-1 beta, to treat children and adults with Cryopyrin-Associated Periodic Syndromes (“CAPS”). In October of 2009, Novartis announced that Ilaris® had been approved in the European Union for CAPS. Canakinumab is also in clinical trials in Type 2 diabetes, chronic obstructive pulmonary disorder, certain forms of gout and systemic juvenile rheumatoid arthritis. In January of 2011, Novartis announced that it had filed for EMA approval of Ilaris® for the treatment and prevention of gout.
|
|
|
·
|
Eli Lilly and Company (“Lilly”) is developing LY2189102, an investigational IL-1 beta antibody, for bi-weekly subcutaneous injection for the treatment of Type 2 diabetes. Lilly announced the initiation of a Phase 2 study in the third quarter of 2009 and has estimated completion of this study in November of 2010.
|
|
|
·
|
In 2008, Biovitrum AB (now called Swedish Orphan Biovitrum, “Biovitrum”) obtained a worldwide exclusive license to Amgen Inc. (“Amgen”)’s Kineret® (anakinra) for its current approved indication. Kineret® is an IL-1 receptor antagonist (IL-1ra) currently marketed to treat rheumatoid arthritis and has been evaluated over the years in multiple IL-1 mediated diseases, including Type 2 diabetes and other indications we are considering for XOMA 052. In addition to other on-going studies, a proof-of-concept clinical trial in the United Kingdom investigating Kineret® in patients with a certain type of myocardial infarction, or heart attack, has been completed. In August of 2010, Biovitrum announced that the FDA had granted orphan drug designation to Kineret® for the treatment of CAPS.
|
|
|
·
|
In February of 2008, Regeneron Pharmaceuticals, Inc. (“Regeneron”) announced it had received marketing approval from the FDA for ARCALYST® (rilonacept) Injection for Subcutaneous Use, an interleukin-1 blocker or IL-1 Trap, for the treatment of CAPS, including Familial Cold Auto-inflammatory Syndrome and Muckle-Wells Syndrome in adults and children 12 and older. In September of 2009, Regeneron announced that rilonacept was approved in the European Union for CAPS. In June of 2010, Regeneron announced positive results of a Phase 3 clinical trial of rilonacept in gout. In March 2011, Regeneron disclosed that it intends to file a supplemental BLA for ARCALYST® for the prevention and treatment of gout.
|
|
|
·
|
Amgen has been developing AMG 108, a fully-human monoclonal antibody that targets inhibition of the action of IL-1. On April 28, 2008, Amgen discussed results from its recently completed Phase 2 study in rheumatoid arthritis. AMG 108 showed statistically significant improvement in the signs and symptoms of rheumatoid arthritis and was well tolerated. In January of 2011, MedImmune, the worldwide biologics unit for AstraZeneca PLC, announced that Amgen granted it rights to develop AMG 108 worldwide except Japan.
|
|
|
·
|
In June of 2009, Cytos Biotechnology AG announced the initiation of an ascending dose Phase 1 study of CYT013-IL1bQb, a therapeutic vaccine targeting IL-1 beta, in Type 2 diabetes and that this study is expected to be completed in the first quarter of 2011.
|
XOMA 3AB
We are also developing XOMA 3AB, a combination, or cocktail, of antibodies designed to neutralize the most potent of botulinum toxins. Other companies are developing other products targeting botulism poisoning and these products may prove more effective than XOMA 3AB. We are aware that:
|
|
·
|
In May of 2006, the U.S. Department of Health & Human Services awarded Cangene Corporation (“Cangene”) a five-year, $362 million contract under Project Bioshield. The contract requires Cangene to manufacture and supply 200,000 doses of an equine heptavalent botulism anti-toxin to treat individuals who have been exposed to the toxins that cause botulism. In May of 2008, Cangene announced significant product delivery under this contract. In March of 2010, this contract was extended for an additional two years, until May of 2013.
|
|
|
·
|
Emergent BioSolutions, Inc. (“Emergent”) is currently in development of a botulism immunoglobulin candidate that may compete with our anti-botulinum neurotoxin monoclonal antibodies.
|
|
|
·
|
We are aware of additional companies that are pursuing biodefense-related antibody products. PharmAthene, Inc. and Human Genome Sciences, Inc. are developing anti-anthrax antibodies. Cangene and Emergent are developing anti-anthrax immune globulin products. These products may compete with our efforts in the areas of other monoclonal antibody-based biodefense products, and the manufacture of antibodies to supply strategic national stockpiles.
|
Manufacturing risks and inefficiencies may adversely affect our ability to manufacture products for ourselves or others.
To the extent we continue to provide manufacturing services for our own benefit or to third parties, we are subject to manufacturing risks. Additionally, unanticipated fluctuations in customer requirements have led and may continue to lead to manufacturing inefficiencies, which if significant could lead to an impairment on our long-lived assets or restructuring activities. We must utilize our manufacturing operations in compliance with regulatory requirements, in sufficient quantities and on a timely basis, while maintaining acceptable product quality and manufacturing costs. Additional resources and changes in our manufacturing processes may be required for each new product, product modification or customer or to meet changing regulatory or third party requirements, and this work may not be successfully or efficiently completed. In addition, to the extent we continue to provide manufacturing services, our fixed costs, such as facility costs, would be expected to increase, as would necessary capital investment in equipment and facilities.
Manufacturing and quality problems may arise in the future to the extent we continue to perform these services for our own benefit or for third parties. Consequently, our development goals or milestones may not be achieved in a timely manner or at a commercially reasonable cost, or at all. In addition, to the extent we continue to make investments to improve our manufacturing operations, our efforts may not yield the improvements that we expect.
Because many of the companies we do business with are also in the biotechnology sector, the volatility of that sector can affect us indirectly as well as directly.
As a biotechnology company that collaborates with other biotech companies, the same factors that affect us directly can also adversely impact us indirectly by affecting the ability of our collaborators, partners and others we do business with to meet their obligations to us and reduce our ability to realize the value of the consideration provided to us by these other companies.
For example, in connection with our licensing transactions relating to our bacterial cell expression technology, we have in the past and may in the future agree to accept equity securities of the licensee in payment of license fees. The future value of these or any other shares we receive is subject both to market risks affecting our ability to realize the value of these shares and more generally to the business and other risks to which the issuer of these shares may be subject.
As we do more business internationally, we will be subject to additional political, economic and regulatory uncertainties.
We may not be able to successfully operate in any foreign market. We believe that, because the pharmaceutical industry is global in nature, international activities will be a significant part of our future business activities and that, when and if we are able to generate income, a substantial portion of that income will be derived from product sales and other activities outside the United States. Foreign regulatory agencies often establish standards different from those in the United States, and an inability to obtain foreign regulatory approvals on a timely basis could put us at a competitive disadvantage or make it uneconomical to proceed with a product or product candidate’s development. International operations and sales may be limited or disrupted by:
|
|
·
|
imposition of government controls,
|
|
|
·
|
export license requirements,
|
|
|
·
|
political or economic instability,
|
|
|
·
|
trade restrictions,
|
|
|
·
|
changes in tariffs,
|
|
|
·
|
restrictions on repatriating profits,
|
|
|
·
|
exchange rate fluctuations,
|
|
|
·
|
withholding and other taxation, and
|
|
|
·
|
difficulties in staffing and managing international operations.
|
If we and our partners are unable to protect our intellectual property, in particular our patent protection for our principal products, product candidates and processes, and prevent its use by third parties, our ability to compete in the market will be harmed, and we may not realize our profit potential.
We rely on patent protection, as well as a combination of copyright, trade secret, and trademark laws to protect our proprietary technology and prevent others from duplicating our products or product candidates. However, these means may afford only limited protection and may not:
|
|
·
|
prevent our competitors from duplicating our products;
|
|
|
·
|
prevent our competitors from gaining access to our proprietary information and technology, or
|
|
|
·
|
permit us to gain or maintain a competitive advantage.
|
Because of the length of time and the expense associated with bringing new products to the marketplace, we and our collaboration and development partners hold and are in the process of applying for a number of patents in the United States and abroad to protect our product candidates and important processes and also have obtained or have the right to obtain exclusive licenses to certain patents and applications filed by others. However, the mere issuance of a patent is not conclusive as to its validity or its enforceability. The United States Federal Courts or equivalent national courts or patent offices elsewhere may invalidate our patents or find them unenforceable. In addition, the laws of foreign countries may not protect our intellectual property rights effectively or to the same extent as the laws of the United States. If our intellectual property rights are not adequately protected, we may not be able to commercialize our technologies, products, or services, and our competitors could commercialize our technologies, which could result in a decrease in our sales and market share that would harm our business and operating results. Specifically, the patent position of biotechnology companies generally is highly uncertain and involves complex legal and factual questions. The legal standards governing the validity of biotechnology patents are in transition, and current defenses as to issued biotechnology patents may not be adequate in the future. Accordingly, there is uncertainty as to:
|
|
·
|
whether any pending or future patent applications held by us will result in an issued patent, or that if patents are issued to us, that such patents will provide meaningful protection against competitors or competitive technologies,
|
|
|
·
|
whether competitors will be able to design around our patents or develop and obtain patent protection for technologies, designs or methods that are more effective than those covered by our patents and patent applications, or
|
|
|
·
|
the extent to which our product candidates could infringe on the intellectual property rights of others, which may lead to costly litigation, result in the payment of substantial damages or royalties, and/or prevent us from using technology that is essential to our business.
|
We have established an extensive portfolio of patents and applications, both United States and foreign, related to our BPI-related product candidates, including novel compositions, their manufacture, formulation, assay and use. We have also established a portfolio of patents, both United States and foreign, related to our bacterial cell expression technology, including claims to novel promoter sequences, secretion signal sequences, compositions and methods for expression and secretion of recombinant proteins from bacteria, including immunoglobulin gene products. Most of the more important European patents in our bacterial cell expression patent portfolio expired in July of 2008.
If certain patents issued to others are upheld or if certain patent applications filed by others issue and are upheld, we may require licenses from others in order to develop and commercialize certain potential products incorporating our technology or we may become involved in litigation to determine the proprietary rights of others. These licenses, if required, may not be available on acceptable terms, and any such litigation may be costly and may have other adverse effects on our business, such as inhibiting our ability to compete in the marketplace and absorbing significant management time.
Due to the uncertainties regarding biotechnology patents, we also have relied and will continue to rely upon trade secrets, know-how and continuing technological advancement to develop and maintain our competitive position. All of our employees have signed confidentiality agreements under which they have agreed not to use or disclose any of our proprietary information. Research and development contracts and relationships between us and our scientific consultants and potential customers provide access to aspects of our know-how that are protected generally under confidentiality agreements. These confidentiality agreements may be breached or may not be enforced by a court. To the extent proprietary information is divulged to competitors or to the public generally, such disclosure may adversely affect our ability to develop or commercialize our products by giving others a competitive advantage or by undermining our patent position.
Litigation regarding intellectual property can be costly and expose us to risks of counterclaims against us.
We may be required to engage in litigation or other proceedings to protect our intellectual property. The cost to us of this litigation, even if resolved in our favor, could be substantial. Such litigation could also divert management’s attention and resources. In addition, if this litigation is resolved against us, our patents may be declared invalid, and we could be held liable for significant damages. In addition, we may be subject to a claim that we are infringing another party’s patent. If such claim is resolved against us, we or our collaborators may be enjoined from developing, manufacturing, selling or importing products, processes or services unless we obtain a license from the other party. Such license may not be available on reasonable terms, thus preventing us from using these products, processes or services and adversely affecting our revenue.
Even if we or our third party collaborators or licensees bring products to market, we may be unable to effectively price our products or obtain adequate reimbursement for sales of our products, which would prevent our products from becoming profitable.
If we or our third party collaborators or licensees succeed in bringing our product candidates to the market, they may not be considered cost-effective, and reimbursement to the patient may not be available or may not be sufficient to allow us to sell our products on a competitive basis. In both the United States and elsewhere, sales of medical products and treatments are dependent, in part, on the availability of reimbursement to the patient from third-party payors, such as government and private insurance plans. Third-party payors are increasingly challenging the prices charged for pharmaceutical products and services. Our business is affected by the efforts of government and third-party payors to contain or reduce the cost of healthcare through various means. In the United States, there have been and will continue to be a number of federal and state proposals to implement government controls on pricing. In addition, the emphasis on managed care in the United States has increased and will continue to increase the pressure on the pricing of pharmaceutical products. We cannot predict whether any legislative or regulatory proposals will be adopted or the effect these proposals or managed care efforts may have on our business.
Healthcare reform measures and other statutory or regulatory changes could adversely affect our business.
In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the healthcare system in ways that could impact our business. In March of 2010, the U.S. Congress enacted and President Obama signed into law the Patient Protection and Affordable Care Act, which includes a number of healthcare reform provisions. Assuming the new law survives recent calls for its repeal, the reforms imposed by the new law would significantly impact the pharmaceutical industry, most likely in the area of pharmaceutical product pricing; however, the full effects of new law cannot be known until these provisions are implemented and the relevant federal and state agencies issue applicable regulations or guidance.
The pharmaceutical and biotechnology industries are subject to extensive regulation, and from time to time legislative bodies and governmental agencies consider changes to such regulations that could have significant impact on industry participants. For example, in light of certain highly-publicized safety issues regarding certain drugs that had received marketing approval, the U.S. Congress has considered various proposals regarding drug safety, including some which would require additional safety studies and monitoring and could make drug development more costly. We are unable to predict what additional legislation or regulation, if any, relating to safety or other aspects of drug development may be enacted in the future or what effect such legislation or regulation would have on our business.
The business and financial condition of pharmaceutical and biotechnology companies are also affected by the efforts of governments, third-party payors and others to contain or reduce the costs of healthcare to consumers. In the United States and various foreign jurisdictions there have been, and we expect that there will continue to be, a number of legislative and regulatory proposals aimed at changing the healthcare system, such as proposals relating to the reimportation of drugs into the U.S. from other countries (where they are then sold at a lower price) and government control of prescription drug pricing. The pendency or approval of such proposals could result in a decrease in our share price or limit our ability to raise capital or to obtain strategic collaborations or licenses.
We are exposed to an increased risk of product liability claims, and a series of related cases is currently pending against us.
The testing, marketing and sales of medical products entails an inherent risk of allegations of product liability. In the event of one or more large, unforeseen awards of damages against us, our product liability insurance may not provide adequate coverage. A significant product liability claim for which we were not covered by insurance would have to be paid from cash or other assets. To the extent we have sufficient insurance coverage, such a claim would result in higher subsequent insurance rates. In addition, product liability claims can have various other ramifications including loss of future sales opportunities, increased costs associated with replacing products, and a negative impact on our goodwill and reputation, which could also adversely affect our business and operating results.
On April 9, 2009, a complaint was filed in the Superior Court of Alameda County, California, in a lawsuit captioned Hedrick et al. v. Genentech, Inc. et al, Case No. 09-446158. The complaint asserts claims against Genentech, us and others for alleged strict liability for failure to warn, strict product liability, negligence, breach of warranty, fraudulent concealment, wrongful death and other claims based on injuries alleged to have occurred as a result of three individuals’ treatment with RAPTIVA®. The complaint seeks unspecified compensatory and punitive damages. Since then, additional complaints have been filed in the same court, bringing the total number of pending cases to fifty-eight. All of the complaints allege the same claims and seek the same types of damages based on injuries alleged to have occurred as a result of the plaintiffs’ treatment with RAPTIVA®. Even though Genentech has agreed to indemnify us in connection with these matters, there can be no assurance that this or other products liability lawsuits will not result in liability to us or that our insurance or contractual arrangements will provide us with adequate protection against such liabilities.
On August 4, 2010, a petition was filed in the District Court of Dallas County, Texas in a case captioned McCall v. Genentech, Inc., et al., No. 10-09544. The defendants filed a notice of removal to the United States District Court for the Northern District of Texas on September 3, 2010. The removed case is captioned McCall v. Genentech, Inc., et al., No. 3:10-cv-01747-B. The parties have fully briefed the Plaintiff’s Motion to Remand and are awaiting a final ruling from the Court. The petition asserts personal injury claims against Genentech, us, and others arising out of the plaintiff’s treatment with RAPTIVA®. The petition alleges claims based on negligence, strict liability, misrepresentation and suppression, conspiracy, and actual and constructive fraud. The petition seeks compensatory damages and punitive damages in an unspecified amount. Even though Genentech has agreed to indemnify us in connection with this matter, there can be no assurance that this or other products liability lawsuits will not result in liability to us or that our insurance or contractual arrangements will provide us with adequate protection against such liabilities.
On January 7, 2011, a complaint was filed in the United States District Court for the Northern District of Texas in a case captioned Massa v. Genentech, Inc., et al., No. 4:11CV70. The Complaint alleges the same claims against Genentech, us and others and seeks the same types of damages as the complaints filed in the Superior Court of Alameda County, California referenced above. Even though Genentech has agreed to indemnify us in connection with this matter, there can be no assurance that this or other products liability lawsuits will not result in liability to us or that our insurance or contractual arrangements will provide us with adequate protection against such liabilities.
The loss of key personnel, including our Chief Executive Officer, could delay or prevent achieving our objectives.
Our research, product development and business efforts could be adversely affected by the loss of one or more key members of our scientific or management staff, particularly our executive officers: Steven B. Engle, our Chairman, Chief Executive Officer and President; Fred Kurland, our Vice President, Finance and Chief Financial Officer; Patrick J. Scannon, M.D., Ph.D., our Executive Vice President and Chief Medical Officer; and Christopher J. Margolin, our Vice President, General Counsel and Secretary. We currently have no key person insurance on any of our employees.
A U.S. holder of our common shares or warrants could be subject to material adverse U.S. federal income tax consequences if we were considered to be a PFIC at any time during the U.S. holder’s holding period.
A non-U.S. corporation generally will be a “passive foreign investment company,” or PFIC, for U.S. federal income tax purposes in any taxable year in which, after applying the relevant look-through rules with respect to the income and assets of its subsidiaries, either 75% or more of its gross income is “passive income” (generally including (without limitation) dividends, interest, annuities and certain royalties and rents not derived in the active conduct of a business) or the average value of its assets that produce passive income or are held for the production of passive income is at least 50% of the total value of its assets. In determining whether we meet the 50% test, cash is considered a passive asset and the total value of our assets generally will be treated as equal to the sum of the aggregate fair market value of our outstanding common shares plus our liabilities. If we own at least 25% (by value) of the stock of another corporation, we will be treated, for purposes of the PFIC tests, as owning our proportionate share of the other corporation’s assets and receiving our proportionate share of the other corporation’s income.
We believe that we were not a PFIC for the 2010 taxable year. However, because PFIC status is determined annually and depends on the composition of a company’s income and assets and the fair market value of its assets (including goodwill), which may be volatile in our industry, there can be no assurance that we will not be considered a PFIC for 2011 or any subsequent year. For example, taking into account our existing cash balances, if the value of our common shares were to decline materially, it is possible that we could become a PFIC in 2011 or a subsequent year. Additionally, due to the complexity of the PFIC provisions and the limited authority available to interpret such provisions, there can be no assurance that our determination regarding our PFIC status could not be successfully challenged by the Internal Revenue Service (“IRS”).
If we were found to be a PFIC for any taxable year in which a U.S. holder (as defined below) held common shares or warrants, certain adverse U.S. federal income tax consequences could apply to such U.S. holder, including a recharacterization of any capital gain recognized on a sale or other disposition of common shares or warrants as ordinary income, ineligibility for any preferential tax rate otherwise applicable to any “qualified dividend income,” a material increase in the amount of tax that such U.S. holder would owe and the possible imposition of interest charges, an imposition of tax earlier than would otherwise be imposed and additional tax form filing requirements.
For purposes of this discussion, the term “U.S. holder” means a beneficial owner of common shares or warrants that is, for U.S. federal income tax purposes, (i) an individual who is a U.S. citizen or resident, (ii) a corporation (or other entity treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States, any state thereof or the District of Columbia, (iii) an estate the income of which is includable in gross income for U.S. income tax purposes regardless of its source, or (iv) a trust, if a U.S. court is able to exercise primary supervision over the administration of the trust and one or more U.S. fiduciaries have the authority to control all substantial decisions of the trust, or if the trust has a valid election in effect under applicable U.S. Treasury Regulations to be treated as a U.S. person. Special rules apply to a U.S. investor who owns our common shares or warrants through an entity treated as a partnership for U.S. federal income tax purposes.
A U.S. holder owning shares in a PFIC (or a corporation that might become a PFIC) might be able to mitigate the adverse tax consequences of PFIC status by making certain elections, including “qualified electing fund” (a “QEF”) or “mark-to-market” elections, if deemed appropriate based on guidance provided by the U.S. holder’s tax advisor. However, it should be noted that (1) the beneficial effect of a QEF election or a mark-to-market election may be substantially diminished if such election is not made from the inception of a U.S. holder’s holding period (a “Year One Election”), (2) neither a QEF election nor a mark-to-market election can be made with respect to warrants, (3) a Year One Election generally cannot be made for any common shares received upon exercise of warrants (“Warrant Shares”) because the holding period of Warrant Shares is deemed, for QEF election and mark-to-market election purposes, to include the holding period of the underlying warrants but the QEF election or mark-to-market election will not be effective until the underlying warrants are exercised, and (4) a QEF election or a mark-to-market election is made on a shareholder-by-shareholder basis and, once made, can only be revoked with the consent of the IRS.
The PFIC rules are very complex, as are the requirements and effects of the various elections designed to mitigate the adverse consequences of the PFIC rules. A U.S. holder should consult its own tax advisor regarding the PFIC rules, including the foregoing limitations on the ability to make a QEF election or a mark-to-market election (or to qualify either such election as a Year One Election), the timing requirements with respect to the various elections and the irrevocability of certain elections (absent the consent of the IRS).
As a result of a recent legislative change, a U.S. holder generally will be required to file IRS Form 8621 if the U.S. holder holds our common shares or warrants in any taxable year in which we are classified as a PFIC (whether or not a QEF or mark-to-market election is made).
Our ability to use our net operating loss carry-forwards and other tax attributes will be substantially limited by Section 382 of the Internal Revenue Code.
Section 382 of the Internal Revenue Code of 1986, as amended, generally limits the ability of a corporation that undergoes an “ownership change” to utilize its net operating loss carry-forwards (“NOLs”) and certain other tax attributes against any taxable income in taxable periods after the ownership change. The amount of taxable income in each taxable year after the ownership change that may be offset by pre-change NOLs and certain other pre-change tax attributes is generally equal to the product of (a) the fair market value of the corporation’s outstanding shares immediately prior to the ownership change and (b) the long-term tax exempt rate (i.e., a rate of interest established by the IRS that fluctuates from month to month). In general, an “ownership change” occurs whenever the percentage of the shares of a corporation owned, directly or indirectly, by “5-percent shareholders” (within the meaning of Section 382 of the Internal Revenue Code) increases by more than 50 percentage points over the lowest percentage of the share of such corporation owned, directly or indirectly, by such “5-percent shareholders” at any time over the preceding three years.
In 2009, we experienced an ownership change under Section 382, which subjects the amount of pre-change NOLs and tax credit carry-forwards that can be utilized to an annual limitation, which will substantially limit the future use of our pre-change NOLs and certain other pre-change tax attributes per year.
Recently proposed legislation, if enacted, could subject us to U.S. federal income taxation as if we were a U.S. corporation.
A bill recently introduced in the House of Representatives provides in certain instances that a corporation that changed its corporate domicile from the United States to a non-U.S. jurisdiction prior to the effective date of the “inversion” rules of Section 7874 of the Code would, for any taxable year beginning on or after the second anniversary of the bill’s enactment, be treated as a U.S. corporation for U.S. federal income taxes purposes if such corporation were managed and controlled primarily in the United States. If this bill were enacted in 2011 in its present form and we were to make no changes to our current management structure, we would likely be treated, beginning in 2014, as a U.S. corporation subject to U.S. federal income taxation on our worldwide income. There can be no assurance that the foregoing bill or another similar legislative proposal will not become law.
Because we are a relatively small biopharmaceutical company with limited resources, we may not be able to attract and retain qualified personnel.
Our success in developing marketable products and achieving a competitive position will depend, in part, on our ability to attract and retain qualified scientific and management personnel, particularly in areas requiring specific technical, scientific or medical expertise. We had approximately 230 employees as of March 8, 2011. We anticipate that we will require additional experienced executive, accounting, research and development, legal, administrative and other personnel in the future. There is intense competition for the services of these personnel, especially in California. Moreover, we expect that the high cost of living in the San Francisco Bay Area, where our headquarters and manufacturing facilities are located, may impair our ability to attract and retain employees in the future. If we do not succeed in attracting new personnel and retaining and motivating existing personnel, our operations may suffer and we may be unable to implement our current initiatives or grow effectively.
Calamities, power shortages or power interruptions at our Berkeley headquarters and manufacturing facility could disrupt our business and adversely affect our operations, and could disrupt the businesses of our customers.
Our principal operations are located in Northern California, including our corporate headquarters and manufacturing facility in Berkeley, California. In addition, many of our collaborators and licensees are located in California. All of these locations are in areas of seismic activity near active earthquake faults. Any earthquake, terrorist attack, fire, power shortage or other calamity affecting our facilities or our customers’ facilities may disrupt our business and could have material adverse effect on our business and results of operations.
We may be subject to increased risks because we are a Bermuda company.
Alleged abuses by certain companies that have changed their legal domicile from jurisdictions within the United States to Bermuda have created an environment where, notwithstanding that we changed our legal domicile in a transaction that was approved by our shareholders and fully taxable to our company under United States law, we may be exposed to various prejudicial actions, including:
|
|
·
|
“blacklisting” of our common shares by certain pension funds,
|
|
|
·
|
legislation restricting certain types of transactions, and
|
|
|
·
|
punitive tax legislation.
|
We do not know whether any of these things will happen, but if implemented one or more of them may have an adverse impact on our future operations or our share price.
It may be difficult to enforce a judgment obtained against us because we are a foreign entity.
We are a Bermuda company. All or a substantial portion of our assets, including substantially all of our intellectual property, may be located outside the United States. As a result, it may be difficult for shareholders and others to enforce in United States courts judgments obtained against us. We have irrevocably agreed that we may be served with process with respect to actions based on offers and sales of securities made hereby in the United States by serving Christopher J. Margolin, c/o XOMA Ltd., 2910 Seventh Street, Berkeley, California 94710, our United States agent appointed for that purpose. Uncertainty exists as to whether Bermuda courts would enforce judgments of United States courts obtained in actions against us or our directors and officers that are predicated upon the civil liability provisions of the United States securities laws or entertain original actions brought in Bermuda against us or such persons predicated upon the United States securities laws. There is no treaty in effect between the United States and Bermuda providing for such enforcement, and there are grounds upon which Bermuda courts may not enforce judgments of United States courts. Certain remedies available under the United States federal securities laws may not be allowed in Bermuda courts as contrary to that nation’s policy.
Our shareholder rights agreement or bye-laws may prevent transactions that could be beneficial to our shareholders and may insulate our management from removal.
In February of 2003, we adopted a new shareholder rights agreement (to replace the shareholder rights agreement that had expired), which could make it considerably more difficult or costly for a person or group to acquire control of us in a transaction that our Board of Directors opposes.
Our bye-laws:
|
|
·
|
require certain procedures to be followed and time periods to be met for any shareholder to propose matters to be considered at annual meetings of shareholders, including nominating directors for election at those meetings;
|
|
|
·
|
authorize our Board of Directors to issue up to 1,000,000 preference shares without shareholder approval and to set the rights, preferences and other designations, including voting rights, of those shares as the Board of Directors may determine; and
|
|
|
·
|
contain provisions, similar to those contained in the Delaware General Corporation Law that may make business combinations with interested shareholders more difficult.
|
These provisions of our shareholders rights agreement and our bye-laws, alone or in combination with each other, may discourage transactions involving actual or potential changes of control, including transactions that otherwise could involve payment of a premium over prevailing market prices to holders of common shares, could limit the ability of shareholders to approve transactions that they may deem to be in their best interests, and could make it considerably more difficult for a potential acquirer to replace management.
|
Item 1B.
|
None.
|
Item 2.
|
Our corporate headquarters and development and manufacturing facilities are located in Berkeley and Emeryville, California. We currently lease five buildings, and space in a sixth building, for which we have a sublease tenant under contract through May of 2014. These buildings house our research and development laboratories, manufacturing facilities and office space. A separate pilot scale manufacturing facility is owned by us. Our building leases expire in the period from 2011 to 2014 and total minimum lease payments due from January of 2011 until expiration of the leases are $13.8 million. We have the option to renew our lease agreements for periods ranging from three to ten years.
On January 15, 2009, we announced a workforce reduction of approximately 42%. As a result, in the second quarter of 2009, we vacated one of our leased buildings and recorded a restructuring charge of $0.5 million primarily related to the net present value of the net future minimum lease payments at the cease-use date, less the estimated future sublease income. Effective December of 2010, we entered into a sublease agreement for this building. The remaining liability related to this lease was $0.2 million and $0.4 million at December 31, 2010 and December 31, 2009, respectively.
Additionally, as a result of the 2009 workforce reduction, we temporarily vacated a building in order to optimize our facility usage. The net book value of fixed assets in the vacant building potentially subject to write-down is approximately $3.5 million as of December 31, 2010. We have determined that there was no impairment of the assets as of December 31, 2010. In 2011, we plan to increase manufacturing and regulatory activities relating to the Behcet’s uveitis indication as part of our license and collaboration agreement with Servier to develop and commercialize XOMA 052. Due to the increased capacity requirements resulting from these activities, we expect to move our pilot scale manufacturing team into this facility in the first half of 2011, at which time we will no longer have any vacant buildings and do not believe impairment assessment will be necessary.
|
Item 3.
|
On April 9, 2009, a complaint was filed in the Superior Court of Alameda County, California, in a lawsuit captioned Hedrick et al. v. Genentech, Inc. et al, Case No. 09-446158. The complaint asserts claims against Genentech, the Company and others for alleged strict liability for failure to warn, strict product liability, negligence, breach of warranty, fraudulent concealment, wrongful death and other claims based on injuries alleged to have occurred as a result of three individuals’ treatment with RAPTIVA®. The complaint seeks unspecified compensatory and punitive damages. Since then, additional complaints have been filed in the same court, bringing the total number of pending cases to fifty eight. All of the complaints allege the same claims and seek the same types of damages based on injuries alleged to have occurred as a result of the plaintiffs' treatment with RAPTIVA®. The Company’s agreement with Genentech provides for an indemnity of XOMA and payment of legal fees by Genentech which the Company believes is applicable to these matters. The Company believes the claims against it to be without merit and intends to defend against them vigorously.
On August 4, 2010, a petition was filed in the District Court of Dallas County, Texas in a case captioned McCall v. Genentech, Inc., et al., No. 10-09544. The defendants filed a Notice of Removal to the United States District Court for the Northern District of Texas on September 3, 2010. The removed case is captioned McCall v. Genentech, Inc., et al., No. 3:10-cv-01747-B. The parties have fully briefed the Plaintiff’s Motion to Remand and are awaiting a final ruling from the Court. The petition asserts personal injury claims against Genentech, the Company, and others arising out of the plaintiff’s treatment with RAPTIVA®. The petition alleges claims based on negligence, strict liability, misrepresentation and suppression, conspiracy, and actual and constructive fraud. The petition seeks compensatory damages and punitive damages in an unspecified amount. The Company’s agreement with Genentech provides for an indemnity of XOMA and payment of legal fees by Genentech which the Company believes is applicable to this matter. The Company believes the claims against it to be without merit and intends to defend against them vigorously.
On January 7, 2011, a complaint was filed in the United States District Court for the Northern District of Texas in a case captioned Massa v. Genentech, Inc., et al., No. 4:11CV70. The complaint alleges the same claims against Genentech, the Company and others and seeks the same types of damages as the complaints filed in the Superior Court of Alameda County, California referenced above. The Company’s agreement with Genentech provides for an indemnity of XOMA and payment of legal fees by Genentech which the Company believes is applicable to this matter. The Company believes the claims against it to be without merit and intends to defend against them vigorously.
|
Item 4.
|
Our executive officers and their respective ages, as of December 31, 2010, and positions are as follows:
|
Name
|
Age
|
Title
|
||
|
Steven B. Engle
|
56
|
Chairman, Chief Executive Officer and President
|
||
|
Patrick J. Scannon, M.D., Ph.D.
|
63
|
Executive Vice President and Chief Medical Officer
|
||
|
Fred Kurland
|
60
|
Vice President, Finance and Chief Financial Officer
|
||
|
Christopher J. Margolin, Esq.
|
64
|
Vice President, General Counsel and Secretary
|
The Board of Directors elects all officers annually. There is no family relationship between or among any of the officers or directors.
Business Experience
Mr. Engle is XOMA's Chairman, Chief Executive Officer and President. He has more than 25 years of executive leadership and biotechnology and pharmaceutical industry experience and, in February of 2010, was elected to the board of directors of the Biotechnology Industry Organization, or BIO. Prior to joining XOMA in 2007, he served as Chairman of the Board and Chief Executive Officer of La Jolla Pharmaceutical Company, a publicly-held biopharmaceutical company focused on the research and development of therapeutic products for autoimmune and antibody-mediated diseases. He joined La Jolla Pharmaceutical Company in 1993, became President and a Director in 1994, Chief Executive Officer in 1995, and Chairman of the Board in 1997. Prior to joining La Jolla, he held executive-level positions at Cygnus Therapeutic Systems, a developer of drug delivery systems, and Micro Power Systems, Inc., a manufacturer of high technology products, including medical devices. He began his professional career with the Strategic Decisions Group and the Stanford Research Institute. Mr. Engle holds an M.S.E.E. and a B.S.E.E. with a focus in biomedical engineering from the University of Texas.
Dr. Scannon is one of our founders and has served as a Director since our formation. Dr. Scannon became Executive Vice President and Chief Scientific Officer in February of 2011. Previously he was our Executive Vice President and Chief Medical Officer beginning in March of 2009 and served as Executive Vice President and Chief Biotechnology Officer from May of 2006 until March of 2009, Chief Scientific and Medical Officer from March of 1993 until May of 2006, Vice Chairman, Scientific and Medical Affairs from April of 1992 to March of 1993 and our President from our formation until April of 1992. In 2007, Dr. Scannon was invited to join the newly formed National Biodefense Science Board, reporting to the Secretary for the Department of Health and Human Services. In 2007, he also became a member of the Board of Directors for Pain Therapeutics, Inc, a biopharmaceutical company. He serves on the Defense Sciences Research Council for the Defense Advanced Research Projects Agency (DARPA) and on the Threat Reduction Advisory Committee for the Department of Defense. From 1979 until 1981, Dr. Scannon was a clinical research scientist at the Letterman Army Institute of Research in San Francisco. A Board-certified internist, Dr. Scannon holds a Ph.D. in organic chemistry from the University of California, Berkeley and an M.D. from the Medical College of Georgia.
Mr. Kurland is our Vice President, Finance and Chief Financial Officer. He joined XOMA on December 29, 2008. Mr. Kurland is responsible for directing the Company’s financial strategy, accounting, financial planning and investor relations functions. He has more than 30 years of experience in biotechnology and pharmaceutical companies including Aviron/MedImmune, Protein Design Labs and Syntex/Roche. Prior to joining XOMA, Mr. Kurland served as Chief Financial Officer of Bayhill Therapeutics, Inc., Corcept Therapeutics Incorporated and Genitope Corporation. From 1998 to 2002, Mr. Kurland served as Senior Vice President and Chief Financial Officer of Aviron, acquired by MedImmune in 2001 and developer of FluMist. From 1996 to 1998, he was Vice President and Chief Financial Officer of Protein Design Labs, Inc., an antibody design company, and from 1995 to 1996, he served as Vice President and Chief Financial Officer of Applied Immune Sciences, Inc. Mr. Kurland also held a number of financial management positions at Syntex Corporation, a pharmaceutical company acquired by Roche, including Vice President and Controller between 1991 and 1995. He received his J.D. and M.B.A. degrees from the University of Chicago and his B.S. degree from Lehigh University.
Mr. Margolin is our Vice President, General Counsel and Secretary. During his time with the Company, Mr. Margolin has been responsible for the legal and intellectual property function and, at various times, the business development, human resources and licensing functions. Prior to joining us in 1991, Mr. Margolin was a corporate attorney holding several different executive legal positions for Raychem Corporation, an international high technology company, for 11 years. From 1975 to 1980, he was a division counsel for TRW Inc. and from 1972 to 1975, he was an associate at the law firm of McCutchen, Black, Verleger and Shea in Los Angeles. Mr. Margolin holds a B.A. from Princeton University, a J.D. from the University of Pennsylvania and an M.B.A. from the University of California, Los Angeles.
PART II
Market for Registrant’s Common Equity
Our common shares trade on The NASDAQ Global Market under the symbol “XOMA.” All references to numbers of common shares and per-share information in this Annual Report have been adjusted retroactively to reflect the Company’s reverse stock split effective August of 2010. The following table sets forth the quarterly range of high and low reported sale prices of our common shares on The NASDAQ Global Market for the periods indicated:
|
Price Range
|
||||||||
|
High
|
Low
|
|||||||
|
2010
|
||||||||
|
First Quarter
|
$ | 11.70 | $ | 6.00 | ||||
|
Second Quarter
|
12.60 | 6.15 | ||||||
|
Third Quarter
|
6.45 | 2.45 | ||||||
|
Fourth Quarter
|
7.48 | 2.24 | ||||||
|
2009
|
||||||||
|
First Quarter
|
$ | 14.10 | $ | 5.55 | ||||
|
Second Quarter
|
20.10 | 6.00 | ||||||
|
Third Quarter
|
16.20 | 10.65 | ||||||
|
Fourth Quarter
|
12.60 | 9.45 | ||||||
On March 8, 2011, there were 2,400 shareholders of record of our common shares, one of which was Cede & Co., a nominee for Depository Trust Company (“DTC”). All of the common shares held by brokerage firms, banks and other financial institutions as nominees for beneficial owners are deposited into participant accounts at DTC and are therefore considered to be held of record by Cede & Co. as one shareholder.
Dividend Policy
We have not paid dividends on our common shares. We currently intend to retain any earnings for use in the development and expansion of our business. We, therefore, do not anticipate paying cash dividends on our common shares in the foreseeable future.
Performance Graph
The following graph compares the five-year cumulative total shareholder return for XOMA common stock with the comparable cumulative return of certain indices. The graph assumes $100 invested on the same date in each of the indices. Returns of the company are not indicative of future performance.
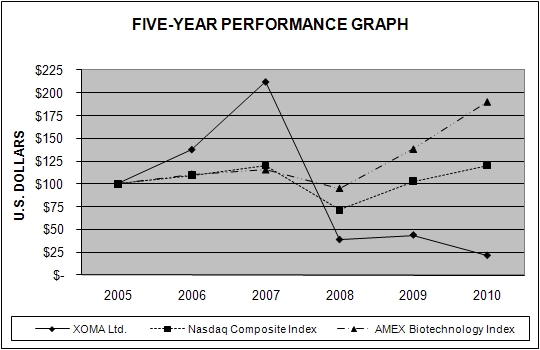
|
As of
December 31,
|
XOMA Ltd.
|
Nasdaq Composite Index
|
AMEX Biotechnology Index
|
|||||||||
|
2005
|
$ | 100.00 | $ | 100.00 | $ | 100.00 | ||||||
|
2006
|
137.50 | 109.52 | 110.77 | |||||||||
|
2007
|
211.88 | 120.27 | 115.51 | |||||||||
|
2008
|
38.75 | 71.51 | 95.04 | |||||||||
|
2009
|
43.75 | 102.89 | 138.36 | |||||||||
|
2010
|
21.38 | 120.29 | 190.57 | |||||||||
|
Item 6.
|
The following table contains our selected financial information including consolidated statement of operations and consolidated balance sheet data for the years 2006 through 2010. The selected financial information has been derived from our audited consolidated financial statements. The selected financial information should be read in conjunction with Item 8: Financial Statements and Supplementary Data and Item 7: Management’s Discussion and Analysis of Financial Condition and Results of Operations included in this Annual Report. The data set forth below is not necessarily indicative of the results of future operations.
|
Year Ended December 31,
|
||||||||||||||||||||
|
2010
|
2009
|
2008
|
2007
|
2006
|
||||||||||||||||
|
(In thousands, except per share amounts)
|
||||||||||||||||||||
|
Consolidated Statement of Operations Data
|
||||||||||||||||||||
|
Total revenues (1)
|
$ | 33,641 | $ | 98,430 | $ | 67,987 | $ | 84,252 | $ | 29,498 | ||||||||||
|
Total operating costs and expenses
|
100,663 | 81,867 | 106,721 | 86,796 | 70,182 | |||||||||||||||
|
Restructuring costs
|
82 | 3,603 | - | - | - | |||||||||||||||
|
(Loss) income from operations
|
(67,104 | ) | 12,960 | (38,734 | ) | (2,544 | ) | (40,684 | ) | |||||||||||
|
Other income (expense), net (2)
|
(1,625 | ) | (6,683 | ) | (6,894 | ) | (9,782 | ) | (11,157 | ) | ||||||||||
|
Net (loss) income before taxes
|
(68,729 | ) | 6,277 | (45,628 | ) | (12,326 | ) | (51,841 | ) | |||||||||||
|
Income tax expense (benefit), net (3)
|
27 | 5,727 | (383 | ) | - | - | ||||||||||||||
|
Net (loss) income
|
$ | (68,756 | ) | $ | 550 | $ | (45,245 | ) | $ | (12,326 | ) | $ | (51,841 | ) | ||||||
|
Basic and diluted net (loss) income per common share
|
$ | (3.69 | ) | $ | 0.05 | $ | (5.11 | ) | $ | (1.45 | ) | $ | (8.10 | ) | ||||||
|
December 31,
|
||||||||||||||||||||
|
2010
|
2009
|
2008
|
2007
|
2006
|
||||||||||||||||
|
(In thousands)
|
||||||||||||||||||||
|
Balance Sheet Data
|
||||||||||||||||||||
|
Cash and cash equivalents
|
$ | 37,304 | $ | 23,909 | $ | 9,513 | $ | 22,500 | $ | 28,002 | ||||||||||
|
Short-term investments
|
- | - | 1,299 | 16,067 | 18,381 | |||||||||||||||
|
Restricted cash
|
- | - | 9,545 | 6,019 | 4,330 | |||||||||||||||
|
Current assets
|
58,880 | 32,152 | 38,704 | 58,088 | 65,888 | |||||||||||||||
|
Working capital
|
23,352 | 13,474 | 11,712 | 34,488 | 43,221 | |||||||||||||||
|
Total assets
|
74,252 | 52,824 | 67,173 | 84,815 | 91,478 | |||||||||||||||
|
Current liabilities
|
35,528 | 18,678 | 26,992 | 23,600 | 22,667 | |||||||||||||||
|
Long-term liabilities (4)
|
15,133 | 16,620 | 71,582 | 60,897 | 106,984 | |||||||||||||||
|
Redeemable convertible preferences shares, at par value
|
1 | 1 | 1 | 1 | 1 | |||||||||||||||
|
Accumulated deficit
|
(853,310 | ) | (784,554 | ) | (785,104 | ) | (739,859 | ) | (727,533 | ) | ||||||||||
|
Total shareholders' equity (net capital deficiency)
|
23,591 | 17,526 | (31,401 | ) | 318 | (38,173 | ) | |||||||||||||
We have paid no dividends in the past five years.
|
(1)
|
2010 includes a non-recurring fee of $4.0 million related to the sale of our CIMZIA® royalty interest to an undisclosed buyer. 2009 includes a non-recurring fee of $28.1 million related to the expansion of our collaboration agreement with Takeda Pharmaceutical Company Limited (“Takeda”) and a non-recurring fee of $25 million related to the sale of our LUCENTIS® royalty interest to Genentech, Inc., a member of the Roche Group (“Genentech”). 2008 includes a non-recurring fee from Novartis AG (“Novartis”) of $13.7 million relating to a restructuring of the existing collaboration agreement. 2007 includes a non-recurring license fee from Pfizer Inc. of $30 million.
|
|
(2)
|
2010 includes a loss associated with the $4.5 million paid in the first quarter of 2010 to the holders of warrants issued in June of 2009, upon modification of the terms.
|
|
(3)
|
2009 includes foreign income tax expense of $5.8 million recognized in connection with the expansion of our existing collaboration with Takeda.
|
|
(4)
|
The balance as of December 31, 2008 includes $50.4 million from our term loan with Goldman Sachs, which we repaid in 2009. In May of 2008, the Company entered into a $55 million amended term loan facility with Goldman Sachs, paying off the remaining balance on the term loan completed in November of 2006. In addition, the outstanding principal on our Novartis note was reduced by $7.5 million due to the restructure of our collaboration with Novartis. In 2007, we eliminated the remaining $44.5 million in convertible debt issued in 2006. In 2006, we exchanged convertible senior notes (issued in 2005) for $60 million of 6.5% Convertible SNAPsSM due 2012 and issued an additional $12 million of 6.5% SNAPsSM to the public for cash.
|
Overview
We are a leader in the discovery, development and manufacture of therapeutic antibodies designed to treat autoimmune, infectious, inflammatory and oncological diseases. Our proprietary development pipeline includes XOMA 052, an antibody that inhibits interleukin-1 beta (“IL-1 beta”) which is expected to advance into entering Phase 3 development for the treatment of Behcet’s uveitis and is in Phase 2 clinical development for Type 2 diabetes with cardiovascular biomarkers; XOMA 3AB, a biodefense anti-botulism product candidate comprised of a combination, or cocktail, of antibodies; and preclinical antibody discovery programs in several indications, including autoimmune, cardio-metabolic, inflammatory, and oncological diseases. We have a fully integrated product development platform, extending from preclinical science, clinical development to scale-up development, and manufacturing.
In December of 2010, we entered into a license and collaboration agreement with Les Laboratoires Servier (“Servier”), to jointly develop and commercialize XOMA 052 in multiple indications. XOMA 052 is designed to inhibit the pro-inflammatory cytokine IL-1 beta that is believed to be a primary trigger of pathologic inflammation in multiple diseases.
Our biodefense initiatives currently include a $65 million multiple-year contract funded by the National Institute of Allergy and Infectious Diseases (“NIAID”), a part of the National Institutes of Health (“NIH”), to support our ongoing development of anti-botulism antibody product candidates toward clinical trials in the treatment of botulism poisoning. This contract is the third that NIAID has awarded us for the development of botulinum antitoxins and brings the program’s total awards to nearly $100 million. We also develop products with premier pharmaceutical companies including Novartis AG (“Novartis”) and Takeda Pharmaceutical Company Limited (“Takeda”).
We have a premier antibody discovery and development platform that incorporates a collection of antibody phage display libraries and proprietary Human Engineering™, affinity maturation, Bacterial Cell Expression (“BCE”) and manufacturing technologies that enhance our ability and that of our collaboration partners to discover and develop new therapeutic antibodies. BCE is a key biotechnology for the discovery and manufacturing of antibodies and other proteins. To date, more than 50 pharmaceutical and biotechnology companies have signed BCE licenses, and a number of licensed product candidates are in clinical development. We continue to develop and commercialize additional antibody-related technologies including proprietary display technologies to enable antibody discovery and optimization. Our technologies have contributed to the success of marketed antibody products, including LUCENTIS® (ranibizumab injection) for wet age-related macular degeneration and CIMZIA® (certolizumab pegol) for rheumatoid arthritis and Crohn’s disease.
Significant Developments in 2010
XOMA 052 Collaboration Agreement
|
|
·
|
In December of 2010, we entered into an agreement with Servier, to jointly develop and commercialize XOMA 052 in multiple indications, which provides for a non-refundable upfront payment of $15 million that was received by us in January of 2011 and a loan of up to €15 million, which converts to approximately $20 million using the December 31, 2010 Euro to US Dollar (“USD”) exchange rate (the “12/31/10 Exchange Rate”). Also, we are eligible to receive a combination of Euro and USD-denominated, development and sales milestones for multiple indications aggregating to a potential maximum of approximately $470 million converted using the 12/31/10 Exchange Rate if we reacquire rights to diabetes and cardiovascular disease indications from Servier in the U.S. and Japan territories, or approximately $770 million converted using the 12/31/10 Exchange Rate if we do not reacquire these rights. In addition, we are eligible to receive tiered royalties up to a mid-teens percentage rate. Further, we retain development and commercialization rights for Behcet's uveitis and other inflammatory and oncology indications in the U.S. and Japan territories, and an option to reacquire rights to diabetes and cardiovascular disease indications from Servier in these territories. Servier will fully fund activities to advance the global clinical development and future commercialization of XOMA 052 in diabetes and cardiovascular related diseases, as well as the first $50 million of future XOMA 052 global clinical development and chemistry and manufacturing controls expenses and 50% of further expenses for the Behcet's uveitis indication, which is expected to advance into Phase 3 development in 2011.
|
XOMA 052 Behcet’s Uveitis
|
|
·
|
During 2010, XOMA announced positive results from a Phase 2 proof-of-concept clinical trial evaluating XOMA 052 in Behcet’s uveitis, demonstrating rapid improvement in vision-threatening disease exacerbations in all seven treated patients despite discontinuation of immunosuppressive drugs such as cyclosporine and/or azathioprine. Follow-up results demonstrated that each of the five patients re-treated with XOMA 052 after they experienced a new uveitis exacerbation responded again to XOMA 052 treatment and maintained their response for several months. The drug appeared to be safe, and no drug-related serious adverse events were reported.
|
|
|
·
|
In August of 2010, we obtained Food and Drug Administration (“FDA”) orphan drug status for XOMA 052 for the treatment of Behcet’s disease. The designation offers a number of potential incentives, which may include, among others, a seven-year period of U.S. marketing exclusivity from the date of marketing authorization, written guidance on the non-clinical and clinical studies needed to obtain marketing approval, and tax credits for certain clinical research. In October of 2010, XOMA 052 was granted orphan drug status by the European Medicines Agency (“EMA”) for the treatment of Behcet’s disease. The designation generally provides EU market exclusivity for up to ten years following approval for the given indication. Other potential benefits include protocol assistance, direct access to centralized marketing authorization procedures and financial incentives.
|
XOMA 052 Type 2 Diabetes
|
|
·
|
During 2010, we completed the patient enrollment in the Phase 2a clinical trial of XOMA 052 in patients with Type 2 diabetes. The primary goal of the 74 patient Phase 2a trial was to gain additional XOMA 052 safety information in Type 2 diabetes patients on a background of stable metformin monotherapy. In January of 2011, we announced that we had conducted an interim review of 3-month data from the Phase 2a trial where XOMA 052 was shown to be well-tolerated with no significant differences in adverse events, lab abnormalities and vital signs between XOMA 052 and placebo and no drug-related adverse events. At the time of this 3-month review, evidence of biological activity was observed including a reduction in high sensitivity C-reactive protein (“HsCRP”) levels and a modest reduction in hemoglobin A1c (“HbA1c”) levels. HsCRP is a biomarker of cardiovascular risk, and HbA1c is a measure indirectly reflecting blood glucose levels as averaged over a period estimated to be 90 to 120 days.
|
|
|
·
|
Also during 2010, we completed the patient enrollment in the Phase 2b clinical trial of XOMA 052 in patients with Type 2 diabetes. The primary goal of the 420 patient Phase 2b trial was to further evaluate the use of multiple dose regimens on the safety, pharmacodynamics and efficacy of XOMA 052 in cardiometabolic and other diseases, and based on positive results in measurements of HbA1c, fasting blood glucose, and HsCRP, to select doses for pivotal Phase 3 studies. We anticipate reporting top line, six month results from the Phase 2b trial by the end of the first quarter of 2011.
|
Biodefense
|
|
·
|
We advanced XOMA 3AB into the pre-Investigational New Drug (“IND”) stage. XOMA 3AB is a multi-antibody product that targets the most potent of the botulinum toxins, Type A. XOMA 3AB along with other anti-botulism antibody products are currently being developed under a $65 million multiple-year contract, under which we reported revenues of $21.4 million in 2010.
|
Financings
|
|
·
|
In February of 2010, we completed an underwritten offering of 2.8 million units, with each unit consisting of one of our common shares and a warrant to purchase 0.45 of a common share, for gross proceeds of approximately $21 million.
|
|
|
·
|
In the first ten months of 2010, we sold 1,396,625 common shares through Wm Smith & Co. (“Wm Smith”), under our At Market Issuance Sales Agreement dated July 14, 2009 (the “2009 ATM Agreement”), for aggregate gross proceeds of $9.3 million, constituting all of the shares available for sale under this agreement. In October of 2010, we entered into a new At Market Issuance Sales Agreement (the “2010 ATM Agreement”) with Wm Smith and McNicoll, Lewis & Vlak LLC (the “Agents”), under which we may sell common shares from time to time through the Agents, as our agents for the offer and sale of the common shares, in an aggregate amount not to exceed the amount that can be sold under our registration statement on Form S-3 (File No. 333-148342) (the “Existing Registration Statement”) filed with the U.S. Securities and Exchange Commission (the “SEC”) on December 26, 2007 and declared effective by the SEC on May 29, 2008. From the inception of the 2010 ATM Agreement through December 31, 2010, we sold a total of 6,739,476 million common shares under this agreement for aggregate gross proceeds of $29.7 million. See Liquidity and Capital Resources – ATM Agreements for a further discussion.
|
|
|
·
|
In July of 2010, we entered into a common share purchase agreement with Azimuth Opportunity, Ltd. (“Azimuth”) pursuant to which we obtained a committed equity line of credit under which we could sell up to $30 million of our registered common shares to Azimuth. In August of 2010, we sold a total of 3,421,407 common shares under this facility for aggregate proceeds of $14.2 million, representing the maximum number of shares that could be sold under this facility. See Liquidity and Capital Resources – Equity Line of Credit for a further discussion.
|
Other
|
|
·
|
In March of 2010, we received a Staff Determination letter from The NASDAQ Stock Market LLC (“NASDAQ”) indicating that we had not regained compliance with the minimum $1.00 per share requirement for continued inclusion on The NASDAQ Global Market, pursuant to NASDAQ Listing Rule 5450(a)(1). On August 18, 2010, the Company effected a reverse split of its common shares in order to regain compliance.
|
|
|
·
|
In August of 2010, we sold our CIMZIA® royalty stream to an undisclosed buyer for gross proceeds of $4.0 million, which included the receipt of royalties of $0.3 million earned in the second quarter of 2010 and an additional one-time, non-refundable payment of $3.7 million. We will no longer receive royalties on sales of CIMZIA®.
|
|
|
·
|
In November of 2010, the Company received approximately $1.0 million resulting from four grants awarded in connection with the Company’s submission of four qualifying therapeutic discovery projects under the Patient Protection and Affordable Care Act of 2010.
|
Critical Accounting Estimates
The accompanying discussion and analysis of our financial condition and results of operations are based upon our consolidated financial statements and the related disclosures, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates, assumptions and judgments that affect the reported amounts in our consolidated financial statements and accompanying notes. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We believe the following policies to be the most critical to an understanding of our financial condition and results of operations because they require us to make estimates, assumptions and judgments about matters that are inherently uncertain.
Revenue Recognition
Effective January 1, 2010, we early adopted the recently revised accounting guidance on revenue recognition for multiple element arrangements on a prospective basis, which requires us to allocate consideration to all deliverables at the inception of the arrangement using the relative selling price method. The relative selling price method establishes the relative selling price of a deliverable using a hierarchy, first through vendor-specific objective evidence (“VSOE”), second through third-party evidence if VSOE is not available and finally, through estimated selling prices if neither VSOE nor third-party evidence is available. Additionally, the revised accounting guidance also refined the criteria for determining when a deliverable should be accounted for as a separate unit of accounting. Based on this guidance, we generally identify separate units of accounting for the multiple element arrangement if the delivered item has value to the customer on a standalone basis. Generally, under the new accounting principle, we will be more likely to separate the units of accounting in multiple element arrangements which may to lead to more accelerated revenue recognition in some cases. Changes in the allocation of the sales price between delivered to undelivered elements might impact the timing of revenue recognition, but would not change the total revenue recognized on any arrangement.
The change in accounting principle for revenue recognition on multiple element arrangements did not have a material impact on our financial results for the year ended December 31, 2010. We anticipate that the effect on the change in accounting principle on subsequent periods will be primarily dependent on the arrangements entered into, the ability to estimate selling prices when VSOE cannot be established and the timing of the delivery of the products and services. Additionally, had the new accounting guidance been applied for the year ended December 31, 2009, there would have been no material impact on the revenue recognized.
License and Collaborative Fees
Revenue from non-refundable license, technology access or other payments under license and collaborative agreements where we have a continuing obligation to perform is recognized as revenue over the expected period of the continuing performance obligation. We estimate the performance period at the inception of the arrangement and reevaluate it each reporting period. This reevaluation may shorten or lengthen the period over which the remaining revenue is recognized. Changes to these estimates are recorded on a prospective basis.
Milestone payments under collaborative and other arrangements are recognized as revenue upon completion of the milestone event, once confirmation is received from the third party and collectability is reasonably assured. This represents the culmination of the earnings process because we have no future performance obligations related to the payment. Milestone payments that require a continuing performance obligation on our part are recognized over the expected period of the continuing performance obligation. Amounts received in advance are recorded as deferred revenue until the related milestone is completed.
Contract Revenue
Contract revenue for research and development involves our providing research and development and manufacturing services to collaborative partners, biodefense contractors or others. Revenue for certain contracts is accounted for by a proportional performance, or output-based, method where performance is based on estimated progress toward elements defined in the contract. The amount of contract revenue and related costs recognized in each accounting period are based on estimates of the proportional performance during the period. Adjustments to estimates based on actual performance are recognized on a prospective basis and do not result in reversal of revenue should the estimate to complete be extended.
In addition, revenue related to certain research and development contracts is billed based on actual costs incurred by XOMA related to the contract, multiplied by full-time equivalent (“FTE”) rates plus a mark-up. The FTE rates are developed based on our best estimates of labor, materials and overhead costs. For certain contracts, such as our government contracts, the FTE rates are agreed upon at the beginning of the contract and are subject to review or audit by the contracting party at any time. Under our contracts with NIAID, a part of the NIH, we bill using NIH provisional rates and thus are subject to future audits at the discretion of NIAID’s contracting office. These audits can result in an adjustment to revenue previously reported.
Up-front fees are recognized ratably over the expected benefit period under the arrangement. Given the uncertainties of research and development collaborations, significant judgment is required to determine the duration of the arrangement. We have $17.1 million of deferred up-front fees related to two research and collaboration agreements that are being amortized over a range of one to five years.
Share-Based Compensation
The valuation of share-based compensation awards is determined at the date of grant using the Black-Scholes option pricing model. This model requires inputs such as the expected term of the option, expected volatility and risk-free interest rate. Further, the forfeiture rate also impacts the amount of aggregate compensation. These inputs are subjective and generally require significant analysis and judgment to develop. To establish an estimate of expected term, we consider the vesting period and contractual period of the award and our historical experience of share option exercises, post-vesting cancellations and volatility. To establish an estimate of forfeiture rate, we consider our historical experience of option forfeitures and terminations. The risk-free rate is based on the yield available on United States Treasury zero-coupon issues. We review our valuation assumptions quarterly and, as a result, it is likely we will change our valuation assumptions used to value share-based awards granted in future periods.
Share-based compensation expense is recognized ratably over the requisite service period. If options are granted that include a performance condition, we estimate the probability of the performance condition being achieved on a quarterly basis. If it is determined that it is probable the performance criteria will be achieved, we estimate an implicit service period from grant date to the most likely date of achievement of the performance criteria and record share-based compensation expense ratably over this implicit service period. These estimates require significant judgment and may change in future periods.
Income Taxes
The application of income tax law and regulations is inherently complex. Interpretations and guidance surrounding income tax laws and regulations change over time. As such, changes in our subjective assumptions and judgments can materially affect amounts recognized in our financial statements.
We account for uncertain tax positions in accordance with Accounting Standards Codification Topic 740, Income Taxes (“ASC 740”). ASC 740 provides for the recognition of deferred tax assets if realization of such assets is more likely than not. Based upon the weight of available evidence, which includes our historical operating performance and carry-back potential, we have determined that total deferred tax assets should be fully offset by a valuation allowance.
Warrant Liabilities
We have issued warrants to purchase our common shares in connection with financing activities. We account for the warrants as a liability at fair value. The fair value of the warrant liability is estimated using the Black-Scholes Model. The Black-Scholes Model requires inputs such as the expected term of the warrants, share price volatility and risk-free interest rate. These inputs are subjective and generally require significant analysis and judgment to develop. For the estimate of the expected term, we use the full remaining contractual term of the warrant. We base our estimate of expected volatility on our historical volatility. These assumptions are reviewed each reporting period and changes in the estimated fair value of the outstanding warrants are recognized in other income (expense).
Results of Operations
Revenue
Total revenue in 2010 was $33.6 million, compared with $98.4 million in 2009 and $68.0 million in 2008 as shown in the table below (in thousands):
|
Year ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
License and collaborative fees
|
$ | 2,182 | $ | 43,822 | $ | 16,366 | ||||||
|
Contract and other revenue
|
27,174 | 25,492 | 30,473 | |||||||||
|
Royalties
|
4,285 | 29,116 | 21,148 | |||||||||
|
Total revenues
|
$ | 33,641 | $ | 98,430 | $ | 67,987 | ||||||
License and Collaborative Fees
License and collaborative fee revenue includes fees and milestone payments related to the out-licensing of our products and technologies. License and collaborative fee revenue in 2010 was $2.2 million, compared with $43.8 million in 2009 and $16.4 million in 2008. The primary components of license and collaboration fee revenue in 2010 were four milestone payments recognized for an aggregate amount of $1.2 million, including one milestone from AVEO Pharmaceuticals, Inc. (“AVEO”) for $0.8 million resulting from AVEO’s initiation of a Phase 2 clinical trial to evaluate its AV-299 antibody. In addition, we recognized $1.0 million in up-front fees and annual maintenance fees relating to various out-licensing arrangements.
The primary components of license and collaborative fee revenue in 2009 were $28.1 million in revenue recognized related to the expansion of our collaboration agreement with Takeda in February of 2009 and $14.1 million in total revenue, including ancillary services provided, related to two antibody discovery collaboration agreements entered into with Arana Therapeutics Limited (“Arana”) and The Chemo-Sero-Therapeutic Research Institute, a Japanese research foundation known as Kaketsuken in September and October of 2009. We also recognized $1.6 million of license and collaborative fee revenue in 2009 related to up-front fees, annual maintenance fees and milestone payments from various out-licensing arrangements.
The primary source of license and collaborative fee revenue in 2008 related to the restructuring of our product development collaboration with Novartis, which involved six development programs including the HCD122 program. Under the restructured agreement, we recognized a collaborative fee of $13.7 million in exchange for giving Novartis control over the HCD122 and LFA102 programs, as well as the right to expand the development of these programs into additional indications outside of oncology. We also recognized $1.7 million in up-front fees and annual maintenance fees relating to various out-licensing arrangements. In addition, we recognized four milestone payments totaling $1.0 million, including two milestone payments from Pfizer, Inc. relating to two different products, including the payment of $0.5 million for the initiation of a Phase 3 clinical trial.
The generation of future revenue related to license fees and collaborative arrangements is dependent on our ability to attract new licensees to our antibody and BCE technologies and new collaboration partners. In connection with the license and collaboration agreement with Servier in December of 2010, we expect to experience an increase from 2010 levels.
Contract and Other Revenue
Contract and other revenue includes agreements where we provide contracted research and development and manufacturing services to our collaboration partners, including Takeda and NIAID. The following table shows the activity in contract and other revenue for the years ended December 31, 2010, 2009, and 2008 (in thousands):
|
Year ended December 31,
|
2009-2010 | 2008-2009 | ||||||||||||||||||
|
2010
|
2009
|
2008
|
Increase (Decrease)
|
Increase (Decrease)
|
||||||||||||||||
|
NIAID 3
|
$ | 21,211 | $ | 5,051 | $ | 4,162 | $ | 16,160 | $ | 889 | ||||||||||
|
Takeda
|
3,568 | 7,549 | 4,369 | (3,981 | ) | 3,180 | ||||||||||||||
|
SRI International
|
1,594 | 331 | - | 1,263 | 331 | |||||||||||||||
|
Merck/Schering-Plough
|
468 | 7,586 | 10,780 | (7,118 | ) | (3,194 | ) | |||||||||||||
|
NIAID 2
|
203 | 1,581 | 1,325 | (1,378 | ) | 256 | ||||||||||||||
|
AVEO
|
79 | 675 | 3,161 | (596 | ) | (2,486 | ) | |||||||||||||
|
Novartis
|
- | 2,459 | 6,602 | (2,459 | ) | (4,143 | ) | |||||||||||||
|
Other
|
51 | 260 | 74 | (209 | ) | 186 | ||||||||||||||
|
Total revenues
|
$ | 27,174 | $ | 25,492 | $ | 30,473 | $ | 1,682 | $ | (4,981 | ) | |||||||||
The 2010 increases in revenue from our NIAID Contract No. HHSN272200800028C (“NIAID 3”) and SRI International contracts is due to increased activity under these contracts. Partially offsetting these increases are decreases in revenue from our Schering-Plough Research Institute, a division of Schering Corporation, now a subsidiary of Merck & Co., Inc. (referred to herein as “Merck/Schering-Plough”) and Takeda contracts in 2010 as a result of the cessation of certain Merck/Schering-Plough programs in 2009 and certain Takeda programs in both 2009 and 2010. Also, the decrease in revenue from our Manufacturing and Technology Transfer Agreement with Novartis in 2010 was due to the completion of the work under this agreement in the third quarter of 2009. In addition, revenue related to our NIAID Contract No. HHSN266200600008C/N01-Al-60008 (“NIAID 2”) decreased in 2010 due to its completion.
The 2009 decrease in revenue under our Merck/Schering-Plough contract was due to the cessation of certain discovery and development programs under our collaboration agreement in 2009. Also, revenue from our Manufacturing and Technology Transfer Agreement with Novartis decreased in 2009 due to the completion of the work under this agreement in the third quarter of 2009. In addition, revenue from our AVEO contract decreased in 2009 as a result of our nearing the end of the contracted service arrangement.
These decreases in contract and other revenue in 2009 were partially offset by the recognition of $2.8 million of previously deferred revenue in the fourth quarter of 2009 related to the cessation of certain discovery and development programs under our collaboration with Takeda, resulting in an increase in contract revenue recognized related to our collaboration with Takeda.
Based on expected levels of revenue generating activity related to our Servier and NIAID 3 contracts, as well as our subcontract awards from SRI International, we expect contract and other revenue to increase in 2011 compared to 2010 levels.
We defer revenue until all requirements under our revenue recognition policy are met. In 2010, we deferred $15.9 million of revenue from contracts including Servier, NIH, Takeda, Merck/Schering-Plough and AVEO, and we recognized $2.8 million in revenue. In 2009, we deferred $16.2 million of revenue from contracts including Takeda, Merck/Schering-Plough and Novartis and recognized $28.4 million in revenue. In 2008, we deferred $17.5 million of revenue from contracts including Merck/Schering-Plough, Takeda and Novartis and recognized $18.4 million in revenue.
The following table shows the activity in deferred revenue for the years ended December 31, 2010, 2009 and 2008 (in thousands):
|
Year ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Beginning deferred revenue
|
$ | 5,008 | $ | 17,213 | $ | 18,064 | ||||||
|
Revenue deferred
|
15,949 | 16,220 | 17,515 | |||||||||
|
Revenue recognized
|
(2,827 | ) | (28,425 | ) | (18,366 | ) | ||||||
|
Ending deferred revenue
|
$ | 18,130 | $ | 5,008 | $ | 17,213 | ||||||
In 2011, we expect a significant portion of the $18.1 million in deferred revenue will be recognized with the remainder to be earned during 2012 through 2015. Future amounts may be affected by additional consideration received, if any, under existing or any future licensing or other collaborative arrangements as well as changes in the estimated period of obligation or services to be provided under the arrangements.
Royalties
Revenue from royalties was $4.3 million in 2010 compared with $29.1 million in 2009 and $21.1 million in 2008. The decrease in royalties in 2010 was primarily due to the sale, during 2009, of our LUCENTIS® royalty interest to Genentech for a total of $25 million, which included the receipt of royalties of $2.7 million recognized in the second quarter of 2009 and an additional one-time, non-refundable payment of $22.3 million in September of 2009. Additionally, the cessation of royalties earned from sales of RAPTIVA® in the second quarter of 2009 further contributed to the decrease in our revenue from royalties. Royalties earned from sales of LUCENTIS® and RAPTIVA® during 2009 were $5.1 million and $1.2 million, respectively, compared to $4.4 million and $6.5 million, respectively, in 2008. We will not receive any further royalties on sales of LUCENTIS® or RAPTIVA®.
Partially offsetting the decreases in revenue from royalties was the sale of our CIMZIA® royalty interest for gross proceeds of $4.0 million in the third quarter of 2010, which included the payment of $0.3 million in royalties received and recognized in the second quarter of 2010. Royalties earned from sales of CIMZIA® were $0.5 million in 2010, compared with $0.5 million in 2009 and $0.1 million in 2008. We will not receive any further royalties on sales of CIMZIA®.
Research and Development Expenses
Biopharmaceutical development includes a series of steps, including in vitro and in vivo preclinical testing, and Phase 1, 2 and 3 clinical studies in humans. Each of these steps is typically more expensive than the previous step, but actual timing and the cost to us depends on the product being tested, the nature of the potential disease indication and the terms of any collaborative arrangements with other companies. After successful conclusion of all of these steps, regulatory filings for approval to market the products must be completed, including approval of manufacturing processes and facilities for the product. Our research and development expenses currently include costs of personnel, supplies, facilities and equipment, consultants, third party costs and other expenses related to preclinical and clinical testing.
Research and development expenses were $77.4 million in 2010, compared with $58.1 million in 2009 and $82.6 million in 2008. The increase in research and development expenses of $19.3 million in 2010, as compared to 2009, was primarily due to increased spending on XOMA 052 related to the Phase 2 clinical program and spending on NIAID 3 due to increased activity under the contract. Partially offsetting these increases in spending were decreases in spending on Merck/Schering-Plough and Takeda-related contract activities due to the cessation of certain discovery and development programs. In addition, there was decreased spending on Novartis-related contract activities due to the completion of work under agreement in the third quarter of 2009.
The decrease in research and development expense of $24.5 million in 2009, as compared to 2008, was primarily a result of our increased focus on cost control. In addition, spending on Novartis and Merck/Schering-Plough/AVEO-related contract activities decreased in 2009 due to our reaching the end of contracted service arrangements, and spending on Merck/Schering-Plough-related contract activities decreased in 2009 due to the cessation of certain discovery and development programs under the collaboration. Spending on XOMA 052 decreased in 2009, as compared to 2008, due to the completion of Phase 1 clinical trial enrollment in the second quarter of 2009 slightly offset by an increase in spending in the fourth quarter of 2009 related to the initiation of the Phase 2 clinical program. In addition, spending on XOMA 629 decreased in 2009, as compared to 2008, due to the Company’s decision to suspend development of this product. These decreases were partially offset by increased spending on preclinical antibody discovery programs in several indications, and on our contracts with NIAID 3, Takeda and SRI International.
Research and development expense in 2008 primarily reflects spending on development of XOMA 052, including Phase 1 clinical trials, and to a lesser extent XOMA 629. In addition, we increased spending on our contracts with Novartis, Merck/Schering-Plough, NIAID 3 and Takeda. Research and development expenses also increased in 2008 related to the preclinical development of several antibodies, XOMA 3AB and upgrades made to our manufacturing plant.
Salaries and related personnel costs are a significant component of research and development expenses. We recorded $29.7 million in research and development salaries and employee-related expenses in 2010, compared with $26.8 million in 2009 and $34.4 million in 2008. Included in these expenses for 2010 were $24.1 million for salaries and benefits, $3.3 million for bonus expense and $2.3 million for share-based compensation, which is a non-cash expense. The increase of $2.9 million in 2010, as compared to 2009, was primarily due to higher salaries and related personnel costs in connection with increased manufacturing activities and work related to NIAID 3.
Included in these expenses for 2009 were $22.2 million for salaries and benefits, $2.4 million for bonus expense and $2.2 million for share-based compensation, which is a non-cash expense, compared with $32.1 million, zero and $2.3 million, respectively, in 2008. The $7.6 million decrease in salaries and employee-related expenses in 2009, as compared to 2008, was due to a decrease in salaries and benefits of $9.9 million due to the workforce reduction announced in January of 2009. In addition, share-based compensation decreased by $0.1 million. Partially offsetting this decrease in research and development personnel expense was an increase in bonus expense in 2009 of $2.4 million. In 2008, the Company did not to pay bonuses in efforts to control spending and manage the Company’s cash balance.
Our research and development activities can be divided into earlier stage programs and later stage programs. Earlier stage programs include molecular biology, process development, pilot-scale production and preclinical testing. Also included in earlier stage programs are costs related to excess manufacturing capacity, which we expect will decrease in 2011 due to the execution of the license and collaboration agreement with Servier, resulting in increased manufacturing capacity requirements. Later stage programs include clinical testing, regulatory affairs and manufacturing clinical supplies. The costs associated with these programs approximate the following (in thousands):
|
Year ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Earlier stage programs
|
$ | 52,323 | $ | 42,961 | $ | 62,872 | ||||||
|
Later stage programs
|
25,090 | 15,170 | 19,704 | |||||||||
|
Total
|
$ | 77,413 | $ | 58,131 | $ | 82,576 | ||||||
Our research and development activities can also be divided into those related to our internal projects and those projects related to collaborative and contract arrangements. The costs related to internal projects versus collaborative and contract arrangements approximate the following (in thousands):
|
Year ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Internal projects
|
$ | 58,065 | $ | 42,206 | $ | 58,468 | ||||||
|
Collaborative and contract arrangements
|
19,348 | 15,925 | 24,108 | |||||||||
|
Total
|
$ | 77,413 | $ | 58,131 | $ | 82,576 | ||||||
In 2010, our largest development program (XOMA 052) accounted for more than 30% but less than 40% of our total research and development expense. In 2010, one development program (NIAID) accounted for more than 20% but less than 30% of our total research and development expense, and in 2009 and 2008, one development program (XOMA 052) accounted for more than 20% but less than 30% of our total research and development expense. In 2009, one development program (NIAID) accounted for more than 10% but less than 20% of our total research and development expense, and in 2008 one development program (Novartis) accounted for more than 10% but less than 20% of our total research and development expense. No development program accounted for more than 30% of our total research and development expense in 2009 or 2008.
We expect our research and development spending in 2011 will increase primarily due to the expected initiation of our Phase 3 clinical program for XOMA 052 for the Behcet's uveitis indication under our license and collaboration agreement with Servier, as well as increased activity under our biodefense contracts.
Future research and development spending may also be impacted by potential new licensing or collaboration arrangements, as well as the termination of existing agreements. Beyond this, the scope and magnitude of future research and development expenses are difficult to predict at this time.
Selling, General and Administrative Expenses
Selling, general and administrative expenses include salaries and related personnel costs, facilities costs and professional fees. In 2010, selling, general and administrative expenses were $23.3 million compared with $23.7 million in 2009 and $24.1 million in 2008. The $0.4 million decrease in selling, general and administrative expenses in 2010 as compared with 2009 was primarily due a net decrease in financing and professional fees of $0.4 million, as well as a decrease in salaries and related personnel costs of $0.4 million. Partially offsetting these decreases was an increase in other expenses of $0.4 million, including an increase in travel-related costs.
The $0.4 million decrease in selling, general and administrative expenses in 2009 as compared with 2008 was primarily related to a decrease in salaries and related personnel costs of $0.6 million, as further discussed below, as well as a decrease in professional fees and other expenses of $1.2 million due to our increased focus on cost control. Partially offsetting these decreases was an increase in fees in 2009 of $1.4 million related to the restructuring negotiations and repayment of the Goldman Sachs term loan.
We recorded salaries and employee-related expenses of $12.3 million in 2010 compared with $12.7 million in 2009 and $13.3 million in 2008. The decrease of $0.4 million in 2010 as compared to 2009 was due to a decrease in salaries and benefits of $1.2 million primarily due to our continued focus on cost controls. Partially offsetting this decrease in selling, general and administrative personnel expense was an increase in bonus expense in 2010 of $0.4 million as compared to 2009, and an increase in share-based compensation of $0.4 million.
The $0.6 million decrease in salaries and employee-related expenses in 2009 as compared to 2008 primarily due to a decrease in salaries and benefits of $1.5 million primarily due to the workforce reduction announced in January of 2009, and an increase in share-based compensation of $0.4 million. Partially offsetting this decrease in selling, general and administrative personnel expense was an increase in bonus expense in 2009 of $1.3 million. In 2008, the Company did not to pay bonuses in efforts to control spending and manage the Company’s cash balance.
We expect selling, general and administrative expenses in 2011 will be comparable to 2010 levels.
Restructuring Charges
In January of 2009, we announced a workforce reduction of approximately 42%. As part of this workforce reduction, we recorded charges of $3.1 million during 2009 related to severance, other termination benefits and outplacement services, which were fully paid by the end of 2009. There were no additional employee-related restructuring charges in connection with this workforce reduction.
As a result of the workforce reduction, in the second quarter of 2009, we vacated one of our leased buildings and recorded a restructuring charge of $0.5 million primarily related to the net present value of the net future minimum lease payments at the cease-use date, less the estimated future sublease income. Effective December of 2010, we entered into a sublease agreement for this building. The remaining liability related to this lease was $0.2 million and $0.4 million at December 31, 2010 and 2009, respectively.
Additionally, as a result of the workforce reduction, we temporarily vacated a building in order to optimize our facility usage. As manufacturing demand increases in the future, we plan to resume operations at this facility. As of December 31, 2010, we performed an analysis of the long-lived assets related to the vacant building, with an approximate net book value of $3.5 million. Based on estimated undiscounted future cash inflows, we have determined that there is no current impairment relating to these assets, and will continue to assess these assets for impairment at each future reporting period.
Other Income (Expense)
Investment and interest income was $16,000 in 2010 compared with $49,000 in 2009 and $0.9 million in 2008. Investment and interest income consists primarily of interest earned on our cash and investment balances. The differences between 2010, 2009 and 2008 balances resulted from varying average cash and investment balances and interest rates.
Interest expense and amortization of debt issuance costs for the Goldman Sachs term loan, to the date of repayment, and Novartis note are shown below for 2010, 2009 and 2008 (in thousands):
|
Year ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Interest expense
|
||||||||||||
| Goldman Sachs term loan | $ | - | $ | 3,932 | $ | 5,095 | ||||||
|
Novartis note
|
354 | 455 | 1,181 | |||||||||
|
Convertible debt
|
- | - | ||||||||||
|
Other
|
31 | 14 | - | |||||||||
|
Total interest expense
|
$ | 385 | $ | 4,401 | $ | 6,276 | ||||||
| Amortization of debt issuance costs | ||||||||||||
|
Goldman Sachs term loan
|
$ | - | $ | 487 | $ | 726 | ||||||
|
Total interest expense
|
$ | 385 | $ | 4,888 | $ | 7,002 | ||||||
The decrease in interest expense in 2010 of $4.5 million as compared to 2009 was due to the repayment in full of the Goldman Sachs term loan facility in September of 2009. In addition, interest expense related to the Novartis note decreased by $0.1 million in 2010 due to a decrease in the average interest rate of this note.
The decrease in interest expense of $2.1 million in 2009 compared to 2008 was due to a decrease in interest expense and amortization of debt issuance costs on the Goldman Sachs term loan of $1.4 million. This decrease was due to the repayment in full of the term loan facility in September of 2009, at which point the remaining debt issuance costs of $1.1 million were recognized as part of the loss on debt extinguishment in our consolidated statement of operations for 2009. In addition, interest expense related to the Novartis note decreased by $0.7 million in 2009 due to a decrease in the average principal balance and interest rate of this note.
Interest expense for 2011 is expected to increase compared to 2010 due to the December 2010 execution of a loan agreement with Servier, to be funded in January of 2011.
Loss on debt extinguishment was $3.6 million in 2009 relating to the repayment of our Goldman Sachs term loan. This loss included a prepayment premium of $2.5 million and the recognition of unamortized debt issuance costs of $1.1 million. In 2008, we recognized a loss on debt extinguishment of $0.7 million reflecting the recognition of the unamortized debt issuance costs related to the original Goldman Sachs term loan, upon refinancing of the loan in May of 2008.
Other income (expense) was ($1.3) million in 2010 compared with $1.8 million in 2009 and ($0.1) million in 2008. The increase in other expense in 2010 as compared to 2009 was primarily due to the loss associated with the $4.5 million paid in the first quarter of 2010 to the holders of warrants issued in June of 2009, upon modification of the terms, partially offset by the change in net gains recognized relating to the revaluation of our warrant liabilities in 2010. This increase in other expense was partially offset by $1.0 million in grants received for four qualifying therapeutic discovery projects under the Patient Protection and Affordable Care Act of 2010. The increase in other income in 2009 as compared to 2008 was primarily related to gains of $1.8 million recognized from the revaluation of our warrants in 2009.
Warrant Liabilities
In February of 2010, we issued warrants to purchase 1,260,000 of XOMA’s common shares in connection with an underwritten offering. We have accounted for the warrants issued in February of 2010 as a liability at fair value as further discussed above in Critical Accounting Estimates: Warrant Liabilities. The fair value of the warrant liability at issuance date was estimated using the Black-Scholes Option Pricing Model (the “Black-Scholes Model”) and we recorded a warrant liability of $4.4 million. We revalued the warrant liability at December 31, 2010 and recorded a decrease in the fair value of $0.9 million as a gain in the other income (expense) line of our consolidated statement of operations. The fair value of the warrant liability was $3.5 million at December 31, 2010. As of December 31, 2010 all of these warrants were outstanding.
In May of 2009, we issued warrants to an institutional investor as part of a registered direct offering. The warrants represented the right to acquire an aggregate of up to 392,157 common shares over a five year period beginning May 15, 2009 at an exercise price of $15.30 per share. In February of 2010, the holders of these warrants agreed to amend the terms of their warrants to remove the Eliminated Adjustment Provisions and the exercise price of these warrants was reduced from $15.30 per share to $0.015 per share.
Prior to amendment, we recorded the warrants issued in May of 2009 as a liability at fair value due to the Eliminated Adjustment Provisions and certain other provisions. At December 31, 2009, the fair value of the warrant liabilities was $2.4 million, estimated using the Monte Carlo Simulation Model (“Simulation Model”). This warrant liability increased to $2.9 million on February 1, 2010 immediately prior to the amendment. This $0.5 million increase was recorded as a loss in other income (expense). Subsequent to amendment of the warrant terms, on February 2, 2010, the fair value of the warrant liability using the Black-Scholes Model was $2.6 million. The $0.3 million decrease in the fair value of the warrant liability was recorded as a gain in other income (expense). In the first quarter of 2010, the holders of these warrants exercised all warrants, acquiring 392,157 common shares for an aggregate exercise price of $5,882.
In June of 2009, we issued warrants to certain institutional investors as part of a separate registered direct offering. The warrants represent the right to acquire an aggregate of up to 347,826 common shares over a five year period beginning December 11, 2009 at an exercise price of $19.50 per share. In February of 2010, the holders of these warrants agreed to amend the terms of their warrants to remove the Eliminated Adjustment Provisions and we made a cash payment of $4.5 million to these warrant holders, which was recorded in other income (expense). The exercise price of these warrants remained unchanged at $19.50 per share. As of December 31, 2010 all of these warrants were outstanding.
Prior to amendment, we recorded the warrants issued in June of 2009 as a liability at fair value due to the Eliminated Adjustment Provisions and certain other provisions. At December 31, 2009, the fair value of the warrant liabilities was $2.4 million, estimated using the Simulation Model. This warrant liability increased to $3.3 million on February 1, 2010 immediately prior to the amendment. This $0.9 million increase was recorded as a loss in other income (expense). We revalued the warrant liability at December 31, 2010 using the Black-Scholes Model and recorded a decrease in the fair value of $2.5 million as a gain in the other income (expense) line of our consolidated statement of operations. The fair value of the warrant liability was $0.8 million at December 31, 2010.
Income Taxes
There was no material income tax expense for the year ended December 31, 2010. We recognized $5.7 million in income tax expense in 2009 compared with an income tax benefit of $0.4 million in 2008. Income tax expense in 2009 is primarily related to $5.8 million of foreign income tax expense recognized in connection with the expansion of our existing collaboration with Takeda signed in February of 2009. We were paid a $29 million expansion fee, of which $5.8 million was withheld for payment to the Japanese taxing authority. We also recognized $0.1 million of income tax benefit for 2009 relating to research and development refundable credits, in addition to the $0.4 million in research and development refundable credits recognized in 2008.
Accounting Standards Codification Topic 740, Income Taxes (“ASC 740”) provides for the recognition of deferred tax assets if realization of such assets is more likely than not. Based upon the weight of available evidence, which includes our historical operating performance and carry-back potential, we have determined that total deferred tax assets should be fully offset by a valuation allowance.
We have recorded cumulative gross deferred tax assets of $214.3 million and $189.9 million at December 31, 2010 and 2009, respectively, principally attributable to the timing of the deduction of certain expenses associated with certain research and development expenses, net operating loss and other carry-forwards. We also recorded corresponding valuation allowances of $214.3 million and $189.9 million at December 31, 2010 and 2009, respectively, to offset these deferred tax assets, as management cannot predict with reasonable certainty that the deferred tax assets to which the valuation allowances relate will be realized.
As of December 31, 2010, we had federal net operating loss carry-forwards of approximately $149.4 million to offset future taxable income. We also had federal research and development tax credit carry-forwards of approximately $9.5 million. In 2009, we experienced an “ownership change” under Section 382 of the Internal Revenue Code, which subjects the amount of federal and state tax carry-forwards that can be utilized to an annual limitation, which will substantially limit our future use of these carry-forwards per year. To the extent we do not utilize our carry-forwards within the applicable statutory carry-forward periods, either because of Section 382 limitations or the lack of sufficient taxable income, the carry-forwards will expire unused.
We did not have unrecognized tax benefits as of December 31, 2010 and do not expect this to change significantly over the next twelve months. In accordance with ASC 740, we will recognize interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense. As of December 31, 2010, we have not accrued interest or penalties related to uncertain tax positions.
Liquidity and Capital Resources
Cash and cash equivalents at December 31, 2010 were $37.3 million compared with $23.9 million at December 31, 2009. Net cash used in operating activities was $52.5 million in 2010, compared with net cash provided by operating activities of $7.4 million in 2009 and net cash used in operating activities of $33.0 million in 2008.
The $60.0 million change in cash provided by operations in 2009 to cash used in operations in 2010 was primarily due to a decrease in revenue receipts for license and collaborative fees and royalties, and an increase in spending on XOMA 052 related to the Phase 2 clinical program. During 2010, we received one-time cash receipts of $3.7 million related to the sale of our CIMZIA® royalty stream and $4.0 million as final payment under our two antibody discovery collaboration agreements entered into with Arana and Kaketsuken. Comparatively, during 2009, we received one-time cash receipts of $23.2 million related to the expansion of our existing collaboration with Takeda and $22.3 million related to the sale of our LUCENTIS® royalty stream to Genentech. In addition, we received $10.0 million in the second half of 2009 related to our two antibody discovery collaboration agreements entered into with Arana and Kaketsuken.
In addition, receivables and related party and other receivables increased by $13.6 million in 2010 primarily due to the $15.0 million up-front fee in connection with the license and collaboration agreement entered into with Servier in December of 2010. These decreases in cash provided by operations were partially offset by an increase in deferred revenue of $13.1 million, primarily related to the license and collaboration agreement entered into with Servier and an increase in the accounts payable and accrued liabilities balance of $2.7 million due to increased research and development expenses and timing of payments.
We expect net cash used in operating activities to decrease in 2011 as a result of cash flows from our license and collaboration agreement with Servier and a reduction in XOMA 052 phase 2 development costs.
The $40.4 million change in cash used in operations in 2008 to cash provided by operations in 2009 was primarily due to the receipt of $23.2 million in the first quarter of 2009 related to the expansion of our existing collaboration with Takeda, the receipt of $22.3 million in the third quarter of 2009 related to the sale of our LUCENTIS® royalty interest to Genentech and the receipt of $10 million in the second half of 2009 related to two antibody discovery collaboration agreements entered into with Arana and Kaketsuken.
Cash used in operations for 2008 consisted of a net loss of $45.2 million offset by non-cash adjustments of $16.1 million, primarily related to depreciation and share-based compensation. In addition, receivables increased by $4.6 million in 2008 primarily related to work performed on the NIAID 3, Novartis, Merck/Schering-Plough and Takeda contracts, offset by a decrease in work performed on the Merck/Schering-Plough/AVEO contract and accrued liabilities decreased by $3.3 million primarily related to the reversal of the 2008 bonus accrual in the fourth quarter when the Company decided it would not pay 2008 bonuses. These decreases in cash were partially offset by an increase in the accounts payable balance of $3.0 million due to the Company paying vendors on longer terms and an increase in other liabilities of $2.1 million related to the NIAID 2 billing adjustment for which a credit was provided to the NIH to be applied to future work performed on the NIAID 2 contract.
Net cash used in investing activities was $0.3 million in 2010, compared with net cash provided by investing activities of $10.6 million in 2009 and $3.2 million in 2008. Cash used in investing activities in 2010 primarily consisted of purchases of fixed assets of $0.3 million.
Net cash provided by investing activities of $10.6 million in 2009 primarily consisted of a decrease in the restricted cash balance of $9.5 million due to use of the funds for the repayment of our Goldman Sachs term loan in September of 2009. In addition, we received proceeds from maturities of investments of $1.3 million. Net cash provided by investing activities of $3.2 million in 2008 consisted of net sales and maturities of investments of $14.8 million, partially offset by the transfer to restricted cash of $3.5 million relating to our term loan facility with Goldman Sachs and purchases of fixed assets of $8.1 million, primarily relating to lab and production equipment.
Net cash provided by financing activities was $66.3 million for 2010, compared with net cash used in financing activities of $3.6 million in 2009 and net cash provided by financing activities of $16.8 million in 2008. Cash provided by financing activities in 2010 related to proceeds received from the issuance of common shares of $70.8 million, including gross proceeds of $21 million from an underwritten offering in February of 2010, $9.3 million from our 2009 ATM Agreement, $14.2 million from our common share purchase agreement with Azimuth in August of 2010, and $29.7 million from our 2010 ATM Agreement. This cash provided by financing activities was partially offset by $4.5 million paid to the holders of warrants issued in June of 2009 upon modification of the terms.
Net cash used in financing activities in 2009 of $3.6 million related to the repayment in full of the Goldman Sachs term loan, including a principal payment of $8.4 million in the second quarter of 2009, repayment of the remaining outstanding balance of $42.0 million in September of 2009, accrued interest to the date of payment of $2.4 million, and payment of a prepayment premium of $2.5 million. This cash used in financing activities was partially offset by proceeds of $49.3 million received from the issuance of common shares in 2009, including gross proceeds of $26.4 million from an equity line of credit in September of 2009, $22 million from two registered direct offerings in May of 2009 and June of 2009, and $2.8 million from our 2009 ATM Agreement.
Net cash provided by financing activities in 2008 of $16.8 million related to the refinancing of our original loan facility with Goldman Sachs in May of 2008, which netted proceeds of approximately $30.9 million, partially offset by a principal payment of $8.2 million against the outstanding balance of the original facility with Goldman Sachs in the first quarter of 2008. In addition, principal payments of $4.6 million on the new Goldman Sachs facility and $8.9 million on our Novartis note were made in the fourth quarter of 2008. We also received proceeds of $7.6 million from the issuance of common shares related to draws made on our equity line of credit with Azimuth.
Equity Line of Credit
In October of 2008, we entered into a common share purchase agreement (the “2008 Purchase Agreement”) with Azimuth, pursuant to which we obtained a committed equity line of credit facility (the “2008 Facility”). From the inception of the 2008 Facility through 2009, we sold a total of 2,815,228 common shares to Azimuth for aggregate gross proceeds of $33.9 million. This included the sale of 2.3 million shares in two transactions in September of 2009. Offering expenses incurred in 2009 related to sales to Azimuth were $0.4 million. At the end of the third quarter of 2009, the 2008 Facility was no longer in effect, and no additional shares can be issued thereunder.
In July of 2010, we entered into a common share purchase agreement (the “2010 Purchase Agreement”) with Azimuth pursuant to which we obtained a committed equity line of credit facility (the “2010 Facility”). In August of 2010, we sold a total of 3,421,407 common shares under the 2010 Facility for aggregate gross proceeds of $14.2 million, representing the maximum number of shares that could be sold under the 2010 Facility. As a result, the 2010 Facility is no longer in effect, and no additional shares can be issued thereunder.
Underwritten Offering
In February of 2010, we completed an underwritten offering of 2.8 million units, with each unit consisting of one of our common shares and a warrant to purchase 0.45 of a common share, for gross proceeds of approximately $21 million, before deducting underwriting discounts and commissions and estimated offering expenses of $1.7 million. The warrants, which represent the right to acquire an aggregate of up to 1.26 million common shares, are exercisable beginning six months and one day after issuance and have a five-year term and an exercise price of $10.50 per share. As of December 31, 2010 all of these warrants were outstanding.
Registered Direct Offerings
In May of 2009, we entered into a definitive agreement with an institutional investor to sell 784,313 units, with each unit consisting of one of our common shares and a warrant to purchase 0.50 of a common share, for gross proceeds of approximately $10 million, before deducting placement agent fees and estimated offering expenses of $0.8 million, in a registered direct offering. In the first quarter of 2010, the holders of these warrants exercised all warrants, acquiring 392,157 common shares for an aggregate exercise price of $5,882.
In June of 2009, we entered into a definitive agreement with certain institutional investors to sell 695,652 units, with each unit consisting of one of our common shares and a warrant to purchase 0.50 of a common share, for gross proceeds of approximately $12 million, before deducting placement agent fees and estimated offering expenses of $0.8 million, in a second registered direct offering. In February of 2010, the holders of these warrants agreed to amend the terms of their warrants to remove the Eliminated Adjustment Provisions and we made a cash payment of $4.5 million to these warrant holders, which was recorded in other income (expense). As of December 31, 2010 all of these warrants were outstanding.
ATM Agreements
In the third quarter of 2009, we entered into the 2009 ATM Agreement, under which we could sell up to 1.7 million of our common shares from time to time through Wm Smith, as our agent for the offer and sale of the common shares. From the inception of the 2009 ATM Agreement through October of 2010, the Company sold a total of 1.7 million common shares through Wm Smith, constituting all of the shares available for sale under the agreement, for aggregate gross proceeds of $12.2 million, including 1.4 million common shares sold in 2010 for aggregate gross proceeds of $9.3 million. Total offering expenses related to these sales were $0.4 million.
In the third quarter of 2010, we entered into the 2010 ATM Agreement, with the Agents, under which we may sell common shares from time to time through the Agents, as our agents for the offer and sale of the common shares, in an aggregate amount not to exceed the amount that can be sold under the Existing Registration Statement. The Agents may sell the common shares by any method permitted by law deemed to be an “at the market” offering as defined in Rule 415 of the Securities Act, including without limitation sales made directly on The NASDAQ Global Market, on any other existing trading market for the common shares or to or through a market maker. The Agents may also sell the common shares in privately negotiated transactions, subject to our prior approval. We will pay the Agents, collectively, a commission equal to 3% of the gross proceeds of the sales price of all common shares sold through them as sales agents under the 2010 ATM Agreement. From the inception of the ATM Agreement through December 31, 2010, we sold a total of 6.7 million common shares under this agreement for aggregate gross proceeds of $29.7 million. Total offering expenses related to these sales were $0.9 million. Subsequent to December 31, 2010 through March 8, 2011, we have sold an additional 796,898 common shares through the Agents pursuant to the 2010 ATM Agreement for aggregate gross proceeds of $4.3 million. Total offering expenses related to these sales were $0.1 million.
Proceeds from the sale of shares under the 2008 Purchase Agreement, the 2010 Purchase Agreement, the 2009 ATM Agreement, the 2010 ATM Agreement, registered direct offerings and other equity offerings are being used to continue development of our XOMA 052 product candidate and for other working capital and general corporate purposes. We also used certain of these proceeds to repay the Goldman Sachs term loan in September of 2009.
* * *
We have incurred significant operating losses and negative cash flows from operations since our inception. At December 31, 2010, we had cash and cash equivalents of $37.3 million. During 2011, we expect to continue using our cash and cash equivalents to fund ongoing operations. Additional licensing, antibody discovery and development collaboration agreements, government funding and financing arrangements may positively impact our cash balances. Based on our cash reserves and anticipated spending levels, revenue from collaborations including the XOMA 052 license and collaboration agreement with Servier, funding from the loan agreement with Servier, biodefense contracts and licensing transactions and other sources of funding the we believe to be available, we estimate that we have sufficient cash resources to meet our anticipated net cash needs through the next twelve months. Any significant revenue shortfalls, increases in planned spending on development programs or more rapid progress of development programs than anticipated, as well as the unavailability of anticipated sources of funding, could shorten this period. If adequate funds are not available, we will be required to delay, reduce the scope of, or eliminate one or more of our product development programs and further reduce personnel-related costs. Progress or setbacks by potentially competing products may also affect our ability to raise new funding on acceptable terms.
Commitments and Contingencies
Schedule of Contractual Obligations
Payments by period due under contractual obligations at December 31, 2010 are as follows (in thousands):
|
Contractual Obligations
|
Total
|
Less than 1 year
|
1 to 3 years
|
3 to 5 years
|
More than 5 years
|
|||||||||||||||
|
Operating leases (a)
|
$ | 13,390 | $ | 5,119 | $ | 7,533 | $ | 738 | $ | - | ||||||||||
|
Debt Obligations(b)
|
||||||||||||||||||||
|
Principal
|
13,694 | - | - | 13,694 | - | |||||||||||||||
|
Interest
|
1,514 | 336 | 673 | 505 | - | |||||||||||||||
|
Total
|
$ | 28,598 | $ | 5,455 | $ | 8,206 | $ | 14,937 | $ | - | ||||||||||
|
(a)
|
Operating leases are net of sublease income of $0.4 million.
|
|
(b)
|
See Item 7A: Quantitative and Qualitative Disclosures about Market Risk and Note 7: Long-Term Debt and Other Arrangements to the accompanying consolidated financial statements for further discussion of our debt obligation.
|
In addition to the above, we have committed to make potential future “milestone” payments to third parties as part of licensing and development programs. Payments under these agreements become due and payable only upon the achievement of certain developmental, regulatory and/or commercial milestones. Because it is uncertain if and when these milestones will be achieved, such contingencies, aggregating up to $97 million (assuming one product per contract meets all milestones) have not been recorded on our consolidated balance sheet. We are also obligated to pay royalties, ranging generally from 1.5% to 14% of the selling price of the licensed component and up to 40% of any sublicense fees to various universities and other research institutions based on future sales or licensing of products that incorporate certain products and technologies developed by those institutions. We are unable to determine precisely when and if our payment obligations under the agreements will become due as these obligations are based on future events, the achievement of which is subject to a significant number of risks and uncertainties.
Although operations are influenced by general economic conditions, we do not believe that inflation had a material impact on financial results for the periods presented. We believe that we are not dependent on materials or other resources that would be significantly impacted by inflation or changing economic conditions in the foreseeable future.
Recent Accounting Pronouncements
Accounting Standards Update No. 2009-13, Revenue Recognition Topic 605: Multiple Deliverable Revenue Arrangements – A Consensus of the FASB Emerging Issues Task Force (“ASU 2009-13”) provides application guidance on whether multiple deliverables exist, how the deliverables should be separated and how the consideration should be allocated to one or more units of accounting. This update establishes a selling price hierarchy for determining the selling price of a deliverable. The selling price used for each deliverable will be based on vendor-specific objective evidence, if available, third-party evidence if vendor-specific objective evidence is not available, or estimated selling price if neither vendor-specific or third-party evidence is available. The new guidance of ASU 2009-13 was adopted by us on a prospective basis effective January 1, 2010 and did not have a material effect on our consolidated financial statements. We entered into only one revenue arrangement subject to the multiple deliverable guidance during 2010. This revenue arrangement with Servier was entered into on December 30, 2010 and the effect of the early adoption of ASU 2009-13 was not material for 2010. We have recognized a total of $0.1 million in license revenue under this agreement during 2010. Had we adopted the new guidance as of January 1, 2009, it would not have affected the amount of revenue recognized in 2009.
In March of 2010, Accounting Standards Codification Topic 605, Revenue Recognition (“ASC 605”) was amended to define a milestone and clarify that the milestone method of revenue recognition is a valid application of the proportional performance model when applied to research or development arrangements. Accordingly, a Company can make an accounting policy election to recognize a payment that is contingent upon the achievement of a substantive milestone in its entirety in the period in which the milestone is achieved. We plan to adopt this guidance as of January 1, 2011 on a prospective basis and do not expect the adoption will have a material effect on our consolidated financial statements.
Subsequent Events
Servier – Cash Receipts and Loan Agreement
In January of 2011, we received a non-refundable upfront cash payment of $15 million in connection with the license and collaboration agreement entered into with Servier in December of 2010. In addition, we also received €15 million, or approximately $20 million when converted using the 12/31/10 Exchange Rate, in connection with the loan agreement entered into with Servier in December of 2010.
The loan is secured by an interest in XOMA’s intellectual property rights to all XOMA 052 indications worldwide, excluding certain rights in the U.S. and Japan territories. Interest is calculated at a floating rate based on a Euro Inter-Bank Offered Rate (“EURIBOR”) and subject to a cap. The interest rate is reset semi-annually in January and July of each year. The interest rate for the initial interest period has been set at 3.22%. Interest is payable semi-annually; however, the loan agreement provides for a deferral of interest payments over a period determined by the parties. During the deferral period, accrued interest will be added to the outstanding principal amount for the purpose of interest calculation for the next six month interest period. After a specified period, all unpaid and accrued interest shall be paid to Servier, and thereafter, all accrued and unpaid interest shall be due and payable at the end of each six-month period. The loan has a final maturity date in 2016; however, after a specified period prior to final maturity, the loan (i) may be repaid at Servier's option, by applying up to a significant percentage of any milestone or royalty payments owed by Servier under our collaboration agreement and (ii) will be repaid by using a significant percentage of any upfront, milestone or royalty payments we receive from any third party collaboration or development partner for rights to XOMA 052 in the U.S. and/or Japan. In addition, the loan becomes immediately due and payable upon certain customary events of default.
2011 ATM Agreement
On February 4, 2011, we entered into an At Market Issuance Sales Agreement (the “2011 ATM Agreement”), with McNicoll, Lewis & Vlak LLC (“MLV”), under which we may sell common shares from time to time through MLV, as our agent for the offer and sale of the common shares, in an aggregate amount not to exceed the amount that can be sold under our registration statement on Form S-3 (File No. 333-172197) filed with the SEC on February 11, 2011, once such registration statement has been declared effective by the SEC. MLV may sell the common shares by any method permitted by law deemed to be an “at the market” offering as defined in Rule 415 of the Securities Act, including without limitation sales made directly on The NASDAQ Global Market, on any other existing trading market for the common shares or to or through a market maker. MLV may also sell the common shares in privately negotiated transactions, subject to our prior approval. We will pay MLV a commission equal to 3% of the gross proceeds of the sales price of all common shares sold through them as sales agent under the 2011 ATM Agreement. As of March 8, 2011, we have not sold any common shares under the 2011 ATM Agreement.
Forward-Looking Information and Cautionary Factors That May Affect Future Results
Certain statements contained herein related to the sufficiency of our cash resources, as well as other statements related to the timing of availability of clinical trial results and the timing of initiation of clinical trials, or that otherwise relate to future periods, are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934. These statements are based on assumptions that may not prove accurate. Actual results could differ materially from those anticipated due to certain risks inherent in the biotechnology industry and for companies engaged in the development of new products in a regulated market. Among other things, the period for which our cash resources are sufficient could be shortened if expenditures are made earlier or in larger amounts than anticipated or are unanticipated, if anticipated revenue or cost sharing arrangements do not materialize, or if funds are not otherwise available on acceptable terms; the results of clinical trials may be delayed or may never become available as a result of complications in the collection or interpretation of statistical data, unavailability of resources, actions or inactions by our present or future collaboration partners, insufficient enrollment in such trials or unanticipated safety issues; and plans to initiate new clinical trials may change depending on availability of resources, actions or inactions by our present or future collaboration partners or unanticipated safety issues. These and other risks, including those related the generally unstable nature of current economic conditions; the results of discovery research and preclinical testing; the timing or results of pending and future clinical trials (including the design and progress of clinical trials; safety and efficacy of the products being tested; action, inaction or delay by the FDA, European or other regulators or their advisory bodies; and analysis or interpretation by, or submission to, these entities or others of scientific data); changes in the status of existing collaborative relationships; the ability of collaborators and other partners to meet their obligations; our ability to meet the demand of the United States government agency with which we have entered our government contracts; competition; market demands for products; scale-up and marketing capabilities; availability of additional licensing or collaboration opportunities; international operations; share price volatility; our financing needs and opportunities; uncertainties regarding the status of biotechnology patents; uncertainties as to the costs of protecting intellectual property; and risks associated with our status as a Bermuda company, are described in more detail in Item 1A: Risk Factors.
Interest Rate Risk
Our exposure to market rate risk for changes in interest rates relates primarily to our investment portfolio and our loan facility. By policy, we make our investments in high quality debt securities, limit the amount of credit exposure to any one issuer and limit duration by restricting the term of the instrument. We generally hold investments to maturity, with a weighted average portfolio period of less than twelve months. However, if the need arose to liquidate such securities before maturity, we may experience losses on liquidation. We do not invest in derivative financial instruments.
We hold interest-bearing instruments that are classified as cash, cash equivalents and short-term investments. Fluctuations in interest rates can affect the principal values and yields of fixed income investments. If interest rates in the general economy were to rise rapidly in a short period of time, our fixed income investments could lose value.
The following table presents the amounts and related weighted average interest rates of our cash and investments at December 31, 2010 and 2009 (in thousands, except interest rate):
|
Maturity
|
Carrying
Amount
(in thousands)
|
Fair Value
(in thousands)
|
Weighted
Average
Interest Rate
|
||||||||||
|
December 31, 2010
|
|||||||||||||
|
Cash and cash equivalents
|
Daily to 90 days
|
$ | 37,304 | $ | 37,304 | 0.09 | % | ||||||
|
December 31, 2009
|
|||||||||||||
|
Cash and cash equivalents
|
Daily to 90 days
|
$ | 23,909 | $ | 23,909 | 0.38 | % | ||||||
As of December 31, 2010, we have an outstanding principal balance on our note with Novartis of $13.7 million, which is due in 2015. The interest rate on this note is charged at a rate of USD six-month LIBOR plus 2%, which was 2.46% at December 31, 2010. No further borrowing is available under this facility.
The variable interest rate related to our long-term debt instrument is based on LIBOR. We estimate that a hypothetical 100 basis point change in interest rates could increase or decrease our interest expense by approximately $0.1 million on an annualized basis.
Foreign Currency Risk
We may hold debt or incur expenses denominated in a foreign currency. The amount of debt held or expenses incurred will be impacted by fluctuations in these foreign currencies. When the U.S. dollar weakens against foreign currencies, the U.S. dollar value of the foreign-currency denominated debt and expense increases, and when the U.S. dollar strengthens against these currencies, the U.S. dollar value of the foreign-currency denominated debt and expense decreases. Consequently, changes in exchange rates may affect our results of operations. We currently have not hedged against our foreign currency risks, however, we will assess the need to hedge against future foreign currency risks when appropriate.
The following consolidated financial statements of the registrant, related notes and report of independent registered public accounting firm are set forth beginning on page F-1 of this report.
|
Report of Independent Registered Public Accounting Firm
|
F-2
|
|
Consolidated Balance Sheets
|
F-3
|
|
Consolidated Statements of Operations
|
F-4
|
|
Consolidated Statements of Shareholders' Equity (Net Capital Deficiency)
|
F-5
|
|
Consolidated Statements of Cash Flows
|
F-6
|
|
Notes to the Consolidated Financial Statements
|
F-7
|
Not applicable.
|
Item 9A.
|
Under the supervision and with the participation of our management, including our Chairman, Chief Executive Officer and President and our Vice President, Finance and Chief Financial Officer, we conducted an evaluation of our disclosure controls and procedures, as such term is defined under Rule 13a-15(e) promulgated under the Securities Exchange Act of 1934, as amended, as of the end of the period covered by this report. Our disclosure controls and procedures are intended to ensure that the information we are required to disclose in the reports that we file or submit under the Securities Exchange Act of 1934 is (i) recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms and (ii) accumulated and communicated to our management, including the Chairman, Chief Executive Officer and President and Vice President, Finance and Chief Financial Officer, as the principal executive and financial officers, respectively, to allow timely decisions regarding required disclosures. Based on this evaluation, our Chairman, Chief Executive Officer and President and our Vice President, Finance and Chief Financial Officer concluded that our disclosure controls and procedures were effective as of the end of the period covered by this report.
There were no changes in our internal controls over financial reporting during 2010 that have materially affected, or are reasonably likely to materially affect, our internal controls over financial accounting.
Management’s Report on Internal Control over Financial Reporting
Management, including our Chairman, Chief Executive Officer and President and our Vice President, Finance and Chief Financial Officer, is responsible for establishing and maintaining adequate internal control over financial reporting (as such term is defined in Exchange Act Rules 13a-159f). The Company’s internal control system was designed to provide reasonable assurance to the Company’s management and board of directors regarding the preparation and fair presentation of published financial statements in accordance with accounting principles generally accepted in the United States.
Management assessed the effectiveness of our internal control over financial reporting as of December 31, 2010. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control—Integrated Framework. Based on our assessment we believe that, as of December 31, 2010, our internal control over financial reporting is effective based on those criteria.
The Company’s internal control over financial reporting as of December 31, 2010, has been audited by Ernst & Young, LLP, the independent registered public accounting firm who also audited the Company’s consolidated financial statements. Ernst & Young’s attestation report on the Company’s internal control over financial reporting follows.
Changes in Internal Control over Financial Reporting
Our management, including our Chairman, Chief Executive Officer and President and our Vice President, Finance and Chief Financial Officer, has evaluated any changes in our internal control over financial reporting that occurred during the quarter ended December 31, 2010, and has concluded that there was no change during such quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Shareholders of XOMA Ltd.:
We have audited XOMA Ltd.’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). XOMA Ltd.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, XOMA Ltd. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of XOMA Ltd. as of December 31, 2010 and 2009 and the related consolidated statements of operations, shareholders’ equity (net capital deficiency) and cash flows for each of the three years in the period ended December 31, 2010, and our report dated March 10, 2011 expressed an unqualified opinion thereon.
/s/ ERNST & YOUNG LLP
Palo Alto, California
March 10, 2011
|
Item 9B.
|
None.
PART III
Certain information regarding our executive officers required by this Item is set forth as a Supplementary Item at the end of Part I of this Form 10-K (pursuant to Instruction 3 to Item 401(b) of Regulation S-K). The Company’s Code of Ethics applies to all employees, officers and directors including the Chairman, Chief Executive Officer and President, and the Vice President, Finance and Chief Financial Officer and Chief Accounting Officer, and is posted on the Company’s website at www.xoma.com. Other information required by this Item will be included in the Company’s proxy statement for the 2011 Annual General Meeting of Shareholders, under the sections labeled “Item 1—Election of Directors” and “Compliance with Section 16(a) of the Securities Exchange Act of 1934”, and is incorporated herein by reference.
|
Item 11.
|
Information required by this Item will be included in the sections labeled “Compensation of Executive Officers”, “Summary Compensation Table”, “Grants of Plan-Based Awards”, “Outstanding Equity Awards as of December 31, 2010”, “Option Exercises and Shares Vested”, “Pension Benefits”, “Non-Qualified Deferred Compensation” and “Compensation of Directors” appearing in our proxy statement for the 2011 Annual General Meeting of Shareholders, and is incorporated herein by reference.
|
Item 12.
|
Information required by this Item will be included in the sections labeled “Share Ownership” and “Equity Compensation Plan Information” appearing in our proxy statement for the 2011 Annual General Meeting of Shareholders, and is incorporated herein by reference.
Information required by this Item will be included in the section labeled “Transactions with Related Persons” appearing in our proxy statement for the 2011 Annual General Meeting of Shareholders, and is incorporated herein by reference.
Information required by this Item will be included in the section labeled “Item 2—Appointment of Independent Registered Public Accounting Firm” appearing in our proxy statement for the 2011 Annual General Meeting of Shareholders, and is incorporated herein by reference.
PART IV
|
|
(a)
|
The following documents are included as part of this Annual Report on Form 10-K:
|
|
(1)
|
Financial Statements:
|
All financial statements of the registrant referred to in Item 8 of this Report on Form 10-K.
|
(2)
|
Financial Statement Schedules:
|
All financial statements schedules have been omitted because the required information is included in the consolidated financial statements or the notes thereto or is not applicable or required.
|
(3)
|
Exhibits:
|
See “Index to Exhibits” on page i of this report.
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on this 10th day of March 2011.
|
XOMA LTD.
|
||
|
By:
|
/s/ STEVEN B. ENGLE
|
|
|
|
Steven B. Engle
|
|
|
|
Chairman of the Board, Chief Executive Officer and President | |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Signature
|
|
Title
|
Date
|
|
|
/s/ Steven B. Engle
|
|
Chairman of the Board, Chief Executive Officer and President
|
March 10, 2011
|
|
|
(Steven B. Engle)
|
(Principal Executive Officer) | |||
|
/s/ Fred Kurland
|
|
Vice President, Finance and Chief Financial Officer
|
March 10, 2011
|
|
|
(Fred Kurland)
|
(Principal Financial Officer and Principal Accounting Officer) | |||
|
/s/ Patrick J. Scannon
|
Executive Vice President, Chief Scientific Officer
|
March 10, 2011
|
||
|
(Patrick J. Scannon, M.D., Ph.D.)
|
||||
|
/s/ W. Denman Van Ness
|
|
Lead Independent Director
|
March 10, 2011
|
|
|
(W. Denman Van Ness)
|
||||
|
/s/ William K. Bowes, Jr.
|
|
Director
|
March 10, 2011
|
|
|
(William K. Bowes, Jr.)
|
||||
|
/s/ Peter Barton Hutt
|
|
Director
|
March 10, 2011
|
|
|
(Peter Barton Hutt)
|
||||
|
/s/ John Varian
|
|
Director
|
March 10, 2011
|
|
|
(John Varian)
|
||||
|
/s/ Timothy P. Walbert
|
|
Director
|
March 10, 2011
|
|
|
(Timothy P. Walbert)
|
||||
|
/s/ Jack L. Wyszomierski
|
Director
|
March 10, 2011
|
||
| Jack L. Wyszomierski |
|
Report of Independent Registered Public Accounting Firm
|
F-2
|
|
Consolidated Balance Sheets
|
F-3
|
|
Consolidated Statements of Operations
|
F-4
|
|
Consolidated Statements of Shareholders' Equity (Net Capital Deficiency)
|
F-5
|
|
Consolidated Statements of Cash Flows
|
F-6
|
|
Notes to the Consolidated Financial Statements
|
F-7
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Shareholders of XOMA Ltd.:
We have audited the accompanying consolidated balance sheets of XOMA Ltd. as of December 31, 2010 and 2009, and the related consolidated statements of operations, shareholders' equity (net capital deficiency), and cash flows for each of the three years in the period ended December 31, 2010. These consolidated financial statements are the responsibility of XOMA Ltd.’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of XOMA Ltd. at December 31, 2010 and 2009, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2010, in conformity with U.S. generally accepted accounting principles.
As discussed in Note 1 to the consolidated financial statements, the Company changed its method of accounting for revenue recognition as a result of the adoption of the amendments to the FASB Accounting Standards Codification resulting from Accounting Standards Update No. 2009-13, Multiple-Deliverable Revenue Arrangements, effective January 1, 2010.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), XOMA Ltd.’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 10, 2011 expressed an unqualified opinion thereon.
/s/ ERNST & YOUNG LLP
Palo Alto, California
March 10, 2011
CONSOLIDATED BALANCE SHEETS
(in thousands, except share and per share amounts)
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
ASSETS
|
||||||||
|
Current assets:
|
||||||||
|
Cash and cash equivalents
|
$ | 37,304 | $ | 23,909 | ||||
|
Trade and other receivables, net
|
20,864 | 7,231 | ||||||
|
Prepaid expenses and other current assets
|
712 | 1,012 | ||||||
|
Total current assets
|
58,880 | 32,152 | ||||||
|
Property and equipment, net
|
14,869 | 20,270 | ||||||
|
Other assets
|
503 | 402 | ||||||
|
Total assets
|
$ | 74,252 | $ | 52,824 | ||||
|
LIABILITIES AND SHAREHOLDERS’ EQUITY
|
||||||||
|
Current liabilities:
|
||||||||
|
Accounts payable
|
$ | 3,581 | $ | 2,942 | ||||
|
Accrued liabilities
|
10,650 | 8,639 | ||||||
|
Deferred revenue
|
17,044 | 2,114 | ||||||
|
Warrant liability
|
4,245 | 4,760 | ||||||
|
Other current liabilities
|
8 | 223 | ||||||
|
Total current liabilities
|
35,528 | 18,678 | ||||||
|
Deferred revenue – long-term
|
1,086 | 2,894 | ||||||
|
Interest bearing obligation – long-term
|
13,694 | 13,341 | ||||||
|
Other long-term liabilities
|
353 | 385 | ||||||
|
Total liabilities
|
50,661 | 35,298 | ||||||
|
Commitments and contingencies (Note 11)
|
||||||||
|
Shareholders’ equity:
|
||||||||
|
Preference shares, $0.05 par value, 1,000,000 shares authorized
|
||||||||
|
Series A, 210,000 designated, no shares issued and outstanding at December 31, 2010 and 2009
|
- | - | ||||||
|
Series B, 8,000 designated, 2,959 shares issued and outstanding at December 31, 2010 and 2009 (aggregate liquidation preference of $29,600)
|
1 | 1 | ||||||
|
Common shares, $0.0075 par value, 46,666,666 shares authorized, 28,491,318 and 13,536,146 shares outstanding at December 31, 2010 and 2009, respectively
|
214 | 101 | ||||||
|
Additional paid-in capital
|
876,686 | 801,978 | ||||||
|
Accumulated deficit
|
(853,310 | ) | (784,554 | ) | ||||
|
Total shareholders’ equity
|
23,591 | 17,526 | ||||||
|
Total liabilities and shareholders’ equity
|
$ | 74,252 | $ | 52,824 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share amounts)
|
Year Ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Revenues:
|
||||||||||||
|
License and collaborative fees
|
$ | 2,182 | $ | 43,822 | $ | 16,366 | ||||||
|
Contract and other revenue
|
27,174 | 25,492 | 30,473 | |||||||||
|
Royalties
|
4,285 | 29,116 | 21,148 | |||||||||
|
Total revenues
|
33,641 | 98,430 | 67,987 | |||||||||
|
Operating expenses:
|
||||||||||||
|
Research and development
|
77,413 | 58,131 | 82,576 | |||||||||
|
Selling, general and administrative
|
23,250 | 23,736 | 24,145 | |||||||||
|
Restructuring
|
82 | 3,603 | - | |||||||||
|
Total operating expenses
|
100,745 | 85,470 | 106,721 | |||||||||
|
(Loss) income from operations
|
(67,104 | ) | 12,960 | (38,734 | ) | |||||||
|
Other income (expense):
|
||||||||||||
|
Investment and interest income
|
16 | 49 | 859 | |||||||||
|
Interest expense
|
(385 | ) | (4,888 | ) | (7,002 | ) | ||||||
|
Loss on debt extinguishment
|
- | (3,645 | ) | (652 | ) | |||||||
|
Other (expense) income
|
(1,256 | ) | 1,801 | (99 | ) | |||||||
|
Net (loss) income before taxes
|
(68,729 | ) | 6,277 | (45,628 | ) | |||||||
|
Income tax (expense) benefit
|
(27 | ) | (5,727 | ) | 383 | |||||||
|
Net (loss) income
|
$ | (68,756 | ) | $ | 550 | $ | (45,245 | ) | ||||
|
Basic and diluted net (loss) income per common share
|
$ | (3.69 | ) | $ | 0.05 | $ | (5.11 | ) | ||||
|
Shares used in computing basic net (loss) income per common share
|
18,613 | 10,993 | 8,862 | |||||||||
|
Shares used in computing diluted net (loss) income per common share
|
18,613 | 11,313 | 8,862 | |||||||||
The accompanying notes are an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENT OF SHAREHOLDERS’ EQUITY
(NET CAPITAL DEFICIENCY)
(in thousands)
|
Preferred Shares
|
Common Shares
|
Paid-In | Accumulated Comprehensive Income | Accumulated | Total Shareholders’ Equity (Net Capital | |||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
(Loss)
|
Deficit
|
Deficiency)
|
|||||||||||||||||||||||||
|
Balance, December 31, 2007
|
3 | $ | 1 | 8,797 | $ | 66 | $ | 740,119 | $ | (9 | ) | $ | (739,859 | ) | $ | 318 | ||||||||||||||||
|
Exercise of share options, contributions to 401(k) and incentive plans
|
─
|
─
|
38 |
─
|
1,389 |
─
|
─
|
1,389 | ||||||||||||||||||||||||
|
Share-based compensation expense under SFAS 123R
|
─
|
─
|
─
|
─
|
4,934 |
─
|
─
|
4,934 | ||||||||||||||||||||||||
|
Sale of shares of common stock
|
─
|
─
|
529 | 4 | 7,192 |
─
|
─
|
7,196 | ||||||||||||||||||||||||
|
Comprehensive income (loss):
|
||||||||||||||||||||||||||||||||
|
Net change in unrealized loss on investments
|
─
|
─
|
─
|
─
|
─
|
7 |
─
|
7 | ||||||||||||||||||||||||
|
Net loss
|
─
|
─
|
─
|
─
|
─
|
─
|
(45,245 | ) | (45,245 | ) | ||||||||||||||||||||||
|
Comprehensive loss
|
─
|
─
|
─
|
─
|
─
|
─
|
─
|
(45,238 | ) | |||||||||||||||||||||||
|
Balance, December 31, 2008
|
3 | 1 | 9,364 | 70 | 753,634 | (2 | ) | (785,104 | ) | (31,401 | ) | |||||||||||||||||||||
|
Exercise of share options, contributions to 401(k) and incentive plans
|
─
|
─
|
135 | 1 | 1,358 |
─
|
─
|
1,359 | ||||||||||||||||||||||||
|
Share-based compensation expense under SFAS 123R
|
─
|
─
|
─
|
─
|
4,395 |
─
|
─
|
4,395 | ||||||||||||||||||||||||
|
Sale of shares of common stock
|
─
|
─
|
4,036 | 30 | 42,591 |
─
|
─
|
42,621 | ||||||||||||||||||||||||
|
Comprehensive income:
|
||||||||||||||||||||||||||||||||
|
Net change in unrealized loss on investments
|
─
|
─
|
─
|
─
|
─
|
2 |
─
|
2 | ||||||||||||||||||||||||
|
Net income
|
─
|
─
|
─
|
─
|
─
|
─
|
550 | 550 | ||||||||||||||||||||||||
|
Comprehensive income
|
─
|
─
|
─
|
─
|
─
|
─
|
─
|
552 | ||||||||||||||||||||||||
|
Balance, December 31, 2009
|
3 | 1 | 13,536 | 101 | 801,978 | - | (784,554 | ) | 17,526 | |||||||||||||||||||||||
|
Exercise of share options, contributions to 401(k) and incentive plans
|
─
|
─
|
94 | 1 | 945 |
─
|
─
|
946 | ||||||||||||||||||||||||
|
Share-based compensation expense under SFAS 123R
|
─
|
─
|
─
|
─
|
4,913 |
─
|
─
|
4,913 | ||||||||||||||||||||||||
|
Sale of shares of common stock
|
─
|
─
|
14,469 | 109 | 66,232 |
─
|
─
|
66,341 | ||||||||||||||||||||||||
|
Exercise of warrants
|
─
|
─
|
392 | 3 | 2,618 |
─
|
─
|
2,621 | ||||||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||||||||||
|
Net loss
|
─
|
─
|
─
|
─
|
─
|
─
|
(68,756 | ) | (68,756 | ) | ||||||||||||||||||||||
|
Comprehensive loss
|
─
|
─
|
─
|
─
|
─
|
─
|
─
|
(68,756 | ) | |||||||||||||||||||||||
|
Balance, December 31, 2010
|
3 | $ | 1 | 28,491 | $ | 214 | $ | 876,686 | $ |
─
|
$ | (853,310 | ) | $ | 23,591 | |||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
|
Year Ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Cash flows from operating activities:
|
||||||||||||
|
Net (loss) income
|
$ | (68,756 | ) | $ | 550 | $ | (45,245 | ) | ||||
|
Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities:
|
||||||||||||
|
Depreciation and amortization
|
5,721 | 6,831 | 6,721 | |||||||||
|
Common shares contribution to 401(k) and management incentive plans
|
905 | 1,198 | 1,008 | |||||||||
|
Share-based compensation expense
|
4,913 | 4,395 | 4,934 | |||||||||
|
Accrued interest on convertible notes and interest bearing obligations
|
353 | (1,116 | ) | 1,921 | ||||||||
|
Revaluation of warrant liability
|
(2,283 | ) | (1,781 | ) | - | |||||||
|
Amortization of discount, premium and debt issuance costs of debt and convertible debt
|
- | 487 | 726 | |||||||||
|
Warrant modification expense
|
4,500 | - | - | |||||||||
|
Loss (gain) on disposal/retirement of property and equipment
|
9 | (15 | ) | 99 | ||||||||
|
Loss on debt extinguishment
|
- | 3,645 | 652 | |||||||||
|
Other non-cash adjustments
|
10 | 27 | - | |||||||||
|
Changes in assets and liabilities:
|
||||||||||||
|
Receivables
|
(13,633 | ) | 9,455 | (4,551 | ) | |||||||
|
Prepaid expenses and other assets
|
199 | 284 | (183 | ) | ||||||||
|
Accounts payable and accrued liabilities
|
2,650 | (2,844 | ) | (290 | ) | |||||||
|
Deferred revenue
|
13,122 | (12,205 | ) | (851 | ) | |||||||
|
Other liabilities
|
(247 | ) | (1,476 | ) | 2,084 | |||||||
|
Net cash (used in) provided by operating activities
|
(52,537 | ) | 7,435 | (32,975 | ) | |||||||
|
Cash flows from investing activities:
|
||||||||||||
|
Proceeds from sales of investments
|
- | - | 9,875 | |||||||||
|
Proceeds from maturities of investments
|
- | 1,300 | 8,099 | |||||||||
|
Purchase of investments
|
- | - | (3,199 | ) | ||||||||
|
Transfer of restricted cash
|
- | 9,545 | (3,526 | ) | ||||||||
|
Purchase of property and equipment
|
(339 | ) | (270 | ) | (8,060 | ) | ||||||
|
Net cash (used in) provided by investing activities
|
(339 | ) | 10,575 | 3,189 | ||||||||
|
Cash flows from financing activities:
|
||||||||||||
|
Proceeds from issuance of long-term debt
|
- | - | 55,000 | |||||||||
|
Principal payments of debt
|
- | (50,394 | ) | (45,779 | ) | |||||||
|
Payment of prepayment premium on repayment of short-term debt
|
- | (2,543 | ) | - | ||||||||
|
Proceeds from issuance of common shares
|
70,771 | 49,323 | 7,578 | |||||||||
|
Payment for modification of warrants
|
(4,500 | ) | - | - | ||||||||
|
Net cash provided by (used in) financing activities
|
66,271 | (3,614 | ) | 16,799 | ||||||||
|
Net increase (decrease) in cash and cash equivalents
|
13,395 | 14,396 | (12,987 | ) | ||||||||
|
Cash and cash equivalents at the beginning of the period
|
23,909 | 9,513 | 22,500 | |||||||||
|
Cash and cash equivalents at the end of the period
|
$ | 37,304 | $ | 23,909 | $ | 9,513 | ||||||
|
Supplemental Cash Flow Information:
|
||||||||||||
|
Cash paid during the year for:
|
||||||||||||
|
Interest
|
$ | - | $ | 5,510 | $ | 4,354 | ||||||
|
Income taxes
|
16 | 5,800 | - | |||||||||
|
Non-cash investing and financing activities:
|
||||||||||||
|
Issuance and Extinguishment of warrant liabilities
|
$ | 1,767 | $ | 6,541 | $ | - | ||||||
|
Interest added to principal balance on Novartis note
|
353 | 462 | 1,183 | |||||||||
|
Debt reduction on Novartis note
|
- | - | 7,500 | |||||||||
The accompanying notes are an integral part of these consolidated financial statements.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
|
1.
|
Description of Business
|
XOMA Ltd. (“XOMA” or the “Company”), a Bermuda company, is a biopharmaceutical company focused on the discovery, development and manufacture of therapeutic antibodies designed to treat inflammatory, autoimmune, infectious and oncological diseases. The Company’s products are presently in various stages of development and most are subject to regulatory approval before they can be commercially launched.
|
2.
|
Basis of Presentation and Significant Accounting Policies
|
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries. All intercompany accounts and transactions have been eliminated in consolidation.
Use of Estimates and Reclassifications
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenue and expenses, and related disclosures. On an on-going basis, management evaluates its estimates, including those related to revenue recognition, research and development expense, long-lived assets, warrant liabilities and share-based compensation. The Company bases its estimates on historical experience and on various other market-specific and other relevant assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ significantly from these estimates, such as the Company’s billing under government contracts. Under the Company’s contracts with the National Institute of Allergy and Infectious Diseases (“NIAID”), a part of the National Institutes of Health (“NIH”), the Company bills using NIH provisional rates and thus are subject to future audits at the discretion of NIAID’s contracting office. These audits can result in an adjustment to revenue previously reported.
Recent Accounting Pronouncements
Accounting Standards Update No. 2009-13, Revenue Recognition Topic 605: Multiple Deliverable Revenue Arrangements – A Consensus of the FASB Emerging Issues Task Force (“ASU 2009-13”) provides application guidance on whether multiple deliverables exist, how the deliverables should be separated and how the consideration should be allocated to one or more units of accounting. This update establishes a selling price hierarchy for determining the selling price of a deliverable. The selling price used for each deliverable will be based on vendor-specific objective evidence, if available, third-party evidence if vendor-specific objective evidence is not available, or estimated selling price if neither vendor-specific or third-party evidence is available. The new guidance of ASU 2009-13 was adopted by the Company on a prospective basis effective January 1, 2010 and did not have a material effect on the Company’s consolidated financial statements. The Company entered into only one revenue arrangement subject to the multiple deliverable guidance during 2010. This revenue arrangement with Servier was entered into on December 30, 2010 and the effect of the early adoption of ASU 2009-13 was not material for 2010. The Company recognized a total of $0.1 million in license revenue under this agreement during 2010. Had the Company adopted the new guidance as of January 1, 2009, it would not have affected the amount of revenue recognized in 2009.
In March of 2010, Accounting Standards Codification Topic 605, Revenue Recognition (“ASC 605”) was amended to define a milestone and clarify that the milestone method of revenue recognition is a valid application of the proportional performance model when applied to research or development arrangements. Accordingly, a Company can make an accounting policy election to recognize a payment that is contingent upon the achievement of a substantive milestone in its entirety in the period in which the milestone is achieved. The Company plans to adopt this guidance as of January 1, 2011 on a prospective basis and does not expect the adoption will have a material effect on the Company’s consolidated financial statements.
Revenue Recognition
Revenue is recognized when the four basic criteria of revenue recognition are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services have been rendered; (3) the fee is fixed and determinable; and (4) collectability is reasonably assured. The determination of criteria (2) is based on management’s judgments regarding whether a continuing performance obligation exists. The determination of criteria (3) and (4) are based on management’s judgments regarding the nature of the fee charged for products or services delivered and the collectability of those fees. Allowances are established for estimated uncollectible amounts, if any.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
The Company recognizes revenue from its license and collaboration arrangements, contract services and royalties. Revenue arrangements with multiple elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered items. The consideration received is allocated among the separate units based on their respective fair values and the applicable revenue recognition criteria are applied to each of the separate units. Advance payments received in excess of amounts earned are classified as deferred revenue until earned.
License and Collaborative Fees
Revenue from non-refundable license, technology access or other payments under license and collaborative agreements where the Company has a continuing obligation to perform is recognized as revenue over the expected period of the continuing performance obligation. The Company estimates the performance period at the inception of the arrangement and reevaluates it each reporting period. This reevaluation may shorten or lengthen the period over which the remaining revenue is recognized. Changes to these estimates are recorded on a prospective basis.
Milestone payments under collaborative and other arrangements are recognized as revenue upon completion of the milestone event, once confirmation is received from the third party and collectability is reasonably assured. This represents the culmination of the earnings process when the Company has no future performance obligations related to the payment. Milestone payments that are not substantive or that require a continuing performance obligation on the part of the Company are recognized over the expected period of the continuing performance obligation. Amounts received in advance are recorded as deferred revenue until the related milestone is completed.
Contract Revenue
Contract revenue for research and development involves the Company providing research and development and manufacturing services to collaborative partners, biodefense contractors or others. Revenue for certain contracts is accounted for by a proportional performance, or output-based, method where performance is based on estimated progress toward elements defined in the contract. The amount of contract revenue and related costs recognized in each accounting period are based on management’s estimates of the proportional performance during the period. Adjustments to estimates based on actual performance are recognized on a prospective basis and do not result in reversal of revenue should the estimate to complete be extended.
Up-front fees are recognized in the same manner as the final deliverable, which is generally ratably over the period of the continuing performance obligation. Given the uncertainties of research and development collaborations, significant judgment is required to determine the duration of the arrangement.
Royalty Revenue
Royalty revenue and royalty receivables are generally recorded in the periods these royalties are earned, in advance of collection. The royalty revenue and receivables in these instances is based upon communication with collaborative partners or licensees, historical information and forecasted sales trends.
Research and Development Expenses
The Company expenses research and development costs as incurred. Research and development expenses consist of direct costs such as salaries and related personnel costs and material and supply costs, and research-related allocated overhead costs, such as facilities costs. In addition, research and development expenses include costs related to clinical trials. Expenses resulting from clinical trials are recorded when incurred based in part on estimates as to the status of the various trials. From time to time, research and development expenses may include up-front fees and milestones paid to collaborative partners for the purchase of rights to in-process research and development. Such amounts are expensed as incurred. Total research and development expenses related to the Company’s collaborative agreements were approximately $19.3 million, $15.9 million and $24.1 million in 2010, 2009 and 2008, respectively.
Share-Based Compensation
Share-based compensation expense is recognized ratably over the requisite service period. If options are granted that include a performance condition, the Company estimates the probability of the performance condition being achieved on a quarterly basis. If it is determined that it is probable the performance criteria will be achieved, the Company estimates an implicit service period from grant date to the most likely date of achievement of the performance criteria and records share-based compensation expense ratably over this implicit service period.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Cash and Cash Equivalents and Short-term Investments
The Company considers all highly liquid debt instruments with maturities of three months or less at the time the Company acquires them to be cash equivalents.
Short-term investments include debt securities classified as available-for-sale. Available-for-sale securities are stated at fair value, with unrealized gains and losses, net of tax, if any, reported in other comprehensive income (loss). The estimate of fair value is based on publicly available market information. Realized gains and losses and declines in value judged to be other-than-temporary on available-for-sale securities are also included in investment and other income. The Company reviews its instruments for other-than-temporary impairment whenever the value of the instrument is less than the amortized cost. The cost of investments sold is based on the specific identification method. Interest and dividends on securities classified as available-for-sale are included in investment and other income.
Property and Equipment and Long-Lived Assets
Property and equipment is stated at cost less depreciation. Equipment depreciation is calculated using the straight-line method over the estimated useful lives of the assets (three to seven years). Leasehold improvements, buildings and building improvements are amortized and depreciated using the straight-line method over the shorter of the lease terms or the useful lives (one to fifteen years).
The Company records impairment losses on long-lived assets used in operations when events and circumstances indicate that the assets might be impaired and the undiscounted cash flows estimated to be generated by those assets in the future are less than the carrying amounts of those assets.
Warrant Liabilities
The Company has issued warrants to purchase its common shares in connection with financing activities. The Company accounts for the warrants as a liability at fair value. The fair value of the warrant liability is estimated using the Black-Scholes Model. The Black-Scholes Model requires inputs such as the expected term of the warrants, share price volatility and risk-free interest rate. These inputs are subjective and generally require significant analysis and judgment to develop. For the estimate of the expected term, the Company uses the full remaining contractual term of the warrant. The Company bases its estimate of expected volatility on its historical volatility. These assumptions are reviewed each reporting period and changes in the estimated fair value of the outstanding warrants are recognized in other income (expense).
In February of 2010, the holders of the May 2009 and June 2009 warrants agreed to amend the terms of their warrants to remove the provisions that would have required a reduction of the warrant exercise price and an increase in the number of shares issuable on exercise of the warrants each time the Company sold common shares at a price less than the exercise price of such warrants (the “Eliminated Adjustment Provisions”). Prior to the amendments, the Company recorded the warrants issued in May and June of 2009 as a liability at fair value due to the Eliminated Adjustment Provisions and certain other provisions, which was estimated using the Monte Carlo Simulation Model (“Simulation Model”).
Income Taxes
The Company accounts for uncertain tax positions in accordance with Accounting Standards Codification Topic 740, Income Taxes (“ASC 740”). ASC 740 provides for the recognition of deferred tax assets if realization of such assets is more likely than not.
Net Income (Loss) per Common Share
Basic net income (loss) per common share is based on the weighted average number of common shares outstanding during the period. Diluted net income (loss) per common share is based on the weighted average number of common shares and other dilutive securities outstanding during the period, provided that including these dilutive securities does not increase the net income (loss) per share.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Potentially dilutive securities are excluded from the calculation of earnings per share if their inclusion is anti-dilutive. The following table shows the total outstanding securities considered anti-dilutive and therefore excluded from the computation of diluted net income (loss) per share (in thousands):
|
December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Options for common shares
|
2,180 | 1,156 | 1,320 | |||||||||
|
Convertible preference shares
|
254 | - | 254 | |||||||||
|
Warrants for common shares
|
1,535 | 740 | - | |||||||||
|
Total
|
3,969 | 1,896 | 1,574 | |||||||||
For the year ended December 31, 2009, the following is a reconciliation of the numerators and denominators of the basic and diluted net income per share (in thousands):
|
Year ended December 31,
|
||||
|
2009
|
||||
|
Numerator
|
||||
|
Net income used for basic and diluted net income per share
|
$ | 550 | ||
|
Denominator
|
||||
|
Weighted average shares outstanding used for basic net income per share
|
10,993 | |||
|
Effect of dilutive share options
|
66 | |||
|
Effect of convertible preference shares
|
254 | |||
|
Weighted average shares outstanding and dilutive securities used for diluted net income per share
|
11,313 | |||
For the years ended December 31, 2010 and 2008, all outstanding common stock equivalents were considered anti-dilutive and therefore the calculations of basic and diluted net loss per share are the same.
|
3.
|
Consolidated Financial Statement Detail
|
Cash and Cash Equivalents
At December 31, 2010 and 2009, cash and cash equivalents consisted of overnight deposits and money market funds and repurchase agreements with maturities of less than 90 days at the date of purchase. Cash and cash equivalent balances were recorded at fair value as follows as of December 31, 2010 and 2009 (in thousands):
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
|
December 31, 2010
|
||||||||||||||||
|
Cost Basis
|
Unrealized Gains
|
Unrealized Losses
|
Estimated Fair Value
|
|||||||||||||
|
Cash
|
$ | 29,536 | $ | - | $ | - | $ | 29,536 | ||||||||
|
Cash equivalents
|
7,768 | - | - | 7,768 | ||||||||||||
|
Total cash and cash equivalents
|
$ | 37,304 | $ | - | $ | - | $ | 37,304 | ||||||||
|
December 31, 2009
|
||||||||||||||||
|
Cost Basis
|
Unrealized Gains
|
Unrealized Losses
|
Estimated Fair Value
|
|||||||||||||
|
Cash
|
$ | 3,065 | $ | - | $ | - | $ | 3,065 | ||||||||
|
Cash equivalents
|
20,844 | - | - | 20,844 | ||||||||||||
|
Total cash and cash equivalents
|
$ | 23,909 | $ | - | $ | - | $ | 23,909 | ||||||||
Receivables
Receivables consisted of the following at December 31, 2010 and 2009 (in thousands):
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Trade receivables, net
|
$ | 20,309 | $ | 6,391 | ||||
|
Other receivables
|
555 | 840 | ||||||
|
Total
|
$ | 20,864 | $ | 7,231 | ||||
Property and Equipment
Property and equipment consisted of the following at December 31, 2010 and 2009 (in thousands):
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Furniture and equipment
|
$ | 31,700 | $ | 31,429 | ||||
|
Buildings, leasehold and building improvements
|
21,463 | 21,463 | ||||||
|
Construction-in-progress
|
203 | 196 | ||||||
|
Land
|
310 | 310 | ||||||
| 53,676 | 53,398 | |||||||
|
Less: Accumulated depreciation and amortization
|
(38,807 | ) | (33,128 | ) | ||||
|
Property and equipment, net
|
$ | 14,869 | $ | 20,270 | ||||
Depreciation and amortization expense was $5.7 million, $6.8 million and $6.7 million for the years ended December 31, 2009, 2009 and 2008, respectively.
Accrued Liabilities
Accrued liabilities consisted of the following at December 31, 2010 and 2009 (in thousands):
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Accrued management incentive compensation
|
$ | 4,982 | $ | 3,681 | ||||
|
Accrued payroll and other benefits
|
2,752 | 2,691 | ||||||
|
Accrued professional fees
|
1,020 | 767 | ||||||
|
Accrued clinical trial costs
|
1,020 | 609 | ||||||
|
Accrued restructuring costs
|
80 | 155 | ||||||
|
Other
|
796 | 736 | ||||||
|
Total
|
$ | 10,650 | $ | 8,639 | ||||
Deferred Revenue
In 2010, the Company deferred $15.9 million of revenue from five contracts including Servier, NIH, Takeda Pharmaceutical Company Limited (“Takeda”), Schering-Plough Research Institute, a division of Schering Corporation, now a subsidiary of Merck & Co., Inc. (referred to herein as “Merck/Schering-Plough”) and AVEO Pharmaceuticals, Inc. (“AVEO”) and recognized $2.8 million in revenue from the five contracts. In 2009, the Company deferred $16.2 million of revenue from five contracts including Takeda, Merck/Schering-Plough, and Novartis and recognized $28.4 million of revenue from the five contracts.
The following table shows the activity in deferred revenue for the years ended December 31, 2010 and 2009 (in thousands):
|
Year ended December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Beginning deferred revenue
|
$ | 5,008 | $ | 17,213 | ||||
|
Revenue deferred
|
15,949 | 16,220 | ||||||
|
Revenue recognized
|
(2,827 | ) | (28,425 | ) | ||||
|
Ending deferred revenue
|
$ | 18,130 | $ | 5,008 | ||||
|
4.
|
Licensing, Collaborative and Other Arrangements
|
Licensing Agreements
XOMA has granted more than 50 licenses to biotechnology and pharmaceutical companies to use the Company’s patented and proprietary technologies relating to bacterial expression of recombinant pharmaceutical products. In exchange, the Company receives license and other fees as well as access to certain of these companies’ antibody display libraries, intellectual property and/or services that complement the Company’s existing development capabilities and support the Company’s own antibody product development pipeline.
Certain of these agreements also provide releases of the licensee companies and their collaborators from claims under the XOMA patents arising from past activities using the companies’ respective technologies to the extent they also used XOMA’s antibody expression technology. Licensees are generally also allowed to use XOMA’s technology in combination with their own technology in future collaborations.
Pfizer
In August of 2007, the Company entered into a license agreement with Pfizer Inc. (“Pfizer”) for non-exclusive, worldwide rights for XOMA’s patented bacterial cell expression technology for research, development and manufacturing of antibody products. Under the terms of the agreement, the Company received a license fee payment of $30 million in 2007. The Company has no further obligations under the license agreement and accordingly, the $30 million was recognized as revenue in 2007.
From 2008 through 2010 the Company received milestone payments relating to five undisclosed product candidates, including a payment of $0.5 million for the initiation of a Phase 3 clinical trial. The Company may also be eligible for additional milestone payments aggregating up to $6.4 million relating to these five product candidates and low single-digit royalties on future sales of all products subject to this license. In addition, the Company may receive potential milestone payments aggregating up to $1.7 million for each additional qualifying product candidate. The Company’s right to milestone payments expires on the later of the expiration of the last-to-expire licensed patent or the tenth anniversary of the effective date. The Company’s right to royalties expires upon the expiration of the last-to-expire licensed patent.. The Company will recognize revenue on milestones when they are achieved and on royalties when the underlying sales occur.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Collaborative and Other Agreements
Servier
In December of 2010, the Company entered into a license and collaboration agreement with Les Laboratoires Servier (“Servier”), to jointly develop and commercialize XOMA 052 in multiple indications, which provides for a non-refundable upfront payment of $15 million that was received by the Company in January of 2011. XOMA 052 is designed to inhibit the pro-inflammatory cytokine IL-1 beta that is believed to be a primary trigger of pathologic inflammation in multiple diseases. Under the terms of the agreement, Servier has worldwide rights to diabetes and cardiovascular disease indications and rights outside the U.S. and Japan to Behcet’s uveitis and other inflammatory and oncology indications. XOMA retains development and commercialization rights for Behcet's uveitis and other inflammatory and oncology indications in the U.S. and Japan, and has an option to reacquire rights to diabetes and cardiovascular disease indications from Servier in these territories (the “Cardiometabolic Indications Option”). Should the Company exercise its Cardiometabolic Indications Option, it will be required to pay Servier an option fee and partially reimburse their incurred development expenses.
The collaboration agreement provides for multiple deliverables which were grouped as follows for accounting purposes: (i) certain intellectual property rights for XOMA 052, as well as product know-how and an initial clinical inventory supply, (ii) development activities, and (iii) manufacturing services, wherein the Company believes have separate stand alone value. Further, the Company believes that each of its license agreements are unique to the targets being developed and companies involved, therefore, neither Vendor Specific Objective Evidence nor Third Party Evidence exist for determining the selling price of the deliverables. The Company determined that the contractually stated amounts are considered to be representative of the estimated selling price, as these values were determined through extensive arms length negotiations with the counterparty and further supported by other term sheets offered to the Company. The first group of deliverables includes certain intellectual property rights for XOMA 052, as well as product know-how and an initial clinical inventory supply. This group of deliverables is considered to have stand-alone value due to the customer’s ability to use the delivered group of items for the intended purpose without the receipt of the development activities. In addition, the manufacturing services are considered to be a contingent deliverable and will be accounted for as a deliverable under a separate arrangement. The $15 million upfront payment will be recognized over the estimated eight month period that the initial group of deliverables will be provided to the Servier. The Company recognized $0.1 million of the upfront payment in 2010. The remaining deliverables shall be recognized using the proportional performance method when those services are rendered, with each deliverable as a separate unit of accounting.
Under this agreement, Servier will fully fund activities to advance the global clinical development and future commercialization of XOMA 052 in diabetes and cardiovascular related diseases. Also, Servier will fund $50 million of future XOMA 052 global clinical development and chemistry and manufacturing controls (“CMC”) expenses and 50% of further expenses for the Behcet’s uveitis indication. XOMA will also be responsible for manufacturing XOMA 052 throughout clinical development and launch.
In addition, under the agreement, the Company is eligible to receive a combination of Euro and USD-denominated, development and sales milestones for multiple indications aggregating to a potential maximum of approximately $470 million converted using the December 31, 2010 Euro to US Dollar (“USD”) exchange rate (the “12/31/10 Exchange Rate”) if XOMA reacquires diabetes and cardiovascular rights in the U.S. and Japan. If XOMA does not reacquire these rights, then the milestone payments aggregate to a potential maximum of approximately $770 million converted using the 12/31/10 Exchange Rate. Milestone payments for which XOMA will be eligible under the agreement include $20 million upon initiation of the first Phase 3 clinical trial for XOMA 052 by Servier in its licensed territory in Type 2 diabetes. Servier’s obligation to pay development and commercialization milestones will continue for so long as Servier is developing or selling products under the agreement.
The Company is also eligible to receive royalties on XOMA 052 sales, which are tiered based on sales levels and range from a mid-single digit to up to a mid-teens percentage rate. The Company’s right to royalties with respect to a particular product and country will continue for so long as such product is sold in such country.
The collaboration will be carried out and managed by committees mutually established by the parties. In general, in the event of any disputes, each party will have decision-making authority over matters relating to its areas of responsibility and territory, but neither party will have unilateral decision-making rights if the decision would have a material adverse impact on the other party’s rights in its territory. The agreement contains customary termination rights relating to matters such as material breach by either party, safety issues and patents. Servier also has a unilateral right to terminate the agreement on a country-by-country basis or in its entirety on 6 months’ notice.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
In December of 2010, the Company also entered into a loan agreement with Servier, which provides for an advance of up to €15 million, or approximately $20 million when converted using the 12/31/10 Exchange Rate. This loan was fully funded in January of 2011. See Note 14: Subsequent Events for additional disclosure of the financing arrangement between the Company and Servier.
NIAID
In September of 2008, the Company announced that it had been awarded a $65 million multiple-year contract funded with federal funds from NIAID, a part of the NIH (Contract No. HHSN272200800028C), to continue development of anti-botulinum antibody product candidates. The contract work is being performed on a cost plus fixed fee basis over a three-year period. The Company is recognizing revenue under the arrangement as the services are performed on a proportional performance basis. In 2010, XOMA performed a comparison of 2009 calculated costs incurred and costs billed to the government under provisional rates. The results of the analysis indicate that the Company incurred $0.9 million of potential billable costs for work performed. This revenue has been deferred and will be recognized upon completion of an NIH audit of XOMA’s 2009 and 2008 actual data. The audit, which was initiated in 2010, will focus on the accuracy of the information the Company used in calculating its 2008 and 2009 incurred costs submissions to allow the NIH auditors a basis to develop their proposed rates. Final rates will be settled through negotiations with the Company. In 2010, the Company recognized revenue of $21.2 million under this contract, compared with $5.1 million in 2009.
In July of 2006, the Company was awarded a $16.3 million contract to produce monoclonal antibodies for the treatment of botulism to protect United States citizens against the harmful effects of botulinum neurotoxins used in bioterrorism. The contract work is being performed on a cost plus fixed fee basis. The original contract was for a three-year period, however the contract was extended into 2010. The Company is recognizing revenue as the services are performed on a proportional performance basis. This work was complete in the third quarter of 2010. In 2010, XOMA performed a comparison of 2009 calculated costs incurred and costs billed to the government under provisional rates. The results of the analysis indicate that the Company incurred $0.3 million of potential billable costs for work performed. This revenue has been deferred and will be recognized upon completion of an NIH audit of XOMA’s 2009 and 2008 actual data. The audit, which was initiated in 2010, will focus on the accuracy of the information the Company used in calculating its 2008 and 2009 incurred costs submissions to allow the NIH auditors a basis to develop their proposed rates. Final rates will be settled through negotiations with the Company. In 2010, the Company recognized revenue of $0.2 million under this contract, compared with $1.6 million in 2009 and $1.3 million in 2008.
SRI International
In the third quarter of 2009, the Company began work on two biodefense subcontract awards from SRI International, including a $2.1 million award to develop novel antibody drugs against the virus that causes SARS and a $2.2 million award to develop a novel antibody, known as F10, that has been shown to neutralize group 1 influenza A viruses, including the H1N1 and H5N1 strains. The subcontract awards are funded through NIAID. The Company will recognize revenue under these arrangements as the related research and development costs are incurred. In 2010, the Company recognized revenue of $1.6 million related to these subcontracts, compared with $0.3 million in 2009.
Takeda
In November of 2006, the Company entered into a fully funded collaboration agreement with Takeda for therapeutic monoclonal antibody discovery and development. Under the agreement, Takeda will make up-front, annual maintenance and milestone payments to the Company, fund its research and development and manufacturing activities for preclinical and early clinical studies and pay royalties on sales of products resulting from the collaboration. Takeda will be responsible for clinical trials and commercialization of drugs after an Investigational New Drug Application (“IND”) submission and is granted the right to manufacture once the product enters into Phase 2 clinical trials. During the collaboration, the Company will discover therapeutic antibodies against targets selected by Takeda. The Company will recognize revenue on the up-front and annual payments on a straight-line basis over the expected term of each target antibody discovery, on the research and development and manufacturing services as they are performed on a time and materials basis, on the milestones when they are achieved and on the royalties when the underlying sales occur. In the first quarter of 2010, the Company received a $1.0 million payment from Takeda for achieving a pre-established, preclinical milestone under one of the discovery and development programs with Takeda. The Company recognized this milestone payment in revenue in the first quarter of 2010. Separately, another discovery and development program with Takeda under this collaboration was discontinued following the analysis of research data. The termination resulted in the recognition of the remaining unamortized balance in deferred revenue of $1.1 million in the first quarter of 2010, as no continuing performance obligation exists. In 2010, the Company recognized revenue of $3.6 million under this agreement, compared with $7.5 million in 2009 and $4.4 million in 2008.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
In February of 2009, the Company expanded its existing collaboration agreement with Takeda to provide Takeda with access to multiple antibody technologies, including a suite of research and development technologies and integrated information and data management systems. The Company may receive milestones of up to $3.25 million per discovery product candidate and low single-digit royalties on future sales of all antibody products subject to this license. The Company’s right to milestone payments expires on the later of the receipt of payment from Takeda of the last amount to be paid under the agreement or the cessation of all research and development activities with respect to all program antibodies, collaboration targets and/or collaboration products. The Company’s right to royalties expires on the later of 10 years from the first commercial sale of such royalty-bearing discovery product, or the expiration of the last-to-expire licensed patent.
Novartis
In November of 2008, the Company restructured its product development collaboration with Novartis entered into in 2004 for the development and commercialization of antibody products for the treatment of cancer. Under the restructured agreement, the Company received $6.2 million in cash and $7.5 million in the form of debt reduction on its existing loan facility with Novartis. In addition, the Company may, in the future, receive milestones and double-digit royalty rates for certain product programs and options to develop or receive royalties on additional programs. In exchange, Novartis received control over certain programs under the original product development collaboration. The Company recognized revenue on the $13.7 million consideration received in November of 2008, as the Company had completed the transfer of the full rights to and materials of the collaboration targets now controlled by Novartis.
Under the original product development collaboration, the Company received initial payments of $10 million in 2004, which were being recognized ratably over five years, the expected term of the agreement, as license and collaborative fees. In February of 2007, the Company announced the parties' mutual exclusivity obligation to conduct antibody discovery, development and commercialization work in oncology had ended. The expiration of this mutual obligation had no impact on the existing collaboration projects which had reached the development stage and the parties continued to collaborate on a non-exclusive basis. The remaining unamortized balance of $4.3 million of the initial collaboration fee of $10 million was recognized in 2007 due to the change in estimate from five years to three years. The Company recognized development expenses relating to the collaboration with Novartis of $4.5 million in 2008.
A loan facility of up to $50 million was available to the Company to fund up to 75% of its share of development expenses incurred beginning in 2005. See Note 7: Long-Term Debt and Other Arrangements for additional disclosure of the financing arrangement between the Company and Novartis.
In December of 2008, the Company entered into a Manufacturing and Technology Transfer Agreement with Novartis, effective July 1, 2008. Under this agreement, XOMA was engaged by Novartis to perform research and development, process development, manufacturing and technology transfer activities with respect to certain product programs under the original product development collaboration. The work performed under this agreement was fully funded by Novartis and completed in the third quarter of 2009. The Company recognized revenue related to this agreement as the research and development and other services were performed on a time and materials basis. In 2009, the Company recognized revenue of $2.5 million related to this agreement, compared with $6.6 million in 2008.
Arana
In September of 2009, the Company entered into an antibody discovery collaboration with Arana Therapeutics Limited, a wholly-owned subsidiary of Cephalon, Inc. (“Arana”), involving multiple proprietary XOMA antibody research and development technologies, including a new antibody phage display library and a suite of integrated information and data management systems. Arana agreed to pay the Company a fee of $6.0 million, of which $4.0 million was received in the third quarter of 2009 and the remaining $2.0 million was received in the third quarter of 2010. The Company may be entitled to future milestone payments, aggregating up to $3 million per product, and low single-digit royalties on product sales. The Company’s right to milestone payments expires on the later of the receipt of payment from Arana of the last amount to be paid under the agreement, the cessation by Arana of the use of all research and development technologies or the cessation by Arana of the exercise of the patent rights granted to them. The Company’s right to royalties expires five years from the first commercial sale of each royalty-bearing product.
Kaketsuken
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
In October of 2009, the Company entered into an antibody discovery collaboration with The Chemo-Sero-Therapeutic Research Institute, a Japanese research foundation known as Kaketsuken, involving multiple proprietary XOMA antibody research and development technologies, including a new antibody phage display library and a suite of integrated information and data management systems. Kaketsuken agreed to pay the Company a fee of $8.0 million, of which $6.0 million was received in the fourth quarter of 2009 and the remaining $2.0 million was received in the fourth quarter of 2010. The Company may be entitled to future milestone payments, aggregating up to $0.2 million per product, and low single-digit royalties on product sales. The Company’s right to milestone payments expires upon the receipt of payment from Kaketsuken of the last amount to be paid pursuant to the agreement. The Company’s right to royalties expires 15 years from the first commercial sale of each royalty-bearing discovery product.
Merck/Schering-Plough/AVEO Pharmaceuticals, Inc. (“AVEO”)
In April of 2006, the Company entered into an agreement with AVEO to utilize XOMA’s HE™ technology to humanize AV-299 under which AVEO paid the Company an up-front license fee and development milestones. Under this agreement the Company created four HE™ versions of the original AV-299, all of which met design goals and from which AVEO selected one as its lead development candidate.
In September of 2006, as a result of the successful humanization of AV-299, the Company entered into a second agreement with AVEO to manufacture and supply AV-299 in support of early clinical trials. Under the agreement, the Company created AV-299 production cell lines, conducted process and assay development and performed Good Manufacturing Practices (“cGMP”) manufacturing activities. AVEO retains all development and commercialization rights to AV-299 and may be required to pay XOMA annual maintenance fees, additional development milestone payments aggregating up to $6.3 million and low single-digit royalties on product sales in the future. The Company’s right to milestone payments expires upon full satisfaction of all financial obligations of AVEO pursuant to the agreement. The Company’s right to royalties expires on the later of 15 years from the first commercial sale of each royalty-bearing product or the expiration of the last-to-expire licensed patent. In the third quarter of 2010, the Company received a $0.8 million milestone payment related to AVEO’s initiation of a Phase 2 clinical trial to evaluate AV-299 for the treatment of non-small cell lung cancer. The Company recognized this milestone payment as revenue in the third quarter of 2010.
In April of 2007, Merck/Schering-Plough entered into a research, development and license agreement with AVEO concerning AV-299 and other anti-HGF molecules, under which AVEO assigned its entire right, title and interest in, to and under its manufacturing agreement with XOMA to Merck/Schering-Plough. In the third quarter of 2010, AVEO regained its worldwide rights from Merck/Schering-Plough to develop and commercialize AV-299 and other anti-HGF molecules. In 2010, the Company recognized revenue of $0.9 million under this agreement, compared with $0.7 million in 2009 and $3.2 million in 2008.
Merck/Schering-Plough
In May of 2006, the Company entered into a fully funded collaboration agreement with the Merck/Schering-Plough for therapeutic monoclonal antibody discovery and development. Under the agreement, Merck/Schering-Plough will make up-front, annual maintenance and milestone payments to the Company, fund the Company’s research and development activities related to the agreement and pay the Company royalties on sales of products resulting from the collaboration. During the collaboration, the Company will discover therapeutic antibodies against targets selected by Merck/Schering-Plough, use the Company’s proprietary Human Engineering™ (“HE™”) technology to humanize antibody candidates generated by hybridoma techniques, perform preclinical studies to support regulatory filings, develop cell lines and production processes and produce antibodies for initial clinical trials. The Company will recognize revenue on the up-front and annual payments on a straight-line basis over the expected term of each target antibody discovery, on the research and development activities as they are performed on a time and materials basis, on the milestones when they are achieved and on the royalties when the underlying sales occur. In 2010, the Company recognized revenue of $0.5 million under this agreement, compared with $7.6 million in 2009 and $10.8 million in 2008. In January of 2011, the Company successfully completed the services to Merck/Schering-Plough and the collaboration agreement in now complete.
UCB
In December of 1998, the Company licensed its bacterial cell expression technology to Celltech Therapeutics Ltd., now UCB Celltech, a branch of UCB, which utilizes this technology in the production of CIMZIA® for the treatment of moderate-to-severe Crohn’s disease and moderate-to-severe rheumatoid arthritis. The license provides for a low single-digit royalty on sales of CIMZIA® in those countries where the bacterial cell expression technology is patented, which includes the U.S. and Canada. In August of 2010, the Company sold its royalty interest in CIMZIA® to an undisclosed buyer for gross proceeds of $4.0 million. In connection with this transaction, XOMA CDRA LLC, a wholly owned bankruptcy-remote entity, was established to hold the rights, title, and interests under the license agreement with UCB. As a bankruptcy-remote entity, XOMA CDRA LLC has a corporate existence, assets, properties, and creditors separate from the Company’s. Accordingly, in calculating the value of its own assets, the Company has not ascribed any value to the assets owned by XOMA CDRA LLC, and the assets of XOMA CDRA LLC will not be available to pay any creditors of the Company. During 2010, including the sale of its royalty interest in CIMZIA®, the Company recognized $4.2 million in revenue compared with $0.5 million in 2009 and $0.1 million in 2008. The Company will no longer receive royalties on sales of CIMZIA®.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Genentech, Inc., a wholly-owned member of the Roche Group (referred to herein as “Genentech”)
In April of 1996, the Company entered into a collaboration agreement with Genentech for the development of RAPTIVA®. In March of 2003, it entered into amended agreements which called for the Company to share in the development costs and to receive a 25% share of future U.S. operating profits and losses and a royalty on sales outside the United States. The amended agreements also called for Genentech to finance the Company’s share of development costs up until first FDA marketing approval via a convertible subordinated loan, and its share of pre-launch marketing and sales costs via an additional commercial loan facility. Under the loan agreement, upon FDA approval of the product, which occurred in October of 2003, the Company elected to pay $29.6 million of the development loan in convertible preference shares, which are convertible into approximately 0.3 million common shares at a price of $116.25 per common share. The commercial loan was repaid in cash in two installments in 2004.
In January of 2005, the Company announced a restructuring of its arrangement with Genentech on RAPTIVA®. Under the restructured arrangement, the Company was entitled to receive mid-single digit royalties on worldwide sales of RAPTIVA® in all indications. The previous cost and profit sharing arrangement for RAPTIVA® in the U.S. was discontinued and Genentech was responsible for all operating and development costs associated with the product. In addition, the Company’s remaining obligation under the development loan was extinguished. In the first half of 2009, RAPTIVA® was withdrawn from the commercial drug markets and royalties ceased.
Genentech utilized the Company’s bacterial cell expression technology under license to develop LUCENTIS® for the treatment of neovascular wet age-related macular degeneration. The Company was entitled to receive a low single-digit royalty on worldwide sales of LUCENTIS®. In the third quarter of 2009, the Company sold its LUCENTIS® royalty interest to Genentech for $25 million, including royalty revenue from the second quarter of 2009. The Company will not receive any further royalties from sales of LUCENTIS®.
The Company recognized royalty revenue related to its agreements with Genentech of $28.6 million in 2009, compared with $21.0 million in 2008.
|
5.
|
Restructuring Charges
|
On January 15, 2009, the Company announced a workforce reduction of approximately 42%. As part of this workforce reduction, the Company recorded a charge of $3.1 million related to severance, other termination benefits and outplacement services, which were fully paid in 2009. The Company does not expect to incur any additional employee-related restructuring charges in connection with this workforce reduction.
.
As a result of the workforce reduction, in the second quarter of 2009, the Company vacated one of its leased buildings and recorded a restructuring charge of $0.5 million primarily related to the net present value of the net future minimum lease payments at the cease-use date, less the estimated future sublease income. Effective December of 2010, the Company entered into a sublease agreement for this building. The remaining liability related to this lease was $0.2 million and $0.4 million at December 31, 2010 and 2009, respectively.
The following table summarizes the restructuring charges and utilization for the years ended December 31, 2010 and 2009 (in thousands):
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
|
Balance as of December 31, 2009
|
Charges
|
Cash Payments
|
Adjustments
|
Balance as of December 31, 2010
|
||||||||||||||||
|
Facilities consolidation
|
$ | 374 | $ | 39 | $ | (202 | ) | $ | 32 | $ | 243 | |||||||||
|
Total
|
$ | 374 | $ | 39 | $ | (202 | ) | $ | 32 | $ | 243 | |||||||||
|
Balance as of December 31, 2008
|
Charges
|
Cash Payments
|
Adjustments
|
Balance as of December 31, 2009
|
||||||||||||||||
|
Employee severance and benefits
|
$ | - | $ | 3,289 | $ | (3,098 | ) | $ | (191 | ) | $ | - | ||||||||
|
Facilities consolidation
|
- | 491 | (124 | ) | 7 | 374 | ||||||||||||||
|
Total
|
$ | - | $ | 3,780 | $ | (3,222 | ) | $ | (184 | ) | $ | 374 | ||||||||
Additionally, as a result of the workforce reduction, the Company temporarily vacated a building in order to optimize its facility usage. As manufacturing demand increases in the future, the Company plans to resume operations at this facility. As of December 31, 2010, the Company performed an analysis of the long-lived assets related to the vacant building, with an approximate net book value of $3.5 million. Based on estimated undiscounted future cash inflows, the Company has determined that there is no current impairment relating to these assets, and will continue to assess these assets for impairment at each future reporting period.
|
6.
|
Fair Value Measurements
|
Effective January 1, 2008, the Company adopted ASC 820, which established a framework for measuring fair value and a fair value hierarchy that prioritizes the inputs used in valuation techniques. ASC 820 describes a fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value which are the following:
Level 1 – Quoted prices in active markets for identical assets or liabilities.
Level 2 – Observable inputs other than quoted prices in active markets for similar assets or liabilities.
Level 3 – Unobservable inputs.
The following tables set forth the Company’s fair value hierarchy for its financial assets (cash equivalents and investments) and liabilities measured at fair value on a recurring basis as of December 31, 2010 and 2009.
Financial assets and liabilities carried at fair value as of December 31, 2010 and 2009 are classified as follows (in thousands):
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
|
Fair Value Measurements at December 31, 2010 Using
|
||||||||||||||||
|
Quoted Prices in Active Markets for Identical Assets
|
Significant Other Observable Inputs
|
Significant Unobservable Inputs
|
||||||||||||||
|
Total
|
(Level 1)
|
(Level 2)
|
(Level 3)
|
|||||||||||||
|
Repurchase agreements
|
$ | 1,428 | $ | 1,428 | $ | - | $ | - | ||||||||
|
Money market funds
|
6,340 | 6,340 | - | - | ||||||||||||
|
Warrant liabilities
|
(4,245 | ) | - | - | (4,245 | ) | ||||||||||
|
Total
|
$ | 3,523 | $ | 7,768 | $ | - | $ | (4,245 | ) | |||||||
|
Fair Value Measurements at December 31, 2009 Using
|
||||||||||||||||
|
Quoted Prices in Active Markets for Identical Assets
|
Significant Other Observable Inputs
|
Significant Unobservable Inputs
|
||||||||||||||
|
Total
|
(Level 1)
|
(Level 2)
|
(Level 3)
|
|||||||||||||
|
Repurchase agreements
|
$ | 6,504 | $ | 6,504 | $ | - | $ | - | ||||||||
|
Money market funds
|
14,340 | 14,340 | - | - | ||||||||||||
|
Warrant liabilities
|
(4,760 | ) | - | - | (4,760 | ) | ||||||||||
|
Total
|
$ | 16,084 | $ | 20,844 | $ | - | $ | (4,760 | ) | |||||||
Due to the unique structure of the secured note agreement with Novartis and since there is no liquid market for this note, there is no practical method to estimate fair value of our long-term debt with Novartis. See Note 7: Long-Term Debt and Other Arrangements for additional disclosure of the financing arrangement between the Company and Novartis.
As discussed in Note 2: Basis of Presentation and Significant Accounting Policies – Significant Accounting Policies, the fair value of the warrant liabilities was determined at December 31, 2010 using the Black-Scholes Model and at December 31, 2009 using the Simulation Model, both of which require inputs such as the expected term of the warrants, share price volatility and risk-free interest rate. These inputs are subjective and generally require significant analysis and judgment to develop.
The fair value of the warrant liabilities was estimated using the following range of assumptions at December 31, 2010 and 2009:
|
December 31, 2010
|
December 31, 2009
|
|||||||
|
Expected volatility
|
93.5 - 94.9 | % | 77.0 - 77.7 | % | ||||
|
Risk-free interest rate
|
2.0 | % | 2.4 - 2.7 | % | ||||
|
Expected term
|
3.9 - 4.1 years
|
4.4 - 5.0 years
|
||||||
The following table provides a summary of changes in the fair value of the Company’s Level 3 financial liabilities for the year ended December 31, 2010 (in thousands):
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
|
Warrant Liabilities
|
||||
|
Balance at December 31, 2008
|
$ | - | ||
|
Initial fair value of warrants
|
6,541 | |||
|
Change in fair value of warrant liabilities included in other income (expense)
|
(1,781 | ) | ||
|
Balance at December 31, 2009
|
4,760 | |||
|
Initial fair value of warrants
|
4,382 | |||
|
Reclassification of warrant liability to equity upon exercise of warrants
|
(2,615 | ) | ||
|
Change in fair value of warrant liabilities included in other income (expense)
|
(2,282 | ) | ||
|
Balance at December 31, 2010
|
$ | 4,245 | ||
|
7.
|
Long-Term Debt and Other Arrangements
|
As of December 31, 2010 and 2009, the Company had long-term debt of $13.7 million and $13.3 million respectively, all of which was under its note with Novartis.
Novartis Note
In May of 2005, the Company executed a secured note agreement with Novartis (then Chiron Corporation), which is due and payable in full in June of 2015. Under the note agreement, the Company borrowed semi-annually to fund up to 75% of the Company’s research and development and commercialization costs under its collaboration arrangement with Novartis, not to exceed $50 million in aggregate principal amount. Interest on the principal amount of the loan accrues at six-month LIBOR plus 2%, which was equal to 2.46% at December 31, 2010, and is payable semi-annually in June and December of each year. Additionally, the interest rate resets in June and December of each year. At the Company’s election, the semi-annual interest payments can be added to the outstanding principal amount, in lieu of a cash payment, as long as the aggregate principal amount does not exceed $50 million. The Company has made this election for all interest payments thus far. Loans under the note agreement are secured by the Company’s interest in its collaboration with Novartis, including any payments owed to it thereunder.
At December 31, 2010 and 2009, the outstanding principal balance under this note agreement was $13.7 million and $13.3 million. Pursuant to the terms of the arrangement as restructured in November of 2008, the Company will not make any additional borrowings under the Novartis note. Accrued interest of $0.4 million, $0.5 million and $1.2 million was added to the principal balance of the loan for the years ended December 31, 2010, 2009 and 2008, respectively.
Goldman Sachs Term Loan
In May of 2008, the Company refinanced its five-year term loan facility with Goldman Sachs, originally entered into in November of 2006, and borrowed the full amount of the facility of $55 million. Interest on this facility was charged at an annual rate equal to the greater of (x) six-month LIBOR or (y) 3.0%, plus 8.5% and was subject to reset on April 1 and October 1 of each year. As of December 31, 2008, the interest rate was 12.3%. The debt was secured by all rights to receive payments due to the Company relating to RAPTIVA®, LUCENTIS® and CIMZIA®. Debt issuance costs under the facility of $2 million were being amortized on a straight-line basis over the five-year life of the loan and were disclosed as current and long-term debt issuance costs on the balance sheet prior to repayment.
In addition, the Company was required to comply with certain covenants including a ratio of royalties collected to interest payable and a requirement that quarterly U.S. and outside-the-U.S. sales of RAPTIVA® and LUCENTIS® exceeded certain specified minimum levels. The Company was in compliance with these covenants as of December 31, 2008, but due to the cessation of royalties from sales of RAPTIVA® related to its market withdrawal in the first half of 2009, the Company was not in compliance with these covenants in the first quarter of 2009.
In September of 2009, the Company fully repaid its term loan facility with Goldman Sachs. Repayment of this loan facility discharged all of the Company’s obligations to the lenders. The Company repaid the outstanding principal balance of $42 million, accrued interest to the date of payment of $2.4 million and a prepayment premium of $2.5 million. In the third quarter of 2009, the Company recorded a loss on repayment of debt of $3.6 million, which included the prepayment premium and the recognition of unamortized debt issuance costs of $1.1 million. This loss was recorded as loss on debt extinguishment in the consolidated statement of operations for the year ended December 31, 2009.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Interest Expense
Interest expense and amortization of debt issuance costs, excluding losses on debt extinguishment, recorded as other expense in the consolidated statement of operations for the year ended December 31, 2010, 2009 and 2008 are shown below (in thousands):
|
Year ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Interest expense
|
||||||||||||
|
Goldman Sachs term loan
|
$ | - | $ | 3,932 | $ | 5,095 | ||||||
|
Novartis note
|
354 | 455 | 1,181 | |||||||||
|
Other
|
31 | 14 | - | |||||||||
|
Total interest expense
|
$ | 385 | $ | 4,401 | $ | 6,276 | ||||||
|
Amortization of debt issuance costs
|
||||||||||||
|
Goldman Sachs term loan
|
$ | - | $ | 487 | $ | 726 | ||||||
|
Total interest expense
|
$ | 385 | $ | 4,888 | $ | 7,002 | ||||||
|
8.
|
Income Taxes
|
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
The total provision for income taxes consists of the following:
|
Year ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Federal income tax provision
|
$ | 27 | $ | (113 | ) | $ | (384 | ) | ||||
|
State income tax provision
|
- | 6 | - | |||||||||
|
Foreign income tax provision
|
- | 5,834 | 1 | |||||||||
|
Total
|
$ | 27 | $ | 5,727 | $ | (383 | ) | |||||
Income tax expense was $27,000 in 2010. Income tax expense in 2009 was primarily related to $5.8 million of foreign income tax expense recognized in connection with the expansion of the Company’s existing collaboration with Takeda in February of 2009. The Company was paid a $29 million expansion fee, of which $5.8 million was withheld for payment to the Japanese taxing authority. The Company also recognized $0.1 million of income tax benefit for 2009 relating to research and development refundable credits, in addition to the $0.4 million in research and development refundable credits recognized in 2008.
The significant components of net deferred tax assets as of December 31, 2010 and 2009 were as follows (in millions):
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Capitalized research and development expenses
|
$ | 65.4 | $ | 65.7 | ||||
|
Net operating loss carryforwards
|
117.4 | 93.3 | ||||||
|
Research and development and other credit carryforwards
|
20.4 | 20.0 | ||||||
|
Other
|
11.1 | 10.9 | ||||||
|
Total deferred tax assets
|
214.3 | 189.9 | ||||||
|
Valuation allowance
|
(214.3 | ) | (189.9 | ) | ||||
|
Net deferred tax assets
|
$ | - | $ | - | ||||
The net increase (decrease) in the valuation allowance was $24.4 million, $(24.8) million and $9.1 million for the years ended December 31, 2010, 2009 and 2008, respectively. Approximately $13.1 million in unutilized federal net operating loss carry-forwards (“NOLs”) expired in 2008, no net operating loss carry-forward expired in 2010 or 2009.
ASC 740 provides for the recognition of deferred tax assets if realization of such assets is more likely than not. Based upon the weight of available evidence, which includes the Company’s historical operating performance and carry-back potential, the Company has determined that total deferred tax assets should be fully offset by a valuation allowance.
As of December 31, 2010, the Company had accumulated federal tax net operating loss carry-forwards of $149.4 million, with expiration dates from 2018 to 2030, federal tax credit carry-forwards of $9.5 million, with expiration dates from 2010 to 2030, state tax net operating loss carry-forwards of $287.4 million, with expiration dates from 2014 to 2030, state tax credit carry-forwards of $16.0 million, without expiration, and foreign tax net operating loss carry-forwards of $399.4 million, without expiration.
In 2009, the Company experienced an “ownership change” under Section 382 of the Internal Revenue Code, which subjects the amount of federal and state tax carry-forwards that can be utilized to an annual limitation, which will substantially limit the Company’s future use of these carry-forwards per year. To the extent that the Company does not utilize its carry-forwards within the applicable statutory carry-forward periods, either because of Section 382 limitations or the lack of sufficient taxable income, the carry-forwards will expire unused.
The Company files income tax returns in the U.S. federal jurisdiction, State of California and Ireland. The Company’s federal income tax returns for tax years 2007 and beyond remain subject to examination by the Internal Revenue Service. The Company’s California and Irish income tax returns of the tax years 2006 and beyond remain subject to examination by the Franchise Tax Board and Irish Revenue Commissioner. In addition, all of the net operating losses and research and development credit carry-forwards that may be used in future years are still subject to adjustment.
The Company did not have unrecognized tax benefits as of December 31, 2010 and does not expect this to change significantly over the next twelve months. The Company will recognize interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense. As of December 31, 2010, the Company has not accrued interest or penalties related to uncertain tax positions.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
|
9.
|
Compensation and Other Benefit Plans
|
The Company grants qualified and non-qualified share options, shares and other share-related awards under various plans to directors, officers, employees and other individuals. Share options are granted at exercise prices of not less than the fair market value of the Company’s common shares on the date of grant. Generally, share options granted to employees fully vest four years from the grant date and expire ten years from the date of the grant or three months from the date of termination of employment (longer in case of death or certain retirements). However, certain options granted to employees vest monthly or immediately, certain options granted to directors vest monthly over one year or three years and certain options may fully vest upon a change of control of the Company or may accelerate based on performance-driven measures. Additionally, the Company has an Employee Share Purchase Plan (“ESPP”) that allows employees to purchase Company shares at a purchase price equal to 95% of the closing price on the exercise date.
Employee Share Purchase Plan
In 1998, the Company’s shareholders approved the 1998 Employee Share Purchase Plan which provides employees of the Company the opportunity to purchase common shares through payroll deductions. Up to 133,333 common shares are authorized for issuance under the Share Purchase Plan. An employee may elect to have payroll deductions made under the Share Purchase Plan for the purchase of common shares in an amount not to exceed 15% of the employee’s compensation.
Effective January 1, 2005, the plan was amended to reduce future offering periods to three months and to change the purchase price per common share to 95% of the closing price of XOMA shares on the exercise date.
In 2010, 2009, and 2008, employees purchased 5,903, 14,735 and 13,027 common shares, respectively, under the Employee Share Purchase Plan. Net payroll deductions under the Share Purchase Plan totaled $41,000, $0.1 million and $0.3 million for 2010, 2009 and 2008, respectively.
Deferred Savings Plan
Under section 401(k) of the Internal Revenue Code of 1986, the Board of Directors adopted, effective June 1, 1987, a tax-qualified deferred compensation plan for employees of the Company. Participants may make contributions which defer up to 50% of their eligible compensation per payroll period, up to a maximum for 2010 of $16,500 (or $22,000 for employees over 50 years of age). The Company may, at its sole discretion, make contributions each plan year, in cash or in the Company’s common shares, in amounts which match up to 50% of the salary deferred by the participants. The expense related to these contributions was $1.0 million, $0.9 million and $1.1 million for the years ended December 31, 2010, 2009 and 2008, respectively, and 100% was paid in common shares in each year.
Share Options
At December 31, 2010, the Company had share-based compensation plans, as described below. The aggregate number of common shares that may be issued under these plans is 3,254,331 shares.
In February of 2009, the Board of Directors approved a company-wide grant of 315,333 share options, of which 304,533 were issued as part of the Company’s annual incentive compensation package. These options vest monthly over four years and include an acceleration clause based on meeting certain performance measures. In 2009 and 2010, management estimated the timing and probability of the achievement of the acceleration event, which occurred in December of 2010. The Company recognized compensation expense of $0.2 million and $0.1 million for the years ended December 31, 2010 and 2009, respectively, in connection with the accelerated awards.
In March of 2010, the Board of Directors of the Company approved a company-wide grant of an aggregate of 865,806 share options. This grant included 856,006 options that were issued as part of the Company’s annual incentive compensation review, of which 596,666 options were granted subject to shareholder approval of an increase in the number of shares available under the Company’s existing share option plans. On July 21, 2010 shareholder approval was obtained at the Company’s annual general meeting of shareholders. A cumulative adjustment of $0.7 million was recorded in the third quarter of 2010 to reflect share-based compensation expense that would have been recorded from grant date to July 20, 2010. The adjustment was based on the fair value of these options at the date of shareholder approval and calculated using the closing share price on that date. The options granted as part of this annual incentive compensation review will vest monthly over four years.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Share Option Plan
Under the Company’s amended 1981 Share Option Plan (“Option Plan”) the Company grants qualified and non-qualified share options to employees and other individuals, as determined by the Board of Directors, at exercise prices of not less than the fair market value of the Company’s common shares on the date of grant. Options granted under the Option Plan may be exercised when vested and generally expire ten years from the date of the grant or three months from the date of termination of employment (longer in case of death or certain retirements). Options granted generally vest over four years. However, certain options may vest monthly or immediately, and certain options fully vest upon a change of control of the Company or may accelerate based on performance-driven measures. The Option Plan will terminate on November 15, 2011.
Up to 2,303,333 shares are authorized for issuance under the Option Plan. As of December 31, 2010, options covering 2,029,352 common shares were outstanding under the Option Plan.
Restricted Share Plan
The Company also has a Restricted Share Plan (“Restricted Plan”) which provides for the issuance of options or grants of common shares to certain employees and other individuals as determined by the Board of Directors at fair market value of the common shares on the grant date. Prior to 2005, options or shares could be granted at not less than 85% of fair market value of the common shares on the grant date. Each option issued under the Restricted Plan will be a non-statutory share option under federal tax laws and will have a term not in excess of ten years from the grant date or three months from the date of termination of employment (longer in the case of death or certain retirements). Options granted generally vest over four years and certain options may fully vest upon a change of control of the Company. The Restricted Plan will terminate on November 15, 2011.
Up to 183,333 shares are authorized for issuance under the Restricted Plan, subject to the condition that not more than 2,486,666 shares are authorized under both the Option Plan and the Restricted Plan. As of December 31, 2010, options covering 83,467 common shares were outstanding under the Restricted Plan.
Directors Share Option Plan and Other Options
In 1992, the shareholders approved a Directors Share Option Plan (“Directors Plan”) which provides for the issuance of options to purchase common shares to non-employee directors of the Company at 100% of the fair market value of the shares on the date of the grant. Up to 106,666 shares are authorized for issuance during the term of the Directors Plan. Options generally vest on the date of grant, or monthly over one year or three years and have a term of up to ten years. As of December 31, 2010, options for 62,867 common shares were outstanding under the Directors Plan.
In addition, in July of 2002, the Company granted a non-qualified fully-vested option to a director to purchase 1,000 common shares at 100% of the fair market value of the shares on the date of grant, which will expire in ten years. This option was not issued as part of the Directors Plan.
In August of 2007, the Company granted a non-qualified option to Steven B. Engle, CEO, to purchase 73,333 common shares at 100% of the fair market value of the shares on the date of grant. The option is subject to the Company’s typical four-year vesting schedule and will expire 10 years from the date of issuance. This option was not issued as part of the Company’s Option Plan or the Restricted Plan.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Share Option Plans Summary
A summary of the status of the Company’s share option plans as of December 31, 2010, 2009 and 2008, and changes during the years ended on those dates is presented below:
|
|
2010
|
2009
|
2008
|
|||||||||||||||||||||
|
Options:
|
Shares
|
Price*
|
Shares
|
Price*
|
Shares
|
Price*
|
||||||||||||||||||
|
Outstanding at beginning of year
|
1,520,102 | $ | 38.40 | 1,320,679 | $ | 48.60 | 740,541 | $ | 54.90 | |||||||||||||||
|
Granted
|
||||||||||||||||||||||||
| (1) | 978,264 | 7.07 | 432,400 | 9.00 | 185,650 | 23.85 | ||||||||||||||||||
| (2) | - | - | - | - | 579,400 | 49.24 | ||||||||||||||||||
|
Exercised
|
(19 | ) | 8.40 | (2,056 | ) | 8.40 | (5,716 | ) | 23.07 | |||||||||||||||
|
Forfeited, expired or cancelled (3)
|
(166,897 | ) | 37.26 | (230,921 | ) | 41.40 | (179,196 | ) | 51.95 | |||||||||||||||
|
Outstanding at end of year
|
2,331,450 | 25.36 | 1,520,102 | 38.40 | 1,320,679 | 48.60 | ||||||||||||||||||
|
Exercisable at end of year
|
1,259,272 | 36.51 | 823,096 | 49.05 | 571,720 | 59.10 | ||||||||||||||||||
|
Weighted average fair value of options granted
|
||||||||||||||||||||||||
| (1) | $ | 3.58 | $ | 5.85 | $ | 14.10 | ||||||||||||||||||
| (2) | - | $ | - | $ | 14.91 | |||||||||||||||||||
* Weighted-average exercise price:
(1) Option price equal to market price on date of grant.
(2) Option price greater than market price on date of grant
(3) The Company adjusts for forfeitures as they occur.
At December 31, 2010, there were 2,240,791 options vested and expected to vest with a weighted-average exercise price of $26.07. The weighted average remaining contractual term of outstanding share options at December 31, 2010 was 7.7 years and the aggregate intrinsic value was $0.1 million. The weighted average remaining contractual term of exercisable share options at December 31, 2010 was 6.8 years and the aggregate intrinsic value was $4,000.
Share-Based Compensation Expense
The Company recognizes compensation expense for all share-based payment awards made to the Company’s employees and directors based on estimated fair values. The valuation of share-based compensation awards is determined at the date of grant using the Black-Scholes option pricing model. This model requires inputs such as the expected term of the option, expected volatility and risk-free interest rate. Further, the forfeiture rate also impacts the amount of aggregate compensation. To establish an estimate of expected term, the Company considers the vesting period and contractual period of the award and its historical experience of share option exercises, post-vesting cancellations and volatility. The estimate of expected volatility is based on the Company’s historical volatility. To establish an estimate of forfeiture rate, the Company considers its historical experience of option forfeitures and terminations. The risk-free rate is based on the yield available on United States Treasury zero-coupon issues.
The following table shows total share-based compensation expense included in the consolidated statements of operations for the years ended December 31, 2010, 2009 and 2008 (in thousands):
|
Year Ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Research and development
|
$ | 2,302 | $ | 2,182 | $ | 2,307 | ||||||
|
Selling, general and administrative
|
2,611 | 2,213 | 2,627 | |||||||||
|
Total share-based compensation expense
|
$ | 4,913 | $ | 4,395 | $ | 4,934 | ||||||
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
There was no capitalized share-based compensation cost as of December 31, 2010 or 2009, and there were no recognized tax benefits related to the Company’s share-based compensation expense during the years ended December 31, 2010 or 2009.
The fair value of share-based awards was estimated using the Black-Scholes model with the following weighted average assumptions for the years ended December 31, 2010, 2009 and 2008:
|
Year Ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Dividend yield
|
0 | % | 0 | % | 0 | % | ||||||
|
Expected volatility
|
79 | % | 75 | % | 65 | % | ||||||
|
Risk-free interest rate
|
1.67 | % | 2.00 | % | 2.84 | % | ||||||
|
Expected term
|
5.3 years
|
5.6 years
|
5.4 years
|
|||||||||
Unvested share option activity for the year ended December 31, 2010 is summarized below:
|
Unvested Number
of Shares
|
Weighted- Average Grant- Date Fair Value
|
|||||||
|
Unvested balance at December 31, 2009
|
696,786 | $ | 20.85 | |||||
|
Granted
|
978,264 | 3.58 | ||||||
|
Vested
|
(574,699 | ) | 8.86 | |||||
|
Forfeited
|
(28,173 | ) | 14.62 | |||||
|
Unvested balance at December 31, 2010
|
1,072,178 | 5.69 | ||||||
At December 31, 2010, there was $4.7 million of unrecognized share-based compensation expense related to unvested share options with a weighted average remaining recognition period of 2.5 years. The estimated fair value of options vested during 2010, 2009 and 2008 was $4.9 million, $4.1 million and $3.6 million, respectively. Total intrinsic value of the options exercised was $6,000 in 2009 and $50,000 million in 2008. Total intrinsic value and total cash received from share option exercises in 2010 was not material.
|
10.
|
Share Capital
|
Reverse Stock Split
All references to numbers of common shares and per-share information in the accompanying financial statements have been adjusted retroactively to reflect the Company’s reverse stock split on August 18, 2010.
Series B Preference Shares
In December of 2003, the Company issued 2,959 of the Series B preference shares to Genentech in repayment of $29.6 million of the outstanding balance under a convertible subordinated debt agreement. Pursuant to the rights of the Series B preference shares, the holders of Series B preference shares will not be entitled to receive any dividends on the Series B preference shares. The Series B preference shares will rank senior with respect to rights on liquidation, winding-up and dissolution of the Company to all classes of common shares. Upon any voluntary or involuntary liquidation, dissolution or winding-up of the Company, holders of Series B preference shares will be entitled to receive $10,000 per share of Series B preference shares before any distribution is made on the common shares. The holder of the Series B preference shares has no voting rights, except as required under Bermuda law.
The holder of Series B preference shares has the right to convert Series B preference shares into common shares at a conversion price equal to $116.25 per common share, subject to adjustment in certain circumstances. Accordingly, the 2,959 issued Series B preference shares are convertible into 254,560 common shares.
The Series B preference shares will be automatically converted into common shares at their then effective conversion rate immediately upon the transfer by the initial holder to any third party which is not an affiliate of such holder. The Company will have the right, at any time and from time to time, to redeem any or all Series B preference shares for cash in an amount equal to the conversion price multiplied by the number of common shares into which each such share of Series B preference shares would then be convertible.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Shareholder Rights Plan
On February 26, 2003, the Company’s Board of Directors unanimously adopted a Shareholder Rights Plan (“Rights Plan”), which was designed to extend the provisions of a similar rights plan originally adopted in October of 1993. Under the Rights Plan, Preference Share Purchase Rights (“Rights”) are authorized and granted at the rate of fifteen Rights for each outstanding common share. Each Right entitles the registered holder of common shares to buy a fraction of a share of the new series of Preference Shares (“Series A Preference Shares”) at an exercise price of $30.00, subject to adjustment. The Rights will be exercisable and will detach from the common share, only if a person or group acquires 20 percent or more of the common shares, announces a tender or exchange offer that if consummated will result in a person or group beneficially owning 20 percent or more of the common shares or if the Board of Directors declares a person or group owning 10 percent or more of the outstanding common shares to be an Adverse Person (as defined in the Rights Plan). Once exercisable, each Right will entitle the holder (other than the acquiring person) to purchase units of Series A Preference Share (or, in certain circumstances, common shares of the acquiring person) with a value of twice the Rights’ exercise price. The Company will generally be entitled to redeem the Rights at $0.015 per Right at any time until the close of business on the tenth day after the Rights become exercisable. The Rights will expire at the close of business on February 26, 2013.
Underwritten Offering
In February of 2010, the Company completed an underwritten offering of 2.8 million units, with each unit consisting of one of the Company’s common shares and a warrant to purchase 0.45 of a common share, for gross proceeds of approximately $21 million. As of December 31, 2010 all of these warrants were outstanding.
Registered Direct Offerings
In May of 2009, the Company entered into a definitive agreement with an institutional investor to sell 784,313 units, with each unit consisting of one of the Company’s common shares and a warrant to purchase 0.50 of a common share, for gross proceeds of approximately $10 million, before deducting placement agent fees and estimated offering expenses of $0.8 million, in a registered direct offering. The investor purchased the units at a price of $12.75 per unit. The warrants, which represent the right to acquire an aggregate of up to 392,157 common shares, were exercisable at any time on or after May 15, 2009 and prior to May 20, 2014 at an exercise price of $15.30 per share. In February of 2010, the holders of these warrants agreed to amend the terms of their warrants to remove the Eliminated Adjustment Provisions and the exercise price of these warrants was reduced from $15.30 per share to $0.015 per share. In the first quarter of 2010, the holders of these warrants exercised all warrants, acquiring 392,157 common shares for an aggregate exercise price of $5,882.
In June of 2009, the Company entered into a definitive agreement with certain institutional investors to sell 695,652 units, with each unit consisting of one of the Company’s common shares and a warrant to purchase 0.50 of a common share, for gross proceeds of approximately $12 million, before deducting placement agent fees and estimated offering expenses of $0.8 million, in a second registered direct offering. The investor purchased the units at a price of $17.25 per unit. The warrants, which represent the right to acquire an aggregate of up to 347,826 common shares, are exercisable at any time on or prior to December 10, 2014 at an exercise price of $19.50 per share. As of December 31, 2010 all of these warrants were outstanding.
ATM Agreement
In the third quarter of 2009, the Company entered into an At Market Issuance Sales Agreement (the “2009 ATM Agreement”), under which the Company could sell up to 1.7 million of its common shares from time to time through Wm Smith, as the agent for the offer and sale of the common shares. Wm Smith could sell these common shares by any method permitted by law deemed to be an “at the market” offering as defined in Rule 415 of the Securities Act of 1933, including but not limited to sales made directly on The NASDAQ Global Market, on any other existing trading market for the common shares or to or through a market maker. Wm Smith could also sell the common shares in privately negotiated transactions, subject to the Company’s approval. The Company paid Wm Smith a commission equal to 3% of the gross proceeds of all common shares sold through it as sales agent under the 2009 ATM Agreement but in no event less than $0.02 per share. Shares sold under the 2009 ATM Agreement were sold pursuant to a prospectus which formed a part of a registration statement declared effective by the U.S. Securities and Exchange Commission (the “SEC”) on May 29, 2008. From the inception of the 2009 ATM Agreement through October of 2010, the Company sold a total of 1.7 million common shares through Wm Smith for aggregate gross proceeds of $12.2 million, including 1.4 million common shares sold in 2010 for aggregate gross proceeds of $9.3 million. Total offering expenses related to these sales from inception to October of 2010 were $0.4 million.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
In the third quarter of 2010, the Company entered into an At Market Issuance Sales Agreement (the “2010 ATM Agreement”), with Wm Smith and McNicoll, Lewis & Vlak LLC (the “Agents”) under which the Company may sell common shares from time to time through the Agents, as its agents for the offer and sale of the common shares, in an aggregate amount not to exceed the amount that can be sold under its registration statement on Form S-3 (File No. 333-148342) filed with the SEC on December 26, 2007. The Agents may sell the common shares by any method permitted by law deemed to be an “at the market” offering as defined in Rule 415 of the Securities Act, including without limitation sales made directly on The NASDAQ Global Market, on any other existing trading market for the common shares or to or through a market maker. The Agents may also sell the common shares in privately negotiated transactions, subject to the Company’s prior approval. The Company will pay the Agents, collectively, a commission equal to 3% of the gross proceeds of the sales price of all common shares sold through them as sales agents under the 2010 ATM Agreement. From the inception of the ATM Agreement through December 31, 2010, the Company sold a total of 6.7 million common shares under this agreement for aggregate gross proceeds of $29.7 million. Total offering expenses related to these sales from inception to December 31, 2010 were $0.9 million. Subsequent to December 31, 2010 through March 8, 2011, the Company has sold an additional 796,898 common shares through the Agents pursuant to the 2010 ATM Agreement for aggregate gross proceeds of $4.3 million. Total offering expenses related to these sales were $0.1 million.
Equity Line of Credit
On October 21, 2008, the Company entered into a common share purchase agreement (the “Purchase Agreement”) with Azimuth Opportunity Ltd. (“Azimuth”), pursuant to which it obtained a committed equity line of credit facility (the “Facility”) under which the Company could sell up to $60 million of its registered common shares to Azimuth over a 24-month period, subject to certain conditions and limitations. The Purchase Agreement required a minimum share price of $1.00 per share to allow the Company to issue shares to Azimuth under the Facility. However, at its election, Azimuth could buy shares below the threshold price at a negotiated discount. The Company was not obligated to utilize any of the $60 million Facility and remained free to enter other financing transactions. Shares under the Facility were sold pursuant to a prospectus which forms a part of a registration statement declared effective by the Securities and Exchange Commission on May 29, 2008. At the end of the third quarter of 2009, the Facility was no longer in effect, and no additional shares could be issued thereunder.
From the inception of the Facility in October of 2008 through December 31, 2009, the Company sold a total of 2,815,228 common shares to Azimuth for aggregate gross proceeds of $33.9 million. This included the sale of 0.3 million shares under the Facility in December of 2008 and 2.3 million shares in two transactions in September of 2009 that Azimuth agreed to purchase notwithstanding that the purchase prices were below the minimum price of $1.00 required by the Purchase Agreement. Under the terms of the Purchase Agreement, the Company negotiated a discount rate (excluding placement agent fees) of 8.86% for the sale in December of 2008 and 8.0% for the sales in September of 2009. Prior to the successful conclusion of negotiations, Azimuth was not obligated to purchase these shares. Offering expenses incurred from inception of the Facility through December 31, 2009 related to sales to Azimuth were $0.7 million.
In July of 2010, the Company entered into a common share purchase agreement (the “Purchase Agreement”) with Azimuth Opportunity Ltd. (“Azimuth”), pursuant to which the Company obtained a committed equity line of credit facility (the “Facility”) under which the Company could sell up to $30 million of its registered common shares to Azimuth over a 12-month period, subject to certain conditions and limitations. The Purchase Agreement provided that the Company could determine, in its sole discretion, the timing, dollar amount and floor price per share of each draw down under the Facility, subject to certain conditions and limitations and that the number and price of shares sold in each draw down were generally to be determined by a contractual formula designed to approximate fair market value, less a discount. The Purchase Agreement also provided that from time to time and in the Company’s sole discretion, it could grant Azimuth the right to exercise one or more options to purchase additional common shares during each draw down pricing period for the amount of shares based upon the maximum option dollar amount and the option threshold price specified by the Company. The Company also agreed to issue 111,111 common shares to Azimuth upon execution of the agreement relating to the Facility, in consideration of Azimuth’s execution and delivery of that agreement. Shares under the Facility and the shares the Company agreed to issue to Azimuth upon execution of the agreement relating to the Facility were sold pursuant to a prospectus which forms a part of a registration statement declared effective by the SEC on May 29, 2008. In August of 2010, the Company sold a total of 3,421,407 common shares under the Facility for aggregate gross proceeds of $14.2 million, representing the maximum number of shares that could be sold under the Facility. As a result, the Facility is no longer in effect, and no additional shares can be issued thereunder.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
|
11.
|
Commitments and Contingencies
|
Collaborative Agreements, Royalties and Milestone Payments
The Company is obligated to pay royalties, ranging generally from 1.5% to 14% of the selling price of the licensed component and up to 40% of any sublicense fees to various universities and other research institutions based on future sales or licensing of products that incorporate certain products and technologies developed by those institutions.
In addition, the Company has committed to make potential future “milestone” payments to third parties as part of licensing and development programs. Payments under these agreements become due and payable only upon the achievement of certain developmental, regulatory and/or commercial milestones. Because it is uncertain if and when these milestones will be achieved, such contingencies, aggregating up to $97 million (assuming one product per contract meets all milestones events) have not been recorded on the consolidated balance sheet. The Company is unable to determine precisely when and if payment obligations under the agreements will become due as these obligations are based on milestone events, the achievement of which is subject to a significant number of risks and uncertainties.
Leases
As of December 31, 2010, the Company leased administrative, research facilities, and office equipment under operating leases expiring on various dates through May of 2014. These leases generally require the Company to pay taxes, insurance, maintenance and minimum lease payments.
The Company estimates future minimum lease commitments to be (in thousands):
|
Operating Leases
|
||||
|
2011
|
5,219 | |||
|
2012
|
4,883 | |||
|
2013
|
2,865 | |||
|
2014
|
785 | |||
|
Thereafter
|
- | |||
|
Minimum lease payments
|
$ | 13,752 | ||
Total rental expense, including other costs required under the Company’s leases, was approximately $5.1 million, $5.2 million and $5.2 million for the years ended December 31, 2010, 2009 and 2008, respectively. Rental expense based on leases allowing for escalated rent payments are recognized on a straight-line basis. The Company is required to restore certain of its leased property to certain conditions in place at the time of lease. The Company believes these costs will not be material to its operations.
As a result of the restructuring in the second quarter of 2009, the Company vacated one of its leased buildings. Effective December of 2010, the Company entered into a sublease agreement for this building through May of 2014. The Company estimates future sublease income to be $0.4 million under this agreement.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
Legal Proceedings
On April 9, 2009, a complaint was filed in the Superior Court of Alameda County, California, in a lawsuit captioned Hedrick et al. v. Genentech, Inc. et al, Case No. 09-446158. The complaint asserts claims against Genentech, the Company and others for alleged strict liability for failure to warn, strict product liability, negligence, breach of warranty, fraudulent concealment, wrongful death and other claims based on injuries alleged to have occurred as a result of three individuals’ treatment with RAPTIVA®. The complaint seeks unspecified compensatory and punitive damages. Since then, additional complaints have been filed in the same court, bringing the total number of pending cases to fifty eight. All of the complaints allege the same claims and seek the same types of damages based on injuries alleged to have occurred as a result of the plaintiffs' treatment with RAPTIVA®. The Company’s agreement with Genentech provides for an indemnity of XOMA and payment of legal fees by Genentech which the Company believes is applicable to this matter. The Company believes the claims against it to be without merit and intends to defend against them vigorously.
On August 4, 2010, a Petition was filed in the District Court of Dallas County, Texas in a case captioned McCall v. Genentech, Inc., et al., No. 10-09544. The defendants filed a Notice of Removal to the United States District Court for the Northern District of Texas on September 3, 2010. The removed case is captioned McCall v. Genentech, Inc., et al., No. 3:10-cv-01747-B. The parties have fully briefed the Plaintiff’s Motion to Remand and are awaiting a final ruling from the Court. The Petition asserts personal injury claims against Genentech, the Company, and others arising out of the plaintiff’s treatment with RAPTIVA®. The Petition alleges claims based on negligence, strict liability, misrepresentation and suppression, conspiracy, and actual and constructive fraud. The Petition seeks compensatory damages and punitive damages in an unspecified amount. The Company’s agreement with Genentech provides for an indemnity of XOMA and payment of legal fees by Genentech which the Company believes is applicable to this matter. The Company believes the claims against it to be without merit and intends to defend against them vigorously.
On January 7, 2011, a Complaint was filed in the United States District Court for the Northern District of Texas in a case captioned Massa v. Genentech, Inc., et al., No. 4:11CV70. The Complaint alleges the same claims and seeks the same types of damages as the Complaints filed in the Superior Court of Alameda County, referenced above. The Complaint asserts personal injury claims against Genentech and the Company arising out of the plaintiff’s treatment with RAPTIVA®. The Company’s agreement with Genentech provides for an indemnity of XOMA and payment of legal fees by Genentech which the Company believes is applicable to this matter. The Company believes the claims against it to be without merit and intends to defend against them vigorously.
|
12.
|
Concentration of Risk, Segment and Geographic Information
|
Concentration of Risk
Cash equivalents and receivables are financial instruments, which potentially subject the Company to concentrations of credit risk, as well as liquidity risk for certain cash equivalents such as money market funds. The Company has not encountered such issues during 2010.
The Company has not experienced any significant credit losses and does not generally require collateral on receivables. In 2010, three customers represented 64%, 13%, and 11% of total revenue and as of December 31, 2010, there were billed receivables of $19.7 million outstanding from two customers representing 72% and 23% of the accounts receivable balance.
In 2009, two customers represented 36% and 29% of total revenue and as of December 31, 2009, there were receivables of $5.7 million outstanding from three customers representing 90% of the accounts receivable balance. In 2008, three customers represented 31%, 30% and 20% of total revenue.
Segment Information
The Company has determined that it operates in one segment as it only reports operating results on an aggregate basis to the chief operating decision maker of the Company. The Company’s property and equipment is held primarily in the United States.
Geographic Information
Revenue attributed to the following countries for each of the three years ended December 31, 2010, 2009 and 2008 was as follows (in thousands):
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
|
Year ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
United States
|
$ | 25,306 | $ | 47,656 | $ | 62,262 | ||||||
|
Europe
|
4,728 | 613 | 1,351 | |||||||||
|
Asia Pacific
|
3,607 | 50,161 | 4,374 | |||||||||
|
Total
|
$ | 33,641 | $ | 98,430 | $ | 67,987 | ||||||
|
13.
|
Subsequent Events
|
Servier – Cash Receipts and Loan Agreement
In January of 2011, the Company received a non-refundable upfront cash payment of $15 million in connection with the license and collaboration agreement entered into with Servier in December of 2010. In addition, the Company also received the full €15 million, or approximately $20 million when converted using the 12/31/10 Exchange Rate, loan in connection with the loan agreement entered into with Servier in December of 2010.
The loan is secured by an interest in XOMA’s intellectual property rights to all XOMA 052 indications worldwide, excluding certain rights in the U.S. and Japan territories. Interest is calculated at a floating rate based on a Euro Inter-Bank Offered Rate (“EURIBOR”) and subject to a cap. The interest rate is reset semi-annually in January and July of each year. The interest rate for the initial interest period has been set at 3.22%. Interest is payable semi-annually; however, the loan agreement provides for a deferral of interest payments over a period determined by the parties. During the deferral period, accrued interest will be added to the outstanding principal amount for the purpose of interest calculation for the next six month interest period. On the repayment commencement date, all unpaid and accrued interest shall be paid to Servier, and thereafter, all accrued and unpaid interest shall be due and payable at the end of each six-month period. The loan has a final maturity date in 2016; however, after a specified period prior to final maturity, the loan (i) may be repaid at Servier's option, by applying up to a significant percentage of any milestone or royalty payments owed by Servier under the Company’s collaboration agreement and (ii) will be repaid by using a significant percentage of any upfront, milestone or royalty payments the Company receives from any third party collaboration or development partner for rights to XOMA 052 in the U.S. and/or Japan. In addition, the loan becomes immediately due and payable upon certain customary events of default.
2011 ATM Agreement
On February 4, 2011, the Company entered into an At Market Issuance Sales Agreement (the “2011 ATM Agreement”), with McNicoll, Lewis & Vlak LLC (“MLV”), under which the Company may sell common shares from time to time through MLV, as its agent for the offer and sale of the common shares, in an aggregate amount not to exceed the amount that can be sold under the Company’s registration statement on Form S-3 (File No. 333-172197) filed with the SEC on February 11, 2011, once such registration statement has been declared effective by the SEC. MLV may sell the common shares by any method permitted by law deemed to be an “at the market” offering as defined in Rule 415 of the Securities Act, including without limitation sales made directly on The NASDAQ Global Market, on any other existing trading market for the common shares or to or through a market maker. MLV may also sell the common shares in privately negotiated transactions, subject to the Company’s prior approval. The Company will pay MLV a commission equal to 3% of the gross proceeds of the sales price of all common shares sold through them as sales agent under the 2011 ATM Agreement. As of March 8, 2011, the Company has not sold any common shares under the 2011 ATM Agreement.
XOMA Ltd.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS - (Continued)
|
14.
|
Quarterly Financial Information (unaudited)
|
The following is a summary of the quarterly results of operations for the years ended December 31, 2010 and 2009:
|
Consolidated Statements of Operations
|
||||||||||||||||
|
Quarter Ended
|
||||||||||||||||
|
March 31
|
June 30
|
September 30
|
December 31
|
|||||||||||||
|
(In thousands, except per share amounts)
|
||||||||||||||||
|
2010
|
||||||||||||||||
|
Total revenues (1)
|
$ | 7,202 | $ | 5,942 | $ | 10,897 | $ | 9,601 | ||||||||
|
Total operating costs and expenses (2)
|
23,140 | 24,372 | 27,542 | 25,691 | ||||||||||||
|
Other income (expense), net
|
(5,847 | ) | 2,866 | 3,013 | (1,657 | ) | ||||||||||
|
Net loss
|
(21,785 | ) | (15,580 | ) | (13,633 | ) | (17,758 | ) | ||||||||
|
Basic net loss per common share
|
$ | (1.36 | ) | $ | (0.93 | ) | $ | (0.69 | ) | $ | (0.84 | ) | ||||
|
Diluted net loss per common share
|
$ | (1.36 | ) | $ | (0.93 | ) | $ | (0.69 | ) | $ | (0.84 | ) | ||||
|
2009
|
||||||||||||||||
|
Total revenues (1)
|
$ | 39,704 | $ | 9,706 | $ | 27,423 | $ | 21,597 | ||||||||
|
Total operating costs and expenses (2)
|
25,930 | 19,474 | 20,643 | 19,423 | ||||||||||||
|
Other expense, net (3)
|
(1,735 | ) | (529 | ) | (4,872 | ) | 453 | |||||||||
|
Net income (loss)
|
6,239 | (10,210 | ) | 1,538 | 2,983 | |||||||||||
|
Basic net income (loss) per common share
|
$ | 0.66 | $ | (1.02 | ) | $ | 0.14 | $ | 0.22 | |||||||
|
Diluted net income (loss) per common share
|
$ | 0.64 | $ | (1.02 | ) | $ | 0.13 | $ | 0.22 | |||||||
|
(1)
|
Revenue in the third quarter of 2010 includes a non-recurring fee of $4.0 million related to the sale of the Company’s CIMZIA® royalty interest to an undisclosed buyer. Revenue in the first quarter of 2009 includes a non-recurring fee of $28.1 million related to the expansion of the Company’s collaboration agreement with Takeda. Revenue in the third quarter of 2009 includes a non-recurring fee of $22.3 million related to the sale of the LUCENTIS® royalty interest to Genentech. Revenue in the fourth quarter of 2009 includes fees of $14.0 million related to two antibody discovery collaborations.
|
|
(2)
|
Operating expenses in the third quarter of 2010 includes increased spending on NIAID 3 due to increased activity under the contract. Operating expenses in the first and second quarters of 2009 include restructuring expense of $3.3 million and $0.3 million, respectively.
|
|
(3)
|
Other expense in the first and fourth quarters of 2010 primarily relates to the revaluation of the warrant liabilities of $5.8 million and $2.5 million, respectively. Other income in the second and third quarters of 2010 primarily relates to the revaluation of the warrant liabilities of $3.0 million and $3.1 million, respectively. Other expense for the third quarter of 2009 includes a loss of $3.6 million on debt extinguishment relating to the repayment of the Goldman Sachs term loan. Other income in the second, third and fourth quarter of 2009 of $1.0 million, $0.2 million and $0.6 million, respectively was recorded relating to the revaluation of the warrant liabilities in 2009.
|
|
Exhibit Number
|
||
|
1.1
|
Underwriting Agreement dated February 2, 2010 (Exhibit 10.1)1
|
|
|
3.1
|
Memorandum of Continuance of XOMA Ltd. (Exhibit 3.4)2
|
|
|
3.2
|
Bye-Laws of XOMA Ltd. (as amended) (Exhibit 3.2)3
|
|
|
4.1
|
Shareholder Rights Agreement dated as of February 26, 2003 by and between XOMA Ltd. and Mellon Investor Services LLC as Rights Agent (Exhibit 4.1)3
|
|
|
Amendment to Shareholder Rights Agreement dated December 21, 2010 between XOMA Ltd. and Wells Fargo Bank, N.A. as Rights Agent*
|
||
|
4.2
|
Resolution Regarding Preferences and Rights of Series A Preference Shares (Exhibit A to Exhibit 4.1)3
|
|
|
4.3
|
Resolution Regarding Preferences and Rights of Series B Preference Shares (Exhibit B to Exhibit 3)4
|
|
|
4.4
|
Indenture between XOMA Ltd. and Wells Fargo Bank, National Association, as trustee, relating to the Company’s 6.50% Convertible SNAPs SM due February 1, 2012 (Exhibit 2)5
|
|
|
4.5
|
Form of Warrant (May 2009 Warrants) (Exhibit 10.2)6
|
|
|
4.5A
|
Form of Amended and Restated Warrant (May 2009 Warrants) (Exhibit 10.5)1
|
|
|
4.6
|
Form of Warrant (June 2009 Warrants) (Exhibit 10.2)7
|
|
|
4.6A
|
Form of Amended and Restated Warrant (June 2009 Warrants) (Exhibit 10.6)1
|
|
|
4.7
|
Form of Warrant (February 2010 Warrants) (Exhibit 10.2)1
|
|
|
10.1
|
1981 Share Option Plan as amended and restated (Exhibit 10.1)8
|
|
|
10.1A
|
Form of Share Option Agreement for 1981 Share Option Plan (Exhibit 10.1A)9
|
|
|
10.2
|
Restricted Share Plan as amended and restated (Exhibit 10.2)8
|
|
|
10.2A
|
Form of Share Option Agreement for Restricted Share Plan (Exhibit 10.2A)9
|
|
|
10.3
|
2007 CEO Share Option Plan (Exhibit 10.7)10
|
|
|
10.4
|
1992 Directors Share Option Plan as amended and restated (Exhibit 10.3)8
|
|
|
10.4A
|
Form of Share Option Agreement for 1992 Directors Share Option Plan (initial grants) (Exhibit 10.3A)9
|
|
|
10.4B
|
Form of Share Option Agreement for 1992 Directors Share Option Plan (subsequent grants) (Exhibit 10.3B)9
|
|
|
10.5
|
2002 Director Share Option Plan (Exhibit 10.10)11
|
|
|
10.6
|
2010 Long Term Incentive and Share Award Plan (Exhibit 10.5)8
|
|
|
10.6A
|
Form of Share Option Agreement for 2010 Long Term Incentive and Share Award Plan (Exhibit 10.5A)8
|
|
|
10.7
|
Management Incentive Compensation Plan as amended and restated (Exhibit 10.3)12
|
|
|
10.7A
|
CEO Incentive Compensation Plan (Exhibit 10.4A)9
|
|
|
10.7B
|
Bonus Compensation Plan (Exhibit 10.4B)9
|
|
10.8
|
1998 Employee Share Purchase Plan as amended and restated (Exhibit 10.4)8
|
|
|
10.9
|
Form of Amended and Restated Indemnification Agreement for Officers (Exhibit 10.6)13
|
|
|
10.9A
|
Form of Amended and Restated Indemnification Agreement for Employee Directors (Exhibit 10.7)13
|
|
|
10.9B
|
Form of Amended and Restated Indemnification Agreement for Non-employee Directors (Exhibit 10.8)13
|
|
|
10.10
|
Amended and Restated Employment Agreement entered into between XOMA (US) LLC and Steven B. Engle, dated as of December 30, 2008 (Exhibit 10.7)14
|
|
|
10.10A
|
Amended and Restated Employment Agreement entered into between XOMA (US) LLC and Patrick J. Scannon, dated as of December 30, 2008 (Exhibit 10.7A)14
|
|
|
10.10B
|
Amended and Restated Employment Agreement entered into between XOMA (US) LLC and Fred Kurland, dated as of December 29, 2008 (Exhibit 10.7B)14
|
|
|
10.10C
|
Amended and Restated Employment Agreement entered into between XOMA (US) LLC and Christopher J. Margolin, dated as of December 30, 2008 (Exhibit 10.7C)14
|
|
|
10.10D
|
Amended and Restated Employment Agreement entered into between XOMA (US) LLC and Charles C. Wells, dated as of December 30, 2008 (Exhibit 10.7D)14
|
|
|
10.11
|
Consulting Agreement effective as of August 3, 2007 between XOMA (US) LLC and John L. Castello (Exhibit 10.8)10
|
|
|
Form of Change of Control Severance Agreement entered into between XOMA Ltd. and certain of its executives, with reference schedule*
|
||
|
10.13
|
Lease of premises at 890 Heinz Street, Berkeley, California dated as of July 22, 1987 (Exhibit 10.12)15
|
|
|
10.14
|
Lease of premises at Building E at Aquatic Park Center, Berkeley, California dated as of July 22, 1987 and amendment thereto dated as of April 21, 1988 (Exhibit 10.13) 15
|
|
|
10.15
|
Lease of premises at Building C at Aquatic Park Center, Berkeley, California dated as of July 22, 1987 and amendment thereto dated as of August 26, 1987 (Exhibit 10.14)15
|
|
|
10.16
|
Letter of Agreement regarding CPI adjustment dates for leases of premises at Buildings C, E and F at Aquatic Park Center, Berkeley, California dated as of July 22, 1987 (Exhibit 10.15)15
|
|
|
10.17
|
Lease of premises at 2910 Seventh Street, Berkeley, California dated March 25, 1992 (Exhibit 10.16)15
|
|
|
10.17A
|
Fifth amendment to lease of premises at 2910 Seventh Street, Berkeley, California dated June 1, 2006
(Exhibit 10.58)16
|
|
|
10.18
|
Lease of premises at 5860 and 5864 Hollis Street, Emeryville, California dated as of November 2, 2001 (with addendum) (Exhibit 10.19)17
|
|
|
10.19
|
Lease of premises at 2850 Seventh Street, Second Floor, Berkeley, California dated as of December 28, 2001 (with addendum and guaranty) (Exhibit 10.20)17
|
|
|
10.20
|
Amended and Restated Research and License Agreement dated September 1, 1993, between the Company and New York University (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission)
(Exhibit 10.28)15
|
|
|
10.20A
|
Third Amendment to License Agreement dated June 12, 1997, between the Company and New York University (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.28A)15
|
|
|
10.20B
|
Fourth Amendment to License Agreement dated December 23, 1998, between the Company and New York University (Exhibit 10.22B)18
|
|
|
10.20C
|
Fifth Amendment to License Agreement dated June 25, 1999, between the Company and New York University (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.21C)19
|
|
|
10.20D
|
Sixth Amendment to License Agreement dated January 25, 2000, between the Company and New York University (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.1)20
|
|
|
10.20E
|
Seventh Amendment to License Agreement by and among New York University, XOMA Technology Limited and XOMA Ireland Limited effective as of November 10, 2004 (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 3)21
|
|
|
10.21
|
Second Amended and Restated Collaboration Agreement dated January 12, 2005, by and between XOMA (US) LLC and Genentech, Inc. (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission)22
|
|
|
10.21A
|
Agreement related to LUCENTIS® License Agreement and RAPTIVA® Collaboration Agreement dated September 9, 2009, by and between XOMA (Bermuda) Ltd., XOMA (US) LLC and Genentech, Inc. (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.18A)23
|
|
|
10.22
|
License Agreement by and between XOMA Ireland Limited and MorphoSys AG, dated as of February 1, 2002 (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.43)24
|
|
|
10.23
|
Amended and Restated License Agreement by and between XOMA Ireland Limited and DYAX Corp., dated as of October 27, 2006 (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission)
(Exhibit 10.32)13
|
|
|
10.24
|
License Agreement by and between XOMA Ireland Limited and Cambridge Antibody Technology Limited, dated as of December 22, 2002 (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.46)3
|
|
|
10.25
|
License Agreement, dated as of December 29, 2003, by and between Diversa Corporation and XOMA Ireland Limited (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 2)25
|
|
|
GSSM License Agreement, effective as of May 2, 2008, by and between Verenium Corporation and XOMA Ireland Limited (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed seperately with the Securities and Exchange Commission)*
|
||
|
10.26
|
Agreement, dated February 27, 2004, by and between Chiron Corporation and XOMA (US) LLC (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.50)26
|
|
|
10.26A
|
Research, Development and Commercialization Agreement, dated as of May 26, 2005, by and between Chiron Corporation and XOMA (US) LLC (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.2)27
|
|
|
10.26B
|
Secured Note Agreement, dated as of May 26, 2005, by and between Chiron Corporation and XOMA (US) LLC (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.3)27
|
|
|
10.26C
|
Amended and Restated Research, Development and Commercialization Agreement, executed November 7, 2008, by and between Novartis Vaccines and Diagnostics, Inc. (formerly Chiron Corporation) and XOMA (US) LLC (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission)28
|
|
|
10.26D
|
Manufacturing and Technology Transfer Agreement, executed December 16, 2008, by and between Novartis Vaccines and Diagnostics, Inc. (formerly Chiron Corporation) and XOMA (US) LLC (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission)28
|
|
10.27
|
Collaboration Agreement, dated as of September 23, 2004, by and between Aphton Corporation and XOMA (US) LLC (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 2)29
|
|
|
10.28
|
Agreement dated March 8, 2005, between XOMA (US) LLC and the National Institute of Allergy and Infectious Diseases (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.53)22
|
|
|
10.28A
|
Agreement dated July 28, 2006, between XOMA (US) LLC and the National Institute of Allergy and Infectious Diseases (Exhibit 10.60)16
|
|
|
10.28B
|
Agreement dated September 15, 2008, between XOMA (US) LLC and the National Institute of Allergy and Infectious Diseases (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission)
(Exhibit 10.39)30
|
|
|
10.28C
|
Second Amendment to Agreement dated September 15, 2008, between XOMA (US) LLC and the National Institute of Allergy and Infectious Diseases (Exhibit 10.24C) 31
|
|
|
10.29
|
License Agreement, effective as of June 20, 2005, by and between Merck & Co., Inc. and XOMA Ireland Limited (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.4)27
|
|
|
10.30
|
Form of Dealer Manager Agreement relating to the Company’s 6.50% Convertible SNAPs SM due February 1, 2012 (Exhibit 1.1)32
|
|
|
10.30A
|
Form of Placement Agreement relating to the Company’s 6.50% Convertible SNAPs SM due February 1, 2012 (Exhibit 1.2)32
|
|
|
10.31
|
Collaboration Agreement dated as of May 22, 2006, by and between Schering Corporation, acting through its Schering-Plough Research Institute division, and XOMA (US) LLC (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.59)16
|
|
|
10.32
|
Collaboration Agreement, dated as of November 1, 2006, between Takeda Pharmaceutical Company Limited and XOMA (US) LLC (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.46)13
|
|
|
10.32A
|
First Amendment to Collaboration Agreement, effective as of February 28, 2007, between Takeda Pharmaceutical Company Limited and XOMA (US) LLC (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.48)33
|
|
|
10.32B
|
Second Amendment to Collaboration Agreement, effective as of February 9, 2009, among Takeda Pharmaceutical Company Limited and XOMA (US) LLC (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission)28
|
|
|
10.33
|
Loan Agreement, dated as of November 9, 2006, between Goldman Sachs Specialty Lending Holdings, Inc., XOMA (US) LLC and XOMA Ltd. (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.47)13
|
|
|
10.33A
|
Amended & Restated Loan Agreement, dated as of May 9, 2008 between Goldman Sachs Specialty Lending Holdings, Inc., XOMA Ltd. and XOMA (US) LLC (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.37)34
|
|
|
10.34
|
License Agreement, effective as of August 27, 2007, by and between Pfizer Inc. and XOMA Ireland Limited (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 2)35
|
|
10.35
|
Common Stock Purchase Agreement, dated as of October 21, 2008, by and between XOMA Ltd. and Azimuth Opportunity Ltd. (Exhibit 10.1)36
|
|
|
10.35A
|
Common Stock Purchase Agreement, dated as of July 23, 2010, by and between XOMA Ltd. and Azimuth Opportunity Ltd. (Exhibit 10.1) 37
|
|
|
10.36
|
Securities Purchase Agreement dated May 15, 2009, between XOMA Ltd. and the investors named therein (Exhibit 10.1)6
|
|
|
10.36A
|
Engagement Letter dated May 15, 2009 (Exhibit 10.3)6
|
|
|
10.36B
|
Securities Purchase Agreement dated June 5, 2009, between XOMA Ltd. and the investors named therein (Exhibit 10.1)7
|
|
|
10.36C
|
Engagement Letter dated June 4, 2009 (Exhibit 10.3)7
|
|
|
10.37
|
Discovery Collaboration Agreement dated September 9, 2009, by and between XOMA Development Corporation and Arana Therapeutics Limited (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.35) 38
|
|
|
10.38
|
At Market Issuance Sales Agreement dated July 14, 2009, between XOMA Ltd. and Wm Smith & Co. (Exhibit 10.36) 23
|
|
|
10.38A
|
At Market Issuance Sales Agreement dated October 26, 2010, between XOMA Ltd. and Wm Smith & Co. and McNicholl, Lewis & Vlak LLC (Exhibit 10.1) 39
|
|
|
10.38B
|
At Market Issuance Sales Agreement dated February 4, 2011, between XOMA Ltd. and McNicholl, Lewis & Vlak LLC (Exhibit 1.2) 40
|
|
|
10.39
|
Discovery Collaboration Agreement dated October 29, 2009, by and between XOMA Development Corporation and The Chemo-Sero-Therapeutic Research Institute (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission)28
|
|
|
10.40
|
Warrant Amendment Agreement dated February 2, 2010 (May 2009 Warrants) (Exhibit 10.3)1
|
|
|
10.40A
|
Form of Warrant Amendment Agreement dated February 2, 2010 (June 2009 Warrants) (Exhibit 10.4)1
|
|
|
10.41
|
Royalty Purchase Agreement, dated as of August 12, 2010, by and among XOMA CDRA LCC, XOMA (US) LLC, XOMA Ltd. and the buyer named therein (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission) (Exhibit 10.38) 31
|
|
|
Collaboration and License Agreement dated as of December 30, 2010, by and between XOMA Ireland Limited, Les Laboratoires Servier and Institut de Recherches Servier (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission)*
|
||
|
Loan Agreement dated as of December 30, 2010, by and between XOMA Ireland Limited and Les Laboratoires Servier (with certain confidential information omitted, which omitted information is the subject of a confidential treatment request and has been filed separately with the Securities and Exchange Commission)*
|
||
|
Subsidiaries of the Company*
|
||
|
Consent of Independent Registered Public Accounting Firm*
|
||
|
Certification of Steven B. Engle, filed pursuant to Section 302 of the Sarbanes-Oxley Act of 2002*
|
||
|
Certification of Fred Kurland, filed pursuant to Section 302 of the Sarbanes-Oxley Act of 2002*
|
||
|
Certification of Steven B. Engle, furnished pursuant to Section 906 of the Sarbanes-Oxley Act of 2002*
|
||
|
Certification of Fred Kurland, furnished pursuant to Section 906 of the Sarbanes-Oxley Act of 2002*
|
||
|
Press Release dated March 10, 2011 furnished herewith
|
||
Footnotes:
|
Footnotes:
|
||
|
*
|
Filed herewith.
|
|
|
1
|
Incorporated by reference to the referenced exhibit to the Company’s Current Report on Form 8-K filed February 2, 2010.
|
|
|
2
|
Incorporated by reference to the referenced exhibit to the Company’s Registration Statement on Form S-4 filed November 27, 1998, as amended.
|
|
|
3
|
Incorporated by reference to the referenced exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2002.
|
|
|
4
|
Incorporated by reference to the referenced exhibit to the Company’s Amendment No. 1 to Form 8-K/A filed April 18, 2003.
|
|
|
5
|
Incorporated by reference to the referenced exhibit to the Company’s Current Report on Form 8-K filed February 13, 2006.
|
|
|
6
|
Incorporated by reference to the referenced exhibit to the Company’s Current Report on Form 8-K filed May 19, 2009.
|
|
|
7
|
Incorporated by reference to the referenced exhibit to the Company’s Current Report on Form 8-K filed June 10, 2009.
|
|
|
8
|
Incorporated by reference to the referenced exhibit to the Company’s Registration Statement on Form S-8 (File No. 333-171429) filed December 27, 2010.
|
|
|
9
|
Incorporated by reference to the referenced exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2007, as amended.
|
|
|
10
|
Incorporated by reference to the referenced exhibit to the Company’s Current Report on Form 8-K filed August 7, 2007.
|
|
|
11
|
Incorporated by reference to the referenced exhibit to the Company’s Registration Statement on Form S-8 (File No. 333-151416) filed June 4, 2008.
|
|
|
12
|
Incorporated by reference to the referenced exhibit to the Company’s Current Report on Form 8-K filed November 6, 2007.
|
|
|
13
|
Incorporated by reference to the referenced exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2006.
|
|
|
14
|
Incorporated by reference to the referenced exhibit to the Company’s Amendment No. 2 to Annual Report on Form 10-K/A for the fiscal year ended December 31, 2009.
|
|
|
15
|
Incorporated by reference to the referenced exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 1997, as amended.
|
|
|
16
|
Incorporated by reference to the referenced exhibit to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2006.
|
|
|
17
|
Incorporated by reference to the referenced exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2001.
|
|
|
18
|
Incorporated by reference to the referenced exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 1998.
|
|
|
19
|
Incorporated by reference to the referenced exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 1999.
|
|
|
20
|
Incorporated by reference to the referenced exhibit to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended March 31, 2000.
|
|
|
21
|
Incorporated by reference to the referenced exhibit to the Company’s Amendment No. 1 on Form 8-K/A filed November 30, 2004.
|
|
|
22
|
Incorporated by reference to the referenced exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2004.
|
|
|
23
|
Incorporated by reference to the referenced exhibit to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended Septemer 30, 2009 filed November 9, 2009.
|
|
|
24
|
Incorporated by reference to the referenced exhibit to Amendment No. 2 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended March 31, 2002 filed on December 12, 2002.
|
|
|
25
|
Incorporated by reference to the referenced exhibit to the Company’s Amendment No. 2 on Form 8-K/A filed March 19, 2004.
|
|
|
26
|
Incorporated by reference to the referenced exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2003.
|
|
|
27
|
Incorporated by reference to the referenced exhibit to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2005.
|
|
|
28
|
Incorporated by reference to the referenced exhibit to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2009.
|
|
|
29
|
Incorporated by reference to the referenced exhibit to the Company’s Amendment No. 1 on Form 8-K/A filed October 26, 2004.
|
|
|
30
|
Incorporated by reference to the referenced exhibit to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2008.
|
|
|
31
|
Incorporated by reference to the referenced exhibit to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2010 filed on November 4, 2010.
|
|
|
32
|
Incorporated by reference to the referenced exhibit to Amendment No. 2 to the Company’s Registration Statement on Form S-4 filed January 11, 2006.
|
|
|
33
|
Incorporated by reference to the referenced exhibit to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended March 31, 2007 filed on March 5, 2010.
|
|
|
34
|
Incorporated by reference to the referenced exhibit to Amendment No. 2 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2008 filed on March 5, 2010.
|
|
|
35
|
Incorporated by reference to the referenced exhibit to the Company’s Current Report on Form 8-K filed September 13, 2007.
|
|
|
36
|
Incorporated by reference to the referenced exhibit to the Company’s Current Report on Form 8-K filed October 22, 2008.
|
|
|
37
|
Incorporated by reference to the referenced exhibit to the Company’s Current Report on Form 8-K filed July 23, 2010.
|
|
|
38
|
Incorporated by reference to the referenced exhibit to Amendment No. 1 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2009 filed on March 5, 2010.
|
|
|
39
|
Incorporated by reference to the referenced exhibit to the Company’s Current Report on Form 8-K filed October 26, 2010.
|
|
|
40
|
Incorporated by reference to the referenced exhibit to the Company’s Registration Statement on Form S-3 (File No. 333-172197) filed February 11, 2011.
|
|