Our lead programs are summarized below. We currently retain global development and commercialization rights to all of our product candidates.
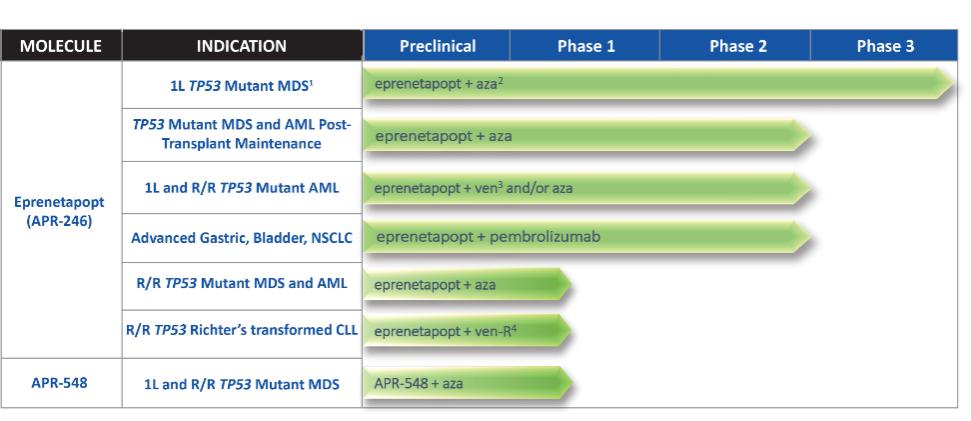
| (1) | Trial did not achieve statistical significance in primary endpoint of complete remission |
| (2) | Azacitidine |
| (3) | Venetoclax |
| (4) | Venetoclax + rituximab |
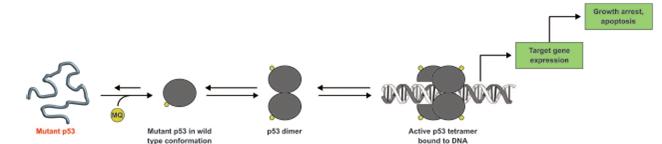
We believe that targeting p53 and thereby reactivating key intrinsic cellular functions has the potential to significantly impact patients’ lives and treatment strategies for a wide variety of cancers. p53 is a tumor suppressor protein that in its normal state functions to sense DNA damage and induce cell cycle arrest, DNA damage repair, senescence and cellular apoptosis. Mutant p53 is an attractive target because it is widely mutated across hematologic and solid tumors and is associated with an aggressive clinical and molecular phenotype. In preclinical studies and clinical trials, mutations in p53 and the apoptotic pathway have been shown to play a key role in cancer genesis, proliferation and resistance to currently marketed therapeutic agents. Many approved and clinical stage oncology drugs are more effective with a functional p53 pathway. Our approach is to restore normal function to p53, thereby re-enabling a cell’s ability to undergo apoptosis. Accordingly, we believe that by targeting p53, our drug candidates may enhance the ability of other anti-cancer therapies to induce cancer cell death. In addition, we believe that our approach may counteract resistance mechanisms that characterize many of the most aggressive cancers. Although we have observed single agent activity in preclinical testing of eprenetapopt, our current clinical program is focused on combination therapy based on the strong additive or synergistic effects we have observed in combination with multiple conventional chemotherapeutic drugs, DNA hypomethylating agents, or HMAs, inhibitors of anti-apoptotic proteins and immuno-oncology checkpoint blockade agents.
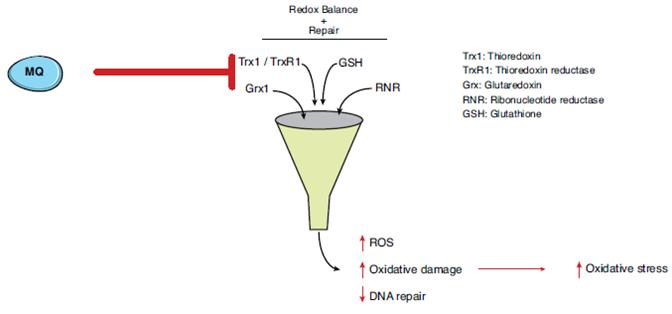
Our lead product candidate, eprenetapopt, is a small molecule that has demonstrated reactivation of mutant p53 in clinical trials. Promising clinical data support the application of eprenetapopt across a variety of hematologic malignancies and other oncologic indications. Eprenetapopt is a pro-drug that is administered intravenously and forms the active moiety, 2-methylene-quinuclidin-3-one, or MQ, under physiological conditions. Eprenetapopt has been shown to induce apoptosis in cancer cells with mutant p53 in Phase 1/2 trials. We believe the mechanism of action of eprenetapopt may provide the basis for its combination with both conventional and novel therapies, such as targeted
7