achieved, to facilitate an accelerated development program for PAX-101 for certain neurologic indications including ASD, ME/CFS and LCS. Based on our pre-IND meeting with the FDA in March 2021 and, in part, on an analysis of the data that we have exclusively licensed from the Ministry of Health, Republic of Malawi and Lwala Hospital (Soroti, Uganda) relative to East African HAT patients treated with suramin, we believe we have created a strong development strategy that we plan to employ in seeking the approval of PAX-101 for the treatment of East African HAT. Based on our prior interactions with the FDA, including our pre-IND and Type B meeting with the FDA, we further believe that an approval, if any, in East African HAT could confer upon us the potential receipt of a priority review voucher (“PRV”) by the FDA, which we could potentially monetize to fund our future clinical programs. We expect further clinical studies of PAX-101 for the treatment of ASD, FXS, FXTAS, and ME/CFS will be required and similar clinical development is needed for PAX-102 to reach the commercial stage. In November 2020, the FDA granted orphan drug designation to PAX-101 for the treatment of East African HAT. However, there can be no assurance that we will receive FDA approval for PAX-101 for the treatment of East African HAT and, even if PAX-101 is approved by the FDA, there can be no assurance that we will receive a PRV. For more information on the PRV process and how we may benefit from it, see the section of this Annual Report on Form 10-K. “Item 1. Business – Governmental Regulation - The Priority Review Voucher Program.”
Development Pipeline
The following table summarizes our current product candidate and indication pipeline.
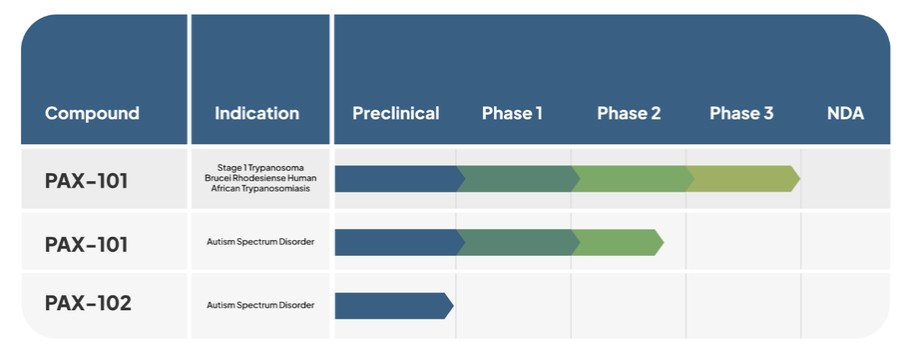
PAX-101 (intravenous suramin)
Our lead product candidate under development is PAX-101, an intravenous formulation of suramin, which we are developing for multiple indications, including East African HAT, ASD, and ME/CFS.
The most advanced indication for which we are developing PAX-101 is for treatment of “Stage 1” Trypanosoma brucei rhodesiense (East African) HAT, the stage of the clinical course of HAT in which the parasite is found in the peripheral circulation, but it has not yet infiltrated the CNS. We maintain exclusively licensed worldwide rights to patient-level data on the use of suramin in the treatment of Stage 1 East African HAT, which we intend to leverage for demonstration of the safety and efficacy of PAX-101. We have met with the FDA in three formal meetings regarding the execution and development of PAX-101 for this indication. Pursuant to those conversations, and to satisfy the FDA’s requirement of demonstrating substantial effectiveness, we conducted a new prospective clinical trial. We completed an analysis and presentation of retrospective data from East African HAT patients previously treated with suramin from 2000 to 2020, for which we have the exclusive license, in July 2023. In addition to these retrospective data, we will also complete preclinical and clinical safety studies to support submission of an NDA for PAX-101’s East African HAT indication. We expect that such work will be completed over the next 10 months, with the intention of filing an NDA in second half of 2024. Additionally, we are completing the development of a proprietary supply chain of drug substance and drug product which will form the basis of our NDA filing. Without establishing this supply chain, we will not be able to submit an NDA for the East African HAT indication. See “Manufacturing” for additional information about our expected timeline for establishing our supply chain. In November 2020, the FDA granted orphan drug designation to PAX-101 for the treatment of East African HAT. It is expected that PAX-101, if approved by the FDA for the East African HAT indication, will qualify for new chemical entity exclusivity (providing sole marketing rights in the United States to the Company with respect to any product that contains suramin for up to seven years), in addition to orphan drug exclusivity,
4