information previously audited by the company’s prior independent registered public accounting firm. Failure to timely file required filings with the SEC could have a material adverse effect on the company.
Nasdaq Notices
As a result of the decline in the trading price for our common stock and the composition of the Board resulting from the Proxy Contest, the company received deficiency letters from Nasdaq relating to non-compliance with Nasdaq’s continued listing requirements (the “Nasdaq Notices”). The Nasdaq Notices identify deficiencies with respect to requirements for (i) minimum Market Value of Listed Securities (“MVLS Requirement”), (ii) minimum Market Value of Publicly Held Shares (“MVPHS Requirement”), (iii) majority of independent directors (“Majority Independent Requirement”), (iv) Audit and Compensation Committee composition (“Committees Requirement”), and (v) minimum closing bid price (“Bid Price Requirement” and, together with the MVLS Requirement, MVPHS Requirement, Majority Independent Requirement, and Committees Requirement, the “Listing Requirements”). Under Nasdaq rules, the company has a cure period of 180 days from the date of each respective Nasdaq Notice to regain compliance with the MVLS Requirement, the MVPHS Requirement and Bid Price Requirement. Such 180-day cure periods expire on July 12, 2022, August 30, 2022 and October 4, 2022, respectively. The Committees Requirement provides a cure period to regain compliance: (i) until the earlier of the company’s next annual stockholders’ meeting or January 5, 2023, or (ii) if the next annual stockholders’ meeting is held before July 5, 2022, then the company must evidence compliance no later than July 5, 2022. The Majority Independent Requirement requires the company to submit to Nasdaq a plan for regaining compliance within 45 days of the date of the Nasdaq Notice, or May 2, 2022. If such plan is accepted, Nasdaq can grant an extension of up to 180 calendar days from the date of the Nasdaq Notice for the Majority Independent Requirement for the company to evidence compliance. While the company believes that it can regain compliance with each of the Listing Requirements, there can be no assurance that the company will be able do so within the prescribed periods, or at all. Our common stock could become subject to delisting from Nasdaq if we fail to regain compliance.
The Duet Platform
We have entered into three licenses, a sponsored research agreement and a clinical research support agreement relating to the Duet Platform with COH, a world-renowned, independent biomedical research and treatment center for cancer and other life-threatening diseases.
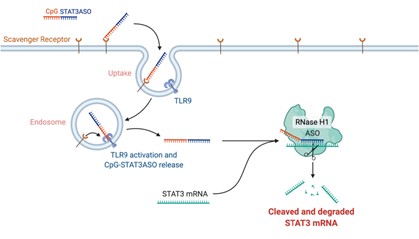
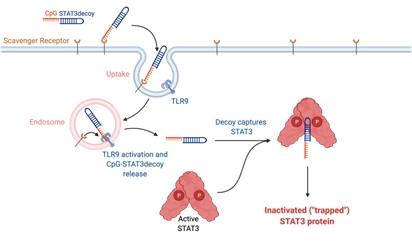
The Duet Platform is comprised of three distinctive, complementary bifunctional oligonucleotides that each consist of a TLR9 agonist (i.e., CpG ODN) linked with a STAT3 inhibitor (Figure 1):
· | RNA silencing | CpG-STAT3siRNA | (“DUET-01”) |
· | Antisense | CpG-STAT3ASO | (“DUET-02”) |
· | DNA-binding inhibitor | CpG-STAT3decoy | (“DUET-03”) |
One of the most sought-after therapeutic targets in cancer is STAT3, an oncogenic transcription factor and a master regulator of immunosuppression. STAT3 is a well-known driver of malignant cell invasion, proliferation, and metastasis in most human cancers. To date, STAT3 has remained undruggable. While numerous STAT3-based therapies have made it into the clinic, there have been no FDA approvals to date. The primary reason STAT3 has remained undruggable is that it’s an intracellular target, making it highly difficult to access. Duet’s approach to make STAT3 druggable is to combine the STAT3 inhibitor with a CpG DNA recognized by the immune receptor, TLR9. Tethering STAT3 to CpG allows for intracellular delivery of the whole molecule and for triggering potent antitumor immune responses.

Figure 1. The Duet Platform. Each molecule in the Duet Platform consists of a TLR9 agonist (i.e., unmethylated CpG sequence) that is chemically linked to a STAT3 inhibitor. The STAT3 inhibitor takes three different forms, as described in the table above.
CpG oligonucleotides serve as a common “danger signal” of bacterial or viral infections detected by TLR9 receptors. Duet links a synthetic CpG oligonucleotide to a STAT3 inhibitor to:
4