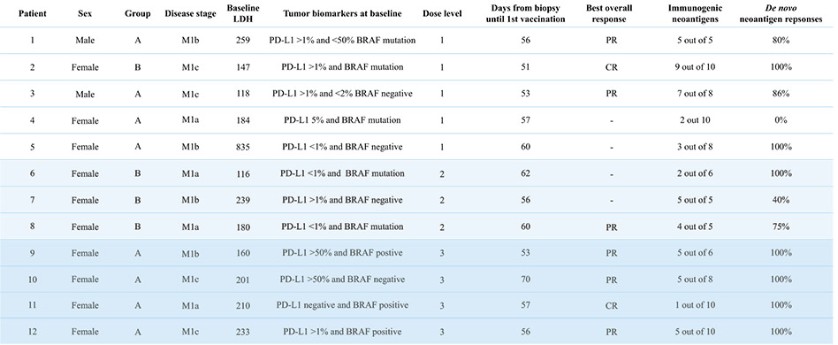
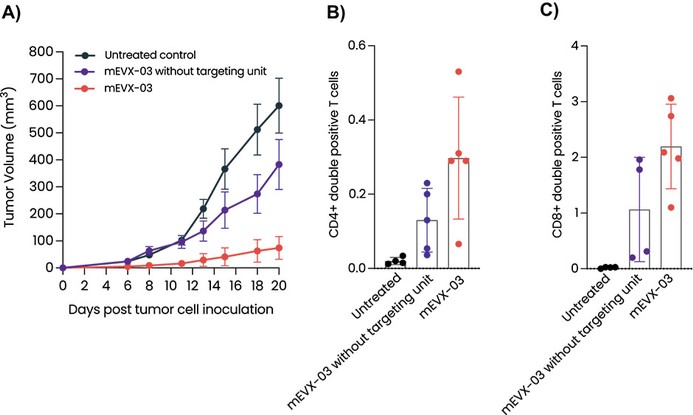
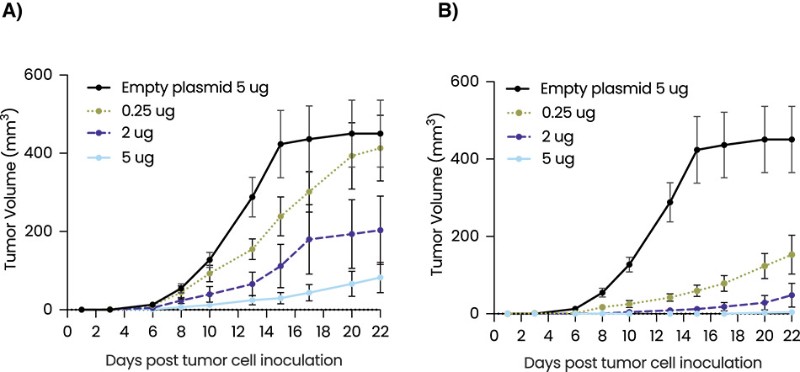
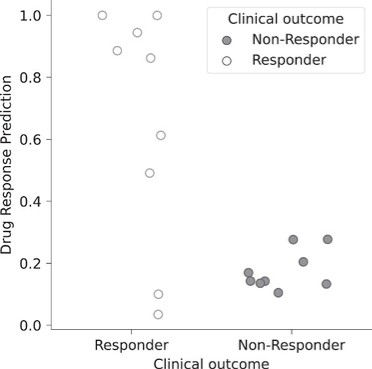
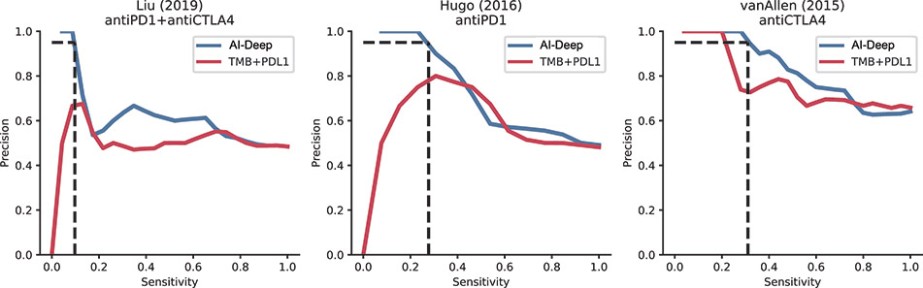
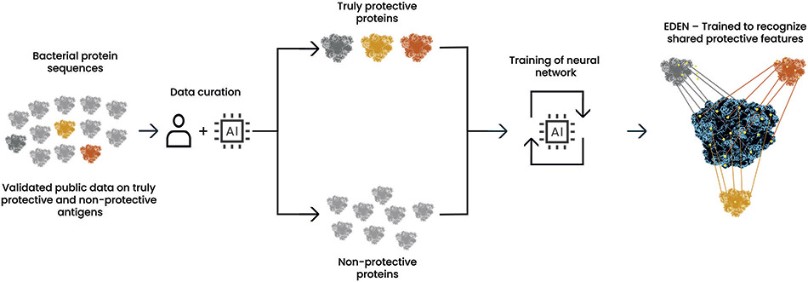
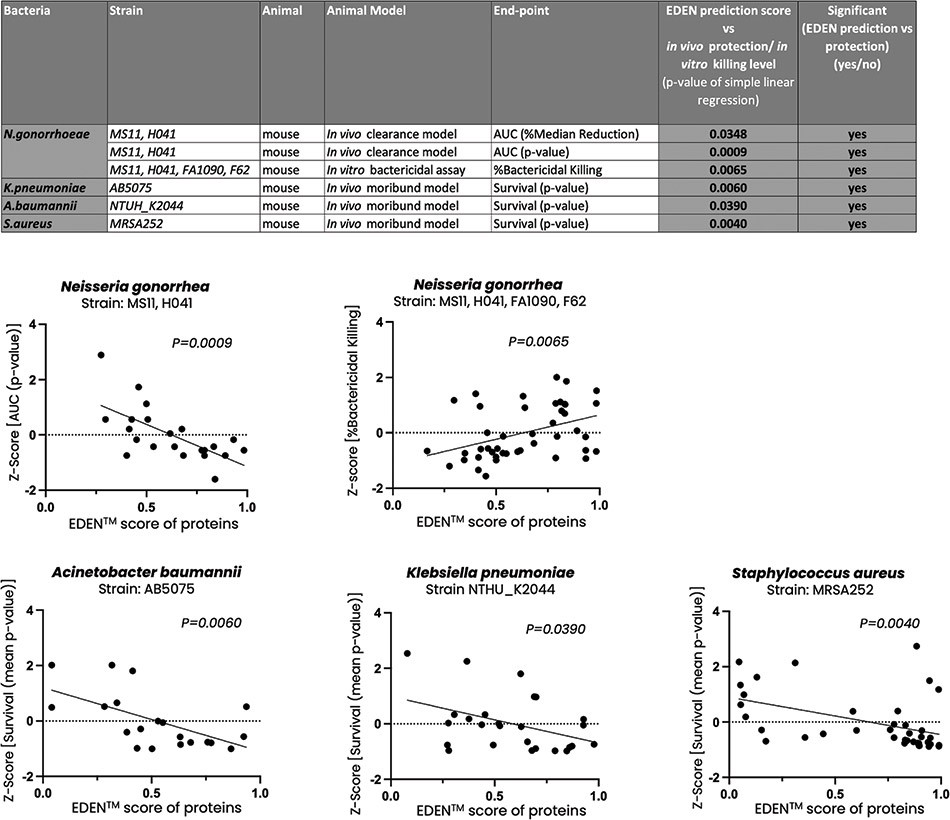
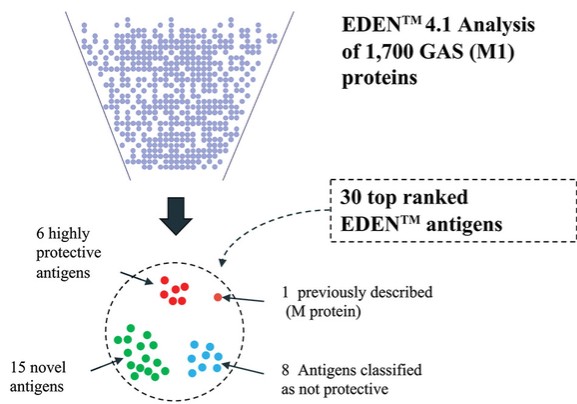
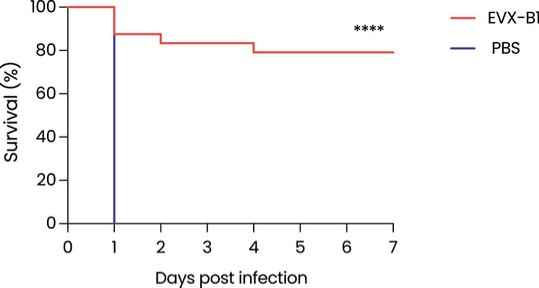
Product Development Pipeline
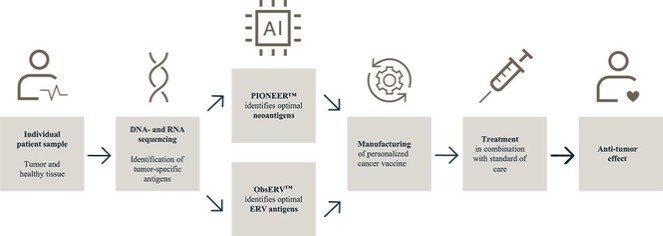
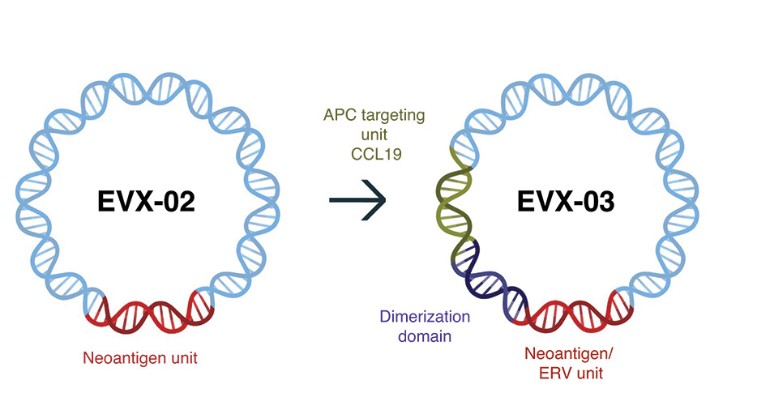
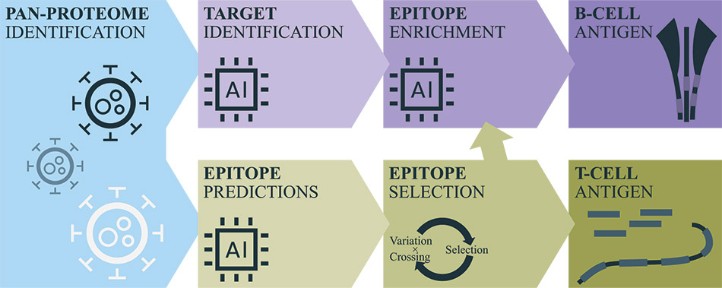
We believe that our AI-identified targets can be delivered using any delivery modality, such as peptides, recombinant proteins, mRNA and our proprietary DNA-targeting technology, and we are building a diverse vaccine pipeline utilizing such different delivery modalities.
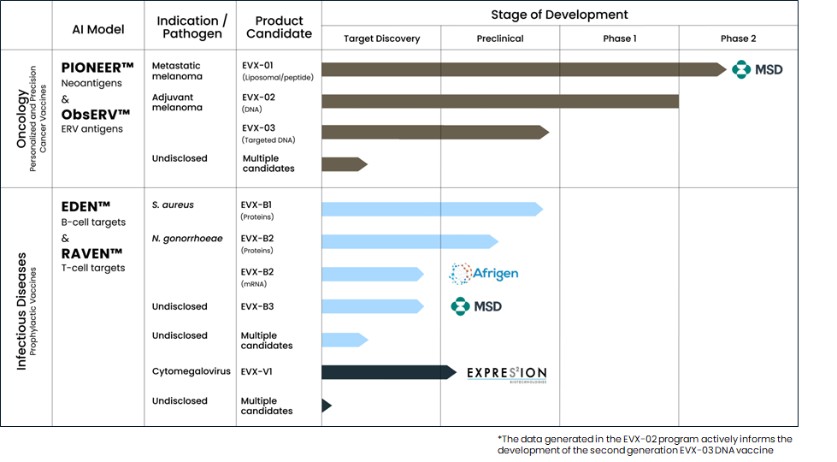
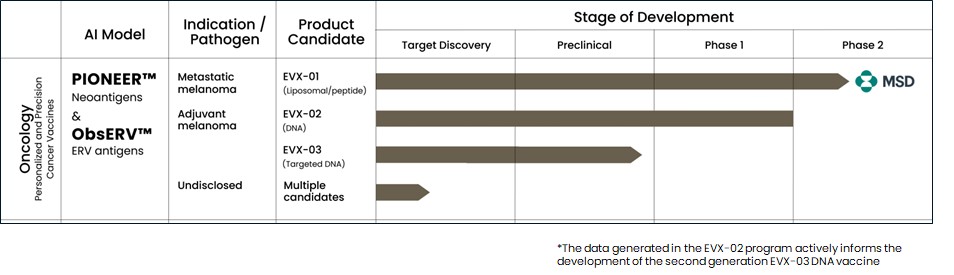
Figure 1: Our AI models and vaccine pipeline.
EVX-01
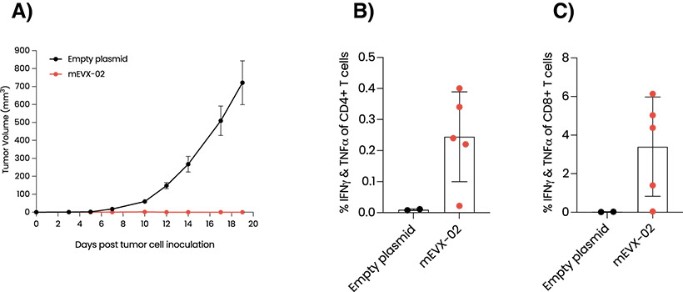
EVX-01 is a novel liposomal, peptide-based cancer vaccine designed to engage a patient’s own immune system to fight their cancer by mounting a targeted response towards the tumor.
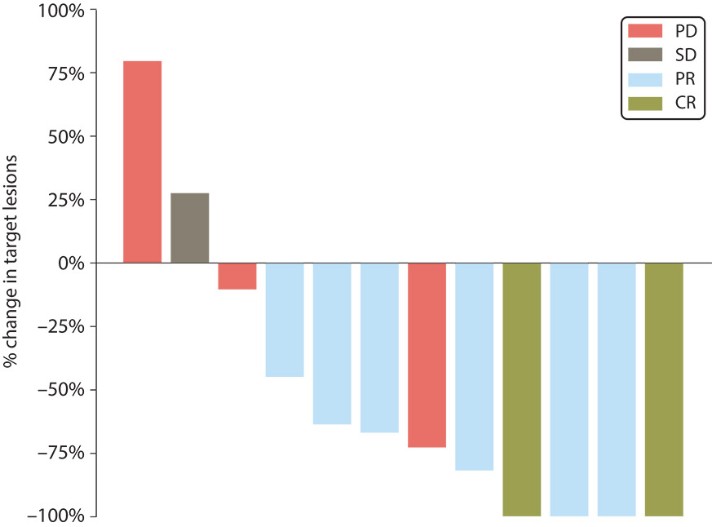
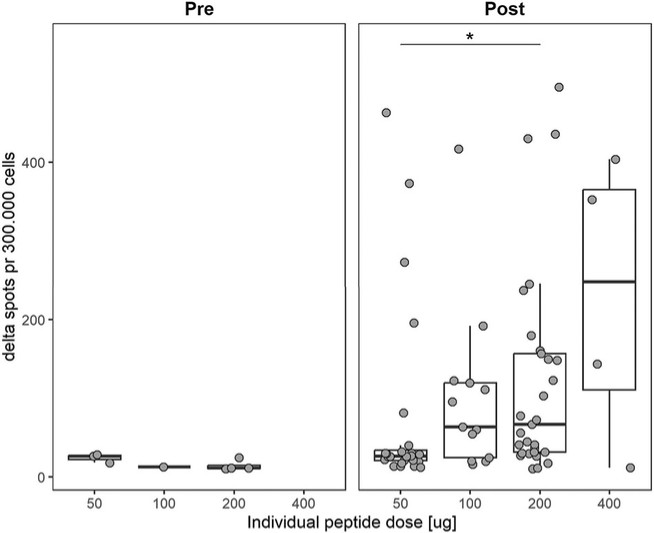
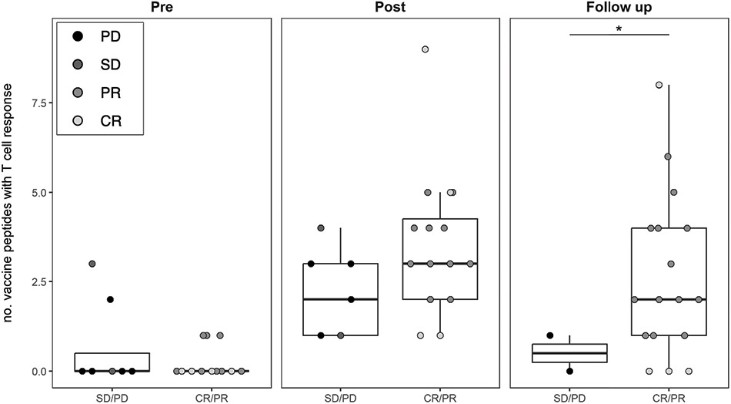
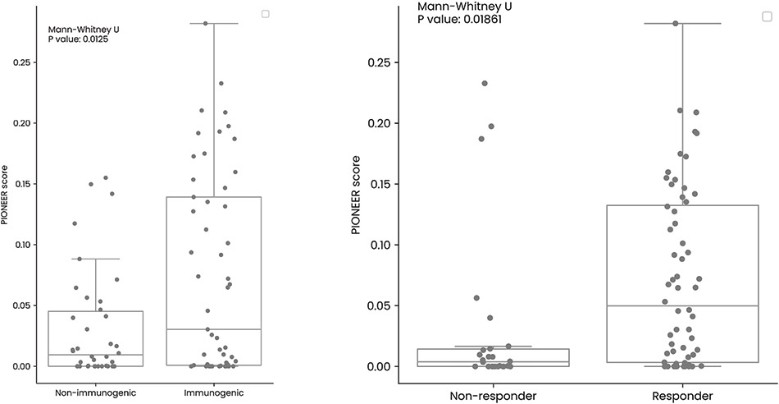
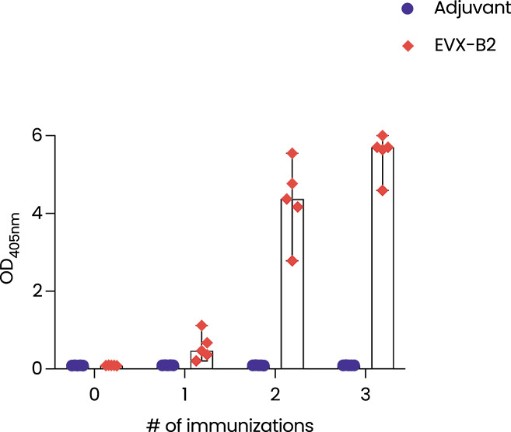
In June 2023 we reported complete clinical data from the Phase 1/2a trial of EVX-01 in metastatic unresectable melanoma demonstrated an overall response rate of 67% across all 12 patients compared with a historical overall response rate of 40% with anti-PD-1 treatment alone. In addition, the data showed induction of neoantigen-specific T cells in 100% of patients and a favorable safety profile.
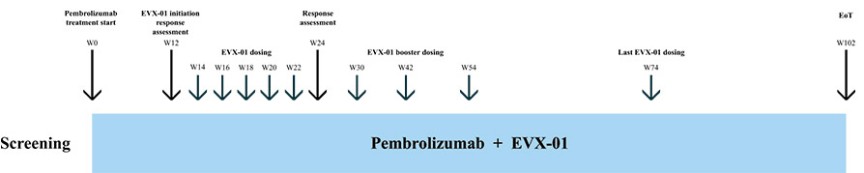
EVX-01 is currently in a Phase 2 global multi-center clinical trial for the treatment of metastatic melanoma and is administered in combination with KEYTRUDA® (pembrolizumab), a humanized anti-human PD-1 monoclonal antibody developed by Merck & Co., Inc., or Merck. A Clinical Trial Collaboration and Supply Agreement, or CTCSA, is in place with MSD International GmbH and MSD International Business GmbH (known collectively as MSD outside the United States and Canada), both of which are subsidiaries of Merck, to evaluate the combination of EVX-01 with MSD’s KEYTRUDA®.
The first patient in the EVX-01 Phase 2 trial was dosed in Australia in September 2022. In November 2022, we submitted an Investigational New Drug Application, or IND, along with a Fast Track designation request to the U.S. Food and Drug Administration, or FDA, for the Phase 2 clinical trial of EVX-01 in combination with KEYTRUDA® for the treatment of patients with metastatic melanoma. On December 22, 2022, the FDA notified us that it had reviewed our IND and determined that we could proceed with our Phase 2 trial. In January 2023, we received Fast Track designation from the FDA for the study.
In addition, we have received approval of our Clinical Trial Applications, or CTAs, for the Phase 2 trial from regulatory authorities in Australia and Italy.
The initial data from five patients from the Phase 2 clinical study were presented at the annual meeting of the Society of Cancer Immunotherapy, or SITC, in San Diego, California November 2023. We believe the data confirmed the previously reported Phase 1/2a findings and further indicated a promising clinical outcome. Full Phase 2 study readout is expected in 2025.
95