v
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 FOR THE TRANSITION PERIOD FROM TO |
Commission File Number
(Exact name of Registrant as specified in its Charter)
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
(Address of principal executive offices) |
(Zip Code) |
Registrant’s telephone number, including area code: (
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
|
|
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES ☐
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐
Indicate by check mark whether the Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the Registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the Registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer |
|
☐ |
|
Accelerated filer |
|
☐ |
|
|
|
|
|||
|
☒ |
|
Smaller reporting company |
|
||
|
|
|
|
|
|
|
Emerging growth company |
|
|
|
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). YES ☐ NO
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the Registrant, based on the closing price of the Registrant's units on The Nasdaq Capital Market on June 30, 2021 was $
The number of shares of the Registrant’s Common Stock outstanding as of March 25, 2022 was
DOCUMENTS INCORPORATED BY REFERENCE
Certain portions of the registrant's definitive proxy statement relating the Company's Annual Meeting of Stockholders, to be filed with the Securities and Exchange Commission within 120 days of the registrant's fiscal year ended December 31, 2021, are incorporated by reference into Part III of this Annual Report on Form 10-K where indicated.
Table of Contents
|
|
Page |
|
|
|
Item 1. |
3 |
|
Item 1A. |
34 |
|
Item 1B. |
73 |
|
Item 2. |
73 |
|
Item 3. |
73 |
|
Item 4. |
73 |
|
|
|
|
|
|
|
Item 5. |
74 |
|
Item 6. |
74 |
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
75 |
Item 7A. |
83 |
|
Item 8. |
F-1 |
|
Item 9. |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
81 |
Item 9A. |
81 |
|
Item 9B. |
82 |
|
Item 9C. |
Disclosure Regarding Foreign Jurisdictions that Prevent Inspections |
82 |
|
|
|
|
|
|
Item 10. |
83 |
|
Item 11. |
83 |
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
83 |
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
83 |
Item 14. |
83 |
|
|
|
|
|
|
|
Item 15. |
84 |
|
Item 16. |
86 |
1
PART I
SPECIAL NOTE REGARDING FORWARD LOOKING STATEMENTS
This Annual Report on Form 10-K (this "Annual Report") contains “forward-looking statements” which are made pursuant to the safe harbor provisions of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. All statements other than statements of historical facts contained in this Annual Report are forward-looking statements. Our forward-looking statements include, but are not limited to, statements regarding our or our management team’s expectations, hopes, beliefs, intentions or strategies regarding the future. In addition, any statements that refer to projections, forecasts or other characterizations of future events or circumstances, including any underlying assumptions, are forward-looking statements. The words “anticipate,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “intends,” “may,” “might,” “plan,” “possible,” “potential,” “predict,” “project,” “should,” “will,” “would” and similar expressions may identify forward-looking statements, but the absence of these words does not mean that a statement is not forward-looking.
The forward-looking statements are based on the current expectations of the Company and its management of and are inherently subject to uncertainties and changes in circumstances and their potential effects and speak only as of the date of such statement. There can be no assurance that future developments will be those that have been anticipated. These forward-looking statements involve a number of risks, uncertainties or other assumptions that may cause actual results or performance to be materially different from those expressed or implied by these forward-looking statements. These risks and uncertainties include, but are not limited to:
2
The forward-looking statements contained in this Form 10-K are based on current expectations and beliefs concerning future developments and their potential effects on us. There can be no assurance that future developments affecting us will be those that we have anticipated. These forward-looking statements involve a number of risks, uncertainties (some of which are beyond our control) or other assumptions that may cause actual results or performance to be materially different from those expressed or implied by these forward-looking statements. These risks and uncertainties include, but are not limited to, those factors described under the heading “Risk Factors.” Should one or more of these risks or uncertainties materialize, or should any of our assumptions prove incorrect, actual results may vary in material respects from those projected in these forward-looking statements. Some of these risks and uncertainties may in the future be amplified by the ongoing COVID-19 pandemic and there may be additional risks that we consider immaterial or which are unknown. It is not possible to predict or identify all such risks. We do not undertake any obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, except as may be required under applicable securities laws.
Item 1. Business.
Overview
Today, the U.S. spends approximately $4 trillion per year on healthcare. About 90% of that spending is for the treatment of chronic diseases. The majority of chronic diseases are caused predominantly by behaviors, including cardiometabolic diseases ("CMDx"), such as diabetes and heart disease. The root causes of CMDx are behaviors relating to diet, physical activity, and other lifestyle factors, yet current treatments are focused on reducing the effects of those diseases rather than addressing the root causes.
Better Therapeutics, Inc. (the "Company", "we", or "us") is developing a platform of FDA-regulated, software-based, PDT candidates for treating diabetes, heart disease, and other cardiometabolic conditions. Our PDTs are designed to deliver a novel form of CBT that can enable changes in neural pathways of the brain so that lasting changes in behavior become possible. We believe addressing the underlying causes of these diseases has the potential to dramatically improve patient health and lower healthcare costs.
In March 2022, we announced primary endpoint data from our pivotal trial of our lead product candidate, BT-001, an investigational PDT platform that is designed to use digitally delivered Nutritional Cognitive Behavioral Therapy ("nCBT") to treat type 2 diabetes.
Inadequacies of the Current Treatment Paradigm
The U.S. has arrived at a massive, worsening, and unsustainable healthcare crisis. The prevalence of CMDx and U.S. healthcare spending have trended upwards over half a century. A crisis this large is not the result of only one factor such as heredity. The advent of digital entertainment, changes in the food we eat, and other social determinants have all played a role.
3
U.S. Healthcare Spending and Prevalence of CMDx
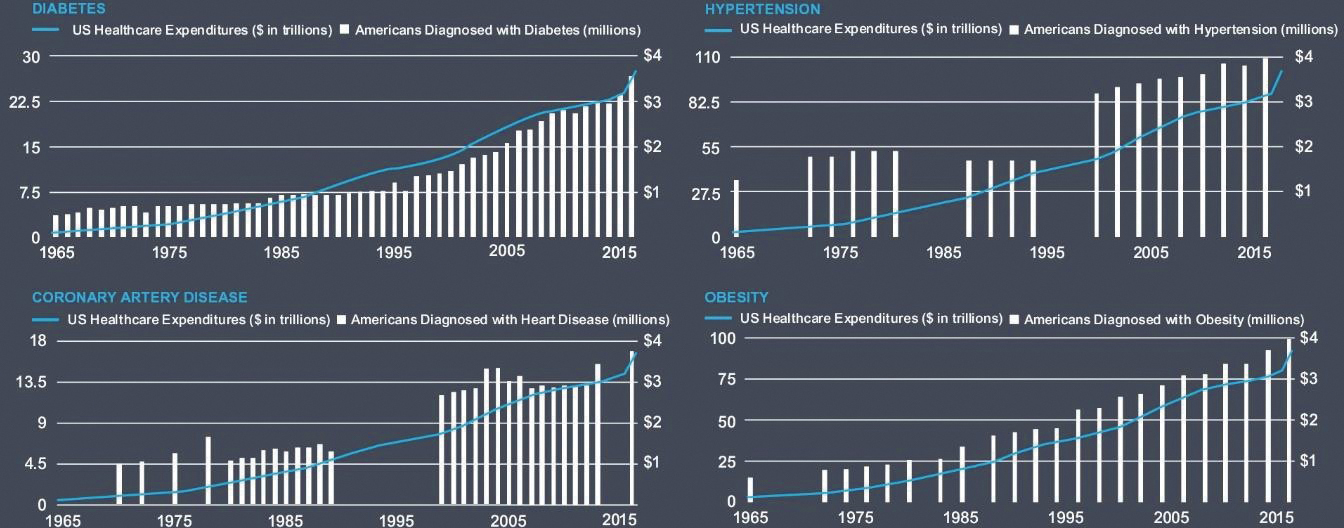
The use of prescription drugs to treat CMDx can provide symptomatic relief and, in some cases, control the progression of disease. However, medications generally do not address root causes, which are predominantly behavioral. There is clear consensus in the scientific and medical community that poor diet, lack of exercise and other lifestyle factors drive the onset, co-morbidity and mortality associated with CMDx. Just three CMDx, type 2 diabetes, hypertension, and hyperlipidemia, account for more than $100 billion in annual prescription drug spending in the United States, none of which addresses root causes.
An estimated 34 million people in the United States have type 2 diabetes. Another estimated 88 million people in the United States have prediabetes, 70% of which are expected to develop into type 2 diabetes during their lifetimes. The annual direct medical costs in the United States for treating type 2 diabetes exceeded $237 billion in 2017, representing an increase of $61 billion since 2012. These costs are forecasted to increase to $472 billion by 2030.
Despite advances in pharmacological treatment, about half of U.S. patients with type 2 diabetes are not achieving glycemic control. Even when adequate glycemic control is achieved via pharmacotherapy, a substantially elevated risk due to all-cause mortality still exists. According to American Diabetes Association, the behavioral determinants of type 2 diabetes are a significant contributor to both poor glycemic control and mortality risk.
The role of behaviors, including dietary pattern and exercise, in the development and progression of type 2 diabetes and other cardiometabolic conditions is well established. These behavioral determinants are resistant to change because they are created and reinforced by strong social norms and culturally reinforced ideas. The use of CBT to directly target these behaviors is a critically important means of achieving high-quality CMDx care. Unfortunately for patients, the U.S. health system is not organized to provide comprehensive CBT at the scale needed. While clinical guidelines consistently recommend that healthcare providers facilitate behavioral changes, they often do not have the ability to provide or prescribe effective behavioral therapy to their patients.
Accordingly, significant unmet needs remain in the therapeutic treatment of CMDx and in the control of associated healthcare spending. We believe that to address this problem, we must focus on root causes and address the near-complete absence to date of behavior-modifying therapeutics for CMDx.
Better Therapeutics, Inc. was incorporated in the State of Delaware on July 31, 2020. We do not own or lease any offices at this time other than a "virtual office" at 548 Market Street, #49404, San Francisco, CA 94104. Our telephone number is (415) 887-2311. Information about us is available on our corporate website at http://www.bettertx.com. Information available on our website is not a part of, and is not incorporated into, this Annual Report. We trade on the Nasdaq Capital Market under the ticker symbol “BTTX.”
4
Our Solution
We have created a platform for the creation of PDTs, essentially software delivered as a mobile application, that is designed to use CBT to address the underlying causes of CMDx.
CBT is a treatment paradigm originally developed for the management of psychiatric conditions such as anxiety and obsessive-compulsive disorder. Traditional CBT aims to correct behavioral responses to a situation that are either non-productive or have adverse effects (maladaptive behaviors) by identifying and changing the core beliefs that produced them. It has since been successfully applied to a wide range of chronic conditions, including CMDx, and has been observed to be generally well-tolerated and to have the potential to provide durable treatment effects, either alone or in combination with other therapies. In current practice, CBT represents a family of therapies that have evolved over several decades and include modalities such as acceptance and commitment therapy, dialectical behavior therapy, and mindfulness-based cognitive therapy.
nCBT, our solution to the crisis described above, is a novel form of behavioral therapy developed by us for patients with type 2 diabetes and other CMDx. nCBT is an adaptation of CBT that is designed specifically to address the cognitive patterns and mental structures that drive dietary patterns and associated lifestyle behaviors.
nCBT builds on traditional CBT by systematically targeting the cognitive structures, behavioral routines, emotional patterns and coping skills that underlie culturally specific eating behaviors. The content and delivery mechanisms of nCBT were developed internally from first principles, leveraging experience from clinician and health coach-patient interactions to distill common maladaptive thinking and beliefs pertaining to diet and lifestyle. It is designed as a digitally delivered therapy so that it can be widely disseminated to large patient populations yet personalized to the individual patient using artificial intelligence (AI)-driven feedback loops.
Our PDTs enable the delivery of nCBT at scale to fill this critical gap in care. To be widely adopted, we believe an effective PDT needs to be prescribed by healthcare providers and reimbursed by payers like a traditional prescription medication. This allows a digital therapeutic to leverage and bolster the trust established in a patient-provider relationship and to provide actionable data back to both provider and patient that can help advance care.
In a pilot study of our lead product candidate, BT-001, we observed that use of BT-001 resulted in a clinically meaningful improvement in glycemic control, based on the generally accepted view that the lowering of HbA1c value by 0.4 is significant. The mean decrease in fasting blood glucose of -22.9 mg/dL corresponds to approximately a 1.0% reduction in hemoglobin A1c, ("A1c"). A1c is a measure of the average blood sugar over a two-to-three-month period. Fasting blood glucose and A1c are both used to diagnose diabetes and to determine whether treatment is effective. An A1c reduction of 1.0% has been associated with a 21% decrease in diabetes related mortality and a 40% reduction in microvascular complications in the UK Prospective Diabetes Study with long-term follow up. Microvascular complications due to diabetes include blindness, damage to nerves in feet that results in pain and numbness, and damage to kidneys that results in chronic kidney disease and failure.
We enrolled the first patient into a potentially pivotal study in April 2021 and completed enrollment of 669 patients in November 2021. The virtual aspects of the trial include recruitment of participants using email and social media and using telemedicine visits. Participants were randomized to receive standard of care with or without BT-001. We announced primary endpoint data from our clinical trial of BT-001 in March 2022. The primary efficacy endpoint was the difference in mean change from baseline in A1c after 90 days of treatment between the two groups and showed highly statistically significant improvement in A1c between the intervention and control groups (-0.4%, p <0.001). Clinically meaningful changes (A1c reductions of 0.4% or more) occurred in 42.7% of the group receiving standard of care and BT-001 versus 25.4% in the group receiving standard of care alone (difference of 17.3%, p <0.001). We believe this demonstrates that the use of BT-001 significantly improved A1c compared to standard of care alone. The six-month trial is ongoing and is expected to be completed in the second quarter of 2022. Given the compelling benefit-to-risk profile of BT-001 and highly statistically significant 0.4% reduction in A1c, we intend to file a de novo classification request with the FDA upon completion of the trial.
5
Our PDTs are designed to be used by patients under the guidance of their primary care provider and may fill an important gap in existing clinical guidelines. Our first PDT candidate, BT-001, is intended to improve glycemic control in adult patients with type 2 diabetes by targeting the behaviors that are root causes, with the potential for patients’ physicians to ultimately reduce or eliminate over time the ongoing need for prescription medications to manage these chronic diseases. With a goal of pursuing commercialization first in type 2 diabetes, we see a compelling opportunity to quickly and efficiently leverage our therapeutics platform to create additional PDTs targeting a broad range of CMDx, and for us to play a significant role in helping reduce the human and monetary costs of CMDx that are currently unsustainable and increasing.
Our Platform
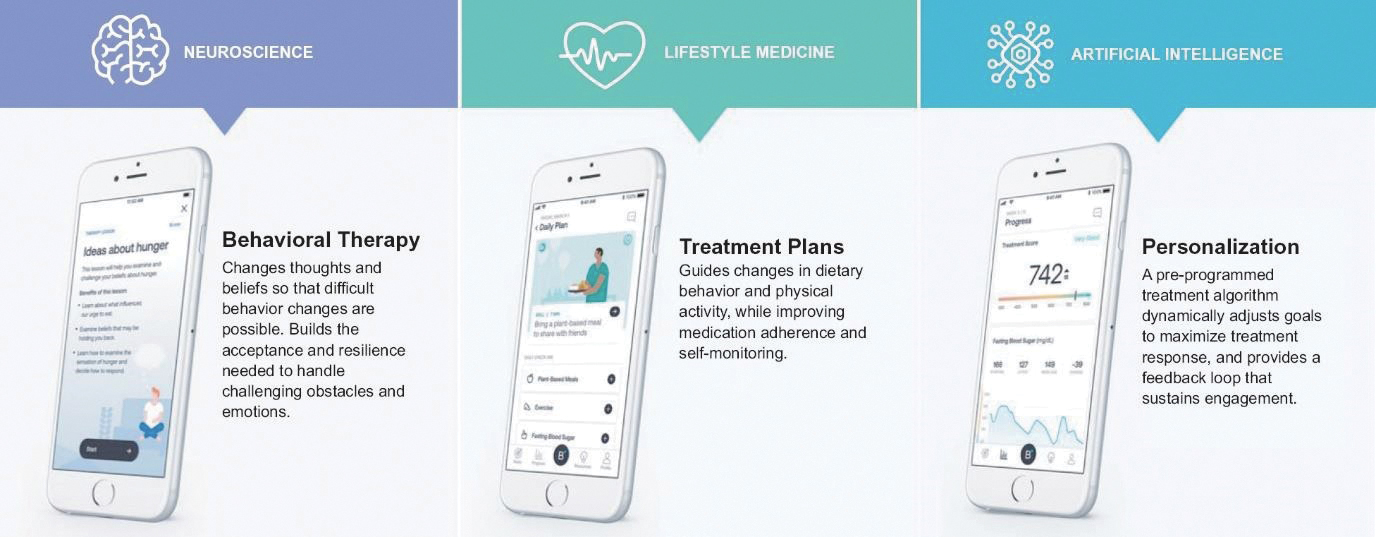
We believe that our platform, if successful in producing an FDA-authorized and marketed product, can support the discovery and development of additional PDTs that can be advanced in clinical development to treat CMDx using nCBT. The platform consists of three integrated components.
Behavioral Therapy. The behavioral therapy components of the platform consist of lessons, skill- building modules, and a mechanism for goal setting. These components deliver nCBT to patients at a pace and sequence that is designed to maximize treatment outcomes on an individual basis. They target the ideas, beliefs, and expectations to help change the neural pathways of the brain, reducing or removing obstacles to making sustained behavioral changes. Our PDT for treating type 2 diabetes, BT-001, consists of 26 therapy lessons, intended to be completed at a rate of about one per week. Each therapy lesson takes 5 to 20 minutes to complete. Associated with each lesson are skill-building modules, enabling practical application of the therapy lesson content in daily life. There are 96 skill-building modules in BT-001, and patients engage in them on a self- directed basis.
Treatment Plans. A daily treatment plan is the primary engagement interface for patients. It guides changes in diet and exercise consistent with daily and weekly goals, encourages adherence to prescribed medications, and enables self-monitoring of disease biometrics. Brief, daily self-reported measures of both behaviors and biometrics serve as inputs to our treatment algorithms.
Personalization. We use AI pre-programmed into our algorithm to adjust goals and personalize treatment plans to each individual patient based on their engagement and inputs. Remotely monitored app-engagement data, self-reported measures, and patient specific health data serve as the primary inputs into our proprietary treatment algorithms. We also use gamification and various feedback mechanisms to reward progress, encourage ongoing use, and visualize the impact of behavior changes made on the primary measures of disease status.
6
Inception, Development and Validation of our Platform
We began development of our platform in 2015, starting with a small number of features thought to be essential for supporting effective and sustained behavior changes based on clinical evidence. Through a cycle of iteration and usability testing, we advanced the platform to a minimal state of readiness, paired the software with board-certified, physician-supervised health coaches, and studied it in various patient populations with CMDx. Those early feasibility studies demonstrated clinical potential comparable to commonly prescribed medications for the treatment of diabetes and hypertension. The data from those earlier studies were peer-reviewed and published in medical journals (see Products; BT-001 and BT-002), and informed further development of a software-only configuration. The first software-only product, BT-001, to emerge from this platform was tested in a pilot study among patients with uncontrolled type 2 diabetes, which demonstrated that use of BT-001 resulted in a clinically meaningful improvement in glycemic control. The data from the pilot study was presented at Endocrine 2020. BT-001 is now being tested in a randomized, controlled clinical trial and based on our primary endpoint data in the clinical trial, we expect to file a de novo classification request with the FDA upon completion of the trial.
In order to establish a comprehensive framework for ongoing product development, we adhere to rigorous product development procedures and processes documented in a commercially scalable Quality Management System (“QMS”). We believe this allows us to employ an agile software development process that results in the highest levels of product innovation while helping ensure consistent product quality and patient safety.
The foundational elements of our QMS are Design Controls and Risk Management Procedures which:
Platform Leverage
Because CMDx share common root causes which our platform is designed to address, we believe we can create products to treat additional CMDx with relatively small changes to one of our existing PDTs. This will greatly reduce product development time and cost. We believe this also means that learnings and improvements on any PDT can be leverageable across the platform. Additionally, because so many CMDx have comorbidities with other CMDx (e.g., patients diagnosed with diabetes are often also diagnosed with heart disease), we can gather data on effectiveness across many diseases with a single study. Finally, we expect to apply to the FDA for authorization for our first product candidate through a de novo classification process. However, we expect to apply for and obtain subsequent products through the 510(k) process. The 510(k) process typically requires a shorter premarket review period but the results from such authorization request processes cannot be guaranteed.
As a result of these efficiencies, we believe we have the potential to develop a portfolio of PDTs for some of the most prevalent diseases in the U.S. at a fraction of the time and cost of traditional therapeutics.
Market Opportunity
In 2016, the direct medical costs due to CMDx potentially addressed by the company’s platform were approximately $490 billion. Approximately 30% of direct medical costs are associated with medications; in type 2 diabetes, the portion associated with medications is approximately 43%. According to the Milken Institute, total direct medical costs by indication in the United States in 2016 were approximately as follows:
7
PRODUCT CANDIDATE DESCRIPTIONS
We currently have five PDT candidates in clinical stages of development:
Our Pipeline

We expect to rapidly develop and, if approved, commercialize multiple product candidates. Our clinical development and regulatory strategy prospectively offer a tempo of related, high-value product launches that, if approved, will be differentiated from a traditional molecular therapeutics company. Unlike traditional therapeutics that require discrete and sequential phase I, II, and III trials, followed by a lengthy regulatory review process, we expect that our PDTs will require a single potentially pivotal trial to generate the data required for submission to the FDA. We believe our potentially pivotal trials can be conducted at a fraction of the cost and time of a new drug trial, and what we believe to be, an expedited FDA review process.
Problem, Solution and Market Opportunity by Product Candidate
BT-001 — Diabetes
Type 2 diabetes is a chronic health condition that results in high levels of blood sugar. It occurs when the body is unable to use insulin properly. Insulin allows blood sugar, which comes mainly from the food we eat, to enter cells to be used for energy. It is highly likely that patients with type 2 diabetes will also develop one or more other medical conditions such as high blood pressure, high cholesterol, heart disease, and/or chronic kidney disease.
Type 2 diabetes is the most common type of diabetes. It was estimated that 34 million adults in the U.S. had type 2 diabetes in 2018. 27 million adults are receiving medical care for type 2 diabetes, but only about 13 million of these patients have well controlled blood sugars. In addition, approximately 88 million U.S. adults have prediabetes, up to 70% of which are expected to develop type 2 diabetes during their lifetime.
8
The American Diabetes Association and American Association of Clinical Endocrinologists and American College of Endocrinology guidelines for the management of type 2 diabetes recommend a) changing behaviors to lower blood sugar, blood pressure, and cholesterol, b) regular monitoring of blood sugar, kidney, heart, blood vessels, eye and nerve function, and c) chronic use of antihyperglycemic medications. Widespread failure to change behavior and the inability of current medications to address root causes of type 2 diabetes has resulted in a massive, growing and unsustainable crisis in the treatment of this disease.
Solution
Under the guidance of a physician, BT-001 is a PDT intended to help patients with type 2 diabetes improve glycemic control. The BT-001 software delivers behavioral therapy to patients via a mobile application that targets behaviors related to improving glycemic control and is intended to reduce A1c. The physician ensures the patient is an appropriate candidate for behavioral therapy, monitors the patient for treatment effects and adjusts concurrent medications as needed.
Market Opportunity
According to the American Diabetes Association (“ADA”), patients diagnosed with diabetes have annual medical costs that are 2.3 times higher than patients without diabetes. The ADA estimated that patients diagnosed with type 2 diabetes incurred average medical costs of $16,750 in 2017, of which about $9,600 was attributed directly to diabetes. Additionally, the ADA estimates total annual drug cost for treating diabetes in 2017 to be approximately $102 billion, which is a four-fold increase since 2007. This includes nearly $15 billion for insulin, $16 billion for other antihyperglycemic agents, and $71 billion for other prescription drugs that can be attributed to higher disease prevalence associated with diabetes.
Clinical Development
Early feasibility study
In 2017, we conducted a 12-week feasibility study in 118 patients with type 2 diabetes. The intervention was delivered by an early version of BT-001 paired with a health coach providing remote support to patients approximately every two weeks by phone. Study participants all had baseline A1c > 6.5% (mean = 8.1%), were mostly female (81%), resided in 38 U.S. states, and had a mean age of 51 years.
After 12-weeks, mean change in A1c was -.8% (p<.001) (this result is considered to be statistically significant), and among those participants with baseline A1c >7.0%, mean change was -1.1% (p<.001) (this result is considered to be statistically significant). Greater glycemic control was observed in those that used BT-001 more often (p=.03) (this result is considered to be statistically significant). The average engagement rate was 4.3 times per day and retention was 86% in this broadly distributed sample.
Data from the study were peer-reviewed and published in Journal of Medical Internet Research Diabetes in 2018.
Key findings of the pilot study
In early 2020, we completed a single-arm, uncontrolled, unblinded pilot study of BT-001, presented the data at Endocrine 2020, and published results in the Journal of the Endocrine Society. In our single-arm pilot study, the addition of the BT-001 treatment regimen to subjects who were, on average, already taking 2.2 oral diabetes medications and continued those medications during the study resulted in an average 1.0% estimated reduction in A1c of participants after 84 days. While the pilot study was not designed as a head-to-head comparison of BT-001 to oral medications, these data compare favorably to historical data published in the Journal of Diabetes Care in August 2010 which suggest an average 0.5% — 1.25% range of A1c reduction from untreated baseline with oral medications alone. The key finding was that the clinical outcomes measured were just as strong using a software-only product as for the earlier software-plus-coaching configuration. In the early feasibility study, the outcomes were attributed to the combination of the early BT-001 software and the remote human intervention delivered by health coaches and behavioral specialists. In contrast, the outcomes found in the pilot could be attributed directly to the use of BT-001 software.
The pilot study involved 80 adults with type 2 diabetes residing in 32 U.S. states who used BT-001 for up to 12 weeks. Participants had a 3-day average fasting blood glucose value of 152 mg/dL or greater, corresponding to a baseline A1c of 7% or greater. On average, participants were 55.7 years old, had a body mass index in the obese range, were taking 2.2 antihyperglycemic medications and were diagnosed with type 2 diabetes 10.4 years prior to the start of the study.
9
Use of BT-001 resulted in clinically meaningful improvement in glycemic control. The mean decrease in fasting blood glucose (or FBG) of -22.9 mg/dL (p<.001) corresponds to approximately a 1.0% reduction in A1c. An A1c reduction of 1.0% has been associated with a 21% decrease in diabetes related mortality and a 40% reduction in microvascular complications in the UK Prospective Diabetes Study, a multisite randomized intervention trial involving 5,102 patients with 20-years of follow up. We believe these results suggest use of BT-001 may be associated with meaningful improvements in glycemic control in a widely distributed treatment population, offers potential as a standalone treatment or when used alongside medications, and is currently conducting further study in its potentially pivotal trial.
We observed a significant dose response (p=.04) (this result is considered to be statistically significant) between the degree of engagement in nCBT content and improvements in glycemic control among adults with type 2 diabetes. This is encouraging because it indicates that digitally delivered behavioral therapy using only software has the potential to treat disease at scale. Reductions in blood glucose were more significant and occurred faster than we had expected. BT-001 allows patients to make behavioral changes at a self-determined pace, which means that for some individuals it might take longer to see blood glucose reductions. In this context, blood sugar control was achieved more rapidly than expected, with 42% of participants achieving a fasting blood glucose less than 152 mg/dL (corresponding to an A1c < 7%, which is commonly regarded as the goal for A1c for most patients with type 2 diabetes) and 16% achieving a fasting blood glucose less than 130 mg/dL (corresponding, on average to an A1c < 6.5%, a much more aggressive goal for A1c) after an average of 65 days. Bi-weekly fasting blood sugars values for participants are displayed in the table below, which suggests a rapid and progressive improvement in blood glucose. We hypothesized that longer duration of use may result in even greater improvements and we plan to study this hypothesis in a randomized controlled trial.
10
Changes in FBG Observed in a Pilot Study of BT-001
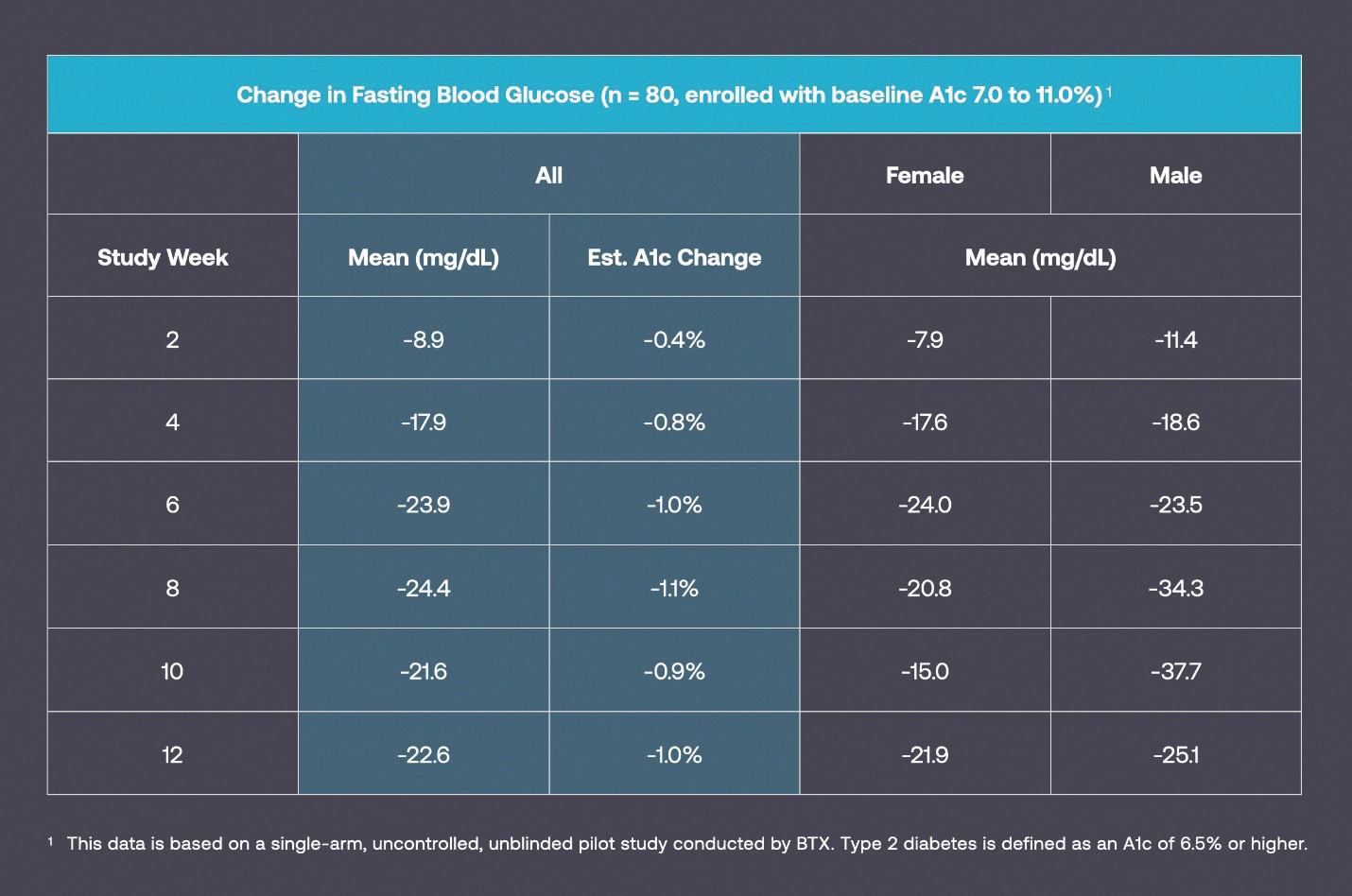
Improvements in blood glucose occurred in participants from across the country and with longstanding diabetes. No serious adverse events were observed in the study period. While it is commonly assumed that only newly diagnosed patients will benefit from behavioral therapy, based on the generally accepted view that the lowering of HbA1c of 0.4 is significant, we were encouraged to see a clinical activity from the usage of BT-001, which is yet to be authorized for marketing by the FDA, in patients who were on average diagnosed with diabetes more than 10 years ago. At baseline, these patients all had poorly controlled diabetes despite taking a mean of 2.2 antihyperglycemic medications. We had geographic diversity with participants from 32 states, including those with increasing prevalence of diabetes (e.g., Florida, Indiana and North Carolina).
11
Potentially Pivotal Trial of BT-001
We screened the first patient into our potentially pivotal unblinded study of BT-001 in February 2021 and completed full enrollment in the fourth quarter of 2021, enrolling a total of 669 patients. The study includes individuals with poorly controlled type 2 diabetes (baseline A1c 7% or above and below 11%) who will each participate for six months. Prior to the start of the study, we discussed core aspects of the design of the trial with the FDA during several formal meeting interactions. During these formal meeting interactions, we aligned with the FDA that an appropriate endpoint is a clinically meaningful change in A1c as determined by the mean change in A1c in the BT-001 group compared to the mean change in the control group. The primary endpoint was evaluated at 90 days, and it will also be evaluated as a secondary endpoint at 180 days. The study is powered to detect a 0.4% or greater change in A1c at 90 days, between BT-001 and control and a statistically significant change (p<0.05) in A1c at 180 days. The study assessed a safety endpoint (the occurrence, relatedness and severity of Adverse Events) at day 90 and will assess a safety endpoint again at day 180. We will use the data from this study to prepare a de novo classification submission to the FDA. We believe a single potentially pivotal trial of BT-001, if successful and its results viewed favorably by the FDA, will be sufficient for the FDA to grant marketing authorization of BT-001 for the treatment of diabetes. We announced primary endpoint data from our clinical trial of BT-001 in March 2022. The primary efficacy endpoint was the difference in mean change from baseline in A1c after 90 days of treatment between the two groups and showed highly statistically significant improvement in A1c between the intervention and control groups (-0.4%, p <0.001). Clinically meaningful changes (A1c reductions of 0.4% or more) occurred in 42.7% of the group receiving standard of care and BT-001 versus 25.4% in the group receiving standard of care alone (difference of 17.3%, p <0.001). We believe this demonstrates that the use of BT-001 significantly improved A1c compared to standard of care alone. The six-month trial is ongoing and is expected to be completed in the second quarter of 2022.
Patients interested in participating in the BT-001 potentially pivotal trial were included if they were between 18 and 75 years old, had a body mass index of 25 kg/m2 or greater, had a stable A1c level and no recent changes in antihyperglycemic medications. Participants were excluded if they use tobacco or other addictive substances, or were taking medications that would interfere with study measures, such as chemotherapy or steroids. Participants with unstable or life-threatening medical illnesses, such as COVID-19, or who are active suicidality were also excluded. The aim of recruitment was to generate a nationally representative sample of adults with type 2 diabetes located in five geographically distinct regions.
Those who passed the run-in period were randomized in a 1-to-1 manner to either a standard of care (“SOC”) group or a standard of care plus BT-001 group. Both groups had blood tests and biometrics collected at 90 days and will have them collected again at180 days, and will be followed closely for adverse events during the entire study period. In addition to A1c levels, participants provided laboratory measures of cholesterol, inflammatory markers, and cardiovascular risk, along with blood pressure and weight at baseline, at day 90 and will do so again at day 180. Participants were also asked to complete standardized surveys to assess changes in depression, quality of life and patient satisfaction at day 90 and will be asked to complete them again at day 180.
Given the compelling benefit-to-risk profile of BT-001 and highly statistically significant 0.4% reduction in A1c observed at day 90 of the clinical trial for BT-001, we intend to file a de novo classification request with the FDA upon completion of the trial. In addition, due to high rates of comorbidity with type 2 diabetes, we anticipate the potentially pivotal data read out from BT-001 will also give us significant pilot data on up to four additional indications including type 2 diabetes with hypertension, hypertension, hyperlipidemia, and hypertriglyceridemia. We have named our PDT’s targeting these conditions, BT-002 and BT-003, respectively. We expect to advance the most promising two of these to potentially pivotal trials in 2022 or early 2023.
BT-002 — Hypertension
Hypertension is a chronic health condition that results in high blood pressure. It occurs when the body is unable to properly regulate the pressure of blood moving through blood vessels. With chronic hypertension, the body’s organs are put under constant stress and are more likely to break down. It is common for patients with longstanding hypertension to develop heart disease, stroke, chronic kidney disease and/or dementia.
Hypertension is one of the most common chronic diseases. In 2017, it was estimated that 108 million U.S. adults have hypertension. Of these patients who are already taking blood pressure lowering medications, approximately 35% still have uncontrolled blood pressure.
12
Guidelines for the management of hypertension recommend a) changing behaviors to lower blood pressure, b) regular monitoring of blood pressure, kidney, and heart function, c) chronic use of antihypertensive medications. Widespread failure to change behavior and the inability of current medications to address root causes of hypertension has resulted in a massive, growing and unsustainable crisis in the treatment of this disease.
Solution
Under the guidance of a physician, BT-002 is a PDT under development to help patients with hypertension improve their blood pressure. The BT-002 software is designed to deliver behavioral therapy to patients via a mobile application that targets behaviors related to achieving blood pressure control and is intended to reduce systolic and diastolic blood pressure.
Market Opportunity
Patients with hypertension are estimated to have nearly triple the prescription drug costs as patients without hypertension. A 2016 study published in the Journal of the American Heart Association concludes the annual prescription drug cost was $2,400 for individuals with hypertension versus only $815 for those without hypertension. For all adults in the United States with hypertension, this represents an estimated annual incremental drug cost for patients with hypertension of $42 billion in 2016.
Clinical Development
A detailed plan for the BT-002 potentially pivotal trial would be refined using blood pressure data obtained from the BT-001 potentially pivotal randomized, controlled trial. We estimate that about one third of participants in the BT-001 potentially pivotal trial will have comorbid hypertension that is poorly controlled at baseline. Since type 2 diabetes and hypertension share common root causes, we expect to see blood pressure improvements in these participants to a degree comparable to BT-002. Because the BT-001 potentially pivotal trial includes measurement of blood pressure along with A1c at every time point, we expect to have 90 and 180 day randomized, controlled data on blood pressure for approximately 200 participants. 90 day data was received in the first quarter of 2022 and we believe it may be sufficient pilot data to allow for planning the BT-002 potentially pivotal trial.
It is anticipated that the BT-002 potentially pivotal trial would evaluate the safety and effectiveness of BT-002 in a nationally representative sample of approximately 500 U.S. adults with hypertension located in 5 geographically distinct regions. Adults, aged 18-75, would be included if their resting blood pressure is poorly controlled (i.e., over 140/90 mmHg). These participants would be randomized in a one-to-one fashion to a control or intervention group. The control group would be provided standard of care treatment. The intervention group would be provided standard of care along with BT-002. The primary outcome measure would be resting systolic blood pressure, measured at 90 days. The secondary outcome measure would be resting systolic blood pressure, measured at 180 days.
BT-003 — Hyperlipidemia
Hyperlipidemia is a chronic health condition that results in high levels of blood cholesterol. It occurs when the body is unable to get rid of harmful types of cholesterol circulating in the blood. Low-density-lipoprotein (LDL) cholesterol is the most common form of harmful cholesterol. A dietary pattern high in unhealthy fats, cholesterol, and refined carbohydrates along with insufficient exercise, are the most common causes of high blood cholesterol. Over time, the presence of too much harmful cholesterol leads to cholesterol build up in the body’s arteries, limiting blood flow. It is very common for patients with longstanding hyperlipidemia to develop one or more other medical conditions caused by cholesterol build-up such as heart disease, stroke, and/or peripheral artery disease.
Hyperlipidemia is one of the most common chronic diseases. It was estimated that 65 million adults in the U.S. had hyperlipidemia in 2016. In 2016, it was estimated that 28 million adults had poorly controlled cholesterol levels.
Guidelines for the management of hyperlipidemia recommend a) changing behaviors to lower harmful cholesterol and raise healthy cholesterol levels, b) regular monitoring of blood cholesterol, blood sugar, and blood pressure, and c) chronic use of cholesterol-lowering medications. Widespread failure to change behavior and the inability of current medications to address root causes of hyperlipidemia has resulted in a massive, growing and unsustainable crisis in the treatment of this disease.
13
Solution
Under the guidance of a physician, BT-003 is a PDT under development to help patients with hyperlipidemia improve cholesterol levels. The BT-003 software is designed to deliver behavioral therapy to patients via a mobile application that targets behaviors related to the control of cholesterol levels and is intended to reduce LDL cholesterol.
Market Opportunity
According to American Heart Association, the annual incremental drug cost for patients with hyperlipidemia was estimated to be $12 billion in 2016. Also, due to updated clinical guidelines which make more aggressive treatment recommendations, an additional 12.3 million more Americans would be treated with cholesterol-lowering medications by 2025, increasing treatment costs by $13.3 billion per year.
Clinical Development
A detailed plan for the BT-003 potentially pivotal trial would be refined using blood cholesterol data obtained from the BT-001 potentially pivotal randomized, controlled trial. We estimate that about one quarter of participants in the BT-001 potentially pivotal trial will have comorbid hyperlipidemia that is poorly controlled at baseline. Since type 2 diabetes and hyperlipidemia share common root causes, we expect to see cholesterol improvements in these participants to a degree comparable to BT-003. Because the BT-001 potentially pivotal trial includes measurement of fasting blood cholesterol along with A1c at every time point, we expect to have 90 and 180 day randomized, controlled data on cholesterol for approximately 140 participants. These data were received in the first quarter of 2022 and we believe it may be sufficient pilot data to allow for planning the BT-003 potentially pivotal trial.
It is anticipated that the BT-003 potentially pivotal trial would evaluate the safety and effectiveness of BT-003 in a nationally representative sample of approximately 500 U.S. adults with hyperlipidemia located in 5 geographically distinct regions. Adults, aged 18-75, would be included if their fasting LDL cholesterol is poorly controlled (i.e., above their risk-adjusted target). These participants would be randomized in a one-to-one fashion to a control or intervention group. The control group would be provided standard of care treatment. The intervention group would be provided standard of care along with BT-003. The primary outcome measure would be fasting LDL cholesterol, measured at 90 days. The secondary outcome measure would be fasting LDL cholesterol, measured at 180 days.
Competitive Advantages
To establish competitive advantage in our target markets, we are building on our early recognition of the potential of PDT’s in CMDx, our focus on treating root causes, and our ability to leverage our platform to accelerate regulatory clearances of subsequent product launches. We believe it has the following advantages over existing and/or potential competitors:
14
Our Strategy
We aspire to change the way CMDx are treated to improve patient health and reduce healthcare spending. We believe our platform technology, first-to-market advantage, intellectual property portfolio, and groundbreaking research will facilitate the achievement of this goal. Our immediate focus is on:
15
Competition
The pharmaceutical, biotechnology and digital health industries are characterized by rapidly advancing technologies, intense competition and an emphasis on proprietary products. While we believe that our technology, development experience and scientific knowledge provide us with competitive advantages, we face potential competition from many different sources, including large pharmaceutical and biotechnology companies, digital health companies, academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for the research, development, manufacturing and commercialization of cardiometabolic therapies. Any products that we successfully develop and commercialize will compete with new therapies that may become available in the future.
We compete in the segments of the pharmaceutical, biotechnology and other related markets that develop therapeutics as treatments for CMDx. There are many other companies that have commercialized and/or are developing such treatments for CMDx including large pharmaceutical and biotechnology companies such as Novo Nordisk, Eli Lilly, Merck, Sanofi, AstraZeneca, and Novartis.
The competitive landscape shown below illustrates our competitors in the market space commonly described as “diabetes tech”, the digital health space focused on addressing problems associated with type 2 diabetes. We believe the competitive landscape is best understood by comparing the primary mechanism of action (behavioral support/intervention or improving medication adherence and tracking); to the business model for patient acquisition (apps marketed direct-to-consumer; tech-enabled healthcare services offered to members of health plans, most often those of self-insured employers; or regulated products prescribed by providers).
16
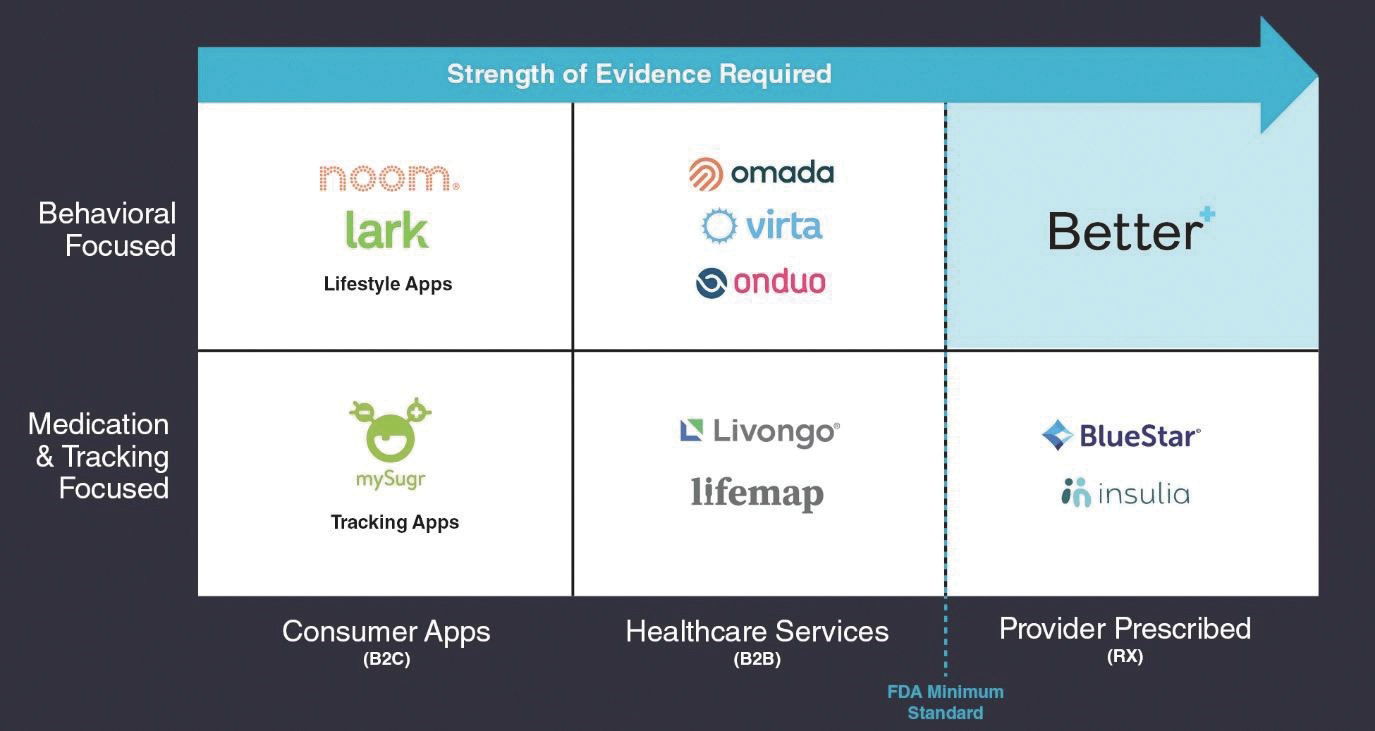
While some solutions have evolved to include elements of various mechanisms such as behavioral support, reminders for medication adherence, or remote monitoring and transmission of biometric data, in our view, each has a primary mechanism for affecting disease and a clearly defined model for acquiring patients or consumers.
To our knowledge, upon completion of our potentially pivotal trial, if successful, and regulatory authorization, BT-001 will be the only regulated PDT with a direct treatment claim for type 2 diabetes that can be prescribed by providers and reimbursed by insurance as a pharmacy benefit, much like a drug. Exploiting this opportunity requires us to generate significant evidence of safety, efficacy and impact on the total cost of care. While many early market entrants (in fact, nearly 360,000 health and wellness apps are now available in Apple’s App Store) are making marketing claims related to the ability to improve type 2 diabetes care and are acquiring patients through their employers or direct-to-consumer advertising, we believe the landscape will change dramatically when new solutions that can be prescribed by providers and covered by insurance become broadly available.
There are a number of companies in the prescription digital therapeutics space but none of these companies have commercialized a prescription digital therapeutic to target a cardiometabolic disease at this time.
Many of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved drugs than we do. Mergers and acquisitions in the pharmaceutical, biotechnology, and digital health industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and enrolling subjects for our clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
We could see a reduction or elimination of our commercial opportunity if our competitors develop and commercialize products that are safer or more effective, are more convenient or are less expensive than any products that we or our collaborators may develop. Our competitors also may obtain FDA or foreign regulatory authorization for their products more rapidly than we may obtain authorization for ours, which could result in our competitors establishing a strong market position before we or our collaborators are able to enter the market. The key competitive factors affecting the success of all of our products, if authorized for marketing, are likely to be their efficacy, safety, convenience, price, and the availability of reimbursement from government and commercial payers.
17
Intellectual Property
Our success depends in part upon our ability to protect our core technology and intellectual property. To protect our intellectual property rights, we rely on patents, trademarks, copyrights and trade secret laws, confidentiality procedures, and employee disclosure and invention assignment agreements. Our intellectual property is critical to our business and we strive to protect it through a variety of approaches, including by obtaining and maintaining patent protection in the United States and internationally for our digital therapeutic platform, novel treatment algorithms and uses thereof, and other inventions that are important to our business. For our digital therapeutic platform, we generally intend to pursue patent protection covering the machine learning aspects and key features of our products, along with the methods of use in treating a wide variety of cardiometabolic disorders and assisting patients and their caregivers in the management of disease. As we continue the development of our product candidates, we intend to identify additional means of obtaining patent protection that would potentially enhance commercial success, including through claims covering additional methods of use as well as subsequent iterations and improvements to our products and use of predictive analytics.
As of December 31, 2021, there are four patent families with national stage applications pending in the U.S., Europe and Canada, for a total of eight pending U.S. and foreign applications, with claims directed to systems encompassing our digital therapeutic platform, and related methods of use in treating cardiometabolic disorders. The statutory expiration for any U.S. and foreign patents issuing from these two patent families will be 2038. There is also a third patent family with national stage applications pending in the U.S., Europe and Canada, with claims directed to methods for predicting health outcomes and managing chronic medications. The statutory expiration for any U.S. and foreign patents issuing in this patent family will be 2039. In addition, there is a fourth patent family represented by a pending U.S. provisional application with claims directed to various implementations of nutritional cognitive behavioral therapy in our digital therapeutic platform. The statutory expiration for any U.S. and foreign patents issuing in this patent family will be 2041.
Government Regulation
Insurance and Coverage
In the United States and markets in other countries, patients generally rely on third-party payers to reimburse all or part of the costs associated with their treatment. Adequate coverage and reimbursement from governmental healthcare programs, such as Medicare and Medicaid, and commercial payers is critical to new product acceptance. Our ability to successfully commercialize our products, if authorized for marketing, will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and other third-party payers, such as private health insurers and health maintenance organizations, decide which treatments they will pay for and establish reimbursement levels. In the United States, the principal decisions about reimbursement for new treatments are typically made by the Centers for Medicare & Medicaid Services (“CMS”), an agency within the U.S. Department of Health and Human Services. CMS decides whether and to what extent a new treatment will be covered and reimbursed under Medicare and private payors tend to follow CMS to a substantial degree. The availability of coverage and extent of reimbursement by governmental and private payers is essential for most patients to be able to afford treatments. Sales of products that we may develop will depend substantially, both domestically and abroad, on the extent to which the costs of our products will be paid by health maintenance, managed care, pharmacy benefit and similar health care management organizations, or reimbursed by government health administration authorities, private health coverage insurers and other third-party payers.
There is also significant uncertainty related to the insurance coverage and reimbursement of newly approved products and coverage may be more limited than the purposes for which the device is authorized by the FDA or comparable foreign regulatory authorities.
Payers consider the following factors in determining reimbursement are based on whether the product is:
18
Each payer determines whether or not it will provide coverage for a treatment, what amount it will pay the manufacturer for the treatment and on what tier of its formulary it will be placed. The position on a payer’s list of covered drugs, biological products, and medical devices, or formulary, generally determines the co-payment that a patient will need to make to obtain the therapy and can strongly influence the adoption of such therapy by patients and physicians. Patients who are prescribed treatments for their conditions and providers prescribing such services generally rely on third-party payers to reimburse all or part of the associated health care costs. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our products. There may be significant delays in obtaining such coverage and reimbursement for newly authorized products, and coverage may be more limited than the purposes for which the product is authorized by the FDA.
In addition, in some foreign countries, the proposed pricing for a prescription device must be approved before it may be lawfully marketed. The requirements governing device pricing vary widely from country to country. For example, the European Union provides options for its Member States to restrict the range of products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost effectiveness of a particular product to currently available therapies. A Member State may approve a specific price for the product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the European Union do not follow price structures of the U.S. and generally prices tend to be significantly lower.
Health Care Laws and Regulations
We are subject to applicable fraud and abuse and other healthcare laws and regulations, including, without limitation, the U.S. federal Anti-Kickback Statute and the U.S. federal False Claims Act (“FCA”), which may constrain the business or financial arrangements and relationships through which we sell, market and distribute our products. In particular, the promotion, sales and marketing of healthcare items and services, as well as certain business arrangements in the healthcare industry (e.g., healthcare providers, physicians and third-party payers), are subject to extensive laws designed to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, structuring and commission(s), certain customer incentive programs and other business arrangements generally. We also may be subject to patient information and privacy and security regulation by both the federal government and the states and foreign jurisdictions in which we conduct our business. The applicable federal, state and foreign healthcare laws and regulations laws that may affect our ability to operate include, but are not limited to:
19
20
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge and may not comply under one or more of such laws, regulations, and guidance. Law enforcement authorities are increasingly focused on enforcing fraud and abuse laws, and it is possible that some of our practices may be challenged under these laws. Efforts to ensure that our current and future business arrangements with third parties, and our business generally, will comply with applicable healthcare laws and regulations will involve substantial costs. If our operations, including our arrangements with physicians and other healthcare providers are found to be in violation of any of such laws or any other governmental regulations that apply to us, we may be subject to penalties, including, without limitation, administrative, civil and criminal penalties, damages, fines, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs (such as Medicare and Medicaid), and imprisonment, as well as additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, any of which could adversely affect our ability to operate our business and our financial results.
Data Privacy and Security Laws
Numerous federal and state laws and regulations govern the collection, use, disclosure, storage and transmission of personally identifiable information, including protected health information. These laws and regulations, including their interpretation by governmental agencies, are subject to frequent change. In addition, in the future, industry requirements or guidance, contractual obligations, and/or legislation at both the federal and the state level may limit, forbid or regulate the use or transmission of health information outside of the United States.
Federal and state consumer protection laws are increasingly being applied by the United States Federal Trade Commission (“FTC”), and states’ attorneys general to regulate the collection, use, storage and disclosure of personal or personally identifiable information, through websites or otherwise, and to regulate the presentation of website content.
There is ongoing concern from privacy advocates, regulators and others regarding data privacy and security issues, and the number of jurisdictions with data privacy and security laws has been increasing. Also, there are ongoing public policy discussions regarding whether the standards for de-identification, anonymization or pseudonymization of health information are sufficient, and the risk of re-identification sufficiently small, to adequately protect patient privacy. We expect that there will continue to be new proposed and amended laws, regulations and industry standards concerning privacy, data protection and information security in the United States, such as the California Consumer Privacy Act (“CCPA”), which went into effect on January 1, 2020 and has been amended several times. Further, a new California privacy law, the California Privacy Rights Act (“CPRA”), was passed by California voters on November 3, 2020. The CPRA will create additional obligations with respect to processing and storing personal information that are scheduled to take effect on January 1, 2023 (with certain provisions having retroactive effect to January 1, 2022). Additionally, a new Virginia privacy law, the Comprehensive Data Protection Act (“VCDPA”), was signed into law on March 2, 2021 and is also scheduled to take effect on January 1, 2023. The VCDPA will impose many similar obligations regarding the processing and storing of personal information as the CCPA and the CPRA. Other U.S. states also are considering omnibus privacy legislation, and industry organizations regularly adopt and advocate for new standards in these areas. While the CCPA, CPRA, and VCDPA contain exceptions for certain activities involving Protected Health Information (“PHI”) already regulated under HIPAA, we cannot yet determine the impact the CCPA, CPRA, VCDPA or other such future laws, regulations and standards may have on our business.
21
Health Care Legislative Reform
In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory changes to the health care system that could impact our ability to sell our products profitably. In particular, in 2010, the ACA was enacted, which, among other things, addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program; extended the Medicaid Drug Rebate program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations; subjected manufacturers to new annual fees and taxes for certain branded prescription drugs; created a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% (increased to 70% pursuant to the Bipartisan Budget Act of 2018, effective as of January 1, 2019) point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; and provided incentives to programs that increase the federal government’s comparative effectiveness research.
Since its enactment, there have been numerous judicial, administrative, executive and legislative challenges to certain aspects of the ACA. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA brought by several states without specifically ruling on the constitutionality of the ACA. Prior to the Supreme Court’s decision, President Biden issued an executive order to initiate a special enrollment period from February 15, 2021 through August 15, 2021 for purposes of obtaining health insurance coverage through the ACA marketplace. The executive order also instructed certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA. It is unclear how other healthcare reform measures of the Biden administration or other efforts, if any, to challenge, repeal or replace the ACA will impact our business.
Other legislative changes have been proposed and adopted in the United States since the ACA was enacted:
22
There has been increasing legislative and enforcement interest in the United States with respect to product pricing practices. Specifically, there have been several recent U.S. Congressional inquiries and proposed federal and state legislation designed to, among other things, bring more transparency to product pricing, reduce the cost of therapies under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. The U.S. Department of Health and Human Services (“HHS”) has already started the process of soliciting feedback on some of these measures and, at the same time, is immediately implementing others under its existing authority. It is unclear what effect such legislative and enforcement interest may have on prescription devices.
We expect that these and other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we received for any approved device, which could have an adverse effect on customers for our product candidates. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payers.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels in the U.S. directed at broadening the availability of healthcare and containing or lowering the cost of healthcare. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our products. Such reforms could have an adverse effect on anticipated revenue from product candidates that we may successfully develop and for which we may obtain regulatory approval and may affect our overall financial condition and ability to develop products. If we, or any third parties we may engage, are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or such third parties are not able to maintain regulatory compliance, our current or any future product candidates we may develop may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability.
FDA Regulation
United States
We are developing medical devices that are subject to extensive and ongoing regulation by the FDA under the Federal Food, Drug, and Cosmetic Act (“FD&C Act”) and its implementing regulations, as well as other federal and state regulatory bodies in the United States and comparable authorities in other countries under other statutes and regulations. The laws and regulations govern, among other things, product design and development, preclinical and clinical testing, manufacturing, packaging, labeling, storage, recordkeeping and reporting, clearance, de novo classification or approval, marketing, distribution, promotion, import and export and post-market surveillance. Failure to comply with applicable requirements may subject a device and/or its manufacturer to a variety of administrative sanctions, such as issuance of warning letters, import detentions, civil monetary penalties and/or judicial sanctions, such as product seizures, injunctions and criminal prosecution.
23
FDA’s Premarket Clearance, De Novo Grant and Approval Requirements
Each digital therapeutic we seek to commercially distribute in the United States will require either a prior de novo classification grant, 510(k) clearance, unless it is exempt, or an approved premarket approval application (“PMA”) from the FDA under its medical device authorities. Generally, if a new device has a predicate that is already on the market under a 510(k) clearance, the FDA will allow that new device to be marketed under a 510(k) clearance; or if there is no legally marketed predicate device and general controls alone or with special controls provide reasonable assurance of safety and effectiveness, the FDA will allow the new device to be marketed under a de novo classification grant; otherwise, a PMA is required. Medical devices are classified into one of three classes — Class I, Class II or Class III — depending on the degree of risk associated with each medical device and the extent of control needed to provide reasonable assurance of safety and effectiveness. Class I devices are deemed to be low risk and are subject to the general controls of the FD&C Act, such as provisions that relate to: adulteration; misbranding; registration and listing; notification, including repair, replacement, or refund; records and reports; and good manufacturing practices. Most Class I devices are classified as exempt from premarket notification under section 510(k) of the FD&C Act, and therefore may be commercially distributed without obtaining 510(k) clearance from the FDA. Class II devices are subject to both general controls and special controls to provide reasonable assurance of safety and effectiveness. Special controls include performance standards, post market surveillance, patient registries and guidance documents. A manufacturer may be required to submit to the FDA a premarket notification requesting permission to commercially distribute some Class II devices. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) device, are placed in Class III. A Class III device cannot be marketed in the United States unless the FDA approves the device after submission of a PMA. However, there are some Class III devices for which FDA has not yet called for a PMA. For these devices, the manufacturer must submit a premarket notification and obtain 510(k) clearance in order to commercially distribute these devices. A premarket notification, de novo classification request and PMA must be accompanied by a user fee, although the fee may be waived under certain circumstances. The FDA can also impose sales, marketing or other restrictions on devices in order to assure that they are used in a safe and effective manner.
510(k) Clearance Pathway
When a 510(k) clearance is required, a manufacturer must submit a premarket notification to the FDA demonstrating that the proposed device is substantially equivalent to a predicate device, which is a previously cleared and legally marketed 510(k) device or a device that was in commercial distribution before May 28, 1976. By regulation, a 510(k) premarket notification must be submitted to the FDA at least 90 days before the manufacturer intends to distribute a device. As a practical matter, clearance often takes significantly longer. To demonstrate substantial equivalence, the manufacturer must show that the proposed device has the same intended use as the predicate device, and it either has the same technological characteristics, or different technological characteristics and the information in the 510(k) premarket notification demonstrates that the device is equally safe and effective and does not raise different questions of safety and effectiveness. The FDA may require further information, including clinical data, to make a determination regarding substantial equivalence. If the FDA determines that the device, or its intended use, is not substantially equivalent to a previously cleared device or use, the FDA will place the device into Class III.
There are three types of 510(k)s: traditional; special; and abbreviated. Special 510(k)s are for devices that are modified and the modification(s) needs a new 510(k) and the method(s) used to evaluate the change(s) are well-established, and the results can be sufficiently reviewed in a summary or risk analysis format. Abbreviated 510(k)s are for devices that conform to a recognized standard. The special and abbreviated 510(k)s are intended to streamline review, and the FDA intends to process special 510(k)s within 30 days of receipt.
24
De Novo Classification
When it is determined that there is no legally marketed predicate device, the de novo process provides a pathway to classify novel medical devices for which general controls alone, or general and special controls, provide reasonable assurance of safety and effectiveness for the intended use. Medical device types that the FDA has not previously classified as Class I, II or III are automatically classified into Class III regardless of the level of risk they pose. The Food and Drug Administration Modernization Act of 1997 (“FDAMA”), established a new route to market for low to moderate risk medical devices that are automatically placed into Class III due to the absence of a predicate device, called the “Request for Evaluation of Automatic Class III Designation,” or the de novo classification procedure. This procedure allows a manufacturer whose novel device is automatically classified into Class III to request down-classification of its medical device into Class I or Class II on the basis that the device presents low or moderate risk, rather than requiring the submission and approval of a PMA application. Prior to the enactment of the FDA Safety and Innovation Act of 2012 (“FDASIA”), a medical device could only be eligible for de novo classification if the manufacturer first submitted a 510(k) premarket notification and received a determination from the FDA that the device was not substantially equivalent. FDASIA streamlined the de novo classification pathway by permitting manufacturers to request de novo classification directly without first submitting a 510(k) premarket notification to the FDA and receiving a not substantially equivalent determination. Under FDASIA, the FDA is required to classify the device within 120 days following receipt of the de novo application. If the manufacturer seeks classification into Class II, the manufacturer must include a draft proposal for special controls that are necessary to provide a reasonable assurance of the safety and effectiveness of the medical device. In addition, the FDA may reject the request for de novo classification if it identifies a legally marketed predicate device that would be appropriate for a 510(k) or determines that the device is not low to moderate risk or that general controls would be inadequate to control the risks and special controls cannot be developed. Devices that are classified into Class I or Class II through a de novo classification request may be marketed and used as predicates for future premarket notification 510(k) submissions.
Premarket Approval Pathway
A PMA must be submitted to the FDA for Class III devices for which the FDA has required a PMA. The PMA process is much more demanding than the 510(k) premarket notification process. A PMA must be supported by extensive data, including but not limited to technical, preclinical, clinical trials, manufacturing and labeling to demonstrate to the FDA’s satisfaction reasonable evidence of safety and effectiveness of the device.
After a PMA is submitted, the FDA has 45 days to determine whether the application is sufficiently complete to permit a substantive review and thus whether the FDA will file the application for review. The FDA has 180 days to review a filed PMA, although the review of an application generally occurs over a significantly longer period of time and can take up to several years. During this review period, the FDA may request additional information or clarification of the information already provided. Also, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. Although the FDA is not bound by the advisory panel decision, the panel’s recommendations are important to the FDA’s overall decision-making process. In addition, the FDA may conduct a preapproval inspection of the manufacturing facility to ensure compliance with the Quality System Regulation (“QSR”). The agency also may inspect one or more clinical sites to assure compliance with FDA’s regulations.
FDA allows applicants to submit discrete sections (modules) of the PMA to FDA for review soon after completing the testing and analysis. FDA intends the modular review approach to provide a mechanism by which applicants may submit preclinical data and manufacturing information for review while still collecting, compiling, and analyzing the clinical data. Therefore, a modular PMA is a compilation of sections or “modules” submitted at different times that together become a complete application. Additionally, the modular approach allows the applicant to potentially resolve any deficiencies noted by FDA earlier in the review process than would occur with a traditional PMA application.
Upon completion of the PMA review, the FDA may: (i) approve the PMA which authorizes commercial marketing with specific prescribing information for one or more indications, which can be more limited than those originally sought; (ii) issue an approvable letter which indicates the FDA’s belief that the PMA is approvable and states what additional information the FDA requires, or the post-approval commitments that must be agreed to prior to approval; (iii) issue a not approvable letter which outlines steps required for approval, but which are typically more onerous than those in an approvable letter, and may require additional clinical trials that are often expensive and time consuming and can delay approval for months or even years; or (iv) deny the application. If the FDA issues an approvable or not approvable letter, the applicant has 180 days to respond, after which the FDA’s review clock is reset.
25
Clinical Trials
Clinical trials are almost always required to support premarket approval, are often required for a de novo classification grant, and are sometimes required for 510(k) clearance. In the United States, for significant risk devices, these trials require submission of an application for an investigational device exemption (“IDE”), to the FDA prior to initiating clinical trials. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE must be approved in advance by the FDA for a specific number of patients at specified study sites. During the trial, the sponsor must comply with the FDA’s IDE requirements for investigator selection, trial monitoring, reporting and recordkeeping. The investigators must obtain patient informed consent, rigorously follow the investigational plan and study protocol, control the disposition of investigational devices and comply with all reporting and recordkeeping requirements. Clinical trials for significant risk devices may not begin until the IDE application is approved by the FDA and the appropriate institutional review boards, ("IRBs"), at the clinical trial sites. An IRB is an appropriately constituted group that has been formally designated to review and monitor medical research involving subjects and which has the authority to approve, require modifications in, or disapprove research to protect the rights, safety and welfare of human research subjects. A nonsignificant risk device does not require FDA approval of an IDE; however, the clinical trial must still be conducted in compliance with various requirements of FDA’s IDE regulations and be approved by an IRB at the clinical trials sites. The FDA or the IRB at each site at which a clinical trial is being performed may withdraw approval of a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the benefits or a failure to comply with FDA or IRB requirements. Even if a trial is completed, the results of clinical testing may not demonstrate the safety and effectiveness of the device, may be equivocal or may otherwise not be sufficient to obtain approval, de novo classification or clearance of the product.
Sponsors of certain clinical trials of devices are required to register with www.clinicaltrials.gov, a public database of clinical trial information. Information related to the device, patient population, study sites and investigators and other aspects of the clinical trial is made public as part of the registration.
Ongoing Regulation by the FDA
Even after a device receives clearance, de novo classification or approval and is placed on the market, numerous regulatory requirements apply. These include:
After a device receives 510(k) clearance or a de novo classification grant, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, will require a new clearance or possibly a PMA. The FDA requires each manufacturer to make this determination initially, but the FDA can review any such decision and can disagree with a manufacturer’s determination. If the FDA disagrees with a determination not to seek a new 510(k) clearance, the FDA may retroactively require a manufacturer to seek 510(k) clearance or possibly a premarket approval. The FDA could also require a manufacturer to cease marketing and distribution and/or recall the modified device until 510(k) clearance or premarket approval is obtained. Also, in these circumstances, manufacturers may be subject to significant regulatory fines and penalties.
26
Some changes to an approved PMA device, including changes in indications, labeling or manufacturing processes or facilities, require submission and FDA approval of a new PMA or PMA supplement, as appropriate, before the change can be implemented. Supplements to a PMA often require the submission of the same type of information required for an original PMA, except that the supplement is generally limited to that information needed to support the proposed change from the device covered by the original PMA. The FDA uses the same procedures and actions in reviewing PMA supplements as it does in reviewing original PMAs.
FDA regulations require manufacturers to register with the FDA and to list the devices they market. Additionally, the California Department of Health Services (“CDHS”), requires manufacturers to register within the state. Following these registrations, the FDA and the CDHS inspect manufacturers on a routine basis for compliance with the QSR and applicable state regulations. These regulations require that companies manufacture their products and maintain related documentation in a prescribed manner with respect to manufacturing, testing and control activities. We are also subject to other federal, state and local laws and regulations relating to safe working conditions, laboratory and manufacturing practices. Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA or state authorities, which may include any of the following sanctions:
The FDA prohibits marketed devices from being marketed for off-label uses and regulates the advertising of certain devices as well. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including substantial monetary penalties and criminal prosecution, including FCA liability for products covered under the federal health care programs.
New government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory clearance, de novo classification or approval of our products under development.
Based on published guidance and four interactions with the FDA, the regulatory pathway for BT-001 will be via a de novo classification request submission. Following the completion of our potentially pivotal study, we intend to prepare and submit to the FDA a de novo application and request for marketing authorization.
Reimbursement Coverage
Despite widespread coverage of medications and digital disease management programs, commercial insurers, Medicare, and Medicaid (collectively “payers”) in the U.S. continue to be challenged with achieving cost effective care for their type 2 diabetes patient populations. It is estimated that type 2 diabetes adds an incremental $10,000 per patient per year or more in direct medical costs of which prescription drugs make up $4,500 per patient per year. Despite these high per patient costs and the considerable resources payers invest in the management of the disease, approximately half of type 2 diabetes patients are not able to achieve glycemic control.
We recently conducted research among eight of the 10 largest payers in the U.S. to gain insight into the willingness to provide reimbursement coverage for BT-001 in type 2 diabetes. Key findings include:
27
Our Payer Research
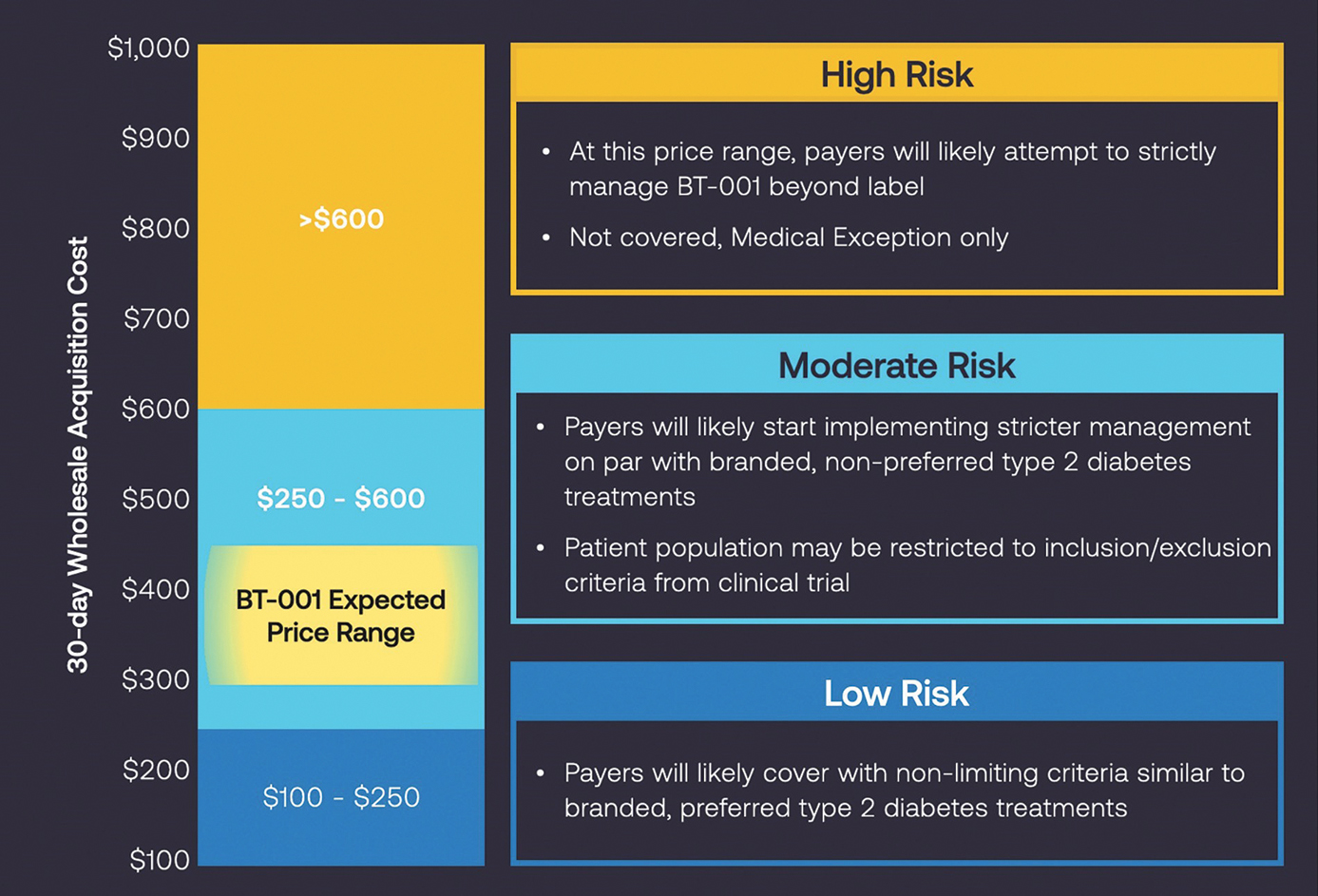
To optimize payer reimbursement coverage at and immediately following launch, we are generating evidence to substantiate the value of BT-001 based on its impact on clinical outcomes, total cost of care and durability of effect. Evidence will be generated from our six-month randomized controlled potentially pivotal trial, and a concurrent one-year real world use study with at least one major U.S. health system. We are conducting such real world evidence study in partnerships with Catalyst Health Network, a clinically integrated network of more than 1,000 health care providers, and Colorado Prevention Center Clinical Research, an affiliate of the University of Colorado Health System.
We also plan to supplement this evidence with an assessment of the total cost of care in our intended patient population using multi-payer claims datasets. To estimate BT-001’s effect on total cost of care, we plan to leverage the totality of evidence related to BT-001 use to create robust cost-effectiveness and budget impact models. We expect to publish these results with reputable organizations like the International Society for Pharmacoeconomics and Outcomes Research and utilize this evidence in the development of our Academy of Managed Care Pharmacy value dossier for submission to formulary review committees. Upon evidence availability, which we anticipate in the first half of 2022, we intend to engage payers to begin reimbursement coverage discussions.
28
Subject to review of final pivotal trial and real-world data, we intend to set pricing for BT-001 at a moderate discount to branded, oral glycemic control medications in order to gain maximum reimbursement coverage. Based on data from our pilot study of BT-001 and an early health economic model we published in the Journal of Medical Internet Research (JMIR) following peer-review, we estimate BT-001 has the potential to demonstrate dominant cost effectiveness and result in net healthcare cost savings of more than $4,500 per patient over a three-year period, primarily through decreased use of medications.
We believe we may be successful in obtaining broad reimbursement coverage for BT-001 because: 1) it addresses an enormous problem — commercial payers and Medicare spend approximately $200 billion each year on type 2 diabetes; 2) these two payer types insure 86% of diabetes patients; 3) it will save payers money; and 4) it fills a gap in existing clinical guidelines and integrates with existing provider workflows.
Sales and Marketing
The intended use at launch for BT-001 would be to improve glycemic control in patients with uncontrolled type 2 diabetes, under the supervision of their physician. This represents a target patient population of about 13 million in the U.S. and $40 billion a year spent on prescription drugs. It is estimated that 86% of type 2 diabetes patients receive regular care from their primary care provider to treat their condition. Within primary care, treatment of type 2 diabetes is concentrated. Based on a review of metformin prescribing data, we estimate 4% (about 17,000) of primary care providers treat about 20% of type 2 diabetes patients (approximately 5 million), suggesting that a relatively small number of primary care providers treat a disproportionate number of diabetes patients.
29
Concentration of Diabetes Patients Among Providers
In recent years, the delivery of primary care services has migrated from solo and small practice settings to larger group practices. Today, an estimated 70% of primary care providers are practicing in large group practice settings. The combination of patients being treated disproportionately by a relatively small number of providers and large-group practice settings creates an opportunity for a focused sales force to engage providers in a cost and time efficient manner to drive awareness and adoption of BT-001 and follow-on products.
We intend to build a primary care sales force of approximately 100, at an annual cost of $30 million during the first-year commercial launch (2023), and scale that organization as widespread reimbursement coverage is achieved and follow-on products are launched.
Go-to-market strategy
As a first mover with a novel class of therapeutic for type 2 diabetes, we have a unique opportunity to raise awareness to PDTs in treating CMDx. Combining advanced targeting analytics with digital marketing, we intend to build awareness among patients and providers of the unique role PDTs can play to improve outcomes by addressing the maladaptive behaviors at the root of type 2 diabetes. We intend to leverage evidence generated from our ongoing potentially pivotal trial and real-world use studies to publish clinical and health outcomes data to showcase BT-001’s benefits. We intend to present these results to key opinion leaders at upcoming congresses and society meetings in 2022. With the evidence generated, we also expect to begin the process of advocating for the incorporation of BT-001 into future consensus guidelines to further integrate its use as a first line PDT for treating type 2 diabetes.
30
At launch, we plan to transition from general awareness-building to branded promotional activities to generate demand for BT-001. These efforts will utilize the full spectrum of our marketing capabilities, including peer-to-peer education and active participation in professional society meetings. We also plan to continue our investment in targeting analytics, as well as digital and non-personal promotion; these will extend the reach of our sales force and help them efficiently and effectively educate primary care providers on BT-001. As payer reimbursement coverage increases, we expect to implement targeted, direct to consumer advertising to further educate patients on the benefits of BT-001 in type 2 diabetes.
Alongside these efforts, we intend to build a medical affairs organization whose primary responsibilities will include engaging thought leaders in scientific discourse, establishing an advisory board of key opinion leaders, creating a primary care-based speaker’s panel and building advocates to support inclusion of BT-001 as part of future type 2 diabetes consensus guidelines. Our medical affairs team will also play a critical, ongoing role in generating and publishing evidence that demonstrates the impact BT-001 and future platform products can have on clinical outcomes, durability of effect and total cost of care.
Integration with the Standard of Care
Type 2 diabetes is a devastating disease that progressively worsens over time and often leads to the development of complex comorbidities, such as hypertension, high cholesterol, heart failure and chronic kidney disease. Lacking the tools to address the maladaptive behaviors that cause disease progression, providers utilize the only treatment options currently available — medications. As a patient’s diabetes worsens, providers typically add multiple medications in an attempt to achieve glycemic control for their patients. By age 65, type 2 diabetes patients are taking an average of five medications for treating diabetes and common comorbidities, while many are failing to achieve glycemic control.
Clinical treatment guidelines from the ADA recommend use of behavioral therapy as a first line treatment on a standalone basis or alongside medications. Despite widespread alignment with these consensus guidelines, there are currently no FDA-regulated treatments available to address this unmet need or practical way for the healthcare system to deliver them. BT-001 represents a unique opportunity for providers to prescribe to their patients FDA-regulated behavioral therapy. Because BT-001 is specifically intended to address the behaviors that are the root causes of their condition, our first-to-market PDT treatment of type 2 diabetes holds out the hope for many of these patients to achieve better glycemic control, reduce or eliminate the need for medications, and avoid insulin therapy altogether.
We believe there are two primary points in the patient journey where prescribing BT-001 would have the greatest clinical impact: 1) upon first diagnosis; and 2) during the immediate period preceding the commencement of insulin, when patient motivation to seek alternative solutions and avoid lifelong insulin injections is greatest.
These two primary points for initiating BT-001 treatment fit easily within existing provider workflows to enable adoption at scale. If approved, BT-001 will be prescription-based and follow the same standard of care for the management of type 2 diabetes. Despite BT-001 being a new product form, we expect it will not negatively impact provider workload.
Partnering
We will progressively increase business development efforts to maximize the value of BT-001 and our platform in non-dilutive ways. We will explore opportunities to partner with pharmaceutical companies marketing traditional drug therapies for CMDx that may benefit from an increase in efficacy and durability when combined with our prescription digital therapeutic. Opportunities may also exist to co-develop novel combination products with a pharmaceutical company operating in the cardiometabolic space.
We intend to commercialize our products in the United States. We will also pursue opportunities to partner with pharmaceutical companies to commercialize our products outside of the United States.
31
Corporate History
Business Combination
Our predecessor, Mountain Crest Acquisition Corp. II (“MCAD”), was incorporated in the State of Delaware on July 31, 2020. On October 28, 2021, we completed our business combination (the “Business Combination”) whereby MCAD acquired all of the issued and outstanding shares of the former Better Therapeutics, Inc. (“Legacy BTX”) in accordance with an agreement and plan of merger (the “Merger Agreement”), dated April 6, 2021, as amended, by and among MCAD, MCAD Merger Sub Inc., a Delaware corporation (“Merger Sub”), and Legacy BTX.
Legacy BTX was formed as a Delaware limited liability company on April 1, 2015 under the name Nutrition Development Group LLC (the “LLC”). The LLC’s name was changed to Farewell LLC on August 18, 2016 and to Better Therapeutics LLC on January 4, 2018. The LLC merged into Legacy BTX, its wholly owned subsidiary, on August 14, 2020 with Legacy BTX surviving the merger.
Pursuant to the Merger Agreement, Merger Sub merged with and into Legacy BTX with Legacy BTX surviving as a wholly-owned subsidiary of MCAD with the new name Better Therapeutics OpCo, Inc. We raised $59 million in funding upon the completion of the Business Combination. In connection with the Business Combination, MCAD was renamed “Better Therapeutics, Inc.”
Human Capital
As of December 31, 2021, we had 44 employees, all of which were full-time employees, including eight in general operations, three in commercial, nine in clinical care and operations, eleven in engineering, ten in product and design, and one in quality and regulatory. None of our employees are represented by a labor union and we believe that our relationships with our employees are good.
We believe that our future success depends upon our continued ability to attract and retain highly skilled employees. We provide our employees with competitive salaries and bonuses, opportunities for equity ownership, development programs that enable continued learning and growth and a robust employment package that promotes well-being across all aspects of their lives, including health care, retirement planning and paid time off. As part of our promotion and retention efforts, we also invest in ongoing development.
Our success is rooted in the diversity of our teams and our commitment to inclusion. We value diversity at all levels and continue to focus on extending our diversity and inclusion initiatives across our entire workforce, from working with managers to develop strategies for building diverse teams to promoting the advancement of leaders from different backgrounds.
Legal Proceedings
We are not currently a party to any material legal proceedings. In the ordinary course of business, we may be subject to legal proceedings, claims and litigation.
Implications of Being an Emerging Growth Company and a Smaller Reporting Company
We are an “emerging growth company” as defined in Section 2(a)(19) of the Securities Act of 1933, as amended (the "Securities Act"), as modified by the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”). As such, we are eligible for and intend to take advantage of certain exemptions from various reporting requirements applicable to other public companies that are not emerging growth companies for as long as we continue to be an emerging growth company, including (i) the exemption from the auditor attestation requirements with respect to internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”), (ii) the exemptions from say-on-pay, say-on- frequency and say-on-golden parachute voting requirements and (iii) reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements.
32
We will remain an emerging growth company until the earlier of: (i) the last day of the fiscal year (a) following the fifth anniversary of the closing of our initial public offering (“IPO”), (b) in which we have total annual gross revenue of at least $1.07 billion, or (c) in which we are deemed to be a “large accelerated filer” under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), which would occur if the market value of our common equity held by non-affiliates exceeds $700.0 million as of the last business day of our most recently completed second fiscal quarter; or (ii) the date on which we have issued more than $1.0 billion in non-convertible debt securities during the prior three-year period.
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This allows an emerging growth company to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to avail ourselves of this extended transition period and, as a result, we may adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-public companies instead of the dates required for other public companies.
Additionally, we are a “smaller reporting company” as defined in Item 10(f)(1) of Regulation S-K. Smaller reporting companies may take advantage of certain reduced disclosure obligations, including, among other things, providing only two years of audited financial statements. We will remain a smaller reporting company until the last day of the fiscal year in which (i) the market value of our ordinary shares held by non-affiliates exceeds $250 million as of the prior June 30, or (ii) our annual revenues exceeded $100 million during such completed fiscal year and the market value of our ordinary shares held by non-affiliates exceeds $700 million as of the prior June 30.
Available Information
We file annual reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, proxy statements and other information with the SEC. Our filings with the SEC are available on the SEC’s website at www.sec.gov. We also maintain a website at http://www.bettertx.com. We make available, free of charge, in the Investor Relations section of our website, documents we file with or furnish to the SEC, including our annual reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and any exhibits and amendments to those reports. We make this information available as soon as reasonably practicable after we electronically file such materials with, or furnish such information to, the SEC. The other information found on our website is not part of this or any other report we file with, or furnish to, the SEC.
33
Item 1A. Risk Factors.
Careful consideration should be given to the following risk factors, in addition to the other information set forth in this Annual Report and in other documents that we file with the SEC, in evaluating the Company and our business. Investing in our common stock involves a high degree of risk. If any of the following risks and uncertainties actually occurs, our business, prospects, financial condition and results of operations could be materially and adversely affected. The risks described below are not intended to be exhaustive and are not the only risks that we face. New risk factors can emerge from time to time, and it is not possible to predict the impact that any factor or combination of factors may have on our business, prospects, financial condition and results of operations.
Summary of Risk Factors
34
35
Risk Related to Our Business
We are a clinical-stage digital therapeutics company with a limited operating history and have incurred significant financial losses since our inception. We anticipate that we will continue to incur significant financial losses for the foreseeable future.
We are a clinical-stage digital therapeutics company with a limited operating history. We were formed in April 2015 and our operations to date have been limited. We have not yet demonstrated an ability to generate revenues, obtain regulatory approvals, manufacture any product on a commercial scale or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization.
We have no products approved for commercial sale and have not generated any revenue from product sales to date, nor do we expect to generate any revenue from product sales for the next few years, if ever. We will continue to incur significant research and development and other expenses related to our preclinical and clinical development and ongoing operations. As a result, we are not profitable and have incurred losses in each period since our inception. Net losses and negative cash flows have had, and will continue to have, an adverse effect on our stockholders’ equity and working capital. Our net loss was $40.3 million for the year ended December 31, 2021. As of December 31, 2021, we had an accumulated deficit of $71.7 million. We expect to continue to incur significant losses for the foreseeable future, and we expect these losses to increase as we continue our research and development of, and seek regulatory approvals for, our product candidates.
We anticipate that our expenses will increase substantially if, and as, we:
Digital therapeutic product development entails substantial upfront capital expenditures and significant risk that any potential product candidate will fail to demonstrate adequate efficacy, gain regulatory approval, secure market access and reimbursement and become commercially viable and therefore any investment in our company is highly speculative. Additionally, our expenses could increase beyond our expectations if we are required by the FDA, or other regulatory authorities to perform clinical trials in addition to those that we currently expect, or if there are any delays in establishing appropriate arrangements for or in completing our clinical trials or the development of any of our product candidates.
You should consider our prospects, factoring in the costs, uncertainties, delays and difficulties frequently encountered by companies in clinical development, especially clinical-stage digital therapeutics companies such as us. Any predictions you make about our future success or viability may not be as accurate as they would otherwise be if we had a longer operating history or a history of successfully developing and commercializing digital therapeutics products. We may encounter unforeseen expenses, difficulties, complications, delays and other known or unknown factors in achieving our business objectives.
36
We have never generated revenue from product sales and may never be profitable.
Our ability to become and remain profitable depends on our ability to generate revenue or execute other business development arrangements. We do not expect to generate significant revenue, if any, unless and until we are able to obtain regulatory approval for, and successfully commercialize the product candidates we are developing or may develop. Successful commercialization will require achievement of many key milestones, including demonstrating safety and efficacy in clinical trials, obtaining regulatory approval for these product candidates, developing, marketing and selling those products for which we may obtain regulatory approval, satisfying any post-marketing requirements and obtaining reimbursement for our products from private insurance or government payers. Because of the uncertainties and risks associated with these activities, we are unable to accurately and precisely predict the timing and amount of revenues, the extent of any further losses or if or when we might achieve profitability. We may never succeed in these activities and, even if we do, we may never generate revenues that are significant enough for us to achieve profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis.
Our failure to become and remain profitable may depress the market price of our common stock and could impair our ability to raise capital, expand our business, diversify our product offerings or continue our operations. If we continue to suffer losses as we have since inception, investors may not receive any return on their investment and may lose their entire investment.
We will need substantial additional funding, and if we are unable to raise capital when needed, we could be forced to delay, reduce or terminate our product discovery and development programs or commercialization efforts.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial amounts to continue the clinical and preclinical development of our product candidates, including the program for our leading product candidate BT-001. We will need to raise additional capital to complete our currently planned clinical trials and any future clinical trials. Other unanticipated costs may arise in the course of our development efforts. If we are able to gain marketing approval for product candidates that we develop, we will require significant additional amounts of funding in order to launch and commercialize such product candidates. We cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of any product candidate we develop and we may need substantial additional funding to complete the development and commercialization of our product candidates.
Our future need for additional funding depends on many factors, including:
We cannot be certain that additional funding will be available on acceptable terms, or at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, reduce or terminate our product development programs or plans for commercialization.
37
We believe that we will be able to fund our operating expenses and capital expenditure requirements into 2023. Our estimate may prove to be wrong, and we could use our available capital resources sooner than we currently expect. Further, changing circumstances, some of which may be beyond our control, could cause us to consume capital significantly faster than we currently anticipate, and we may need to seek additional funds sooner than planned.
Due to the significant resources required for the development of our pipeline, and depending on our ability to access capital, we must prioritize the development of certain product candidates over others. We may fail to expend our limited resources on product candidates or indications that may have been more profitable or for which there is a greater likelihood of success.
We currently have one clinical-stage product candidate as well as several other product candidates that are at various earlier stages of development. We seek to maintain a process of prioritization and resource allocation to maintain an optimal balance between aggressively pursuing our more advanced clinical-stage product candidate, BT-001, and ensuring the development of additional potential product candidates. Due to the significant resources required for the development of our product candidates, we must decide which product candidates to pursue and advance and the amount of resources to allocate to each.
Our decisions concerning the allocation of research, development, collaboration, management and financial resources toward particular product candidates or therapeutic areas may not lead to the development of any viable commercial products and may divert resources away from better opportunities. If we make incorrect determinations regarding the viability or market potential of any of our product candidates or misread trends in the pharmaceutical industry, in particular for cardiometabolic disorders, our business, financial condition, and results of operations could be materially adversely affected. As a result, we may fail to capitalize on viable commercial products or profitable market opportunities, be required to forego or delay pursuit of opportunities with other product candidates or other diseases and disease pathways that may later prove to have greater commercial potential than those we choose to pursue, or relinquish valuable rights to such product candidates through collaboration, licensing, or other royalty arrangements in cases in which it would have been advantageous for us to invest additional resources to retain sole development and commercialization rights.
Raising additional capital may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
We expect our expenses to increase in connection with our planned operations. Unless and until we can generate a substantial amount of revenue from our product candidates, we expect to finance our future cash needs through public or private equity offerings, debt financings, collaborations, licensing arrangements or other sources, or any combination of the foregoing. In addition, we may seek additional capital due to favorable market conditions or strategic considerations, even if we believe that we have sufficient funds for our current or future operating plans.
To the extent that we raise additional capital through the sale of common stock, convertible securities or other equity securities, your ownership interest may be diluted, and the terms of these securities could include liquidation or other preferences and anti-dilution protections that could adversely affect your rights as a common stockholder. In addition, debt financing, if available, may result in fixed payment obligations and may involve agreements that include restrictive covenants that limit our ability to take specific actions, such as incurring additional debt, making capital expenditures, creating liens, redeeming stock or declaring dividends, that could adversely impact our ability to conduct our business. In addition, securing financing could require a substantial amount of time and attention from our management and may divert a disproportionate amount of their attention away from day-to-day activities, which may adversely affect management’s ability to oversee the development of our product candidates.
If we raise additional capital through collaborations or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise additional capital when needed, we may be required to delay, reduce or terminate our product discovery and development programs or commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourself.
38
The amount of our future losses is uncertain and our quarterly and annual operating results may fluctuate significantly or fall below the expectations of investors or securities analysts, each of which may cause our stock price to fluctuate or decline.
Our quarterly and annual operating results may fluctuate significantly in the future due to a variety of factors, many of which are outside of our control and may be difficult to predict, including the following:
The cumulative effects of these factors could result in large fluctuations and unpredictability in our quarterly and annual operating results. As a result, comparing our operating results on a period-to-period basis may not be meaningful. This variability and unpredictability could also result in our failing to meet the expectations of industry or financial analysts or investors for any period. If our operating results or revenue fall below the expectations of analysts or investors or below any forecasts we may provide to the market, or if the forecasts we provide to the market are below the expectations of analysts or investors, the price of our common stock could decline substantially. Such a stock price decline could occur even when we have met any previously publicly stated guidance we may provide.
Our business is highly dependent on the success of our lead product candidate, BT-001. If we are unable to successfully complete clinical development, obtain regulatory approval for or commercialize BT-001, or if we experience delays in doing so, our business will be materially harmed.
To date, we as an organization have not completed any clinical trials or development of any product candidates. Our future success and ability to generate revenue from our lead product candidates, is dependent on our ability to successfully develop, obtain regulatory approval for and commercialize BT-001. We completed enrollment in our potentially pivotal clinical trial for BT-001 in November 2021 and announced primary endpoint data in March 2022. If BT-001 encounters efficacy problems, development delays or regulatory issues or other problems, the development plans for our other product candidates and business would be materially harmed.
We may not have the financial resources to continue development of our product candidates if BT-001 experiences any issues that delay or prevent regulatory approval of, or our ability to commercialize, BT-001, including:
39
The failure of our products, if approved, to achieve and maintain market acceptance would cause our business, financial condition and results of operation to be materially and adversely affected.
Our current business strategy is highly dependent on our products potentially achieving FDA authorization for commercial distribution and maintaining market acceptance. Market acceptance and adoption of our products depends on educating people with cardiometabolic conditions, as well as payers, health plans and government entities, as to the distinct features, clinical impact, cost savings, and other benefits of our products. If we are not successful in demonstrating to physicians who treat potential patients the benefits of our products, if approved, or if we are not able to achieve the support of insurance carriers for our products, our business, financial condition and results of operation would be materially and adversely affected.
In addition, our products may be perceived by patients and healthcare providers to be more complicated or less effective than traditional approaches, and people may be unwilling to change their current health regimens. Moreover, we believe that healthcare providers tend to be slow to change their medical treatment practices because of perceived liability risks arising from the use of new products and the uncertainty of third-party reimbursement. Accordingly, healthcare providers may not recommend our products until there is sufficient evidence to convince them to alter their current approach.
Competitive products may reduce or eliminate the commercial opportunity for our product candidates, if approved. If our competitors develop technologies or product candidates more rapidly than we do, or their technologies or product candidates are more effective or safer than ours, our ability to develop and successfully commercialize our product candidates may be adversely affected.
The clinical and commercial landscapes for the treatment of cardiometabolic diseases are highly competitive and subject to rapid and significant technological change. We face competition with respect to our indications for our product candidates from major pharmaceutical companies, specialty pharmaceutical companies, biotechnology companies and potentially other technology companies. There are a number of large pharmaceutical and biotechnology companies that currently market and sell drugs or are pursuing the development of drug candidates for the treatment of the indications that we are pursuing.
Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization. In addition, technology companies are increasingly exploring the potential for digital products to manage and treat cardiometabolic diseases that could compete with our product candidates, if approved.
40
Our competitors may have significantly greater financial resources, established presence in the market, expertise in research and development, manufacturing, preclinical and clinical testing, obtaining regulatory approvals and reimbursement and marketing approved products than we do. Accordingly, our competitors may be more successful than we may be in obtaining regulatory approval for therapies and achieving widespread market acceptance. Our competitors’ products may be more effective, or more effectively marketed and sold, than any product candidate we may commercialize and may render our therapies obsolete or non-competitive before we can recover development and commercialization expenses. If any of our product candidates, including BT-001, is approved, it could compete with a range of therapeutic treatments that are in development.
If we obtain approval for any of our product candidates, we may face competition based on many different factors, including the efficacy, safety and tolerability of our products, the ease with which our products can be administered, the timing and scope of regulatory approvals for these products, the availability and cost of manufacturing, marketing and sales capabilities, price, reimbursement coverage and patent position. Existing and future competing products could present superior treatment alternatives, including being more effective, safer, less expensive or marketed and sold more effectively than any product we may develop. Competitive products may make any product we develop obsolete or noncompetitive before we recover the expense of developing and commercializing our product candidates. Such competitors could also recruit our employees, which could negatively impact our level of expertise and our ability to execute our business plan.
In addition, our competitors may obtain patent protection or FDA approval and commercialize products more rapidly than we do, which may impact future approvals or sales of any of our product candidates that receive regulatory approval. If the FDA approves the commercial sale of any of our product candidates, we will also be competing with respect to marketing capabilities and manufacturing efficiency. We expect competition among products will be based on product efficacy and safety, the timing and scope of regulatory approvals, marketing and sales capabilities, product price, reimbursement coverage by government and private third-party payers, regulatory exclusivities and patent position. Our profitability and financial position will suffer if our product candidates receive regulatory approval but cannot compete effectively in the marketplace.
Additionally, mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller and other early-stage companies may also prove to be significant competitors, particularly as the develop disruptive therapies through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites, as well as in acquiring technologies complementary to, or necessary for, our programs.
Acquisitions and investments could result in operating difficulties, dilution and other harmful consequences that may adversely impact our business, results of operations and financial condition.
We may in the future make acquisitions to add complementary companies, products, technologies, or revenue. These transactions could be material to our results of operations and financial condition. We may also evaluate and enter into discussions regarding a wide array of potential strategic transactions. The identification of suitable acquisition candidates can be difficult, time-consuming and costly, and we may not be able to complete acquisitions on favorable terms, if at all. The process of integrating an acquired company, business or technology may create unforeseen operating difficulties and expenditures. The areas where we face risks include:
41
Future acquisitions could also result in expenditures of significant cash, dilutive issuances of our equity securities, the incurrence of debt, restrictions on our business, contingent liabilities, amortization expenses or write-offs of goodwill, any of which could harm our financial condition. In addition, any acquisitions we announce could be viewed negatively by patients.
Additionally, competition within our industry for acquisitions of business, technologies and assets may become intense. Even if we are able to identify an acquisition that we would like to consummate, we may not be able to complete the acquisition on commercially reasonable terms or the target may be acquired by another company. We may enter into negotiations for acquisitions that are not ultimately consummated.
Those negotiations could result in diversion of management time and significant out-of-pocket costs. If we fail to evaluate and execute acquisitions successfully, we may not be able to realize the benefits of these acquisitions, and our operating results could be harmed. If we are unable to successfully address any of these risks, our business, financial condition or operating results could be harmed.
If we are unable to develop our sales, marketing and distribution capability on our own or through collaborations with marketing partners, we will not be successful in commercializing our product candidates, if approved.
We currently have no marketing, sales or distribution capabilities. We intend to establish a sales and marketing organization, to commercialize our product candidates, if approved. These efforts will require substantial additional resources, some or all of which may be incurred in advance of any approval of the product candidate. Any failure or delay in the development of our sales, marketing and distribution capabilities would adversely impact the commercialization of our product candidates, if approved.
Factors that may inhibit our efforts to commercialize our product candidates, if approved, include:
42
With respect to our existing and future product candidates, we may choose to collaborate with third parties that have direct sales forces and established distribution systems to serve as an alternative to our own sales force and distribution systems. Our future product revenue may be lower than if we directly marketed or sold our product candidates, if approved. In addition, any revenue we receive will depend in whole or in part upon the efforts of these third parties, which may not be successful and are generally not within our control. If we are not successful in commercializing any approved products, our future product revenue will suffer and we may incur significant additional losses.
If we are unable to achieve widespread acceptance of our products, if approved, our revenue growth could be slower than we expect, and our business may be adversely affected.
We expect to generate revenue from physicians prescription of our products, if approved, for patients. As a result, widespread acceptance, prescription and use of our products, if approved, is critical to our future growth and success. If the market fails to grow or grows more slowly than we currently anticipate, demand for our products, if approved, could be negatively affected and our revenue may grow more slowly than we expect and our business may be adversely affected. Demand for our products, if approved, is affected by a number of factors, many of which are beyond our control. Some of these potential factors include:
Any failure to offer high-quality patient support may adversely affect our relationships with our existing and prospective patients, and in turn our business, results of operations and financial condition.
In implementing and using our products, our patients will depend on our patient support to resolve issues in a timely manner. We may be unable to respond quickly enough to accommodate short-term increases in demand for patient support. Increased patient demand for support could increase costs and adversely affect our results of operations and financial condition. Any failure to maintain high-quality patient support, or a market perception that we do not maintain high-quality patient support, could adversely affect patient satisfaction or the willingness of physicians to prescribe our products, and in turn our business, results of operations, and financial condition.
If we fail to effectively manage our growth, we may be unable to execute our business plan, adequately address competitive challenges or maintain our corporate culture, and our business, financial condition and results of operations would be harmed.
The growth and expansion of our business creates significant challenges for our management, operational and financial resources. To effectively manage our growth, we must continue to improve our operational, financial and management processes and systems and to effectively expand, train and manage our employee base. As our organization continues to grow and we are required to implement more complex organizational management structures, we may find it increasingly difficult to maintain the benefits of our corporate culture. This could negatively affect our business performance.
43
We may in the future enter into collaborations, in-licensing arrangements, joint ventures, or strategic alliances with third parties that may not result in the development of commercially viable products or the generation of significant future revenues.
In the ordinary course of our business, we may enter into collaborations, in-licensing arrangements, joint ventures, or strategic alliances to develop proposed products and to pursue new markets.
In the future, proposing, negotiating, and implementing collaborations, in-licensing arrangements, joint ventures, strategic alliances, or partnerships may be a lengthy and complex process. Other companies, including those with substantially greater financial, marketing, sales, technology or other business resources, may compete with us for these opportunities or arrangements. We may not identify, secure or complete any such transactions or arrangements in a timely manner, on a cost-effective basis, on acceptable terms or at all, and may not realize the anticipated benefits of any such transaction or arrangement.
Additionally, with respect to current and future collaborations, we may not be in a position to exercise sole decision-making authority regarding the transaction or arrangement, which could create the potential risk of creating impasses on decisions, and our collaborators may have economic or business interests or goals that are, or that may become, inconsistent with our business interests or goals.
It is possible that conflicts may arise with our collaborators, such as conflicts concerning the achievement of performance milestones, or the interpretation of significant terms under any agreement, such as those related to financial obligations or the ownership or control of intellectual property developed during the collaboration. If any conflicts arise with our current or future collaborators, they may act in their self-interest, which may be adverse to our best interest, and they may breach their obligations to us. In addition, we have limited control over the amount and timing of resources that our current collaborators or any future collaborators devote to our collaborators’ or our future products. Disputes between us and our collaborators may result in litigation or arbitration which would increase our expenses and divert the attention of our management. Further, these transactions and arrangements are contractual in nature and may be terminated or dissolved under the terms of the applicable agreements and, in such event, we may not continue to have rights to the products relating to such transaction or arrangement or may need to purchase such rights at a premium.
We could suffer disruptions, outages, defects, and other performance and quality problems with our platform or with the cloud and internet infrastructure on which it relies.
Our business depends on our platform to be available without disruption. We have experienced, and may in the future experience, disruptions, outages, defects, and other performance and quality problems with our platform. We have also experienced, and may in the future experience, disruptions, outages, defects, and other performance and quality problems with the cloud and internet infrastructure on which our platform relies. These problems can be caused by a variety of factors, including introductions of new functionality, vulnerabilities and defects in proprietary and open source software, human error or misconduct, capacity constraints, design limitations, or denial of service attacks or other security-related incidents.
Further, if our contractual and other business relationships with our cloud service providers are terminated, suspended, or suffer a material change to which we are unable to adapt, such as the elimination of services or features on which we depend, we could be unable to provide our platform and could experience significant delays and incur additional expense in transitioning patients to a different cloud service provider.
Any disruptions, outages, defects, and other performance and quality problems with our platform or with the cloud and internet infrastructure on which it relies, or any material change in our contractual and other business relationships with our cloud services providers, could result in reduced use of our platform, increased expenses, including service credit obligations, and harm to our brand and reputation, any of which could have a material adverse effect on our business, financial condition, and results of operations.
We depend on our senior management team, and the loss of one or more of our executive officers or key employees or an inability to attract and retain highly skilled employees could adversely affect our business.
Our success depends largely upon the continued services of our key executive officers. These executive officers are at-will employees and therefore they may terminate employment with us at any time with no advance notice. We rely on our leadership team in the areas of operations, clinical and software development, information security, marketing, compliance and general and administrative functions. From time to time, there may be changes in our executive management team resulting from the hiring or departure of executives, which could disrupt our business.
44
The loss of one or more of the members of our senior management team, or other key employees, could harm our business. The replacement of one or more of our executive officers or other key employees would likely involve significant time and costs and may significantly delay or prevent the achievement of our business objectives.
To continue to execute our growth strategy, we also must attract and retain highly skilled personnel. Competition is intense for qualified professionals. We may not be successful in continuing to attract and retain qualified personnel. We have from time to time in the past experienced, and we expect to continue to experience in the future, difficulty in hiring and retaining highly skilled personnel with appropriate qualifications. The pool of qualified personnel with experience working in the healthcare market is limited overall. In addition, many of the companies with which we compete for experienced personnel have greater resources than we have.
Additionally, our success is dependent on our ability to evolve our culture, align our talent with our business needs, engage our employees and inspire our employees to be open to change and innovate. Our business would be adversely affected if we fail to adequately plan for succession of our executives and senior management, or if we fail to effectively recruit, integrate, retain and develop key talent and/or align our talent with our business needs, in light of the current rapidly changing environment.
Our business could be disrupted by catastrophic events and man-made problems, such as power disruptions, data security breaches, and terrorism.
Our platform and the cloud-based infrastructure on which our platform relies are vulnerable to damage or interruption from the occurrence of any catastrophic event, including earthquake, fire, flood, tsunami, or other weather event, power loss, telecommunications failure, software or hardware malfunction, cyber- attack, war, terrorist attack, incident of mass violence or disease, such as the COVID-19 pandemic, and similar events, which could result in lengthy interruptions in access to our platform. In addition, acts of terrorism, including malicious internet-based activity, could cause disruptions to the internet or the economy as a whole. Even with our disaster recovery arrangements, access to our platform could be interrupted. If our systems were to fail or be negatively impacted as a result of a natural disaster or other event, our ability to deliver our platform and products to our patients would be impaired or we could lose critical data. If we are unable to develop adequate plans to ensure that our business functions continue to operate during and after a disaster, and successfully execute on those plans in the event of a disaster or emergency, our business, financial condition, and results of operations would be harmed.
We have implemented a disaster recovery program that allows us to move website traffic to a backup data center in the event of a catastrophe. This allows us the ability to move traffic in the event of a problem, and the ability to recover in a short period of time. However, to the extent our disaster recovery program does not effectively support the movement of traffic in a timely or complete manner in the event of a catastrophe, our business and results of operations may be harmed.
We do not carry business interruption insurance sufficient to compensate us for the potentially significant losses, including the potential harm to our business, financial condition and results of operations that may result from interruptions in access to our platform as a result of system failures.
Our Loan Agreement with Hercules Capital contains restrictions that limit our flexibility in operating our business.
In August 2021, we entered into a loan and security agreement (the “Loan Agreement”) with Hercules Capital, Inc. (“Hercules Capital”) as agent and lender. The Loan Agreement provides for an up to $50.0 million senior secured term loan facility (the “Term Loan Facility”). The Loan Agreement is secured by a lien on substantially all of our assets, including, but not limited to, shares of our subsidiaries, our current and future intellectual property, insurance, trade and intercompany receivables, inventory and equipment and contract rights. The Loan Agreement requires us to meet specified minimum cash requirements, as described below, and contains various affirmative and negative covenants that limit our ability to engage in specified types of transactions. These covenants, which are each subject to customary exceptions, limit our ability to, without Hercules Capital’s prior written consent, effect any of the following, among other things:
45
Our board of directors (the "Board") or management team could believe that taking any one of these actions would be in our best interests and the best interests of our stockholders. If that were the case and if we were unable to complete any of these actions because Hercules Capital does not provide its consent, that could adversely impact our business, financial condition and results of operations.
In addition, on or after July 1, 2023, we are required to maintain a minimum aggregate balance of $10.0 million in cash in one or more controlled accounts. Such requirement terminates if we reach certain valuation requirements. These accounts are required to be maintained as cash collateral accounts securing our obligations under the Loan Agreement. While such requirements apply under the Loan Agreement, our ability to use the cash amounts held in these controlled accounts in the operation of our business will be limited.
On October 28, 2021, we drew down on $10 million of the Term Loan Facility. Our ability to draw on the remaining Term Loan Facility is contingent on our compliance with the covenants described above and certain other covenants and milestones. Even if we meet these conditions, we may elect not to draw on the remaining Term Loan Facility.
In the event of a default under the Loan Agreement, including, among other things, our failure to make any payment when due or our failure to comply with any provision of the Loan Agreement, subject to customary grace periods, Hercules Capital could elect to declare all amounts outstanding to be immediately due and payable and terminate all commitments to extend further credit. If we are unable to repay the amounts due under the Loan Agreement, Hercules Capital could proceed against the collateral granted to it to secure this indebtedness, which could have an adverse effect on our business, financial condition and results of operations.
Hercules Capital interests as a lender may not always be aligned with our interests. If our interests come into conflict with those of Hercules Capital, including in the event of a default under the Loan Agreement, Hercules Capital may choose to act in its self-interest, which could adversely affect the success of our current and future collaborative efforts with Hercules Capital.
Risks Related to our Intellectual Property and Potential Litigation
We may be subject to legal proceedings and litigation, including intellectual property and privacy disputes, which are costly to defend and could materially harm our business and results of operations.
We may be party to lawsuits and legal proceedings in the normal course of business. These matters are often expensive and disruptive to normal business operations. We may face allegations, lawsuits and regulatory inquiries, audits and investigations regarding data privacy, security, labor and employment, consumer protection and intellectual property infringement, including claims related to privacy, patents, publicity, trademarks, copyrights and other rights. A portion of the technologies we use incorporates open source software, and we may face claims claiming ownership of open source software or patents related to that software, rights to our intellectual property or breach of open source license terms, including a demand to release material portions of our source code or otherwise seeking to enforce the terms of the applicable open source license. We may also face allegations or litigation related to our acquisitions, securities issuances or business practices, including public disclosures about our business. Litigation and regulatory proceedings, and particularly the patent infringement and class action matters we could face, may be protracted and expensive, and the results are difficult to predict. Certain of these matters may include speculative claims for substantial or indeterminate amounts of damages and include claims for injunctive relief. Additionally, our litigation costs could be significant. Adverse outcomes with respect to litigation or any of these legal proceedings may result in significant settlement costs or judgments, penalties and fines, or require us to modify our products or require us to stop offering certain products, all of which could negatively impact our revenue growth. We may also become subject to periodic audits, which would likely increase our regulatory compliance costs and may require us to change our business practices, which could negatively impact our revenue growth. Managing legal proceedings, litigation and audits, even if we achieve favorable outcomes, is time-consuming and diverts management’s attention from our business.
46
The results of regulatory proceedings, litigation, claims, and audits cannot be predicted with certainty, and determining reserves for pending litigation and other legal, regulatory and audit matters requires significant judgment. There can be no assurance that our expectations will prove correct, and even if these matters are resolved in our favor or without significant cash settlements, these matters, and the time and resources necessary to litigate or resolve them, could harm our reputation, business, financial condition, results of operations and the market price of our common stock.
Furthermore, our business exposes us to potential product liability claims if our products fail to properly perform due to undetected errors or similar problems. There can be no assurance that, despite testing we undertake, errors will not be found in new products after commencement of commercial use. In addition, the misuse of our products, or the failure of patients to adhere to operating guidelines, could cause significant harm to patients, including death, which could result in product liability claims. Product liability lawsuits and claims, with or without merit, could cause us to incur substantial costs, and could place a significant strain on our financial resources, divert the attention of management from our core business, harm our reputation and adversely affect our ability to attract and retain patients, any of which could have a material adverse effect on our business, financial condition and results of operations.
Although we maintain third-party product liability insurance coverage, it is possible that claims against us may exceed the coverage limits of our insurance policies. Even if any product liability loss is covered by an insurance policy, these policies typically have substantial deductibles for which we are responsible.
Product liability claims in excess of applicable insurance coverage could have a material adverse effect on our business, financial condition and results of operations. In addition, any product liability claim brought against us, with or without merit, could result in an increase of our product liability insurance premiums. Insurance coverage varies in cost and can be difficult to obtain, and we cannot guarantee that we will be able to obtain insurance coverage in the future on terms acceptable to us or at all.
Failure to protect or enforce our intellectual property rights could harm our business and results of operations.
We believe that our intellectual property is an essential asset of our business. If we do not adequately protect our intellectual property, our brand and reputation could be harmed and competitors may be able to use our technologies and erode or negate any competitive advantage we may have, which could materially harm our business, negatively affect our position in the marketplace, limit our ability to commercialize our platform and delay or render impossible our achievement of profitability. A failure to protect our intellectual property in a cost-effective and meaningful manner could have a material adverse effect on our ability to compete. We regard the protection of our trade secrets, copyrights, trademarks, trade dress, databases, domain names and patents as critical to our success. We strive to protect our intellectual property rights by relying on federal, state and common law rights and other rights provided under foreign laws. These laws are subject to change at any time and could further restrict our ability to protect or enforce our intellectual property rights. In addition, the existing laws of certain foreign countries in which we operate may not protect our intellectual property rights to the same extent as do the laws of the United States. We also have a practice of entering into confidentiality and invention assignment agreements with our employees and contractors, and often enter into confidentiality agreements with parties with whom we conduct business in order to limit access to, and disclosure and use of, our proprietary information. In addition, from time to time we make our technology and other intellectual property available to others under license agreements, including open source license agreements and trademark licenses under agreements with any development collaborators for the purpose of co-branding or co-marketing our products or services. However, these contractual arrangements and the other steps we have taken to protect our intellectual property rights may not prevent the misappropriation of our proprietary information, infringement of our intellectual property rights, disclosure of trade secrets and other proprietary information, or deter independent development of similar or competing technologies, duplication of our technologies or efforts to design around our patents by others, and may not provide an adequate remedy in the event of such misappropriation or infringement.
47
Obtaining and maintaining effective intellectual property rights is expensive, including the costs of defending our rights. We make business decisions about when to seek patent protection for a particular technology and when to rely upon trade secret protection, and the approach we select may ultimately prove to be inadequate. We are seeking to protect certain of our intellectual property rights through filing applications for copyrights, trademarks, patents and domain names in a number of jurisdictions, a process that is expensive and may not be successful in all jurisdictions. We are continuing to monitor and evaluate our intellectual property protection in various jurisdictions as we expand our business. Even in cases where we seek patent protection, there is no assurance that the resulting patents will effectively protect every significant feature of our products, technology, or proprietary information, or provide us with any competitive advantages. Moreover, we cannot guarantee that any of our pending patent applications will issue or be approved. The United States Patent and Trademark Office, or the USPTO, also requires compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process and after a patent has issued. There are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If this occurs, our competitors might be able to enter the market, which would have a material adverse effect on our business. Even where we have intellectual property rights, they may later be found to be unenforceable or have a limited scope of enforceability. In addition, we may not seek to pursue such protection in every jurisdiction. In particular, we believe it is important to maintain, protect and enhance our brands. Accordingly, we pursue the registration of domain names and our trademarks and service marks in the United States and in some jurisdictions outside of the United States.
Third parties may challenge our use of our trademarks, oppose our trademark applications or otherwise impede our efforts to protect our intellectual property in certain jurisdictions. In the event that we are unable to register our trademarks in certain jurisdictions, we could be forced to rebrand our products, which could result in loss of brand recognition and could require us to devote resources to advertising and marketing new brands. We have already and may, over time, increase our investment in protecting innovations through investments in patents and similar rights, and this process is expensive and time-consuming.
In order to protect our intellectual property rights, we may be required to spend significant resources to monitor and protect these rights. We may not always detect infringement of our intellectual property rights, and defending or enforcing our intellectual property rights, even if successfully detected, prosecuted, enjoined or remedied, could result in the expenditure of significant financial and managerial resources.
Litigation may be necessary to enforce our intellectual property rights, protect our proprietary rights or determine the validity and scope of proprietary rights claimed by others. Any litigation of this nature, regardless of outcome or merit, could result in substantial costs and diversion of management and technical resources, any of which could adversely affect our business and results of operations. We may also incur significant costs in enforcing our trademarks against those who attempt to imitate our brand and other valuable trademarks and service marks. Furthermore, our efforts to enforce our intellectual property rights may be met with defenses, counterclaims, countersuits and adversarial proceedings such as oppositions, inter partes review, post-grant review, re-examination or other post-issuance proceedings, that attack the validity and enforceability of our intellectual property rights. An adverse determination of any litigation proceedings could put our patents at risk of being invalidated or interpreted narrowly and could put our related pending patent applications at risk of not issuing. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential or sensitive information could be compromised by disclosure in the event of litigation. In addition, during the course of litigation there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
If we fail to maintain, protect and enhance our intellectual property rights, our business, results of operations and financial condition may be harmed and the market price of our common stock could decline.
48
Risks Related to Discovery and Development
Our current product candidates are in various stages of development. Our product candidates may fail in development or suffer delays that adversely affect their commercial viability. If we fail to obtain or maintain FDA de novo classification or clearance to market and sell our BT-001 digital therapeutic, or if such classification or clearance is delayed, our business will be materially harmed.
The process of seeking regulatory de novo classification or clearance to market a medical device is expensive and time consuming. There can be no assurance that marketing authorization will be granted. If we are not successful in obtaining timely de novo classification granting marketing authorization of our BT- 001 digital therapeutic, or any of our other product candidates, we may never be able to generate significant revenue and may be forced to cease operations. Specifically, we hope to pursue additional regulatory marketing clearances for our BT-001 digital therapeutic for additional uses if our first de novo classification is granted. The FDA de novo classification process requires an applicant to demonstrate the safety and efficacy based, in part, on extensive data, including, but not limited to preclinical, clinical trial, technical, manufacturing and labeling data. The FDA regulatory clearance process requires an applicant to demonstrate the device to be marketed is as safe and effective, that is, substantially equivalent, to a legally marketed device and the de novo classification process requires an applicant to demonstrate the safety and effectiveness of a new device. The FDA can delay, limit or deny de novo classification or clearance of a device for many reasons, including:
Obtaining de novo classification and clearance from the FDA or any foreign regulatory authority could result in unexpected and significant costs for us and consume management’s time and other resources. The FDA could ask us to supplement our submissions, collect additional non-clinical data, conduct additional clinical trials, prepare additional manufacturing data or information or engage in other time-consuming actions, or it could simply deny our applications. In addition, if granted marketing authorization, we will be required to obtain additional FDA approvals or clearances prior to making certain modification to our devices, and the FDA may revoke the approval or clearance or impose other restrictions if post-market data demonstrates safety issues or lack of efficacy. If we are unable to obtain and maintain the necessary regulatory authorizations and clearances to market our products, our financial condition may be adversely affected, and our ability to grow domestically and internationally would likely be limited. Additionally, even if authorized or cleared for marketing, our BT-001 digital therapeutic may not receive marketing authorization for the indications that are necessary or desirable for successful commercialization or profitability.
We are substantially dependent on the FDA’s de novo classification of our BT-001 digital therapeutic, as well as market acceptance in the United States of BT-001, and our failure to receive FDA de novo classification of our BT- 001 digital therapeutic or the failure to gain such market acceptance for it would negatively impact our business.
Since our inception, we have devoted substantially all of our efforts to the development of our BT-001 digital therapeutic application that we believe, if granted de novo classification, will serve as the basis for future marketing clearances for additional uses in other indications. We have not yet received de novo classification from the FDA to market and sell our BT-001 digital therapeutic in the United States. However, we will incur costs, including costs to build our sales force, in anticipation of potential FDA de novo classification being granted. If we are unable to obtain the necessary grant from the FDA to market and sell our BT-001 digital therapeutic in the United States and then to achieve significant market acceptance in the United States, our results of operations will be adversely affected as the United States is expected to be the principal market for our BT-001, if authorized. Further, because we have incurred costs prospectively in advance of FDA de novo classification, we would be unable to recoup these costs if the BT-001 is not granted marketing authorization by the FDA or if it is granted de novo classification but fails to obtain market acceptance. We have other digital therapeutic candidates in development that depend on marketing clearance to be obtained under FDA’s 510(k) clearance pathway, enabled by the de novo classification of our first BT- 001 product candidate; thus, if we are unsuccessful in obtaining de novo classification of our initial BT-001 digital therapeutic, we would need to seek de novo classification for the next BT-001 digital therapeutic indication we seek to market. Unexpected or serious complications or other unforeseen negative effects related to the development or market acceptance of any BT-001 digital therapeutic we seek to market could materially and adversely affect our business.
49
The clinical trial process required to obtain marketing authorizations for our product candidates is lengthy and expensive with uncertain outcomes. If clinical trials of any of our digital therapeutic applications in development fail to produce results necessary to support regulatory marketing authorization or clearance in the United States or, with respect to our current or future products, elsewhere, we will be unable to commercialize these products and may incur additional costs or experience delays in completing, or ultimately be unable to complete, the commercialization of those products.
We are currently conducting a virtual clinical trial and plan to seek de novo classification for our BT-001 digital therapeutic application for the treatment of type 2 diabetes. The virtual aspects of the trial include recruitment of participants using email and social media and conducting the study using telemedicine visits. In order to obtain de novo classification, we must obtain clinical data demonstrating the safety and efficacy of the product candidate. Conducting clinical trials is a complex and expensive process, can take many years, and outcomes are inherently uncertain. We incur substantial expense for, and devote significant time to, clinical trials but cannot be certain that the trials will ever result in commercial revenue. We may experience significant setbacks in clinical trials, even after earlier clinical trials showed promising results, and failure can occur at any time during the clinical development process. Any of our products may malfunction or may produce undesirable adverse effects that could cause us, IRBs, or regulatory authorities to interrupt, delay or halt clinical trials. We, IRBs, the FDA, or another regulatory authority may suspend or terminate clinical trials at any time to avoid exposing trial participants to unacceptable health risks. Successful results of earlier pilot studies are not necessarily indicative of future clinical trial results, and predecessor pilot study or clinical trial results may not be replicated in subsequent clinical trials.
Moreover, interim results or topline results may be subject to change upon full review of the data from a clinical trial. Additionally, the FDA may disagree with our interpretation of the data from our pilot studies and clinical trials, or may find the clinical trial design, conduct or results inadequate to demonstrate safety or efficacy, and may require us to pursue additional clinical trials, which could further delay the de novo classification grant or clearance of our product candidates. The data we collect from our pilot studies and clinical trials may not be sufficient to support FDA de novo classification or clearance, and if we are unable to demonstrate the safety and efficacy of our future products in our clinical trials, we will be unable to obtain the regulatory authorizations we need to commercialize our products.
In addition, we may estimate and publicly announce the anticipated timing of the accomplishment of various clinical, regulatory and other product development goals, which are often referred to as milestones. These milestones could include: the submission to the FDA of a meeting request to discuss product development pathways or submission IDE, if applicable, to commence clinical trials of our product candidates; the enrollment of patients in clinical trials; the release of data from clinical trials; the obtainment of the right to affix the CE mark in the European Union. The actual timing of these milestones could vary dramatically compared to our estimates, in some cases for reasons beyond our control. We cannot assure you that we will meet our projected milestones and if we do not meet these milestones as publicly announced, the commercialization of our products may be delayed and, as a result, our stock price may decline.
Clinical trials are necessary to support de novo classification requests and certain 510(k) premarket notifications and may be necessary to support subsequent 510(k) submissions for modified versions of any digital therapeutic devices for which we obtain marketing authorization. This requires the enrollment of large numbers of suitable subjects, which may be difficult to identify, recruit and maintain as participants in the clinical trial. Adverse outcomes in our potentially pivotal trials or post-approval studies could also result in restrictions on or withdrawal of marketing clearances we obtain. We will likely need to conduct additional clinical studies in the future for the authorization of the use of our products in some foreign countries. Clinical testing is difficult to design and implement, can take many years, can be expensive and carries uncertain outcomes. The initiation and completion of any of these trials may be prevented, delayed, or halted for numerous reasons. We may experience a number of events during the conduct of our clinical trials that could adversely affect the costs, timing or successful completion, including:
50
Clinical trials must be conducted in accordance with the applicable laws and regulations of the FDA and other applicable regulatory authorities’ legal requirements, regulations or guidelines, and are subject to oversight by these governmental agencies and IRBs at the medical institutions where the clinical trials are conducted. We may in the future have to terminate a clinical trial site or investigator which is found through our clinical trial monitoring activities to be noncompliant with our clinical trial protocols or with applicable laws, regulations, requirements and guidelines for the conduct of our clinical trials.
Furthermore, we rely on clinical trial sites to ensure the proper and timely conduct of our clinical trials and while we have agreements governing their committed activities, we have limited influence over their actual performance. We depend on our CROs to support the conduct of our clinical trials in compliance with good clinical practice ("GCP"), requirements. To the extent our CROs fail to help oversee the conduct the study in compliance with GCP standards or are delayed for a significant time in the execution of the trial, including achieving full enrollment, we may be affected by increased costs, program delays or both. In addition, clinical trials that are conducted in countries outside the United States may subject us to further delays and expenses as a result of increased shipment costs, additional regulatory requirements and the engagement of non-U.S. CROs, as well as expose us to risks associated with clinical investigators who are unknown to the FDA, and different standards of diagnosis, screening and medical care.
51
Failure can occur at any stage of clinical testing. Our clinical trials may produce negative or inconclusive results or may demonstrate a lack of effect of our product candidates. We may decide, or regulators may require us, to conduct additional clinical and non-clinical testing in addition to those we have planned. Our failure to adequately demonstrate the safety and effectiveness of any product candidates we may develop or may develop in the future would prevent receipt of regulatory marketing authorization and, ultimately, the commercialization of that product or indication for use. Even if our future products are granted de novo classification or cleared in the United States, commercialization of our products in foreign countries would require marketing authorization by regulatory authorities in those countries.
Marketing authorization procedures vary among jurisdictions and can involve requirements and administrative review periods different from, and greater than, those in the United States, including the conduct of additional pilot studies or clinical trials. Any of these occurrences could have an adverse effect on our business, financial condition and results of operations.
Enrollment and retention of patients in clinical trials is an expensive and time-consuming process and could be made more difficult or rendered impossible by multiple factors outside our control.
We may encounter delays or difficulties in enrolling, or be unable to enroll, a sufficient number of patients to complete any of our clinical trials on our current timelines, or at all, and even once enrolled, we may be unable to retain a sufficient number of patients to complete any of our trials. Slow enrollment in our clinical trials may lead to delays in our development timelines and milestones.
Patient enrollment in clinical trials and completion of patient follow-up depend on many factors, including the size of the patient population, the nature of the trial protocol, the ability of patients to continue to receive medical care, the eligibility criteria for the clinical trial, patient compliance, competing clinical trials and clinicians’ and patients’ perceptions as to the potential advantages of the product being studied in relation to other available therapies, including any new treatments that may be approved for the indications we are investigating. For example, patients may be discouraged from enrolling in our clinical trials if the trial protocol requires them to undergo extensive post-treatment procedures or follow-up to assess the safety and efficacy of a product candidate, or they may be persuaded to participate in contemporaneous clinical trials of a competitor’s product candidate. In addition, patients participating in our clinical trials may drop out before completion of the trial or experience adverse medical events unrelated to our products. Delays in patient enrollment or failure of patients to continue to participate in a clinical trial may delay commencement or completion of the clinical trial, cause an increase in the costs of the clinical trial and delays, make our data more difficult to interpret, affect the powering of our trial, or result in the failure of the clinical trial.
Delays or failures in planned patient enrollment or retention may result in increased costs, program delays or both, which could have a harmful effect on our ability to develop our product candidates, or could render further development impossible. In addition, we rely on clinical trial sites to ensure timely conduct of our clinical trials and, while we have entered into agreements governing their services, we are limited in our ability to compel their actual performance.
Interim, “topline,” and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to confirmation, audit, and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose preliminary or topline data from our pilot studies and clinical trials, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study or trial. We also make assumptions, estimations, calculations, and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the topline or preliminary results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, topline data should be viewed with caution until the final data are available. From time to time, we may also disclose interim data from our clinical trials. Interim or preliminary data from clinical trials are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment and treatment continues and more patient data become available or as patients from our clinical trials continue other treatments for their disease. Adverse differences between preliminary or interim data and final data could significantly harm our business prospects. Further, disclosure of interim data by us or by our competitors could result in volatility in the price of our common stock.
52
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the potential of the particular program, the likelihood of marketing authorization or clearance or commercialization of the particular product candidate, the commercial success of any product for which we may have already obtained authorization or clearance, and our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is derived from information that is typically extensive, and you or others may not agree with what we determine is material or otherwise appropriate information to include in our disclosure.
If the interim, topline, or preliminary data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, our product candidates may be harmed, which could harm our business, operating results, prospects or financial condition.
If patients or physicians are not willing to change current practices to adopt our BT-001 digital therapeutic, if granted authorization for marketing, our future product candidates may fail to gain increased market acceptance, and our business will be adversely affected.
Our primary strategy to grow our revenue is to drive the adoption of our BT-001 digital therapeutic, if granted marketing authorization, by physicians to assist their patients in improving glycemic control by lowering HbA1c. Physicians may choose not to adopt our digital therapeutic products for a number of reasons, including:
We focus our sales, marketing and training efforts primarily on primary care physicians. However, physicians from other disciplines, such as endocrinologists, as well as other medical professionals, such as nurse practitioners and physician assistants, are often the initial point of contact for patients with diabetes management needs. We believe that educating physicians in these disciplines and other medical professionals about the clinical merits, patient benefits and safety profile of our digital therapeutic products is an element of increasing product adoption. If additional primary care physicians or other medical professionals do not appreciate and recommend the benefits of our digital therapeutic for any reason, including those listed above, our ability to execute our growth strategy will be impaired, and our business may be adversely affected.
In addition, patients may not be able to adopt or may choose not to adopt our digital therapeutic if, among other potential reasons, they are worried about potential adverse effects of use of our digital therapeutic or they are unable to obtain adequate third-party coverage or reimbursement.
Our long-term growth depends on our ability to enhance our digital therapeutic products, expand our indications and develop and commercialize additional products once granted marketing authorization and clearance.
It is important to our business strategy that we continue to enhance our BT-001 digital therapeutic with additional functionalities and, in the future, additional indications, as well as develop and introduce new products. Developing products is expensive and time-consuming and could divert management’s attention away from our core business. The success of any new product offering or product enhancements will depend on several factors, including our ability to:
53
If we are not successful in expanding our indications and developing and commercializing new products and product enhancements, our ability to increase our revenue may be impaired, which could have a material adverse effect on our business, financial condition and results of operations.
Our product candidates represent novel and innovative potential therapeutic areas, and negative perception of any product candidate that we develop could adversely affect our ability to conduct our business, obtain regulatory approvals or identify alternate regulatory pathways to market for such product candidate.
Certain of our product candidates are considered relatively new and novel therapeutic approaches. Our and their success will depend upon physicians who specialize in the treatment of diseases targeted by our and their product candidates prescribing potential treatments that involve the use of our and their product candidates in lieu of, or in addition to, existing treatments with which they are more familiar and for which greater clinical data may be available. Access will also depend on consumer acceptance and adoption of products that are commercialized. In addition, responses by the U.S., state or foreign governments to negative public perception or ethical concerns may result in new legislation or regulations that could limit our ability to develop or commercialize any product candidates, obtain or maintain regulatory approval, identify alternate regulatory pathways to market or otherwise achieve profitability.
For example, in the United States, no prescription digital therapeutic candidates designed to deliver cognitive behavioral therapy for treating diabetes, heart disease, and other cardiometabolic conditions have been approved. We are developing a platform of FDA-regulated, software-based, prescription digital therapeutic candidates for treating such conditions through a novel form of cognitive behavioral therapy. The FDA may lack experience in evaluating the safety and efficacy of product candidates based on cognitive behavioral therapy, which could result in a longer than expected regulatory review process, increase expected development costs and delay or prevent potential commercialization of product candidates.
Risks Related to Government Regulation
Our products and operations are subject to extensive government regulation and oversight both in the United States and abroad, and our failure to comply with applicable requirements could harm our business.
We and our products are subject to extensive regulation in the United States and elsewhere, including by the FDA and its foreign counterparts. The FDA and foreign regulatory agencies regulate, among other things, with respect to medical devices: design, development and manufacturing; testing, labeling, content and language of instructions for use; clinical trials; product safety; premarket clearance and approval; establishment registration and device listing; marketing, sales and distribution; complaint handling; record keeping procedures; advertising and promotion; recalls and field safety corrective actions; post-market surveillance, including reporting of deaths or serious injuries and malfunctions that, if they were to recur, could lead to death or serious injury; post-market approval studies; and product import and export.
The regulations to which we are subject are complex and have tended to become more stringent over time. Regulatory changes could result in restrictions on our ability to carry on or expand our operations, higher than anticipated costs or lower than anticipated sales. The FDA enforces these regulatory requirements through periodic unannounced inspections. We do not know whether we will pass any future FDA inspections or those conducted by foreign regulatory agencies. Failure to comply with applicable regulations could jeopardize our ability to sell our products and result in enforcement actions such as: warning letters; fines; injunctions; civil penalties; termination of distribution; recalls or seizures of products; delays in the introduction of products into the market; total or partial suspension of production; refusal to grant future clearances or approvals; withdrawals or suspensions of current marketing authorizations, resulting in prohibitions on the sale and distribution of our products; and in the most serious cases, criminal penalties.
54
We may not receive the necessary de novo classification grant for our BT-001 digital therapeutic or clearances for future expanded indications of our BT-001 digital therapeutic product candidate, and failure to timely obtain these regulatory authorizations would adversely affect our ability to grow our business.
Our strategy is dependent on the initial de novo classification by FDA of our BT-001 digital therapeutic granting its ability for marketing in the United States. In the United States, before we can market a new medical device, or a new use of, new claim for or significant modification to an existing products, we must first receive either clearance under Section 510(k) of the FD&C Act, or grant under the de novo classification process added under the FDAMA, or PMA, from the FDA, unless an exemption applies.
The de novo classification process, which is the development pathway required based on discussions with the FDA for our BT-001 digital therapeutic for our current planned use in treatment of type 2 diabetes, provides a pathway to classify novel medical devices for which general controls alone, or general and special controls, provide reasonable assurance of safety and effectiveness for the intended use, but for which there is no legally marketed predicate device. A de novo classification is a risk-based classification process where devices that are classified into Class I or Class II through a de novo classification request may be marketed and used as predicates for future premarket notification 510(k) submissions.
In the 510(k) clearance process, before a device may be marketed, the FDA must determine that a proposed device is “substantially equivalent” to a legally-marketed “predicate” device, which includes a device that has been previously cleared through the 510(k) process, a device that was legally marketed prior to May 28, 1976 (pre-amendments device), a device that was originally on the United States market pursuant to an approved PMA and later down-classified, or a 510(k)-exempt device. To be “substantially equivalent,” the proposed device must have the same intended use as the predicate device, and either have the same technological characteristics as the predicate device or have different technological characteristics and not raise different questions of safety or effectiveness than the predicate device. Clinical data are sometimes required to support substantial equivalence demonstrations. We plan to pursue the 510(k) clearance process for the addition of expanded indications for our BT-001 digital therapeutic.
Where the de novo classification or 510(k) clearance pathways are not available for medical devices, and where no policy of enforcement discretion exists enabling a manufacturer to market a medical device without obtaining premarket authorization, the process of obtaining PMA approval may apply, which is the most rigorous product development pathway for seeking marketing approval for a medical device. In review of a PMA application, the FDA must determine that a proposed device is safe and effective for its intended use based, in part, on extensive data, including, but not limited to pre-clinical, clinical trial, technical, manufacturing and labeling data beyond that which is required to support a de novo classification request or 510(k) clearance submission. The PMA process is typically required for devices that are deemed to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices.
Modifications to products that are approved through a PMA application generally require FDA approval. Similarly, certain modifications made to products cleared through a 510(k) or the de novo classification process may require a new 510(k) clearance or a new de novo classification request. Both the PMA approval, de novo classification, and the 510(k) clearance processes can be expensive, lengthy and uncertain. The FDA’s 510(k) clearance process usually takes from three to 12 months, but can last longer, while the de novo classification request process is usually longer and requires a clinical trial. The process of obtaining a PMA is much more costly and uncertain than the de novo or 510(k) clearance processes and generally takes from one to three years, or even longer, from the time the application is filed with the FDA. In addition, a PMA generally requires the performance of one or more clinical trials. Despite the time, effort and cost, a device may not be approved, granted or cleared by the FDA. Any delay or failure to obtain necessary regulatory approvals could harm our business. Furthermore, even if we are granted regulatory authorizations, clearances or approvals, they may include significant limitations on the indicated uses for the device, which may limit the market for the device.
In the United States, we are currently developing our BT-001 digital therapeutic through the de novo classification pathway. Any modification to our BT-001 digital therapeutic that has not been previously authorized may require us to submit a 510(k) premarket clearance application or de novo classification request prior to implementing the change. If the FDA requires us to go through a lengthier, more rigorous examination for future products or modifications to existing products than we had expected, product introductions or modifications could be delayed or canceled, which could adversely affect our ability to grow our business.
The FDA can delay, limit or deny de novo classification, clearance or approval of a device for many reasons, including:
55
In addition, the FDA may change its policies, adopt additional regulations or revise existing regulations, or take other actions, which may prevent or delay de novo classification, clearance or approval of our future products under development or impact our ability to modify our currently cleared products on a timely basis. Such policy or regulatory changes could impose additional requirements upon us that could delay our ability to obtain new authorizations, increase the costs of compliance or restrict our ability to maintain any authorizations we may successfully obtain.
We may market digital products for uses under current FDA enforcement discretion or outside of the current definition of a “medical device” in the United States.
Currently, the FDA’s regulatory framework permits the marketing of certain digital applications and products outside of the FDA’s active regulation under its device authorities or, in other cases, completely outside FDA regulation if the product uses do not meet the definition of a “medical device.” From time to time, we may develop and commercialize products that we determine fall within the current areas of FDA enforcement discretion or outside the definition of a medical device, but the FDA may not agree with our determination. If the FDA disagrees with any such determinations that we make, we may be required to cease further marketing or distribution of those products until such time as we obtain any required premarket authorization, clearance or approval for those products and we may be subject to receiving an FDA untitled letter or warning letter for such product marketing and distribution activities, amongst other potential enforcement mechanisms available to the FDA.
Failure to comply with post-marketing regulatory requirements could subject us to enforcement actions, including substantial penalties, and might require us to recall or withdraw a products from the market.
After de novo classification, if granted, for our BT-001 digital therapeutic product candidate, we will be subject to ongoing and pervasive regulatory requirements governing, among other things, the manufacture, marketing, labeling, sale, promotion, advertising, medical device reporting, registration, distribution, and listing of devices. For example, we must submit reports to the FDA, for certain adverse events. Failure to submit such reports, or failure to submit the reports in a timely manner, could result in enforcement action by the FDA. Following its review of these medical device adverse event reports, the FDA might ask for additional information or initiate further investigation.
In addition, our digital therapeutics may become subject to post-market study requirements. Any failure to conduct the required studies in accordance with an IRB, and informed consent requirements, or adverse findings in these studies, could also be grounds for modification or withdrawal of marketing authorization for any product we may commercialize.
The FDA and the FTC, also regulate the advertising and promotion of our products and services to ensure that the claims we make are consistent with our regulatory authorizations, that there is adequate and reasonable data to substantiate the claims and that our promotional labeling and advertising is neither false nor misleading. If the FDA or FTC determines that any of our advertising or promotional claims are misleading, not substantiated or not permissible, we may be subject to enforcement actions, including warning letters, and we may be required to revise our promotional claims and make other corrections or restitutions.
56
The regulations to which we are subject are complex and have become more stringent over time. Regulatory changes could result in restrictions on our ability to continue or expand our operations, higher than anticipated costs, or lower than anticipated sales. Even after we have obtained the proper regulatory authorization to market a device, we have ongoing responsibilities under FDA regulations and applicable foreign laws and regulations. The FDA, state and foreign regulatory authorities have broad enforcement powers. Our failure to comply with applicable regulatory requirements could result in enforcement action by the FDA, state or foreign regulatory authorities, which may include any of the following sanctions:
Any of these sanctions could result in higher than anticipated costs or lower than anticipated sales and have a material adverse effect on our reputation, business, financial condition and results of operations.
If treatment guidelines for diabetes patient management change or the standard of care evolves, we may need to redesign and seek new marketing authorization from the FDA for one or more of our product candidates.
If treatment guidelines for diabetes patient management change or the standard of care for this or any other conditions in which we seek to develop digital therapeutics evolves, we may need to redesign the applicable product or product candidates we market or seek to develop and may need to seek and obtain new de novo classifications, clearances or approvals from the FDA and the equivalent from foreign regulatory authorities. If treatment guidelines or the standards of care change so that different treatments become desirable, the clinical utility of one or more of our products could be diminished and our business could be adversely affected.
The misuse or off-label use of our products may harm our reputation in the marketplace, result in injuries that lead to product liability suits or result in costly investigations, fines or sanctions by regulatory bodies if we are deemed to have engaged in the promotion of these uses, any of which could be costly to our business.
Although our products, if authorized for marketing, are marketed for the specific therapeutic uses for which the devices were designed and our personnel will be trained to not promote our products for uses outside of the FDA-approved indications for use, known as “off-label uses,” we cannot, however, prevent a physician from using our products in ways, when in the physician’s independent professional medical judgment, he or she deems it appropriate. There may be increased risk of injury to patients if primary care physicians attempt to use our products off-label. Furthermore, the use of our products for indications other than those authorized, cleared or approved by the FDA or authorized by any foreign regulatory body may not effectively treat such conditions, which could harm our reputation in the marketplace among primary care physicians and patients.
57
If following authorization of our BT-001 digital therapeutic or any other product candidates we may commercialize the FDA or any foreign regulatory body determines that our promotional materials or training constitute promotion of an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance or imposition of an untitled letter or warning letter, injunction, seizure, civil fine or criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action under other regulatory authority, such as false claims laws for any products for which we obtain government reimbursement, if they consider our business activities to constitute promotion of an off-label use, which could result in significant penalties, including, but not limited to, criminal, civil and administrative penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs and the curtailment of our operations.
In addition, physicians may misuse our products with their patients if they are not adequately trained, potentially leading to injury and an increased risk of product liability. If our products are misused, we may become subject to costly litigation by our patients or their patients. As described above, product liability claims could divert management’s attention from our core business, be expensive to defend and result in sizeable damage awards against us that may not be covered by insurance.
Our products may cause or contribute to adverse medical events or be subject to failures or malfunctions that we are required to report to the FDA, and if we fail to do so, we would be subject to sanctions that could harm our reputation, business, financial condition and results of operations. The discovery of serious safety issues with our products, or a recall of our products either voluntarily or at the direction of the FDA or another governmental authority, could have a negative impact on us.
We are subject to the FDA’s medical device reporting regulations and similar foreign regulations, which require us to report to the FDA when we receive or become aware of information that reasonably suggests that one or more of our products may have caused or contributed to a death or serious injury or malfunctioned in a way that, if the malfunction were to recur, it could cause or contribute to a death or serious injury. The timing of our obligation to report is triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events of which we become aware within the prescribed timeframe. We may also fail to recognize that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of the product. If we fail to comply with our reporting obligations, the FDA could take action, including warning letters, untitled letters, administrative actions, criminal prosecution, imposition of civil monetary penalties, revocation of our device authorization, seizure of our products or delay in clearance or approval of future products.
The FDA and foreign regulatory bodies have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture of a product or in the event that a product poses an unacceptable risk to health. The FDA’s authority to require a recall must be based on a finding that there is reasonable probability that the device could cause serious injury or death. We may also choose to voluntarily recall a product if any material deficiency is found. A government- mandated or voluntary recall by us could occur as a result of an unacceptable risk to health, component failures, malfunctions, manufacturing defects, labeling or design deficiencies, packaging defects or other deficiencies or failures to comply with applicable regulations. Product defects or other errors may occur in the future.
58
Depending on the corrective action we take to redress a product’s deficiencies or defects, the FDA may require, or we may decide, that we will need to obtain new authorizations, clearance or approvals for the device before we may market or distribute the corrected device. Seeking such authorizations, clearances or approvals may delay our ability to replace the recalled devices in a timely manner. Moreover, if we do not adequately address problems associated with our devices, we may face additional regulatory enforcement action, including FDA warning letters, product seizure, injunctions, administrative penalties or civil or criminal fines.
Companies are required to maintain certain records of recalls and corrections, even if they are not reportable to the FDA. We may initiate voluntary withdrawals or corrections for our products in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, it could require us to report those actions as recalls and we may be subject to enforcement action. A future recall announcement could harm our reputation with patients, potentially lead to product liability claims against us and negatively affect our sales. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require the dedication of our time and capital, distract management from operating our business and may harm our reputation and financial results.
In the event we seek to market our products in international markets, if we do not obtain and maintain international regulatory registrations or approvals for our products, we will be unable to market and sell our products outside of the United States.
Sales of our products outside of the United States are subject to foreign regulatory requirements that vary widely from country to country. In addition, the FDA regulates exports of medical devices from the United States. While the regulations of some countries may not impose barriers to marketing and selling our products or only require notification, others require that we obtain the marketing authorization of a specified regulatory body. Complying with foreign regulatory requirements, including obtaining registrations or marketing authorizations, can be expensive and time-consuming, and we may not receive regulatory authorizations, clearances or approvals in each country in which we may plan to market our products or we may be unable to do so on a timely basis. The time required to obtain registrations or marketing authorizations, if required by other countries, may be longer than that required for FDA de novo classification, clearance or approval, and requirements for such registrations and marketing authorizations may significantly differ from FDA requirements. If we modify our products, we may need to apply for additional regulatory authorizations before we are permitted to sell the modified product. In addition, we may not continue to meet the quality and safety standards required to maintain the authorizations that we have received. If we are unable to maintain our authorizations in a particular country, we will no longer be able to sell the applicable product in that country.
Regulatory de novo classification, clearance or approval by the FDA does not ensure registration or marketing authorization by regulatory authorities in other countries, and registration or marketing authorization by one or more foreign regulatory authorities does not ensure registration or marketing authorization by regulatory authorities in other foreign countries or by the FDA. However, a failure or delay in obtaining registration or marketing authorization in one country may have a negative effect on the regulatory process in others.
59
Risks Related to Healthcare Laws and Regulation
The insurance coverage and reimbursement status of newly-approved products is uncertain. Failure to obtain or maintain adequate coverage and reimbursement for any of our product candidates, if approved, could limit our ability to market those products and decrease our ability to generate revenue.
In the United States and markets in other countries, patients generally rely on third-party payers to reimburse all or part of the costs associated with their treatment. Adequate coverage and reimbursement from governmental healthcare programs, such as Medicare and Medicaid, and commercial payers is critical to new product acceptance. Our ability to successfully commercialize our product candidates will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and other third-party payers, such as private health insurers and health maintenance organizations, decide which products they will pay for and establish reimbursement levels. In the United States, the principal decisions about reimbursement for new products are typically made by CMS, an agency within the U.S. Department of Health and Human Services. CMS decides whether and to what extent a new medicine will be covered and reimbursed under Medicare and private payors tend to follow CMS to a substantial degree. The availability of coverage and extent of reimbursement by governmental and private payers is essential for most patients to be able to afford treatments. Sales of product candidates that we may identify will depend substantially, both domestically and abroad, on the extent to which the costs of our product candidates will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities, private health coverage insurers and other third-party payers. If coverage and adequate reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize our product candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize a sufficient return on our investment.
There is also significant uncertainty related to the insurance coverage and reimbursement of newly approved products and coverage may be more limited than the purposes for which the medicine is approved by the FDA or comparable foreign regulatory authorities.
Factors payers consider in determining reimbursement are based on whether the product is:
Each payer determines whether or not it will provide coverage for a treatment, what amount it will pay the manufacturer for the treatment and on what tier of its formulary it will be placed. The position on a payer’s list of covered drugs, biological products, and medical devices, or formulary, generally determines the co-payment that a patient will need to make to obtain the therapy and can strongly influence the adoption of such therapy by patients and physicians. Patients who are prescribed treatments for their conditions and providers prescribing such services generally rely on third-party payers to reimburse all or part of the associated healthcare costs. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our products. There may be significant delays in obtaining such coverage and reimbursement for newly approved products, and coverage may be more limited than the purposes for which the product is approved by the FDA.
Moreover, eligibility for coverage and reimbursement does not imply that a product will be paid for in all cases or at a rate that covers our costs, including research, development, intellectual property, manufacture, sale and distribution expenses. Interim reimbursement levels for new products, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement rates may vary according to the use of the product and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost products and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory discounts or rebates required by government healthcare programs or private payers, by any future laws limiting product prices.
60
Third-party payers have attempted to control costs by limiting coverage and the amount of reimbursement for particular products. We cannot be sure that coverage and reimbursement will be available for any product that we commercialize and, if reimbursement is available, what the level of reimbursement will be. Inadequate coverage and reimbursement may impact the demand for, or the price of, any product for which we obtain marketing approval. If coverage and adequate reimbursement are not available, or are available only at limited levels, we may not be able to successfully commercialize our product candidates.
In addition, in some foreign countries, the proposed pricing for a prescription device must be approved before it may be lawfully marketed. The requirements governing product pricing vary widely from country to country. For example, the European Union provides options for its Member States to restrict the range of products for which their national health insurance systems provide reimbursement and to control the prices of products for human use. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost effectiveness of a particular product to currently available therapies. A Member State may approve a specific price for the products or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for products will allow favorable reimbursement and pricing arrangements for any of our product candidates. Historically, products launched in the European Union do not follow price structures of the U.S. and generally prices tend to be significantly lower.
We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws health information privacy and security laws, and other health care laws and regulations. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
We are subject to applicable fraud and abuse and other healthcare laws and regulations, including, without limitation, the U.S. federal Anti-Kickback Statute and the FCA, which may constrain the business or financial arrangements and relationships through which we sell, market and distribute our products. In particular, the promotion, sales and marketing of healthcare items and services, as well as certain business arrangements in the healthcare industry (e.g., healthcare providers, physicians and third-party payers), are subject to extensive laws designed to prevent fraud, kickbacks, self- dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, structuring and commission(s), certain customer incentive programs and other business arrangements generally. We also may be subject to patient information and privacy and security regulation by both the federal government and the states and foreign jurisdictions in which we conduct our business. The applicable federal, state and foreign healthcare laws and regulations laws that may affect our ability to operate include, but are not limited to:
61
62
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge and may not comply under one or more of such laws, regulations, and guidance. Law enforcement authorities are increasingly focused on enforcing fraud and abuse laws, and it is possible that some of our practices may be challenged under these laws. Efforts to ensure that our current and future business arrangements with third parties, and our business generally, will comply with applicable healthcare laws and regulations will involve substantial costs. If our operations, including our arrangements with physicians and other healthcare providers are found to be in violation of any of such laws or any other governmental regulations that apply to us, we may be subject to penalties, including, without limitation, administrative, civil and criminal penalties, damages, fines, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs (such as Medicare and Medicaid), and imprisonment, as well as additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, any of which could adversely affect our ability to operate our business and our financial results.
We are subject to data privacy and security laws and regulations governing our collection, use, disclosure, or storage of personally identifiable information, including protected health information and payment card data, which may impose restrictions on us and our operations and subject us to penalties if we are unable to fully comply with such laws.
Numerous federal and state laws and regulations govern the collection, use, disclosure, storage and transmission of personally identifiable information, including protected health information. These laws and regulations, including their interpretation by governmental agencies, are subject to frequent change and could have a negative impact on our business. In addition, in the future, industry requirements or guidance, contractual obligations, and/or legislation at both the federal and the state level may limit, forbid or regulate the use or transmission of health information outside of the United States. These varying interpretations can create complex compliance issues for us and our partners and potentially expose us to additional expense, adverse publicity and liability, any of which could adversely affect our business.
Federal and state consumer protection laws are increasingly being applied by the FTC, and states’ attorneys general to regulate the collection, use, storage and disclosure of personal or personally identifiable information, through websites or otherwise, and to regulate the presentation of website content.
The security measures that we and our third-party vendors and subcontractors have in place to ensure compliance with privacy and data protection laws may not protect our facilities and systems from security breaches, acts of vandalism or theft, computer viruses, misplaced or lost data, programming and human errors or other similar events. Even though we provide for appropriate protections through our agreements with our third party vendors, we still have limited control over their actions and practices. A breach of privacy or security of personally identifiable health information may result in an enforcement action, including criminal and civil liability, against us. We are not able to predict the extent of the impact such incidents may have on our business. Enforcement actions against us could be costly and could interrupt regular operations, which may adversely affect our business. While we have not received any notices of violation of the applicable privacy and data protection laws and believe we are in compliance with such laws, there can be no assurance that we will not receive such notices in the future.
There is ongoing concern from privacy advocates, regulators and others regarding data privacy and security issues, and the number of jurisdictions with data privacy and security laws has been increasing. Also, there are ongoing public policy discussions regarding whether the standards for de-identification, anonymization or pseudonymization of health information are sufficient, and the risk of re-identification sufficiently small, to adequately protect patient privacy. We expect that there will continue to be new proposed and amended laws, regulations and industry standards concerning privacy, data protection and information security in the United States, such as the CCPA. Further, the CPRA was passed by California voters on November 3, 2020. The CPRA will create additional obligations with respect to processing and storing personal information that are scheduled to take effect on January 1, 2023 (with certain provisions having retroactive effect to January 1, 2022). Other U.S. states also are considering omnibus privacy legislation and industry organizations regularly adopt and advocate for new standards in these areas. While the CCPA and CPRA contains an exceptions for certain activities involving PHI under HIPAA, we cannot yet determine the impact the CCPA, CPRA or other such future laws, regulations and standards may have on our business.
63
Future laws, regulations, standards, obligations amendments, and changes in the interpretation of existing laws, regulations, standards and obligations could impair our or our clients’ ability to collect, use or disclose information relating to patients or consumers, including information derived therefrom, which could decrease demand for our Platform, increase our costs and impair our ability to maintain and grow our client base and increase our revenue. Accordingly, we may find it necessary or desirable to fundamentally change our business activities and practices or to expend significant resources to modify our software or platform and otherwise adapt to these changes.
Further, our patients may expect us to comply with more stringent privacy and data security requirements than those imposed by laws, regulations or self-regulatory requirements, and we may be obligated contractually to comply with additional or different standards relating to our handling or protection of data.
Any failure or perceived failure by us to comply with federal or state laws or regulations, industry standards or other legal obligations, or any actual or suspected privacy or security incident, whether or not resulting in unauthorized access to, or acquisition, release or transfer of personally identifiable information or other data, may result in governmental enforcement actions and prosecutions, private litigation, fines and penalties or adverse publicity and could cause our clients to lose trust in us, which could have an adverse effect on our reputation and business. We may be unable to make such changes and modifications in a commercially reasonable manner or at all, and our ability to develop new products could be limited. Any of these developments could harm our business, financial condition and results of operations. Privacy and data security concerns, whether valid or not valid, may inhibit retention of our Platform by existing clients or adoption of our Platform by new clients.
Healthcare legislative reform measures and constraints on national budget social security systems may have a material adverse effect on our business and results of operations.
In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory changes to the health care system that could impact our ability to sell our products profitably. In particular, in 2010, the ACA, was enacted, which, among other things, addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program; extended the Medicaid Drug Rebate program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations; subjected manufacturers to new annual fees and taxes for certain branded prescription drugs; created a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% (increased to 70% pursuant to the Bipartisan Budget Act of 2018, effective as of January 1, 2019) point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; and provided incentives to programs that increase the federal government’s comparative effectiveness research.
Since its enactment, there have been judicial, Congressional and executive challenges to certain aspects of the ACA. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA brought by several states without specifically ruling on the constitutionality of the ACA. Prior to the Supreme Court’s decision, President Biden issued an executive order to initiate a special enrollment period from February 15, 2021 through August 15, 2021 for purposes of obtaining health insurance coverage through the ACA marketplace. The executive order also instructed certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA. It is unclear how other healthcare reform measures of the Biden administration or other efforts, if any, to challenge, repeal or replace the ACA will impact our business.
Other legislative changes have been proposed and adopted in the United States since the Affordable Care Act was enacted. In August 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers up to 2% per fiscal year, and, due to subsequent legislative amendments, will remain in effect through 2030 unless additional Congressional action is taken. Pursuant to the Coronavirus Aid, Relief, and Economic Security Act, also known as the CARES Act, as well as subsequent legislation, these reductions have been suspended from May 1, 2020 through March 31, 2022 due to the ongoing COVID-19 pandemic. Following the temporary suspension, a 1% payment reduction will occur beginning April 1, 2022 through June 30, 2022, and the 2% payment reduction will resume on July 1, 2022.
64
There has been increasing legislative and enforcement interest in the United States with respect to product pricing practices. Specifically, there have been several recent U.S. Congressional inquiries and proposed federal and state legislation designed to, among other things, bring more transparency to product pricing, reduce the cost of products under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. The HHS has already started the process of soliciting feedback on some of these measures and, at the same time, is immediately implementing others under its existing authority. It is unclear what effect such legislative and enforcement interest may have on prescription devices.
We expect that these and other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved device, which could have an adverse effect on patients for our product candidates. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payers.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels in the U.S. directed at broadening the availability of healthcare and containing or lowering the cost of healthcare. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our products. Such reforms could have an adverse effect on anticipated revenue from products that we may successfully develop and for which we may obtain regulatory approval and may affect our overall financial condition and ability to develop product candidates. If we or any third parties we may engage are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or such third parties are not able to maintain regulatory compliance, our current or any future product candidates we may develop may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability.
Our employees, independent contractors, consultants, commercial collaborators, principal investigators, vendors and other agents may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements.
We are exposed to the risk that our employees, independent contractors, consultants, commercial collaborators, principal investigators, vendors and other agents may engage in fraudulent conduct or other illegal activity. Misconduct by these parties could include intentional, reckless or negligent conduct or disclosure of unauthorized activities to us that violates applicable regulations, including those laws requiring the reporting of true, complete and accurate information to regulatory agencies, manufacturing standards and U.S. federal and state healthcare laws and regulations. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. We could face liability under the U.S. federal Anti-Kickback Statute and similar U.S. state laws. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, referrals, customer incentive programs and other business arrangements. Misconduct by these parties could also involve the improper use of individually identifiable information, including, without limitation, information obtained in the course of clinical trials, which could result in significant regulatory sanctions and serious harm to our reputation. Further, should violations include promotion of unapproved (off-label) uses one or more of our products, we could face significant regulatory sanctions for unlawful promotion, as well as substantial penalties under the FCA, and similar state laws. Similar concerns could exist in jurisdictions outside of the United States as well. It is not always possible to identify and deter misconduct by employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. The precautions we take to detect and prevent misconduct may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, imprisonment, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of noncompliance with these laws, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business, financial condition and results of operations.
65
Risks Related to Our Legal and Regulatory Environment
Failure to comply with anti-bribery, anti-corruption and anti-money laundering laws could subject us to penalties and other adverse consequences.
We are subject to the U.S. Foreign Corrupt Practices Act (the "FCPA") and other anti-corruption, anti-bribery, and anti-money laundering laws in the jurisdictions in which we do business, both domestic and abroad. These laws generally prohibit us and our employees from improperly influencing government officials or commercial parties in order to obtain or retain business, direct business to any person or gain any improper advantage. The FCPA and similar applicable anti-bribery and anti-corruption laws also prohibit our third-party business partners, representatives and agents from engaging in corruption and bribery. We and our third-party business partners, representatives and agents may have direct or indirect interactions with officials and employees of government agencies or state-owned or affiliated entities. We may be held liable for the corrupt or other illegal activities of these third-party business partners and intermediaries, our employees, representatives, contractors, partners and agents, even if we do not explicitly authorize such activities. These laws also require that we keep accurate books and records and maintain internal controls and compliance procedures designed to prevent any such actions. While we have policies and procedures to address compliance with such laws, we cannot assure you that our employees and agents will not take actions in violation of our policies or applicable law, for which we may be ultimately held responsible. Our exposure for violating these laws will increase as we expand internationally and as we commence sales and operations in foreign jurisdictions. Any violation of the FCPA or other applicable anti-bribery, anti-corruption laws and anti- money laundering laws could result in whistleblower complaints, adverse media coverage, investigations, imposition of significant legal fees, loss of export privileges, severe criminal or civil sanctions or suspension or debarment from U.S. government contracts, substantial diversion of management’s attention, drop in stock price or overall adverse consequences to our business, all of which may have an adverse effect on our reputation, business, financial condition, and results of operations.
Federal, state and local employment-related laws and regulations could increase our cost of doing business and subject us to fines and lawsuits.
Our operations are subject to a variety of federal, state and local employment-related laws and regulations, including, but not limited to, the U.S. Fair Labor Standards Act, which governs such matters as minimum wages, the Family Medical Leave Act, overtime pay, compensable time, recordkeeping and other working conditions, Title VII of the Civil Rights Act, the Employee Retirement Income Security Act, the Americans with Disabilities Act, the National Labor Relations Act, regulations of the Equal Employment Opportunity Commission, regulations of the Office of Civil Rights, regulations of the Department of Labor (DOL), regulations of state attorneys general, federal and state wage and hour laws, and a variety of similar laws enacted by the federal and state governments that govern these and other employment-related matters. As our employees are located in a number of states, compliance with these evolving federal, state and local laws and regulations could substantially increase our cost of doing business while failure to do so could subject us to fines and lawsuits. We are currently subject to employee-related legal proceedings in the ordinary course of business. While we believe that we have adequate reserves for those losses that we believe are probable and can be reasonably estimated, the ultimate results of legal proceedings and claims cannot be predicted with certainty.
Risks Related to the recently completed Business Combination
Management’s focus and resources may be diverted from operational matters and other strategic opportunities as a result of the recently completed Business Combination.
The Business Combination may place a significant burden on our management and other internal resources. The diversion of management’s attention and any difficulties encountered in the transition process could harm our financial condition, results of operations and prospects. In addition, uncertainty about the effect of the Business Combination on our systems, employees, customers, partners, and other third parties, including regulators, may have an adverse effect on us. These uncertainties may impair our ability to attract, retain and motivate key personnel for a period of time after the completion of the Business Combination.
66
We will incur significant increased expenses and administrative burdens as a public company, which could have an adverse effect on our business, financial condition and results of operations.
As a public company, we will face increased legal, accounting, administrative and other costs and expenses as a public company that we did not incur as a private company. The Sarbanes-Oxley Act, including the requirements of Section 404, as well as rules and regulations subsequently implemented by the SEC, the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010 and the rules and regulations promulgated and to be promulgated thereunder, the ("PCAOB") and the securities exchanges, impose additional reporting and other obligations on public companies. Compliance with public company requirements will increase costs and make certain activities more time-consuming. A number of those requirements will require us to carry out activities we have not done previously. In addition, additional expenses associated with SEC reporting requirements will be incurred. Furthermore, if any issues in complying with those requirements are identified (for example, if the auditors identify a material weakness or significant deficiency in the internal control over financial reporting), we could incur additional costs rectifying those issues, and the existence of those issues could adversely affect our reputation or investor perceptions of it. It may also be more expensive to obtain director and officer liability insurance. Risks associated with our status as a public company may make it more difficult to attract and retain qualified persons to serve on our Board or as executive officers. The additional reporting and other obligations imposed by these rules and regulations will increase legal and financial compliance costs and the costs of related legal, accounting and administrative activities. These increased costs will require us to divert a significant amount of money that could otherwise be used to expand the business and achieve strategic objectives. Advocacy efforts by stockholders and third parties may also prompt additional changes in governance and reporting requirements, which could further increase costs.
We qualify as an “emerging growth company” and as a “smaller reporting company”, and if we take advantage of certain exemptions from disclosure requirements available to emerging growth companies or smaller reporting companies, which could make our securities less attractive to investors and may make it more difficult to compare our performance to the performance of other public companies.
We qualify as an “emerging growth company” as defined in Section 2(a)(19) of the Securities Act, as modified by the JOBS Act. As such, we are eligible for and intend to take advantage of certain exemptions from various reporting requirements applicable to other public companies that are not emerging growth companies for as long as we continue to be an emerging growth company, including (i) the exemption from the auditor attestation requirements with respect to internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act, (ii) the exemptions from say-on-pay, say-on-frequency and say-on-golden parachute voting requirements and (iii) reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements. We will remain an emerging growth company until the earliest of (i) the last day of the fiscal year in which the market value of the shares of our common stock that are held by non-affiliates exceeds $700 million as of June 30 of that fiscal year, (ii) the last day of the fiscal year in which we have total annual gross revenue of $1.07 billion or more during such fiscal year, (iii) the date on which we have issued more than $1 billion in non-convertible debt in the prior three-year period or (iv) the last day of the fiscal year following the fifth anniversary of the date of the first sale of our common stocks in our IPO. In addition, Section 107 of the JOBS Act also provides that an emerging growth company can take advantage of the exemption from complying with new or revised accounting standards provided in Section 7(a)(2)(B) of the Securities Act as long as we are an emerging growth company. An emerging growth company can therefore delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to avail ourselves of this exemption from new or revised accounting standards and, therefore, we may not be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies. Investors may find our common stock less attractive because we will rely on these exemptions, which may result in a less active trading market for our common stock and its stock price may be more volatile.
We are also a “smaller reporting company,” meaning that the market value of our stock held by non-affiliates is less than $700 million as of the prior June 30 and our annual revenue is less than $100 million during the most recently completed fiscal year. We may continue to be a smaller reporting company if either (i) the market value of our stock held by non-affiliates is less than $250 million as of the prior June 30 or (ii) our annual revenue is less than $100 million during the most recently completed fiscal year and the market value of our stock held by non-affiliates is less than $700 million as of the prior June 30. If we are a smaller reporting company at the time we cease to be an emerging growth company, we may continue to rely on exemptions from certain disclosure requirements that are available to smaller reporting companies. Specifically, as a smaller reporting company we may choose to present only the two most recent fiscal years of audited financial statements in this Annual Report and take advantage of reduced disclosure obligations regarding executive compensation.
If we fail to maintain an effective system of disclosure controls and internal control over financial reporting, our ability to produce timely and accurate financial statements or comply with applicable regulations could be impaired.
67
As a public company, we are subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act and the rules and regulations of the applicable listing standards of The Nasdaq Capital Market ("Nasdaq"). We expect that the requirements of these rules and regulations will continue to increase our legal, accounting and financial compliance costs, make some activities more difficult, time-consuming and costly and place significant strain on our personnel, systems and resources. The Sarbanes-Oxley Act requires, among other things, that we maintain effective disclosure controls and procedures and internal control over financial reporting. We are continuing to develop and refine our disclosure controls and other procedures that are designed to ensure that information required to be disclosed by us in the reports that we will file with the SEC is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms and that information required to be disclosed in reports under the Exchange Act is accumulated and communicated to our principal executive and financial officers. We are also continuing to improve our internal control over financial reporting, which includes hiring additional accounting and financial personnel to implement such processes and controls. In order to maintain and improve the effectiveness of our disclosure controls and procedures and internal control over financial reporting, we have expended, and anticipate that we will continue to expend, significant resources, including accounting-related costs and significant management oversight. If any of these new or improved controls and systems do not perform as expected, we may experience material weaknesses in our controls.
Our current controls and any new controls that we develop may become inadequate because of changes in conditions in our business. Further, weaknesses in our disclosure controls and internal control over financial reporting may be discovered in the future. Any failure to develop or maintain effective controls or any difficulties encountered in their implementation or improvement could harm our results of operations or cause us to fail to meet our reporting obligations and may result in a restatement of our financial statements for prior periods. Any failure to implement and maintain effective internal control over financial reporting also could adversely affect the results of periodic management evaluations and annual independent registered public accounting firm attestation reports regarding the effectiveness of our internal control over financial reporting that we will eventually be required to include in our periodic reports that will be filed with the SEC. Ineffective disclosure controls and procedures and internal control over financial reporting could also cause investors to lose confidence in our reported financial and other information, which would likely have a negative effect on the trading price of our common stock. In addition, if we are unable to continue to meet these requirements, we may not be able to remain listed on Nasdaq. We are not currently required to comply with the SEC rules that implement Section 404 of the Sarbanes-Oxley Act and are therefore not required to make a formal assessment of the effectiveness of our internal control over financial reporting for that purpose. As a public company, we are required to provide an annual management report on the effectiveness of our internal control over financial reporting commencing with our second annual report on Form 10-K.
Our independent registered public accounting firm is not required to formally attest to the effectiveness of our internal control over financial reporting until after we are no longer an “emerging growth company” as defined in the JOBS Act. At such time, our independent registered public accounting firm may issue a report that is adverse in the event it is not satisfied with the level at which our internal control over financial reporting is documented, designed or operating. Any failure to maintain effective disclosure controls and internal control over financial reporting could have an adverse effect on our business and results of operations and could cause a decline in the price of our common stock.
68
If we fail to establish and maintain effective internal control over financial reporting, we may not be able to accurately report our financial results, which may cause investors to lose confidence in our reported financial information and may lead to a decline in the market price of our stock.
Pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, we are required to furnish a report by our management on our internal control over financial reporting. However, while we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed period, we are engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that we will not be able to conclude, within the prescribed timeframe or at all, that our internal control over financial reporting is effective as required by Section 404. We identified a material weakness our internal control over financial reporting related to the inaccurate accounting for the value of shares to be issued to the underwriter at the closing of our IPO as well as inaccurate accounting for certain accrued expenses and prepaid expenses and the Company’s restatement of its financial statements to reclassify all redeemable equity instruments to temporary equity from permanent equity. Up to and including the third fiscal quarter of 2021, our disclosure controls and procedures were not effective. We have implemented a remediation plan, described under Part I, Item 4, Evaluation of Disclosure Controls and Procedures of our Form 10-Q for the third quarter of 2021, to remediate the material weakness but can give no assurance that the measures we have taken will prevent any future material weaknesses or deficiencies in internal control over financial reporting. Even though we believe we have strengthened our controls and procedures, in the future those controls and procedures may not be adequate to prevent or identify irregularities or errors or to facilitate the fair presentation of our financial statements.
Risks Related to Our Organizational Structure
Our executive chairman of the board of directors, David Perry, and our chief executive officer, president and director, Kevin Appelbaum, together will have significant influence over the company.
As of December 31, 2021, Mr. Perry and Mr. Appelbaum own, collectively, approximately 56.4% of the outstanding shares of our common stock. As long as such persons each own or control a significant percentage of outstanding voting power, they have the ability to strongly influence all corporate actions requiring stockholder approval, including the election and removal of directors and the size of our board of directors, any amendment of our certificate of incorporation or bylaws, or the approval of any merger or other significant corporate transaction, including a sale of substantially all of our assets. Some of these persons or entities may have interests different than yours.
Delaware law and our governing documents contain certain provisions, including anti-takeover provisions, that limit the ability of stockholders to take certain actions and could delay or discourage takeover attempts that stockholders may consider favorable.
Our governing documents and the Delaware General Corporation Law (“DGCL”), contain provisions that could have the effect of rendering more difficult, delaying, or preventing an acquisition deemed undesirable by the Board and therefore depress the trading price of our common stock. These provisions could also make it difficult for stockholders to take certain actions, including electing directors who are not nominated by the current members of the Board or taking other corporate actions, including effecting changes in our management. Among other things, our governing documents include provisions regarding:
69
These provisions, alone or together, could delay or prevent hostile takeovers and changes in control or changes in the Board or management.
Our amended and restated bylaws designate specific courts as the exclusive forum for certain litigation that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us.
Pursuant to our amended and restated bylaws, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware is the sole and exclusive forum for state law claims for (1) any derivative action or proceeding brought on our behalf; (2) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers, or other employees to us or our stockholders; (3) any action asserting a claim arising pursuant to any provision of the Delaware General Corporation Law or our certificate of incorporation or bylaws (including the interpretation, validity or enforceability thereof); or (4) any action asserting a claim governed by the internal affairs doctrine. We refer to this provision in our bylaws as the Delaware Forum Provision. The Delaware Forum Provision will not apply to any causes of action arising under the Securities Act or the Exchange Act. Our amended and restated bylaws further provide that unless we consent in writing to the selection of an alternative forum, the federal district courts of the United States shall be the sole and exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act. We refer to this provision in our bylaws as the Federal Forum Provision. In addition, our amended and restated bylaws provide that any person or entity purchasing or otherwise acquiring any interest in shares of our capital stock is deemed to have notice of and consented to the Delaware Forum Provision and the Federal Forum Provision; provided, however, that stockholders cannot and will not be deemed to have waived our compliance with the U.S. federal securities laws and the rules and regulations thereunder.
The Delaware Forum Provision and the Federal Forum Provision in our bylaws may impose additional litigation costs on stockholders in pursuing any such claims. Additionally, these forum selection clauses may limit our stockholders’ ability to bring a claim in a judicial forum that they find favorable for disputes with us or our directors, officers or employees, which may discourage the filing of lawsuits against us and our directors, officers and employees, even though an action, if successful, might benefit our stockholders. In addition, while the Delaware Supreme Court ruled in March 2020 that federal forum selection provisions purporting to require claims under the Securities Act be brought in federal court are “facially valid” under Delaware law, there is uncertainty as to whether other courts will enforce our Federal Forum Provision. If the Federal Forum Provision is found to be unenforceable, we may incur additional costs associated with resolving such matters. The Federal Forum Provision may also impose additional litigation costs on stockholders who assert that the provision is not enforceable or invalid. The Court of Chancery of the State of Delaware and the federal district courts of the United States may also reach different judgments or results than would other courts, including courts where a stockholder considering an action may be located or would otherwise choose to bring the action, and such judgments may be more or less favorable to us than our stockholders.
70
Risks Related to Our Common Stock
Unstable market and economic conditions may have series adverse consequences on our business, financial condition and stock price.
As widely reported, global credit and financial markets have experienced extreme volatility and disruptions in the past several years, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in the rate of inflation, increases in unemployment rates and uncertainty about economic stability, including most recently in connection with the ongoing and evolving COVID-19 pandemic. There can be no assurance that further deterioration in credit and financial markets and confidence in economic conditions will not occur. Our general business strategy may be adversely affected by any such economic downturn, volatile business environment or continued unpredictable and unstable market conditions. Our business could be also be impacted by volatility caused by geopolitical events, such as the conflict in Ukraine. If the current equity and credit markets deteriorate, or do not improve, it may make any necessary debt or equity financing more difficult, more costly, and more dilutive. Furthermore, our stock price may decline due in part to the volatility of the stock market and the general economic downturn.
Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and stock price and could require us to delay, scale back or discontinue the development and commercialization of one or more of our product candidates.
There can be no assurance that we will be able to comply with the continued listing standards of Nasdaq.
If Nasdaq delists our shares of common stock from trading on its exchange for failure to meet Nasdaq’s listing standards, we and our stockholders could face significant material adverse consequences including:
The price of our common stock may be volatile.
The price of our common stock may fluctuate due to a variety of factors, including:
71
These market and industry factors may materially reduce the market price of our common stock regardless of our operating performance.
Reports published by analysts, including projections in those reports that differ from our actual results, could adversely affect the price and trading volume of our common shares.
Securities research analysts may establish and publish their own periodic projections for us. These projections may vary widely and may not accurately predict the results we actually achieve. Our share price may decline if our actual results do not match the projections of these securities research analysts. Similarly, if one or more of the analysts who write reports on us downgrades our stock or publishes inaccurate or unfavorable research about our business, our share price could decline. If one or more of these analysts ceases coverage of us or fails to publish reports on us regularly, our share price or trading volume could decline.
A significant portion of our total outstanding shares are restricted from immediate resale but may be sold into the market in the near future. This could cause the market price of our common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market could occur at any time. These sales, or the perception in the market that the holders of a large number of shares intend to sell shares, could reduce the market price of our common stock. Although certain stockholders will be subject to certain restrictions regarding the transfer of our common stock, these shares may be sold after the expiration of the lock-up. As restrictions on resale end and the registration statements are available for use, the market price of our common stock could decline if the holders of currently restricted shares sell them or are perceived by the market as intending to sell them.
Our issuance of additional capital stock in connection with financings, acquisitions, investments, our stock incentive plans or otherwise will dilute all other stockholders.
We expect to issue additional capital stock in the future that will result in dilution to all other stockholders. We expect to grant equity awards to employees, directors, and consultants under our stock incentive plans. We may also raise capital through equity financings in the future. As part of our business strategy, we may acquire or make investments in complementary companies, products, or technologies and issue equity securities to pay for any such acquisition or investment. Any such issuances of additional capital stock may cause stockholders to experience significant dilution of their ownership interests and the per share value of our common stock to decline.
Because we have no current plans to pay cash dividends on our common stock, you may not receive any return on investment unless you sell your common stock for a price greater than that which you paid for it.
We have no current plans to pay cash dividends on our common stock. The declaration, amount and payment of any future dividends will be at the sole discretion of our board of directors. Our board of directors may take into account general and economic conditions, our financial condition and operating results, our available cash, current and anticipated cash needs, capital requirements, contractual, legal, tax and regulatory restrictions, implications on the payment of dividends by us to our stockholders or by our subsidiary to us and such other factors as our board of directors may deem relevant. In addition, the terms of our loan agreement with Hercules Capital restrict our ability to pay cash dividends. Accordingly, we may not pay any dividends on our common stock in the foreseeable future.
Future offerings of debt or equity securities by us may adversely affect the market price of our common stock.
In the future, we may attempt to obtain financing or to further increase our capital resources by issuing additional shares of our common stock or offering debt or other equity securities, including commercial paper, medium-term notes, senior or subordinated notes, debt securities convertible into equity or shares of preferred stock. Future acquisitions could require substantial additional capital in excess of cash from operations. We would expect to obtain the capital required for acquisitions through a combination of additional issuances of equity, corporate indebtedness and/or cash from operations.
72
Issuing additional shares of our common stock or other equity securities or securities convertible into equity may dilute the economic and voting rights of our existing stockholders or reduce the market price of our common stock or both. Upon liquidation, holders of such debt securities and preferred shares, if issued, and lenders with respect to other borrowings would receive a distribution of our available assets prior to the holders of our common stock. Debt securities convertible into equity could be subject to adjustments in the conversion ratio pursuant to which certain events may increase the number of equity securities issuable upon conversion. Preferred shares, if issued, could have a preference with respect to liquidating distributions or a preference with respect to dividend payments that could limit our ability to pay dividends to the holders of our common stock. Our decision to issue securities in any future offering will depend on market conditions and other factors beyond our control, which may adversely affect the amount, timing and nature of our future offerings.
Item 1B. Unresolved Staff Comments.
Not Applicable.
Item 2. Properties.
We do not own or lease any real property. We run a virtual office model and our business mailing address is 548 Market Street, #49404, San Francisco, CA 94104.
Item 3. Legal Proceedings.
We are not currently a party to any material legal proceedings, nor are ware aware of any pending or threatened litigation. In the ordinary course of business, we may be subject to legal proceedings, claims and litigation.
Item 4. Mine Safety Disclosures.
Not Applicable
73
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information for Common Stock
Our common stock is listed for trading on The Nasdaq Capital Market under the symbol BTTX.
Holders of Record
As of March 25, 2022 there were approximately 67 stockholders of record of our common stock.
Dividend Policy
We currently intend to retain all available funds and any future earnings to fund the growth and development of our business. We have never declared or paid any cash dividends on our capital stock. We do not intend to pay cash dividends to our stockholders in the foreseeable future. In addition, the terms of our loan agreement with Hercules Capital preclude us from paying dividends. Investors should not purchase our common stock with the expectation of receiving cash dividends.
Any future determination to declare dividends will be made at the discretion of our board of directors and will depend on our financial condition, operating results, capital requirements, general business conditions, and other factors that our board of directors may deem relevant.
Securities Authorized for Issuance Under Equity Compensation Plans
The information required by Item 5 of Form 10-K regarding equity compensation plans is incorporated herein by reference to Item 12 of Part III of this Annual Report.
Recent Sales of Unregistered Securities
None.
Purchases of Equity Securities by the Issuer and Affiliated Persons
None.
Use of Proceeds
Of the gross proceeds received from the IPO and the full exercise of the underwriters' option to purchase additional units, $57.5 million was placed in MCAD's trust account. The net proceeds of the IPO were applied to fund the Business Combination and related expenses.
Item 6. [Reserved]
74
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our consolidated financial statements and related notes, included in Item 8 of this Annual Report. Unless otherwise specified all dollar amounts are in U.S. Dollars. Except for per share amounts, all amounts are in thousands, unless otherwise noted.
Overview
Our mission is to address unmet needs for treatment of cardiometabolic diseases such as diabetes and heart disease. The U.S. spends approximately $4.0 trillion per year on healthcare, and approximately 90% of that spending is for the treatment of chronic diseases. Most chronic diseases are caused predominantly by behaviors, including cardiometabolic diseases such as diabetes and heart disease. The root causes of cardiometabolic diseases are behaviors relating to diet, physical activity, and other lifestyle factors, yet current treatments are focused on reducing the effects of those diseases rather than addressing the root causes.
In response to addressing the root causes of cardiometabolic diseases, we developed a proprietary platform for the development of FDA-regulated, software-based, PDTs for treating diabetes, heart disease, and other cardiometabolic conditions. Our PDTs deliver a novel form of cognitive behavioral therapy that enables changes in neural pathways of the brain so that lasting changes in behavior become possible. In November 2021, we completed enrolling patients in a pivotal study of our lead product candidate for the treatment of patients with type 2 diabetes, BT-001, designed to support a regulatory submission for marketing authorization from the FDA. Participants were randomized to receive standard of care with or without BT-001. We announced primary endpoint data from our clinical trial of BT-001 in March 2022. The primary efficacy endpoint was the difference in mean change from baseline in A1c after 90 days of treatment between the two groups and showed highly statistically significant improvement in A1c between the intervention and control groups (-0.4%, p <0.001). Clinically meaningful changes (A1c reductions of 0.4% or more) occurred in 42.7% of the group receiving standard of care and BT-001 versus 25.4% in the group receiving standard of care alone (difference of 17.3%, p <0.001). We believe this demonstrates that the use of BT-001 significantly improved A1c compared to standard of care alone. The unique characteristics of prescription digital therapeutics and CMDx, may make it possible for us to launch multiple products now in development for the treatment of other CMDx over the next few years.
We are building a fully integrated PDTs company focused on treating the root causes of cardiometabolic diseases. Our therapeutics are intended to fill a known gap in the treatment of cardiometabolic diseases and integrate within the existing healthcare system. We expect primary care providers to prescribe our therapeutics and insurers to reimburse them much like they would a drug, and for the patient to remain in the care of their provider while using them.
Financial Overview
Since our inception in 2015, we have focused substantially all of our resources on conducting research and development activities, including discovery and preclinical studies, establishing and maintaining our intellectual property, hiring personnel, raising capital and providing general and administrative support for these operations. We have recorded revenue from a pilot program with a private health insurance provider to provide a digital therapeutic program that includes a mobile app. We have funded our operations to date primarily from the issuance of convertible notes and simple agreements for future equity ("SAFEs"), the issuance and sale of our preferred units, borrowing on our term loan agreement and funding from the merger with MCAD.
We have incurred net losses in each year since inception. Our net losses were $40,335 and $6,387 for the twelve months ended December 31, 2021 and 2020, respectively. As of December 31, 2021 and December 31, 2020, we had an accumulated deficit of $71,743 and $31,408, respectively. Substantially all of our net losses have resulted from costs incurred in connection with our research and development programs and from general and administrative costs associated with our operations. We expect to continue to incur significant expenses and increasing operating losses over at least the next several years. We expect our expenses will increase substantially in connection with our ongoing activities, as we:
75
We were initially formed as a limited liability company under the laws of the State of Delaware and converted to a Delaware Corporation in August 2020. In connection with our conversion to a Delaware corporation, each of our outstanding shares of the members of the limited liability company was converted into shares of capital stock. On the date of conversion, the following conversions of limited liability shares took place: (i) each Series Seed convertible preferred unit converted into one share of Series Seed convertible preferred stock, (ii) each Series A convertible preferred unit converted into one share of Series A convertible preferred stock, (iii) each of the common units issued by the limited liability company (each a "Common Unit") was converted into one share of common stock, and (iv) each outstanding convertible note converted into a SAFE with a corresponding investment balance as the converted convertible notes.
On April 6, 2021, the Company entered into a merger agreement with MCAD, a special purpose acquisition company. In connection with the merger agreement, MCAD entered into subscription agreements (the “Subscription Agreements”) dated as of April 6, 2020, with certain institutional and accredited investors, pursuant to which, among other things, MCAD agreed to issue and sell, in a private placement immediately prior to the closing of the Business Combination, an aggregate of 5,000,000 shares of Common Stock for $10.00 per share (the “PIPE Shares”). On October 28, 2021, we completed the merger with MCAD. We raised $59 million in funding upon the completion of the merger with MCAD. Under the merger Agreement, MCAD acquired all of the outstanding shares of Legacy BTX in exchange for 15,174,729 shares of MCAD. In connection with the merger, MCAD was renamed Better Therapeutics, Inc.
Impact of COVID-19
In March 2020, the World Health Organization declared COVID-19 a global pandemic. The ongoing COVID-19 pandemic has not had a significant impact on our operations. Management is unable to estimate the future financial effects, if any, to our business as a result of COVID-19 because of the high level of uncertainties and unpredictable outcomes of this disease.
We are continuing to evaluate the impact of the ongoing COVID-19 pandemic, including the emergence of new variants of COVID-19, on our business and are taking proactive measures to protect the health and safety of our employees, as well as to maintain business continuity. Based on guidance issued by federal, state and local authorities, we transitioned to a fully remote work model for our employees, effective July 2020. We believe that the measures we are implementing are appropriate, reflecting both regulatory and public health guidance, to maintain business continuity. We will continue to closely monitor and seek to comply with guidance from governmental authorities and adjust our activities as appropriate.
The ultimate impact of the ongoing COVID-19 pandemic or a similar health epidemic is highly uncertain and subject to change. We do not yet know the full extent of potential delays or impacts on our business, our clinical trial, healthcare systems or the global economy as a whole. However, these effects could harm our operations, and we will continue to monitor the ongoing COVID-19 pandemic closely.
Components of Results of Operations
Revenue
Since our inception in 2015, we have recognized an immaterial amount of revenue resulting from a pilot program with a private health insurer. We expect that our primary sources of revenue will be through reimbursement coverage for our treatments by commercial insurers, Medicare, and Medicaid in the U.S. and our near-term plan is to obtain broad reimbursement coverage for our first PDT for treating type 2 diabetes, BT-001. We expect to be successful in obtaining a broad reimbursement coverage through demonstrating and generating a comprehensive set of evidence to substantiate the value of BT-001 based on its impact on clinical outcomes, total cost of care, and durability of effect. Obtaining a broad reimbursement coverage and timing of obtaining such coverage for BT-001 and our other product candidates is highly uncertain. As a result, the timing and the amount of revenue we expect to recognize from monetizing our product candidates may vary based on various factors.
We also may explore opportunities to partner with pharmaceutical companies marketing traditional drug therapies for cardiometabolic diseases that may benefit from an increase in efficacy and durability when combined with our prescription digital therapeutic.
76
Operating Expenses
We classify operating expenses into three main categories: (i) research and development (ii) sales and marketing and (iii) general and administrative.
Research and Development
Our research and development expenses consist of external and internal expenses incurred in connection with our research activities and development programs. These expenses include external expenses, including expenses associated with contract research organizations engaged to manage and conduct clinical trials; and other research and development expenses associated with software development and licenses, and other external development spend. Additionally, our research and development expenses include internal personnel expenses, including expenses for salaries, benefits and stock-based compensation, and allocation of certain overhead expenses.
Research and development costs incurred to develop software and our platform for internal use are capitalized and separately presented on the balance sheet as capitalized software development costs. Costs incurred during the preliminary planning and evaluation stage of the project are expensed as incurred. Costs incurred during the application development stage of the project are capitalized. To date, the majority of these expenses have been incurred to advance our lead product candidate, BT-001.
We expect our research and development expenses to increase substantially for the foreseeable future as we continue to invest in research and development activities related to developing our platform and our product candidates, as our product candidates advance into later stages of development, and as we continue to conduct clinical trials. The successful development of our platform and our product candidates is highly uncertain. As a result, we are unable to determine the duration and completion costs of our research and development projects or when and to what extent we will generate revenue from the commercialization and sale of any of our product candidates.
Sales and Marketing
Sales and marketing expenses consist primarily of advertising and public relations costs and consulting services. We expect our sales and marketing expenses to increase for the foreseeable future as we prepare to prepare for commercialization of BT-001. Our sales and marketing efforts are expected to focus on targeting patients and primary care physicians through general awareness and branded promotional activities. We expect to incur significant investments in building a primary care sales force, and our plan and expectation is to have recruited and deployed such sales force during the first year of commercialization of our initial product candidate.
General and Administrative
General and administrative expenses consist primarily of personnel-related costs and professional services including legal, recruiting, audit and accounting services. Personnel-related costs consist of salaries, benefits, and stock-based compensation. We expect our general and administrative expenses to increase for the foreseeable future due to anticipated increases in headcount to advance our product candidates and as a result of operating as a public company, including expenses related to compliance with the rules and regulations of the SEC, additional insurance expenses, investor relations activities and other administrative and professional services.
Interest Expense, Net
Interest expense, net primarily consists of interest expense related to convertible notes and long-term debt entered into in 2021.
77
Results of Operations
Comparisons of the Years Ended December 31, 2021 and 2020
The following table summarizes our results of operations for the periods presented:
|
|
Twelve Months Ended, December 31, |
|
|||||||||||||
|
|
2021 |
|
|
2020 |
|
|
$ Change |
|
|
% Change |
|
||||
Revenue |
|
$ |
— |
|
|
$ |
8 |
|
|
$ |
(8 |
) |
|
N/M |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Research and development |
|
|
19,436 |
|
|
|
3,660 |
|
|
|
15,776 |
|
|
|
431 |
% |
Sales and marketing |
|
|
2,336 |
|
|
|
216 |
|
|
|
2,120 |
|
|
|
981 |
% |
General and administrative |
|
|
8,788 |
|
|
|
2,455 |
|
|
|
6,333 |
|
|
|
258 |
% |
Total operating expenses |
|
$ |
30,560 |
|
|
$ |
6,331 |
|
|
$ |
24,229 |
|
|
|
383 |
% |
Loss from operations |
|
|
(30,560 |
) |
|
|
(6,323 |
) |
|
|
(24,237 |
) |
|
|
383 |
% |
Interest expense, net |
|
|
(185 |
) |
|
|
(100 |
) |
|
|
(85 |
) |
|
|
85 |
% |
Gain on loan forgiveness |
|
|
647 |
|
|
|
- |
|
|
|
647 |
|
|
|
100 |
% |
Change in fair value of SAFEs |
|
|
(10,390 |
) |
|
|
189 |
|
|
|
(10,579 |
) |
|
N/M |
|
|
Loss before provision for income taxes |
|
|
(40,488 |
) |
|
|
(6,234 |
) |
|
|
(34,254 |
) |
|
|
549 |
% |
Provision for (benefit from) income taxes |
|
|
(153 |
) |
|
|
153 |
|
|
|
(306 |
) |
|
|
-200 |
% |
Net loss |
|
$ |
(40,335 |
) |
|
$ |
(6,387 |
) |
|
$ |
(33,948 |
) |
|
|
532 |
% |
N/M — The percentage change is not meaningful
Research and Development Expenses
Research and development expenses were $19,436 for the year ended December 31, 2021, compared to $3,660 for the year ended December 31, 2020, representing an increase of $15,776. The increase was primarily due to a $11,218 increase in costs incurred for clinical trials in 2021 and a $4,268 increase, net of capitalized costs, in personnel related costs as additional full-time personnel were hired within clinical research, product design and engineering.
Sales and Marketing Expenses
Sales and marketing expenses were $2,336 for the year ended December 31, 2021, compared to $216 for the year ended December 31, 2020, representing an increase of $2,120. The overall increase in sales and marketing expenses was primarily related to an increase of $583 in consulting fees, $691 increase in personnel related expenses and $374 for marketing and advertising related expenses as we prepare for product commercialization.
General and Administrative Expenses
General and administrative expenses were $8,788 for the year ended December 31, 2021, compared to $2,455 for the year ended December 31, 2020, representing an increase of $6,333. The overall increase in general and administrative expenses was primarily related to an increase of $3,075 in personnel related costs, an increase of $1,631 in outside services including consulting, audit, and legal fees related to the business combination and an increase in business related insurance of $751.
Interest Expense, Net
Interest expense, net was $185 for the year ended December 31, 2021, compared to $100 for the year ended December 31, 2020, representing an increase of $85. The increase in interest expense, net was the result of interest expense incurred on the new secured term loan agreement with Hercules Capital.
Change in Fair Value of SAFEs
The expense related to the change in fair value of our SAFEs was $10,390 for the year ended December 31, 2021, compared to a gain of $189 for the year ended December 31, 2020. The increase in expense was the result of the issuance and subsequent change in fair value of the SAFEs during the year ended December 31, 2021.
Gain on Loan Forgiveness
On May 9, 2020 (the “Origination Date”), the Company received $640 in aggregate loan proceeds (the “PPP Loan”) from Celtic Bank Corporation (the “Lender”) pursuant to the Paycheck Protection Program established under the Coronavirus Aid, Relief, and Economic Security Act of 2020 (the CARES Act"). In May 2021, the Company received approval of loan forgiveness and recorded a gain on loan forgiveness of $647.
78
Liquidity and Capital Resources
Since our inception through September 30, 2021, our operations had been financed primarily by the sale of convertible promissory notes, sale of SAFEs and the sale and issuance of Series Seed and Series A preferred units, which has resulted in net proceeds of approximately $46,132.
On April 6, 2021, we entered into a merger agreement with MCAD. In connection with the merger agreement, MCAD entered into Subscription Agreements with certain institutional and accredited investors, pursuant to which, among other things, MCAD agreed to issue and sell, in a private placement immediately prior to the closing of the Business Combination, an aggregate of 5,000,000 PIPE Shares. On October 28, 2021, we completed the merger with MCAD. We raised $59,000 in funding upon the completion of the merger with MCAD. Under the merger Agreement, MCAD acquired all of the outstanding shares of Legacy BTX in exchange for 15,174,729 shares of MCAD.
On August 18, 2021, we entered into a $50,000 secured term loan agreement with Hercules Capital. The term loan has a maturity date of August 1, 2025, which can be extended to February 1, 2026, and is secured by substantially all of our assets. Payments due for the term loan are interest-only until March 1, 2023 (subject to extension to September 1, 2023 or September 1, 2024 upon the achievement of certain milestones), after which principal shall be repaid in equal monthly installments. Interest is payable monthly in arrears. The outstanding principal bears interest at the greater of (a) 8.95% or (b) 8.95% plus the prime rate minus 3.25%. Prepayment of the outstanding principal is permitted under the secured term loan agreement and subject to certain prepayment fees. The Company incurred $518 of debt issuance costs related to the borrowings under the secured term loan agreement. Debt issuance costs are being amortized through the maturity date of the secured term loan and are reported as a direct reduction of long-term debt on the balance sheet. Amortization expense, included in interest expense, net on the accompanying statements of operations and comprehensive loss totaled $23 and zero for the years ended December 31, 2021 and 2020, respectively. In addition, we will be required to pay an end of term charge of the greater of (a) $893 and (b) 5.95% of the aggregate outstanding principal upon repayment of the loan. The secured term loan agreement contains customary representations, warranties, non-financial covenants, and events of default. We are permitted to borrow the loans in four tranches based on the completion of certain milestones which include, as set forth more fully in the secured term loan agreement: (i) $15,000 upon the closing of the Business Combination, (ii) $10,000 when we achieve certain positive clinical trial results sufficient to submit a de-novo classification request with respect to BT-001, (iii) $10,000 when we have received FDA approval for such marketing of BT-001 for the improvement of glycemic control and initiated a pivotal trial for a new indication in people with type 2 diabetes and received, prior to March 15, 2023, net cash proceeds of at least $40,000 from equity financings, and (iv) $15,000 on or before June 15, 2023, subject to Hercules Capital' approval. In October 2021, we borrowed $10,000 under the secured term loan agreement.
Our primary use of cash is to fund operating expenses, which consist of research and development expenses related to our lead product candidate, BT-001, and preclinical programs, and to a lesser extent, general and administrative expenses. Cash used to fund operating expenses is impacted by the timing of when we pay these expenses, as reflected in the change in our outstanding accounts payable and accrued expenses.
We have incurred negative cash flows from operating activities and investing activities and significant losses from operations in the past. We expect to continue to incur operating losses at least for the next 12 months due to the investments that we intend to make in our business and, as a result, we may require additional capital resources to grow our business.
We expect to incur substantial expenses in the foreseeable future for the development and potential commercialization of our product candidates and ongoing internal research and development programs. At this time, we cannot reasonably estimate the nature, timing or aggregate amount of costs for our development, potential commercialization, and internal research and development programs. However, in order to complete our planned product development, and to complete the process of obtaining regulatory authorization or clearance for our product candidates, as well as to build the sales, marketing and distribution infrastructure that we believe will be necessary to commercialize our product candidates, if approved, we may require substantial additional funding in the future. In the event that additional financing is required from outside sources, we may not be able to raise it on terms acceptable to us, or at all. If we are unable to raise additional capital when desired, our business, results of operations, and financial condition would be adversely affected. These factors raise substantial doubt regarding the Company's ability to continue as a going concern.
79
Summary Statement of Cash Flows
The following table sets forth the primary sources and uses of cash and cash equivalents for the periods presented below:
|
|
Year Ended |
|
|
Year Ended |
|
||
|
|
December 31, |
|
|
December 31, |
|
||
|
|
2021 |
|
|
2010 |
|
||
Cash used in operating activities. |
|
$ |
(30,818 |
) |
|
$ |
(5,774 |
) |
Cash used in investing activities |
|
|
(1,071 |
) |
|
|
(2,305 |
) |
Cash provided by financing activities |
|
|
72,332 |
|
|
|
7,445 |
|
Net increase (decrease) in cash and cash equivalents |
|
$ |
40,443 |
|
|
$ |
(634 |
) |
Cash Used in Operating Activities
In 2021, cash used in operating activities was $30,818 which consisted of a net loss of $40,335 and a net change of $2,339 in our net operating assets and liabilities partially offset by $11,856 in non-cash charges. The net change in our operating assets and liabilities was primarily due a net increase in prepaid expenses and other assets of $4,613 partially offset by a net increase in accounts payable and accrued expenses of $2,274. The non-cash charges of $11,856 consisted of share-based compensation expense, deferred income taxes, depreciation and amortization expense, loss on the change in fair value of SAFEs and gain on PPP Loan forgiveness.
In 2020, net cash used in operating activities was $5,774 which consisted of a net loss of $6,387 partially offset by a net change of $306 in our net operating assets and liabilities and $307 in non-cash charges. The net change in our operating assets and liabilities was primarily due a net decrease in accounts payable and accrued expenses of $252 and a net decrease in prepaid expenses and other assets of $54. The non-cash charges of $307 consisted of share-based compensation expense, deferred income taxes, depreciation expense, loss on the write-off of property and equipment and change in fair value of SAFEs.
Cash Used in Investing Activities
In 2021, cash used in investing activities was $1,071 and was primarily related to capitalized internal-use software costs.
In 2020, cash used in investing activities was $2,305 and was primarily related to capitalized internal-use software costs.
Cash Provided by Financing Activities
In 2021, cash provided by financing activities was $72,332 consisting of $44,174 in net proceeds from the business combination and PIPE investment, $18,675 in net proceeds from the issuance of SAFEs and $9,482 in net proceeds from the issuance of long-term debt.
In 2020, cash provided by financing activities was $7,445 consisting of $3,650 in net proceeds from the issuance of convertible notes, $3,155 in net proceeds from the issuance of SAFEs and $640 from proceeds from the Payroll Protection Program note.
Contractual Obligations and Commitments
Contractual obligations are cash amounts that we are obligated to pay as part of certain contracts that we have entered into during the normal course of business. We terminated our lease on August 31, 2020, and as such, we do not have any contractual obligations and other commitments as of December 31, 2021.
Off-Balance Sheet Arrangements
Since the date of our incorporation, we have not engaged in any off-balance sheet arrangements.
80
Reclassification
Certain prior year amounts have been reclassified for consistency with the current year presentation. These reclassifications had no effect on the reported results of operations. An adjustment has been made to the Statement of Operations and Comprehensive Loss for fiscal year ended December 31, 2020 to reclassify $682 thousand of cost of sales into research and development expense to align with industry standards. This change in classification does not affect previously reported net loss in the Statement of Operations and Comprehensive Loss.
Critical Accounting Policies, Significant Judgments and Use of Estimates
Our financial statements have been prepared in accordance with generally accepted accounting principles in the United States. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities and expenses, as well as the related disclosure of contingent assets and liabilities as of the date of the financial statements. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources.
Actual results may differ from these estimates under different assumptions or conditions. We believe that the accounting policies discussed below are critical to understanding our historical and future performance, as these policies relate to the more significant areas involving management’s judgments and estimates.
While our significant accounting policies are described in the notes to our financial statements, we believe that the following critical accounting policies are most important to understanding and evaluating our reported financial results.
Simple Agreements for Future Equity (“SAFE”)
We classify SAFEs as contingently redeemable liabilities under ASC 480 as a result of certain redemption provisions which may result in the SAFEs being redeemed for cash or other assets upon a liquidity event with such events not solely within our control. Additionally, the SAFEs are settleable into a variable-number of shares of preferred stock, at a stated discount, upon a preferred stock financing event. As further discussed in footnote 9 of our financial statements for the period ending on December 31, 2020, we have determined that our preferred stock is contingently redeemable upon certain events not solely within our control. As a result, the SAFEs would potentially be settled in contingently redeemable shares with redemption of such shares being outside of our control.
The SAFEs are measured and recognized at fair value using a Monte Carlo valuation approach and are subject to re-measurement at each balance sheet date. The Monte Carlo valuation approach takes into consideration the probability of various events, including liquidity events and equity financing events, and places a value for each event. The fair value of SAFEs was determined to be $0 and $11,740 as of December 31, 2021 and December 31, 2020, respectively.
At the end of each reporting period, changes in fair value during the period are recognized and presented as a financial statement line item in the consolidated statements of operations and comprehensive loss.
Share-Based Compensation Expense
We account for share-based compensation expense by measuring and recognizing compensation expense for all share-based awards made to employees and non-employees based on estimated grant-date fair values.
Excluding performance-based stock awards, we recognize compensation costs on a straight-line basis over the requisite service period of the employee and non-employee, which is generally the option vesting term of four years. For performance-based awards, share-based compensation expense will be recognized when it is probable that the performance criteria will be achieved. We recognize actual forfeitures by reducing the share-based compensation expense in the same period as the forfeitures occur.
We estimate the fair value of stock options granted to employees and non-employees using the Black-Scholes option-pricing valuation model. The Black-Scholes model requires the input of subjective assumptions, including fair value of the underlying profit interest unit or stock award, expected term, expected volatility, risk-free interest rate, and expected dividend yield, which are described in greater detail below. Estimating the fair value of stock options and profit interest units as of the grant date using the Black-Scholes option pricing model is affected by assumptions regarding several complex variables. Changes in the assumptions can materially affect the fair value and ultimately how much share-based compensation expense is recognized. These inputs are subjective and generally require significant analysis and judgment to develop. These inputs are as follows:
81
We will continue to use judgment in evaluating the expected volatility, expected terms, and interest rates utilized for our stock-based compensation expense calculations on a prospective basis.
Prior to our conversion into a Delaware corporation in August 2020, we had granted profit interest units to employees and non-employees. In August 2020, in conjunction with the conversion of the company to a Delaware corporation, the profits interest units were converted to common stock of Legacy BTX, and the common stock issued in exchange for the profit interest units continue to be subject to the same vesting conditions as the previously granted profit interest. We accounted for the conversion of profit interest units into common stock as a modification under ASC 718.
The profits interest units were common units with a profits interest distribution threshold and give the holder a right to share in the appreciation in the value of Legacy BTX and share in any distributions of profits. The profit interest unit awards generally vest over four years and automatically in full upon a sale of the business. The grantees had the right to retain vested units upon termination of employment or when non-employees ceasing to provide services or goods to us. Prior to the conversion, we had not made distributions to the holders of the profits interest units.
Determination of Fair Value of Profit Interests and Common Stock
As there has been no public market for our profit interests or common stock prior to the date of the business combination, the estimated fair value of our common stock has been determined by our Board as of the date of each stock award grant, with input from management, considering contemporaneous independent third-party valuations of our profit interests and common stock, and the Board’s assessment of additional objective and subjective factors that it believed were relevant and which may have changed from the date of the most recent valuation through the date of the grant. These independent third-party valuations were performed in accordance with the guidance outlined in the American Institute of Certified Public Accountants’ Accounting and Valuation Guide, Valuation of Privately-Held-Company Equity Securities Issued as Compensation, or the Practice Aid. The methodology to determine the fair value of our common stock included estimating the fair value of the enterprise using a market approach, which estimates the fair value of a company by including an estimation of the value of the business based on guideline public companies under a number of different scenarios. The assumptions used to determine the estimated fair value of our common stock are based on numerous objective and subjective factors, combined with management judgment, including external market conditions affecting the pharmaceutical and biotechnology industry and trends within the industry; our stage of development; the rights, preferences and privileges of our redeemable convertible preferred stock relative to those of our common stock; the prices at which we sold shares of our redeemable convertible preferred stock; our financial condition and operating results, including our levels of available capital resources; the progress of our research and development efforts and business strategy; the timing and probability of future financings; equity market conditions affecting comparable public companies; general U.S. market conditions; and the lack of marketability of our common stock.
82
The Practice Aid identifies various available methods for allocating enterprise value across classes and series of capital stock to determine the estimated fair value of common stock at each valuation date. Given the absence of a public trading market of our common stock, the Board considered numerous subjective and objective factors to determine the best estimate of fair value of our profit interests and common stock underlying the stock options granted to our employees and non-employees.
The grant date fair value of our common stock was determined using the Option Pricing Method ("OPM"). Under the OPM, shares are valued by creating a series of call options with exercise prices based on the liquidation preferences and conversion terms of each equity class. The estimated fair values of the preferred and common stock are inferred by analyzing these options. This method is appropriate to use when the range of possible future outcomes is so difficult to predict that estimates would be highly speculative, and dissolution or liquidation is not imminent.
Application of the OPM involves the use of estimates, judgment, and assumptions that are highly complex and subjective, such as those regarding time from valuation date to the option or incentive unit expiration, volatility of the underlying stock or incentive unit, and an assumption for a discount for lack of marketability. Changes in any or all of these estimates and assumptions, or the relationships between those assumptions, impact our valuations as of each valuation date and may have a material impact on the valuation of common stock. The assumptions underlying these valuations represent our management’s best estimate, which involve inherent uncertainties and the application of management judgment. As a result, if factors or expected outcomes change and we use significantly different assumptions or estimates, our stock-based compensation expense could be materially different.
Now that the Business Combination is completed, we determine the fair value of our common stock based on the closing price of our common stock on the date of grant.
JOBS Act
We are an “emerging growth company” as defined in the JOBS Act. The JOBS Act permits emerging growth companies to take advantage of an extended transition period to comply with new or revised accounting standards, delaying the adoption of these accounting standards until they would apply to private companies. We have elected to use this extended transition period under the JOBS Act until the earlier of the date we (i) are no longer an emerging growth company or (ii) affirmatively and irrevocably opt out of the extended transition period provided in the JOBS Act. As a result, our financial statements may not be comparable to companies that comply with new or revised accounting pronouncements as of public company effective dates.
We could be an emerging growth company until the last day of the fiscal year ending after the fifth anniversary of our IPO, although circumstances could cause us to lose that status earlier, including if we become a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act or if we have total annual gross revenue of $1.07 billion or more during any fiscal year before that time, in which cases we would no longer be an emerging growth company as of the following December 31 or, if we issue more than $1.0 billion in non-convertible debt during any three year period before that time, we would cease to be an emerging growth company immediately.
Recently Adopted Accounting Pronouncements
See Note 2 to our annual financial statements for the year ended December 31, 2021.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Interest Rate Risk
Our cash and cash equivalents as of December 31, 2021 consisted of readily available checking funds and a money market account. Our cash and cash equivalents as of December 30, 2020 consisted of readily available checking funds. We do not believe that our cash or cash equivalents have significant risk of default or illiquidity. While we believe our cash and cash equivalents do not contain excessive risk, we cannot provide absolute assurance that in the future any investment will not be subject to adverse changes in market value. In addition, we maintain significant amounts of cash and cash equivalents at one financial institution that is in excess of federally insured limits.
83
Additionally, on August 14, 2020, upon the conversion of Legacy BTX to a Delaware corporation, Legacy BTX's convertible promissory notes and accrued interest were exchanged for an equivalent amount of SAFE agreements. As of December 31, 2020 we did not have any interest rate risk. As of December 31, 2021 we have interest rate risk related to the Hercules Debt. As the prime interest rate increases, our interest rate will increase. A hypothetical 1% increase in the prime rate will result in a $100 increase in interest expense.
Effects of Inflation
Inflation generally affects us by increasing our cost of labor and clinical trial costs. We do not believe that inflation had a material effect on our financial statements.
Effects of Exchange Rate Fluctuations
We do not believe that exchange rate fluctuations had a significant impact on our results of operations for any periods presented herein.
84
Item 8. Financial Statements and Supplementary Data.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
F-1
Report of Independent Registered Public Accounting Firm
The Shareholders and Board of Directors
Better Therapeutics, Inc.
San Francisco, California
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Better Therapeutics, Inc. (the Company) as of December 31, 2021 and 2020, the related consolidated statements of operations and comprehensive loss, convertible preferred stock and stockholders'/members' equity (deficit), and cash flows for the years then ended, and the related notes to the financial statements (collectively, the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and 2020, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The accompanying financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has suffered recurring losses from operations and has a substantial accumulated deficit. This raises substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters also are described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/
We have served as the Company's auditor since 2021.
March 28, 2022
F-2
BETTER THERAPEUTICS, INC.
BALANCE SHEETS
(in thousands, except share data)
|
|
December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
ASSETS |
|
|
|
|
|
|
||
Current assets: |
|
|
|
|
|
|
||
Cash and cash equivalents |
|
$ |
|
|
$ |
|
||
Prepaid expenses |
|
|
|
|
|
|
||
Other current assets |
|
|
|
|
|
|
||
Total current assets |
|
|
|
|
|
|
||
Capitalized software development costs, net |
|
|
|
|
|
|
||
Property and equipment, net |
|
|
|
|
|
|
||
Other long-term assets |
|
|
|
|
|
|
||
Total Assets |
|
$ |
|
|
$ |
|
||
LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT), AS ADJUSTED |
|
|
|
|
|
|
||
Current liabilities: |
|
|
|
|
|
|
||
Accounts payable |
|
$ |
|
|
$ |
|
||
Accrued payroll |
|
|
|
|
|
|
||
Other accrued expenses |
|
|
|
|
|
|
||
Total current liabilities |
|
|
|
|
|
|
||
Long-term debt, net of debt issuance costs |
|
|
|
|
|
|
||
Deferred tax liability |
|
|
|
|
|
|
||
Simple Agreements for Future Equity |
|
|
|
|
|
|
||
Total liabilities |
|
|
|
|
|
|
||
(Note 17) |
|
|
|
|
|
|
||
Stockholders' equity (deficit) |
|
|
|
|
|
|
||
Common stock, $ |
|
|
|
|
|
|
||
Additional paid-in capital |
|
|
|
|
|
|
||
Accumulated deficit |
|
|
( |
) |
|
|
( |
) |
Total Stockholders' Equity (Deficit) |
|
|
|
|
|
( |
) |
|
Total Liabilities and Stockholders’ Equity (Deficit) |
|
$ |
|
|
$ |
|
||
The accompanying notes are an integral part of these financial statements.
F-3
BETTER THERAPEUTICS, INC.
STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(in thousands, except share and per share data)
|
|
Years Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Revenue |
|
$ |
— |
|
|
$ |
|
|
Operating expenses: |
|
|
|
|
|
|
||
Research and development |
|
|
|
|
|
|
||
Sales and marketing |
|
|
|
|
|
|
||
General and administrative |
|
|
|
|
|
|
||
Total operating expenses |
|
|
|
|
|
|
||
Loss from operations |
|
|
( |
) |
|
|
( |
) |
Interest expense, net |
|
|
( |
) |
|
|
( |
) |
Gain on Loan Forgiveness |
|
|
|
|
|
— |
|
|
Change in fair value of SAFEs |
|
|
( |
) |
|
|
|
|
Loss before provision for (benefit from) income taxes |
|
|
( |
) |
|
|
( |
) |
Provision for (benefit from) income taxes |
|
|
( |
) |
|
|
|
|
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
Cumulative preferred dividends allocated to Series A Preferred Shareholders |
|
|
— |
|
|
|
( |
) |
Net loss attributable to common shareholders, basic and diluted |
|
$ |
( |
) |
|
$ |
( |
) |
Net loss per share attributable to common shareholders, basic and diluted |
|
$ |
( |
) |
|
$ |
( |
) |
Weighted-average shares used in computing net loss per share |
|
|
|
|
|
|
||
F-4
BETTER THERAPEUTICS, INC.
(in thousands, except share data)
|
|
Series Seed |
|
|
Series A |
|
|
Series Seed |
|
|
Series A |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
|||||||||||||||||||||||||||
|
|
Convertible |
|
|
Convertible |
|
|
Convertible |
|
|
Convertible |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional |
|
|
|
|
|
Accumulate |
|
|||||||||||||||||||||||||||
|
|
Preferred Units |
|
|
Preferred Units |
|
|
Preferred Stock |
|
|
Preferred Stock |
|
|
|
Common Units |
|
|
Common Stock |
|
|
Paid-in |
|
|
Accumulated |
|
|
Stockholder |
|
|||||||||||||||||||||||||||||||||
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Equity (Deficit) |
|
|||||||||||||||
Balance as of December |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
$ |
— |
|
|
|
|
|
|
$ |
|
|
|
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
( |
) |
||||||
Retroactive application of the recapitalization due to the business combination: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
Preferred Stock |
|
|
( |
) |
|
$ |
( |
) |
|
|
( |
) |
|
$ |
( |
) |
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
$ |
— |
|
|
|
|
— |
|
|
$ |
— |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
||||
Common Stock |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||||||||||
Balance as of December 31, 2019, as adjusted |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|||
Net Loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Issuance and conversion of profits interest units to common stock |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|||||||||||
Conversion of profits interest units to restricted stock |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|||||||||||
Share-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Balance as of December 31, 2020, as adjusted |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|||
Net Loss |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
Forfeiture of restricted stock |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
Issuance of common stock in connection with business combination, net of issuance costs of $ |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
|
||||
Exercise of Common Stock |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
Share based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Balance as of December 31, 2021 |
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
$ |
— |
|
|
|
|
— |
|
|
$ |
— |
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
The accompanying notes are an integral part of these financial statements.
F-5
BETTER THERAPEUTICS, INC.
STATEMENTS OF CASH FLOWS
(in thousands)
|
|
Years Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
CASH FLOWS FROM OPERATING ACTIVITIES |
|
|
|
|
|
|
||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
||
Depreciation and amortization |
|
|
|
|
|
|
||
Change in fair value of SAFEs |
|
|
|
|
|
( |
) |
|
Loss on write-off of property and equipment |
|
|
|
|
|
|
||
Share-based compensation expense |
|
|
|
|
|
|
||
Deferred income taxes |
|
|
( |
) |
|
|
|
|
Gain on loan forgiveness |
|
|
( |
) |
|
|
— |
|
Changes in operating assets and liabilities |
|
|
|
|
|
|
||
Prepaid expenses and other assets |
|
|
( |
) |
|
|
|
|
Accounts payable |
|
|
|
|
|
|
||
Accrued expenses and other liabilities |
|
|
|
|
|
|
||
Net cash used in operating activities |
|
|
( |
) |
|
|
( |
) |
CASH FLOWS FROM INVESTING ACTIVITIES |
|
|
|
|
|
|
||
Purchase of property and equipment |
|
|
( |
) |
|
|
( |
) |
Capitalized internal-use software costs |
|
|
( |
) |
|
|
( |
) |
Net cash used in investing activities |
|
|
( |
) |
|
|
( |
) |
CASH FLOWS FROM FINANCING ACTIVITIES |
|
|
|
|
|
|
||
Proceeds from payroll protection program note |
|
|
|
|
|
|
||
Proceeds from issuance of convertible notes |
|
|
|
|
|
|
||
Proceeds from issuance of SAFE notes |
|
|
|
|
|
|
||
Proceeds from business combination and PIPE Investment |
|
|
|
|
|
|
||
Payment of costs directly attributable to the issuance of common stock in connection with business combination and PIPE investment |
|
|
( |
) |
|
|
|
|
Proceeds from issuance of long-term debt |
|
|
|
|
|
|
||
Debt issuance costs |
|
|
( |
) |
|
|
|
|
Proceeds from exercise of stock options |
|
|
|
|
|
|
||
Net cash provided by financing activities |
|
|
|
|
|
|
||
Net change in cash and cash equivalents |
|
|
|
|
|
( |
) |
|
Cash and cash equivalents, beginning of period |
|
|
|
|
|
|
||
Cash and cash equivalents, end of period |
|
$ |
|
|
$ |
|
||
Supplemental disclosures of cash flow information: |
|
|
|
|
|
|
||
Cash paid for Interest |
|
$ |
|
|
$ |
|
||
Supplemental disclosures of noncash investing and financing activities |
|
|
|
|
|
|
||
Conversion of convertible notes to SAFE notes |
|
$ |
|
|
$ |
|
||
The accompanying notes are an integral part of these financial statements.
F-6
BETTER THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS
Better Therapeutics Inc. (“we”, “us”, “the Company”, or “Better”), a Delaware corporation, was founded in April 2015 as Nutrition Development Group, LLC. In August 2016, we changed our name to Farewell LLC and in January 2018 we changed our name to Better Therapeutics LLC. On August 14, 2020, we converted to a Delaware corporation. As a result of the conversion to a Delaware corporation, as discussed below, all common units, Series Seed Preferred Units and Series A Preferred Units converted to an equivalent number of common stock, Series Seed Preferred Stock and Series A Preferred Stock. In addition, all outstanding profits interest units were converted to common stock, and all outstanding convertible promissory notes were converted to simple agreements for future equity (“SAFEs”). On October 28, 2021, Mountain Crest Acquisition Corp. II, a Delaware corporation ("MCAD") merged with and into Better Therapeutics with Better Therapeutics surviving as a wholly-owned subsidiary of the Company with the new name Better Therapeutics, Inc. MCAD consummated the acquisition of all the issued and outstanding shares of the former Better Therapeutics, Inc ("Legacy BTX"). Accordingly, for accounting purposes, the financial statements of the combined entity represent a continuation of the financial statements of Better with the business combination being treated as the equivalent of Better Therapeutics issuing stock for the net assets of MCAD, accompanied by a recapitalization. The net assets of MCAD are stated at fair value with no goodwill or other intangible assets recorded. Operations prior to the merger are those of Better Therapeutics.
As a result of the Business Combination, the shares and corresponding capital amounts and loss per share related to Legacy BTX's outstanding convertible preferred stock and common stock prior to the Business Combination have been retroactively restated to reflect the Exchange Ratio established in the Merger Agreement. For additional information on the Business Combination, refer to Note 3 of these financial statements.
Better Therapeutics has developed a platform of FDA-regulated, software-based, Prescription Digital Therapeutics ("PDTs") for treating diabetes, heart disease, and other cardiometabolic conditions. Our PDTs deliver a novel form of cognitive behavioral therapy that enables changes in neural pathways of the brain so that lasting changes in behavior become possible. Addressing the underlying causes of these diseases has the potential to dramatically improve patient health and lower healthcare costs. Our current clinical development candidates are intended to treat cardiometabolic diseases, including type 2 diabetes, hypertension, hyperlipidemia, non-alcoholic fatty liver disease ("NAFLD":, non-alcoholic steatohepatitis ("NASH") and chronic kidney disease ("CKD"). Our headquarters are in San Francisco, California.
The Company is in the development stage and our activities have consisted principally of raising capital and preforming research and development.
Liquidity and Capital Resources
Since inception we have incurred significant losses from operations. As of December 31, 2021, we had cash of $
We incurred a net loss of $
We have primarily funded our operations through the sale of preferred stock, convertible notes, SAFEs and funding from the merger with MCAD. The continued execution of our long-term business plan will require us to explore financing options such as issuance of equity or debt instruments. While we have historically been successful in obtaining equity financing, there can be no assurance that such additional financing, if necessary, will be available or, if available, that such financings can be obtained on satisfactory terms. These factors raise substantial doubt regarding the Company's ability to continue as a going concern.
F-7
At this time, there is significant uncertainty relating to the ongoing COVID-19 pandemic and the impact of related responses. Any impact of COVID-19 on our business, results of operations and financial condition will largely depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the geographic spread of the disease, the duration of the pandemic, travel restrictions and social distancing in the United States and other countries, business closures or business disruptions, the ultimate impact on financial markets and the global economy, and the effectiveness of actions taken in the United States and other countries to contain and treat the disease.
Basis of Presentation
The financial statements and accompanying notes have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”). Amounts are presented in thousands except share and per share information.
Reclassification
Certain prior year amounts have been reclassified for consistency with the current year presentation. An adjustment has been made to the Statement of Operations and Comprehensive Loss for fiscal year ended December 31, 2020 to reclassify $
Comprehensive Loss
For the years ended December 31, 2021 and 2020, there was no difference between comprehensive loss and net loss.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make certain estimates, judgments, and assumptions that affect the reported amounts of assets and liabilities and the related disclosures at the date of the financial statements, as well as the reported amounts of revenue and expenses during the periods presented. The estimates and assumptions used in the accompanying financial statements are based upon management’s evaluation of the relevant facts and circumstances. Such estimates, judgments, and assumptions include estimated costs for capitalized internal-use software, fair values of stock-based awards, valuation allowance for deferred tax assets and fair value of SAFEs. Actual results could be different from these estimates. To the extent there are material differences between these estimates, judgments, or assumptions and actual results, our financial statements will be affected.
Emerging Growth Company Status
We are an emerging growth company, as defined in the JOBS Act. The JOBS Act provides that an emerging growth company can take advantage of the extended transition period for complying with new or revised accounting standards. Thus, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to avail ourselves of this extended transition period and, as a result, we do not adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other public companies until required by private company accounting standards.
Concentration of Risk
Financial instruments that potentially subject us to credit risk consist principally of cash and cash equivalents. We maintain our cash primarily with domestic financial institutions of high credit quality, which may exceed federal deposit insurance corporation limits. We invest our cash equivalents in highly rated money market funds. We have not experienced any losses in such accounts. We believe we are not exposed to any significant credit risk on cash and cash equivalents and perform periodic evaluations of the credit standing of such institutions.
F-8
Fair Value Measurements
The carrying value of our financial instruments, including cash equivalents, accounts payable, accrued liabilities and notes payable approximates fair value due to their short-term nature. The Company’s investment portfolio consists of money market funds, which are carried at fair value. The company has determined the carrying value to be equal to the fair value and has classified these investments as Level 1 financial instruments.
We measure financial assets and liabilities at fair value at each reporting period using a fair value hierarchy that requires the use of observable inputs and minimizes the use of unobservable inputs. We define fair value as the price that would be received from selling an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. Fair value is estimated by applying the following hierarchy, which prioritizes the inputs used to measure fair value into three levels and bases the categorization within the hierarchy upon the lowest level of input that is available and significant to the fair value measurement:
Certain SAFEs are classified as Level 3 financial instruments. The balance of the SAFEs are
Property and Equipment, Net
Property and equipment, net, are stated at cost, less accumulated depreciation. Depreciation is calculated using the straight-line method over the estimated useful lives of the assets, which are generally to
Useful lives for property and equipment are as follows:
Property and Equipment |
Estimated Useful Life |
|
Computer, equipment and software |
|
|
Furniture and fixtures |
|
Capitalized Internal-Use Software Costs
Costs incurred to develop software and our platform for internal use consist primarily of direct employee-related and third-party contractor costs and are accounted for pursuant to ASC 350-40, Internal Use Software. Costs incurred during the preliminary planning and evaluation stage of the project are expensed as incurred. Costs incurred during the application development stage of the project are capitalized and amortized over an estimated useful life of
Impairment of Long-Lived Assets
We review long-lived assets for impairment when circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of these assets is measured by a comparison of the carrying amounts to the sum of the future undiscounted cash flows the assets are expected to generate over the remaining useful lives of the assets. If a long-lived asset fails a recoverability test, we measure the amount by which the carrying value of the asset exceeds its fair value. There were no events or changes in business circumstances during the years ended December 31, 2021 and 2020 that indicated the carrying amounts of any long-lived assets were not fully recoverable.
F-9
Advertising Expense
We recognize advertising expenses as they are incurred, and such costs are included in sales and marketing expense in the statements of operations. During the years ended December 31, 2021 and 2020, advertising expense totaled $
Equity-Based Compensation
We account for equity-based compensation arrangements granted to employees in accordance with ASC 718, “Compensation: Stock Compensation”, by measuring the grant date fair value of the award and recognizing the resulting expense over the period during which the employee is required to perform service in exchange for the award. Equity-based compensation expense is only recognized for awards subject to performance conditions if it is probable that the performance condition will be achieved.
We account for equity-based compensation arrangements issued to non-employees using the fair value approach prescribed by ASU 2018-07, “Compensation-Stock Compensation (ASC 718): Improvements to Non-employee Share-Based Payment Accounting”. The value of non-employee equity-based compensation is measured at the grant date using a fair value-based measure.
We estimate the fair value of each equity-based award on the date of grant using the Black-Scholes option-pricing model. The determination of the fair value of each stock award using this option-pricing model is affected by our assumptions regarding a number of complex and subjective variables. These variables include, but are not limited to, the fair value of the common stock at the date of grant, the expected term of the awards, the expected stock price volatility over the term of the awards, risk-free interest rate, and dividend yield as follows:
Fair Value of Common Stock — Prior to the business combination given the absence of a public trading market, our board of directors considered numerous objective and subjective factors to determine the fair value of our common stock at each grant date. These factors included but were not limited to (i) contemporaneous third-party valuations of common stock; (ii) the prices for our redeemable convertible preferred stock sold to outside investors; (iii) the rights and preferences of redeemable convertible preferred stock relative to common stock; (iv) the lack of marketability of our common stock; (v)developments in the business; and (vi) the likelihood of achieving a liquidity event given prevailing market conditions. Subsequent to the business combination we determined the fair value of common stock based on the closing price of our common stock on the date of the grant.
Expected Term — The expected term represents the period that the equity-based awards are expected to be outstanding. We determine the expected term using the simplified method. The simplified method deems the term to be the average of the time-to-vesting and the contractual life of the options. For stock options granted to non-employees, the expected term equals the remaining contractual term of the option from the vesting date.
Expected Volatility — As we had no trading history for our common stock when we granted our option awards prior to the Business Combination, the expected volatility was estimated by taking the average historic price volatility for industry peers, consisting of several public companies in our industry that are either similar in size, stage, or financial leverage, over a period equivalent to the expected term of the awards. Due to our limited trading history, we will continue to determine expected volatility using estimate of industry peers.
Risk-Free Interest Rate — The risk-free interest rate is calculated using the average of the published interest rates of U.S. Treasury zero-coupon issues with maturities that are commensurate with the expected term.
Dividend Yield — The dividend yield assumption is
We account for forfeitures when they occur. For awards forfeited before completion of the requisite service period, previously recognized compensation cost is reversed in the period the award is forfeited.
F-10
Income Taxes
Prior to August 14, 2020, Legacy BTX was a limited liability company taxed as a partnership. The income and losses of the Legacy BTX flowed directly through to the members of the partnership. Accordingly,
We account for income taxes using the asset and liability method under which deferred tax assets and liabilities are determined based on the differences between the financial reporting and tax bases of assets and liabilities with consideration given to net operating losses and tax credit carryforwards. Deferred tax assets and liabilities are measured using the enacted tax rates that are expected to be in effect when the differences are expected to reverse.
We assess the likelihood that deferred tax assets will be recovered from future taxable income and a valuation allowance is established when necessary to reduce deferred tax assets to the amounts more likely than not expected to be realized. We adopted Accounting Standards Update (“ASU”) No. 2015-17, Income Taxes — Balance Sheet Classification of Deferred Taxes, and classified our deferred income taxes as non-current in the balance sheets.
We recognize and measure uncertain tax positions using a two-step approach. The first step is to evaluate the tax position taken or expected to be taken by determining if the weight of available evidence indicates that it is more likely than not that the tax position will be sustained in an audit, after resolution of any related appeals or litigation processes. The second step is to measure the tax benefit as the largest amount that is more than 50% likely to be realized upon ultimate settlement. Significant judgment is required to evaluate uncertain tax positions. We evaluate our uncertain tax positions on a regular basis. Our evaluations are based on a number of factors, including changes in facts and circumstances, changes in tax law, correspondence with tax authorities during the course of the audit, and effective settlement of audit issues.
Net Loss Per Share Attributable to Common Stockholders
Basic and diluted net loss per share attributable to common stock is presented in conformity with the two-class method required for participating securities. Under the two-class method, the net loss attributable to common stock is not allocated to the preferred stock as the holders of our convertible preferred stock did not have a contractual obligation to share in our losses. Under the two-class method, net loss is attributed to common stock and participating securities based on their participation rights. Basic net loss per share attributable to common stock is computed by dividing the net loss attributable to common stock by the weighted-average number of shares of common stock outstanding during the period. Cumulative dividends attributable to participating securities are subtracted from net loss in determining net loss attributable to common stockholders. As we have reported net losses for all periods presented, all potentially dilutive securities are antidilutive and, accordingly, basic net loss per share equals diluted net loss per share.
Revenue Recognition
On January 1, 2020, we adopted the requirements of Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers (Topic 606) (“ASC 606”). ASC 606 establishes a principle for recognizing revenue upon the transfer of promised goods or services to customers, in an amount that reflects the expected consideration received in exchange for those goods or services. The adoption of ASC 606 also requires the adoption of ASC Subtopic 340-40, Other Assets and Deferred Costs-Contracts with Customers, which provides for the deferral of certain incremental costs of obtaining a contract with a customer. Collectively, references to ASC 606 used herein refer to both ASC 606 and Subtopic 340-40. The core principle of ASC 606 is to recognize revenue to depict the transfer of promised goods or services to clients in an amount that reflects the consideration the entity expects to be entitled in exchange for those goods or services. This principle is achieved through applying the following five-step approach:
F-11
Our historical revenue is derived from pilot agreements with customers to provide a digital therapeutic program that includes mobile apps and health coaching services. Clients are private health insurance providers that have contracted with us to offer our solution as a free benefit offering to their covered population.
The monthly fees are recognized as earned based on the end user’s health outcomes and app usage. These pilot agreements ended during 2020.
Segment Reporting
We operate as
Recent Accounting Pronouncements
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842), which modifies lease accounting for lessees to increase transparency and comparability by recording lease assets and liabilities for operating leases and disclosing key information about leasing arrangements. In July 2018, the FASB issued ASU No. 2018-10, Codification Improvements to Topic 842, Leases, and ASU No. 2018-11, Leases (Topic 842), Targeted Improvements, which affect certain aspects of the previously issued guidance. In December 2018, the FASB issued ASU No. 2018-20, Narrow-Scope Improvements for Lessor, Leases (Topic 842), which provides guidance on sales tax and other taxes collected from lessees. In December 2019, the FASB issued ASU No. 2019-01, Codification Improvements to Topic 842, Leases, which affect certain aspects of the previously issued guidance. Amendments include an additional transition method that allows entities to apply the new standard on the adoption date and recognize a cumulative effect adjustment to the opening balance of retained earnings, as well as a new practical expedient for lessors. Under the JOBS Act, we have elected to avail ourselves of the extended transition period and, as a result, we will adopt this standard on January 1, 2022 and it will not have a material impact on our financial statements.
In June 2018, the FASB issued ASU No. 2018-07, Improvements to Nonemployee Share-Based Payment Accounting. The standard simplifies the accounting for share-based payments granted to nonemployees for goods and services and aligns most of the guidance on such payments to the nonemployees with the requirements for share-based payments granted to employees. We
In August 2018, the FASB issued ASU 2018-13, Fair Value Measurement (Topic 820): Disclosure Framework-Changes to the Disclosure Requirements for Fair Value Measurement, which eliminates, adds and modifies certain disclosure requirements for fair value measurements as part of the FASB’s disclosure framework project. The new standard is effective for fiscal years beginning after December 15, 2019, with early adoption permitted, including interim reporting periods within those fiscal years. Our
In December 2019, the FASB issued ASU No. 2019-12, Simplifying the Accounting for Income Taxes (Topic 740). This ASU simplifies the accounting for income taxes by, among other things, eliminating certain existing exceptions related to the general approach in ASC 740 relating to franchise taxes, reducing complexity in the interim-period accounting for year-to-date loss limitations and changes in tax laws, and clarifying the accounting for transactions outside of business combination that result in a step-up in the tax basis of goodwill. The transition requirements are primarily prospective, and the effective date is January 1, 2021. Our
F-12
In August 2020, the FASB issued ASU 2020-06, Debt — Debt with Conversion and Other Options (ASC 470-20) and Derivatives and Hedging — Contracts in Entity’s Own Equity (ASC 815-40). ASU 2020-06 simplifies the accounting for certain financial instruments with characteristics of liabilities and equity, including convertible instruments and contracts on an entity’s own equity. The ASU 2020-06 is part of the FASB’s simplification initiative, which aims to reduce unnecessary complexity in GAAP. This ASU’s amendments are effective for fiscal years beginning after December 15, 2023, and interim periods within those fiscal years. The Company is currently evaluating the impact ASU 2020-06 will have on its financial statements.
On April 6, 2021, the Company entered into a merger agreement with MCAD, a special purpose acquisition company. In connection with the merger agreement, MCAD entered into subscription agreements (the “Subscription Agreements”) dated as of
On October 28, 2021, pursuant to the terms of the merger agreement, we completed the merger with MCAD. We raised $
We accounted for the business combination as a reverse recapitalization, which is the equivalent of Legacy BTX issuing stock for the net assets of MCAD, accompanied by a recapitalization, with MCAD treated as the acquired company for accounting purposes. The determination of MCAD as the “acquired” company for accounting purposes was primarily based on the fact that subsequent to the business combination, Legacy BTX has a majority of the voting power of the combined company, Legacy BTX will comprise all of the ongoing operations of the combined entity, a majority of the governing body of the combined company and Legacy BTXs' senior management will comprise all of the senior management of the combined company. The net assets of MCAD were stated at historical cost with no goodwill or other intangible assets recorded. Reported results from operations included herein prior to the business combination are those of Legacy BTX. The shares and corresponding capital amounts and loss per share related to Legacy BTXs' outstanding redeemable convertible preferred stock, redeemable convertible common stock and common stock prior to the business combination have been retroactively restated to reflect the exchange ratio established in the business combination of , or the Exchange Ratio.
In connection with the business combination, we incurred underwriting fees and other costs considered direct and incremental to the transaction totaling $
PIPE Financing (Private Placement)
Concurrent with the execution of the Business Combination Agreement, we entered into subscription agreement with MCAD. Pursuant to the Subscription Agreements, each PIPE Investor subscribed for and purchased, and MCAD issued and sold to such investors an aggregate of
Summary of Net Proceeds
The following table summarizes the elements of the net proceeds from the business combination as of December 31, 2021:
|
Recapitalization |
|
|
Cash - MCAD cash and cash held in trust |
$ |
|
|
Cash - Proceeds from PIPE Investment |
|
|
|
Less: underwriting fees and other offering costs |
|
( |
) |
Net proceeds from business combination |
$ |
|
|
|
|
|
|
In addition to the net proceeds disclosed above, we also assumed $
F-13
Summary of Shares Issued
The following table summarizes the number of shares of common stock outstanding immediately following the consummation of the business transaction:
|
Number of Shares |
|
|
MCAD shares and rights outstanding prior to the business combination |
|
|
|
Less: redemptions of MCAD shares prior to the business combination |
|
( |
) |
Common stock of MCAD |
|
|
|
Shares issued pursuant to the PIPE including transaction related shares |
|
|
|
Business combination and PIPE financing shares |
|
|
|
Conversion of Legacy BTX SAFEs to Common Stock |
|
|
|
Conversion of Legacy BTX Preferred Seed A to Common Stock |
|
|
|
Conversion of Legacy BTX Preferred Series A to Common Stock |
|
|
|
Conversion of Legacy BTX Common Stock into new common stock |
|
|
|
Total shares of Better Therapeutics Common Stock outstanding immediately following the business combination |
|
|
|
|
|
|
|
Property and equipment consisted of the follow:
|
|
December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Computer, equipment and software |
|
$ |
|
|
$ |
|
||
Furniture and fixtures |
|
|
|
|
|
|
||
Property and equipment |
|
|
|
|
|
|
||
Less: accumulated depreciation |
|
|
( |
) |
|
|
( |
) |
Property and equipment, net |
|
$ |
|
|
$ |
|
||
Depreciation expense for the years ended December 31, 2021 and 2020 was $
Capitalized internal use software and accumulated amortization were as follows:
|
December 31, |
|
|||||
|
2021 |
|
|
2020 |
|
||
Gross carrying amount |
$ |
|
|
$ |
|
||
Accumulated amortization |
|
( |
) |
|
|
— |
|
Capitalized internal-use software, net |
$ |
|
|
$ |
|
||
|
|
|
|
|
|
||
We have recorded amortization expense related to capitalized internal-use software of $
F-14
|
December 31, |
|
|||||
|
2021 |
|
|
2020 |
|
||
Due to service providers |
$ |
|
|
$ |
— |
|
|
Due to professionals |
|
|
|
|
— |
|
|
Other |
|
|
|
|
|
||
Accrued interest |
|
|
|
|
|
||
Other accrued liabilities |
$ |
|
|
$ |
|
||
|
|
|
|
|
|
||
In 2019, Legacy BTX issued $
On May 9, 2020 (the “Origination Date”), the Company received $
On August 18, 2021, we entered into a $
F-15
Future payments on long-term debt as of December 31, 2021 are as follows:
Fiscal year ending December 31, |
|
Amount |
|
|
|
2022 |
|
$ |
— |
|
|
2023 |
|
|
|
|
|
2024 |
|
|
|
|
|
2025 |
|
|
|
|
|
|
|
|
|
|
|
Unamortized debt issuance costs |
|
|
( |
) |
|
|
|
$ |
|
|
|
|
|
|
|
|
|
On August 14, 2020, upon the conversion of Legacy BTX to a Delaware corporation, $
The SAFEs include a provision allowing for cash redemption upon the occurrence of a change of control, the occurrence of which is outside the control of the Company. Therefore, the SAFEs are classified as marked-to-market liabilities pursuant to ASC 480 in other long-term liabilities.
On October 28, 2021 in connection with the business combination all SAFEs were converted to common stock.
The SAFEs were marked to fair value during 2021 resulting in a change in fair value reported as loss of $
On May 4, 2015, Legacy BTX entered into a Series Seed Preferred Unit Purchase Agreement to issue Series Seed Preferred Units to an investor for cash. The Company issued
On December 2, 2015, Legacy BTX entered into a Series A Preferred Unit Purchase Agreement to issue Series A Preferred Units to investors for cash. The Company issued
On August 14, 2020, the Company changed the corporate structure to a Delaware corporation. Upon the change in the corporate structure, each of the Series Seed Preferred Units and Series A Preferred Units were canceled and converted into a corresponding number of shares of Series Seed Preferred Stock and Series A Preferred Stock, $
F-16
In October 2021, in connection with the Business Combination all outstanding shares of Series Seed Preferred Stock and Series A Preferred Stock were converted into common stock. The total number of shares of preferred stock authorized and issued as of December 31, 2021 is
The conversion of these preferred shares has been retroactively restated to reflect the Exchange Ratio established in the Merger Agreement on the Statement of Convertible Preferred Stock and Stockholders'/Members' Equity (Deficit).
Common Stock
The Company has retroactively adjusted the shares issued and outstanding prior to April 6, 2021 to give effect to the conversion ratio established in the Merger Agreement to determine the number of shares of common stock into which they were converted.
On August 14, 2020, Legacy BTX changed the corporate structure to a Delaware corporation. Upon the change in the corporate structure, each of the Common Units were canceled and converted into a corresponding number of shares of Common Stock with a par value of $
On October 28, 2021 in connection with the Business Combination all existing outstanding shares of common stock were exchanged for new shares at a conversion ratio of
The Company measures and reports certain financial instruments as assets and liabilities at fair value on a recurring basis.
|
|
December 31, 2020 |
|
|||||||||||||
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Total |
|
||||
Liabilities |
|
|
|
|
|
|
|
|
|
|
|
|
||||
SAFE Agreements |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
The Company’s SAFE agreements issued are recorded at fair value in our balance sheet. The fair value of the Company’s SAFE agreements is based on significant inputs not observable in the market which cause the instrument to be classified as a Level 3 measurements with the fair value hierarchy. The valuation uses assumptions and estimates in a discounted cash flow model, under a future as-if converted value in a qualified round of financing and if paid out in a change in control, with payouts discounted to their present value using a discount rate that reflected the risk of the payoff to the holder. The future as-if converted values were estimated based on the fixed discount to the next round based on Management’s estimate of the next round timing, and the Monte Carlo method which was used to estimate the future enterprise value of the Company at a change in control event and the expected payment to the SAFE holders at each simulated enterprise value. The rate of return for the next financing event and liquidation event scenarios was
On October 28, 2021 in connection with the Business Combination all SAFEs were converted to common stock.
Changes in the fair value of the SAFE agreements are recognized in the statement of operations and comprehensive loss. The fair value of the Company’s SAFE agreements was
F-17
Series Seed Preferred Stock, Series A Preferred Stock, and common stock are participating securities in the calculation of loss per share as they participate in undistributed earnings on an as-if-converted basis. Basic and diluted earnings per share was the same for each period presented as the inclusion of all potential common stock outstanding would have been anti-dilutive.
The following table sets forth the computation of basic and diluted loss (in thousands, except for share and per share amounts):
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Net Loss |
|
$ |
( |
) |
|
$ |
( |
) |
Less: Cumulative preferred dividends allocated to Series A |
|
|
— |
|
|
|
( |
) |
Net loss attributable to common stockholders, basic and diluted |
|
|
( |
) |
|
|
( |
) |
Weighted-average shares of common stock outstanding |
|
|
|
|
|
|
||
Less: weighted-average shares of common stock subject to vesting |
|
|
( |
) |
|
|
( |
) |
Weighted-average shares of common stock outstanding used in the calculation of basic and diluted net loss per share attributable to shareholders |
|
|
|
|
|
|
||
Loss per share attributable to common shareholders, basic |
|
$ |
( |
) |
|
$ |
( |
) |
The following potentially dilutive securities have been excluded from the computation of diluted weighted average shares outstanding, as they would be antidilutive:
|
|
For the Year Ended |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
SAFE agreements |
|
|
— |
|
|
|
|
|
Stock Options |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
In August 2020, we adopted the Better Therapeutics, Inc. 2020 Stock Option and Grant Plan (the “2020 Plan”) to grant equity-based incentives to officers, directors, consultants and employees. The equity-based incentives include Incentive Stock Options, Non-Qualified Stock Options, Restricted Stock Awards, Unrestricted Stock Awards, and Restricted Stock Units. A total of
In October 2021, we adopted the Better Therapeutics OpCo. Inc., 2021 Stock Option and Grant Plan (the "2021 Plan") to grant equity based incentive to officers, directors, consultants and employees. The equity-based incentives include, Incentive Stock Options, Non-Qualified Stock Options, Stock appreciation rights, Restricted Stock Awards, Restricted Stock Units, Unrestricted Stock Awards, Cash-based Awards and Dividend Equivalent Rights. A total of
Stock Options
F-18
|
|
Options Outstanding |
|
|||||||||||||
|
|
|
|
|
|
|
|
Weighted |
|
|
|
|
||||
|
|
Shares |
|
|
Weighted- |
|
|
Average |
|
|
|
|
||||
|
|
Subject to |
|
|
Average |
|
|
Remaining |
|
|
Aggregate |
|
||||
|
|
Options |
|
|
Exercise |
|
|
Contractual |
|
|
Intrinsic |
|
||||
|
|
Outstanding |
|
|
Price |
|
|
Life (Years) |
|
|
Value |
|
||||
Balance as of December 31, 2020 |
|
|
|
|
$ |
|
|
|
|
|
|
— |
|
|||
Conversion of options due to business combination |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
Balance as of January 1, 2021, as converted |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
Granted |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Exercised |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
Forfeited |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
Balance as of December 31, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
Aggregate intrinsic value represents the difference between the exercise price and the fair value of the shares underlying common stock.
The weighted-average grant date fair value of stock options granted to employees during the years ended December 31, 2021 was $
The fair value of each option award granted to employees is estimated on the grant date using the Black- Scholes option pricing model. The Black-Scholes option pricing model requires the input of subjective assumptions, including the fair value of the underlying common stock, the expected term of the option, the expected volatility of the price of our common stock, risk-free interest rates, and the dividend yield of our common stock. The assumptions used to determine the fair value of the option awards represent our best estimates. These estimates involve inherent uncertainties and the application of our judgment. The related stock-based compensation expense is recognized on a straight-line basis over the requisite service period of the awards, which is generally
The Black-Scholes option pricing model assumptions used in evaluating our awards to employees are as follows:
|
|
Year Ended |
|
|
|
|
December 31, |
|
|
|
|
2021 |
|
|
Expected Term (Years) |
|
|
|
|
Expected Volatility |
|
|
% |
|
Risk-free interest rate |
|
|
% |
|
Dividend Yield |
|
|
|
|
Restricted Stock
The Company issued
Stock-based compensation expense for time-based restricted stock awards of $
F-19
Equity-Based Compensation Expense
Equity-based compensation expense in the statement of operations is summarized as follows:
|
|
Year Ended December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Research and development |
|
$ |
|
|
$ |
|
||
Sales and marketing |
|
|
|
|
|
- |
|
|
General and administrative |
|
|
|
|
|
|
||
Total equity-based compensation expense |
|
$ |
|
|
$ |
|
||
For the period ended December 31, 2021 and 2020, $
We recorded an income tax benefit of $
|
|
December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Current: |
|
|
|
|
|
|
||
Federal |
|
$ |
|
|
$ |
|
||
State |
|
|
( |
) |
|
|
|
|
Total current |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
||
Deferred: |
|
|
|
|
|
|
||
Federal |
|
|
( |
) |
|
|
|
|
State |
|
|
|
|
|
|
||
Total deferred |
|
|
( |
) |
|
|
|
|
Total provision for income taxes |
|
$ |
( |
) |
|
$ |
|
|
The reconciliation of federal statutory income tax rate to our effective income tax rate is as follows:
|
|
Year Ended |
|
|||||
|
|
December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Expected income tax benefit at the federal statutory rate |
|
$ |
( |
) |
|
$ |
( |
) |
State taxes, net of federal benefit |
|
|
( |
) |
|
|
( |
) |
Research and development credit, net |
|
|
— |
|
|
|
( |
) |
Deferred tax on conversion to a corporation |
|
|
— |
|
|
|
|
|
Non-deductible items |
|
|
|
|
|
|
||
Partnership loss |
|
|
— |
|
|
|
|
|
Other |
|
|
— |
|
|
|
|
|
Change in valuation allowance |
|
|
|
|
|
|
||
Total |
|
$ |
( |
) |
|
$ |
|
|
F-20
Significant components of our deferred tax assets are summarized as follows:
|
|
December 31, |
|
|||||
|
|
2021 |
|
|
2020 |
|
||
Deferred tax assets: |
|
|
|
|
|
|
||
Federal and state new operating loss carryforwards |
|
$ |
|
|
$ |
|
||
Research and development tax credits |
|
|
|
|
|
|
||
Depreciation and amortization |
|
|
|
|
|
|
||
Stock based compensation |
|
|
|
|
|
— |
|
|
Accruals and reserves |
|
|
|
|
|
|
||
Gross deferred tax assets |
|
$ |
|
|
|
|
||
Less Valuation allowance |
|
|
( |
) |
|
|
( |
) |
Net deferred tax assets |
|
$ |
|
|
$ |
|
||
Deferred tax liabilities: |
|
|
|
|
|
|
||
Capitalized internal use software |
|
|
( |
) |
|
|
( |
) |
Net deferred tax liabilities |
|
|
( |
) |
|
|
( |
) |
Net deferred tax liability |
|
$ |
— |
|
|
$ |
( |
) |
As of December 31, 2021, we had $
Management regularly assesses the ability to realize deferred tax assets recorded based upon the weight of available evidence, including such factors as recent earnings history and expected future taxable income on a jurisdiction-by-jurisdiction basis. In the event that the Company changes its determination as to the amount of realizable deferred tax assets, the Company will adjust its valuation allowance with a corresponding impact to the provision for income taxes in the period in which such determination is made. The Company’s management believes that, based on a number of factors, it is more likely than not, that all or some portion of the deferred tax assets will not be realized; and accordingly, for the year ended December 31, 2021, the Company has provided a valuation allowance against the Company’s U.S. net deferred tax assets. The net change in the valuation allowance for the year ended December 31, 2021 was an increase of $
The Internal Revenue Code of 1986, as amended, imposes restrictions on the utilization of net operating losses in the event of an “ownership change” of a corporation. Accordingly, a company’s ability to use net operating losses may be limited as prescribed under Internal Revenue Code Section 382 (“IRC Section 382”). Events which may cause limitations in the amount of the net operating losses that the Company may use in any one year include, but are not limited to, a cumulative ownership change of more than
On March 27, 2020, the Coronavirus Aid, Relief, and Economic Security Act (“CARES Act”) was passed into law. The CARES Act includes several significant business tax provisions including modification to the taxable income limitation for utilization of net operating losses incurred in 2019 and 2020, an increase to the limitation on deductibility of certain business interest expense, bonus depreciation for purchases of qualified improvement property and special deductions on certain corporate charitable contributions. The Company analyzed the provisions of the CARES Act and determined there was
Uncertain Tax Positions
We are required to inventory, evaluate, and measure all uncertain tax positions taken or to be taken on tax returns and to record liabilities for the amount of such positions that may not be sustained, or may only partially be sustained, upon examination by the relevant taxing authorities.
F-21
The following is a summary of the changes in the Company’s gross unrecognized tax benefits:
|
|
December 31, |
|
|
|
|
2020 |
|
|
Balance as of December 31, 2020 |
|
$ |
|
|
Increase related to tax position taken |
|
|
— |
|
Balance as of December 31, 2021 |
|
$ |
|
|
As of December 31, 2021, the total amount of gross unrecognized tax benefits was $
We file federal and state income tax returns in the U.S. For U.S. federal and state income tax purposes, the statute of limitations currently remains open for all years due to our NOL carryforwards. We are not currently under examination in any jurisdiction.
Operating Leases
We entered into an operating lease agreement for our office. We recognized the operating lease costs on a straight-line basis over the term of each agreement, considering provisions such as free or escalating base monthly rental payments or deferred payment terms. We record rent expense associated with operating lease obligations in operating expenses in the statements of operations. In August 2020, we negotiated a termination settlement of this office lease for $
Legal Matters
From time to time, we become involved in claims, vendor disputes and other legal matters arising in the ordinary course of business. We investigate these claims as they arise. Although claims are inherently unpredictable, we are currently not aware of any matters that, if determined adversely to us, would individually or taken together have a material adverse effect on our business, results of operations, financial position or cash flows. We record liabilities for legal and other contingencies when losses are probable and estimable.
Although the results of litigation and claims are inherently unpredictable, we have not recorded an accrual for such contingencies as we believe that there was not at least a reasonable possibility that we had incurred a material loss with respect to such loss contingencies as of December 31, 2021 and 2020.
In 2019, the Company issued $
In March 2021, Andy Armanino, the former chief executive of officer of Armanino LLP and close relative to the current chief executive officer of Armanino LLP joined the Company's board of directors. The Company used Armanino LLP for tax, valuation, and outsourced accounting services. During the years ended December 31, 2021 and 2020 the Company incurred $
F-22
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure.
On November 19, 2021, the Audit Committee of the Board (the “Audit Committee”) approved the dismissal of Marcum LLP (“Marcum”) as our independent registered public accounting firm, effective as of the filing on November 22, 2021 of our Quarterly Report on Form 10-Q for the quarter ended September 30, 2021.
On November 19, 2021, the Audit Committee approved the engagement of Elliott Davis, LLC (“Elliott Davis”) as our independent registered public accounting firm for the fiscal year ending December 31, 2021. That engagement was effective as of the filing on November 22, 2021 of our Quarterly Report on Form 10-Q for the quarter ended September 30, 2021.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our reports under the Securities Exchange Act of 1934, as amended, is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s ("SEC") rules and forms, and that such information is accumulated and communicated to our management, including our chief executive officer and chief financial officer, as appropriate, to allow timely decisions regarding required disclosure. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefit of controls must be considered relative to their costs. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management necessarily is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
As of the end of the period covered by the report, we carried out an evaluation, under the supervision and with the participation of our management, including our chief executive officer and chief financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)). Based on that evaluation, our chief executive officer and chief financial officer concluded that, as of December 31, 2021 our disclosure controls and procedures were effective at the reasonable assurance level.
MANAGEMENT’S REPORT ON INTERNAL CONTROL OVER FINANCIAL REPORTING
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Because of its inherent limitations, internal control over financial reporting may not prevent or detect all misstatements. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Our management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2021. The framework used by management in making that assessment was the criteria set forth in the document entitled “Internal Control – Integrated Framework 2013” issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on that assessment, our Chief Executive Officer and Chief Financial Officer has determined and concluded that, as of December 31, 2021, the Company’s internal control over financial reporting were effective.
As of June 30, 2021, we identified material weaknesses in our internal control over financial reporting related to the inaccurate accounting for the value of shares to be issued to the underwriters at the closing of our IPO, as well as inaccurate accounting for certain accrued expenses and prepaid expenses. We have implemented a remediation plan to remediate the material weakness.
This Annual Report does not include an attestation report of the Company’s registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by the Company’s registered public accounting firm pursuant to the permanent exemption of the Securities and Exchange Commission that permit the Company to provide only management’s report in this Annual Report.
Changes in Internal Control over Financial Reporting
There has been no change in our internal control over financial reporting that occurred during the most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
81
Item 9B. Other Information.
None.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not Applicable.
82
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
The information required by this Item 10 will be set forth in the “Directors,” “Executive Officers,” “Corporate Governance” and “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” sections of our definitive proxy statement relating to our 2022 annual meeting of shareholders (the “Proxy Statement”), which will be filed with the SEC within 120 days after the end of the year covered by this Annual Report.
Item 11. Executive Compensation.
The information required by this Item 11 will be set forth in the “Executive and Director Compensation” section of the Proxy Statement, which will be filed with the SEC within 120 days after the end of the year covered by this Annual Report.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required by this Item 12 will be set forth in the “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” section of the Proxy Statement, which will be filed with the SEC within 120 days after the end of the year covered by this Annual Report.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this Item 13 will be set forth in the “Certain Relationships and Related Person Transactions” and “Corporate Governance” sections of the Proxy Statement, which will be filed with the SEC within 120 days after the end of the year covered by this Annual Report.
Item 14. Principal Accounting Fees and Services.
Our independent registered accounting firm is Elliot Davis, LLC, Greenville, South Carolina, PCAOB ID Number
83
PART IV
Item 15. Exhibits, Financial Statement Schedules.
(2) Financial statement schedules have been omitted because they are either not required or not applicable or the information is included in the consolidated financial statements or the notes thereto.
(3) The exhibits required by Item 601 of Regulation S-K and Item 15(b) of this Annual Report are listed in the Exhibit Index below. The exhibits listed in the Exhibit Index are incorporated by reference herein.
(b) Exhibits
84
Exhibit Index
Exhibit Number |
|
Description |
2.1+ |
|
|
3.1 |
|
|
3.2 |
|
|
4.1 |
|
|
4.2 |
|
|
4.3 |
|
|
4.4* |
|
|
10.1† |
|
|
10.2† |
|
|
10.3† |
|
|
10.4† |
|
|
10.5† |
|
|
10.6† |
|
|
10.7† |
|
|
10.8† |
|
|
10.9† |
|
|
10.10† |
|
|
10.11† |
|
|
10.12† |
|
|
10.13† |
|
85
10.14† |
|
|
10.15 |
|
|
10.16 |
|
|
21.1 |
|
|
23.1* |
|
Consent of Elliott Davis, LLC, independent registered public accounting firm. |
31.1* |
|
|
31.2* |
|
|
32.1** |
|
|
32.2** |
|
|
101.INS |
|
Inline XBRL Instance Document – the instance document does not appear in the Interactive Data File because XBRL tags are embedded within the Inline XBRL document. |
101.SCH |
|
Inline XBRL Taxonomy Extension Schema Document |
101.CAL |
|
Inline XBRL Taxonomy Extension Calculation Linkbase Document |
101.DEF |
|
Inline XBRL Taxonomy Extension Definition Linkbase Document |
101.LAB |
|
Inline XBRL Taxonomy Extension Label Linkbase Document |
101.PRE |
|
Inline XBRL Taxonomy Extension Presentation Linkbase Document |
104 |
|
Cover Page Interactive Data File (embedded within the Inline XBRL document) |
* Filed herewith.
** Furnished herewith. This certification is being furnished solely to accompany this report pursuant to 18 U.S.C. Section 1350, and is not being filed for purposes of Section 18 of the Exchange Act of 1934, as amended, and is not to be incorporated by reference into any filings of the Company, whether made before or after the date hereof, regardless of any general incorporation language in such filing.
+ Certain schedules and exhibits to this agreement have been omitted pursuant to Item 601(b)(2) of Regulation S-K. A copy of any omitted schedule and/or exhibit will be furnished to the SEC upon request.
† Management contract or compensation plan or arrangement.
* Filed herewith.
(c) Financial Statement Schedules
No financial statements have been submitted because they are not required or are not applicable or because the information required is included in the consolidated financial statements or the notes thereto.
Item 16. Form 10-K Summary
None.
86
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
BETTER THERAPEUTICS, INC. |
|
|
|
|
|
Date: March 28, 2022 |
|
By: |
/s/Kevin J. Appelbaum |
|
|
|
Kevin J. Appelbaum |
|
|
|
Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Report has been signed below by the following persons on behalf of the Registrant in the capacities and on the dates indicated.
Name |
|
Title |
|
Date |
|
|
|
|
|
/s/Kevin J. Appelbaum |
|
Director, President and Chief Executive Officer |
|
March 28, 2022 |
Kevin J. Appelbaum |
|
(Principal Executive Officer) |
|
|
|
|
|
|
|
/s/David P. Perry |
|
Executive Chairman and Director |
|
March 28, 2022 |
David P. Perry |
|
|
|
|
|
|
|
|
|
/s/Mark Heinen |
|
Head of Finance and Interim Chief Financial Officer |
|
March 28, 2022 |
Mark Heinen |
|
(Principal Financial Officer and Principal Accounting Officer) |
|
|
|
|
|
|
|
/s/Richard Carmona |
|
Director |
|
March 28, 2022 |
Richard Carmona |
|
|
|
|
|
|
|
|
|
/s/Geoffrey Parker |
|
Director |
|
March 28, 2022 |
Geoffrey Parker |
|
|
|
|
|
|
|
|
|
/s/Andrew Armanino |
|
Director |
|
March 28, 2022 |
Andrew Armanino |
|
|
|
|
|
|
|
|
|
/s/Risa Lavisso-Mourey |
|
Director |
|
March 28, 2022 |
Risa Lavizzo-Mourey |
|
|
|
|
|
|
|
|
|
/s/Suying Liu |
|
Director |
|
March 28, 2022 |
Suying Liu |
|
|
|
|
|
|
|
|
|
/s/Elder Granger |
|
Director |
|
March 28, 2022 |
Elder Granger |
|
|
|
|
87