UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form
| ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For
the fiscal year ended
| TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ________to_________
Commission
file number
(Exact name of registrant as specified in its charter)
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s
telephone number, including area code:
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of exchange on which registered | ||
| The
|
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes
☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes
☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ | |
| ☒ | Smaller reporting company | |||
| Emerging growth company |
If
an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate
by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered
public accounting firm that prepared or issued its audit report.
If
securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant
in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate
by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐
The
aggregate market value of the voting and non-voting common equity held by non-affiliates as of June 30, 2023, was $
As of March 28, 2024, there were shares of common stock, par value $0.0001, issued and outstanding.
Mira Pharmaceuticals, Inc.
Annual Report on Form 10-K
For the fiscal year ended December 31, 2023
TABLE OF CONTENTS
Unless we have indicated otherwise, or the context otherwise requires, references in this Report to “MIRA,” the “Company,” “we,” “us” and “our” or similar terms refer to Mira Pharmaceuticals, Inc., a Florida corporation.
From time to time, we may use our website, our Facebook page at https://www.facebook.com/people/Mira-Pharmaceuticals-Inc/100087641460083, our Twitter at https://twitter.com/PharmaMira and on our LinkedIn account at www.linkedin.com/company/mira-pharmaceuticals-inc to distribute material information. Our financial and other material information is routinely posted to and accessible on the Investors section of our website, available at www.mirapharmaceuticals.com. Investors are encouraged to review the Investors section of our website because we may post material information on that site that is not otherwise disseminated by us. However, information that is contained in and can be accessed through our website, our Facebook page, our Twitter posts and our LinkedIn posts are not incorporated into, and does not form a part of, this Annual Report.
| i |
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K (this “Report”) contains forward-looking statements (as defined in Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act) that reflect our current expectations and views of future events. In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “expect,” “plan,” “anticipate,” “could,” “intend,” “target,” “project,” “contemplate,” “believe,” “estimate,” “predict,” “potential”, or “continue” or the negative of these terms or other similar expressions. In particular, statements about our pre-clinical and clinical trials and expectations regarding such trials, the markets in which we operate, including growth of such markets, and our expectations, beliefs, plans, strategies, objectives, prospects, assumptions, or future events or performance contained in this Report generally under the headings “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business” are forward-looking statements.
We have based these forward-looking statements on our current expectations, assumptions, estimates and projections. While we believe these expectations, assumptions, estimates, and projections are reasonable, such forward-looking statements are only predictions and involve known and unknown risks and uncertainties, many of which are beyond our control. These and other important factors, including those discussed in this Report under the headings “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business,” may cause our actual results, performance, or achievements to differ materially from any future results, performance or achievements expressed or implied by these forward-looking statements, or could affect our share price. Important factors that could cause actual results or events to differ materially from those expressed in forward-looking statements include, but are not limited to, the following:
| ● | our ability to obtain and maintain regulatory approval of our product candidates; | |
| ● | our ability to successfully commercialize and market our product candidates, if approved; | |
| ● | our ability to contract with third-party suppliers, manufacturers and other service providers and their ability to perform adequately; | |
| ● | the potential market size, opportunity, and growth potential for our product candidates, if approved; | |
| ● | our ability to obtain additional funding for our operations and development activities; | |
| ● | the accuracy of our estimates regarding expenses, capital requirements and needs for additional financing; | |
| ● | the initiation, timing, progress and results of our pre-clinical studies and clinical trials, and our research and development programs; | |
| ● | the timing of anticipated regulatory filings; | |
| ● | the timing of availability of data from our clinical trials; | |
| ● | our future expenses, capital requirements, need for additional financing, and the period over which we believe that our existing cash and cash equivalents will be sufficient to fund our operating expenses and capital expenditure requirements; | |
| ● | our ability to retain the continued service of our key professionals and to identify, hire and retain additional qualified professionals; | |
| ● | our ability to advance product candidates into, and successfully complete, clinical trials; | |
| ● | our ability to recruit and enroll suitable patients in our clinical trials; | |
| ● | the timing or likelihood of the accomplishment of various scientific, clinical, regulatory, and other product development objectives; |
| 1 |
| ● | the pricing and reimbursement of our product candidates, if approved; | |
| ● | the rate and degree of market acceptance of our product candidates, if approved; | |
| ● | the implementation of our business model and strategic plans for our business, product candidates, and technology; | |
| ● | the scope of protection we are able to establish and maintain for intellectual property rights covering our product candidates and technology; | |
| ● | developments relating to our competitors and our industry; | |
| ● | the development of major public health concerns and the future impact of such concerns on our clinical trials, business operations and funding requirements; and | |
| ● | other risks and factors listed under “Risk Factors” and elsewhere in this Report. |
Given the risks and uncertainties set forth in this Report, you are cautioned not to place undue reliance on such forward-looking statements. The forward-looking statements contained in this Report are not guarantees of future performance and our actual results of operations, financial condition, and liquidity, and the development of the industry in which we operate, may differ materially from the forward-looking statements contained in this Report. In addition, even if our results of operations, financial condition and liquidity, and events in the industry in which we operate, are consistent with the forward-looking statements contained in this Report, they may not be predictive of results or developments in future periods.
Any forward-looking statement that we make in this Report speaks only as of the date of such statement. Except as required by federal securities laws, we do not undertake any obligation to update or revise, or to publicly announce any update or revision to, any of the forward-looking statements, whether as a result of new information, future events or otherwise, after the date of this Report.
Summary of Principal Risks
Our business is subject to numerous risks and uncertainties that represent challenges that we face in connection with the implementation of our strategy and the growth of our business. In particular, the following are the principal risks which could cause a decline in the price of shares of our common stock:
| ● | We are a development-stage, pre-clinical biotechnology company that has no revenues and has incurred losses since our inception. We expect to incur losses for the foreseeable future and may never be able to generate revenues or achieve or maintain profitability. | |
| ● | Our limited operating history may make it difficult for you to evaluate the success of our business to date and to assess our future viability. | |
| ● | Our losses from operations and negative cash flows of December 31, 2023 raise substantial doubt about our ability to continue as a going concern absent obtaining adequate new debt or equity financings. | |
| ● | The report of our independent registered accounting firm on our audited financial statements for the fiscal year ended December 31, 2023 contains an explanatory paragraph relating to our ability to continue as a going concern. | |
| ● | We are dependent on the success of our product candidates, some of which may not receive regulatory approval or be successfully commercialized. | |
| ● | We will need additional funds to complete the further development of our business plan, and there is no assurance that additional financing will be available or will be available on terms acceptable to us. |
| 2 |
| ● | Certain of our executive officers will not be employed by us on a full-time basis. | |
| ● | We face risks related to health, pandemics, epidemics, and outbreaks could significantly disrupt our pre-clinical studies and clinical trials, commercialization efforts, supply chain, regulatory and clinical development activities, and other business operations, in addition to the impact of a global economic slowdown. | |
| ● | Results of pre-clinical studies and future early clinical trials are not necessarily predictive indicators of future results. | |
| ● | We may fail to expand our anticipated outsourced manufacturing capability in time to meet market demand for our products and product candidates, and the FDA may refuse to accept the facilities of our contract manufacturers as being suitable to produce our products and product candidates. Any problems in our manufacturing process could have a material adverse effect on our business, results of operations and financial condition | |
| ● | Our future success will largely depend on the success of our product candidates, which development will require significant capital resources and years of clinical development effort | |
| ● | There is a high rate of failure for drug candidates proceeding through clinical trials | |
| ● | We rely on, and expect to continue to rely on, third parties to conduct clinical trials for our product candidates. If these third parties do not successfully carry out their contractual duties, comply with regulatory requirements or meet expected deadlines, we may not be able to obtain marketing approval for or commercialize our product candidates, and our business could be substantially harmed | |
| ● | We rely on third parties to manufacture our clinical product supplies, and we intend to rely on third parties for at least a portion of the manufacturing process of our product candidates, if approved. Our business could be harmed if those third parties fail to provide us with sufficient quantities of product or fail to do so at acceptable quality levels or prices or fail to maintain or achieve satisfactory regulatory compliance | |
| ● | Even if any of our product candidates receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors, and others in the medical community necessary for commercial success | |
| ● | If we are unable to obtain and maintain intellectual property protection for our technology and products, or if the scope of the intellectual property protection obtained is not sufficiently broad, our competitors could commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be impaired | |
| ● | Certain recent initial public offerings of companies with relatively small public floats comparable to our anticipated public float have experienced extreme volatility that was seemingly unrelated to the underlying performance of the respective company, and our securities may potentially experience rapid and substantial price volatility, which may make it difficult for prospective investors to assess the value of our securities |
| 3 |
PART I
ITEM 1. Description of Business
Overview
We are a pre-clinical-stage pharmaceutical development company with two neuroscience programs targeting a broad range of neurologic and neuropsychiatric disorders. We hold exclusive license rights in the U.S., Canada and Mexico for Ketamir-2, a novel, patent pending oral ketamine analog under pre-clinical investigation to potentially deliver ultra-rapid antidepressant effects, providing hope for individuals battling treatment-resistant depression (or TRD), major depressive disorder with suicidal ideation (or MDSI) and potentially post-traumatic stress disorder (or PTSD).
Additionally, our novel oral pharmaceutical marijuana molecule, MIRA-55, is being studied for its potential to alleviate neuropathic pain, as well as anxiety and cognitive decline, symptoms commonly associated with early-stage dementia. MIRA-55, if approved by the U.S. Food and Drug Administration (or FDA), could mark a significant advancement in addressing various neuropsychiatric, inflammatory, and neurologic diseases and disorders.
The U.S. Drug Enforcement Administration (DEA)’s scientific review of Ketamir-2 concluded that it would not be considered a controlled substance or listed chemical under the Controlled Substances Act (CSA) and its governing regulations. Additionally, we have submitted the required paperwork for MIRA-55 to be evaluated by the DEA.
We were incorporated under the laws of the State of Florida in September 2020 and commenced substantive operations, including our pharmaceutical development program, in late 2020.
Our Product Candidates in Development
KETAMIR-2
Major Depressive Disorder (or MDD) is a significant global health concern, affecting over 264 million people worldwide and ranking among the leading causes of disability according to the World Health Organization. In the United States alone, it impacts nearly 21.1 million adults, accounting for about 8.3 % of the adult population in 2-2021 according to data form the National Institutes of Health. This widespread mental health disorder not only undermines the quality of life and daily functioning of individuals but also imposes a substantial economic burden, with costs in the U.S. amounting to tens of billions of dollars annually. MDD is also a major risk factor for suicide, a leading cause of death globally, highlighting its profound impact on public health and the urgent need for effective treatment and management strategies. If approved by the FDA, Ketamir-2 may potentially provide antidepressant therapeutic effects.
Despite the fact that antidepressants have been on the market for decades, with imipramine being the first FDA-approved antidepressant in 1959, the need for a rapid-acting antidepressant that can help patients with Treatment-Resistant Depression (or TRD) using a novel mechanism of action (e.g. not a monoamine reuptake inhibitor) has been growing. In 2019, ketamine was introduced but required by the FDA to utilize a Risk Evaluation and Mitigation Strategy (REMS) because of its: (1) poor oral availability requiring intravenous (or IV) or intranasal (or IN) administration, (2) ability to cause side effects including dissociation, sedation and acute hypertension, and (3) potential abuse liability.
| 4 |
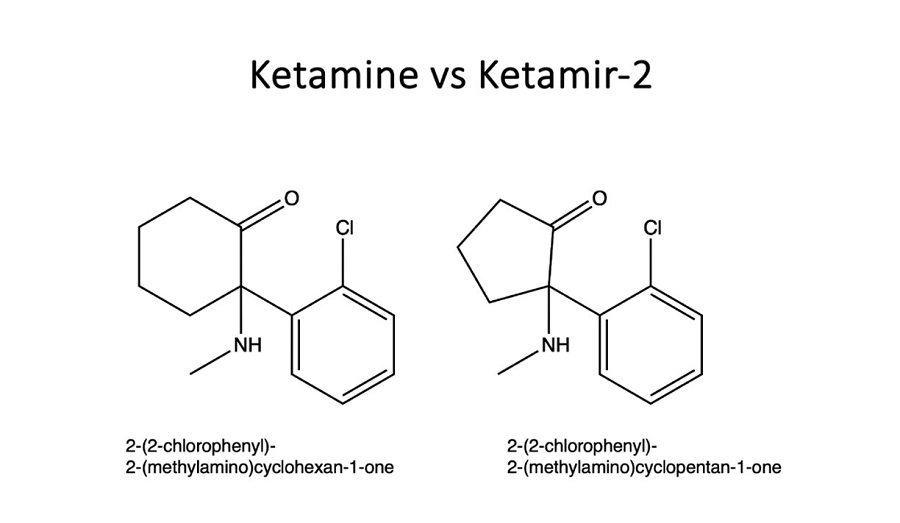
Ketamir-2 is a new chemical entity, an analog of ketamine that is designed to potentially preserve the same rapid antidepressant response but with improved bioavailability. It may also have decreased side effects, and decreased abuse liability, though such conclusions are within the sole authority of the FDA. This combination is intended to potentially facilitate safer and less cumbersome dosing requirements, with the goal of obtaining an orally administered pill that can be taken at home.
Figure: Chemical structures of ketamine and Ketamir-2 for comparison purposes.
The DEA conducted a scientific review of the Ketamir-2 in 2023 in accordance with the definitions within the CSA and its implementing regulations. Based on this review, DEA determined that” Ketamir-2 is “not a controlled substances or listed chemical under the CSA.”
Mechanism of Action of Ketamir-2
Ketamir-2’s mechanism of action (or MOA) as a rapid acting antidepressant is the same as ketamine’s, based on the fact that the two share a common inhibitory effect on the N-methyl-D-aspartate (or NMDA) receptor, a type of glutamate receptor that is believed to be integral to the antidepressant effects of both of these ketamine and Ketamir-2. In fact, Ketamir-2 and ketamine differ in less than 2% in their antagonist activity at the GRIN1/GRIN2B receptor subunit of the NMDA receptor (based in in silico analysis, see below). This subunit combination is prominently linked to neuroplasticity, believed to be a key factor in depression and the action of antidepressants such as ketamine. GRIN2B-containing NMDA receptors are implicated in synaptic plasticity changes associated with depression and its treatment.
Ketamine’s mechanism of action (or MOA) as a rapidly acting antidepressant is multifaceted and distinct from traditional antidepressants like selective serotonin reuptake inhibitors (or SSRIs) and tricyclic antidepressants. While ketamine has shown promise as a rapid-acting antidepressant, especially in treatment-resistant depression, its use is limited due to potential side effects and abuse potential that Ketamir-2 has been targeted to minimize. Moreover, whereas ketamine has a poor oral bioavailability and must therefore be given IV or IN, Ketamir-2 has a much better bioavailability suggesting it may be appropriate for oral use.
| 5 |
The following is a detailed synopsis of the MOAs of both Ketamir-2 and ketamine:
| 1. | NMDA Receptor Antagonism: Ketamine primarily acts as a non-competitive antagonist of the NMDA. By inhibiting these receptors, ketamine modulates the release of the neurotransmitter glutamate. This modulation leads to an increase in glutamatergic signaling via activation of AMPA receptors, another type of glutamate receptor. This enhanced signaling is believed to play a crucial role in ketamine’s rapid antidepressant effects. | |
| 2. | mTOR Pathway Activation: Ketamine activates the mammalian target of rapamycin (or mTOR) pathway, a key regulator of cell growth and survival. This activation is linked to increased synaptogenesis in the prefrontal cortex. The mTOR pathway plays a significant role in neural plasticity and has been implicated in the pathophysiology of depression. | |
| 3. | Effects on GABAergic System: Recent research indicates that ketamine may also affect the gamma-aminobutyric acid (GABAergic) system, which is responsible for inhibitory neurotransmission in the brain. Alterations in GABAergic signaling have been associated with mood disorders. | |
| 4. | BDNF Release and Synaptogenesis: The increased glutamatergic transmission leads to the activation of downstream pathways that result in the release of Brain-Derived Neurotrophic Factor (BDNF). BDNF is crucial for neuroplasticity – the brain’s ability to reorganize and form new neural connections. Studies suggest that this increase in BDNF and subsequent synaptogenesis (formation of new synapses) in brain areas like the prefrontal cortex is a key factor in the antidepressant effects of ketamine. | |
| 5. | Anti-inflammatory Effects: Depression is increasingly linked with chronic inflammation. Ketamine has been shown to have anti-inflammatory properties, which might contribute to its antidepressant effects. | |
| 6. | Neuroendocrine Regulation: Ketamine may influence the hypothalamic-pituitary-adrenal (HPA) axis, which is often dysregulated in depression. By modulating this axis, ketamine could exert additional antidepressant effects. | |
| 7. | Rapid Onset of Action: Unlike traditional antidepressants, which typically take weeks to exert their effects, ketamine’s impact on mood can be noticed within hours of administration. This rapid action is especially beneficial in acute management of severe depression and suicidal ideation. |
In summary, while Ketamir-2’s and ketamine’s antidepressant MOA are still being studied and explored, current evidence suggests a complex and involved synergistic action on various neural pathways, primarily through the modulation of glutamatergic neurotransmission, enhancement of neuroplasticity, and potentially through anti-inflammatory and neuroendocrine mechanisms. Both drugs rapid onset and efficacy in treatment-resistant cases make them potentially valuable tools in psychiatry, but the potentially improved side effect profile and oral bioavailability are what differentiate Ketamir-2 and ketamine as described below.
Ketamir-2 Clinical Development Program
The clinical development plan for Ketamir-2 involves a series of methodically structured phases, starting with IND-enabling studies and progressing through Phase 1 and Phase 2 clinical trials. These trials aim to establish the safety, efficacy, and optimal use of Ketamir-2 in treating psychiatric conditions like TRD, Major Depressive Disorder with Suicidal Ideation (MDSI), and potentially PTSD. The strategy underscores patient safety while evaluating Ketamir-2’s therapeutic benefits and risks. The successful development of Ketamir-2 could significantly impact the treatment landscape for depression, offering a novel approach that addresses the shortcomings of current therapies.
Initially, the development process begins with completion of all necessary IND-enabling studies. These preclinical studies, encompassing pharmacokinetics, pharmacodynamics, toxicology, and safety pharmacology, are crucial for ensuring that the investigational drug meets regulatory standards. The successful completion of these studies allows for the submission of an Investigational New Drug (IND) application to the FDA, specifically targeting TRD. We anticipate that we will submit our IND for Ketamir-2 by the end of 2024. See the section below titled “Research and Testing to Date – Ketamir-2” for more information.
Upon FDA acceptance of our Ketamir-2 IND, our plan progresses to Phase 1 clinical trials. These trials are designed to assess the safety and tolerability of Ketamir-2 in healthy volunteers. They are typically randomized, double-blind, and placebo-controlled, and aim to determine the appropriate dosing while closely monitoring for adverse effects. Key to this phase is the collection of pharmacokinetic and pharmacodynamic data, which guides the dosing strategies for subsequent trials.
| 6 |
Phase 1: Safety and Dosage Determination in Healthy Volunteers
| 1. | Study Design: |
| ○ | A randomized, double-blind, placebo-controlled trial. | |
| ○ | Primary objective: Assess safety and tolerability of Ketamir-2. | |
| ○ | Secondary objectives: Determine pharmacokinetics and pharmacodynamics. |
| 2. | Participant Selection: |
| ○ | Enroll healthy volunteers, ensuring a diverse demographic representation. | |
| ○ | Exclude individuals with a history of psychiatric illness, substance abuse, or significant medical conditions. |
| 3. | Dosing and Administration: |
| ○ | Start with a low dose, escalating gradually to higher doses. | |
| ○ | Monitor participants closely for adverse effects. |
| 4. | Outcome Measures: |
| ○ | Safety assessments: Vital signs, laboratory tests, ECG, adverse event monitoring. | |
| ○ | PK/PD assessments: Blood sampling for drug levels, brain imaging for receptor binding (if feasible). |
Following the establishment of safety and initial dosing parameters in Phase 1, the development plan moves into Phase 2. This phase involves trials with patients diagnosed with TRD. The primary goal here is to evaluate the optimal dose and tolerability of Ketamir-2 in this specific patient population. Additionally, these trials provide preliminary data on the efficacy of Ketamir-2 for the treatment of TRD. Safety remains a priority, with close monitoring for any adverse events and detailed assessments using depression rating scales.
Phase 2: Dose, Tolerability, and Early Efficacy in TRD
| 1. | Study Design: |
| ○ | A randomized, controlled trial with TRD patients. | |
| ○ | Primary objective: Evaluate the optimal dose and tolerability. | |
| ○ | Secondary objective: Obtain preliminary efficacy data. |
| 2. | Participant Selection: |
| ○ | Enroll patients diagnosed with TRD. | |
| ○ | Utilize standardized diagnostic criteria and severity scales. |
| 3. | Dosing Regimen: |
| ○ | Implement a dose range based on Phase 1 findings. | |
| ○ | Consider flexible dosing or fixed-dose regimen based on safety and tolerability data. |
| 4. | Outcome Measures: |
| ○ | Tolerability assessment: Adverse event monitoring, patient-reported outcomes. | |
| ○ | Efficacy assessment: Depression rating scales (e.g., HDRS, MADRS). |
As the development of Ketamir-2 progresses, there is potential to expand its indications. One such area is MDSI, where Ketamir-2’s application could be particularly beneficial given ketamine’s established efficacy in this domain. This would involve designing a trial specifically targeting MDSI, with a focus on the rapid onset of action and short-term safety considerations.
Furthermore, given the emerging research suggesting ketamine’s therapeutic potential in PTSD, a similar approach could be considered for Ketamir-2. Developing a trial protocol for PTSD treatment requires a careful balance, considering the complexity of the disorder, potential comorbidities, and the need for robust safety and efficacy data.
| 7 |
Pursuing Additional INDs:
| 1. | Major Depressive Disorder with Suicidal Ideation (MDSI): |
| ○ | Following successful Phase 2 outcomes, pursue an IND for MDSI, leveraging existing data and research on ketamine. | |
| ○ | Design a trial specifically targeting MDSI, focusing on rapid onset of action and short-term safety. |
| 2. | Post-Traumatic Stress Disorder (PTSD): |
| ○ | Based on early research suggesting ketamine’s efficacy in PTSD, consider developing a clinical trial protocol for Ketamir-2 in PTSD. | |
| ○ | Prioritize safety and efficacy, given the complex nature of PTSD and potential comorbidities. |
In summary, the clinical development plan for Ketamir-2 is a meticulous, multi-phase strategy that prioritizes patient safety while exploring the drug’s potential in treating complex psychiatric conditions. Each phase is carefully designed to address specific research questions and regulatory requirements, ensuring a thorough evaluation of Ketamir-2’s therapeutic benefits and risks.
Manufacture of Product for Pre-Clinical and Clinical Development Activities
Recipharm Israel LTD, a leading global contract development and manufacturing organization (or CMDO), is currently developing a large-scale synthesis protocol for us and will be supplying quantities of Ketamir-2 and MIRA-55 needed for our pre-clinical and clinical development activities. We previously utilized Curia Global as our CMDO and are currently in discussions with other partners to have Ketamir-2 and MIRA-55 formulated into solid oral dosage forms for clinical trials.
We also utilize Frontage Laboratories and Pharmaseed LTD to conduct preclinical studies on Ketamir-2.
MIRA1A
In early February 2024, we made a significant discovery during the manufacturing and scale-up process of our patented molecule known as “MIRA1a,” which we believed was the molecule used in our pre-clinical trials and had been synthesized by contract manufacturer. Through this process, we identified a novel and improved version of the molecule, which we call MIRA-55.
As part of our due diligence and subsequent testing, which began in late 2023, we discovered that the pre-clinical studies we conducted, previously attributed to MIRA1a, were in fact performed on MIRA-55. Following this revelation, in early March 2024, we promptly filed a provisional patent for MIRA-55, which encompasses all pre-clinical studies disclosed in our two registration statements on Form S-1, declared effective on August 2, 2023, and December 27, 2023 (File Nos. 333-273024 and 333-276118, respectively). If such patent is issued, we would own the patent rights to both MIRA1a and MIRA-55.
Moreover, based on our pre-clinical analyses to date, we believe that MIRA-55 is an improvement over MIRA1a in that it displays enhanced potency and potential for efficacy.
Based on our discoveries to date, we decided to advance MIRA-55 as our lead compound for our oral pharmaceutical marijuana drug candidate while still retaining our rights to MIRA1a. As such, we do not intend to move MIRA1a forward as of the date of this Report.
| 8 |
MIRA-55
Our objective is to develop and commercialize new treatment options for neuropsychiatric, inflammatory, and neurologic diseases and disorders. Cannabinoids are a class of chemical compounds that are naturally occurring and are primarily found in cannabis plant extracts. The two major cannabinoids found in cannabis plant extracts include tetrahydrocannabinol, a compound that is the main psychoactive ingredient of cannabis (or THC) and cannabidiol, the second most prevalent active ingredient in cannabis which does not have psychoactive properties (or CBD). These compounds bind to CB1 and CB2 cannabinoid receptors, which are found throughout the body. Specifically, CB1 receptors are concentrated in the central nervous system (or CNS), while CB2 receptors are found mostly in peripheral organs and are associated with the immune system. When the chemical compounds bind to these cannabinoid receptors, the process elicits certain physiological responses. Physiological responses to cannabinoids may vary among individuals. Some of the effects of cannabinoids have been shown to impact nervous system functions, immune responses, muscular motor functions, gastrointestinal maintenance, blood sugar management, and the integrity of ocular functions. Based on pre-clinical testing, our product candidate, MIRA-55, appears to have a strong selectivity for CB2 versus CB1, and is designed to minimize the risk of psychoactive adverse events associated with CB1 activation.
Mechanism of Action of MIRA-55
We believe that the effects of MIRA-55 at the cannabinoid receptors CB1 and CB2 is predicted to account for the majority of its potential therapeutic effects, especially as it relates to its anti-anxiety, anti-pain and anti-inflammatory properties. For example, the difference in the dose-response effects of MIRA-55 compared with THC on CB1 receptors appears to coincide with its improved therapeutic profile. If approved by the FDA, MIRA-55 may potentially provide therapeutic effects and enhanced cognition for anxiety, pain and inflammation.
THC has been demonstrated to have biphasic physiological effects (meaning effects in two phases), which have been described for over 40 years: at low levels THC has positive effects while high doses cause the opposite, undesirable symptoms. Examples of biphasic effects at low versus high levels of THC include the anti-anxiety versus pro-anxiety effects, respectively. Through pre-clinical test, we obtained the following dose-response effects for MIRA-55 and THC at the CB1 receptor (see below). In contrast to THC, which displays an initial maximally stimulatory and then inhibitory response at CB1, MIRA-55 appears to act as a monophasic partial agonist (meaning it has a lower intrinsic activity than full agonists) in that it creates a stimulation throughout its dose range, achieving a moderate activation of the CB1 even at high doses. We believe that this accounts for the potential broad therapeutic efficacy of MIRA-55 and the observed absence of negative symptoms even at maximal doses of the drug.
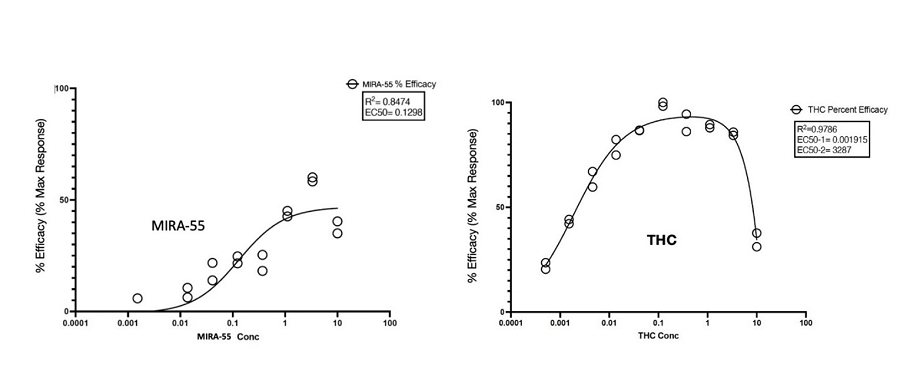
Figure: Compound activity with the selected GPCR Biosensor Assays: THC vs MIRA-55 agonist activity at the CB1 Receptor.
| 9 |
In pharmacology, “efficacy” or “Emax” refers to the maximum response that can be achieved with a drug or agent. It represents the extent or magnitude of the response produced by the drug once it has bound to its target, typically referred to as a receptor. The binding between a drug and its receptor is characterized by affinity, which quantifies the strength of their interaction. Efficacy, however, assesses the action or effect of the drug following binding to the receptor.
The dose-response curve is a commonly used graph in pharmacology that depicts the relationship between the effect of a drug and its dosage. The X-axis represents the increasing doses of the drug, while the Y-axis represents the response produced by the drug. In the case of the figure above, the term “% Efficacy” on the Y-axis refers to the maximum response that can be achieved with the agonist (MIRA-55 or THC) in relation to its ability to activate GPCR receptors (specifically CB1 receptors). GPCRs are G-protein-coupled receptors that form a large group of proteins which are expressed on the cell surface of eukaryotic cells to detect molecules outside the cell and activate cellular response.
The data presented in the figure above has been normalized to the maximal and minimal responses observed in the presence of a control compound and vehicle, respectively. This normalization allows for a standardized comparison of the agonist’s efficacy.
MIRA-55 Pre-clinical Developments and Studies
As of the date of this Report, we completed several pre-clinical studies of MIRA-55, including, but not limited to, radio-ligand binding assay, elevated plus maze (or EPM) model of anxiety and hot plate model thermal sensitivity testing.
We have studied the effects of acute administration of MIRA-55 on anxiety-related phenotypes in mice to model human conditions. An intraperitoneal injection of Placebo [PBO] (e.g. saline) or MIRA-55 (e.g. 50mg/kg = Treatment) was administered to C57Bl/6 mice (n=5/group) that were 8-12 weeks old. Thirty minutes following injection, mice were tested in anxiety related measures using the Elevated Plus Maze (EPM). The EPM is a widely used pre-clinical behavioral assay for rodents and it has been validated to assess the anti-anxiety effects of pharmacological agents. If determined and approved by the FDA or other regulatory agencies, MIRA-55 appears to have anti-anxiety effects at doses that lacked side effects of sedation or intoxication in mice. The EPM is a test measuring anxiety in rodents as a screening test for putative anxiolytic compounds and as a general research tool in neurobiological anxiety research such as Generalized Anxiety Disorder (or GAD) or Post-Traumatic Stress Disorder (or PTSD). The model is based on the animal’s aversion to open spaces which are present in the open arms (Open Arm) of the maze. Anti-anxiety effects of test agents are demonstrated by an increase in the percentage of time spent in the Open Arm with treatment compared to placebo. The total distance traveled is a measure of the overall level of arousal and mobility of the mice undergoing testing on the EPM and is used to rule out any sedating or intoxicating effects of the test agent.
Pre-clinical studies also have shown the potential of MIRA-55 for relieving pain. A number of clinically approved pharmacological agents used to treat pain, including opioids, have been demonstrated to delay or ameliorate the onset of heat sensitivity upon paw exposure of mice to heat. Thirty minutes after treatment with either a placebo (control) or MIRA-55, mice were placed on a heated plate to measure the time it took for each mouse to lift its paw in response to the mild pain they felt from the heat. Mice treated with pain alleviating drugs took significantly longer to become bothered by the heat and to lift their paws. Similarly, mice treated with MIRA-55 statistically took significantly more time to lift their legs, indicating MIRA-55’s potential effectiveness as a possible treatment for pain in this model. If approved by the FDA, MIRA-55 may potentially provide therapeutic effects for pain control.
MIRA-55 is a CB2 agonist which may also be an optimal treatment for neurodegenerative diseases associated with neuroinflammation caused by microglial activation. CB2 agonism has been shown in pre-clinical studies to regulate neuroinflammatory processes, reducing the neuronal damage characteristic of degeneration. We believe there may be a strong rationale for CB2 agonism in neurodegenerative diseases, given increased CB2 expression in patients with these diseases as well as preliminary results from animal models. We see potential for a potent CB2 agonist to treat a range of neurodegenerative diseases. MIRA-55, through its robust activity at CB2 compared to CB1, was designed to minimize the risk of psychotropic adverse events associated with CB1 activation. If approved by the FDA, MIRA-55 may potentially provide therapeutic effects for neurodegenerative and neuroinflammatory illnesses.
| 10 |
Our pre-clinical development program for MIRA-55 has included a variety of testing. Summarized below are the tests we have completed. Our interpretation of results derived from pre-clinical data or our conclusions based on our pre-clinical data may prove inaccurate and are not necessarily predictive indicators of future results. See the section below titled “Research and Testing to Date – MIRA-55” for more information.
Our MIRA-55 Clinical Development Program
Following the pre-clinical development plan outlined above, we plan to submit to the FDA an Investigational New Drug application (or IND) focused on investigating MIRA-55 for the treatment of anxiety and cognitive decline in elderly patients.
We expect that our first IND application submission relating to MIRA-55 for the treatment of elderly patients suffering from anxiety with some cognitive decline is currently planned for the end of the second quarter of 2025, as we believe this is a patient population with unmet needs. If allowed to proceed by the FDA, a Phase I trial will be initiated 30 days post-IND submission. We expect that our second IND for MIRA-55 will focus on investigating MIRA-55 for the treatment of neuropathic pain.
All development plans depend on FDA acceptance of our IND applications. As appropriate and pursuant to discussions with the FDA, we may periodically adjust the timeline for certain filings and associated clinical trials. It is important to note that the process for conducting clinical trials is uncertain and there is no assurance that our clinical development activities will meet the planned timelines set forth above.
Our Market Opportunity and Market Advantage
Ketamir-2
Ketamir-2’s market opportunity and market advantage was analyzed by IQVIA who were contracted to perform an independent Market Characterization and Drug Valuation Analysis. TRD and MDSI indications represent areas of high unmet medical need, with significant disease burden and limited effective treatments available. Ketamir-2’s formulation as a once-daily oral medication addresses shortcomings in existing treatments, such as route of administration (RoA) and time to effectiveness.
The market opportunity for Ketamir-2 is substantial. Based on the IQVIA analysis, the U.S. has a large patient pool looking for effective treatments, with diagnosed prevalence rates of 3.1% for MDSI and 2.4% for TRD, translating to total addressable populations of 4.9 million and 3.8 million patients respectively. Based on total estimates of MDSI and TRD together, this represents a Total Diagnosed Prevalence rate of 12.3 million patients and, assuming a Treatment Rate of 65%, the Total Addressable Population is 8.7 million patients This represents a significant market, especially considering the current limitations and side effects associated with existing treatments.
| 11 |

Figure: Estimates by IQVIA of the total addressable populations affected with MDSI and TRD.
Ketamir-2’s market advantage lies in its novel profile and potential to address these unmet needs. As a synthetic ketamine derivative, we believe it potentially offers an improved mechanism to treat disease, building on the success of existing marketed therapeutics but with differences to the base molecules that potentially reduce unwanted side effects. Ketamir-2’s oral formulation is being developed to potentially not require health care professional supervision, potentially improving patient compliance and ease of use.

Figure: Summary of assessment by IQVIA of valuation of Ketamir-2, including the background, commercial opportunity, and drivers of valuation assessment.
| 12 |
The projected addressable market for Ketamir-2 are promising. If approved by the FDA and deemed safe, peak annual net sales in the U.S. are estimated to potentially reach approximately $3 billion across both MDSI and TRD, with a base case eNPV (expected net present value) of around $92 million. In the high case scenario, the unadjusted peak revenue opportunity could go up to about $7.8 billion by 2035, with the eNPV potentially reaching $324 million. The estimated patient pool for Ketamir-2 treatment may reach approximately 0.2 million patients in the U.S. by 2036. The NPV (net present value) ranges from approximately $270 million to $4.6 billion, with the base case being around $1.4 billion.
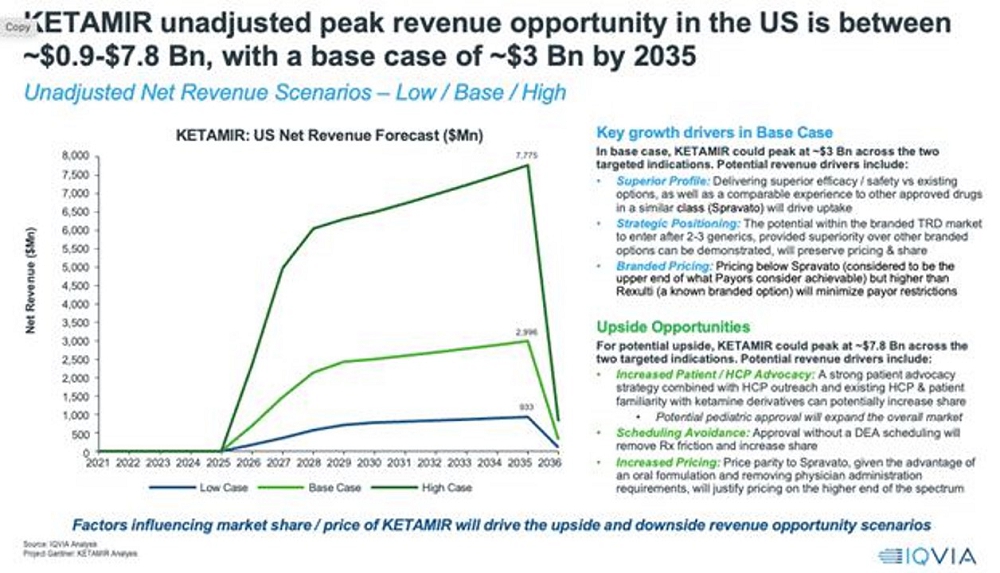
Figure: Actual valuation of Ketamir-2 over time, including base and peak revenue opportunities in the US.
These estimates are based on several key assumptions, including the market share Ketamir-2 might achieve, the years to peak sales, gross price per dose, and the Probability of Technical & Regulatory Success (PTRS). Feedback from key opinion leaders (KOLs) and payors suggests that there is a significant unmet need in behavioral health, particularly for treatments like Ketamir-2 with fewer adverse effects and more consistent outcomes. However, issues such as pricing, insurance coverage, and potential DEA scheduling are important considerations that could affect Ketamir-2’s market penetration.
As such, we believe Ketamir-2 presents a significant market opportunity in the treatment of TRD and MDSI, with the potential in the PTSD market, offering a novel approach with potential advantages over existing therapies in terms of efficacy, safety, and patient compliance. The financial outlook is positive, contingent upon successful market penetration and realization of its therapeutic potential.
MIRA-55
MIRA-55, if approved, will compete in three key overlapping growth markets: the anxiety, cognitive decline (CNS/dementia), and neuropathic pain markets where multiple products with varying safety and efficacy profiles are already on the market. MIRA-55 competes at the intersection of these three markets given the target patient profile for MIRA-55.
| 13 |
MIRA-55 will compete primarily within the CNS market that encapsulates anxiety, dementia, other pain, Alzheimer’s, migraines and related conditions. Based on the market size of the CNS opportunity as set forth in IQVIA’s Global Use of Medicines 2023 analysis (the “IQVIA Report”), we estimate that by 2027, the U.S. CNS market will be worth $48 billion, growing between two and five percent during the period from 2023 to 2027. Within that market opportunity, anxiety is worth between approximately $10 billion and $15 billion in annual sales. If approved by the FDA, MIRA-55 may potentially provide therapeutic effects for anxiety, dementia and pain.
Anxiety and pain are expected to grow approximately five percent over the same period according to the IQVIA Report, while Alzheimer’s is expected to grow approximately twelve percent. This is critical given MIRA-55’s focus on early-stage patients with dementia, as according to the Alzheimer’s Association 2023 Alzheimer’s Disease Facts and Figures analysis (the “Alzheimer Association”), 500,000 new Alzheimer cases emerge in the U.S. each year. According to the Alzheimer Association, about 60 to 80 percent of Alzheimer cases evolve into dementia. Thus, Alzheimer case directions are an important signal and gateway for MIRA-55-related opportunities in dementia. Based on that epidemiology, the U.S. Center for Disease Control (“CDC”) estimates that approximately 5.8 million Americans are living with Alzheimer’s, with that number expected to grow to 14 million by 2060 (“CDC Alzheimer”).
MIRA-55’s other key market will be the neuropathic pain market. Developing targeted and efficient therapies for neuropathic pain stands as a priority to address this common source of suffering and morbidity. Innovative strategies are under exploration to tackle the distinctive challenges posed by this type of pain. According to the International Association for The Study of Pain, neuropathic pain affects approximately 7-10% of the world’s population. Examples include diabetic peripheral neuropathy, postherpetic neuralgia, and multiple sclerosis related neuropathy.
Our initial focus will be a dual path: potentially winning in traditional markets as well as the marijuana analog markets using a safe, effective and, if determined by the FDA, an FDA-approved treatment option since safety and efficacy determinations are in the exclusive purview of the FDA. According to Grandview Research, today, legal medical marijuana is a $11.6 billion industry whereas legal recreational marijuana is a $26.9 billion industry. Both are sub-sets of the traditional pain and anxiety markets. However, in many patient populations, non-U.S. legal, and cultural settings, marijuana may not be the first or a viable option for treatment of neurological disorders. As a result, these patients will typically use non-steroidal anti-inflammatory drugs (NSAIDs) or various mood management drugs, opening them up to a range of non-ideal outcomes. The objective of MIRA-55 is to offer physicians and patients an approved, viable synthetic option. Thus, if approved by the FDA, we believe that MIRA-55 may potentially provide a preferred alternative in such patient populations, as it is not derived from the marijuana plant.
MIRA-55 is being developed as the first manufactured prescription drug to potentially target the CB1 and CB2 receptors for neuropathic pain and anxiety without the impurities of marijuana or its side effects, such as increased appetite and paranoia. MIRA-55 has demonstrated the ability to rapidly and significantly improve cognitive performance with acute use—i.e. doubling cognitive performance after a single dose in normal mice MIRA-55 is a novel synthetic cannabinoid analog directed at potentially treating patients with dementia associated cognitive decline and anxiety diagnoses. Unlike other cannabinoids in the market, MIRA-55 is not derived from plants. Plants generate alkaloids as a defense mechanism, and it has been speculated that plant-derived cannabinoids have adverse side effects in humans.
Furthermore, in animal studies conducted by us, MIRA-55 has preliminarily demonstrated more than 30-fold increased CB2 activation compared to CBD.
| 14 |
Our Strategy
Ketamir-2
The goal is to continue develop Ketamir-2 as an orally administered medication with potentially fewer side effects, free from the restrictions such as those imposed by ketamine’s REMS, to fill the current clinical need for a rapid acting antidepressant to manage TRD and MDSI in patients who are able to take Ketamir-2 at home. The strategic plan for Ketamir-2’s development encompasses several critical stages, from scaling up manufacturing to exploring effective exit strategies.
| ● | Scaling Up Manufacturing at Recipharm. The first step for us is to scale up the manufacturing process of Ketamir-2. Small-scale synthesis has already been achieved at third-party vendor Recipharm, which will be essential for refining the process and identifying potential challenges. This will be followed by a phase of process optimization, focusing on improving yield, purity, and cost-efficiency. We will then transition to pilot-scale production to validate the manufacturing process under real-world conditions. Once the process is established, large-scale manufacturing can commence, ensuring compliance with Good Manufacturing Practices (GMP) standards. Integral to this stage is the development of a robust supply chain strategy to manage the consistent availability of raw materials and distribution. | |
| ● | IND-Enabling Research. Prior to IND submission, we must conduct comprehensive IND-enabling research, which we will undertake through third-party vendors. This includes pharmacokinetics/pharmacodynamics (PK/PD) studies to understand how Ketamir-2 is absorbed, distributed, metabolized, and excreted, along with its mechanism of action. Toxicology and tolerability studies are also crucial, encompassing both acute and chronic toxicology assessments in relevant animal models, including 7 and 28-day studies in rats and dogs. Additionally, the development of a stable and effective formulation for Ketamir-2’s oral administration is necessary. | |
| ● | IND Submission for TRD Indication. With the data from preclinical studies in hand, we will prepare and submit an IND to the FDA. This submission will include all preclinical data and a proposed plan for clinical trials. A well-thought-out regulatory strategy is essential to address potential queries and concerns from regulatory bodies. | |
| ● | Clinical Trials - Phase 1 and 2. Upon IND acceptance, clinical development will proceed with Phase 1 trials, focusing on assessing the safety, tolerability, and optimal dosing in a small group of healthy volunteers. This will be followed by Phase 2 trials, where the efficacy of Ketamir-2 will be evaluated in a larger group of patients, along with further safety assessments. | |
| ● | Potential Collaborations. We could focus on partnerships and licensing for later clinical development and commercialization of Ketamir-2. We can explore partnerships with larger pharmaceutical companies for further development and commercialization of Ketamir-2. Licensing agreements can also be considered, allowing other companies to market Ketamir-2 in different regions or for varied indications. While such relationship could offload a significant amount of the financial and operational burden associated with these activities, such a relationship would likely lead to loss of autonomy and come with high expectations for returns. The following options could be considered depending on the available opportunities: |
| 1. | Strategic Partnerships and Collaborations: this can involve partnering with larger pharmaceutical companies, which brings the benefit of their extensive resources, global market reach, and regulatory expertise. However, such partnerships often mean sharing profits and relinquishing some control over the drug. Collaborations with biotechnology firms in similar therapeutic areas can also be beneficial, offering synergistic research efforts and niche expertise, though these firms may not provide as much financial support as larger pharma companies. | |
| 2. | Licensing Agreements. We could choose to license the drug to another company for further development and commercialization. Out-licensing can provide an immediate capital infusion and reduce the risk and investment required for later-stage trials. However, this often leads to losing direct control over the development and commercialization processes. Co-development and co-marketing deals are another form of licensing where the development, marketing, and commercialization responsibilities are shared, which can combine strengths and reduce individual risks but requires aligned objectives and effective collaboration. |
| ● | Additional Funding and Investment. Seeking additional capital through debt or equity financing could be a way to fund Phase 3 trials and marketing efforts but could dilute shareholders’ equity. Undertaking Phase 3 clinical trials and, if approved, marketing activities would require substantial investments and expertise which we currently do not have and would need to develop. |
| 15 |
Additionally, the potential for an acquisition from a larger pharmaceutical company remains a viable exit strategy, especially if Ketamir-2 demonstrates substantial promise.
Throughout this process, it is crucial for us to maintain a robust intellectual property strategy, regularly assess the antidepressant market landscape, especially for TRD, and engage with key stakeholders. Implementing a risk management plan is also essential to navigate potential development and commercialization challenges. This strategic plan must be adaptable, capable of responding to new data, regulatory feedback, and changes in the market. Regular assessments and checkpoints will ensure the project aligns with our strategic goals and the evolving landscape of pharmaceutical development.
MIRA-55
Our goal is to develop therapeutics targeting well-characterized CB1 and CB2 receptors with optimized pharmacological properties to transform the lives of patients with neurological diseases. Key elements of our strategy to achieve this goal include:
| ● | Advance our MIRA-55 through clinical development and approval. Our product candidate, MIRA-55, is in pre-clinical studies. Existing treatment options for neuropsychiatric disorders and neurological diseases have significant limitations, and, if approved, we believe MIRA-55 would represent a major therapeutic advancement for patients. | |
| ● | Continue pre-clinical development of MIRA-55 across a range of CNS diseases associated with neurodegeneration and progress into clinical development. MIRA-55 is currently in IND-enabling studies for neurobehavioral disorders such as dementia, PTSD, neuropathic pain, as well as neurodegenerative diseases. We believe MIRA-55 may have potential in several diseases associated with neuroinflammation. | |
| ● | Identify additional product candidates and expand current candidates into additional neurological diseases. We see potential for our current product candidate to be evaluated in clinical trials outside of its initial indications and will evaluate additional indications to maximize the potential of our drug development program. Our current product focus is on targets that are well characterized in neurological diseases but for which there are limitations with currently available therapies. We also plan to continue to identify and develop additional novel product candidates that align with our focus. | |
| ● | Explore strategic collaborations to maximize the value of our product candidates. We plan to explore collaborations opportunistically to maximize the value of our product candidates. We intend to retain significant economic and commercial rights to our programs in key geographic areas that are core to our long-term strategy. |
Competition
Ketamir-2
The principle competitor of Ketamir-2 is ketamine or ketamine analogs. Ketamine, originally known as a dissociative anesthetic, has emerged as a significant breakthrough in the treatment of depression, particularly due to its rapid-acting antidepressant properties. The FDA approved in 2019 esketamine delivered intranasal, developed by Janssen with the brand name Spravato. This has opened new avenues in psychiatric treatment, especially for patients who do not respond to traditional antidepressants, have depression with suicidal ideation, or require rapid antidepressant responses.
In contrast to most novel antidepressants, which are multi-billion dollar drugs annually, for 2023 Janssen reported $683 million in revenue from Spravato. We believe the primary reason for Spravato’s revenue performance versus other antidepressants is because Spravato’s REMS requires Spravato to be patient administered but clinician observed for 2 hours, with the patient unable to drive for the rest of the day. As described further below, we believe this presents challenges for both patients and clinicians, which has restricted the use of this form of ketamine from patients who would benefit from this treatment (e.g. those with TRD and MDSI). Ketamir-2, if ultimately FDA approved without the requirement of a REMS, could potentially avoid these challenges.
| 16 |
Niche Filled by ketamine
| 1. | Treatment-Resistant Depression: Ketamine has shown efficacy in cases where conventional antidepressants fail, addressing a significant gap in mental health treatment. | |
| 2. | Rapid Onset of Action: Unlike traditional antidepressants that may take weeks to show effects, ketamine can produce noticeable antidepressant effects within hours to days, providing immediate relief in acute cases of depression. | |
| 3. | Suicidality: It has shown promise in rapidly reducing suicidal thoughts, which is crucial in acute psychiatric emergencies. |
Limitations of ketamine Due to Side Effects
| 1. | Psychotomimetic Effects: Ketamine can induce dissociative symptoms, hallucinations, and other psychotomimetic effects, limiting its use to controlled settings. | |
| 2. | Potential for Abuse: Given its history as a recreational drug, there are concerns about its potential for abuse and addiction. | |
| 3. | Short Duration of Effect: The antidepressant effect of ketamine can be transient, requiring repeated administrations, which may increase the risk of side effects. | |
| 4. | Physical Side Effects: These may include increased heart rate, elevated blood pressure, nausea, and dizziness. |
Requirements of ketamine under the REMS (Risk Evaluation and Mitigation Strategy)
The use of ketamine, especially Esketamine (a nasal spray form of ketamine approved for treatment-resistant depression), is regulated under the Risk Evaluation and Mitigation Strategy (REMS) program to ensure safe use:
| 1. | Healthcare Setting Administration: Esketamine must be administered in a certified healthcare setting under the supervision of a healthcare provider. | |
| 2. | Patient Monitoring: Patients must be monitored for at least two hours after administration due to the risk of sedation and dissociation. | |
| 3. | Restricted Distribution: The drug is not available for take-home use and can only be dispensed to healthcare facilities and pharmacies enrolled in the REMS program. | |
| 4. | Patient Education and Consent: Patients must be informed about the risks and provide written consent. | |
| 5. | Follow-up and Reporting: Healthcare providers are required to report any serious adverse effects and ensure follow-up to monitor the patient’s response to treatment. |
Ketamine’s possible role as a rapid-acting antidepressant could fill a crucial niche in the management of treatment-resistant depression and acute suicidality. However, its potential use is tempered by significant side effects and the stringent requirements of the REMS program, which necessitate careful patient selection and monitoring to optimize safety and efficacy.
| 17 |
The finding of up to 80% oral bioavailability with the potential for decreased abuse liability (e.g. because of the lack of opiate agonist activity) and potentially decreased side effects (e.g. fewer dissociative experiences and less hypertension) puts Ketamir-2 in a situation to potentially offer the same antidepressant effects but with fewer restrictions, perhaps even permitting patients to take it orally at home.
MIRA-55
We are subject to competition from pharmaceutical and biotechnology companies and academic and research institutions. We believe our future success will depend, in large part, on our ability to maintain a first mover advantage and competitive lead in our industry.
Competition arises mainly from two sources, traditional cell-based in vitro culture approaches and traditional in vivo animal models and testing. We also face future competition from companies developing cannabinoid therapies, as summarized in the table below:

Sativex (delta-9-tetrahydrocannibinol and cannabidiol in the EU) is an oromucosal spray indicated as treatment for symptom improvement in adult patients with moderate to severe spasticity due to multiple sclerosis (MS) who have not responded adequately to other anti-spasticity medication and who demonstrate clinically significant improvement in spasticity related symptoms during an initial trial of therapy. Sativex is not assigned a schedule in the U.S. by the DEA as it is not approved but is a Class B controlled drug under the Misuse of Drugs Act 1971 and is placed in Schedule 4 to the Misuse of Drug Regulations 2001 in the United Kingdom.
Marinol (dronabinol) is an oral cannabinoid indicated in adults for the treatment of: Anorexia associated with weight loss in patients with AIDS and nausea and vomiting associated with cancer chemotherapy in patients who have failed to respond adequately to conventional antiemetic treatments. Marinol is a Schedule III controlled substance.
Cesamet (Nabilone) is a synthetic cannabinoid for oral administration that are indicated for the treatment of the nausea and vomiting associated with cancer chemotherapy in patients who have failed to respond adequately to conventional antiemetic treatments. Cesamet contains nabilone, which is a controlled in Schedule II of the Controlled Substances Act (CSA).
| 18 |
Research and Testing to Date
Ketamir-2
Preclinical Research Findings
In Silico Analysis of Targets of Ketamir-2 vs ketamine
In silico analysis, referring to computer-based techniques, has become an integral part of pharmaceutical research and development.7 This approach utilizes computational methods to analyze and predict the properties and behaviors of pharmaceutical compounds. The use of in silico analysis is especially crucial in the early stages of drug development, as it aids in identifying potential drug targets and elucidating differences between a new drug and its parent compound. By analyzing large datasets, such as genomic, proteomic, and metabolomic data, researchers can predict how different compounds might interact with various biological targets. This approach helps in understanding the mechanism of action of new drugs and can significantly reduce the time and cost associated with experimental screening. InSilico Trials was contracted to provide a comparison between targets of Ketamir-2 vs ketamine employing their target identification protocol. The following characterize some of the unique targets that are predicted to interact with either Ketamir-2 or ketamine, thereby differentiating one drug from the next.
Ketamir-2 selective target:
BRD4, or Bromodomain-containing protein 4, is a member of the bromodomain and extra-terminal (BET) family of proteins and has been implicated in the regulation of gene expression, particularly those involved in cell cycle progression and inflammatory responses.8 In the context of depression, research has started to explore the role of BRD4 and its potential impact.
| 1. | BRD4 and Neuroinflammation: Inflammation is increasingly recognized as a significant factor in the pathophysiology of depression. BRD4 has been found to regulate the expression of inflammatory genes. Its inhibition, therefore, might reduce neuroinflammation, which is thought to contribute to depressive symptoms. | |
| 2. | Gene Expression Regulation: BRD4 influences the transcription of genes involved in mood regulation and stress response. Dysregulation of these genes can contribute to the development of depression.9 | |
| 3. | Pharmacological Target: BRD4 is a target for new pharmacological interventions in depression. Inhibitors of BRD4, such as JQ1, have shown promise in preclinical studies for their antidepressant effects. These compounds can modulate the expression of genes associated with mood and stress response. | |
| 4. | Epigenetic Mechanisms: As an epigenetic regulator, BRD4’s role in modifying the expression of genes without changing the DNA sequence might be crucial in understanding the long-term impact of environmental factors on depression.10 | |
| 5. | Animal Studies: Research in animal models has provided some evidence that modulation of BRD4 activity can influence behaviors related to depression. However, translating these findings to human depression is complex and requires more research. | |
| 6. | Thus, while BRD4 is not traditionally associated with depression like neurotransmitter systems (e.g., serotonin or dopamine), emerging evidence suggests that it plays a role in the disease’s pathophysiology. Its involvement in regulating gene expression, particularly related to inflammation and stress response, positions it as a potential target for novel antidepressant therapies. |
| 19 |
Ketamine selective targets:
Alpha-2a adrenergic receptor: Alpha-2a adrenergic receptors are G protein-coupled receptors (GPCRs) involved in the modulation of neurotransmitter release. They are generally thought to be inhibitory, reducing the release of norepinephrine when activated, which can lead to various physiological effects.
| ● | Cardiovascular Effects: Alpha-2a receptors play a role in cardiovascular regulation, which might explain some of the blood pressure and heart rate changes seen with ketamine. | |
| ● | Sedation: Activation of these receptors can lead to sedative effects, which is consistent with the tranquilizing effects that ketamine can produce. |
Sigma Opioid Receptor: Ketamine is known for its dissociative anesthetic properties, which are primarily attributed to its antagonism of the N-methyl-D-aspartate (NMDA) receptor. However, the sigma receptors, particularly the sigma-1 receptor, have also been implicated in the psychotomimetic and dissociative effects of ketamine. Here’s how ketamine’s interaction with sigma opioid receptors might contribute to its dissociative side effects:
| ● | Cognitive and Perceptual Processes: Activation of has been linked to modulating cognitive and perceptual processes, which could be associated with the dissociative effects experienced during ketamine administration. | |
| ● | Modulation of NMDA Receptor Activity: Sigma-1 receptors are known to interact with NMDA receptors, and this interaction might enhance or modulate the dissociative effects of ketamine, which primarily acts as an NMDA receptor antagonist. |
Mu-Opioid Receptor: The Mu-opioid receptor (MOR) is one of the principal targets within the central nervous system for endogenous opioids like endorphins and enkephalins, as well as for exogenous opioid analgesics such as morphine and fentanyl. Activation of MOR typically results in analgesic effects, reduced gastrointestinal motility, respiratory depression, and can influence the reward system in the brain, which is associated with the pleasurable sensations or euphoria. Activation of the MOR by ketamine could contribute to side effects related to its abuse liability:
| ● | Euphoria and Reward: MOR activation is heavily implicated in the reward pathway and can produce euphoria. This effect is a key driver of the abuse potential of opioids. | |
| ● | Tolerance and Dependence: Chronic activation of the MOR leads to tolerance (the need for increasing doses to achieve the same effect) and physical dependence, contributing to the cycle of abuse. | |
| ● | Sedation: MOR activation can also result in sedation, which might contribute to the overall sedative effects of ketamine, particularly at higher doses. |
| ○ | Ketamir-2 is a newly synthesized compound analogous to ketamine. In a virtual screen, aimed at identifying potential interaction sites, no opioid receptor binding was found for Ketamir. Testing its potential agonist or antagonist activities on mu-opioid receptors, it was found that Ketamir has no mu-opioid antagonist activity. It was found that Ketamir has some minimal agonist activity, but at high concentrations, which are outside its therapeutic range. In this activity, it is several folds lower than ketamine, which is believed to exert its dependency function through this activity. | |
| ○ | This much lower activity of Ketamir-2 MOP agonist activity suggests that Ketamir-2 may have less addictive properties, thus potentially improving its safety profile. |
Bioavailability:
The Caco-2 cell model, originating from a human colorectal adenocarcinoma cell line, plays a significant role in pharmaceutical research for estimating the intestinal absorption and indirectly the bioavailability of drugs. Bioavailability, the proportion of a drug that enters the systemic circulation when introduced into the body, is crucial for determining a drug’s effectiveness. Traditionally, bioavailability is determined through in vivo studies, including human and animal trials, as well as in vitro models like the Caco-2 cell model and in silico computational approaches.
The Caco-2 model involves culturing cells that differentiate into a monolayer mimicking the intestinal epithelium, complete with tight junctions and microvilli. This model is pivotal in permeability studies to assess how well drugs can pass through the intestinal barrier and in understanding both active and passive drug transport mechanisms. While primarily used for estimating drug absorption, the Caco-2 model also serves to predict potential drug-drug interactions within the gastrointestinal system.
| 20 |
The Caco-2 model offers a high-throughput, cost-effective, and human-relevant system, making it a preferred choice for initial screening of multiple compounds. In pharmaceutical research, the Caco-2 model often serves as an initial study to predict the absorption properties of new drugs and is typically validated against clinical data once that becomes available. It plays a crucial role in the early stages of drug development, influencing decisions on which compounds to advance.
CaCO-2 cells are human epithelial colorectal adenocarcinoma cells that are widely used as an in vitro model of the intestinal barrier. The CaCO-2 assay is employed to study the absorption and transport of orally administered drugs across the intestinal epithelium. The assay evaluates the permeability of a drug from the apical (AP) side, representative of the intestinal lumen, to the basolateral (BL) side, representative of the blood side, and vice versa.
The bidirectional transport assays conducted with CaCO-2 cells can provide the following insights about two different drugs:
| 1. | Absorption Potential: The AP to BL (A→B) transport rate can indicate a drug’s ability to be absorbed through the intestines into systemic circulation. Higher transport rates suggest better absorption potential. |
| 2. | Efflux Ratio: By comparing the BL to AP (B→A) transport rate with the A→B transport rate, one can determine the efflux ratio. If the efflux ratio is significantly greater than 1, this implies that there are active efflux mechanisms, such as P-glycoprotein, that are pumping the drug back into the intestinal lumen, thus reducing its absorption. |
| 3. | Permeability Classification: The transport rates can be used to classify the drugs according to their permeability. High permeability drugs are absorbed more completely and are likely to have a more reliable and faster onset of action. |
| 4. | Influence of Efflux and Influx Transporters: Differences in the AB-BA values between two drugs can indicate the involvement of different efflux or influx transporters, suggesting that the drugs have different affinities for these transporters. |
| 5. | Impact of Metabolism: If a drug is extensively metabolized by the intestinal wall before reaching systemic circulation, this will be reflected in a low A→B permeability. |
| 6. | Predicting Oral Bioavailability: Generally, drugs that exhibit high permeability in CaCO-2 assays are expected to have good oral bioavailability, although this is not always the case due to other factors such as solubility and first-pass metabolism. |
In summary, the CaCO-2 intestinal absorption (AB-BA) assay is a valuable tool for predicting the intestinal absorption and oral bioavailability of drugs. Differences in the assay results between two drugs can provide important information about their absorption characteristics, potential interactions with transporters, overall oral bioavailability, and possible drug-drug interactions.
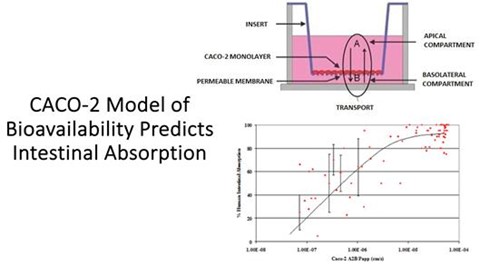
Figure: Model of the CaCO-2 model of drug intestinal absorption and how well it correlates with actual measures of human intestinal absorption.
| 21 |
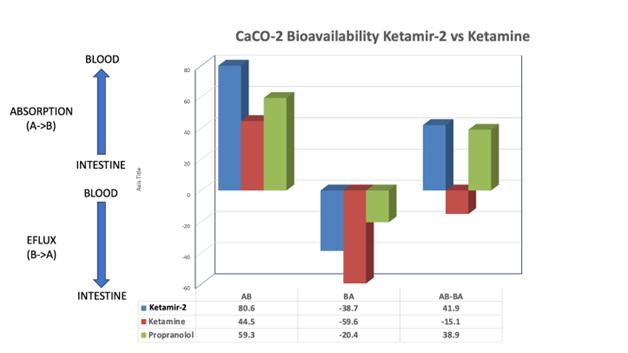
Figure: Data obtained from the CaCO-2 model of intestinal absorption. Propranolol, a commonly prescribed beta-blocker that is taken orally and used to treat hypertension, is included as a positive control. The intestinal absorption (AB), Intestinal efflux (BA) and net absorption (AB-BA) are shown.
As can be seen in in the figure above, the absorption from the intestinal lumen into the blood that is 80% greater (80.6 vs 44.5), the rate of efflux back into the intestinal lumen that is 35% less (-38.7 vs -59.6), and the net absorption (AB-BA) rate is 3.77 fold greater [(41.9+15.1)/15.1=3.77], respectively. Since the reported oral bioavailability of ketamine has been reported to be between 16-30% (average of 23%), then the predicted oral bioavailability of Ketamir-2 could be as high as 87% (i.e. Ketamir-2’s oral bioavailability is 3.77 fold greater than ketamine’s = 23%*3.77=87%).
This is just an approximation, and when sufficient Ketamir-2 has been synthesized to do in vivo animal initially and then human Pharmacokinetic (PK) studies, it will be possible to get a more precise estimate of Ketamir-2’s Oral Bioavailability compared to ketamine by testing and calculating the area under the concentration-time curve (AUCoral) for oral dosing divided by the AUC for IV dosing (i.e. AUCoral/AUCiv). But based on the available preliminary estimates, it appears highly likely that the oral bioavailability of Ketamir-2 in humans is going to be substantially larger than that of ketamine. Orally available Ketamir-2, as opposed to IV or IN ketamine, would be much easier to be patient self-delivered at home, thereby improving on the ease and availability of this rapid acting antidepressant for TRD & MDSI.
MIRA-55
Eurofins DiscoverX has developed a panel of cell lines stably expressing non-tagged GPCRs that signal through cyclic adenosine monophosphate, a messenger used for intracellular signal transduction in many different organisms (or cAMP). Hit Hunter® cAMP assays are specialized tests that track the activation of a type of cell receptor known as GPCR. GPCRs play a crucial role in how cells respond to external signals, and they are activated through two pathways: Gi and Gs secondary messenger signaling. These pathways are like internal communication systems in cells that relay signals from the outside to trigger specific responses inside the cell. The assay is conducted in a straightforward, uniform manner without the need for image-based analysis. This method uses a technology developed by DiscoverX called Enzyme Fragment Complementation (or EFC). In EFC, fragments of an enzyme, specifically β-galactosidase (β-Gal), are brought together to become functional only when the GPCR is activated. β-Galactosidase, the enzyme used as a functional reporter in this assay, is typically inactive in fragmented form and becomes active when the fragments reassemble, indicating the activation of the GPCR. In this case, the GPCR target was CB1 receptor. Compounds were tested in agonist and antagonist mode with the requested GPCR Biosensor Assays. For agonist assays, data was normalized to the maximal and minimal response observed in the presence of control ligand and vehicle. This Eurofins DiscoverX system was used to test THC vs MIRA-55 agonist activity at the CB1 receptor.
| 22 |
Unlike CB1 receptors that mediate many of the psychotropic effects of cannabinoids on the CNS, CB2 receptors are predominantly present on cells of the immune system. Based on preliminary results of our GPCR biosensor assays, the CB2 receptor agonistic effects of MIRA-55 are 8-fold more potent than THC and 30-fold more potent than CBD.
The study regarding the ability of MIRA-55 vs THC vs CBD to activate CB2Receptors and alter intracellular cAMP levels was performed by the CRO Eurofins DiscoverX.
As can be seen in the table below, the EC50 (i.e. concentration required to induce a half maximal response) for MIRA-55 was 8 times more potent than THC and at least 30 times more potent that CBD—i.e. it only took 1 uM of MIRA-55 to induce the same response that required 8 uM of THC and >30 uM of CBD.
| Compound Name | Assay Name | Assay Format | Assay Target | Result Type | EC50 | Unit | ||||||
| MIRA-55 | cAMP | Agonist | CNR2/CB2 | EC50 | 1.008462 | uM | ||||||
| THC | cAMP | Agonist | CNR2/CB2 | EC50 | 8.209884 | uM | ||||||
| CBD | cAMP | Agonist | CNR2/CB2 | EC50 | >30 | uM |
Figure: The foregoing measurements were performed as follows:
DiscoverX has developed a panel of cell lines that stably express non-tagged GPCRs (G-protein coupled receptors) capable of signaling through cAMP. The Hit Hunter® assay platform is used to investigate the functionality and response of these GPCRs.
In the case of the CB2 receptor, which is a GPCR involved in various physiological processes and has potential therapeutic implications, the Hit Hunter® assay can be employed to study the effects of drug agonists on CB2 receptor activity.
To measure the half maximal response (EC50) of CB2 receptor activation by a drug agonist that leads to a decrease in cAMP levels, an alternative approach may be required. One common method involves using forskolin, an activator of adenylate cyclase, to stimulate cAMP production. Forskolin bypasses the GPCR signaling and directly activates adenylate cyclase, resulting in increased cAMP levels.
In the presence of forskolin, the drug agonist at the CB2 receptor can then be tested at various concentrations to determine its ability to inhibit the forskolin-induced cAMP production. The drug’s concentration that leads to a 50% reduction in forskolin-stimulated cAMP levels can be considered the half maximal response or EC50.
Completed Pre-Clinical Tests*
| ● | EPM model of anxiety | |
| ● | Thermal Sensitivity Model of Pain | |
| ● | Context Fear Conditioning Model of Cognition—Test of learning and memory. | |
| ● | Rat Psychomotor Vigilance Test (“PVT”) of Cognition—Test of attention. |
* These were non-human studies that were not powered for statistical significance and as such, no p-values are available.
| ● | EPM Model of Anxiety Test: |
| ● | Method: We studied the effect of acute administration of MIRA-55 on anxiety-related phenotypes in mice to model human conditions. |
| ■ | An intraperitoneal (i.p.) injection of Placebo (e.g. saline) or MIRA-55 (e.g. 50mg/kg = Treatment) was administered to C57Bl/6 mice (n=5/group) that were 8-12 weeks old | |
| ■ | 30 minutes following injection, mice were tested in anxiety related measures using EPM |
| 23 |
| ● | Outcome: The following chart demonstrates MIRA-55’s anti-anxiety effects: |
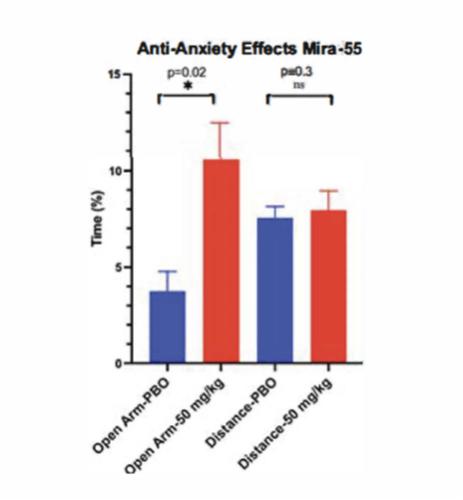
Figure: Effects of MIRA-55 vs Placebo Treatment on Mouse Behavior in the Elevated Plus Maze.
| 24 |
EPM is a widely used behavioral test to assess anxiety-like behavior in rodents. Typically, rodents tend to avoid open spaces due to their natural aversion to potentially dangerous areas. Therefore, spending more time in the open arms of the maze indicates decreased anxiety-like behavior. Similarly, the total distance travelled can reflect general locomotor activity and exploratory behavior, which can be influenced by the state of anxiety and the effect of drugs.
The EPM apparatus consists of two open arms and two enclosed arms elevated above the floor. Blue Bars represent the percentage of time spent in the open arms by mice in the placebo and drug-treated groups. Green Bars show the total distance travelled by mice in both groups during the EPM test.
| ● | Thermal Sensitivity Model of Pain: |
| ● | Method: We studied the potential for pain reduction in pre-clinical models of heat tolerance using a hot plate methodology. | |
| ● | Outcome: MIRA-55 provided significantly delayed thermal sensitivity and enhanced pain tolerance. |
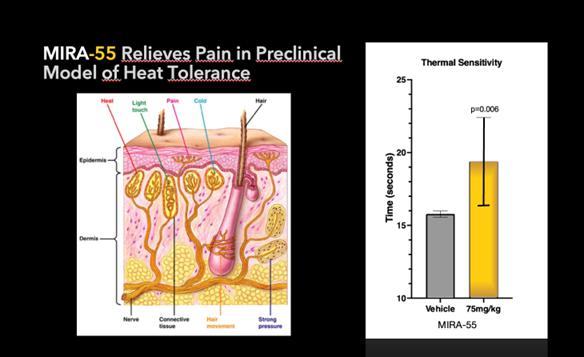
Figure: In this thermal sensitivity test, mice are placed on a heated metal plate (e.g. 52-55 degrees Celsius). The time taken for the mouse to show a pain response - licking or shaking of the paws, jumping, or trying to escape from the hot plate - is measured. This time interval is known as the “hot-plate latency”. A longer latency is indicative of reduced pain sensation or a higher pain tolerance.
The Thermal Sensitivity Model of Pain in mice is a widely used experimental approach to study nociception, which is the perception of pain. In this model, thermal stimuli are applied to the hind paws of mice to assess their sensitivity to heat-induced pain. The procedure typically involves placing the mouse on a temperature-controlled surface, such as a hot plate or a radiant heat source. The temperature is gradually increased, and the response of the mouse is measured, such as the latency to withdraw its paw from the heat source. The withdrawal latency is considered an indicator of pain sensitivity, with shorter latencies indicating greater sensitivity. By comparing the response of normal mice to that of mice with altered pain sensitivity, such as genetically modified mice or mice treated with analgesic drugs, researchers can gain insights into the mechanisms underlying pain perception and potential therapeutic interventions. The Thermal Sensitivity Model of Pain in mice provides a controlled and reproducible method for studying thermal nociception, allowing researchers to investigate the effects of various genetic, pharmacological, and environmental factors on pain sensitivity. This model has contributed significantly to our understanding of pain pathways and the development of novel analgesic treatments.
As performed at Johns Hopkins, in our thermal sensitivity test, which measured sensitivity to thermal pain, MIRA-55 significantly increased the time it took mice to lift their legs in comparison to placebo (p=0.006) at 75mg/kg. This indicates that MIRA-55 has an analgesic effect and may be a potential treatment for pain. Each group (i.e. placebo and 75 mg/kg) was comprised of 9 mice, for a total of 18 mice.
| 25 |
The issue of how to test the effect of MIRA-55 on cognition was complicated by the following:1) MIRA-55 has anti-anxiety (i.e. anxiolytic) effects, 2) anxiolytics can potentially improve cognitive assessment outcomes by reducing anxiety levels that may otherwise hinder cognitive functioning. Thus, in commonly performed tests of cognition in mice, such as novel object recognition and Morris water maze, anxiolytic medications can indirectly result in improved performance by decreasing anxiety rather than by directly improving cognition. In order to separate assessments of the impact of MIRA-55 on cognitive performance from its demonstrated anti-anxiety effects, we employed a model of context fear conditioning wherein we dosed the mice after training. Context fear conditioning in mice is a behavioral paradigm used to measure cognitive processes related to associative learning and memory. Associative learning, where an individual learns to associate specific stimuli or contexts with particular outcomes, in this case the mice associate being in a specific chamber with receiving a mild foot shock that occurs during training the day before testing. This process of forming associations between stimuli, actions, and consequences is involved in numerous skills and behaviors in everyday life: it underlies learning new skills, developing habits, and acquiring knowledge through experiences and conditioning. The use of associating the chamber with the foot shock on day one, means that when the mice are returned to the chamber on day 2 a measure of how much freezing they do corresponds to a read out of how well they can recall the experiences they had during training on day 1 (i.e. the greater the freezing, the better the recollection of the association between the chamber and food shock). Since the mice are given MIRA-55 AFTER training that takes place on day 1, and only before testing on day 2, there is no concern about the anxiolytic effects of MIRA-55 on learning during training, but rather this model tests MIRA-55’s effects on performance only—which in this case represents memory (i.e. the ability to recognize and recall the chamber where they had previously been shocked) and to translate that into an associated behavior (i.e. freezing). As published in the Journal of Neuropharmacology in 2023, THC and cannabis impair context fear conditioning, both when given prior to training (because of its anti-anxiety effects) and when given prior to testing (because of its cognitive impairing effects). As demonstrated in the figure below, MIRA-55 resulted a dramatic effect on cognitive performance in the context fear conditioning model: as shown in B, the second panel from the left, the percentage of time spent freezing—that is a demonstration of their memory and association—in the mice who received MIRA-55 at a dose of 75 mg/kg was more than twice that of those who received 0 mg/kg=placebo (i.e. 55% vs 25%, p<0.0001). Thus, MIRA-55 doubled the cognitive performance of the mice compared to placebo. This degree of improvement in cognitive performance in healthy mice dosed just prior to testing and after learning has not been demonstrated with any cannabinoid compound previously.
| ● | Trace Fear Conditioning Model of Cognition: |
| ● | Method: We studied the potential for improving recall in healthy mice using a fear conditioning model. | |
| ● | Outcome: MIRA-55 sharply improves cognitive recall as dosage rises. |
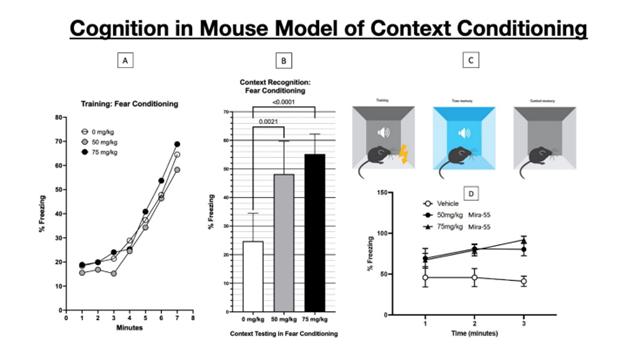
Figure: The Contextual Fear Conditioning Model of Cognition in mice is an experimental paradigm used to study associative learning and memory processes. It focuses on the ability of mice to form an association between a specific environmental context and an aversive stimulus, which leads to the acquisition and subsequent retrieval of contextual memories. During the acquisition phase of the model, mice are exposed to a distinct context, such as a particular chamber or environment. In this context, they receive an aversive stimulus, typically a mild foot shock. The presentation of the foot shock creates an association between the contextual cues and the aversive experience. Following the acquisition phase, the mice undergo a testing phase to assess their memory of the association between the context where they received the foot shock and the memory of the aversive stimulus. They are returned to the same context where the conditioning took place and their behavioral responses, particularly fear-related behaviors such as freezing or defensive reactions, are measured.
These behavioral responses serve as indicators of the mice’s ability to retrieve the associative memory formed during the acquisition phase. The Contextual Fear Conditioning Model of Cognition in mice has been widely used in neuroscience research to explore the mechanisms of associative learning, memory formation, and the neural circuits involved in fear-related associations. It has contributed to our understanding of how animals, including humans, learn to associate environmental cues with aversive experiences, and has implications for understanding and treating conditions related to associative learning, memory deficits, and emotional disorders.
As performed at Johns Hopkins, in the Contextual Fear Conditioning Model the data shows that during training (in the absence of any treatment) the mice learned as indicated by increased freezing over time. The following day, 30 minutes after MIRA-55 administration, the mice were tested in the context test, which showed significantly increased % freezing (p=<0.0001) in females given 50mg/kg or 75mg/kg MIRA-55. The experiments were conducted with 10 mice in each group (placebo, 50 or 75 mg/kg MIRA-55) for a total of 30 mice.
In the context conditioning figure above, mice learn to associate the neutral context (the chamber) with the aversive stimulus (the foot shock), leading to a conditioned fear response (freezing). This is indicated by ‘freezing’ behavior - a fear-related response in mice characterized by immobility except for respiratory movements.
A timeline of the experimental procedure, indicating acclimatization, training (conditioning), and testing phases is shown above. Panel A, the left-most panel, shows that on day 1 the pairing of a neutral context (the conditioning chamber shown in panel C) with an aversive stimulus (a mild foot shock). With successive foot shocks the mice show increasing amounts of freezing, since they instinctively freeze in anticipation of being shocked. Panel B, titled “Context Recognition: Fear Conditioning,” shows the percentage freezing the mice did on day 2 after receiving placebo or MIRA-55 just prior to being placed in the same chamber they had been shocked on day 1. Since mice freeze in anticipation of receiving a shock, the relative amount of freezing in those mice given 0 mg/kg (placebo) vs either 50 or 75 mg/kg MIRA-55 is a readout of (i.e. proportional to) how well the mice recalled that the chamber they were returned to was the one in which they had been shocked. As shown in panel B, the mice who received 75 mg/kg of MIRA-55 right before being placed into the chamber showed 200% of the freezing than did the mice who received placebo (55% vs 25%, respectively. Panel D, in the lower right corner of the figure, shows that at 1 min after being placed in the chamber on day 2, the mice that got vehicle (=0 mg/kg MIRA-55), relative to those that got MIRA-55, have much less freezing, and in fact have less freezing over time. The mice given MIRA-55 start off with better recognition and recall of the chamber (demonstrated as increased freezing) at 1 minute and increase the association of the chamber with the prior shocks (because they increase freezing over time).
| 26 |
Because MIRA-55 is an anxiolytic, we decided to test whether it could impair cognitive function. We therefore sought to determine if MIRA-55 could impair attention—a different aspect of cognition than memory, recall and associative learning, and one that is affected negatively by sedating compounds (e.g. THC, Cannabis, benzodiazepine, etc.) and positively by stimulants (e.g. caffeine, nicotine, amphetamine) In order to assess whether MIRA-55 affected attention as compared to THC required a different testing model—Psychomotor Vigilance Test (PVT). The rat Psychomotor Vigilance Test (rPVT) is a widely used method to measure sustained attention in rodents. In the rPVT model, rats are trained to respond to a visual stimulus by pressing a lever, with shorter reaction times indicative of better attentional performance. Mice with longer reaction times or higher variability in response times may be considered to have attention deficits or altered vigilance. Data is shown as percentage accuracy at pressing the lever within the allowed reaction time vs dose of drug used. In the figure below, it can be seen that at doses of THC that impair attention, MIRA-55 had no negative effects on attention (i.e. their accuracy at pressing a lever at the right amount of time after receiving a trained cue was not impaired at all).
| ● | Rat PVT of Cognition |
| ● | Method: We performed a PVT to evaluate simple reaction time. | |
| ● | Outcome: MIRA-55 does not impair cognition. At 3 mg/kg and 10 mg/kg MIRA-55 causes minimal impairment in rat PVT whereas THC has a clear negative effect even at these low doses. |
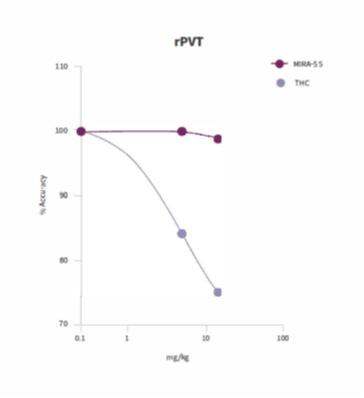
Figure: Comparison of MIRA-55 versus THC on Psychomotor Vigilance Test (PVT) Performance in Rats. The figure displays the percentage accuracy of rats in the Psychomotor Vigilance Test (PVT) following administration of MIRA-55 (blue) or THC (red). The y-axis represents the percentage accuracy (% Accuracy), indicating the proportion of correct responses in the PVT task. The x-axis represents the treatment condition, with increasing amount of compound being given to the rats before testing. The data shows that rats treated with MIRA-55 exhibited no decrease in percentage accuracy compared to the THC group (p < 0.05). The results indicate that administration of MIRA-55 had no negative impact on attention performance in the PVT task, as evidenced by the maintenance of 100% accuracy across the dosage range, compared to THC that impaired attention leading to decreased accuracy more and more with increasing dosages.
| 27 |
The Psychomotor Vigilance Test (PVT) is a behavioral test used in rats to assess attention and speed of response, providing insights into their vigilance and cognitive performance. It is based on the measurement of reaction times to visual stimuli, typically presented in a simple reaction time task paradigm.
In the PVT, rats are typically placed in an operant chamber or testing apparatus equipped with a visual stimulus, such as a light or LED. The rats are trained to perform a specific response, such as pressing a lever or nose-poking, when the visual stimulus appears. The timing of the visual stimuli is randomized to prevent predictability and maintain the animals’ attention.
During the test, the rats are required to pay attention to the visual stimuli and respond as quickly as possible when they appear. The reaction time, which represents the time it takes for the rat to initiate the response upon stimulus presentation, is recorded. This measure reflects the speed of response and can provide an indication of the rat’s attentional state and ability to sustain attention over time. By analyzing the reaction time data, researchers can evaluate the rat’s attentional performance, including measures such as mean reaction time, variability in response times, and the occurrence of lapses or errors. The PVT has been widely used to investigate the effects of different manipulations, such as pharmacological interventions that cause sedation, sleep deprivation, or experimental treatments, on attention, alertness, and cognitive performance in rats.
Therefore, the combination of cognitive assessments demonstrated the following: despite having anxiolytic effects, 1) MIRA-55 significantly improved associative learning, memory and recall in the context fear conditioning model, and 2) MIRA-55 had no negative effects on attention at doses that THC showed significant impairment. This is the first time a cannabinoid has been shown to enhance (rather than inhibit) cognition when given to normal healthy mice after training but before testing, demonstrating a specific cognitive improvement as a direct effect on the brain that is independent of indirect effects—such as with acute administration by decreasing anxiety or with long term administration by having anti-inflammatory effects in neurodegenerative diseases.
Due to further optimization of the manufacturing process, our pre-clinical work in 2024 will include the conduct of several other pre-clinical studies and initiation of a 7-day maximum tolerated dose study of MIRA-55 in rats and dogs.
| Status | Planned Activity | ||
| Drug Substance Preparation | ● ● ● |
Analytical Development and qualification NonGMP Production Refinement and optimization GLP/GMP Production Refinement | |
| Testing | ● ● ● ● ● ● ● ● ● |
Acute toxicity study mice Genotoxicity studies MTD/7D DRF Dog MTD/7D DRF Rat Dog 28-day Toxicology Rat 28-day Toxicology Cardiovascular Study Dog (Telemetry) Respiratory Study Rat hERG (Manual Patch-Clamp) Neurobehavioral Evaluation Rats Neurobehavioral Evaluation Mice |
We further plan on neurobehavioral evaluation of orally and intraperitoneally administered MIRA-55 in rats and mice, respiratory evaluation of orally administered MIRA-55 in rats, and in vitro testing for effects of MIRA-55 on hERG (the human Ether-à-go-go-Related Gene) channel currents. The hERG is an early in vitro assay required by the FDA to alert companies of any potential cardiac abnormalities by the product before proceeding with dose studies in humans. hERG is a gene that codes for a protein known as the alpha subunit of a potassium ion channel. This ion channel (sometimes simply denoted as ‘hERG’) is best known for its contribution to the electrical activity of the heart: the hERG channel mediates the repolarizing current in the cardiac action potential, which helps coordinate the heart’s beating. When this channel’s ability to conduct electrical current across the cell membrane is inhibited or compromised, either by application of drugs or by rare mutations in some individuals, it can result in a potentially fatal disorder called long QT syndrome.
| 28 |
Testing is anticipated to conclude in the first quarter of 2025. Additionally, a 28-day toxicology analysis for dogs and rats is expected to begin at the end of the fourth quarter of 2024 and continue through the first quarter of 2025.
We have started the analytical development and manufacturing of MIRA-55 as of January 2023. By the third quarter of 2024, we anticipate our suppliers will be developing MIRA-55 at scale and manufactured under GLP/cGMP conditions, expanding on earlier non-GMP volumes of MIRA-55 for use in our initial testing programs. We plan to work closely with our suppliers to generate sufficient volumes of cGMP-grade MIRA-55 materials for the planned pre-clinical toxicity programs, expanded animal testing and human trials expected to be performed in 2025, subject to FDA approval.
Regulation
The FDA and comparable regulatory authorities in state and local jurisdictions impose substantial and burdensome requirements upon companies involved in the clinical development, manufacture, marketing, and distribution of drugs. These agencies and other federal, state, and local entities regulate, among other things, the research and development, testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion, distribution, post-approval monitoring and reporting, sampling and export and import of our drug candidates.
U.S. Government Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and its implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA’s refusal to approve pending New Drug Applications (or NDAs), withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| ● | completion of pre-clinical laboratory tests, animal studies and formulation studies in compliance with the FDA’s good laboratory practice (“GLP”) regulations; | |
| ● | submission to the FDA of an IND application, which must become effective before human clinical trials may begin; |
| ● | approval by an independent Institutional Review Board (“IRB”), at each clinical site before each trial may be initiated; | |
| ● | performance of adequate and well-controlled human clinical trials in accordance with good clinical practices (“GCP”) requirements to establish the safety and efficacy of the proposed drug product for each indication; | |
| ● | demonstration that the API and finished product are manufactured under well controlled (eventually cGMP) conditions and meet all applicable standards of identity, strength, quality, and purity; | |
| ● | submission to the FDA of an NDA; | |
| ● | satisfactory completion of an FDA advisory committee review, if applicable; | |
| ● | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with cGMP requirements and to assure that the facilities, methods, and controls are adequate to preserve the drug’s identity, strength, quality, and purity; | |
| ● | FDA review and approval of the NDA, including consideration of the views of any FDA advisory committee, prior to commercial marketing or sale of the drug in the United States; and | |
| ● | compliance with any post-approval requirements, including the potential requirement to implement a Risk Evaluation and Mitigation Strategy (“REMS”) or to conduct a post-approval study. |
| 29 |
Pre-clinical studies
Before testing any drug or biological product candidate in humans, the product candidate must undergo rigorous pre-clinical testing. The pre-clinical developmental stage generally involves laboratory evaluations of drug chemistry, formulation, and stability, as well as studies to evaluate toxicity in animals, to assess the potential for adverse events (“AEs”) and, in some cases, to establish a rationale for therapeutic use. The conduct of pre-clinical studies is subject to federal regulations and requirements, including GLP regulations for safety/toxicology studies. An IND sponsor must submit the results of the pre-clinical studies, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND.
An IND is a request for authorization from the FDA to ship an investigation product and then administer it to humans and must be allowed to proceed by the FDA before human clinical trials may begin. Some long-term pre-clinical testing, such as animal tests of reproductive AEs and carcinogenicity, may continue after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions before that time related to one or more proposed clinical trials and places the trial on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical trials
The clinical stage of development involves the administration of the investigational product to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by, or under control of, the trial sponsor, in accordance with GCPs, which include the requirement that all research patients provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters to be used to monitor subject safety and assess efficacy. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Furthermore, each clinical trial must be reviewed and approved by an IRB for each institution at which the clinical trial will be conducted to ensure that the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. There also are requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries. Information about most clinical trials must be submitted within specific timeframes for publication on the www.clinicaltrials.gov website. Information related to the product, patient population, phase of investigation, study sites and investigators and other aspects of the clinical trial is made public as part of the registration of the clinical trial. Sponsors are also obligated to disclose the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed in some cases for up to two years after the date of completion of the trial. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined:
| ● | Phase I clinical trials generally involve a small number of healthy volunteers or disease-affected patients who are initially exposed to a single dose and then multiple doses of the product candidate. The primary purpose of these clinical trials is to assess the metabolism, pharmacologic action, side effect tolerability and safety of the drug. | |
| ● | Phase II clinical trials involve studies in disease-affected patients to determine the dose required to produce the desired benefits. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected, possible adverse effects and safety risks are identified, and a preliminary evaluation of efficacy is conducted. | |
| ● | Phase III clinical trials generally involve a larger number of patients at multiple sites and are designed to provide the data necessary to demonstrate the effectiveness of the product for its intended use, its safety in use and to establish the overall benefit/risk relationship of the product and provide an adequate basis for product approval. These trials may include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to mimic the actual use of a product during marketing. |
Post-approval trials, sometimes referred to as Phase IV clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow up. In certain instances, the FDA may mandate the performance of Phase IV clinical trials as a condition of approval of an NDA or a Biologics License Application (“BLA”).
| 30 |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if significant adverse events (“SAEs”) occur. The FDA or the sponsor may suspend or terminate a clinical trial at any time, or the FDA may impose other sanctions on various grounds, including a finding that the research patients are being exposed to an unacceptable health risk. Similarly, an IRB can refuse, suspend, or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Concurrently with clinical trials, companies usually complete additional pre-clinical studies and must also develop additional information about the physical characteristics of the drug or biological product as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency, and purity of the final biological product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the biological product candidate does not undergo unacceptable deterioration over its shelf life.
Marketing Approval
Assuming successful completion of the required clinical testing, the results of the pre-clinical studies and clinical trials, together with detailed information relating to the product’s chemistry, manufacture, controls, and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications. In most cases, the submission of an NDA is subject to a substantial application user fee.
The review process typically takes twelve months from the date the NDA is submitted to the FDA. The FDA conducts a preliminary review of all NDAs within the first 60 days after submission to determine whether they are sufficiently complete to permit substantive review before accepting them for “filing.” The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information and may be subject to an additional application user fee. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged, or held meets standards designed to assure the product’s continued safety, quality and purity. Under the current guidelines in effect in the Prescription Drug User Fee Act (PDUFA), the FDA has a goal to review and act on the submission within ten months from the completion of the preliminary review of a standard NDA for a new molecular entity.
The FDA also may require submission of a REMS plan to ensure that the benefits of the drug outweigh its risks. The REMS plan could include medication guides, physician communication plans, assessment plans, and/or elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA may inspect one or more clinical trial sites to assure compliance with GCP requirements.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter generally contains a statement of specific conditions that must be met in order to secure final approval of the NDA and may require additional clinical trials or pre-clinical studies in order for FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
| 31 |
Post-approval requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There also are continuing annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data.
Intellectual Property
KETAMIR-2
We license the U.S., Canadian, and Mexican patent rights for the use of KETAMIR-2 in human applications from MIRALOGX. MIRALOGX filed international application no. PCT/US2024/018594 under the Patent Cooperation Treaty (PCT) on March 6, 2024, titled, ANTIDEPRESSANT COMPOUNDS, PHARMACEUTICAL COMPOSITIONS, AND METHODS OF TREATING DEPRESSION AND OTHER DISORDERS, and in due course intends to enter the national phase in the United States, Canada and Mexico, among other countries. These applications, if granted and subject to payment of patent maintenance fees, would offer protection extending through at least March 6, 2044. The patent rights for KETAMIR-2 outside of the United States, Canada, and Mexico are not included in our current patent rights.
Our license from MIRALOGX is set forth in the Exclusive License Agreement, dated November 15, 2023, pursuant to which the licensed field of use includes therapeutic treatments and other medical or health uses in humans, and related preclinical studies and activities conducted in furtherance of obtaining regulatory approval for and commercialization of human therapeutic treatments and uses (the “MIRALOGX License Agreement”). “Licensed Product” is defined as a drug product containing as an active agent 2-(2-chlorophenyl)-2-(methylamino)cyclopentan-1-one or a pharmaceutically acceptable salt or ester thereof. We also have the right to grant corresponding sublicenses under the licensed patent rights. The MIRALOGX License Agreement provides for the payment to MIRALOGX of an 8% royalty (payable quarterly) on our net sales of Licensed Products by us or our sublicensees and on non-royalty bearing milestone revenue, with the royalty obligation ceasing upon the later of the expiration of the last-to-expire licensed patent. The agreement also provides for an up-front Cost Reimbursement of $100,000 payable to MIRALOGX to cover the already-incurred costs associated with the patent rights. The Cost Reimbursement is the only payment made to date under the agreement. MIRALOGX may terminate the agreement upon insolvency, an uncured breach including the failure to make any payment owed under the agreement or the failure to use commercially reasonable efforts to develop the licensed product, or upon a default of the November 15, 2023 Promissory Note and Loan Agreement. The MIRALOGX License Agreement provides that MIRALOGX will have sole control over the filing, prosecution, maintenance, and management of the licensed patent rights, provided that we will be responsible for the cost of prosecuting and maintaining the licensed patents. The agreement grants to us the primary right, but not the obligation, to enforce the licensed patent rights.
Besides relying on patents, we also rely on trade secrets, proprietary know-how and continuing innovation to develop and maintain our competitive position, especially when we do not believe that patent protection is appropriate or can be obtained. We seek protection of these trade secrets, proprietary know-how and any continuing innovation, in part, through confidentiality and proprietary information agreements. However, these agreements may not provide meaningful protection for, or adequate remedies to protect, our technology in the event of unauthorized use or disclosure of information. Furthermore, our trade secrets may otherwise become known to, or be independently developed by, our competitors. We intend to seek appropriate patent protection for technology in our research and development programs, where applicable, and their uses by filing patent applications in the United States and other selected countries. We intend for these patent applications to cover, where possible, claims for compositions of matter, medical uses, processes for preparation and formulations.
| 32 |
MIRA-55
We have a pending provisional patent application directed to MIRA-55, a structure that was synthesized and isolated during the research and development of MIRA1a titled “Synthetic Cannabinoid Analogs, Pharmaceutical Compositions and Methods of Treating Anxiety and Other Disorders”. The Company intends to pursue domestic and foreign filings based on the provisional application to seek global patent protection for MIRA-55.
MIRA1a
The U.S. Patent 10,787,675 B2, titled “Purified Synthetic Marijuana and Methods of Treatment by Administering Same,” which covers the MIRA1a compound per se as a racemic mixture, an isolated R-enantiomer, or an isolated S-enantiomer, as well as pharmaceutical formulations of the compound, was assigned to our Company by SRQ Patent Holdings II, LLC (“SRQ”) in December 2021. This patent also covers MIRA1a in methods of treating Alzheimer’s disease, anxiety, depression, and addictions. Subject to payment of patent maintenance fees, the ‘675 patent offers protection extending through at least February 11, 2039. According to the assignment and royalty agreement, we owe 8% in royalty revenue on net sales price and royalty revenue and 8% of milestone payment revenue to SRQ.
The royalties shall continue, in each country on a product-by-product and country-by-country basis until the later of i) the date of expiration of the last to expire patent included within the Innovation, or ii) the date of expiration of the last strategic partnership/licensing agreement including the Innovation.
We currently have no plans to develop the MIRA1a compound for approval and commercialization in or outside of the United States. See “Risk Factors— Risks Related to Our Intellectual Property— We own the rights associated with our patents in the United States, but we do not own the rights to patents covering MIRA1a in foreign jurisdictions.”
Properties
Our corporate headquarters and executive offices were in Baltimore, Maryland, which lease expires on April 30, 2024. Our current business address is 1200 Brickell Avenue, Suite 1950 #1183, Miami, Florida 32183, which is a virtual office.
Employees
As of March 28, 2024, we had three employees and various consultants providing support. None of our employees are represented by a labor union or are covered by a collective bargaining agreement. We consider our relationship with our employees to be satisfactory.
Legal Proceedings
From time to time, we may be named in claims arising in the ordinary course of business. Currently, no legal proceedings, government actions, administrative actions, investigations, or claims are pending against us or involve us that, in the opinion of our management, could reasonably be expected to have a material adverse effect on our business and financial condition.
We anticipate that we will expend significant financial and managerial resources in the defense of our intellectual property rights in the future if we believe that our rights have been violated. We also anticipate that we will expend significant financial and managerial resources to defend against claims that our products and services infringe upon the intellectual property rights of third parties.
Corporation Information
Our corporate headquarters is located at 1200 Brickell Avenue, Suite 1950 #1183, Miami, Florida 32183. Our telephone number is 786-432-9792.
Our principal website address is www.mirapharmaceuticals.com. The information contained on, or that can be accessed through, our website is deemed not to be incorporated in this Report or to be part of this Report. You should not consider information contained on our website to be part of this Report.
| 33 |
ITEM 1A. Risk Factors
RISK FACTORS
Investing in shares of our common stock is very speculative and involves a high degree of risk. You should carefully consider the risks and uncertainties described below, the section of this Report entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and related notes included elsewhere in this Report before investing in shares of our common stock. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties that we are unaware of, or that we currently believe are not material, may also become important factors that affect us. If any of the following risks occur, our business, operating results and prospects could be materially harmed. In that event, the price of our common stock could decline, and you could lose part or all of your investment.
Risks Related to Our Operations and Financial Condition
We are an early development-stage company with no revenues. As such, our losses from operations and negative cash flows as of December 31, 2023 raise substantial doubt about our ability to continue as a going concern absent obtaining adequate new debt or equity financings.
As a very early development-stage enterprise that is focused on the development of a pre-clinical pharmaceutical product, we have generated no revenue and have an accumulated deficit of $21.3 million through December 31, 2023, and $9.3 million through December 31, 2022. We have concluded that substantial doubt exists about our ability to continue as a going concern for the 12 months following the issuance of the financial statements included in this Annual Report on Form 10-K. As of the issuance date of these financial statements, we believe that we have sufficient resources available to support our development activities and business operations and timely satisfy our obligations as they come due into the fourth quarter of 2024. We do not have sufficient cash and cash equivalents as of the date of filing this Annual Report on Form 10-K to support our operations for at least the 12 months following the issuance of the financial statements.
To alleviate the conditions that raise substantial doubt about our ability to continue as a going concern, we plan to secure additional capital, potentially through a combination of public or private equity offerings and strategic transactions, including potential alliances and drug product collaborations, however, none of these alternatives are committed at this time. There can be no assurance that we will be successful in obtaining sufficient funding on terms acceptable to us to fund continuing operations, if at all, identify and enter into any strategic transactions that will provide the capital that we will require or achieve the other strategies to alleviate the conditions that raise substantial doubt about our ability to continue as a going concern. If none of these alternatives are available, or if available, are not available on satisfactory terms, we will not have sufficient cash resources and liquidity to fund our business operations for at least the 12 months following the date the financial statements are issued. The failure to obtain sufficient capital on acceptable terms when needed may require us to delay, limit, or eliminate the development of business opportunities and our ability to achieve our business objectives and our competitiveness, and our business, financial condition, and results of operations will be materially adversely affected. In addition, the perception that we may not be able to continue as a going concern may cause others to choose not to deal with us due to concerns about our ability to meet our contractual obligations.
The report of our independent registered accounting firm on our audited financial statements for the fiscal year ended December 31, 2023 contains an explanatory paragraph relating to our ability to continue as a going concern.
The auditor’s opinion on our audited financial statements for the year ended December 31, 2023 includes an explanatory paragraph stating that we have incurred recurring losses from operations that raise substantial doubt about our ability to continue as a going concern. While we believe that we will be able to obtain the capital we need to continue our operations, there can be no assurances that we will be successful in these efforts or will be able to resolve our liquidity issues or eliminate our operating losses. If we are unable to obtain sufficient funding, we would need to significantly reduce our operating plans and curtail some or all of our development efforts. Accordingly, our business, prospects, financial condition, and results of operations will be materially and adversely affected, and we may be unable to continue as a going concern. If we seek additional financing to fund our business activities in the future and there remains substantial doubt about our ability to continue as a going concern, investors or other financing sources may be unwilling to provide additional funding on commercially reasonable terms or at all.
| 34 |
Because we have a limited operating history, you may not be able to accurately evaluate our operations.
We have had limited operations to date. Therefore, we have a limited operating history upon which to evaluate the merits of investing in our company. Potential investors should be aware of the difficulties normally encountered by new companies and the high rate of failure of such enterprises. The likelihood of success must be considered in light of the problems, expenses, difficulties, complications, and delays encountered in connection with the operations that we plan to undertake. These potential problems include, but are not limited to, unanticipated problems relating to the ability to generate sufficient cash flow to operate our business, and additional costs and expenses that may exceed current estimates. We expect to continue to incur significant losses into the foreseeable future. We recognize that if the effectiveness of our business plan is not forthcoming, we will not be able to continue business operations. There is no history upon which to base any assumption as to the likelihood that we will prove successful, and it is doubtful that we will generate any operating revenues or ever achieve profitable operations. If we are unsuccessful in addressing these risks, our business will most likely fail.
We have significant and increasing liquidity needs and will require additional funding.
Our operations have consumed substantial amounts of cash since inception. For the year ended December 31, 2023, we reported a net operating cash outflow of $4.5 million and a net cash inflow from investing activities of $8.8 million. For the year ended December 31, 2022, we reported a net operating cash outflow of $5.6 million and a net cash inflow from investing activities of $3.1 million.
Research and development, and general and administrative expenses, and cash used for operations will continue to be significant and may increase substantially in the future in connection with new research and development initiatives and continued product commercialization efforts. We may need to raise additional capital to fund our operations, continue to conduct clinical trials to support potential regulatory approval of marketing applications and to fund commercialization of our products.
The amount and timing of our future funding requirements will depend on many factors, including, but not limited to:
| ● | the timing of FDA approval, if any; | |
| ● | the DEA continuing to classify Ketamir-2 and MIRA1a as a substance not subject to CSA; | |
| ● | the DEA granting the classification of MIRA-55 as a substance not subject to CSA; | |
| ● | the timing and amount of revenue from sales of our products, or revenue from grants or other sources; | |
| ● | the rate of progress and cost of our clinical trials and other product development programs; | |
| ● | costs of establishing or outsourcing sales, marketing, and distribution capabilities; | |
| ● | costs and timing of completion of expanded in-house manufacturing facilities as well as any outsourced commercial manufacturing supply arrangements for our product candidates; | |
| ● | costs of filing, prosecuting, defending, and enforcing any patent claims and other intellectual property rights associated with our product candidates; | |
| ● | costs of operating as a U.S. public company; | |
| ● | the effect of competing technological and market developments; | |
| ● | personnel, facilities, and equipment requirements; and | |
| ● | the terms and timing of any additional collaborative, licensing, co-promotion, or other arrangements that we may establish. |
| 35 |
While we expect to fund our future capital requirements from a number of sources including existing cash balances, future cash flows from operations and the proceeds from further public offerings, we cannot assure you that any of these funding sources will be available to us on favorable terms, or at all. Further, even if we can raise funds from all of the above sources, the amounts raised may not be sufficient to meet our future capital requirements.
Operating results may vary significantly in future periods.
Our operating and financial results are likely to fluctuate significantly in the future. Our operating and financial results are unpredictable and may fluctuate, for among other reasons, due to:
| ● | our achievement of product development objectives and milestones; | |
| ● | clinical trial enrollment and expenses; | |
| ● | research and development expenses; and | |
| ● | the timing and nature of contract manufacturing and contract research payments. |
In addition, a high portion of our costs are determined on an annual basis, due in part to our significant research and development costs. Thus, increases in our costs could disproportionately affect financial results in a quarter. Other factors, including non-cash expenses associated with financing activity, could also lead to fluctuations in our results of operations. Because of these factors, our operating and financial results in one or more future quarters may fail to meet the expectations of securities analysts or investors, which could cause our share price to decline.
We have yet to generate revenues or achieve a profit and may not generate revenue or achieve a profit for many years, if at all.
We have not yet produced any revenues or profit and may not for many years, if at all. Our ability to generate revenue is dependent on the receipt of regulatory approval of our product candidates, which will take years to achieve and may not be obtained. We therefore cannot assure you we will be able to ever generate sufficient revenue to pay for our expenses or achieve profitability. Our ability to continue as a going concern in the future is dependent upon raising capital from financing transactions and keeping operating expenses below our revenue levels in order to achieve positive cash flows, none of which can be assured.
Conflicts of interest may arise between us and MIRALOGX.
MIRALOGX licenses us the patent pending rights to KETAMIR-2. MIRALOGX is a separate intellectual property development company owned by the Bay Shore Trust. The Bay Shore Trust is also our largest stockholder. The interests of MIRALOGX are 100% owned by the Bay Shore Trust. Our relationship with MIRALOGX and the Bay Shore Trust may create, or may create the appearance of, conflicts of interest when we are faced with decisions that could have different implications for MIRALOGX than the decisions have for us. Furthermore, in light of the license agreement that we have with MIRALOGX, if a dispute were to arise between MIRALOGX and us relating to our past or future relationship with MIRALOGX or with respect to intellectual property matters, these potential conflicts of interest may make it more difficult for us to favorably resolve such disputes.
| 36 |
Certain of our executive officers will not be employed by us on a full-time basis.
Erez Aminov, our Chairman and Chief Executive Officer, will not be employed by our company on full-time basis. As provided in his respective employment agreement with our company, Mr. Aminov is expected to devote approximately fifty percent (50%) of his business time to the affairs of our company. Because this officer will not work full time for our company, instances may occur where he may not be immediately available to provide solutions to problems or address concerns that arise in the course of us conducting our business and thus adversely affect our business. In addition, he can become subject to conflicts of interest because he devotes part of his working time to other business endeavors and has responsibilities to other entities. Although such officer is aware of his duty and accountability to our company and to applicable laws and policies relating to corporate opportunity and conflicts of interest, such conflicts of interest may include deciding how much time to devote to our affairs, as well as what business opportunities should be presented to us.
Risks Relating to Our Business and Our Industry
Our future viability will largely depend on the positive development of Ketamir-2 and MIRA-55, and any future product candidates, which development will require significant capital resources and years of clinical development effort.
We currently have no drug products on the market, and all of our drug development projects are in a pre-clinical stage of development. Our business depends almost entirely on the successful pre-clinical and clinical development, FDA regulatory approval, and commercialization of our product candidates, principally Ketamir-2 and MIRA-55. Investors need to be aware that substantial additional investments including pre-clinical and clinical development and FDA regulatory submission and approval efforts will be required before we are permitted to undertake clinical studies and market and commercialize our product candidates, if ever. It may be several years before we can commence clinical trials, if ever. Any clinical trial will be subject to extensive and rigorous review and regulation by numerous government authorities in the United States and other jurisdictions where we intend, if approved, to market our product candidates. Before obtaining regulatory approvals for any of our product candidates, we must demonstrate through pre-clinical testing and clinical trials that the product candidate is safe and effective for its specific application. This process can take many years and may include post- marketing studies and surveillance, which would require the expenditure of substantial resources. Of the large number of drugs in development for approval in the United States (and the rest of the world), only a small percentage will successfully complete the FDA regulatory approval financing to fund our planned research, development, and clinical programs, we cannot assure you that any of our product candidates will be successfully developed or commercialized.
We may be unable to formulate or scale up any or all of our product candidates. There is no guarantee that any of the product candidates will be or are able to be manufactured or produced in a manner to meet the FDA’s criteria for product stability, content uniformity and all other criteria necessary for product approval in the United States and other markets. Any of our product candidates may fail to achieve their specified endpoints in clinical trials.
Furthermore, product candidates may not be approved even if they achieve their specified endpoints in clinical trials. The FDA may disagree with our trial design and our interpretation of data from clinical trials or may change the requirements for approval even after it has reviewed and commented on the design for our clinical trials. The FDA may also approve a drug for fewer or more limited indications than we request or may grant approval contingent on the performance of costly post-approval clinical trials (i.e., Phase IV trials). In addition, the FDA may not approve the labeling claims that we believe are necessary or desirable for the successful commercialization of our product candidates.
If we are unable to obtain regulatory approval for Ketamir-2 and MIRA-55 within the timeline we anticipate, we will not be able to execute our business strategy effectively and our ability to substantially grow our revenues will be limited, which would have a material adverse impact on our long-term business, results of operations, financial condition, and prospects.
| 37 |
We are dependent on our current and future product candidates, some of which may not receive regulatory approval or be successfully commercialized.
Our ability to progress our plan will depend on our ability to clinically develop, gain regulatory approval for and ultimately commercialize our product candidates. Our ability to successfully commercialize our product candidates will depend on, among other things, our ability to:
| ● | successfully complete pre-clinical and other nonclinical studies and clinical trials in a manner that allows us to progress our studies; | |
| ● | receive IND acceptance and regulatory approvals from the FDA; | |
| ● | produce, through a validated process, in manufacturing facilities inspected and approved by regulatory authorities, including the FDA, sufficiently large quantities of product candidates to permit successful commercialization; | |
| ● | obtain reimbursement from payers such as government health care programs and insurance companies and achieve commercially attractive levels of pricing; | |
| ● | secure acceptance of our product candidates from physicians, health care payers, patients, and the medical community; | |
| ● | create positive publicity surrounding our product candidates; | |
| ● | manage our spending as costs and expenses increase due to clinical trials and commercialization; and | |
| ● | obtain and enforce sufficient intellectual property for our product candidates. |
Our failure or delay with respect to any of the factors above could have a material adverse effect on our business, results of operations and financial condition.
Impact of global tensions may increase uncertainty of our future operations.
The global tensions arising from the Palestine-Israel war and the war in Ukraine may result in disruptions in the broader global economic environment. The uncertain nature, magnitude, and duration of hostilities stemming from such conflicts, including the potential effects of sanctions and countersanctions, or retaliatory cyber-attacks on the world economy and markets, have contributed to increased market volatility and uncertainty, which could have an adverse impact on macroeconomic factors that affect our business and operations, such as pre-clinical study issues, manufacturer delays or shipping delays.
Moreover, the conflict between Palestine and Israel could impact future business decisions to locate potential clinical trials in Israel. It is not possible to predict the short and long-term implications of military conflicts or wars or geopolitical tensions which could include further sanctions, uncertainty about economic and political stability, increases in inflation rate and energy prices, cyber-attacks, supply chain challenges and adverse effects on currency exchange rates and financial markets.
Results of pre-clinical studies and earlier clinical trials are not necessarily predictive indicators of future results.
Any positive results from future pre-clinical testing of our product candidates and potential future clinical trials may not necessarily be predictive of the results from Phase 1, Phase 2 or Phase 3 clinical trials. In addition, our interpretation of results derived from clinical data or our conclusions based on our pre-clinical data may prove inaccurate. Frequently, pharmaceutical and biotechnology companies have suffered significant setbacks in clinical trials after achieving positive results in pre-clinical testing and early phase clinical trials, and we cannot be certain that we will not face similar setbacks. These setbacks may be caused by the fact that pre-clinical and clinical data can be susceptible to varying interpretations and analyses. Furthermore, certain product candidates may perform satisfactorily in pre-clinical studies and clinical trials, but nonetheless fail to obtain FDA approval or appropriate approvals by the appropriate regulatory authorities in other countries. If we fail to produce positive results in our clinical trials for our product candidates, the development timeline and regulatory approval and commercialization prospects for them and as a result our business and financial prospects, would be materially adversely affected.
| 38 |
We have limited marketing experience, and we do not anticipate at this time establishing a sales force or distribution and reimbursement capabilities, and we may not be able to successfully commercialize any of our product candidates if they are approved in the future.
If regulatory approval of our products is ever obtained, our ability to generate revenues ultimately depends on our ability to sell our approved products and secure adequate third-party reimbursement. We currently have limited experience in marketing and selling our products. We currently do not have any products approved for sale in the United States or in any other country.
The commercial success of our product candidates will not even be possible for the foreseeable future and will depend on a number of factors beyond our control, including the willingness of physicians to prescribe our products to patients, payers’ willingness and ability to pay for the drugs, the level of pricing achieved, patients’ response to our drugs and the ability of our marketing partners to generate sales. There can be no guarantee that we will be able to establish or maintain the personnel, systems, arrangements and capabilities necessary to successfully commercialize Ketamir-2 and MIRA-55 or any product candidate approved by the FDA in the future. If we fail to establish or maintain successful marketing, sales and reimbursement capabilities or fail to enter into successful marketing arrangements with third parties, our product revenues may suffer.
Should we later determine it is in our best interest to develop a sales force we may be unable to effectively train and equip our sales force, therefore our ability to successfully commercialize our products may be harmed.
We will be required to expend significant time and resources to train our sales force to be credible, persuasive and compliant with applicable laws in marketing Ketamir-2 and MIRA-55 or our other product candidates to physicians for their approved uses. In addition, we must continue to train our sales force to ensure that a consistent and appropriate message about Ketamir-2 and MIRA-55 or our other product candidates are being delivered to our potential customers. If we are unable to effectively train our sales force and equip them with effective materials, including medical and sales literature to help them inform and educate potential customers about the benefits of Ketamir-2 and MIRA-55 and our product candidates and its proper administration, our efforts to successfully commercialize Ketamir-2 and MIRA-55 and our product candidates could be jeopardized, which would negatively impact our ability to generate product revenues.
We will need to further increase the size and complexity of our organization in the future, and we may experience difficulties in managing our growth and executing our growth strategy.
Our management and personnel, systems, and facilities currently in place may not be adequate to support our business plan and future growth. As a result, we may need to further expand certain areas of our organization.
Our need to effectively manage our operations, growth and various projects requires that we:
| ● | continue to improve our operational, financial, management and regulatory compliance controls and reporting systems and procedures; | |
| ● | attract and retain enough talented employees; | |
| ● | manage our clinical trials effectively; | |
| ● | manage our external manufacturing operations with contract research organizations effectively and in a cost-effective manner; | |
| ● | manage our development efforts effectively while carrying out our contractual obligations to contractors and other third parties; and |
| 39 |
In addition, we may utilize the services of part-time outside consultants and contractors to perform several tasks for us, including tasks related to compliance programs, clinical trial management, regulatory affairs, formulation development and other drug development functions. Our growth strategy may entail expanding our use of consultants and contractors to implement these and other tasks going forward. If we are not able to effectively expand our organization by hiring new employees and expanding our use of consultants and contractors, we may be unable to successfully implement the tasks necessary to effectively execute on our planned research, development, manufacturing, and commercialization activities and, accordingly, may not achieve our research, development and commercialization goals.
Our product candidates, if approved, may be unable to achieve the expected market acceptance and, consequently, limit our ability to generate revenue from new products.
Even when product development is successful and regulatory approval has been obtained, our ability to generate sufficient revenue depends on the acceptance of our products by physicians and patients. We cannot assure you that our product candidates will achieve the expected level of market acceptance and revenue if and when they obtain the requisite regulatory approvals. The market acceptance of any product depends on a number of factors, including the indication statement and warnings required by regulatory authorities in the product label. Market acceptance can also be influenced by continued demonstration of efficacy and safety in commercial use, physicians’ willingness to prescribe the product, reimbursement from third-party payers such as government health care programs and private third-party payers, the price of the product, the nature of any post-approval risk, management activities mandated by regulatory authorities, competition, and marketing and distribution support. Further, an ineffective or inefficient distribution model at launch may lead to the inability to fulfill demand, and consequently a loss of revenue. Any factors preventing or limiting the market acceptance of our products could have a material adverse effect on our business, results of operations and financial condition.
If the price for any future approved products decreases or if government and other third-party payers do not provide coverage and adequate reimbursement levels, our revenue and prospects for profitability will suffer.
Patients who are prescribed medicine for the treatment of their conditions generally rely on third-party payers to reimburse all or part of the costs associated with their prescription drugs. Reimbursement systems in international markets vary significantly by country and by region, and reimbursement approvals generally must be obtained on a country-by-country basis. Coverage and adequate reimbursement from governmental healthcare programs, such as Medicare and Medicaid, and commercial payers is critical to new product acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower-cost therapeutic alternatives are already available or subsequently become available. Even if we obtain coverage for products we may market, the resulting reimbursement payment rates may require co-payments that patients find unacceptably high. Patients may not use our products if coverage is not provided, or reimbursement is inadequate to cover a significant portion of its cost.
In addition, the market for our products will depend significantly on access to third-party payers’ drug formularies or lists of medications for which third-party payers provide coverage and reimbursement. The industry competition to be included in such formularies often leads to downward pricing pressures on pharmaceutical companies. Also, third-party payers may refuse to include a particular branded drug in their formularies or otherwise restrict patient access to a branded drug when a less costly generic equivalent or other alternative is available, even if not approved for the indications for which our products are approved.
Third-party payers or governmental or commercial entities are developing increasingly sophisticated methods of controlling healthcare costs. The current environment is putting pressure on companies to price products below what they may feel is appropriate. Selling our products at less than an optimized price could impact our revenues and overall success as a company. It will be difficult to determine the optimized price for our products. In addition, in the U.S., no uniform policy of coverage and reimbursement for drug products exists among third-party payers. Therefore, coverage and reimbursement for our products may differ significantly from payer to payer. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payer separately, with no assurance that coverage will be obtained. If we are unable to obtain coverage of, and adequate payment levels for, products we may market to third-party payers, physicians may limit how much or under what circumstances they will prescribe or administer them, and patients may decline to purchase them. This in turn could affect our ability to successfully commercialize products we may market, and thereby adversely impact our profitability, results of operations, financial condition, and future success.
| 40 |
In addition, where we have chosen to collaborate with a third party on product candidate development and commercialization, our partner may elect to reduce the price of our products in order to increase the likelihood of obtaining reimbursement approvals. In many countries, products cannot be commercially launched until reimbursement is approved and the negotiation process in some countries can exceed 12 months. In addition, pricing and reimbursement decisions in certain countries can be affected by decisions taken in other countries, which can lead to mandatory price reductions and/or additional reimbursement restrictions across a number of other countries, which may thereby adversely affect our sales and profitability. In the event that countries impose prices that are not sufficient to allow us or our partners to generate a profit, our partners may refuse to launch the product in such countries or withdraw the product from the market, which would adversely affect sales and profitability. Events, such as price decreases, government mandated rebates or unfavorable reimbursement decisions, could affect the pricing and reimbursement of Ketamir-2 and MIRA-55 and our other product candidates and could have a material adverse effect on our business, reputation, results of operations and financial condition.
We expect to face intense competition, often from companies with greater resources and experience than we have.
Demand for ketamine analogs like Ketamir-2 and synthetic cannabinoids such as MIRA-55 and will likely be dependent on a number of social, political, legislative, and economic factors that are beyond our control. While we believe that there will be a demand for such drugs, and that the demand will grow, there is no assurance that such demand will happen, that we will benefit from any demand or that our business, in fact, will ever generate revenues from our drug development programs or become profitable.
The emerging markets for product candidates like ours and related medical research and development is and will likely remain competitive. The development and commercialization of drugs and medicines is highly competitive. We compete with a variety of multinational pharmaceutical companies and specialized biotechnology companies, as well as products and processes being developed by universities and other research institutions. Many of our competitors have developed, are developing, or will develop drugs and processes which may be competitive with our drug candidates. Competitive therapeutic treatments include those that have already been approved by medicines regulators and accepted by the medical community and any new treatments that may enter the market. For some of our drug development programs / areas of therapeutic interest, other treatment options are currently available, under development, and may become commercially available in the future. If any of our product candidates are approved for the diseases and conditions we are currently pursuing, they may compete with a range of medicines or therapeutic treatments that are either in development, will be developed in the future or currently marketed.
Established companies may have a competitive advantage over us due to their size and experiences, financial resources, and institutional networks. Many of our competitors may have significantly greater financial, technical, and human resources than we do. Due to these factors, our competitors may have an advantage in marketing their approved drugs and may obtain regulatory approval of their drug candidates before we are able to, which may limit our ability to develop or commercialize our drug candidates. Our competitors may also develop drugs / medicines that are safer, more effective, more widely used and less expensive than ours. These advantages could materially impact our ability to develop and, if approved, commercialize our product candidates successfully. Furthermore, some of these competitors may make acquisitions or establish collaborative relationships among themselves or with third parties to increase their ability to rapidly gain market share.
Our product candidates may compete with other synthetic cannabinoids, as well as with cannabinoid or cannabis-based drugs, in addition to competing with state-licensed medical and recreational marijuana, in markets where the recreational and/or medical use of marijuana is legal. There is continuing support in the U.S. for further state legalization of marijuana. In markets where recreational and/or medical marijuana is not legal, our product candidates, once approved by regulators, may compete with marijuana or marijuana-based products purchased in the illegal drug market. This may or may not affect the commercial price that we may be able to achieve for our synthetic regulatory-approved medicines, should they be approved by the FDA.
| 41 |
Moreover, as generic versions of drug products enter the market, the price for such medicines may be expected to decline rapidly and substantially. Even if we are the first to obtain FDA approval of one of our product candidates, the future potential approval of generics could adversely affect the price we are able to charge, and the profitability of our product(s) will likely decline.
Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in more resources being concentrated among a smaller number of our competitors. Smaller and other early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
These companies may compete with us in recruiting and retaining qualified scientific, management and commercial personnel, utilizing contract manufacturing facilities or contract research organizations (CROs), or establishing clinical trial sites and subject registration for clinical trials, as well as in acquiring technologies complementary to our research projects.
Product shipment delays could have a material adverse effect on our business, results of operations and financial condition.
The shipment, import and export of Ketamir-2 and MIRA-55 and our other product candidates require import and export licenses. In the U.S., FDA, U.S. Customs and Border Protection and the DEA, and in other countries similar regulatory authorities, regulate the import and export of pharmaceutical products that contain controlled substances. Specifically, the import and export process require the issuance of import and export licenses by the relevant controlled substance authority in both the importing and exporting country. We may not be granted, or if granted, maintain, such licenses from the authorities in certain countries. Even if we obtain the relevant licenses, shipments of Ketamir-2 and MIRA-55 and our product candidates may be held up in transit, which could cause significant delays and may lead to product batches being stored outside required temperature ranges. Inappropriate storage may damage the product shipment resulting in a partial or total loss of revenue from one or more shipments of Ketamir-2 and MIRA-55 or our other product candidates. A partial or total loss of revenue from one or more shipments of Ketamir-2 and MIRA-55 or our other product candidates could have a material adverse effect on our business, results of operations and financial condition. Even though the DEA has confirmed in writing that it conducted a scientific review of the chemical structure of MIRA1a and Ketamir-2 in accordance with the definitions within the CSA and its implementing regulations and determined that MIRA1a and Ketamir-2 is not a controlled substance or listed chemical, there is no assurance that the DEA may not change its position. We have filed the necessary requirements with the DEA to review MIRA-55, however, there can be no assurance that the DEA will conclude that MIRA-55 is not a controlled substance or listed chemical.
The manufacture of our product candidates is complex and uncertain, and until we develop a validated manufacturing process, we may encounter difficulties in supplying our planned and future clinical trials. If we encounter such difficulties, or fail to meet quality standards, our ability to meet clinical timelines and expand our development strategy could be impacted.
The processes involved in manufacturing Ketamir-2, MIRA-55 and other product candidates are complex, expensive, highly regulated and subject to multiple risks and uncertainties. We have been faced with issues such as this in the initial synthesis of MIRA-55 (which we initially believed was based on our patented MIRA1a molecule).
In addition, as product candidates are developed through early to late-stage clinical trials and then to approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods, are modified along the way to optimize the scale, process and results. Any changes to the manufacturing processes carry the risk that they will not achieve these intended objectives, or that the product candidates may not meet the rigorous quality standards necessary for use in our pre-clinical or clinical trials.
Also, if planned or future manufacturing of Ketamir-2, MIRA-55 or other product candidates fails to meet the quality standards for use in our pre-clinical or clinical trials, or the active drug substance does not meet our quality specifications, it could impact our timelines and limit our development strategy. For example, and as discussed above, in the first quarter of 2024, we concluded that during the manufacturing and scale-up process of MIRA1a, the intended MIRA1a compound was in fact synthesized as MIRA-55.
| 42 |
Moreover, our contract manufacturing organizations (“CMOs”) or contract development and manufacturing organization (“CDMOs”) may be unable to successfully increase the manufacturing scale for our product candidates in a timely or cost-effective manner and may experience delays due to limited manufacturing capacity. In addition, quality issues may arise during manufacturing activities. If our CMOs or CDMOs are unable to successfully manufacture our product candidates in sufficient quantity in a timely manner or produce active drug substances that do not meet our quality specifications, our planned pre-clinical or clinical trials may be delayed or modified.
We may fail to expand our manufacturing capability in time to meet market demand for our products and product candidates, and the FDA may refuse to accept our facilities or those of our contract manufacturers as being suitable for the production of our products and product candidates. Any problems in our manufacturing process could have a material adverse effect on our business, results of operations and financial condition.
Before we can begin commercial manufacture of any product candidates for sale in the U.S., we must obtain FDA regulatory approval for the product, which requires a successful FDA inspection of our manufacturing facilities and those of our contract manufacturers, processes, and quality systems in addition to other product-related approvals. Although we may successfully navigate this pre-approval inspection process as it relates in the U.S., pharmaceutical manufacturing facilities are continuously subject to post-approval inspection by the FDA and foreign regulatory authorities. Due to the complexity of the processes used to manufacture our product candidates, we may be unable to initially or continue to pass federal, state or international regulatory inspections in a cost-effective manner. If we are unable to comply with manufacturing regulations, we may be subject to fines, unanticipated compliance expenses, recall or seizure of any approved products, total or partial suspension of production and/or enforcement actions, including injunctions, and criminal or civil prosecution. These possible sanctions would adversely affect our business, results of operations and financial condition.
Business interruptions could delay us in the process of developing our product candidates and could disrupt our product sales.
Our research and development activities are conducted through outside contractors and manufacturers. Loss of our contracted manufacturing facilities, stored inventory or laboratory facilities through fire, theft or other causes, or loss of our raw material, could have an adverse effect on our ability to continue product development activities and to conduct our business. Failure to supply our partners with commercial product may lead to adverse consequences, including the right of partners to take over responsibility for product supply. We currently do not have insurance coverage to compensate us for such business interruptions. Our contract manufacturers and suppliers provide that in their separate operations; however, such coverage may prove insufficient to fully compensate us for the damage to our business resulting from any significant property or casualty loss to those facilities.
If product liability lawsuits are successfully brought against us, we will incur substantial liabilities and may be required to limit the commercialization of Ketamir-2 and MIRA-55 and our product candidates.
Although we have never had any product liability claims or lawsuits brought against us, we face potential product liability exposure related to the testing of our product candidates in human clinical trials. We may face exposure to claims by an even greater number of persons when we begin to market and distribute our products commercially in the U.S., Europe and elsewhere. Now, and in the future, an individual may bring a liability claim against us alleging that Ketamir-2, MIRA-55 or one of our other product candidates caused an injury. While we continue to take what we believe are appropriate precautions, we may be unable to avoid significant liability if any product liability lawsuit is brought against us. Large judgments have been awarded in class action or individual lawsuits based on drugs that had unanticipated side effects. If we cannot successfully defend ourselves against product liability claims, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| ● | decreased demand for Ketamir-2, MIRA-55 or our other product candidates if such product candidates are approved; | |
| ● | injury to our reputation; | |
| ● | withdrawal of clinical trial participants; | |
| ● | costs of related litigation; | |
| ● | substantial monetary awards to patients and others; | |
| ● | increased cost of liability insurance; | |
| ● | loss of revenue; and | |
| ● | the inability to successfully commercialize our products. |
| 43 |
Counterfeit versions of our products could harm our business.
Counterfeiting activities and the presence of counterfeit products in a number of markets and over the Internet continue to be a challenge for maintaining a safe drug supply for the pharmaceutical industry. Counterfeit products are frequently unsafe or ineffective and can be life-threatening. To distributors and users, counterfeit products may be visually indistinguishable from the authentic version. Reports of adverse reactions to counterfeit drugs along with increased levels of counterfeiting could be mistakenly attributed to the authentic product, affect patient confidence in the authentic product and harm the business of companies such as ours. If our products were to be the subject of counterfeits, we could incur reputational and financial harm.
We depend upon our key personnel and our ability to attract and retain employees.
Our future growth and success depend on our ability to recruit, retain, manage, and motivate our employees. The inability to hire or retain experienced management personnel could adversely affect our ability to execute our business plan and harm our operating results. Due to the specialized scientific and managerial nature of our business, we rely heavily on our ability to attract and retain qualified scientific, technical, and managerial personnel. The competition for qualified personnel in the pharmaceutical field is intense. Due to this intense competition, we may be unable to continue to attract and retain the qualified personnel necessary for the development of our business or to recruit suitable replacement personnel.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with FDA or foreign regulations, provide accurate information to FDA or other regulatory authorities, comply with applicable manufacturing standards, comply with other foreign, federal, and state laws and regulations, report information or data accurately or disclose unauthorized activities to us. Employee misconduct could also involve the improper use of information, including information obtained during clinical trials, or illegal appropriation of drug products, which could result in government investigations and serious harm to our reputation. The precautions we take to detect and prevent these prohibited activities may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
We are subject to the U.S. Foreign Corrupt Practices Act and other anti-corruption laws, as well as export control laws, customs laws, sanctions laws and other laws governing our operations. If we fail to comply with these laws, we could be subject to civil or criminal penalties, other remedial measures, and legal expenses, which could adversely affect our business, results of operations and financial condition.
Our operations are subject to anti-corruption laws, including the U.S. Foreign Corrupt Practices Act of 1977, as amended (the “FCPA”), and other anti-corruption laws that apply in countries where we do business. The FCPA and these other laws generally prohibit us and our employees and intermediaries from bribing, being bribed or making other prohibited payments to government officials or other persons to obtain or retain business or gain some other business advantage. We and our commercial partners operate in a number of jurisdictions that pose a high risk of potential FCPA violations, and we participate in collaborations and relationships with third parties whose actions could potentially subject us to liability under the FCPA or local anti-corruption laws. In addition, we cannot predict the nature, scope, or effect of future regulatory requirements to which our international operations might be subject or the manner in which existing laws might be administered or interpreted.
We are also subject to other laws and regulations governing our international operations, including regulations administered by the government of the U.S. and other countries in which we operate or plan to operate, including applicable export control regulations, economic sanctions on countries and persons, customs requirements, and currency exchange regulations, (collectively referred to as the “Trade Control laws”).
| 44 |
However, there is no assurance that we will be completely effective in ensuring our compliance with all applicable anti-corruption laws, including the FCPA or other legal requirements, including Trade Control laws. If we are not in compliance with the FCPA and other anti-corruption laws or Trade Control laws, we may be subject to criminal and civil penalties, disgorgement and other sanctions and remedial measures, and legal expenses, which could have an adverse impact on our business, financial condition, results of operations and liquidity, as well as our reputation. Likewise, any investigation of any potential violations of the FCPA, other anti-corruption laws or Trade Control laws by the U.S. or other authorities could also have an adverse impact on our reputation, our business, results of operations and financial condition.
Our proprietary information, or that of our suppliers and business partners, may be lost or we may suffer security breaches.
In the ordinary course of our business, we will collect and store sensitive data, including valuable and commercially sensitive intellectual property, clinical trial data, our proprietary business information and that of our suppliers and business partners, and personally identifiable information of our clinical trial subjects and employees, on our networks, and with our third-party cloud service providers. The secure processing, maintenance and transmission of this information is critical to our operations. Despite our security measures, our information technology and infrastructure, and that of our third parties, may be vulnerable to attacks by hackers or breached due to employee error, malfeasance, or other disruptions. Any breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost, or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, regulatory penalties, disrupt our operations, damage our reputation, and cause a loss of confidence in our products and our ability to conduct clinical trials, which could adversely affect our business and reputation and lead to delays in gaining regulatory approvals for Ketamir-2 and MIRA-55 or other product candidates.
Failure of our information technology systems, including cybersecurity attacks or other data security incidents, could significantly disrupt the operation of our business.
Our business is increasingly dependent on critical, complex, and interdependent information technology (“IT”) systems, including internet-based systems, some of which are managed or hosted by third parties, to support business processes as well as internal and external communications. The size and complexity of our IT systems make us potentially vulnerable to IT system breakdowns, malicious intrusion, and computer viruses, which may result in the impairment of our ability to operate our business effectively.
We are continuously evaluating and, where appropriate, enhancing our IT systems to address our planned growth, including to support our planned manufacturing operations. There are inherent costs and risks associated with implementing the enhancements to our IT systems, including potential delays in access to, or errors in, critical business and financial information, substantial capital expenditures, additional administrative time and operating expenses, retention of sufficiently skilled personnel to implement and operate the enhanced systems, demands on management time, and costs of delays or difficulties in transitioning to the enhanced systems, any of which could harm our business and results of operations. In addition, the implementation of enhancements to our IT systems may not result in productivity improvements at a level that outweighs the costs of implementation, or at all. In addition, our systems and the systems of our third-party providers and collaborators are potentially vulnerable to data security breaches which may expose sensitive data to unauthorized persons or to the public. Such data security breaches could lead to the loss of confidential information, trade secrets or other intellectual property, could lead to the public exposure of personal information (including personally identifiable information or individually identifiable health information) of our employees, clinical trial patients, customers, business partners, and others, could lead to potential identity theft, or could lead to reputational harm. Data security breaches could also result in loss of clinical trial data or damage to the integrity of that data. In addition, the increased use of social media by our employees and contractors could result in inadvertent disclosure of sensitive data or personal information, including but not limited to, confidential information, trade secrets and other intellectual property.
| 45 |
Any such disruption or security breach, as well as any action by us or our employees or contractors that might be inconsistent with the rapidly evolving data privacy and security laws and regulations applicable within the United States and elsewhere where we conduct business, could result in enforcement actions by U.S. states, the U.S. federal government or foreign governments, liability or sanctions under data privacy laws, including healthcare laws such as HIPAA, that protect certain types of sensitive information, regulatory penalties, other legal proceedings such as but not limited to private litigation, the incurrence of significant remediation costs, disruptions to our development programs, business operations and collaborations, diversion of management efforts and damage to our reputation, which could harm our business and operations. Because of the rapidly moving nature of technology and the increasing sophistication of cybersecurity threats, our measures to prevent, respond to and minimize such risks may be unsuccessful.
Security breaches, loss of data and other disruptions could compromise sensitive information related to our business, prevent us from accessing critical information or expose us to liability, which could adversely affect our business and our reputation.
In the ordinary course of our business, we, our vendors, and our third-party cloud service providers may collect and store sensitive data, including legally protected patient health information, credit card information, personally identifiable information about our employees and patients, intellectual property, and proprietary business information. We manage and maintain our applications and data utilizing cloud-based and on-site systems. These applications and data encompass a wide variety of business-critical information including research and development information, commercial information and business and financial information.
The secure processing, storage, maintenance, and transmission of this critical information is vital to our operations and business strategy, and we devote significant resources to protecting such information. Although we take measures to protect sensitive information from unauthorized access or disclosure, our information technology and infrastructure may be vulnerable to attacks by hackers, or viruses, breaches, or interruptions due to employee error, malfeasance or other disruptions, or lapses in compliance with privacy and security mandates. Any such virus, breach or interruption could compromise our networks and the information stored there could be accessed by unauthorized parties, publicly disclosed, lost or stolen. We have measures in place that are designed to prevent, and if necessary to detect and respond to such security incidents, breaches of privacy, and security mandates. However, in the future, any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, such as HIPAA in the United States and the General Data Protection Regulation in the European Union, or GDPR, government enforcement actions and regulatory penalties. Unauthorized access, loss or dissemination could also disrupt our operations, including our ability to process samples, provide test results, share and monitor safety data, bill payers or patients, provide customer support services, conduct research and development activities, process and prepare company financial information, manage various general and administrative aspects of our business and may damage our reputation, any of which could adversely affect our business, financial condition and results of operations.
Legislative or regulatory reform of the health care system in the U.S. may affect our ability to profitably sell our products, if approved.
Our ability to commercialize our future products successfully, alone or with collaborators, will depend in part on the extent to which coverage and reimbursement for the products will be available from government and health administration authorities, private health insurers and other third-party payers. The continuing efforts of the U.S. government, insurance companies, managed care organizations and other payers for health care services to contain or reduce health care costs may adversely affect our ability to set prices for our products which we believe are fair, and our ability to generate revenues and achieve and maintain profitability.
Specifically, in the U.S., there have been a number of legislative and regulatory proposals to change the health care system in ways that could affect our ability to sell our products profitably. For example, certain states in the U.S. are proposing legislation mandating publicly funded health program coverage of medical cannabis. In addition, the 2010 Affordable Care Act, or the ACA, substantially changed the way healthcare is financed by both governmental and private insurers. Both Congress and the U.S. President have already taken some actions that are intended to significantly limit the ACA, and we expect efforts to further modify or repeal the ACA to continue. The success and potential effects of these efforts to repeal or modify the ACA are not clear.
| 46 |
We expect additional federal and state legislative proposals for health care reform, which could limit the prices that can be charged for the products we develop and may limit our commercial opportunity.
The continuing efforts of government and other third-party payers to contain or reduce the costs of health care through various means may limit our commercial opportunity. It will be time-consuming and expensive for us to go through the process of seeking coverage and reimbursement from Medicare, Medicaid, and other governmental health programs and from private payers. Our products may not be considered cost-effective, and government and third-party private health insurance coverage and reimbursement may not be available to patients for any of our future products or sufficient to allow us to sell our products on a competitive and profitable basis. Our results of operations could be adversely affected by ACA, changes to the ACA, and by other health care reforms that may be enacted or adopted in the future. In addition, increasing emphasis on managed care in the U.S. will continue to put downward pressure on the pricing of pharmaceutical products. Cost-control initiatives could decrease the price that we or any potential collaborators could receive for any of our future products and could adversely affect our ability to generate revenue in the U.S. market and maintain profitability.
We may acquire other companies which could divert our management’s attention, result in additional dilution to our shareholders and otherwise disrupt our operations and harm our operating results.
We may in the future seek to acquire businesses, products, or technologies that we believe could complement or expand our product offerings, enhance our technical capabilities or otherwise offer growth opportunities. The pursuit of potential acquisitions may divert the attention of management and cause us to incur various expenses in identifying, investigating, and pursuing suitable acquisitions, whether or not they are consummated. If we acquire additional businesses, we may not be able to integrate the acquired personnel, operations and technologies successfully, effectively manage the combined business following the acquisition or realize anticipated cost savings or synergies. We also may not achieve the anticipated benefits from the acquired business due to a number of factors, including:
| ● | incurrence of acquisition-related costs; | |
| ● | diversion of management’s attention from other business concerns; |
| ● | unanticipated costs or liabilities associated with the acquisition; | |
| ● | harm to our existing business relationships with collaboration partners as a result of the acquisition; | |
| ● | harm to our brand and reputation; | |
| ● | the potential loss of key employees; | |
| ● | use of resources that are needed in other parts of our business; and | |
| ● | use of substantial portions of our available cash to consummate the acquisition. |
In the future, if our acquisitions do not yield expected returns, we may be required to take charges to our operating results arising from the impairment assessment process. Acquisitions may also result in dilutive issuances of equity securities or the incurrence of debt, which could adversely affect our operating results. In addition, if an acquired business fails to meet our expectations, our business, results of operations and financial condition may be adversely affected.
| 47 |
Risks Related to Development and Regulatory Approval of Our Product Candidates
Clinical trials for our product candidates are expensive, time-consuming, uncertain, and susceptible to change, delay or termination. The results of clinical trials are open to differing interpretations.
Clinical trials are expensive, time consuming and difficult to design and implement. Regulatory agencies may analyze or interpret the results differently than us. Even if the results of our clinical trials are favorable, the clinical trials for a number of our product candidates are expected to continue for several years and may take significantly longer to complete. In addition, we, the FDA, or other regulatory authorities, including state and local authorities, or an Institutional Review Board, or IRB, with respect to a trial at its institution, may suspend, delay or terminate our clinical trials at any time, require us to conduct additional clinical trials, require a particular clinical trial to continue for a longer duration than originally planned, require a change to our development plans such that we conduct clinical trials for a product candidate in a different order, e.g., in a step-wise fashion rather than running two trials of the same product candidate in parallel, or the DEA could suspend or terminate the registrations and quota allotments we require in order to procure and handle controlled substances, for various reasons, including:
| ● | lack of effectiveness of any product candidate during clinical trials; | |
| ● | discovery of serious or unexpected toxicities or side effects experienced by trial participants or other safety issues, such as drug interactions, including those which cause confounding changes to the levels of other concomitant medications; | |
| ● | slower than expected rates of subject recruitment and enrollment rates in clinical trials; | |
| ● | difficulty in retaining subjects who have initiated a clinical trial but may withdraw at any time due to adverse side effects from the therapy, insufficient efficacy, fatigue with the clinical trial process or for any other reason; | |
| ● | delays or inability in manufacturing or obtaining sufficient quantities of materials for use in clinical trials due to regulatory and manufacturing constraints; | |
| ● | inadequacy of or changes in our manufacturing process or product formulation; |
| ● | delays in obtaining regulatory authorization to commence a trial, including “clinical holds” or delays requiring suspension or termination of a trial by a regulatory agency, such as the FDA, before or after a trial is commenced; | |
| ● | changes in applicable regulatory policies and regulation, including changes to requirements imposed on the extent, nature, or timing of studies; | |
| ● | delays or failure in reaching agreement on acceptable terms in clinical trial contracts or protocols with prospective clinical trial sites; | |
| ● | uncertainty regarding proper dosing; | |
| ● | delay or failure to supply product for use in clinical trials which conforms to regulatory specification; | |
| ● | unfavorable results from ongoing pre-clinical studies and clinical trials; | |
| ● | failure of our contract research organizations, or CROs, or other third-party contractors to comply with all contractual requirements or to perform their services in a timely or acceptable manner; | |
| ● | failure by us, our employees, our CROs or their employees to comply with all applicable FDA or other regulatory requirements relating to the conduct of clinical trials or the handling, storage, security, and recordkeeping; | |
| ● | scheduling conflicts with participating clinicians and clinical institutions; | |
| ● | failure to design appropriate clinical trial protocols; | |
| ● | regulatory concerns with cannabinoid products generally and the potential for abuse; | |
| ● | insufficient data to support regulatory approval; | |
| ● | inability or unwillingness of medical investigators to follow our clinical protocols; or | |
| ● | difficulty in maintaining contact with patients during or after treatment, which may result in incomplete data. |
Any of the foregoing could have a material adverse effect on our business, results of operations and financial condition.
| 48 |
Clinical trials of synthetic cannabinoid drug candidates and ketamine analogs are novel with very limited or non-existing history; we face a significant risk that the trials will not result in commercially viable drugs and treatments.
At present, there is only a very limited documented clinical trial history from which we can derive any scientific conclusions for our product candidates or prove that our present assumptions for the current and planned research are scientifically compelling. The active pharmaceutical ingredient (or API) content shown in INDs can vary from one IND to another – hence it is not necessarily possible to extrapolate results from studies with one product and predict efficacy of safety with another product containing a similar API and different source. Whilst the principal synthetic cannabinoid component may be similar, the APIs may differ in terms of minor cannabinoid content, impurity profiles or degradant profiles. While we are encouraged by the results of clinical trials by others (where they exist), there can be no assurance that any pre-clinical study or clinical trial will result in in commercially viable drugs or treatments.
Clinical trials are expensive, time consuming and difficult to design and implement. We, as well as the regulatory authorities may suspend, delay or terminate our clinical trials at any time, may require us, for various reasons, to conduct additional clinical trials, or may require a particular clinical trial to continue for a longer duration than originally planned, including, among others:
| ● | lack of effectiveness of any API, formulation, or delivery system during clinical trials; |
| ● | discovery of serious or unexpected toxicities or side effects experienced by trial participants or other safety issues; | |
| ● | slower than expected rates of subject recruitment and enrollment rates in clinical trials; | |
| ● | delays or inability in manufacturing or obtaining sufficient quantities of GMP-grade materials for use in clinical trials due to regulatory and manufacturing constraints; | |
| ● | delays in obtaining regulatory authorization to commence a trial, including Institutional Review Board (“IRB”) approvals or DEA approvals, licenses required for obtaining and using synthetic cannabinoids or cannabinoid-like substances for research, either before or after a trial is commenced; | |
| ● | unfavorable results from ongoing pre-clinical studies and clinical trials; | |
| ● | patients or investigators failing to comply with clinical trial protocols; | |
| ● | patients failing to return for post-treatment follow-up at the expected rate; | |
| ● | sites participating in an ongoing clinical trial withdraw, requiring us to engage new sites; | |
| ● | third-party clinical investigators decline to participate in our clinical trials, do not perform the clinical trials on the anticipated schedule, or act in ways inconsistent with the established investigator agreement, clinical trial protocol, good clinical practices, and other IRB requirements; | |
| ● | third-party entities do not perform data collection and analysis in a timely or accurate manner or at all; or | |
| ● | regulatory inspections of our clinical trials require us to undertake corrective action or suspend or terminate our clinical trials. |
Any of the foregoing could have a material adverse effect on our business, results of operations and financial condition.
| 49 |
Any failure by us to comply with existing regulations could harm our reputation and operating results.
We are subject to extensive regulation by U.S. federal and state governments in each of the markets where we have product candidates progressing through the approval process.
We must also adhere to all regulatory requirements including FDA’s Good Laboratory Practice, Good Clinical Practice, and current Good Manufacturing Practices requirements (“cGMP”) pharmacovigilance requirements, advertising, and promotion restrictions, reporting and recordkeeping requirements. If we or our suppliers fail to comply with applicable regulations, including FDA pre-or post-approval cGMP requirements, then FDA could sanction us. Even if a drug is FDA-approved, regulatory authorities may impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-marketing trials. Ketamir-2 and MIRA-55, and any of our product candidates that may be approved in the U.S. in the future, will be subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage, distribution, import, export, advertising, promotion, sampling, recordkeeping and submission of safety and other post-market information, including both federal and state requirements in the U.S. In addition, manufacturers and manufacturers’ facilities are required to comply with extensive FDA requirements, including ensuring that quality control and manufacturing procedures conform to GMP. As such, we, and our contract manufacturers (in the event contract manufacturers are appointed in the future) are subject to continual review and periodic inspections to assess compliance with GMP. Accordingly, we and others with whom we work must continue to spend time, money, and effort in all areas of regulatory compliance, including manufacturing, production, quality control and quality assurance. We will also be required to report certain adverse reactions and production problems, if any, to the FDA, and to comply with requirements concerning advertising and promotion for our products. Promotional communications with respect to prescription drugs are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved label.
If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, or disagrees with the promotion, marketing or labeling of the product, it may impose restrictions on that product or us, including requiring withdrawal of the product from the market. If we fail to comply with applicable regulatory requirements, a regulatory agency or enforcement authority may:
| ● | issue untitled or warning letters; | |
| ● | seek to enjoin our activities; | |
| ● | impose civil or criminal penalties; | |
| ● | suspend regulatory approval; | |
| ● | suspend any of our ongoing clinical trials; | |
| ● | refuse to approve pending applications or supplements to approved applications submitted by us; | |
| ● | impose restrictions on our operations, including by requiring us to enter into a Corporate Integrity Agreement or closing our contract manufacturers’ facilities, if any; or | |
| ● | seize or detain products or require a product recall. |
In addition, any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. Any failure to comply with ongoing regulatory requirements may significantly and adversely affect our ability to commercialize and generate revenue from our product candidates. If regulatory sanctions are applied or if regulatory approval is withdrawn, the value of our business and our operating results may be adversely affected.
Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business and damage our reputation. We expend significant resources on compliance efforts and such expenses are unpredictable and might adversely affect our results. Changing laws, regulations and standards might also create uncertainty, higher expenses and increase insurance costs. As a result, we intend to invest all reasonably necessary resources to comply with evolving standards, and this investment might result in increased management and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities.
| 50 |
We are subject to federal and state healthcare laws and regulations and implementation of or changes to such healthcare laws and regulations could adversely affect our business and results of operations.
In the United States, there have been a number of legislative and regulatory proposals to change the healthcare system in ways that could impact our ability to sell our product candidates. If we are found to be in violation of any of these laws or any other federal or state regulations, we may be subject to administrative, civil and/or criminal penalties, damages, fines, individual imprisonment, exclusion from federal health care programs and the restructuring of our operations. Any of these could have a material adverse effect on our business and financial results. Since many of these laws have not been fully interpreted by the courts, there is an increased risk that we may be found in violation of one or more of their provisions. Any action against us for violation of these laws, even if we ultimately are successful in our defense, will cause us to incur significant legal expenses and divert our management’s attention away from the operation of our business.
We expect that the ACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we may receive for any approved product. There have been judicial challenges to certain aspects of the ACA and numerous legislative attempts to repeal and/or replace the ACA in whole or in part, and we expect there will be additional challenges and amendments to the ACA in the future. At this time, the full effect that the ACA will have on our business in the future remains unclear. An expansion in the government’s role in the U.S. healthcare industry may cause general downward pressure on the prices of prescription drug products, lower reimbursements, or any other product for which we obtain regulatory approval, reduce product utilization, and adversely affect our business and results of operations. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize products for which we may receive regulatory approval.
The regulatory approval processes with the FDA are lengthy and inherently unpredictable.
We are not permitted to market our drug candidates as medicines in the United States or other countries until we receive approval of a New Drug Application (“NDA”) from the FDA or in any foreign countries until we receive the approval from the regulatory authorities of such countries. Prior to submitting an NDA to the FDA for approval of our drug candidates we will need to have completed our pre-clinical studies and clinical trials and demonstrate that our products meet all applicable standards of identity, strength, quality, and purity throughout their expiration date. Successfully completing any clinical program and obtaining approval of an NDA is a complex, lengthy, expensive, and uncertain process, and the FDA (or other country medicines regulatory body) may delay, limit, or deny approval of product candidates for many reasons, including, among others, because:
| ● | an inability to demonstrate that our product candidates are safe and effective in treating patients to the satisfaction of the FDA; | |
| ● | results of clinical trials that may not meet the level of statistical or clinical significance required by the FDA; | |
| ● | disagreements with the FDA with respect to the number, design, size, conduct or implementation of clinical trials; | |
| ● | requirements by the FDA to conduct additional clinical trials; | |
| ● | disapproval by the FDA of certain formulations, labeling or specifications of product candidates; | |
| ● | findings by the FDA that the data from pre-clinical studies and clinical trials are insufficient; | |
| ● | findings by the FDA that our API or finished products do not meet all applicable standards of identity, strength, quality, and purity; | |
| ● | the FDA may disagree with the interpretation of data from pre-clinical studies and clinical trials; and | |
| ● | the FDA may change their approval policies or adopt new regulations. |
Any of these factors, many of which are beyond our control, could increase development time and / or costs or jeopardize our ability to obtain regulatory approval for our drug candidates.
| 51 |
There is a high rate of failure for drug candidates proceeding through clinical trials.
Generally, there is a high rate of failure for drug candidates proceeding through clinical trials. We may suffer significant setbacks in our clinical trials similar to the experience of a number of other companies in the pharmaceutical and biotechnology industries, even after receiving promising results in earlier trials. Further, even if we view the results of a clinical trial to be positive, FDA may disagree with our interpretation of the data. In the event that we obtain negative results from clinical trials for product candidates or other problems related to potential chemistry, manufacturing and control issues or other hurdles occur and our product candidates are not approved, we may not be able to generate sufficient revenue or obtain financing to continue our operations, our ability to execute on our current business plan may be materially impaired, our reputation in the industry and in the investment community might be significantly damaged and the price of our common stock could decrease significantly. In addition, our inability to properly design, commence and complete clinical trials may negatively impact the timing and results of our clinical trials and ability to seek approvals for our drug candidates.
If we are found in violation of federal or state “fraud and abuse” laws, we may be required to pay a penalty and/or be suspended from participation in federal or state health care programs, which may adversely affect our business, financial condition, and results of operations.
In the United States, we are subject to various federal and state health care “fraud and abuse” laws, including anti-kickback laws, false claims laws and other laws intended to reduce fraud and abuse in federal and state health care programs, which could affect us particularly upon successful commercialization of our products in the U.S. The Medicare and Medicaid Patient Protection Act of 1987, or federal Anti-Kickback Statute, makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf), to knowingly and willfully solicit, receive, offer or pay any remuneration that is intended to induce the referral of business, including the purchase, order or prescription of a particular drug for which payment may be made under a federal health care program, such as Medicare or Medicaid. Under federal law, some arrangements, known as safe harbors, are deemed not to violate the federal Anti-Kickback Statute. Although we seek to structure our business arrangements in compliance with all applicable requirements, it is often difficult to determine precisely how the law will be applied in specific circumstances. Accordingly, it is possible that our practices may be challenged under the federal Anti-Kickback Statute and Federal False Claims Act. Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including fines and/or exclusion or suspension from federal and state health care programs such as Medicare and Medicaid and debarment from contracting with the U.S. government. In addition, private individuals have the ability to bring actions on behalf of the government under the federal False Claims Act as well as under the false claims laws of several states.
Many states have adopted laws similar to the federal anti-kickback statute, some of which apply to the referral of patients for health care services reimbursed by any source, not just governmental payers. There are ambiguities as to what is required to comply with these state requirements and if we fail to comply with an applicable state law requirement, we could be subject to penalties.
Neither the government nor the courts have provided definitive guidance on the application of fraud and abuse laws to our business. Law enforcement authorities are increasingly focused on enforcing these laws, and it is possible that some of our practices may be challenged under these laws. While we believe we have structured our business arrangements to comply with these laws, it is possible that the government could allege violations of, or convict us of violating, these laws. If we are found in violation of one of these laws, we could be required to pay a penalty and could be suspended or excluded from participation in federal or state health care programs, and our business, results of operations and financial condition may be adversely affected.
| 52 |
Serious adverse events or other safety risks could require us to abandon development and preclude, delay or limit approval of our product candidates, limit the scope of any approved label or market acceptance, or cause the recall or loss of marketing approval of products that are already marketed.
If any of our product candidates prior to or after any approval for commercial sale, cause serious or unexpected side effects, or are associated with other safety risks such as misuse, abuse or diversion, a number of potentially significant negative consequences could result, including:
| ● | regulatory authorities may interrupt, delay or halt clinical trials; | |
| ● | regulatory authorities may deny regulatory approval of our product candidates; | |
| ● | regulatory authorities may require certain labeling statements, such as warnings or contraindications or limitations on the indications for use, and/or impose restrictions on distribution in the form of a REMS in connection with approval or post-approval; |
| ● | regulatory authorities may withdraw their approval, require more onerous labeling statements, impose a more restrictive Risk Evaluation and Mitigation Strategy (“REMS”), or require us to recall any product that is approved; | |
| ● | we may be required to change the way the product is administered or conduct additional clinical trials; | |
| ● | our relationships with our collaboration partners may suffer; | |
| ● | we could be sued and held liable for harm caused to patients; or | |
| ● | our reputation may suffer. The reputational risk is heightened with respect to those of our product candidates that are being developed for pediatric indications. |
We may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to participants or if preliminary data demonstrate that our product candidates are unlikely to receive regulatory approval or unlikely to be successfully commercialized. Following receipt of approval for commercial sale of a product we may voluntarily withdraw or recall that product from the market if at any time we believe that its use, or a person’s exposure to it, may cause adverse health consequences or death. To date we have not withdrawn, recalled, or taken any other action, voluntary or mandatory, to remove an approved product from the market. In addition, regulatory agencies, IRBs, or data safety monitoring boards may at any time recommend the temporary or permanent discontinuation of our clinical trials or request that we cease using investigators in the clinical trials if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements, or that they present an unacceptable safety risk to participants. Although we have never been asked by a regulatory agency, IRB, or data safety monitoring board to discontinue a clinical trial temporarily or permanently, if we elect or are forced to suspend or terminate a clinical trial of any of our product candidates, the commercial prospects for that product will be harmed and our ability to generate product revenue from that product may be delayed or eliminated. Furthermore, any of these events may result in labeling statements such as warnings or contraindications. In addition, such events or labeling could prevent us or our partners from achieving or maintaining market acceptance of the affected product and could substantially increase the costs of commercializing our product candidates and impair our ability to generate revenue from the commercialization of these products either by us or by our collaboration partners.
| 53 |
Risks Related to Our Reliance Upon Third Parties
Our existing collaboration arrangements and any that we may enter into in the future may not be successful, which could adversely affect our ability to develop and commercialize our product candidates.
We may seek additional collaboration arrangements with pharmaceutical or biotechnology companies for the development or commercialization of our product candidates. We may, with respect to our product candidates, enter into new arrangements on a selective basis depending on the merits of retaining commercialization rights for ourselves as compared to entering into selective collaboration arrangements with leading pharmaceutical or biotechnology companies for each product candidate, both in the U.S. and internationally. To the extent that we decide to enter into collaboration agreements, we will face significant competition in seeking appropriate collaborators and the terms of any collaboration or other arrangements that we may establish may not be favorable to us.
Any existing or future collaboration that we enter may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborators generally have significant discretion in determining the efforts and resources that they will apply to these collaborations. Disagreements between parties to a collaboration arrangement regarding development, intellectual property, regulatory or commercialization matters can lead to delays in the development process or commercialization of the applicable product candidate and, in some cases, termination of the collaboration arrangement. These disagreements can be difficult to resolve if neither of the parties has final decision-making authority. Any such termination or expiration could harm our business reputation and may adversely affect us financially.
We depend on a limited number of suppliers for materials and components required to manufacture our product candidates. The loss of these suppliers, or their failure to supply us on a timely basis, could cause delays in our current and future capacity and adversely affect our business.
We depend on a limited number of suppliers for the materials and components required to manufacture our product candidates. As a result, we may not be able to obtain sufficient quantities of critical materials and components in the future. A delay or interruption by our suppliers may also harm our business, results of operations and financial condition. In addition, the lead time needed to establish a relationship with a new supplier can be lengthy, and we may experience delays in meeting demand in the event we must switch to a new supplier. The time and effort to qualify for and, in some cases, obtain regulatory approval for a new supplier could result in additional costs, diversion of resources or reduced manufacturing yields, any of which would negatively impact our operating results. Our dependence on single-source suppliers exposes us to numerous risks, including the following: our suppliers may cease or reduce production or deliveries, raise prices or renegotiate terms; our suppliers may become insolvent or cease trading; we may be unable to locate a suitable replacement supplier on acceptable terms or on a timely basis, or at all; and delays caused by supply issues may harm our reputation, frustrate our customers and cause them to turn to our competitors for future needs.
We maintain our cash at financial institutions, at times in balances that exceed federally insured limits. The failure of financial institutions could adversely affect our ability to pay operational expenses or make other payments.
Our cash held in non-interest-bearing and interest-bearing accounts can at times exceed the Federal Deposit Insurance Corporation (“FDIC”) insurance limits. If such banking institutions were to fail, we could lose all or a portion of those amounts held in excess of such insurance limitations. In addition, even if account holders are ultimately made whole with respect to a future bank failure, account holders’ access to their accounts and assets held in their accounts may be substantially delayed. Any material loss that we may experience in the future or inability for a material time period to access our cash and cash equivalents could have an adverse effect on our ability to pay our operational expenses or make other payments, which could adversely affect our business.
| 54 |
We rely on, and expect to continue to rely on, third parties to conduct clinical trials for our product candidates. If these third parties do not successfully carry out their contractual duties, comply with regulatory requirements or meet expected deadlines, we may not be able to obtain marketing approval for or commercialize our product candidates, and our business could be substantially harmed.
We have agreements with third-party CROs to operationalize, provide monitors for and to manage data for our ongoing clinical trials. We rely heavily on these parties for the execution of clinical trials and control only certain aspects of their activities. As a result, we have less direct control over the start-up, conduct, timing and competition of these clinical trials, and the management of data developed through the clinical trials than would be the case if we were relying entirely upon our own staff. Communicating with outside parties can also be challenging, potentially leading to mistakes as well as difficulties in coordinating activities. However, we remain responsible for the conduct of these trials and are subject to enforcement which may include civil and criminal liabilities for any violations of FDA rules and regulations and the comparable foreign regulatory provisions during the conduct of our clinical trials. Outside parties may:
| ● | have staffing difficulties; | |
| ● | fail to comply with contractual obligations; | |
| ● | Devote inadequate resources to our clinical trials; | |
| ● | Experience regulatory compliance issues; | |
| ● | Undergo changes in priorities or become financially distressed; or | |
| ● | Form more favorable relationships with other entities, some of which may be our competitors. |
These factors, among others, may materially adversely affect the willingness or ability of third parties to conduct our clinical trials and may subject us to unexpected cost increases that are beyond our control. Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on CROs does not relieve us of our regulatory responsibilities. We and our CROs are required to comply with GCPs, which are guidelines enforced by the FDA, the competent authorities of the EU member states and equivalent competent authorities in foreign jurisdictions for any products in clinical development. The FDA and foreign regulatory authorities enforce these regulations and GCP guidelines through periodic inspections of clinical trial sponsors principal investigators, and trial sites, and IRBs. If we or our CROs fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or other equivalent competent authorities in foreign jurisdictions may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, the FDA or foreign regulatory authorities will determine that any of our clinical trials comply with GCPs. In addition, our clinical trials must be conducted with products produced under current Good Manufacturing Practices, or cGMPs and similar foreign requirements. Our failure or the failure of our CROs to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process and could also subject us to enforcement action up to and including civil and criminal penalties.
If any of our relationships with these third-party CROs terminate, we may not be able to enter into arrangements with alternative CROs. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain are compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, any such clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize, Ketamir-2, MIRA-55 or our other product candidates. As a result, our financial results and the commercial prospects for Ketamir-2 MIRA-55 or our other product candidates would be harmed, our costs could increase and our ability to generate revenue could be delayed.
| 55 |
We rely and expect to continue to rely on third parties to manufacture our clinical product supplies and clinical candidates, and we may rely on third parties for at least a portion of the manufacturing process of our product candidates, if approved. Our business could be harmed if those third parties fail to provide us with sufficient quantities of product supplies or product candidates or fail to do so at acceptable quality levels or prices.
We do not currently own any facility that may be used as a clinical-scale manufacturing and processing facility, and we rely on outside vendors and collaborators to manufacture supplies and process our product candidates. For certain of our components or product candidates, we rely on single suppliers or manufacturers to supply or manufacture, but we plan to expand the number of suppliers and manufacturers as we advance our product candidates through clinical development. Our product candidates are not yet manufactured or processed on a commercial scale and we may remain unable to do so for any of our product candidates. Although in the future we may develop our own manufacturing facilities, we may also continue to use third parties as part of our manufacturing processes and may, in any event, never be successful in developing our own manufacturing facilities. Our anticipated reliance on third-party manufacturers exposes us to the following risks:
| ● | We may be unable to identify manufacturers on acceptable terms or at all because the number of potential manufacturers is limited and the FDA must inspect any manufacturers for current cGMP. | |
| ● | Non-compliance of our third-party manufacturers with requirements of our marketing application(s). In addition, a new manufacturer would have to be educated in, or develop substantially equivalent processes for, the production of our product candidates. | |
| ● | Third-party manufacturers may have little or no experience with our product candidates, and therefore may require a significant amount of support from us in order to implement and maintain the infrastructure and processes required to manufacture our product candidates. | |
| ● | Third-party manufacturers might be unable to timely manufacture our product candidates or produce the quantity and quality required to meet our clinical and commercial needs, if any. | |
| ● | Third-party manufacturers may not be able to execute our manufacturing procedures and other logistical support requirements appropriately. | |
| ● | Third-party manufacturers may not perform as agreed, may not devote sufficient resources to our product candidates or may not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store, and distribute our products, if any. | |
| ● | Manufacturers are subject to ongoing periodic unannounced inspection by the FDA and corresponding state or foreign agencies to ensure strict compliance with cGMP and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards. | |
| ● | We may not own, or may have to share, the intellectual property rights to any improvements made by our third-party manufacturers in the manufacturing processes for our product candidates. | |
| ● | Our third-party manufacturers could breach or terminate their agreements with us, and we may be required to pay fees upon suspension or termination of the agreement even if the manufacturers do not deliver adequate supply of the product candidates or their components. | |
| ● | Raw materials and components used in the manufacturing processes, particularly those for which we have no other source or supplier, may not be available or may not be suitable or acceptable for use due to factors beyond our control. | |
| ● | Our third-party manufacturers may have unacceptable or inconsistent product quality success rates and yields, and we have no direct control over their ability to maintain adequate quality control, quality assurance and qualified personnel. |
| 56 |
Each of these risks could delay or prevent the completion of our clinical trials or the approval of any of our product candidates by the FDA, result in higher costs or adversely impact commercialization of our product candidates. In addition, we will rely on third parties to perform certain specification tests on our product candidates prior to delivery to patients. If these tests are not appropriately done and test data are not reliable, patients could be put at risk of serious harm and the FDA could place significant restrictions on our company until deficiencies are remedied. Furthermore, our or a third party’s failure to execute on our manufacturing requirements, to do so on commercially reasonable terms or to comply with cGMP could adversely affect our business in a number of ways, including:
| ● | An inability to initiate or continue clinical trials of our product candidates under development; | |
| ● | Delay in submitting regulatory applications, or receiving marketing approvals, for our product candidates; | |
| ● | Loss of the cooperation of future collaborators; | |
| ● | Subjecting third-party manufacturing facilities or our manufacturing facilities to additional inspections by regulatory authorities; | |
| ● | Requirements to cease development or to recall batches of our product candidates; and | |
| ● | In the event of approval to market and commercialize our product candidates, an inability to meet commercial demands for our product or any other future product candidates. |
If any CMO or CDMO with whom we contract fails to perform its obligations, we may be forced to enter into an agreement with a different CMO or CDMO, which we may not be able to do on reasonable terms, if at all. In such scenario, our clinical trials supply could be delayed significantly as we establish alternative supply sources. In some cases, the technical skills required to manufacture our products or product candidates may be unique or proprietary to the original CMO or CDMO and we may have difficulty, or there may be contractual restrictions prohibiting us from, transferring such skills to a back-up or alternate supplier, or we may be unable to transfer such skills at all. In addition, if we are required to change CMOs or CDMOs for any reason, we will be required to verify that the new CMO or CDMO maintains facilities and procedures that comply with quality standards and with all applicable regulations. We will also need to verify, such as through a manufacturing comparability study, that any new manufacturing process will produce our product candidate according to the specifications previously submitted to the FDA or another regulatory authority. The delays associated with the verification of a new CMO or CDMO could negatively affect our ability to develop product candidates or commercialize our products in a timely manner or within budget. In addition, changes in manufacturers often involve changes in manufacturing procedures and processes, which could require that we conduct bridging studies between our prior clinical supply used in our clinical trials and that of any new manufacturer. We may be unsuccessful in demonstrating the comparability of clinical supplies which could require the conduct of additional clinical trials.
We rely on, and expect to continue to rely on, third parties to conduct clinical trials for our product candidates. If these third parties do not successfully carry out their contractual duties, comply with regulatory requirements or meet expected deadlines, we may not be able to obtain marketing approval for or commercialize our product candidates, and our business could be substantially harmed.
We are dependent on third parties to conduct our clinical trials and preclinical and nonclinical studies. Specifically, we rely on, and intend to continue to rely on, medical institutions, clinical investigators, contract research organizations, or CROs, and consultants to conduct nonclinical studies and clinical trials, in each case in accordance with our study protocols and applicable regulatory requirements. These CROs, investigators and other third parties play a significant role in the conduct and timing of these studies or trials and the subsequent collection and analysis of data. Though we expect to carefully manage our relationships with our CROs, investigators and other third parties, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects. Further, while we have and will have agreements governing the activities of our third-party contractors, we have limited influence over their actual performance. Nevertheless, we are responsible for ensuring that each of our clinical trials is conducted in accordance with the applicable protocol and legal, regulatory and scientific standards and requirements, and our reliance on our CROs and other third parties does not relieve us of our regulatory responsibilities. In addition, we and our CROs are required to comply with GLP and GCP requirements, as applicable, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities related to the conduct of nonclinical studies and clinical trials, respectively. Regulatory authorities enforce GCPs through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of our CROs or trial sites fail to comply with applicable GLP or GCP or other requirements, the collected nonclinical data or the clinical data generated in our clinical trials may be deemed unreliable, and the FDA or comparable foreign regulatory authorities may require us to perform additional nonclinical studies or clinical trials before approving our marketing applications, if ever. Furthermore, our clinical trials must be conducted with materials manufactured in accordance with cGMP regulations. Failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
| 57 |
There is a risk that our CROs, investigators or other third parties will be unable to devote adequate time and resources to such trials or studies or perform as contractually required. If any of these third parties fail to meet expected deadlines, adhere to our clinical protocols or meet regulatory requirements, or otherwise perform in a substandard manner, our clinical trials may be extended, delayed or terminated. In addition, many of the third parties with whom we contract may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials or other development activities that could harm our competitive position. In addition, principal investigators for our clinical trials are expected to serve as scientific advisors or consultants to us from time to time and may receive cash or equity compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, or the FDA concludes that the financial relationship may have affected the interpretation of the study, the integrity of the data generated at the applicable clinical trial site may be questioned and the utility of the clinical trial itself may be jeopardized, which could result in the delay or rejection by the FDA of any NDA we submit. Any such delay or rejection could prevent us from receiving regulatory approval for, or commercializing, TELOIR-1 and any future product candidates.
Our CROs have the right to terminate their agreements with us in the event of an uncured material breach and under other specified circumstances. If any of our relationships with these third parties terminate, we may not be able to enter into arrangements with alternative third parties on commercially reasonable terms, in a timely manner or at all. Switching or adding CROs, investigators and other third parties involves additional cost and requires our management’s time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can materially impact our ability to meet our desired clinical development timelines. Though we work to carefully manage our relationships with our CROs, investigators and other third parties, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
Risks Related to Our Intellectual Property
We may not be able to adequately protect our product candidates or our proprietary technology in the marketplace.
Our success will depend, in part, on our ability to obtain patents, protect our trade secrets and operate without infringing on the proprietary rights of others. We may rely upon a combination of patents, trade secret protection (i.e., know-how), trademarks, licenses, and confidentiality agreements to protect the intellectual property of our product candidates. The strengths of patents in the pharmaceutical field involve complex legal and scientific questions and can be uncertain. Where appropriate, we seek patent protection for certain aspects of our products and technology. However, patent protection for naturally occurring compounds is exceedingly difficult to obtain, defend and enforce. Filing, prosecuting and defending patents throughout the world would be prohibitively expensive, so our policy is to look to patent technologies with commercial potential in jurisdictions with significant commercial opportunities. However, patent protection may not be available for some of the products or technology we are developing. If we must spend significant time and money protecting, defending, or enforcing our patents, designing around patents held by others or licensing, potentially for large fees, patents or other proprietary rights held by others, our business, results of operations and financial condition may be harmed. We may not develop additional proprietary products that are patentable.
| 58 |
The patent positions of pharmaceutical products are complex and uncertain. The scope and extent of patent protection for our product candidates are particularly uncertain. To date, our principal product candidates have been based on specific formulations of certain previously known cannabinoids found in nature in the cannabis sativa plant. While we have sought patent protection, where appropriate, directed to, among other things, composition-of-matter for our specific formulations, their methods of use, and methods of manufacture, we do not have and will not be able to obtain composition of matter protection on these previously known cannabinoids per se. We anticipate that the products we develop in the future will continue to be based on the same or other naturally occurring compounds, as well as additional synthetic compounds we may discover. Although we have sought and expect to continue to seek patent protection for our product candidates, their methods of use, and methods of manufacture, any, or all of them may not be subject to effective patent protection. If any of our products are approved and marketed for an indication for which we do not have an issued patent, our ability to use our patents to prevent a competitor from commercializing a non-branded version of our commercial products for that non-patented indication could be significantly impaired or even eliminated.
Publication of information related to our product candidates by us, or others may prevent us from obtaining or enforcing patents relating to these products and product candidates. Furthermore, others may independently develop similar products, may duplicate our products, or may design around our patent rights. In addition, any of our issued patents may be opposed and/or declared invalid or unenforceable. If we fail to adequately protect our intellectual property, we may face competition from companies who attempt to create a generic product to compete with our product candidates. We may also face competition from companies who develop a substantially similar product to one of our product candidates that is not covered by any of our patents.
If third parties claim that our intellectual property, products, processes, or anything else used by us infringes upon their intellectual property, our operating profits could be adversely affected.
There is a substantial amount of litigation, both within and outside the U.S., involving patent and other intellectual property rights in the pharmaceutical industry. We may, from time to time, be notified of claims that we are infringing upon patents, trademarks, copyrights, or other intellectual property rights owned by third parties, and we cannot provide assurances that other companies will not, in the future, pursue such infringement claims against us, our commercial partners or any third-party proprietary technologies we have licensed. If we were found to infringe upon a patent or other intellectual property right, or if we failed to obtain or renew a license under a patent or other intellectual property right from a third party, or if a third party that we were licensing technologies from was found to infringe upon a patent or other intellectual property rights of another third party, we may be required to pay damages, including damages of up to three times the damages found or assessed, if the infringement is found to be willful, suspend the manufacture of certain products or reengineer or rebrand our products, if feasible, or we may be unable to enter certain new product markets. Any such claims could also be expensive and time consuming to defend and divert management’s attention and resources. Our competitive position could suffer as a result. In addition, if we have declined or failed to enter into a valid non-disclosure or assignment agreement for any reason, we may not own the invention or our intellectual property, and our products may not be adequately protected. Thus, we cannot guarantee that our product candidates, or our commercialization thereof, does not and will not infringe any third party’s intellectual property.
| 59 |
If we are unable to obtain and maintain intellectual property protection for our technology and products, or if the scope of the intellectual property protection obtained is not sufficiently broad, our competitors could commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be impaired.
Our success depends in large part on our ability to obtain and maintain patent protection in relevant countries with respect to our proprietary technology and products. We seek to protect our proprietary position by filing patent applications in the United States and internationally that are related to our novel technologies and product candidates. This patent portfolio includes issued patents and pending patent applications covering pharmaceutical compositions and methods of use.
The patent prosecution process is expensive and time-consuming, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. We may choose not to seek patent protection for certain innovations and may choose not to pursue patent protection in certain jurisdictions, and under the laws of certain jurisdictions, patents or other intellectual property rights may be unavailable or limited in scope. It is also possible that we will fail to identify patentable aspects of our discovery and nonclinical development output before it is too late to obtain patent protection. Moreover, in some circumstances, we may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the patents, covering technology that we license from third parties. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation. In addition, the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. For example, India and China do not allow patents for methods of treating the human body. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our owned or licensed patents or pending patent applications, or that we were the first to file for patent protection of such inventions. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. Our pending and future patent applications may not result in patents being issued which protect our technology or products, in whole or in part, or which effectively prevent others from commercializing competitive technologies and products. Changes in either the patent laws or interpretation of the patent laws in the EU, the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection.
The risks described pertaining to our patents and other intellectual property rights also apply to the intellectual property rights that we license, and any failure to obtain, maintain and enforce these rights could have a material adverse effect on our business. In some cases, we may not have control over the prosecution, maintenance or enforcement of the patents that we license, and our licensors may fail to take the steps that we believe are necessary or desirable in order to obtain, maintain and enforce the licensed patents. Any inability on our part to protect adequately our intellectual property may have a material adverse effect on our business, operating results and financial position.
The USPTO and various non-U.S. governmental patent agencies require compliance with several procedural, documentary, fee payment and other similar provisions during the patent application process. In certain situations, non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to enter the market and this circumstance would have a material adverse effect on our business.
In addition, we acquired rights to Ketomir-2 through a license agreement with MIRALOGX and may in the future enter into other license agreements with third parties for other intellectual property rights or assets. These license agreements may impose various diligence, milestone payment, royalty, and other obligations on us. If we fail to comply with our obligations under these agreements, or we are subject to a bankruptcy, we may be required to make certain payments to the licensor, we may lose the exclusivity of our license, or the licensor may have the right to terminate the license, in which event we would not be able to develop or market products covered by the license. Additionally, the milestone and other payments associated with these licenses will make it less profitable for us to develop our drug candidates than if we had developed the licensed technology internally.
| 60 |
In some cases, patent prosecution of our licensed technology may be controlled solely by the licensor. If our licensors fail to obtain and maintain patent or other protection for the proprietary intellectual property we license from them, we could lose our rights to the intellectual property or our exclusivity with respect to those rights, and our competitors could market competing products using the intellectual property. In certain cases, we may control the prosecution of patents resulting from licensed technology. In the event we breach any of our obligations related to such prosecution, we may incur significant liability to our licensing partners. If disputes over intellectual property and other rights that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates.
We have no patent protection for MIRA-55, which could adversely impact MIRA-55’s potential competitive position.
We have no issued patents relating to MIRA-55 and our patent application for MIRA-55 may not result in an issued patent. While we attempt to protect our proprietary information as trade secrets through certain agreements with our employees, consultants, agents and other organizations to which we disclose our proprietary information, we cannot give assurance that these agreements will provide effective protection for our proprietary information in the event of unauthorized use or disclosure of such information. If other products similar to MIRA-55 are approved and marketed, we may be unable to prevent them from competing with MIRA-55 in MIRA-55’s potential marketplace. We expect that the presence of one or more competing products could reduce our potential market share and could negatively impact potential price levels and third-party reimbursement for MIRA-55, any of which would materially affect our business.
Risks Relating to the Ownership of our Common Stock
Because of the speculative nature of investment risk, you may lose your entire investment.
An investment in our securities carries a high degree of risk and should be considered as a speculative investment. We have a limited operating history, no revenues, have not paid dividends, and are unlikely to pay dividends in the immediate or near future. The likelihood of our success must be considered in light of the problems, expenses, difficulties, complications and delays frequently encountered in connection with the establishment of any business. An investment in our securities may result in the loss of an investor’s entire investment. Only potential investors who are experienced in high-risk investments and who can afford to lose their entire investment should consider an investment in our securities.
Certain of our founding stockholders, plus our existing officers and directors, control a substantial interest in us and thus may influence certain actions requiring stockholder vote.
Our founding stockholders, which include the Bay Shore Trust, and MIRALOGX, collectively own in excess of 30% of our issued and outstanding common stock. Our officers and directors also own shares of our common stock. Therefore, these entities and individuals could influence the outcome of matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions.
Sales of a significant number of shares of our common stock in the public markets, or the perception that such sales could occur, could depress the market price of our common stock.
Sales of a significant number of shares of our common stock in the public markets, or the perception that such sales could occur as a result of our utilization of a universal shelf registration statement or otherwise could depress the market price of our common stock and impair our ability to raise capital through the sale of additional equity securities. Notably, a large number of shares of our common stock held by Bay Shore Trust and MIRALOGX have been registered for public resale and could be sold in the public market, depressing our stock price. Moreover, we cannot in general predict the effect that future sales of our common stock or the market perception that we are permitted to sell a significant number of our securities would have on the market price of our common stock.
| 61 |
The requirements of being a public company may strain our resources, divert management’s attention and affect our ability to attract and retain executive management and qualified board members.
As a reporting issuer, we are subject to the reporting requirements of applicable securities legislation of the jurisdiction in which we are a reporting issuer, the listing requirements of Nasdaq and other applicable securities rules and regulations. Compliance with these rules and regulations increase our legal and financial compliance costs, make some activities more difficult, time-consuming or costly and increase demand on its systems and resources. Applicable securities laws require us to, among other things, file certain annual and quarterly reports with respect to its business and results of operations. In addition, applicable securities laws require us to, among other things, maintain effective disclosure controls and procedures and internal control over financial reporting.
In order to maintain and, if required, improve its disclosure controls and procedures and internal control over financial reporting to meet this standard, significant resources and management oversight are required and, as a result, management’s attention may be diverted from other business concerns, which could harm our business and results of operations. To comply with these requirements, we may need to hire more employees in the future or engage outside consultants, which will increase its costs and expenses.
In addition, changing laws, regulations and standards relating to corporate governance and public disclosure are creating uncertainty for public companies, increasing legal and financial compliance costs and making some activities more time consuming. These laws, regulations and standards are subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We intend to continue to invest resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of management’s time and attention from revenue-generating activities to compliance activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to their application and practice, regulatory authorities may initiate legal proceedings against us, which could adversely affect our business and financial results.
As a public company subject to these rules and regulations, it may be more expensive to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These factors could also make it more difficult for us to attract and retain qualified members of the Board, particularly to serve on the Audit Committee and Compensation Committee, and qualified executive officers.
We are an “emerging growth company,” and any decision on our part to comply only with certain reduced reporting and disclosure requirements applicable to emerging growth companies could make shares of our common stock less attractive to investors.
We are an “emerging growth company,” as defined in Section 2(a) of the Securities Act. For as long as we continue to be an emerging growth company, we may choose to take advantage of exemptions from various reporting requirements applicable to other public companies that are not emerging growth companies, including, but not limited to, not being required to have our independent registered public accounting firm audit our internal control over financial reporting under Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We could be an emerging growth company until the fifth anniversary of the fiscal year end date following the completion of our initial public offering, however, our status would change more quickly if we have more than US$1.235 billion in annual revenue, if the market value of our shares of common stock held by non-affiliates equals or exceeds US$700 million as of June 30 of any year, or we issue more than US$1.0 billion of non-convertible debt over a three-year period before the end of that period.
| 62 |
Investors could find our shares less attractive if we choose to rely on these exemptions. If some investors find shares less attractive as a result of any choice to reduce future disclosure, there may be a less active trading market for our shares and our share price may be more volatile.
For as long as we are an “emerging growth company”, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal controls over financial reporting pursuant to Section 404. We could be an “emerging growth company” until the fifth anniversary of the fiscal year end date following the completion of our initial public offering. An independent assessment of the effectiveness of our internal controls could detect problems that our management’s assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation.
If we identify material weaknesses in our internal control over financial reporting, or if we are unable to comply with the requirements of Section 404 in a timely manner or assert that our internal control over financial reporting is effective, or if our independent registered public accounting firm is unable to express an opinion as to the effectiveness of our internal control over financial reporting when required, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our securities could be negatively affected, and we could become subject to investigations by the stock exchange on which our securities are listed, the SEC, or other regulatory authorities, which could require additional financial and management resources.
Additionally, we are a “smaller reporting company” as defined in Item 10(f)(1) of Regulation S-K. Smaller reporting companies may take advantage of certain reduced disclosure obligations, including, among other things, providing only two years of audited financial statements. We will remain a smaller reporting company until the last day of any fiscal year for so long as either: (i) the market value of our shares of common stock held by non-affiliates does not equal or exceed $250 million as of the prior June 30th; or (ii) our annual revenues did not equal or exceed $100 million during such completed fiscal year. To the extent we take advantage of such reduced disclosure obligations, it may also make the comparison of our financial statements with other public companies difficult or impossible.
If we fail to maintain compliance with Nasdaq Listing Rules, our shares may be delisted from Nasdaq, which would result in a limited trading market for our shares and make obtaining future debt or equity financing more difficult for the us.
Our common stock is listed on the Nasdaq Capital Market under the symbol “MIRA”. However, there is no assurance that we will be able to continue to maintain our compliance with the Nasdaq continued listing requirements. If we fail to do so, our securities may lose their status on Nasdaq and they would likely be traded on the over-the-counter markets, including the Pink Sheets market. As a result, selling our securities could be more difficult because smaller quantities of shares or warrants would likely be bought and sold, transactions could be delayed, and security analysts’ coverage of us may be reduced. In addition, in the event our securities are delisted, broker dealers would bear certain regulatory burdens which may discourage broker dealers from effecting transactions in the securities and further limit the liquidity of the securities. These factors could result in lower prices and larger spreads in the bid and ask prices for the securities. Such delisting from Nasdaq and continued or further declines in the share price of the securities could also greatly impair our ability to raise additional necessary capital through equity or debt financing and could significantly increase the ownership dilution to shareholders caused by our issuing equity in financing or other transactions.
If our shares were to be delisted from Nasdaq, they may become subject to the SEC’s “penny stock” rules.
Delisting from Nasdaq may cause our securities to become subject to the SEC’s “penny stock” rules. The SEC generally defines a penny stock as an equity security that has a market price of less than $5.00 per share or an exercise price of less than $5.00 per share, subject to certain exemptions. One such exemption is to be listed on Nasdaq. Therefore, if shares of our common stock were to be delisted from Nasdaq, our securities could become subject to the SEC’s “penny stock” rules. These rules require, among other things, that any broker engaging in a purchase or sale of our securities provide its customers with: (i) a risk disclosure document, (ii) disclosure of market quotations, if any, (iii) disclosure of the compensation of the broker and its salespersons in the transaction, and (iv) monthly account statements showing the market values of our securities held in the customer’s accounts. A broker would be required to provide the bid and offer quotations and compensation information before effecting the transaction. This information must be contained in the customer’s confirmation. Generally, brokers are less willing to effect transactions in penny stocks due to these additional delivery requirements. These requirements may make it more difficult for shareholders to purchase or sell the shares of our common stock. Since the broker, not us, prepares this information, we would not be able to assure that such information is accurate, complete or current.
| 63 |
Some provisions of Florida law and our amended and restated articles of incorporation and amended and restated bylaws may have anti-takeover effects that could discourage an acquisition of us by others, even if an acquisition would be beneficial to our shareholders and may prevent attempts by our shareholders to replace or remove our current management.
Our status as a Florida corporation and the anti-takeover provisions of the Florida Business Corporation Act, which we sometimes refer to as the FBCA, may discourage, delay or prevent a change in control even if a change in control would be beneficial to our shareholders.
The control share acquisition statute, Section 607.0902 of the FBCA, generally provides that in the event a person acquires voting shares of the company in excess of 20% of the voting power of all of our issued and outstanding shares, such acquired shares will not have any voting rights unless such rights are restored by the holders of a majority of the votes of each class or series entitled to vote separately, excluding shares held by the person acquiring the control shares or any of our officers or employees who are also directors of the company. Certain acquisitions of shares are exempt from these rules, such as shares acquired pursuant to the laws of intestate succession or pursuant to a gift or testamentary transfer, pursuant to a merger or share exchange effected in compliance with the FBCA if we are a party to the agreement, or pursuant to an acquisition of our shares if the acquisition has been approved by our board of directors before the acquisition. The control share acquisition statute generally applies to any “issuing public corporation,” which means a Florida corporation which has:
| ● | One hundred or more shareholders; | |
| ● | Its principal place of business, its principal office, or substantial assets within Florida; and | |
| ● | Either (i) more than 10% of its shareholders are resident in Florida; (ii) more than 10% of its shares are owned by residents of Florida; or (iii) one thousand shareholders are resident in Florida. |
The affiliated transaction (or so-called “business combination”) statute, Section 607.0901 of the FBCA, provides that we may not engage in certain mergers, consolidations, sales of assets, issuances of stock, reclassifications, recapitalizations, and other affiliated transactions with any “interested shareholder” for a period of three years following the time that such shareholder became an interested shareholder, unless:
| ● | Prior to the time that such shareholder became an interested shareholder, our board of directors approved either the affiliated transaction or the transaction which resulted in the shareholder becoming an interested shareholder; or; | |
| ● | Upon consummation of the transaction that resulted in the shareholder becoming an interested shareholder, the interested shareholder owned at least 85% of our voting shares outstanding at the time the transaction commenced; or | |
| ● | At or subsequent to the time that such shareholder became an interested shareholder, the affiliated transaction is approved by our board of directors and authorized at an annual or special meeting of shareholders, and not by written consent, by the affirmative vote of at least two-thirds of the outstanding voting shares which are not owned by the interested shareholder. |
An “interested shareholder” is generally defined as any person who is the beneficial owner of more than 15% of our outstanding voting shares. Currently, Bay Shore Trust would be considered an “interested shareholder.”
| 64 |
The voting requirements set forth above do not apply to a particular affiliated transaction if one or more conditions are met, including, but not limited to, the following: if the affiliated transaction has been approved by a majority of our disinterested directors; if we have not had more than 300 shareholders of record at any time during the three years preceding the date the affiliated transaction is announced; if the interested shareholder has been the beneficial owner of at least 80% of our outstanding voting shares for at least three years preceding the date the affiliated transaction is announced; or if the consideration to be paid to the holders of each class or series of voting shares in the affiliated transaction meets certain requirements of the statute with respect to form and amount, among other things.
Both the control share acquisition statute and the affiliated transactions statute may have the effect of discouraging or preventing certain change of control or takeover transactions involving us.
In addition, our amended and restated articles of incorporation and amended and restated bylaws contain provisions that may make it more difficult for a third party to acquire us or increase the cost of acquiring us, even if doing so would benefit our shareholders, including transactions in which shareholders might otherwise receive a premium for their shares. These provisions include:
| ● | nothing in our amended and restated articles of incorporation precludes future issuances without shareholder approval of the authorized but unissued shares of our common stock; | |
| ● | advance notice procedures apply for shareholders to nominate candidates for election as directors or to bring matters before an annual meeting of shareholders; | |
| ● | a special meeting of shareholders can only be called by our chairman of the board of directors, our chief executive officer, our president (in the absence of a chief executive officer), a majority of our board of directors or the holders of 10% or more of all of our votes entitled to be cast on any issue proposed to be considered at the special meeting of shareholders; | |
| ● | no provision in our amended and restated articles of incorporation or amended and restated bylaws provides for cumulative voting, which limits the ability of minority shareholders to elect director candidates; | |
| ● | directors will only be able to be removed for cause; | |
| ● | our amended and restated articles of incorporation authorizes undesignated preferred stock, the terms of which may be established and shares of which may be issued, without the approval of the holders of our capital stock; and | |
| ● | certain litigation against us can only be brought in Florida. |
These provisions could discourage, delay or prevent a transaction involving a change in control of our company. These provisions could also discourage proxy contests and make it more difficult for you and other shareholders to elect directors of your choosing and cause us to take corporate actions other than those you desire. See “Description of Capital Stock.”
Our amended and restated bylaws designates the state courts located within the state of Florida as the exclusive forum for substantially all disputes between us and our shareholders and the federal district courts as the exclusive forum for Securities Act claims, which could limit our shareholders’ ability to obtain a favorable judicial forum for disputes with us.
Our amended and restated bylaws provide that, unless we consent in writing to the selection of an alternative forum, the sole and exclusive forum for (i) any derivative action or proceeding brought on our behalf, (ii) any action asserting a claim of breach of a fiduciary duty owed by any of our current or former directors, officers or other employees to us or our shareholders, (iii) any action arising pursuant to any provision of the FBCA, our amended and restated articles of incorporation or our amended and restated bylaws, or (iv) any other action asserting a claim that is governed by the internal affairs doctrine shall be a state court located within the state of Florida (or, if a state court located within the state of Florida does not have jurisdiction, the federal district court for the Middle District of Florida); provided that, the exclusive forum provision will not apply to suits brought to enforce any liability or duty created by the Exchange Act, or to any claim for which the federal courts have exclusive jurisdiction. Our amended and restated bylaws also provide that, unless we consent in writing to the selection of an alternative forum, the U.S. federal district courts shall be the exclusive forum for the resolution of any claims arising under the Securities Act. Under the Securities Act, federal and state courts have concurrent jurisdiction over all suits brought to enforce any duty or liability created by the Securities Act, and investors cannot waive compliance with the federal securities laws and the rules and regulations thereunder. Accordingly, there is uncertainty as to whether a court would enforce such a forum selection provision as written in connection with claims arising under the Securities Act.
| 65 |
By becoming a shareholder in our company, you will be deemed to have notice of and have consented to the provisions of our amended and restated bylaws related to choice of forum. The choice of forum provisions in our amended and restated bylaws may limit our shareholders’ ability to obtain a favorable judicial forum for disputes with us. Additionally, the enforceability of choice of forum provisions in other companies’ governing documents has been challenged in legal proceedings, and it is possible that, in connection with any applicable action brought against us, a court could find the choice of forum provisions contained in our amended and restated bylaws to be inapplicable or unenforceable in such action. If so, we may incur additional costs associated with resolving such action in other jurisdictions, which could harm our business, results of operations, and financial condition.
Securities or industry analysts may not regularly publish reports on us, which could cause the price of our securities or trading volumes to decline.
The trading market for our securities could be influenced by research and reports that industry and/or securities analysts may publish us, our business, the market or our competitors. We do not have any control over these analysts and cannot be assured that such analysts will cover us or provide favorable coverage. If any of the analysts who may cover our business change their recommendation regarding our securities adversely, or provide more favorable relative recommendations about our competitors, the price of our securities would likely decline. If any analysts who may cover our business were to cease coverage or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause the price of our securities or trading volumes to decline.
We will likely conduct further offerings of our equity securities in the future, in which case your proportionate interest may become diluted.
We will likely be required to conduct equity offerings in the future to finance our current projects or to finance subsequent projects that we decide to undertake. If our common stock shares are issued in return for additional funds, the price per share could be lower than that paid by our current shareholders. We anticipate continuing to rely on equity sales of our common stock shares in order to fund our business operations. If we issue additional common stock shares or securities convertible into shares of our common stock, your percentage interest in us could become diluted.
We may issue shares of preferred stock in the future, which could make it difficult for another company to acquire us or could otherwise adversely affect holders of our common stock, which could depress the price of our common stock.
Our certificate of incorporation authorizes us to issue one or more series of preferred stock. Our board of directors will have the authority to determine the preferences, limitations and relative rights of the shares of preferred stock and to fix the number of shares constituting any series and the designation of such series, without any further vote or action by our shareholders. Our preferred stock could be issued with voting, liquidation, dividend and other rights superior to the rights of our common stock. The potential issuance of preferred stock may delay or prevent a change in control of us, discouraging bids for our common stock at a premium to the market price, and materially adversely affect the market price and the voting and other rights of the holders of our common stock.
We have never declared or paid any cash dividends or distributions on our capital stock. We do not anticipate paying any cash dividends on our common stock in the foreseeable future.
We have never declared or paid any cash dividends or distributions on our capital stock. We currently intend to retain our future earnings, if any, to support operations and to finance expansion and therefore we do not anticipate paying any cash dividends on our common stock in the foreseeable future.
| 66 |
The declaration, payment and amount of any future dividends will be made at the discretion of the board of directors, and will depend upon, among other things, the results of our operations, cash flows and financial condition, operating and capital requirements, and other factors as the board of directors considers relevant. There is no assurance that future dividends will be paid, and, if dividends are paid, there is no assurance with respect to the amount of any such dividend.
Item 1B. Unresolved Staff Comments.
None.
Item 1C. Cybersecurity.
Cybersecurity Risk Management and Strategy
We recognize the importance of assessing, identifying, and managing material risks associated with cybersecurity threats, as such term is defined in Item 106(a) of Regulation S-K. These risks include, among other things: operational risks, intellectual property theft, fraud, extortion, harm to employees or customers and violation of data privacy or security laws.
Identifying and assessing cybersecurity risk is integrated into our overall risk management systems and processes. Cybersecurity risks related to our business, technical operations, privacy and compliance issues are identified and addressed through a multi-faceted approach including third party assessments, internal IT Audit, IT security, governance, risk and compliance reviews. To defend, detect and respond to cybersecurity incidents, we, among other things: conduct proactive privacy and cybersecurity reviews of systems and applications, audit applicable data policies, conduct employee training, monitor emerging laws and regulations related to data protection and information security and implement appropriate changes.
Our risk management program also assesses third party risks, and we perform third-party risk management to identify and mitigate risks from third parties such as vendors, suppliers, and other business partners associated with our use of third-party service providers. Cybersecurity risks are evaluated when determining the selection and oversight of applicable third-party service providers and potential fourth-party risks when handling and/or processing our employee, business or customer data.
To date, we have not identified any cybersecurity threats or past incidents that have had, or are likely to have, a material impact on our company’s operations, business strategy, financial performance, or results of operations.
Cybersecurity Governance
To manage our cybersecurity governance, we use Coalition Control, a cyber risk management platform that combines insurance, technology, and services from Coalition and its partners into an online experience. It allows us to detect, assess, and mitigate cyber risks proactively. Coalition Control monitors and detects risks across our entire external digital footprint, including assets, apps, services, and data leaks. The tool shows us where any potential vulnerabilities are identified and how to fix them.
Our CFO is responsible for the day-to-day oversight of cybersecurity risks, and who utilizes the Coalition management platform for such risk management. Our CFO keeps the Board apprised of ongoing cybersecurity risk mitigation and any breaches if presented.
Item 2. Description of Property.
Refer to Note 2, “Leases” to our consolidated financial statements included in Part IV of this Report on Form 10-K, which is incorporated into this item by reference.
Item 3. Legal Proceedings.
None
Item 4. Mine Safety Disclosures.
Not applicable.
| 67 |
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information
Our common stock began trading August 3, 2023, on The Nasdaq Capital Market under the symbol “MIRA.”
Holders of Common Stock
As of March 28, 2024, we had approximately 91 holders of record of our common stock. No cash dividends have been paid on the common stock to date. We currently intend to retain earnings for further business development and do not expect to pay cash dividends in the foreseeable future.
Securities Authorized for Issuance Under Equity Compensation Plans
See Item 12. - Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Unregistered Sales of Equity Securities and Use of Proceeds
None
Issuer Purchases of Equity Securities
None
Item 6. Reserved
| 68 |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis provide information which our management believes is relevant to an assessment and understanding of our results of operations and financial condition. You should read the following discussion and analysis of our results of operations and financial condition together with our financial statements and related notes and other information included elsewhere in this Report.
In addition to historical financial information, this discussion contains forward-looking statements based upon our current expectations that involve risks and uncertainties. Our actual results could differ materially from such forward-looking statements as a result of various factors, including those set forth under “Risk Factors” and “Cautionary Note Regarding Forward-Looking Statements” included elsewhere in this Report. Additionally, our historical results are not necessarily indicative of the results that may be expected for any period in the future.
Overview
We are a pre-clinical-stage pharmaceutical development company with two neuroscience programs targeting a broad range of neurologic and neuropsychiatric disorders. We have an exclusive licensing agreement for Ketamir-2, a unique, patent pending novel oral ketamine analog under investigation to potentially deliver ultra-rapid antidepressant effects, providing hope for individuals battling treatment-resistant depression (TRD), major depressive disorder with suicidal ideation (MDSI) and potentially post-traumatic stress disorder (PTSD).
Additionally, our novel oral pharmaceutical marijuana, MIRA-55, is currently under investigation for treating adult patients suffering from anxiety and cognitive decline, often associated with early-stage dementia. MIRA-55, if approved by the FDA, could mark a significant advancement in addressing various neuropsychiatric, inflammatory, and neurologic diseases and disorders.
The U.S. Drug Enforcement Administration (DEA)’s scientific review of Ketamir-2 concluded that it would not be considered a controlled substance or listed chemical under the Controlled Substances Act (CSA) and its governing regulations. Additionally, we have filed the required paperwork for MIRA-55 to be evaluated by the U.S. DEA.
We had net losses of $12 million and $7.1 million for the year ended December 31, 2023 and December 31, 2022, respectively.
Recent Developments
In early February 2024, we made a significant discovery during the manufacturing and scale-up process of our patented molecule known as “MIRA1a,” which we had been utilizing with a contract manufacturer. Through this process, we identified a novel and improved version of the molecule, MIRA-55. MIRA-55 exhibits enhanced potency and holds promise for improved efficacy compared to MIRA1a.
As part of our due diligence and subsequent testing, we discovered that the pre-clinical studies we conducted, previously attributed to MIRA1a, were in fact performed on MIRA-55. Following this revelation, we promptly filed a provisional patent for MIRA-55, which encompasses all pre-clinical studies disclosed in our two registration statements on Form S-1, declared effective on August 2, 2023 and December 27, 2023 (File Nos. 333-273024 and 333-276118, respectively).
Moreover, based on our pre-clinical analyses to date, we believe that MIRA-55 is an improvement over MIRA1a in that it displays enhanced potency and potential for efficacy. In early March 2024, we filed a provisional patent application for MIRA-55, aiming for global patent protection. If such patent is issued, we would own the patent rights to both MIRA1a and MIRA-55.
Based on our discoveries to date, we have decided to advance MIRA-55 as our lead compound for our oral pharmaceutical marijuana drug candidate while still retaining our rights to MIRA1a.
| 69 |
Reverse Stock Split
Effective June 28, 2023, we completed a 1-for-5 reverse stock split of our outstanding common stock. Unless otherwise noted, the share and per share information in this Report reflects the reverse stock split.
Components of our Results of Operations
Research and Development Expenses
Research and development expenses represent costs incurred to conduct research and development of our product candidate. We recognize all research and development costs as they are incurred. Research and development expenses consist primarily of the following:
| ● | salaries and benefits; | |
| ● | contracted research and manufacturing; |
| ● | consulting arrangements; and | |
| ● | other expenses incurred to advance our research and development activities. |
Our operating expenses have historically been the costs associated with our patent prosecution and initial investment in pre-clinical research and development activities. We expect research and development expenses will increase in the future as we advance Ketamir-2 and MIRA-55 into and through clinical trials and pursue regulatory approvals, which will require a significant investment in costs of clinical trials, regulatory support, and contract manufacturing. In addition, we will evaluate opportunities to acquire or in-license additional product candidates and technologies, which may result in higher research and development expenses due to license fee and/or milestone payments, as well as added clinical development costs.
The process of conducting clinical trials necessary to obtain regulatory approval is costly and time consuming. We may never succeed in timely development and achieving regulatory approval for our product candidates. The probability of success of our product candidates may be affected by numerous factors, including clinical data, competition, manufacturing capability and commercial viability. As a result, we are unable to determine the duration and completion costs of our development projects or when and to what extent we will generate revenue from the commercialization and sale of our product candidates.
General and Administrative Expenses
General and administrative expenses consist of employee-related expenses, including salaries, benefits, and travel, and other administrative functions, as well as fees paid for legal, accounting and tax services, consulting fees and facilities costs not otherwise included in research and development expense. Legal costs include general corporate legal fees and patent costs. We expect to incur additional expenses as a result of becoming a public company, including expenses related to compliance with the rules and regulations of the SEC and Nasdaq, additional insurance, investor relations and other administrative expenses and professional services.
Interest expense
Interest expense, net consists of accrued interest on a related party line of credit, net of earned interest income.
Results of Operations for the year ended December 31, 2023 and 2022
Year Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Revenues | $ | - | $ | - | ||||
| Operating costs: | ||||||||
| General and administrative expenses | 6,499,537 | 2,992,125 | ||||||
| Related party travel costs | 453,550 | 1,704,350 | ||||||
| Research and development expenses | 1,572,963 | 2,351,465 | ||||||
| Total operating costs | 8,526,049 | 7,047,940 | ||||||
| Interest expense, net | (3,456,294 | ) | (10,250 | ) | ||||
| Net loss attributable to common stockholders | $ | (11,982,343 | ) | $ | (7,058,190 | ) | ||
| Basic and diluted loss per share | $ | (0.64 | ) | $ | (0.40 | ) | ||
| Weighted average common stock shares outstanding | 18,566,158 | 17,566,533 | ||||||
| 70 |
General and Administrative Expenses. We incurred $6.5 million and $2.9 million in general and administrative expenses during the year ended December 31, 2023 and December 31, 2022, respectively. General and administrative expenses are composed primarily of compensation, insurance, professional fees, stock-based compensation, administration and other related costs. The increase is primarily due to an increase in stock-based compensation, debt issuance costs, and compensation related to the IPO efforts of the executive team.
Related Party Travel Costs. We incurred $0.4 million and $1.7 million in related party travel costs during the year ended December 31, 2023 and December 31, 2022 respectively. Related party travel costs consisted of a lease and use of an airplane with an entity under common control. The decrease in related party travel costs in 2023 is due to the termination of the lease in March 2023.
Interest expense. We incurred $3.5 million, net in interest expense and interest income during the year ended December 31, 2023, and $0.01 million interest expense during the year ended December 31, 2022, respectively. Interest expense during 2023 included $2.8 million of write-off of unamortized deferred financing costs, $0.44 million of debt issuance costs and $0.02 million of interest income. The remaining 2023 and 2022 interest expense consists of accrued interest on a related party line of credit.
Research and Development Expenses. During the year ended December 31, 2023, we incurred $1.6 million in research and development expenses, which were primarily related to initial payments for toxicology studies, consultants and stock compensation. We incurred $2.4 million in research and development expenses during the three months ended December 31, 2022, relating to initial payment for toxicology study costs. Research and development expenses include pre-clinical, toxicology and consultant expenses. Major components of research and development expenses during the year ended December 31, 2023 are as follows:
| R&D Category | Expense | |||
| R&D consultants | $ | 0.25 million | ||
| R&D research | $ | 0.37 million | ||
| R&D toxicology | $ | 0.21 million | ||
| R&D stock compensation | $ | 0.74 million |
Liquidity and Capital Resources
Since our inception in September 2020, we have financed our operations primarily through an unsecured line of credit with a major shareholder and an affiliated company and through a private placement of shares of our common stock that occurred during the fourth quarter 2021 and during 2022. We intend to finance our clinical development programs and working capital needs from existing cash, potential new sources of debt and equity financing, including the proceeds from our completed IPO in August 2023. We may enter into new licensing and commercial partnership agreements.
On April 28, 2023, we entered into a Promissory Note and Loan Agreement with the Bay Shore Trust, a trust established by our founder, and under which various of his family members are beneficiaries (the “Bay Shore Trust”). Under this Promissory Note and Loan Agreement (the “Bay Shore Note”), we have the right to borrow up to an aggregate of $5,000,000 from the Bay Shore Trust at any time up to the second anniversary of the issuance of the Bay Shore Note or, if earlier, upon the completion of our initial public offering. Our right to borrow funds under the Bay Shore Note is subject to the absence of a material adverse change in our assets, operations, or prospects. The Bay Share Note, together with accrued interest, will become due and payable on the second anniversary of the issuance of the note, provided that it may be prepaid at any time without penalty. The Bay Shore Note will accrue interest at a rate equal 7% per annum, simple interest, during the first year that the note is outstanding and 10% per annum, simple interest, thereafter. The Bay Shore Note is unsecured. As of December 31, 2023, the Bay Shore Note was paid in full except for an unpaid interest balance of $.01 million. In consideration of the loan facility provided by the Bay Shore Trust, we issued to the Bay Shore Trust a common stock purchase warrant on April 28, 2023, giving the Bay Shore Trust the right to purchase up to 1,000,000 shares of common stock at an exercise price of $5.00 per share, which warrant will expire five years after the date of grant.
| 71 |
Since January 1, 2023, MIRALOGX, LLC, an intellectual property development and holding company owned by Bay Shore Trust (“MIRALOGX”), has advanced funds on behalf of Bay Shore Trust to our company in order to fund operating activities. The total amount advanced and outstanding from MIRALOGX was $1.6 million immediately prior to being consolidated into the Bay Shore Note in 2023, and such amounts become a part of the outstanding balance of the Bay Shore Note, which as of December 31, 2023, is $0.
On July 20, 2023, we entered into a conversion agreement with the Bay Shore Trust under which the Bay Shore Trust agreed to convert, upon the completion of our initial public offering, $1,100,190 of the outstanding principal balance of the Bay Shore Note into shares of our common stock at a conversion price equal to our initial public offering price, which resulted in the issuance of 157,170 shares to the Bay Shore Trust upon the completion of our initial public offering (the “Bay Shore Trust Conversion Agreement”).
In August 2023, we completed our IPO of common stock selling 1,275,000 shares at an offering price of $7.00 per share, resulting in gross proceeds of $8.9 million. Net proceeds received after underwriting fees and offering expenses were $8.1 million. We raised $3.2 million in 2022. Substantially all our equity capital had been raised at $1.00 per share (pre-reverse split).
We used $3.4 million in operating activities during the year ended December 31, 2023, compared to $5.6 million in operating activities during the year ended December 31, 2022.
We have incurred significant losses and negative cash flows from operations since inception and expect to incur additional losses until such time that we can generate significant revenue and profit. We had negative cash flow from operations of approximately $3.4 million for the year ended December 31, 2023 and an accumulated deficit of approximately $21.2 million as of December 31, 2023. As of December 31, 2023, we had cash and cash equivalents of approximately $4.6 million and working capital of $4.4 million. We currently expect that our cash and cash equivalents be sufficient to fund our operations, development plans, and capital expenditures through at least the fourth quarter of 2024.
We did not have any material non-cancellable contractual obligations as of December 31, 2023.
Cash Flows
The following table provides information regarding our cash flows for the periods presented:
| Year ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Net cash provided by (used in): | ||||||||
| Operating activities | $ | (4,532,403 | ) | $ | (5,604,759 | ) | ||
| Financing activities | 8,783,991 | 3,146,185 | ||||||
| Net change in cash | $ | 4,251,588 | $ | (2,458,574 | ) | |||
| 72 |
Net Cash Used in Operating Activities
The cash used in operating activities resulted primarily from our net losses, stock-based compensation expense, amortization of debt issuance costs and changes in components of accounts payable and accrued liabilities.
For the year ended December 31, 2023, operating activities used $4.5 million of cash, primarily due to a net loss of $12 million, a $0.6 million change in accounts payable, accrued and prepaid expenses, offset by $2.5 million in stock-based compensation expense, $0.7 million in amortization of debt issuance costs, $3.5 million of interest expense, and $1.1 million of repayments under related party line of credit. Interest expense, net was primarily composed of warrant expense and line of credit expense, offset by interest income. Accounts payable, accrued and prepaid expenses was primarily composed of research and development payables, consultant costs, insurance costs and investor relations expenses.
For the year ended December 31, 2022, operating activities used $5.6 million of cash, primarily due to a net loss of $7.1 million, a $0.06 million change in accounts payable, accrued and prepaid expenses, offset by $1.3 million in stock-based compensation expense. Accounts payable, accrued and prepaid expenses was primarily composed of research and development payables, consultant costs, insurance costs and investor relations expenses.
Net Cash Provided by Financing Activities
For the year ended December 31, 2023, financing activities provided $8.8 million of cash, resulting primarily from $7.7 million in proceeds from sale of common stock, less offering costs and $2.1 million in advances from related party line of credit, offset by $1.1 million of repayments under related party line of credit.
For the year ended December 31, 2022, financing activities provided $3.1 million of cash, resulting primarily from $2.9 million in proceeds from sale of common stock, less offering costs, offset by $0.16 million of repayments under related party line of credit.
We currently anticipate that we will seek to monetize our product candidates, Ketamir-2 and MIRA-55, at the end of our planned Phase 2 studies. Prior to that time, we anticipate that additional capital may be required to support ongoing activities and further phases of development. Should that be required, our available capital may be consumed more rapidly than currently anticipated, resulting in the need for additional funding. In addition, there can be no assurance that additional funding, when and if required, will be available at commercially favorable terms, if at all.
Accordingly, we may need to raise additional capital, which may be available to us through a variety of sources, including:
| ● | public equity markets; | |
| ● | private equity financings; | |
| ● | commercialization agreements and collaborative arrangements; | |
| ● | sale of product royalty; | |
| ● | grants and new license revenues; | |
| ● | bank loans; and | |
| ● | public or private debt. |
Additional funding, capital, or loans (including, without limitation, milestone, or other payments from potential commercialization agreements) may be unavailable on favorable terms, if at all. If adequate funds are not available, we may be required to significantly reduce or refocus our operations or to obtain funds through arrangements that may require us to relinquish rights to certain technologies and drug formulations or potential markets, any of which could have a material adverse effect on us, our financial condition, and our results of operations. To the extent that additional capital is raised through the sale of equity or convertible debt securities or exercise of warrants and options, the issuance of such securities would result in ownership dilution to existing stockholders.
| 73 |
If we are unable to attract additional funds on commercially acceptable terms, it may adversely affect our ability to achieve our development and commercialization goals, which could have a material and adverse effect on our business, results of operations and financial condition.
We believe that we have sufficient resources available to support our development activities and business operations and timely satisfy our obligations as they become due into the fourth quarter of 2024. We do not have sufficient cash and cash equivalents as of the date of filing this Annual Report on Form 10-K to support our operations for at least the 12 months following the date the financial statements are issued. These conditions raise substantial doubt about our ability to continue as a going concern through 12 months after the date that the financial statements are issued.
To alleviate the conditions that raise substantial doubt about our ability to continue as a going concern, we plan to secure additional capital, potentially through a combination of public or private equity offerings and strategic transactions, including potential alliances and drug product collaborations; however, none of these alternatives are committed at this time. There can be no assurance that we will be successful in obtaining sufficient funding on terms acceptable to us to fund continuing operations, if at all, identify and enter into any strategic transactions that will provide the capital that we will require or achieve the other strategies to alleviate the conditions that raise substantial doubt about our ability to continue as a going concern. If none of these alternatives are available, or if available, are not available on satisfactory terms, we will not have sufficient cash resources and liquidity to fund our business operations for at least the 12 months following the date the financial statements are issued. The failure to obtain sufficient capital on acceptable terms when needed may require us to delay, limit, or eliminate the development of business opportunities and our ability to achieve our business objectives and our competitiveness, and our business, financial condition, and results of operations will be materially adversely affected. In addition, the perception that we may not be able to continue as a going concern may cause others to choose not to deal with us due to concerns about our ability to meet our contractual obligations.
The accompanying financial statements have been prepared on a going concern basis, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business, and do not include any adjustments relating to recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might be necessary should we be unable to continue as a going concern.
Recently Issued and Adopted Accounting Pronouncements
A description of recently issued and adopted accounting pronouncements that may potentially impact our financial position and results of operations is disclosed in Note 8 to our financial statements appearing at the end of this Report.
Off-Balance Sheet Arrangements
During the periods presented, we did not have, nor do we currently have, any off-balance sheet arrangements as defined under SEC rules.
Summary of Critical Accounting Policies
Income taxes
We are a C corporation. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amount of existing assets and liabilities and their respective tax bases. Deferred tax assets are recognized for temporary differences that will result in deductible amounts in future years and for loss carryovers. A valuation allowance is recognized regarding deferred tax assets, if any, if it is more likely than not that some portion of the deferred tax asset will not be realized.
| 74 |
Research and development expenses
Research and development costs are expensed in the period in which they are incurred and include the expenses paid to third parties, such as contract research organizations and consultants, who conduct research and development activities on our behalf. Patent-related costs, including registration costs, documentation costs and other legal fees associated with the application, are expensed in the period in which they are incurred.
Use of estimates
The preparation of financial statements in accordance with generally accepted accounting principles in the United States of America requires our company’s management to make estimates and assumptions that affect the reported amounts of assets and liabilities, and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results may differ from such estimates and such differences could be material.
Stock-based compensation
We account for stock-based compensation under the provisions of FASB ASC 718, “Compensation - Stock Compensation”, which requires the measurement and recognition of compensation expense for all stock-based awards made to employees, directors and consultants based on estimated fair values on the grant date. We estimate the fair value of stock-based awards on the date of grant using the Black-Scholes model. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods using the straight-line method. We have elected to account for forfeiture of stock-based awards as they occur.
Emerging Growth Company Election
We are an “emerging growth company” as defined in Section 2(a) of the Securities Act and have elected to take advantage of the benefits of the extended transition period for new or revised financial accounting standards. We expect to continue to take advantage of the benefits of the extended transition period, although we may decide to early adopt such new or revised accounting standards to the extent permitted by such standards. We expect to use this extended transition period for complying with new or revised accounting standards that have different effective dates for public and non-public companies until the earlier of the date we (i) are no longer an emerging growth company or (ii) affirmatively and irrevocably opt out of the extended transition period provided in the JOBS Act. This may make it difficult or impossible to compare our financial results with the financial results of another public company that is either not an emerging growth company or is an emerging growth company that has chosen not to take advantage of the extended transition period exemptions because of the potential differences in accounting standards used.
In addition, we intend to rely on the other exemptions and reduced reporting requirements provided by the JOBS Act. Subject to certain conditions set forth in the JOBS Act and compliance with applicable laws, if, as an emerging growth company, we rely on such exemptions, we are not required to, among other things: (a) provide an auditor’s attestation report on our system of internal control over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act of 2002; (b) provide all of the compensation disclosures that may be required of non-emerging growth public companies under the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010; (c) comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements (auditor discussion and analysis); and (d) disclose certain executive compensation-related items such as the correlation between executive compensation and performance and comparisons of the Chief Executive Officer’s compensation to median employee compensation.
We will remain an emerging growth company under the JOBS Act until the earliest of (a) December 31, 2028, (b) the last date of our fiscal year in which we had total annual gross revenue of at least $1.07 billion, (c) the date on which we are deemed to be a “large accelerated filer” under the rules of the SEC or (d) the date on which we have issued more than $1.0 billion in non-convertible debt securities during the previous three years.
| 75 |
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Smaller reporting companies are not required to provide the information required by this item.
Item 8. Financial Statements and Supplementary Data.
Our
Consolidated Financial Statements and Notes thereto and the report of Cherry Bekaert, our independent registered public accounting firm
(PCAOB ID:
Item 9. Changes In and Disagreements With Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
We have established disclosure controls and procedures designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and is accumulated and communicated to management, including the principal executive officer (our Chief Executive Officer) and principal financial officer (our Chief Financial Officer), to allow timely decisions regarding required disclosure.
Our management, under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of the end of the period covered by this Annual Report on Form 10-K. Management recognizes that any disclosure controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives. Our disclosure controls and procedures have been designed to provide reasonable assurance of achieving their objectives. Based on such evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were not effective at the end of fiscal year 2023.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the year ended December 31, 2023 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| 76 |
Management’s Report on Internal Control Over Financial Reporting
This annual report does not include a report of management's assessment regarding internal control over financial reporting or an attestation report of the company's registered public accounting firm due to a transition period established by rules of the Securities and Exchange Commission for newly public companies.
Item 9B. Other Information.
None.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
| 77 |
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
March 2024 Changes to Our Management and Board of Directors
Restructuring of the Board of Directors
On March 9, 2024, after a series of discussions between our board of directors (the “Board”) and senior management regarding the need to have additional scientific expertise among the members of the Board, Ms. Talhia Tuck, Mr. Brad Kroenig and Mr. Hugh McColl, each voluntarily resigned from the Board, effective immediately. This action allowed the remaining members of the Board to appoint new members of the Board, as discussed below. The resignations of Ms. Tuck, Mr. Kroenig, and Mr. McColl were not the result of any disagreement with our company on any matter relating to its operations, policies or practices.
Also on March 9, 2024, Dr. Chris Chapman notified the Board and senior company management of his resignation both as Executive Chairman and as an employee of our company, effective immediately, citing his desire to focus his time on his role as Chairman and Chief Executive Officer of Telomir Pharmaceuticals, Inc., given the recent initial public offering of that company. Dr. Chapman’s resignation was not the result of any disagreement with our company on any matter relating to its operations, policies or practices.
On March 13, 2024, the remaining members of the Board (Erez Aminov and Michael Jerman) unanimously approved the appointment of (i) Mr. Aminov, our Chief Executive Officer, as Chairman of the Board and (ii) Dr. Matthew P. Del Giudice, Dr. Denil N. Shekhat and Mr. Edward MacPherson as members of the Board, to fill the vacancies on the Board occasioned by the resignations from the Board described above, for a term expiring at our 2024 annual meeting of shareholders.
Resignation of Chief Science Officer
We are focused on strengthening our clinical and regulatory development expertise with a view towards a future IND for one of our product candidates. As part of this development, on March 7, 2024, following discussions with our management, Adam Kaplin, M.D., Ph.D. resigned from his position as President and Chief Scientific Officer of the company to pursue other business endeavors, effective immediately. As described under “Key Consultants” below, in light of Mr. Kaplin’s resignation, we expanded the role of an existing consultant to assist in clinical and regulatory affairs.
Current Directors and Executive Officers
Our directors and executive officers and their ages as of the date of this Report are as follows:
| Name | Age | Position | ||
| Erez Aminov | 46 | Chief Executive Officer and Chairman | ||
| Michelle Yanez | 52 | Chief Financial Officer, Secretary and Treasurer | ||
| Michael Jerman | 40 | Director | ||
| Matthew Paul Del Giudice, M.D. | 42 | Director | ||
| Denil Nanji Shekhat, M.D. | 43 | Director | ||
| Edward MacPherson | 36 | Director |
The following is a brief biography of each of our current executive officers and directors:
Erez Aminov has served as a director and our Chief Executive Officer since April 2023 and our Chairman since March 2024. Mr. Aminov is an experienced biotechnology consultant and investor and initially joined our as a consultant in 2022. Mr. Aminov’s experience in the biotech consulting sector began in 2021 when he founded Locate Venture Corp. in September 2021. Locate Venture is a strategy and investment consulting firm focused on advancing and supporting early-stage biotech startups. Prior to founding Locate Venture Corp., from February 2015 to September 2020, Mr. Aminov served as the President of Finds4less Inc., a global distributor of electronics and gaming products. In this role, Mr. Aminov provided strategic oversight and direction for all aspects of the company’s operations, while also spearheading new business development initiatives to capitalize on emerging market opportunities. Mr. Aminov’s more than two decades of experience includes experience with the biotech industry’s particular challenges, including creating strategic alliances and guiding startups toward growth and prosperity. Mr. Aminov earned a B.A. in Accounting from Touro University in New York. We believe that Mr. Aminov is qualified to serve as one of our directors based on his finance and investment experience, particularly with early stage life sciences companies.
| 78 |
Michelle Yanez, MBA has served as our Chief Financial Officer since April 2023, prior to which she served as our Corporate Controller since May 2022. Ms. Yanez is a senior financial executive with over 25 years of experience in public and privately held biotech, pharmaceutical, and life science companies. Ms. Yanez’ experience includes a broad range of responsibilities in a highly complex and regulated market. She also brings deep corporate governance experience through her work with corporate boards, including audit and finance committees. Since May 2022, Ms. Yanez is part-time Corporate Controller at Telomir Pharmaceuticals, Inc., a publicly traded pre-clinical-stage pharmaceutical company, focusing on the development and commercialization of therapeutic treatment for human stem cells (Nasdaq: TELO). From May 2002 until its acquisition in April 2022, Ms. Yanez held various positions, including the Director of Financial Reporting, of BioDelivery Sciences International, Inc. (Nasdaq: BDSI). In her role, she led financial offerings, managed due diligence for product acquisitions and financings and managed finance documents and filings for the tender offer, leading to the acquisition of BioDelivery Sciences in April 2022. Ms. Yanez also serves as a non-employee director of Inhibitor Therapeutics, Inc. (OTCQB: INTI), a publicly traded pharmaceutical development company focused on therapeutics for certain cancers and non-cancerous proliferation disorders, since December 2022. Ms. Yanez is a member of the Institute of Management Accountants and a member of the SEC Professionals Group. Ms. Yanez received her MBA degree cum laude from Rutgers Business School.
Michael Jerman, CPA joined our company as a director in December 2023. He also serves as a member of the board of directors of Inhibitor Therapeutics, Inc. (OTC:INTI). Mr. Jerman has served as the managing partner at Hollywell Partners, a professional accounting and finance consulting firm, since May 2019, and has provided chief financial officer and other services to multiple private equity-backed companies in the energy, SaaS, and manufacturing industries. Prior to his role with Hollywell Partners, he was a Director with PwC in the US and UK from January 2007 to August of 2019 and was a Captain with the United States Air Force from July 2003 to June 2015. He has led global public and private client engagements in the industries of retail and consumer, energy, utilities and mining, and transportation and logistics. Mr. Jerman has significant experience in client equity and debt offerings, business combinations inclusive of public listing and reporting requirements, initial valuations and ongoing goodwill impairment analyses, share-based awards, restructuring, and global taxes, as well as stakeholder management, specifically with board and management presentation experience to include annual and quarterly requirements, fee negotiations, technical accounting and finance discussions, and fraud and non-compliance investigations. Mr. Jerman has specialized in rapid project mobilization and deployment of skilled resources for emergency issues, design, and implementation of small to large scale assurance requirements and advisory projects. Mr. Jerman’s additional experience includes leading PwC’s data acquisition methods and tools, client acquisitions and systems implementations to include new SOX-compliant control plan implementations across multiple systems, leading co-sourced internal audit projects, and time spent driving PwC’s lean efficiency initiatives. Mr. Jerman was a member of the PwC national office within the SEC PCAOB quality group supporting Europe and the EMEA regions with complex accounting and audit consultations. He earned a B.S. in accounting from the University of South Florida, an M.S. in accounting from the University of Tampa, and an M.B.A. from the University of Oxford.
Dr. Matthew Paul Del Giudice joined our company as a director in March 2024. Dr. Del Giudice has practiced as a radiologist since 2014. He currently serves as a general overnight emergency radiologist at the Cleveland Clinic and as a real estate investor with Comfort Living, LLC. Prior to joining the Cleveland Clinic, from March 2021 to May 2022, Dr. Del Giudice was a general radiologist with Radiology and Imaging Specialists in Lakeland, Florida. From July 2015 to February 2021, Dr. Del Giudice was a radiologist with Radiology Partners Phoenix, and from July 2014 to June 2015, he practiced as a musculoskeletal radiologist at the University of Arizona Health Sciences Center – Tucson. Dr. Del Giudice received his B.S. from the University of Illinois at Urbana-Champaign, his M.D. from Loyola University Stritch School of Medicine, completed his radiology residency at Loyola University Medical Center, and his musculoskeletal radiology fellowship at the University of Arizona Health Sciences Center – Tucson. Dr. Del Giudice is licensed to practice medicine in Florida and Ohio.
| 79 |
Dr. Denil Nanji Shekhat joined our company as a director in March 2024. Dr. Shekhat has practiced as a radiologist since 2014 and currently practices at DNS Teleradiology in Wellington, Florida. Prior to starting DNS Teleradiology, Dr. Shekhat was a musculoskeletal specialist for Radiology Associates of Florida/ Radiology Partners from July 2018 to December 2023. From July 2015 to August 2018, Dr. Shekhat practiced as a general and musculoskeletal radiologist with Bethesda Radiology Associates. Dr. Shekhat received his B.A. in economics from Bowdoin College, his M.D. from the University of Tennessee Health Science Center, College of Medicine, completed his radiology residency at Baptist Memorial Hospital and his musculoskeletal radiology fellowship at the University of Arizona. Dr. Shekhat is currently licensed to practice medicine in Florida.
Edward MacPherson joined our company as a director in March 2024. Mr. MacPerson currently serves as Chief Growth Officer for Power Digital, an industry leading digital marketing agency. Prior to joining Power Digital, from May 2016 to December 2023, he served as CEO and Head of Growth for Endrock Growth & Analytics, a company he founded and sold to Power Digital. Prior to founding Endrock Growth & Analytics, Mr. MacPherson held senior marketing and leadership positions at sunglass maker Prive Revaux (March 2018 to April 2020), curated meal company Menud (October 2014 to April 2018) and Rejuvenetics, LLC, a distributor of health and wellness products (December 2012 to March 2016). Mr. Macpherson holds a BA in Economics from Gettysburg College.
Key Consultants
On March 13, 2024, we entered into an Amended and Restated Consulting Agreement with Angel Pharmaceutical Consulting & Technologies Ltd., an Israeli consulting firm (“APCT”). All services provided to our company by APCT (which began in October 2023) are provided directly by Dr. Itzchak Angel, who shall be our Chief Scientific Advisor. Dr. Angel has over 30 years of experience in the pharmaceutical industry, guiding strategic drug and business development initiatives in both large and emerging companies.
Dr. Angel has served as Head of Pharmacology of Synthelabo (Paris, France, now Sanofi) for numerous years, where he was instrumental in the development and bringing into the market of several drugs such as Xatral (Alfuzosin), Ambien (Zolpidem) and Mizollen (Mizolastine). He formerly served as President and Chief Executive Officer of stem-cell company Accellta (Haifa, Israel) and Vice President for Research and Development at Proteologics Ltd, and at D-Pharm Biopharmaceuticals (Rehovot, Israel) where he developed several neurology compounds (stroke, Alzheimer’s and Parkinson’s Disease) into advanced clinical development and was involved in submitting numerous INDs of drugs under development. Dr. Angel is the author of more than 100 book chapters, papers, and abstracts as well as the named inventor of a number of pharmaceutical patents. Dr. Angel received his B.S. and M.Sc. in Biology from Tel-Aviv University, Israel, cum laude in 1979, and received Ph.D. cum laude from the Hamburg University, Germany in 1982.
As part of his consulting services, Dr. Angel shall assist our company with (i) pharmaceutical regulatory affairs, toxicology, drug research and pre-clinical and clinical testing, (ii) outsourcing and helping our company in managing third party vendors and (iii) working with our company in our interactions with regulatory bodies.
Board Composition
Our business and affairs are managed under the direction of our board of directors, which currently consists of five members. The number of directors is determined by our board of directors, subject to the terms of our amended and restated articles of incorporation and bylaws that. Our directors are elected for one-year terms.
Family Relationships
There are no family relationships among any of our directors and executive officers.
| 80 |
Director Independence
Our board of directors has undertaken a review of the independence of each director. Based on information provided by each director concerning his or her background, employment, and affiliations, our board of directors has determined that Michael Jerman, Dr. Matthew Del Giudice, Dr. Denil Shekhat and Edward MacPherson do not have any relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and are independent directors under the Nasdaq Listing Rules.
In making these determinations, our board of directors considered the current and prior relationships that each non-employee director has with our company and all other facts and circumstances our board of directors deemed relevant in determining their independence, including the transactions described in the section of this Report titled “Item 13. Certain Relationships and Related Party Transactions.”
Committees of the Board of Directors
Our board of directors has established an audit committee, a compensation committee, and a nominating and corporate governance committee. The functions of these committees are described below. Members will serve on these committees until their resignation or until otherwise determined by our board of directors. Our board of directors may establish other committees as it deems necessary or appropriate from time to time.
Audit Committee
Our audit committee consists of Michael Jerman, Dr. Denil Shekhat and Edward MacPherson, with Michael Jerman serving as the chair of the audit committee. Each member of the committee meets the requirements for independence under the listing standards of Nasdaq and SEC rules and regulations, including Rule 10A-3(b)(1) under the Exchange Act. Each member of our audit committee also meets the financial literacy requirements of the listing standards of Nasdaq. In addition, our board of directors has determined that Michael Jerman is an audit committee financial expert within the meaning of Item 407(d) of Regulation S-K under the Securities Act.
The audit committee’s main purpose is to oversee our corporate accounting and financial reporting process. Our audit committee is responsible for, among other things:
| ● | selecting a qualified firm to serve as the independent registered public accounting firm to audit our financial statements; | |
| ● | helping to ensure the independence and performance of the independent registered public accounting firm; | |
| ● | discussing the scope and results of the audit with the independent registered public accounting firm, and reviewing, with management and the independent registered public accounting firm, our interim and year-end results of operations; | |
| ● | developing procedures for employees to submit concerns anonymously about questionable accounting or audit matters; | |
| ● | reviewing our policies on risk assessment and risk management; | |
| ● | reviewing related party transactions; |
| ● | reviewing and pre-approving, as required, all audit and all permissible non-audit services to be performed by the independent registered public accounting firm; and | |
| ● | assisting our board of directors in monitoring the performance of our internal audit function. |
Our audit committee operates under a written charter that satisfies the applicable rules and regulations of the SEC and the listing standards of Nasdaq, a copy of which is available on our website at www.mirapharmaceuticals.com.
| 81 |
Compensation Committee
Our compensation committee consists of Dr. Denil Shekhat and Edward MacPherson, with Dr. Denil Shekhat serving as the chair of the compensation committee. Each member of the committee meets the requirements for independence under the listing standards of Nasdaq and SEC rules and regulations. Each member of our compensation committee is also a non-employee director, as defined pursuant to Rule 16b-3 promulgated under the Exchange Act, or Rule 16b-3. In arriving at these determinations, our board of directors examined all factors relevant to determining whether any compensation committee member has a relationship to us that is material to that member’s ability to be independent from management in connection with carrying out such member’s duties as a compensation committee member.
The compensation committee’s main purpose is to review and recommend policies relating to compensation and benefits of our officers and employees. Our compensation committee is responsible for, among other things:
| ● | reviewing, approving, and determining, or making recommendations to our board of directors regarding, the compensation and compensation arrangements of our executive officers; | |
| ● | administering our equity compensation plans; |
| ● | reviewing and approving, or making recommendations to our board of directors regarding, incentive compensation and equity compensation plans; and | |
| ● | establishing and reviewing general policies relating to compensation and benefits of our employees. |
Our compensation committee operates under a written charter that satisfies the applicable rules and regulations of the SEC and the listing standards of Nasdaq, a copy of which is available on our website.
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee consists of Dr. Matthew DelGuidice and Dr. Denil Shekhat with Dr. Matthew DelGuidice serving as the chair of the nominating and corporate governance committee. Each member of the committee meets the requirements for independence under the listing standards of Nasdaq and SEC rules and regulations.
Our nominating and corporate governance committee is responsible for, among other things:
| ● | identifying, evaluating, and selecting, or making recommendations to our board of directors regarding, nominees for election to our board of directors and its committees; | |
| ● | developing and overseeing the annual evaluation of our board of directors and of its committees; | |
| ● | considering and making recommendations to our board of directors regarding the composition of our board of directors and its committees; | |
| ● | overseeing our corporate governance practices; and | |
| ● | making recommendations to our board of directors regarding corporate governance guidelines. |
Our nominating and corporate governance committee operates under a written charter that satisfies the applicable listing standards of Nasdaq, a copy of which is available on our website.
Compensation Committee Interlocks and Insider Participation
None of the members of our compensation committee is a current or former executive officer or employee of our company. None of our executive officers serves as a member of the compensation committee of any entity that has one or more executive officers serving on our compensation committee.
| 82 |
Risk Oversight
One of the key functions of our board of directors is informed oversight of our risk management process. Our board of directors administers this oversight function directly through our board of directors as a whole, and through various standing committees of our board of directors that address risks inherent in their respective areas of oversight. In particular, our board of directors is responsible for monitoring and assessing strategic risk exposure, including risks associated with cybersecurity and data protection, and our audit committee has the responsibility to consider our major financial risk exposures and the steps our management has taken to monitor and control these exposures, including guidelines and policies to govern the process by which risk assessment and management is undertaken. Our audit committee will review legal, regulatory, and compliance matters that could have a significant impact on our financial statements. Our nominating and corporate governance committee will monitor the effectiveness of our corporate governance practices, including whether they are successful in preventing illegal or improper liability-creating conduct. Our compensation committee will assess and monitor whether any of our compensation policies and programs has the potential to encourage excessive risk taking. While each committee is responsible for evaluating certain risks and overseeing the management of such risks, our entire board of directors will be regularly informed through committee reports about such risks.
Board Diversity
Our nominating and corporate governance committee is responsible for reviewing with the board of directors, on an annual basis, the appropriate characteristics, skills, and experience required for the board of directors as a whole and its individual members. Although our board of directors does not have a formal written diversity policy with respect to the evaluation of director candidates, in its evaluation of director candidates, our nominating and corporate governance committee will consider factors including, without limitation, issues of character, integrity, judgment, potential conflicts of interest, other commitments, and diversity, and with respect to diversity, such factors as gender, race, ethnicity, experience, and area of expertise, as well as other individual qualities and attributes that contribute to the total diversity of viewpoints and experience represented on the board of directors.
The nominating and corporate governance committee will ensure compliance with the new rule by Nasdaq for board diversity (the “Nasdaq Diversity Rule”), on or before the date required under the Nasdaq Diversity Rule. The Nasdaq Diversity Rule requires, assuming our shares of common stock are listed on the Nasdaq Capital Market and that we are a smaller reporting company, that we will have at least two directors serving on our board of directors, at least one of which identifies as female and the second of which identifies as female, underrepresented minority or LGBTQ+, by December 31, 2026, unless our board of directors is comprised of five or less directors.
Code of Business Conduct and Ethics
Our board of directors has adopted a code of business conduct and ethics applicable to all of our directors, officers (including our principal executive officer, principal financial officer, and principal accounting officer) and all global employees in accordance with applicable federal securities laws and corporate governance rules of the Nasdaq Capital Market. Our code of business conduct and ethics is available on our website. Any amendments to the code of business conduct and ethics, or waivers of its requirements, will, if required, be disclosed on our website.
Insider Trading Policy
Our board of directors has adopted an insider trading policy filed hereto as Exhibit 19.1 and is incorporated herein by this reference.
Corporate Governance Guidelines
Our board of directors has adopted corporate governance guidelines, a copy of which is available on our website.
| 83 |
Director Compensation
We did not provide any cash compensation to any of our directors during the year ended December 31, 2023 in their capacity as directors. However, on April 28, 2023, each non-employee director was granted an additional option to purchase up to 10,000 shares of our common stock under the 2022 Omnibus Plan. Each such option was immediately vested in full upon grant and has a 10-year term.
Certain of our former directors have received option grants as a result of their service to our company in a non-director capacity. Prior to his appointment as Executive Chairman, Dr. Chapman was a party to a consulting agreement with our company entered into in April 2022 and was granted additional options in his capacity as a consultant on June 15, 2022. Dr. Chapman also received employee related grants in April 2023 and August 2023. Mr. Kroenig previously provided consulting services to our company in 2022 and received an additional option grant on June 15, 2022, under which he has the right to purchase up to 10,000 shares of our common stock. Upon his appointment as our General Counsel, Mr. Christos Nicholoudis was granted an option to purchase shares of our common of 15,000 shares in April 2023, and 10,000 shares in August 2023.
Item 11. Executive Compensation
This section discusses the material components of the executive compensation program for the following persons: (i) all persons serving as our principal executive officers during 2023 and (ii) the most highly compensated of our other executive officers who received compensation during 2023 of at least $100,000 and who were executive officers on December 31, 2023. We refer to these persons as our “named executive officers” elsewhere in this Report. Our “named executive officers” and their positions are as follows:
| ● | Erez Aminov, Chief Executive Officer and Chairman; | |
| ● | Michelle Yanez, MBA, Chief Financial Officer, Secretary and Treasurer and; | |
| ● | Adam Kaplin, MD, PhD, former President and Chief Scientific Officer; |
In April 2023, Mr. Aminov succeeded Mr. Uzonwanne as our Chief Executive Officer, and Ms. Yanez succeeded Mr. McNulty as our Chief Financial Officer.
Summary Compensation Table
The following table shows the compensation paid by us during the 2023 and 2022 fiscal years to our named executive officers.
| Name and principal position | Year | Salary ($) | Bonus ($) | Stock Awards ($) | Option Awards ($) (6) | Non-Equity Incentive Plan Compensation ($) | Nonqualified Deferred Compensation Earnings ($) | All Other Compensation ($) | Total ($) | |||||||||||||||||||||||||||
| Erez Aminov, | 2023 | 83,333 | 208,006 (1) | - | 1,368,600 | - | - | 5,625 | (2) | 1,665,564 | ||||||||||||||||||||||||||
| CEO | 2022 | - | - | - | - | - | - | - | - | |||||||||||||||||||||||||||
| Michelle Yanez, | 2023 | 165,000 | 88,475 | (1) | - | 282,215 | - | - | 5,934 | (2) | 541,624 | |||||||||||||||||||||||||
| CFO | 2022 | 110,000 | - | 36,950 | - | - | 6,071 | (2) | 153,021 | |||||||||||||||||||||||||||
| Adam Kaplin, | 2023 | 50,000 | - | - | 149,600 | - | - | - | 199,600 | |||||||||||||||||||||||||||
| former President & CSO | 2022 | - | 50,001 | (3) | - | 739,000 | - | - | - | 789,001 | ||||||||||||||||||||||||||
| Jude Uzonwanne, | 2023 | 75,000 | - | - | - | - | - | 6,569 | (2) | 81,569 | ||||||||||||||||||||||||||
| former CEO | 2022 | 125,000 | 50,000 | (4) | - | 739,000 | (5) | - | - | 8,385 | (2) | 922,385 | ||||||||||||||||||||||||
| Jim McNulty, | 2023 | 154,000 | - | - | - | - | - | - | 154,000 | |||||||||||||||||||||||||||
| former CFO | 2022 | 266,869 | 100,000 | (3) | - | - | - | - | - | 366,869 | ||||||||||||||||||||||||||
| (1) | The amounts represent IPO bonuses paid in 2023. | |
| (2) | Amount represents health insurance premiums paid. | |
| (3) | The amounts represent milestone payments pursuant to prior employment agreements. | |
| (4) | The bonus represents a paid sign-on amount. | |
| (5) | Of these 2022 option grants, 75% were cancelled and non-exercisable as of April 2023, pursuant to the termination of Mr. Uzonwanne. | |
| (6) | The reported amounts represent the aggregate grant date fair value of the awards computed in accordance with Financial Accounting Standards Board Account Standards Codification Topic 718, Stock Compensation, as modified or supplemented, or FASB ASC Topic 718. The assumptions used in calculating the grant date fair value of the stock options reported in this column are set forth in Note 8 to our Consolidated Financial Statements for the year ended December 31, 2022 included in this Report. In April 2023, we entered into an agreement with Mr. Uzonwanne in which the number of shares subject to his option agreement was reduced from 200,000 to 40,000. |
Narrative Disclosure to Summary Compensation Table
Employment Agreements
Except as set forth below, we currently have no written employment agreements with any of our named executive officers.
| 84 |
Erez Aminov
Effective April 28, 2023, we entered into an employment agreement with Mr. Aminov, as amended on August 28, 2023, pursuant to which Mr. Aminov will serve as our Chief Executive Officer. Under his employment agreement, as amended, Mr. Aminov has agreed to devote at least 50% of his business time to the affairs of the Company. Mr. Aminov’s employment agreement provides that his employment will be on an at-will basis and can be terminated by either Mr. Aminov or our company at any time and for any reason. Under the agreement, Mr. Aminov will receive a base salary of $0.2 million per year, effective August 1, 2023. In the event that Mr. Aminov’s employment is terminated by our company without “Cause” or is terminated by Mr. Aminov for “Good Reason”, Mr. Aminov will be entitled to severance compensation in the form of salary continuation for a period of three months (subject to Mr. Aminov executing and delivering a customary general release in favor of the company). “Cause” is defined in the agreement to include dishonesty, misappropriation, willful misconduct, breach of the agreement, and other customary matters. “Good Reason” is defined to include a material adverse change in Mr. Aminov’s compensation or duties and level of responsibility. The employment agreement also contains customary confidentiality and invention-assignment covenants to which Mr. Aminov is subject.
On August 17, 2023, Mr. Aminov received a $0.1 million cash bonus net of federal, state, local and income taxes related to the successful completion of the IPO.
In March 2024, Mr. Aminov assumed the role of Chairman and on March 25, 2024, the Compensation Committee of the Board of Directors approved an increase to Mr. Aminov’s base salary of $0.8 million, bringing his total annual base salary to $0.28 million.
Michelle Yanez
On April 28, 2023, we entered into an employment agreement with Ms. Yanez pursuant to which Ms. Yanez will serve as our Chief Financial Officer on a full-time basis. Ms. Yanez’s employment agreement provides that her employment will be on an at-will basis and can be terminated by either Ms. Yanez or our company at any time and for any reason. Under the agreement, Ms. Yanez will receive an initial base salary of $0.17 per year. In the event that her employment is terminated by our company without “Cause” or is terminated by Ms. Yanez for “Good Reason”, Ms. Yanez will be entitled to severance compensation in the form of salary continuation for a period of three months (subject to Ms. Yanez executing and delivering a customary general release in favor of the company). “Cause” is defined in the agreement to include dishonesty, misappropriation, willful misconduct, breach of the agreement, and other customary matters. “Good Reason” is defined to include a material adverse change in Ms. Yanez’s compensation or duties and level of responsibility. The employment agreement also contains customary confidentiality and invention-assignment covenants to which Ms. Yanez is subject.
On August 17, 2023, Ms. Yanez received a $0.05 million cash bonus net of federal, state, local and income taxes related to the successful completion of the IPO. On March 25, 2024, the Compensation Committee of the Board of Directors approved an increase in Ms. Yanez’s base salary of $0.06 million, bringing her annual base salary to $0.23 million.
Chris Chapman
On April 28, 2023, we entered into an employment agreement with Dr. Chapman, as amended on August 28, 2023, and October 13, 2023, pursuant to which Dr. Chapman served as our Executive Chairman. Dr. Chapman’s employment agreement, as amended, provided that his employment would be on a part-time basis whereby Dr. Chapman would devote time and effort to the business and affairs of the company on an as needed basis, and it further provides that such employment would be on an at-will basis and could be terminated by either Dr. Chapman or our company at any time and for any reason. Under the agreement, Dr. Chapman would receive a base salary of $0.05 million per year for a period of 90 days following the October 13, 2023 amendment, and following the 90-day period, Dr. Chapman’s base salary will increase to $0.15 million. In the event that Dr. Chapman’s employment is terminated by our company without “Cause” or is terminated by Dr. Chapman for “Good Reason”, Dr. Chapman would be entitled to severance compensation in the form of salary continuation for a period of three months (subject to Dr. Chapman executing and delivering a customary general release in favor of the company). “Cause” is defined in the agreement to include dishonesty, misappropriation, willful misconduct, breach of the agreement, and other customary matters. “Good Reason” is defined to include a material adverse change in Dr. Chapman’s compensation or duties and level of responsibility. The employment agreement also contains customary confidentiality and invention-assignment covenants to which Dr. Chapman is subject.
On August 17, 2023, Dr. Chapman received a $0.05 million cash bonus net of federal, state, local and income taxes related to the successful completion of the IPO.
On March 9, 2024, Dr. Chapman resigned from our company as Executive Chairman, and as an employee.
| 85 |
Consulting Relationship with Adam Kaplin
Dr. Kaplin was a paid non-employee consultant to our company under which he provided services and consultation on an as-needed basis. Dr. Kaplin was paid $0.01 million a month for his services. We do not currently have a written consulting agreement with Dr. Kaplin.
Grants of Plan-Based Awards in 2023
| Estimated Future Payouts Under Non-Equity Incentive Plan Awards | Estimated Future Payouts Under Equity Incentive Plan Awards | All Other Stock Awards: Number of Shares of Stocks or | All Other Option Awards: Number of Securities Underlying | Exercise or Base Price of Option | Closing stock price on Award | Grant Date Fair Value of Stock and | ||||||||||||||||||||||||||||||||||||||||
| Name | Grant Date (1) | Threshold ($) | Target ($) | Maximum ($) | Threshold (#) | Target (#) | Maximum (#) | Units (#) | Options (#) | Awards ($/Sh) | date ($/Sh) | Option Awards | ||||||||||||||||||||||||||||||||||
| Erez Aminov, CEO | 4/28/2023 | - | - | - | - | - | - | - | 150,000 | (2) | $ | 5.00 | - | (3) | $ | 112,200 | ||||||||||||||||||||||||||||||
| 8/17/2023 | - | - | - | - | - | - | - | 150,000 | (4) | $ | 6.50 | $ | 6.50 | $ | 807,600 | |||||||||||||||||||||||||||||||
| Michelle Yanez, CFO | 4/28/2023 | - | - | - | - | - | - | 46,667 | (2) | $ | 5.00 | - | (3) | $ | 174,535 | |||||||||||||||||||||||||||||||
| 8/17/2023 | - | - | - | - | - | - | 20,000 | (4) | $ | 6.50 | $ | 6.50 | $ | 107,680 | ||||||||||||||||||||||||||||||||
| Adam Kaplin, former President & CSO | 4/28/2023 | - | - | - | - | - | - | - | 40,000 | (2) | $ | 5.00 | - | (3) | $ | 149,600 | ||||||||||||||||||||||||||||||
| Jude Uzonwanne, former CEO | - | - | - | - | - | - | - | - | - | - | $ | - | ||||||||||||||||||||||||||||||||||
| James McNulty, former CFO | - | - | - | - | - | - | - | - | - | - | $ | - | ||||||||||||||||||||||||||||||||||
| (1) | The “Grant Date” represents the date on which the Compensation Committee of the Board took action to grant the applicable award. | |
| (2) | The stock awards disclosed in this item consist of options, as issued under our 2022 Omnibus Incentive Plan, which vest ratably in thirds beginning April 2023. | |
| (3) | There was no closing stock price for our common stock since our IPO did not occur until August 2023. | |
| (4) | The stock awards disclosed in this item consist of options, as issued under our 2022 Omnibus Incentive Plan, which vested 100% at grant. |
Outstanding equity awards
The following table summarizes outstanding unexercised options held by each of our named executive officers, as of December 31, 2023.
| 86 |
| OPTION AWARDS | STOCK AWARDS | |||||||||||||||||||||||||||||||||
| Name | Number of Securities Underlying Unexercised Options (#) Exercisable | Number of Securities Underlying Unexercised Options (#) Unexercisable | Equity Incentive Plan Awards: Number of Securities Underlying Unexercised Unearned Options (#) | Options Exercise Prices ($) | Option Expiration Date | Number of Shares or Units of Stock That Have Not Vested (#) | Market Value of Shares or Units of Stock That Have Not Vested ($) | Equity Incentive Plan Awards: Number of Unearned Shares, Units or Other Rights That Have Not Vested (#) | Equity Incentive Plan Awards: Market or Payout Value of Unearned Shares, Units or Other Rights That Have Not vested (#) | |||||||||||||||||||||||||
| Erez Aminov | 150,000 | - | - | $ | 6.50 | 8/16/33 | - | - | - | - | ||||||||||||||||||||||||
| 50,000 | 100,000 | - | $ | 5.00 | 4/27/33 | - | - | - | - | |||||||||||||||||||||||||
| Michelle Yanez | 20,000 | - | - | $ | 6.50 | 8/16/33 | - | - | - | - | ||||||||||||||||||||||||
| 15,556 | 31,111 | - | $ | 5.00 | 4/27/33 | - | - | - | - | |||||||||||||||||||||||||
| 6,667 | 3,333 | - | $ | 5.00 | 6/14/32 | - | - | - | - | |||||||||||||||||||||||||
| Adam Kaplin | 13,334 | 26,666 | - | $ | 5.00 | 4/27/33 | - | - | - | - | ||||||||||||||||||||||||
| 100,000 | 100,000 | - | $ | 5.00 | 6/14/32 | - | - | - | - | |||||||||||||||||||||||||
| Jude Uzonwanne | 50,000 | - | - | $ | 5.00 | 6/14/32 | - | - | - | - | ||||||||||||||||||||||||
| James McNulty | - | - | - | - | - | - | - | |||||||||||||||||||||||||||
Option Exercises and Stock Vested
No stock options were exercised by our executive officers during the year ended December 31, 2023:
2022 Omnibus Incentive Plan
Our board of directors has adopted, and our stockholders have approved, our 2022 Omnibus Incentive Plan, or the 2022 Omnibus Plan. The 2022 Omnibus Plan authorizes the grant of incentive stock options, within the meaning of Section 422 of the Internal Revenue Code, to our employees and any of our parent and subsidiary corporations’ employees, and the grant of nonstatutory stock options, restricted stock, restricted stock units, stock appreciation rights, performance units and performance shares to our employees, directors, and consultants and any of our future subsidiary corporations’ employees and consultants. The following is a summary of certain terms and conditions of the 2022 Omnibus Plan. This summary is qualified in its entirety by reference to the 2022 Omnibus Plan attached as an exhibit to this Report. You are encouraged to read the full text of the 2022 Omnibus Plan.
As of December 31, 2023, there are options to purchase an aggregate of 1,210,001 shares of our common stock outstanding under the 2022 Omnibus Plan.
| 87 |
Administration
The 2022 Omnibus Plan is administered by our board of directors or our compensation committee, or any other committee or subcommittee or one or more of our officers to whom authority has been delegated (collectively, the “Administrator”). The Administrator has the authority to interpret the 2022 Omnibus Plan and award agreements entered into with respect to the 2022 Omnibus Plan; to make, change and rescind rules and regulations relating to the 2022 Omnibus Plan; to make changes to, or reconcile any inconsistency in, the 2022 Omnibus Plan or any award agreement covering an award; and to take any other actions needed to administer the 2022 Omnibus Plan.
Eligibility
The Administrator may designate any of the following as a participant under the 2022 Omnibus Plan: any officer or employee, or individuals engaged to become an officer or employee, of our company or our affiliates; and consultants of our company or our affiliates, and our directors, including our non-employee directors.
Types of Awards
The 2022 Omnibus Plan permits the Administrator to grant stock options, stock appreciation rights (“SARs”), performance shares, performance units, shares of common stock, restricted stock, restricted stock units (“RSUs”), cash incentive awards, dividend equivalent units, or any other type of award permitted under the 2022 Omnibus Plan. The Administrator may grant any type of award to any participant it selects, but only our employees or our subsidiaries’ employees may receive grants of incentive stock options within the meaning of Section 422 of the Internal Revenue Code. Awards may be granted alone or in addition to, in tandem with, or (subject to the repricing prohibition described below) in substitution for any other award (or any other award granted under another plan of our company or any affiliate, including the plan of an acquired entity).
Shares Reserved Under the 2022 Omnibus Incentive Plan
The 2022 Omnibus Plan provides that 2,000,000 shares of our common stock are reserved for issuance under the 2022 Omnibus Plan, all of which may be issued pursuant to the exercise of incentive stock options. The number of shares available for issuance under our 2022 Omnibus Plan will also include an annual increase on the first day of each fiscal year equal to the lesser of:
| ● | 200,000 shares; | |
| ● | 1.0% of the outstanding shares of all class of our common stock as of the last day of the immediately preceding fiscal year; or | |
| ● | such other amount as our board of directors may determine. |
The number of shares reserved for issuance under the 2022 Omnibus Plan will be reduced on the date of the grant of any award by the maximum number of shares, if any, with respect to which such award is granted. However, an award that may be settled solely in cash will not deplete the 2022 Omnibus Plan’s share reserve at the time the award is granted. If (a) an award expires, is canceled, or terminates without issuance of shares or is settled in cash, (b) the Administrator determines that the shares granted under an award will not be issuable because the conditions for issuance will not be satisfied, (c) shares are forfeited under an award, (d) shares are issued under any award and we reacquire them pursuant to our reserved rights upon the issuance of the shares, (e) shares are tendered or withheld in payment of the exercise price of an option or as a result of the net settlement of outstanding stock appreciation rights or (f) shares are tendered or withheld to satisfy federal, state or local tax withholding obligations, then those shares are added back to the reserve and may again be used for new awards under the 2022 Omnibus Plan. However, shares added back to the reserve pursuant to clauses (d), (e) or (f) in the preceding sentence may not be issued pursuant to incentive stock options.
Options
The Administrator may grant stock options and determine all terms and conditions of each stock option, which include the number of stock options granted, whether a stock option is to be an incentive stock option or non-qualified stock option, and the grant date for the stock option. However, the exercise price per share of common stock may never be less than the fair market value of a share of common stock on the date of grant and the expiration date may not be later than 10 years after the date of grant. Stock options will be exercisable and vest at such times and be subject to such restrictions and conditions as are determined by the Administrator, including with respect to the manner of payment of the exercise price of such stock options.
| 88 |
Stock Appreciation Rights
The Administrator may grant SARs, which represent the right of a participant to receive cash in an amount, or common stock with a fair market value, equal to the appreciation of the fair market value of a share of common stock during a specified period of time. The 2022 Omnibus Plan provides that the Administrator will determine all terms and conditions of each SAR, including, among other things: (a) whether the SAR is granted independently of a stock option or relates to a stock option, (b) the grant price, which may never be less than the fair market value of our common stock as determined on the date of grant, (c) a term that must be no later than 10 years after the date of grant, and (d) whether the SAR will settle in cash, common stock or a combination of the two.
Performance and Stock Awards
The Administrator may grant awards of shares of common stock, restricted stock, RSUs, performance shares or performance units. Restricted stock means shares of common stock that are subject to a risk of forfeiture or restrictions on transfer, which may lapse upon the achievement or partial achievement of performance goals (as described below) or upon the completion of a period of service. An RSU grants the participant the right to receive cash or shares of common stock the value of which is equal to the fair market value of one share of common stock, to the extent performance goals are achieved or upon the completion of a period of service. Performance shares give the participant the right to receive shares of common stock to the extent performance goals are achieved. Performance units give the participant the right to receive cash or shares of common stock valued in relation to a unit that has a designated dollar value or the value of which is equal to the fair market value of one or more shares of common stock, to the extent performance goals are achieved.
The Administrator will determine all terms and conditions of the awards including (a) whether performance goals must be achieved for the participant to realize any portion of the benefit provided under the award, (b) the length of the vesting or performance period and, if different, the date that payment of the benefit will be made, (c) with respect to performance units, whether to measure the value of each unit in relation to a designated dollar value or the fair market value of one or more shares of common stock, and (d) with respect to performance shares, performance units, and RSUs, whether the awards will settle in cash, in shares of common stock (including restricted stock), or in a combination of the two.
Cash Incentive Awards
The Administrator may grant cash incentive awards. An incentive award is the right to receive a cash payment to the extent one or more performance goals are achieved. The Administrator will determine all terms and conditions of a cash incentive award, including, but not limited to, the performance goals (described below), the performance period, the potential amount payable, and the timing of payment. While the 2022 Omnibus Plan permits cash incentive awards to be granted under the 2022 Omnibus Plan, we may also make cash incentive awards outside of the 2022 Omnibus Plan.
Performance Goals
For purposes of the 2022 Omnibus Plan, the Administrator may establish objective or subjective performance goals which may apply to any performance award. Such performance goals may include, but are not limited to, one or more of the following measures with respect to our company or any one or more of our subsidiaries, affiliates, or other business units: net sales; cost of sales; gross income; gross revenue; revenue; operating income; earnings before taxes; earnings before interest and taxes; earnings before interest, taxes, depreciation and amortization; earnings before interest, taxes, depreciation, amortization and exception items; income from continuing operations; net income; earnings per share; diluted earnings per share; total stockholder return; fair market value of a share of common stock; cash flow; net cash provided by operating activities; net cash provided by operating activities less net cash used in investing activities; ratio of debt to debt plus equity; return on stockholder equity; return on invested capital; return on average total capital employed; return on net capital employed; return on assets; return on net assets employed before interest and taxes; operating working capital; average accounts receivable (calculated by taking the average of accounts receivable at the end of each month); average inventories (calculated by taking the average of inventories at the end of each month); economic value added; succession planning; manufacturing return on assets; manufacturing margin; and customer satisfaction. Performance goals may also relate to a participant’s individual performance. The Administrator reserves the right to adjust any performance goals or modify the manner of measuring or evaluating a performance goal.
| 89 |
Dividend Equivalent Units
The Administrator may grant dividend equivalent units. A dividend equivalent unit gives the participant the right to receive a payment, in cash or shares of common stock, equal to the cash dividends or other distributions that we pay with respect to a share of common stock. We determine all terms and conditions of a dividend equivalent unit award, except that dividend equivalent units may not be granted in connection with a stock option or SAR, and dividend equivalent unit awards granted in connection with another award cannot provide for payment until the date such award vests or is earned, as applicable.
Other Stock-Based Awards
The Administrator may grant to any participant shares of unrestricted stock as a replacement for other compensation to which such participant is entitled, such as in payment of director fees, in lieu of cash compensation, in exchange for cancellation of a compensation right or as a bonus.
Transferability
Awards are not transferable, including to any financial institution, other than by will or the laws of descent and distribution, unless the Administrator allows a participant to (a) designate in writing a beneficiary to exercise the award or receive payment under the award after the participant’s death, (b) transfer an award to a former spouse as required by a domestic relations order incident to a divorce, or (c) transfer an award without receiving any consideration.
Adjustments
If (a) we are involved in a merger or other transaction in which our shares of common stock are changed or exchanged; (b) we subdivide or combine shares of common stock or declare a dividend payable in shares of common stock, other securities, or other property (other than stock purchase rights issued pursuant to a stockholder rights agreement); (c) we effect a cash dividend that exceeds 10% of the fair market value of a share of common stock or any other dividend or distribution in the form of cash or a repurchase of shares of common stock that our board of directors determines is special or extraordinary, or that is in connection with a recapitalization or reorganization; or (d) any other event occurs that in the Administrator’s judgment requires an adjustment to prevent dilution or enlargement of the benefits intended to be made available under the 2022 Omnibus Plan, then the Administrator will, in a manner it deems equitable, adjust any or all of (1) the number and type of shares subject to the 2022 Omnibus Plan and which may, after the event, be made the subject of awards; (2) the number and type of shares of common stock subject to outstanding awards; (3) the grant, purchase, or exercise price with respect to any award; and (4) the performance goals of an award. In any such case, the Administrator may also provide for a cash payment to the holder of an outstanding award in exchange for the cancellation of all or a portion of the award, subject to the terms of the 2022 Omnibus Plan.
The Administrator may, in connection with any merger, consolidation, acquisition of property or stock, or reorganization, authorize the issuance or assumption of awards upon terms and conditions we deem appropriate without affecting the number of shares of common stock otherwise reserved or available under the 2022 Omnibus Plan.
| 90 |
Change of Control
Upon a change of control (as defined in the 2022 Omnibus Plan), the successor or surviving corporation may agree to assume some or all outstanding awards or replace them with the same type of award with similar terms and conditions, without the consent of any participant, subject to the following requirements:
| ● | Each award that is assumed must be appropriately adjusted, immediately after such change of control, to apply to the number and class of securities that would have been issuable to a participant upon the consummation of such change of control had the award been exercised, vested, or earned immediately prior to such change of control, and other appropriate adjustment to the terms and conditions of the award may be made. | |
| ● | If the securities to which the awards relate after the change of control are not listed and traded on a national securities exchange, then (a) each participant must be provided the option to elect to receive, in lieu of the issuance of such securities, cash in an amount equal to the fair value of the securities that would have otherwise been issued, and (b) no reduction may be taken to reflect a discount for lack of marketability, minority, or any similar consideration, for purposes of determining the fair value of such securities. | |
| ● | If a participant is terminated from employment without cause, or due to death or disability, or the participant resigns employment for good reason (as defined in any award or other agreement between the participant and our company or an affiliate) within two years following the change of control, then upon such termination, all of the participant’s awards in effect on the date of such termination will vest in full or be deemed earned in full. |
If the purchaser, successor, or surviving entity does not assume the awards or issue replacement awards, then immediately prior to the change of control date, unless the Administrator otherwise determines:
| ● | Each stock option or SAR then held by a participant will become immediately and fully vested, and all stock options and SARs will be cancelled on the change of control date in exchange for a cash payment equal to the excess of the change of control price of the shares of common stock over the purchase or grant price of such shares under the award. | |
| ● | Unvested restricted stock and RSUs (that are not performance awards) will vest in full. | |
| ● | All performance shares, performance units and cash incentive awards for which the performance period has expired will be paid based on actual performance, and all such awards for which the performance period has not expired will be cancelled in exchange for a cash payment equal to the amount that would have been due under such awards, valued assuming achievement of target performance goals at the time of the change of control, prorated based on the number of full months elapsed in the performance period. | |
| ● | All unvested dividend equivalent units will vest (to the same extent as the award granted in tandem with such units) and be paid. | |
| ● | All other unvested awards will vest and any amounts payable will be paid in cash. |
Term of Plan
Unless earlier terminated by our board of directors, the 2022 Omnibus Plan will terminate on, and no further awards may be granted, after the tenth (10th) anniversary of its effective date.
Termination and Amendment of Plan
| 91 |
Our board of directors or the Administrator may amend, alter, suspend, discontinue, or terminate the 2022 Omnibus Plan at any time, subject to the following limitations:
| ● | Our board of directors must approve any amendment to the 2022 Omnibus Plan if we determine such approval is required by prior action of our board of directors, applicable corporate law, or any other applicable law; | |
| ● | Stockholders must approve any amendment to the 2022 Omnibus Plan, which may include an amendment to materially increase the number of shares reserved under the 2022 Omnibus Plan, if we determine that such approval is required by Section 16 of the Exchange Act, the Code, the listing requirements of any principal securities exchange or market on which the shares are then traded, or any other applicable law; and | |
| ● | Stockholders must approve any amendment to the 2022 Omnibus Plan that would diminish the protections afforded by the participant award limits or repricing and backdating prohibitions. |
Amendment, Modification, Cancellation and Disgorgement of Awards
Subject to the requirements of the 2022 Omnibus Plan, the Administrator may modify or amend any award or waive any restrictions or conditions applicable to any award or the exercise of the award, or amend, modify, or cancel any terms and conditions applicable to any award, in each case, by mutual agreement of the Administrator and the participant or any other person that may have an interest in the award, so long as any such action does not increase the number of shares of common stock issuable under the 2022 Omnibus Plan.
We do not need to obtain participant (or other interested party) consent for any such action (a) that is permitted pursuant to the adjustment provisions of the 2022 Omnibus Plan; (b) to the extent we deem the action necessary to comply with any applicable law or the listing requirements of any principal securities exchange or market on which our common stock is then traded; (c) to the extent we deem the action is necessary to preserve favorable accounting or tax treatment of any award for us; or (d) to the extent we determine that such action does not materially and adversely affect the value of an award or that such action is in the best interest of the affected participant or any other person as may then have an interest in the award.
The Administrator can cause a participant to forfeit any award, and require the participant to disgorge any gains attributable to the award, if the participant engages in any action constituting, as determined by the Administrator in its discretion, cause for termination, or a breach of a material company policy, any award agreement or any other agreement between the participant and us or one of our affiliates concerning noncompetition, nonsolicitation, confidentiality, trade secrets, intellectual property, nondisparagement or similar obligations.
Any awards granted under the 2022 Omnibus Plan, and any shares of common stock issued or cash paid under an award, will be subject to recoupment our Compensation Recovery Policy (as described below), or any recoupment or similar requirement otherwise made applicable by law, regulation or listing standards to us, or that may be provided for in any cash or equity award granted by us.
| 92 |
Compensation of Directors
The following table sets forth all compensation paid to our Board members during the year ended December 31, 2023:
| Name | Fees Earned or Paid in Cash ($) (1) | Stock Awards ($) | Option Awards ($) (7) | Non-Equity Incentive Plan Compensation ($) | Change in Pension Value and Nonqualified Deferred Compensation Earnings ($) | All Other Compensation ($) | Total ($) | |||||||||||||||||||||
| Chris Chapman, PhD. (2) | 128,629 | - | 493,600 | - | - | - | 622,229 | |||||||||||||||||||||
| Mike Jerman | - | - | - | - | - | - | - | |||||||||||||||||||||
| Talhia Tuck(3) | - | - | 35,150 | - | - | - | 35,150 | |||||||||||||||||||||
| Brad Kroenig(3) | - | - | 35,150 | - | - | - | 35,150 | |||||||||||||||||||||
| Hugh McColl III(3) | - | - | 35,150 | - | - | - | 35,150 | |||||||||||||||||||||
| Christos Nicholoudis, Esq. (4) | 74,085 | - | 109,940 | - | - | - | 184,025 | |||||||||||||||||||||
Dave Vorhoff, former director (5) | - | - | 35,150 | - | - | - | 35,150 | |||||||||||||||||||||
| Brian Daly, former director (6) | - | - | - | - | - | - | - | |||||||||||||||||||||
| (1) | Cash payments made to Dr. Chapman and Mr. Nicholoudis are related to their employment agreements, respectively. | |
| (2) | On March 9, 2024, Dr. Chapman resigned from our Company as Executive Chairman and as an employee. | |
| (3) | On March 9, 2024, Ms. Tuck, Mr. Kroenig and Mr. McColl resigned from our Company as members of the Board of Directors. | |
| (4) | On January 15, 2024, Mr. Nicholoudis resigned from our Company as General Counsel and as a member of the Board of Directors. | |
| (5) | On October 19, 2023, Mr. Vorhoff resigned from our Company as a member of the Board of Directors. | |
| (6) | On December 15, 2023, Mr. Daly resigned from our Company as a member of the Board of Directors. | |
| (7) | The reported amounts represent the aggregate grant date fair value of the awards computed in accordance with Financial Accounting Standards Board Account Standards Codification Topic 718, Stock Compensation, as modified or supplemented, or FASB ASC Topic 718. The assumptions used in calculating the grant date fair value of the stock options reported in this column are set forth in Note 8 to our Consolidated Financial Statements for the year ended December 31, 2022 included in this Report. |
Compensation Recovery Policy
On October 2, 2023, our Board of Directors adopted a policy (commonly known as a “clawback” policy) which provides for the recovery of erroneously awarded incentive compensation to certain of our officers in the event that we are required to prepare an accounting restatement due to material noncompliance by us with any financial reporting requirements under the federal securities laws. This policy is designed to comply with Section 10D of the Securities Exchange Act of 1934, as amended, related rules and the listing standards of Nasdaq Stock Market or any other securities exchange on which our shares are listed in the future. The policy is administered by our Board of Directors or, if so designated by the Board of Directors, the Compensation Committee. Any determinations made by the Board shall be final and binding on all affected individuals.
The individuals covered by this policy (the “Covered Officers”) are any current or former employee who is or was identified as our president, principal financial officer, principal accounting officer (or if there is no such accounting officer, the controller), any vice-president in charge of a principal business unit, division, or function (such as sales, administration, or finance), any other officer who performs a significant policy-making function, or any other person (including any executive officer of our subsidiaries or affiliates) who performs similar significant policy-making functions for us.
The policy covers our recoupment of “Incentive-Based Compensation” (as defined in the policy) received by a person after beginning service as a Covered Executive and who served as a Covered Officer at any time during the performance period for that Incentive Compensation. In the event we are required to prepare an accounting restatement, the policy requires us to recover, reasonably promptly, any excess incentive compensation (as determined by our Board of Directors or Compensation Committee) received by any Covered Officer during the three completed fiscal years immediately preceding the date on which we are required to prepare such accounting restatement. The foregoing description of our Compensation Recovery Policy does not purport to be complete and is qualified in its entirety by the terms and conditions of such policy, a copy of which is filed as an exhibit to this Report and is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The following table sets forth, as of the date of this Report, the ownership of our securities by: (i) each of our directors, (ii) all persons who, to our knowledge, are the beneficial owners of more than 5% of the outstanding shares of common stock, (iii) each of the executive officers, and (iv) all of our directors and executive officers, as a group. Each person named in this table has sole investment power and sole voting power with respect to the shares of common stock set forth opposite such person’s name, except as otherwise indicated.
| 93 |
| Name and Address of Beneficial Owner | Amount and Nature of Beneficial Ownership | Percentage of Class as of March 28 2024 | ||||||
| Directors and Executive Officers (1) | ||||||||
| Erez Aminov | 623,500 | 4.15 | % | |||||
| Michelle Yanez | 57,779 | * | ||||||
| Michael Jerman | 25,000 | * | ||||||
| Matthew Del Giudice | 25,000 | * | ||||||
| Denil Nanji Shekhat | 25,000 | * | ||||||
| Edward MacPherson | 25,000 | * | ||||||
| All current directors and officers as a group (6 persons) (2) | 781,279 | 5.21 | % | |||||
| 5% Stockholders | ||||||||
| Brian McNulty(3) | 5,110,270 | 34.57 | % | |||||
*Represents beneficial ownership of less than 1%
| (1) | Unless otherwise denoted, the address of each noted person is 1200 Brickell Avenue, Suite 1950 #1183, Miami, Florida 33131. | |
| (2) | Includes shares subject to options granted under our 2022 Omnibus Plan that are exercisable as of the Beneficial Ownership Date or within 60 days of the Beneficial Ownership Date held as follows: Mr. Aminov, 250,000 shares and Ms. Yanez, 57,779 shares, Mr. Jerman, 25,000 shares, Dr. Del Guidice, 25,000 shares, Dr. Shekhat, 25,000 shares, Mr. MacPherson, 25,000 shares, and all current officers and directors as a group, 407,779 shares. Excludes shares subject to options granted under our 2022 Omnibus Plan that are not exercisable within 60 days of the Beneficial Ownership Date. |
| (3) | Includes (i) 10,000 shares held directly by Mr. McNulty, (ii) 2,740,270 shares held by the Bay Shore Trust, (iii) 660,000 shares held by the Celeste J Williams Lifetime QTIP Trust, (iv) 1,000,000 shares issuable pursuant to warrants held by the Bay Shore Trust that are immediately exercisable, and (v) 700,000 shares issuable pursuant to warrants held by MIRALOGX LLC, that are immediately exercisable. As trustee of the Bay Shore Trust and the Celeste J Williams Lifetime QTIP Trust, Mr. McNulty has sole voting and dispositive power over the shares held by each trust, and, as a result is deemed to have beneficial ownership (as determined under Section 13(d) of the Exchange Act) of the securities held by the trusts. The address for MIRALOGX LLC and the Bay Shore Trust is 900 West Platt Street, Suite 200, Tampa, Florida, 33606. |
DELINQUENT SECTION 16(A) REPORTS
Section 16(a) of the Exchange Act requires directors and executive officers, and persons who own more than 10% of the Company’s common stock, to report to the SEC their initial ownership of the Company’s common stock and any subsequent changes in that ownership. Specific due dates for these reports have been established by the SEC and we are required to disclose in this Annual Report on Form 10-K any late filings or failures to file.
Based solely on review of the copies of such reports furnished to us and written representations from reporting persons that no other reports were required during the fiscal year ended December 31, 2023, we believe that, during the 2023 fiscal year, all of the Company’s directors and executive officers complied with all Section 16(a) filing requirements applicable to them, with the exception of one late filing By the Bay Shore Trust, which was required to be filed on November 22, 2023, but was filed on December 27, 2023.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table indicates shares of common stock authorized for issuance under our 2022 Omnibus Plan as of December 31, 2023:
Plan category | Number of securities to be issued upon exercise of outstanding options and warrants | Weighted- average exercise price of outstanding options and warrants | Number of securities remaining available for future issuance | |||||||||
| Equity compensation plans approved by security holders | 2,973,571 | $ | 4.45 | 789,999 | ||||||||
| Equity compensation plans not approved by security holders | - | - | - | |||||||||
| Total | 2,973,571 | $ | 4.45 | 789,999 | ||||||||
| 94 |
Item 13. Certain Relationships and Related Transactions, and Director Independence.
The following is a description of transactions within the last two years to which we have been a party, in which the amount involved exceeded or will exceed $120,000, and in which any of our executive officers, directors or holders of more than 5% of our voting securities, or an immediate family member thereof, had or will have a direct or indirect material interest. We believe the terms obtained or consideration that we paid or received, as applicable, in connection with the transactions described below were comparable to terms available or amounts that would be paid or received, as applicable, in arm’s-length transactions with unrelated third parties.
Line of Credit and Promissory Note with the Bay Shore Trust
On April 28, 2023, we entered into the Bay Shore Note with the Bay Shore Trust, under which we have the right to borrow up to an aggregate of $5,000,000 from the Bay Shore Trust at any time up to the second anniversary of the issuance of the Bay Shore Note or, if earlier, upon the completion of our initial public offering. Our right to borrow funds under the Bay Shore Note is subject to the absence of a material adverse change in our assets, operations, or prospects. The Bay Share Note, together with accrued interest, will become due and payable on the second anniversary of the issuance of the note, provided that it may be prepaid at any time without penalty. The Bay Shore Note will accrue interest at a rate equal 7% per annum, simple interest, during the first year that the note is outstanding and 10% per annum, simple interest, thereafter. The Bay Shore Note is unsecured. As of June 30, 2023, the Bay Shore Note had an outstanding principal balance of $1.8 million and accrued and unpaid interest of $0.04 million. Under the Bay Shore Trust Conversion Agreement, the Bay Shore Trust agreed to convert, upon the completion of our initial public offering, $1,100,190 of the outstanding principal balance of the Bay Shore Note into shares of our common stock at a conversion price equal to our initial public offering price, which resulted in the issuance of 157,170 shares to the Bay Shore Trust upon the completion of our initial public offering. The note was paid off as of December 31, 2023.
In consideration of the loan facility provided by the Bay Shore Trust, we issued to the Bay Shore Trust a common stock purchase warrant on April 28, 2023 giving the Bay Shore Trust the right to purchase up to 1,000,000 shares of common stock at an exercise price of $5.00 per share, which warrant will expire five years after the date of grant. Pursuant to a registration rights agreement, we have granted to Bay Shore Trust the right to require us, at any time after one year following our initial public offering, to register for resale the shares issuable upon the exercise of the warrant, with such registration rights being in the form of demand and “piggyback” registration rights that are subject to customary limitations and restrictions. Upon issuance, the warrant met the criteria to be classified as equity based on an analysis under Accounting Standards Codification (480) ASC 480, “Distinguishing Liabilities from Equity” and was measured at fair value, resulting in an initial fair value of approximately $3.5 million upon issuance of the warrant using Black-Scholes valuation techniques.
Transactions with MIRALOGX LLC
Since January 1, 2023, MIRALOGX has advanced funds on behalf of Bay Shore Trust to our company in order to fund operating activities. The total amount advanced and outstanding from MIRALOGX was $1.6 million immediately prior to being consolidated into the Bay Shore Note on June 30, 2023, and such amounts became a part of the outstanding balance of the Bay Shore Note as of June 30, 2023 and are payable under the terms of the Bay Shore Note.
We are also a party to an Agreement for Shared Lease Costs, dated April 1, 2023, with MIRALOGX under which we have agreed to pay our pro rata share of the operating usage costs owing by MIRALOGX under an aircraft lease agreement between MIRALOGX and Supera Aviation I LLC (“Supera Aviation”) based on our usage of the leased aircraft each month. No amounts are payable by us under this agreement unless and to the extent we choose to utilize the leased aircraft. As such, we discontinued the use of the aircraft in March 2023. Prior to entering into this agreement, we were a party to an aircraft lease agreement with Supera Aviation from April 20, 2021, through March 31, 2023. We paid Supera Aviation an aggregate of $0.5 million during the first quarter of 2023 and $1.7 million in 2022. Supera Aviation is a company owned by Starwood Trust.
| 95 |
On November 15, 2023, we entered into an exclusive license agreement in with MIRALOGX to develop and commercialize a drug product containing 2-(2-chlorophenyl)-2-(methylamino) cyclopentan-1-one (sometimes referred to by the Parties as “M209” or “KETAMIR-2”) as an active agent in North America. The exclusive license in the license agreement includes our right to sublicense the licensed intellectual property. Pursuant to the terms of the license agreement, and subject to the conditions set forth therein, we paid MIRALOGX a one-time, nonrefundable payment of $100,000 upon the signing of the Agreement and will be obligated to pay quarterly royalty payments on sales of the Product in the Territory of 8% of net sales and 8% of other revenue (such as milestone or sublicense payments) from licensed products. Also, in consideration of License Agreement, we issued to MIRALOGX a common stock purchase warrant to purchase up to 700,000 shares of our common stock. The MIRALOGX Warrants are exercisable, in whole or in part, any time prior to November 15, 2028, at a cash exercise price of $2.00 per share.
On November 15, 2023, we entered into a promissory note and loan agreement with MIRALOGX. Pursuant to the loan agreement, we may borrow up to $3.0 million from MIRALOGX to fund the development of licensed products under the license agreement. Together with any advance request, we will deliver to the Lender a budget for the requested advance. The budget may only include costs directly associated with preparing an IND application for KETAMIR-2, exclusive of personnel costs. Any advances made by the Lender to us pursuant to this note may be repaid by us (together with any and all interest accrued thereon) at any time without penalty or premium in accordance with the terms hereof. Amounts repaid hereunder may not be reborrowed. The loan agreement has a one-year term, and all outstanding principal and accrued but unpaid interest must be repaid in full on November 15, 2023. Interest on the amounts borrowed under the loan agreement accrues at an annual fixed rate of 8%. We may prepay all or a portion of the outstanding principal and accrued unpaid interest under the loan agreement at any time without a prepayment fee.
Consulting and Employment Agreements with Dr. Chris Chapman
On April 1, 2022, we entered into a Consulting Agreement with Dr. Chapman pursuant to which he provided regulatory and drug development consulting services to the Company on an as-requested basis. Pursuant to the Consulting Agreement, he was to be paid a one-time fee of $100,000 upon the completion of our initial public offering (of which $50,000 was prepaid in in the first quarter of 2022) plus a monthly fee of $20,000 thereafter. The monthly fee was to begin upon the completion of our initial public offering. He was also reimbursed for reasonable out-of-pocket expenses incurred in connection with his duties under the Consulting Agreement. The agreement had a term of one year with an automatic one-year extension, provided that either party could terminate the agreement without cause upon 30-days prior written notice.
In his capacity as a consultant, Dr. Chapman was also granted on June 15, 2022, an option to purchase up to 200,000 shares of our common stock at an exercise price of $5.00 per share. Upon Dr. Chapman becoming Executive Chairman, received additional compensation in that capacity, and his employment agreement replaced his Consulting Agreement. See “Executive Compensation” above. Dr. Chapman resigned his positions with our company on March 9, 2024.
Review and Approval of Related Party Transactions
Our board of directors has adopted a written policy regarding the review and approval of related party transactions. Our audit committee charter provides that the audit committee shall review and approve or disapprove any related party transactions, which are transactions between us and related persons in which the aggregate amount involved exceeds or may be expected to exceed the lessor of $120,000 or one percent of the average of our total assets at year end for the last two completed fiscal years and in which a related person has or will have a direct or indirect material interest. Our policy regarding transactions between us and related persons provides that a related person is defined as a director, executive officer, nominee for director or greater than 5% beneficial owner of our common stock, in each case since the beginning of the most recently completed year, and any of their immediate family members.
| 96 |
Certain of the foregoing disclosures are summaries of certain provisions of our related party agreements and are qualified in their entirety by reference to all of the provisions of such agreements. Because these descriptions are only summaries of the applicable agreements, they do not necessarily contain all of the information that you may find useful. Copies of certain of the agreements have been filed as exhibits to this Report and are available electronically on the website of the SEC at www.sec.gov.
As a matter of corporate governance policy, we have not and will not make loans to officers or loan guarantees available to “promoters” as that term is commonly understood by the SEC and state securities authorities.
All future transactions between us and our officers, directors or five percent stockholders, and respective affiliates will be on terms no less favorable than could be obtained from unaffiliated third parties and will be approved by a majority of our independent directors who do not have an interest in the transactions and who had access, at our expense, to our legal counsel or independent legal counsel.
Item 14. Principal Accountant Fees and Services.
Audit Fees.
The aggregate fees billed by Cherry Bekaert LLP for professional services rendered for the audit of our annual financial statements, review of the financial information included in our Forms 10-Q for the respective periods and other required filings with the SEC for the years ended December 31, 2023 and December 31, 2022 totaled $0.06 million and $0.05 million, respectively.
The above amounts include interim procedures and audit fees, as well as attendance at audit committee meetings.
Audit-Related Fees.
The aggregate fees billed by Cherry Bekaert LLP for audit-related fees for the years ended December 31, 2023 and 2022 were $0.1 million and $0.01 million, respectively. The fees were provided in consideration of services consisting of review and update procedures associated with registration statements and other SEC filings.
Tax Fees.
The aggregate fees billed by Cherry Bekaert LLP for professional services rendered for tax compliance for the years ended December 31, 2023 were $0.02 million. There were no such fees incurred in 2022. The fees were provided in consideration of services consisting of preparation of tax returns and related tax advice.
All Other Fees. None
The Audit Committee of our board of directors has established its pre-approval policies and procedures, pursuant to which the Audit Committee approved the foregoing audit and non-audit services provided by Cherry Bekaert LLP in 2023. Consistent with the Audit Committee’s responsibility for engaging our independent auditors, all audit and permitted non-audit services require pre-approval by the Audit Committee. The full Audit Committee approves proposed services and fee estimates for these services. The Audit Committee chairperson has been designated by the Audit Committee to approve any audit-related services arising during the year that were not pre-approved by the Audit Committee. Any non-audit service must be approved by the full Audit Committee. Services approved by the Audit Committee chairperson are communicated to the full Audit Committee at its next regular meeting and the Audit Committee reviews services and fees for the fiscal year at each such meeting. Pursuant to these procedures, the Audit Committee approved the foregoing services provided by Cherry Bekaert LLP.
| 97 |
PART IV
Item 15. Exhibits, Financial Statement Schedules.
The information called for by this Item is incorporated herein by reference to the Exhibit Index in this Form 10-K.
| 98 |
+ Denotes management contract or compensatory plan or arrangement.
| * | Filed herewith |
| ** | Furnished herewith |
# A signed original of this written statement required by Section 906 has been provided to the Company and will be retained by the Company and furnished to the Securities and Exchange Commission or its staff upon request.
Item 16. Form 10-K
Summary None.
| 99 |
MIRA PHARMACEUTICALS, INC.
INDEX TO FINANCIAL STATEMENTS
| F-1 |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders
MIRA Pharmaceuticals, Inc.
Tampa, Florida
Opinion on the Financial Statements
We have audited the accompanying balance sheets of MIRA Pharmaceuticals, Inc. (the “Company”) as of December 31, 2023 and 2022, and the related statements of operations, stockholders’ equity (deficit) and cash flows for the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The accompanying financial statements have been prepared assuming the Company will be able to continue as a going concern. As discussed in Note 2 to the financial statements, the Company has incurred recurring net losses and negative operating cash flows since inception. These factors, among others, raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
We have served as the Company’s auditor since 2022.
/s/
April 1, 2024
| F-2 |
MIRA PHARMACEUTICALS, INC.
BALANCE SHEETS
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash | $ | $ | ||||||
| Deferred offering costs | ||||||||
| Other receivables | ||||||||
| Prepaid expenses | ||||||||
| Total current assets | ||||||||
| Operating lease, right of use assets | ||||||||
| Related party operating lease, right of use assets | ||||||||
| Related party accounts receivable | ||||||||
| Total assets | $ | $ | ||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) | ||||||||
| Current liabilities: | ||||||||
| Trade accounts payable and accrued liabilities | $ | $ | ||||||
| Related party accounts payable | ||||||||
| Related party line of credit | ||||||||
| Related party accrued interest | ||||||||
| Current portion of operating lease liabilities | ||||||||
| Related party current portion of operating lease liabilities | ||||||||
| Total current liabilities | ||||||||
| Non-current operating lease liabilities | ||||||||
| Total liabilities | ||||||||
| Stockholders’ Deficit | ||||||||
| Preferred Stock, $ par value, shares authorized and issued or outstanding. | ||||||||
| Common Stock, $ par value; shares authorized, and shares issued and outstanding at December 31, 2023 and December 31, 2022, respectively. | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| Total stockholders’ equity (deficit) | ( | ) | ||||||
| Total liabilities and stockholders’ equity (deficit) | $ | $ | ||||||
See notes to consolidated financial statements
| F-3 |
MIRA PHARMACEUTICALS, INC.
STATEMENTS OF OPERATIONS
| Year Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Revenues | $ | $ | ||||||
| Operating costs: | ||||||||
| General and administrative expenses | ||||||||
| Related party travel costs | ||||||||
| Research and development expenses | ||||||||
| Total operating costs | ||||||||
| Interest expense, net | ( | ) | ( | ) | ||||
| Net loss attributable to common stockholders | $ | ( | ) | $ | ( | ) | ||
| Basic and diluted loss per share | $ | ) | $ | ) | ||||
| Weighted average common stock shares outstanding | ||||||||
See notes to consolidated financial statements
| F-4 |
MIRA PHARMACEUTICALS, INC.
STATEMENTS OF STOCKHOLDERS’ EQUITY
| Common Stock | Additional Paid-In | Stock Subscription | Accumulated | Total Stockholders’ | ||||||||||||||||||||
| Shares | Amount | Capital | Receivable | Deficit | Equity | |||||||||||||||||||
| Balances, January 1, 2022 | $ | $ | $ | $ | ( | ) | $ | |||||||||||||||||
| Sale of common stock | ||||||||||||||||||||||||
| Stock-based compensation | - | |||||||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||||||
| Balances, December 31, 2022 | $ | $ | $ | $ | ( | ) | $ | ( | ) | |||||||||||||||
| Common Stock | Additional Paid-In | Stock Subscription | Accumulated | Total Stockholders’ | ||||||||||||||||||||
| Shares | Amount | Capital | Receivable | Deficit | Deficit | |||||||||||||||||||
| Balances, January 1, 2023 | $ | $ | $ | $ | ( | ) | $ | ( | ) | |||||||||||||||
| Stock-based compensation | - | ( | ) | |||||||||||||||||||||
| Issuance of common stock at IPO, net | ||||||||||||||||||||||||
| Issuance of common stock conversion of debt | ||||||||||||||||||||||||
| Issuance of common stock | ||||||||||||||||||||||||
| Issuance of Warrants | - | |||||||||||||||||||||||
| Net loss | - | ( | ) | ( | ) | |||||||||||||||||||
| Balances, December 31, 2023 | $ | $ | $ | $ | ( | ) | $ | |||||||||||||||||
See notes to consolidated financial statements
| F-5 |
MIRA PHARMACEUTICALS, INC.
STATEMENTS OF CASH FLOWS
| Year Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Cash flows from Operating activities | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Adjustments to reconcile net loss to net cash from operations | ||||||||
| Interest expense | ||||||||
| Amortization of debt issuance costs | ||||||||
| Stock-based compensation expense | ||||||||
| Non-cash investor relations fees | ||||||||
| Change in operating assets and liabilities: | ||||||||
| Right of use lease, net | ( | ) | ||||||
| Accounts payable and accrued expenses | ( | ) | ||||||
| Prepaid expenses | ( | ) | ||||||
| Accounts receivable | ( | ) | ||||||
Related party line of credit | ||||||||
| Net cash flows used in operating activities | ( | ) | ( | ) | ||||
| Financing activities: | ||||||||
| Advances (to) from affiliates | ( | ) | ||||||
| Advances received from related party line of credit | ||||||||
| Deferred offering costs | ( | ) | ||||||
| Repayments under related party line of credit | ( | ) | ( | ) | ||||
| Proceeds from sale of common stock, less offering costs | ||||||||
| Net cash flows provided by financing activities | ||||||||
| Net change in cash | ( | ) | ||||||
| Cash, beginning of year | ||||||||
| Cash, end of period | $ | $ | ||||||
| Cash paid for interest | ||||||||
See notes to consolidated financial statements
| F-6 |
MIRA PHARMACEUTICALS, INC.
SUPPLEMENTAL CASH FLOW INFORMATION
Non-cash Financing and Investing Activities:
The
Company recorded the fair value of a total of shares of common stock issued to Bay Shore Trust during the year ended December
31, 2023 which totaled approximately $
On
November 15, 2023, the Company entered a warrant agreement and recorded the fair value of a total of shares of common stock issued
to MIRALOGX, LLC which totaled $
The
Company recorded the fair value of a total of shares of common stock issued to Bay Shore Trust during the year ended December
31, 2023 totaling approximately $
The
Company recorded the fair value of a total of shares of common stock issued to the MZ Group during the year ended December 31,
2023 totaling $
The
Company recorded a right of use asset and a corresponding liability in the amount of $
See notes to consolidated financial statements
| F-7 |
MIRA PHARMACEUTICALS, INC.
NOTES TO THE FINANCIAL STATEMENTS
Note 1. Description of business and summary of significant accounting policies:
Overview
MIRA Pharmaceuticals, Inc. (“MIRA” or the “Company” and formerly known as MIRA1a Therapeutics, Inc.) is a pre-clinical-stage pharmaceutical development company with two neuroscience programs targeting a broad range of neurologic and neuropsychiatric disorders. The Company has an exclusive licensing agreement for Ketamir-2, a unique, patent pending novel oral ketamine analog under investigation to potentially deliver ultra-rapid antidepressant effects, providing hope for individuals battling treatment-resistant depression (TRD) and major depressive disorder with suicidal ideation (MDSI). The Company’s novel oral pharmaceutical marijuana, MIRA-55, is currently under investigation for treating adult patients suffering from anxiety and cognitive decline, often associated with early-stage dementia. MIRA-55, if approved by the FDA, could mark a significant advancement in addressing various neuropsychiatric, inflammatory, and neurologic diseases and disorders.
The U.S. Drug Enforcement Administration (DEA)’s scientific review of Ketamir-2 concluded that it would not be considered a controlled substance or listed chemical under the Controlled Substances Act (CSA) and its governing regulations. Additionally, we have submitted the required paperwork for MIRA-55 to be evaluated by the DEA.
The Company was organized as a Florida corporation in September 2020 and commenced substantive operations in late 2020, at which time the Company commenced its pharmaceutical development program.
The accounting and reporting policies of the Company conform to accounting principles generally accepted in the United States of America (“GAAP”).
As used herein, the Company’s Common Stock, par value $ per share, is referred to as the “Common Stock” and the Company’s preferred stock, par value $ per share, is referred to as the “Preferred Stock”.
Operating updates
In early February 2024, we made a significant discovery during the manufacturing and scale-up process of our patented molecule known as “MIRA1a,” which we had been utilizing with a contract manufacturer. Through this process, we identified a novel and improved version of the molecule, MIRA-55. MIRA-55 exhibits enhanced potency and holds promise for improved efficacy compared to MIRA1a.
As part of our due diligence and subsequent testing, we discovered that the pre-clinical studies we conducted, previously attributed to MIRA1a, were in fact performed on MIRA-55. Following this revelation, we promptly filed a provisional patent for MIRA-55, which encompasses all pre-clinical studies disclosed in our two registration statements on Form S-1, declared effective on August 2, 2023 and December 27, 2023 (File Nos. 333-273024 and 333-276118, respectively).
Moreover, based on our pre-clinical analyses to date, we believe that MIRA-55 is an improvement over MIRA1a in that it displays enhanced potency and potential for efficacy. In early March 2024, we filed a provisional patent application for MIRA-55, aiming for global patent protection. If such patent is issued, we would own the patent rights to both MIRA1a and MIRA-55.
Additional testing is required to confirm our preliminary beliefs. However, based on our discoveries to date, the Company has decided to advance MIRA-55 as our lead compound for our oral pharmaceutical marijuana drug candidate while still retaining our rights to MIRA1a. As such, we do not intend to move MIRA1a forward as of the date of this Report.
| F-8 |
Initial public offering
On
August 7, 2023, the Company closed its initial public offering consisting of shares at a price of $ per share for approximately
$
The shares were offered and sold pursuant to the Company’s Registration Statement on Form S-1, as amended (File No. 333-273024), originally filed with the Securities and Exchange Commission (the “SEC”) on June 29, 2023 (the “Registration Statement”) and the final quarterly report filed with the Commission pursuant to Rule 424(b)(4) of the Securities Act of 1933, as amended. The Registration Statement was declared effective by the Commission on August 2, 2023. The common stock began trading on The Nasdaq Capital Market on August 3, 2023 under the symbol “MIRA”. The closing of the IPO occurred on August 7, 2023.
As of the completion of the IPO, among other things, certain of the Company’s then-outstanding convertible debt was converted into shares of common stock. See Note 5 for more information.
Revenue recognition
The Company currently has no source of revenue. Miscellaneous income, including interest, is recognized when earned by the Company.
Income taxes
The Company is taxed as a C corporation. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amount of existing assets and liabilities and their respective tax bases. Deferred tax assets are recognized for temporary differences that will result in deductible amounts in future years and for loss carryovers. A valuation allowance is recognized regarding deferred tax assets, if any, if it is more likely than not that some portion of the deferred tax asset will not be realized.
Research and development expenses
Research and development costs are expensed in the period in which they are incurred and include the expenses paid to third parties, such as contract research organizations and consultants, who conduct research and development activities on behalf of the Company. Patent-related costs, including registration costs, documentation costs and other legal fees associated with the application, are expensed in the period in which they are incurred.
| F-9 |
General and administrative expense
General and administrative expenses are primarily comprised of personnel costs, marketing expenses, amortization, insurance expenses, professional services fees, travel and office expenses, and stock-based compensation.
Advertising expenses
The Company expenses advertising costs when incurred. Advertising expense for the years ended December 31, 2023 and 2022 is as follows:
| December 31, 2023 | December 31, 2022 | |||||||
| Advertising expenses | $ | $ | ||||||
Leases
The Company accounts for leases under the provisions of FASB ASC Topic 842, “Leases”, which requires the Company to recognize right-to-use (ROU) assets and lease liabilities for operating leases on the balance sheet.
Use of estimates
The preparation of financial statements in accordance with generally accepted accounting principles in the United States of America requires the Company’s management to make estimates and assumptions that affect the reported amounts of assets and liabilities, and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results may differ from such estimates and such differences could be material.
Cash
The Company maintains cash balances with financial institutions that management believes are of high credit quality. The Company’s cash account at times may exceed federally insured limits. The Company has not experienced any losses in such accounts. The Company believes it is not exposed to any significant credit risk from its cash account.
Stock-based compensation
The Company accounts for stock-based compensation under the provisions of FASB ASC 718, “Compensation - Stock Compensation”, which requires the measurement and recognition of compensation expense for all stock-based awards made to employees, directors and consultants based on estimated fair values on the grant date. The Company estimates the fair value of stock-based awards on the date of grant using the Black-Scholes model. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods using the straight-line method. The Company has elected to account for forfeiture of stock-based awards as they occur.
Segment information
ASC Topic 280, “Disclosures about Segments of an Enterprise and Related Information,” established standards for the way that public business enterprises report information about operating segments in annual financial statements and requires those enterprises to report selected information about operating segments in interim financial reports issued to stockholders. Management has determined that the Company operates in one business segment, which is the research and development of neuroscience drug candidates.
| F-10 |
Recent accounting pronouncements not yet adopted
In December 2023, the FASB issued Accounting Standards Update No. 2023-09, “Income Taxes (Topic 740): Improvements to Income Tax Disclosures” (“ASU 2023-09”), which modifies the rules on income tax disclosures to require entities to disclose (1) specific categories in the rate reconciliation, (2) the income or loss from continuing operations before income tax expense or benefit (separated between domestic and foreign) and (3) income tax expense or benefit from continuing operations (separated by federal, state and foreign). ASU 2023-09 also requires entities to disclose their income tax payments to international, federal, state and local jurisdictions, among other changes. The guidance is effective for annual periods beginning after December 15, 2024. Early adoption is permitted for annual financial statements that have not yet been issued or made available for issuance. ASU 2023-09 should be applied on a prospective basis, but retrospective application is permitted. The Company is currently evaluating the potential impact of adopting this new guidance on its financial statements and related disclosures.
Management has considered all other recent accounting pronouncements that are issued, but not effective, and it does not believe that they will have a significant impact on the Company’s results of operations or financial position.
Change in accounting principle
In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842), which supersedes existing guidance for accounting for leases under Topic 840, Leases. The FASB also subsequently issued additional ASUs which amend and clarify Topic 842. The most significant change in the new leasing guidance is the requirement to recognize right-to-use (ROU) assets and lease liabilities for operating leases on the balance sheet.
The
Company adopted these ASUs effective January 1, 2022 using the modified retrospective approach. As a result of adopting these ASUs, the
Company recorded ROU assets and lease liabilities of approximately $
Fair value of financial instruments
The Company measures the fair value of financial instruments in accordance with GAAP which defines fair value, establishes a framework for measuring fair value, and expands disclosures about fair value measurements.
GAAP defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. GAAP also establishes a fair value hierarchy, which requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. The Company considers the carrying amount of deferred offering costs to approximate fair value due to short-term nature of this instrument. GAAP describes three levels of inputs that may be used to measure fair value:
Level 1 – quoted prices in active markets for identical assets or liabilities.
Level 2 – quoted prices for similar assets and liabilities in active markets or inputs that are observable.
Level 3 – inputs that are unobservable (for example cash flow modeling inputs based on assumptions).
Contingencies
In the normal course of business, the Company may be subject to loss contingencies, such as legal proceedings, amounts arising from contractual arrangements and claims arising out of the Company’s business that cover a wide range of matters, including, among others, government investigations, shareholder lawsuits, and tax matters. In accordance with ASC Topic 450, Accounting for Contingencies, (ASC 450), the Company records accruals for such loss contingencies when it is probable that a liability will be incurred, and the amount of loss can be reasonably estimated. The Company, in accordance with this guidance, does not recognize gain contingencies until realized or realizable.
| F-11 |
Note 2. Liquidity and capital resources:
In
accordance with Accounting Standards Codification 205-40, Going Concern, the Company has evaluated whether there are conditions
and events, considered in the aggregate, that raise substantial doubt about the Company’s ability to continue as a going concern
within one year after the date the financial statements are issued. As of December 31, 2023, the Company had cash of approximately $
Historically, the Company has been primarily engaged in developing MIRA-55. During these activities, the Company sustained substantial losses. The Company’s ability to fund ongoing operations and future clinical trials required for FDA approval is dependent on the Company’s ability to obtain significant additional external funding in the near term. Since inception, the Company financed its operations through the sale of Common Stock, the IPO and related party financings. Additional sources of financing may be sought by the Company. The Company expects to be able to fund operations through the fourth quarter of 2024, with available borrowings on the loan agreement (Note 4). Additional financing will be needed by the Company to fund its operations after such date to complete clinical developments and to commercially develop its product candidate. However, there can be no assurance that any fundraising will be achieved on commercially reasonable terms, if at all.
The Company expects to continue to generate losses in the foreseeable future. The Company’s liquidity needs will be determined largely by the budgeted operational expenditures incurred in regard to the progression of its product candidates. Management believes that the Company has sufficient resources available to support its development activities and business operations and timely satisfy its obligations as they become due into the fourth quarter of 2024. The Company does not have sufficient cash and cash equivalents as of the date of filing this Annual Report on Form 10-K to support its operations for at least the 12 months following the date the financial statements are issued. These conditions raise substantial doubt about the Company’s ability to continue as a going concern through 12 months after the date the financial statements are issued.
To alleviate the conditions that raise substantial doubt about the Company’s ability to continue as a going concern, the Company plans to secure additional capital, potentially through a combination of public or private equity offerings and strategic transactions, including potential alliances and drug product collaborations; however, none of these alternatives are committed at this time. There can be no assurance that the Company will be successful in obtaining sufficient funding on terms acceptable to it to fund continuing operations, if at all, identify and enter into any strategic transactions that will provide the capital that it will require or achieve the other strategies to alleviate the conditions that raise substantial doubt about the Company’s ability to continue as a going concern. If none of these alternatives are available, or if available, are not available on satisfactory terms, the Company will not have sufficient cash resources and liquidity to fund its business operations for at least the 12 months following the date the financial statements are issued. The failure to obtain sufficient capital on acceptable terms when needed may require the Company to delay, limit, or eliminate the development of business opportunities and its ability to achieve its business objectives and its competitiveness, and its business, financial condition, and results of operations will be materially adversely affected. In addition, the perception that the Company may not be able to continue as a going concern may cause others to choose not to deal with it due to concerns about its ability to meet its contractual obligations.
The accompanying financial statements have been prepared on a going concern basis, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business, and do not include any adjustments relating to recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might be necessary should the Company be unable to continue as a going concern.
| F-12 |
Note 3 Accounts payable and accrued liabilities:
The following table represents the components of accounts payable and accrued liabilities as of:
| December 31, 2023 | December 31, 2022 | |||||||
| Trade accounts payable | $ | $ | ||||||
| Accrued other | ||||||||
| $ | $ | |||||||
Note 4. License agreement, related party:
MIRALOGX
On November 15, 2023, the Company and MIRALOGX, LLC, a Florida limited liability company (“MIRALOGX”), entered into an exclusive license agreement (the “License Agreement”) to develop and commercialize a drug product containing 2-(2- chlorophenyl)-2-(methylamino) cyclopentan-1-one (sometimes referred to by the Parties as “M209” or “KETAMIR-2”) (“the Product”) as an active agent in North America. (the “Territory”). The exclusive license in the License Agreement includes the right of the Company to sublicense the licensed intellectual property.
Pursuant
to the terms of the License Agreement, and subject to the conditions set forth therein, the Company paid MIRALOGX a one-time, nonrefundable
payment of $
Also,
in consideration of License Agreement, the Company issued to MIRALOGX a Common Stock Purchase Warrant to purchase up to shares
of the Company’s common stock (the “MIRALOGX Warrants”). The MIRALOGX Warrants are exercisable, in whole or in part,
any time prior to November 15, 2028 at a cash exercise price of $
The Company and MIRALOGX have made customary representations and warranties in the License Agreement and have agreed to certain other customary covenants, including confidentiality, cooperation, and indemnity provisions. Either party may terminate the License Agreement for cause if the other party materially breaches or defaults in the performance of its obligations, and, if curable, such material breach remains uncured for 120 days. Unless earlier terminated, the License Agreement will continue in effect until the last to expire of the Patent Rights (the “Term”), unless earlier terminated.
The Company and MIRALOGX have the same founder.
| F-13 |
Note 5. Debt, related party:
MIRALOGX
On November 15, 2023, the Company entered into a Promissory Note and Loan Agreement (the “Loan Agreement”) with MIRALOGX.
Pursuant
to the Loan Agreement, the Company may borrow up to $
Together with any Advance Request, the Company shall deliver to the Lender a budget for the requested Advance (the “Budget”). The Budget may only include costs directly associated with preparing an Investigational New Drug (“IND”) application for KETAMIR-2, exclusive of personnel costs. Any Advances made by the Lender to the Company pursuant to this Note may be repaid by the Company (together with any and all interest accrued thereon) at any time without penalty or premium in accordance with the terms hereof. Amounts repaid hereunder may not be reborrowed.
The
Loan Agreement has a one-year term, and all outstanding principal and accrued but unpaid interest must be repaid in full on November
15, 2024. Interest on the amounts borrowed under the Loan Agreement accrues at an annual fixed rate of
Bay Shore Trust
In
May 2021, the Company entered into a revolving credit facility which allowed for borrowings of up to $
In
April 2023, the Company entered into a Promissory Note and Loan Agreement with the Bay Shore Trust, a trust established by a shareholder
of the Company. Under this Promissory Note and Loan Agreement (the “Bay Shore Note”), the Company has the right to borrow
up to an aggregate of $
The
Bay Shore Note replaced the revolving credit facility that the Company entered into with Starwood Trust, a separate trust established
by a shareholder of the Company, in May 2021 and pursuant to which the Company had an outstanding principal balance of $
In consideration of the loan facility provided by the Bay Shore Trust, in April 2023, the Company issued to the Bay Shore Trust a common stock purchase warrant giving the Bay Shore Trust the right to purchase up to shares of common stock at an exercise price of $ per share, which warrant will expire five years after the date of grant. Pursuant to a registration rights agreement, the Company has granted to Bay Shore Trust the right to require the Company, at any time after one year following the Company’s IPO, to register for resale the shares issuable upon the exercise of the warrant, with such registration rights being in the form of demand and “piggyback” registration rights that are subject to customary limitations and restrictions. See Note 8 for additional details related to these warrants.
| F-14 |
On
July 20, 2023, the Company entered into a conversion agreement with the Bay Shore Trust under which the Bay Shore Trust had agreed
to convert, upon the completion of the IPO, $
Note 6. Related party transactions:
Due from Related Party – As of the year ended December 31, 2023, the Company paid $
Due to Related Party – Amounts due to related parties as of December 31, 2023 and December 31, 2022, are recorded as related
party accounts payable, in the accompanying balance sheets. As of December 31, 2022, amounts due to related parties totaled $
Travel
expenses – In April 2021, the Company entered into an airplane lease with an entity under common control that the Company incurs
approximately $
License agreement - See Note 4.
Line of credit - See Note 5.
Note 7. Leases:
The
Company’s corporate headquarters was in Baltimore, Maryland, which includes a lease for office space. This lease began in November
2021 and was amended in April 2023. This space is approximately
The
Company had leased an office in Tampa, Florida, for its finance and general operations, which began in March 2022 for 37 months. On December
1, 2023, the Company formally terminated the lease with the landlord. There is a remaining deposit due from the landlord to the Company
of $
The Company also leased a jet (Note 5) from a related party, which terminated on March 31 2023.
Variable lease costs
Variable lease costs primarily include utilities, property taxes, and other operating costs that are passed on from the lessor. Variable lease costs related to the aircraft include usage expenses, which includes pilot expenses, jet fuel and general flight expenses.
| F-15 |
The components of lease expense were as follows:
| Year Ended December 31, | ||||||||
| Lease Costs | 2023 | 2022 | ||||||
| Operating Lease Cost | ||||||||
| Operating Lease | $ | $ | ||||||
| Variable Lease Costs | ||||||||
| Total Lease Cost | $ | $ | ||||||
Supplemental cash flow information related to leases were as follows:
| Year Ended December 31, | ||||||||
| Other Lease Information | 2023 | 2022 | ||||||
| Cash paid for amounts included in the measurement of lease liabilities | ||||||||
| Operating cash flows from operating leases | $ | $ | ||||||
| Year Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Lease Term and Discount | ||||||||
| Weighted Average remaining lease term | ||||||||
| Weighted Average discount rate | % | % | ||||||
Maturity of Lease Liabilities
Future minimum lease payments under non-cancellable leases as of December 31, 2023 were as follows:
Maturity of Lease Liabilities
| December 31, 2023 | ||||
| 2024 | $ | |||
| 2025 | ||||
| Total Lease payments | ||||
| Less: Interest | ( | ) | ||
| Present Value of Lease Liabilities | $ | |||
| F-16 |
On April 1, 2023 the Company entered into an Agreement For Shared Lease Costs with MIRALOGX, LLC, (the “Shared Agreement”) who is a related party for the jet usage. Under the Shared Agreement, the Company agrees to make monthly contributions or payments in accordance with its monthly use of shared aircraft toward rent payments. However, the Company has not used the aircraft after the termination of the lease and there are no minimum payments due without usage.
Note 8. Income taxes:
The significant components of the Company’s net deferred tax assets are as follows as of December 31:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Deferred tax assets | ||||||||
| Net operating loss carry-forward | $ | $ | ||||||
| Section 174 Qualified Research Expenditures | ||||||||
| Stock compensation | ||||||||
| ROU liability | ||||||||
| Other | ||||||||
| Less: valuation allowance | ( | ) | ( | ) | ||||
| Deferred tax liabilities | ||||||||
| ROU asset | ( | ) | ( | ) | ||||
| Total net deferred tax asset | $ | $ | ||||||
Beginning
in 2022, in accordance with Internal Revenue Code Section 174, Qualified Research Expenditures are capitalized for tax purposes and amortized
over a period of five years. Accordingly, for income tax purposes, the Company has recorded a deferred tax asset totaling approximately
$
The components of the provision for income taxes consist of the following:
| 2023 | 2022 | |||||||
| Deferred tax: | ||||||||
| Deferred | ( | ) | ( | ) | ||||
| Change in valuation allowance | ||||||||
| Total deferred | ||||||||
| Total provision for income taxes | $ | $ | ||||||
ASC
Topic 740 requires that a deferred tax amount be reduced by a valuation allowance if, based on the weight of available evidence it is
more likely than not (a likelihood of more than 50%) that some portion or all of the deferred tax assets will not be realized. The valuation
allowance should be sufficient to reduce the deferred tax asset to the amount that is more likely than not to be realized. The Company
has recorded a full valuation allowance against its deferred tax assets generated by net operating loss carryforwards as it has determined
that such amounts may not be recognizable, given the historical losses of the Company to date. As of December 31, 2023, the Company has
a cumulative federal net operating loss carryforward of approximately $
| F-17 |
Note 9. Stockholders’ equity:
Capital stock
The
Company has the authority to issue
Reverse stock-split
Stock issuances
At
IPO in August 2023, shares of the Company’s common stock were issued at a price of $ per share which resulted in
gross proceeds of $
Additionally,
the Company issued its investor relations firm $
During
the year ended December 31, 2022, the Company sold million shares of Common Stock at $ per share, net of offering costs of $
2022 Omnibus Incentive Plan
In June 2022, the Company’s Board of Directors adopted, and its stockholders approved, the Company’s 2022 Omnibus Incentive Plan, as amended and restated in August 2023, (“2022 Omnibus Plan”). The 2022 Omnibus Plan authorizes the grant of incentive stock options, within the meaning of Section 422 of the Internal Revenue Code, to the Company’s employees and any of its parent and subsidiary corporations’ employees, and for the grant of nonstatutory stock options, restricted stock, restricted stock units, stock appreciation rights, performance units and performance shares to the Company’s employees, directors, and consultants and any of its future subsidiary corporations’ employees and consultants.
The 2022 Omnibus Plan provides that shares of the Company’s Common Stock are reserved for issuance under the 2022 Omnibus Plan, all of which may be issued pursuant to the exercise of incentive stock options.
Stock-based compensation
The fair value of each option award is estimated on the grant date using the Black-Scholes valuation model that uses assumptions for expected volatility, expected dividends, expected term, and the risk-free interest rate. Expected price volatility is based on the historical volatilities of a peer group as the Company does not have a trading history for its shares prior to its IPO. Industry peers consist of several public companies in the biotech industry similar to the Company in size, stage of life cycle and product indications. The Company intends to continue to consistently apply this process using the same or similar public companies until a sufficient amount of historical information regarding the volatility of the Company’s own stock price becomes available, or unless circumstances change such that the identified companies are no longer similar to the Company, in which case, more suitable companies whose share prices are publicly available would be utilized in the calculation.
Expected term of options granted is derived using the “simplified method” which computes expected term as the average of the sum of the vesting term plus contract term. The risk-free rate is based on the 5-year U.S. Treasury yield curve in effect at the time of grant. The Company recognizes forfeitures as they occur.
| F-18 |
During
the year ended December 31, 2023, a total of options to purchase Common Stock, with an aggregate fair market value of approximately
$
As of December 31, 2023, there was approximately $ million of unrecognized compensation cost related to unvested share-based compensation awards granted. These costs will be expensed over the next two years.
| Number of Shares | Weighted Average Exercise Price Per Share | Aggregate Intrinsic Value | ||||||||||
| Outstanding as January 1, 2022 | $ | $ | ||||||||||
| Options granted | $ | |||||||||||
| Outstanding as December 31, 2022 | $ | $ | ||||||||||
| Options granted | $ | |||||||||||
| Forfeitures | ( | ) | $ | |||||||||
| Outstanding as December 31, 2023 | $ | $ | ||||||||||
| Range of Exercise Prices | Number Outstanding | Weighted Average Remaining Contractual Life (Years) | Weighted Average Exercise Price | Number Exercisable | Aggregate Intrinsic Price | |||||||||||||||||
| $ | - | $ | $ | |||||||||||||||||||
| $ | - | $ | ||||||||||||||||||||
| $ | ||||||||||||||||||||||
| Expected price volatility | - | % | ||
| Risk-free interest rate | - | % | ||
| Weighted average fair values (grants post-split) | $ | - $ | ||
| Weighted average expected life in years | - years | |||
| Dividend yield | - |
On March 25, 2024, a total of options to purchase Common Stock, with an aggregate fair market value of approximately $ million were granted to the Company’s Independent Board of Directors. These option vest as follows: (i) 50% on date of grant and (ii) 50% at 1 year anniversary at date of grant.
On March 26, 2024, a total of options to purchase Common Stock were granted to the Company’s executive officers. These option vest as follows: (i) 50% six months from the date of grant and (ii) 50% at 1 year anniversary at date of grant.
Both aforementioned option grants have a term of 10 years from the grant date.
Warrants
MIRALOGX warrants
The
Company issued to MIRALOGX a common stock purchase warrant on November 15, 2023 giving MIRALOGX the right to purchase up to shares
of common stock at an exercise price of $
| F-19 |
The
fair value of the warrants were estimated on the grant date using the Black-Scholes valuation model and level 3 inputs based on assumptions
for expected volatility, expected dividends, expected term, and the risk-free interest rate, which resulted in $
Bay Shore Trust warrants
In
consideration of the line of credit provided by the Bay Shore Trust, the Company issued to the Bay Shore Trust a common stock
purchase warrant on April 28, 2023 giving the Bay Shore Trust the right to purchase up to
shares of common stock at an exercise price of $
The
fair value of the warrants were estimated on the grant date using the Black-Scholes valuation model and level 3 inputs based on assumptions
for expected volatility, expected dividends, expected term, and the risk-free interest rate, which resulted in $
Subsequent
to the IPO, the Bay Shore Trust line of credit was paid in full early, of $
Underwriter warrants
In
connection with the IPO, the Company issued
| Expected price volatility | % | |||
| Risk-free interest rate | % | |||
| Weighted average fair values | $ | |||
| Weighted average expected life in years | years | |||
| Dividend yield |
Earnings Per Share
During the year ended December 31, 2023 and 2022, outstanding stock options and warrants of and , respectively, were not included in the computation of diluted earnings per share, because to do so would have had an antidilutive effect.
| F-20 |
Note 10. Employment Agreements:
Erez Aminov
On
April 28, 2023, the Company entered into an employment agreement with Mr. Erez Aminov pursuant to which Mr. Aminov serves as the Company’s
Chief Executive Officer on a full-time basis. Mr. Aminov’s employment agreement provides that his employment will be on an at-will
basis and can be terminated by either Mr. Aminov or the Company at any time and for any reason. Under the agreement, Mr. Aminov will
receive an initial base salary of $
On
August 17, 2023, Mr. Aminov received a $
On
August 28, 2023, the Company amended Mr. Aminov’s employment agreement to increase his yearly compensation from its current
amount of $
Michelle Yanez
On
April 28, 2023, the Company entered into an employment agreement with Ms. Michelle Yanez pursuant to which Ms. Yanez serves as the Company’s
Chief Financial Officer on a full-time basis. Ms. Yanez’s employment agreement provides that her employment will be on an at-will
basis and can be terminated by either Ms. Yanez or the company at any time and for any reason. Under the agreement, Ms. Yanez will receive
an initial base salary of $
On
August 17, 2023, Ms. Yanez received a $
Chris Chapman
On
April 28, 2023, the Company entered into an employment agreement with Dr. Chris Chapman pursuant to which Dr. Chapman served as the Company’s
Executive Chairman. Dr. Chapman’s employment agreement provided that his employment will be on a part-time basis whereby Dr. Chapman
would devote 50% of his full business time and effort to the business and affairs of the company, and it further provided that such employment
would be on an at-will basis and could be terminated by either Dr. Chapman or the company at any time and for any reason. Under the agreement,
Dr. Chapman would receive an initial base salary of $
On
August 17, 2023, Dr. Chapman received a $
On August 28, 2023, the Company amended Dr. Chapman’s employment agreement to indicate that he works part-time on an as needed basis for the Corporation, rather than fifty percent (50%) of the time, effective August 1st, 2023.
On
October 13, 2023, the Company amended Dr. Chapman’s employment agreement to reflect a temporary reduction in his compensation
from $
| F-21 |
Christos Nicholoudis
On
April 28, 2023, the Company entered into an employment agreement with Christos Nicholoudis pursuant to which Mr. Nicholoudis served as
the Company’s General Counsel. Under the agreement, Mr. Nicholoudis receive an initial base salary of $
On
August 17, 2023, Mr. Nicholoudis received a $
Note 11. Subsequent Events:
Section 16(b) disgorgement
In
January 2024, the Company recorded a related party receivable of $
Restructuring of the Board of Directors
On March 9, 2024, after a series of discussions between our board of directors (the “Board”) and senior management regarding the need to have additional scientific expertise among the members of the Board, Ms. Talhia Tuck, Mr. Brad Kroenig and Mr. Hugh McColl, each voluntarily resigned from the Board, effective immediately. This action allowed the remaining members of the Board to appoint new members of the Board, as discussed below. The resignations of Ms. Tuck, Mr. Kroenig, and Mr. McColl were not the result of any disagreement with our company on any matter relating to its operations, policies or practices.
Also on March 9, 2024, Dr. Chris Chapman notified the Board and senior company management of his resignation both as Executive Chairman and as an employee of our company, effective immediately, citing his desire to focus his time on his role as Chairman and Chief Executive Officer of Telomir Pharmaceuticals, Inc., given the recent initial public offering of that company. Dr. Chapman’s resignation was not the result of any disagreement with our company on any matter relating to its operations, policies or practices.
On March 13, 2024, the remaining members of the Board (Erez Aminov and Michael Jerman) unanimously approved the appointment of (i) Mr. Aminov, our Chief Executive Officer, as Chairman of the Board and (ii) Dr. Matthew P. Del Giudice, Dr. Denil N. Shekhat and Mr. Edward MacPherson as members of the Board, to fill the vacancies on the Board occasioned by the resignations from the Board described above, for a term expiring at our 2024 annual meeting of shareholders.
Resignation of Chief Science Officer
We are focused on strengthening our clinical and regulatory development expertise with a view towards a future IND for one of our product candidates. As part of this development, on March 7, 2024, following discussions with our management, Adam Kaplin, M.D., Ph.D. resigned from his position as President and Chief Scientific Officer of the company to pursue other business endeavors, effective immediately. As described under “Key Consultants” below, in light of Mr. Kaplin’s resignation, we expanded the role of an existing consultant to assist in clinical and regulatory affairs.
| F-22 |
SIGNATURES
In accordance with Section 13 or 15(d) of the Exchange Act, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| MIRA PHARMACEUTICALS, INC. | ||
| Date: April 1, 2024 | By: | /S/ Erez Aminov |
| Name: | Erez Aminov | |
| Title: | Chief Executive Officer | |
| (Principal Executive Officer) | ||
| By: | /S/ Michelle Yanez | |
| Name: | Michelle Yanez | |
| Title: | Chief Financial Officer | |
| (Principal Financial Officer) | ||
In accordance with the Exchange Act, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Person | Capacity | Date | ||
| /s/ Erez Aminov | Chief Executive Officer and Chairman | April 1, 2024 | ||
| Erez Aminov | ||||
| /s/ Michelle Yanez | Chief Financial Officer | April 1, 2024 | ||
| Michelle Yanez | ||||
| /s/ Michael Jerman | Director | April 1, 2024 | ||
| Michael Jerman | ||||
| /s/ Matthew Del Giudice | Director | April 1, 2024 | ||
| Matthew Del Giudice | ||||
| /s/ Denil Shekhat | Director | April 1, 2024 | ||
| Denil Shekhat | ||||
| /s/ Edward MacPherson | Director | April 1, 2024 | ||
| Edward MacPherson |
| 100 |