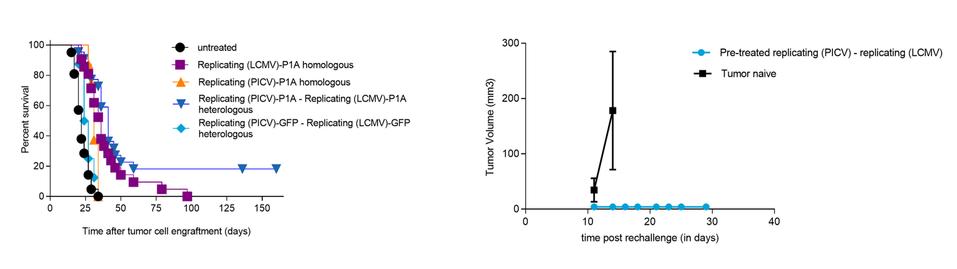
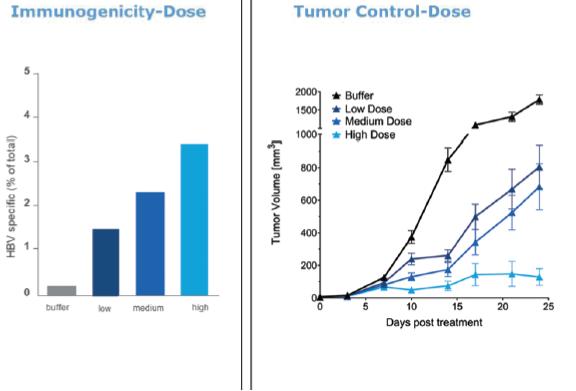
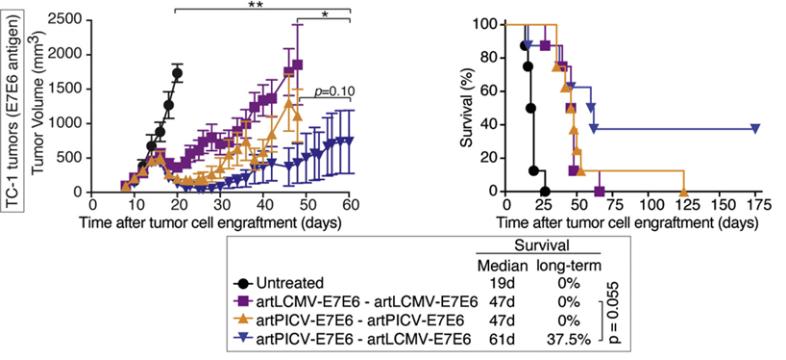
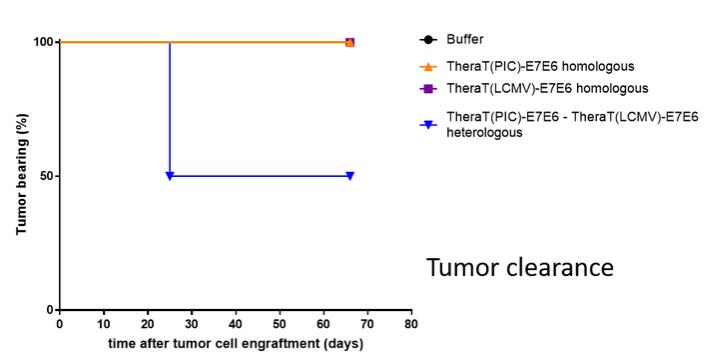
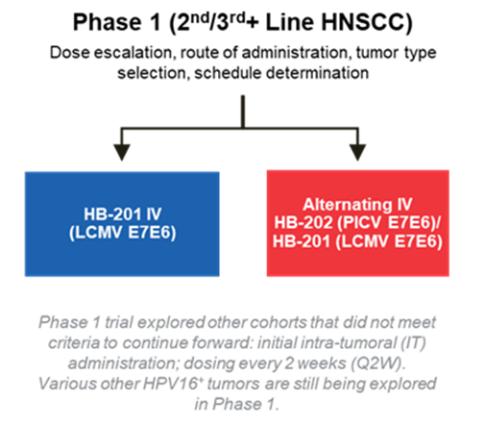
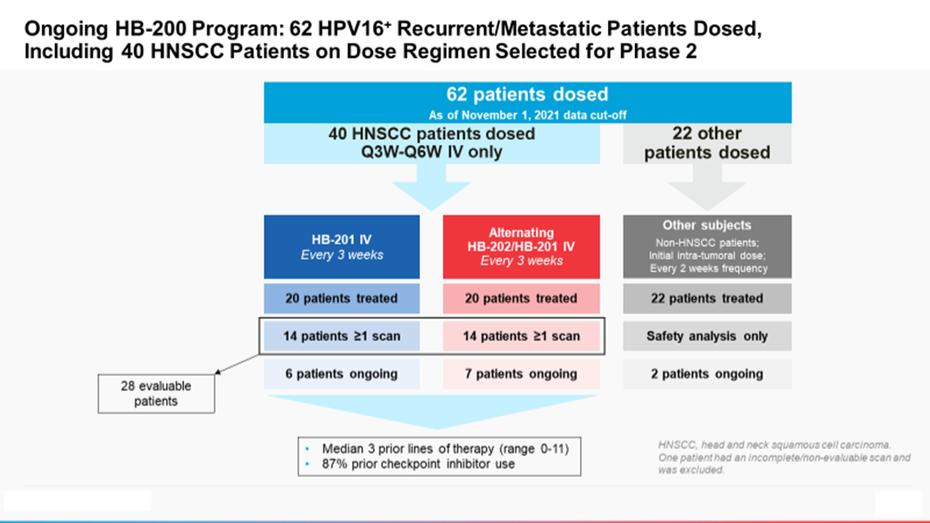
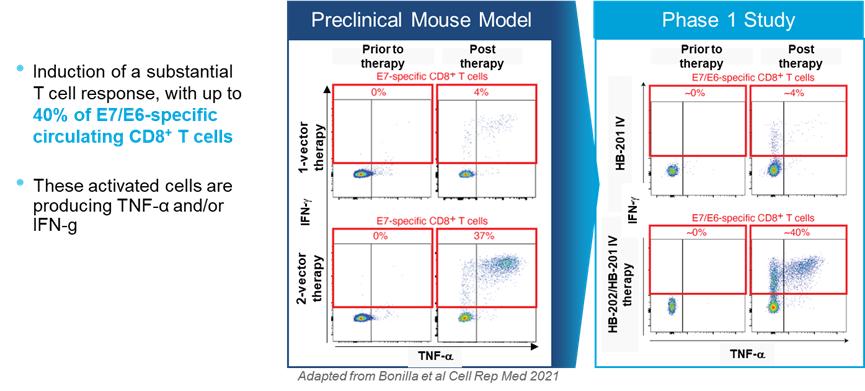
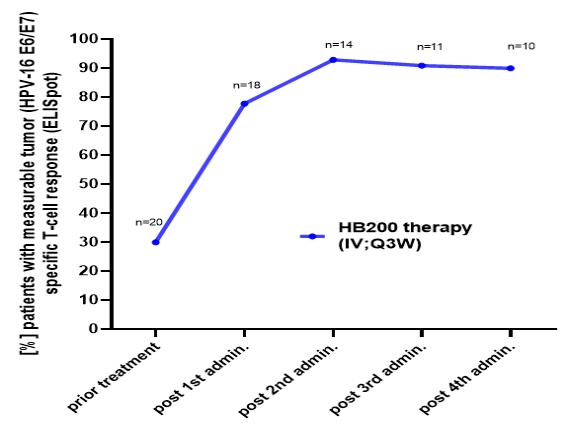
Intellectual Property
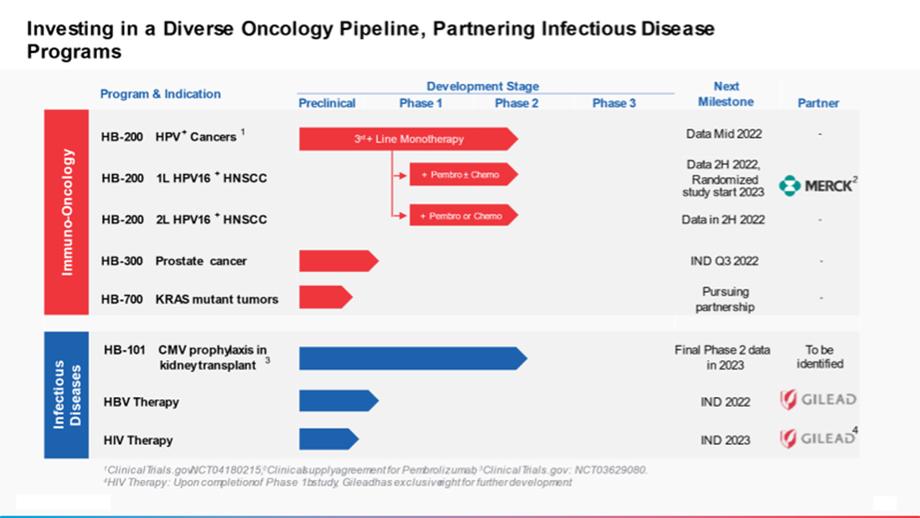
Our success depends, in part, on our ability to obtain and maintain intellectual property protection for our product candidates, technology and knowhow, to defend and enforce our intellectual property rights, in particular, our patent rights, to preserve the confidentiality of our knowhow and trade secrets, and to operate without infringing the proprietary rights of others. We seek to protect our product candidates and technologies by, among other methods, filing U.S. and foreign patent applications related to our proprietary technology, inventions and improvements that are important to the development of our business. We also rely on trade secrets, knowhow, continuing technological innovation and in-licensing of third-party intellectual property to develop and maintain our proprietary position. We, or our collaborators and licensors, file patent applications directed to our key product candidates in an effort to establish intellectual property positions to protect our product candidates as well as uses of our product candidates for the prevention and/or treatment of diseases.
As of February 24, 2022, we are the owner or exclusive licensee to nine issued U.S. patents and ten pending U.S. patent applications, two pending international Patent Cooperation Treaty (PCT) applications, five pending U.S. provisional patent applications, and 105 issued foreign patents and approximately 78 foreign patent applications. These patents and patent applications are related to our technologies concerned with the arenavirus-based immunization systems, non-replicating and replicating, our product candidates and various development programs, which are directed to the use of these immunization systems for the treatment and/or prevention of various infectious diseases or cancer, and certain clinical uses of our current or future product candidates in oncology. The issued patents and pending patent applications contain claims directed to various aspects of our work, including compositions of matter, methods of treatment and prevention, methods of producing certain compositions, and use of our product candidates in combination with certain other therapeutics.
Non-Replicating Technology Portfolio
Our patent portfolio related to our non-replicating technology includes a patent family exclusively licensed to us from the University of Zurich. This patent family includes four patents granted in the United States and patents granted in Europe (validated in Austria, Belgium, Czech Republic, Denmark, France, Germany, Ireland, Italy, Netherlands, Poland, Spain, Sweden, Switzerland and the United Kingdom), Canada, China, India, Hong Kong and Japan. This patent family also includes pending applications in the United States, Europe, China, Hong Kong and India. The granted patents and pending applications related to our non-replicating technology are expected to expire no earlier than 2028, not giving effect to any potential patent term extensions and patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other governmental fees. Our non-replicating technology is being employed or may be employed in one or more of the product candidates or programs described herein.
Replicating Technology Portfolio
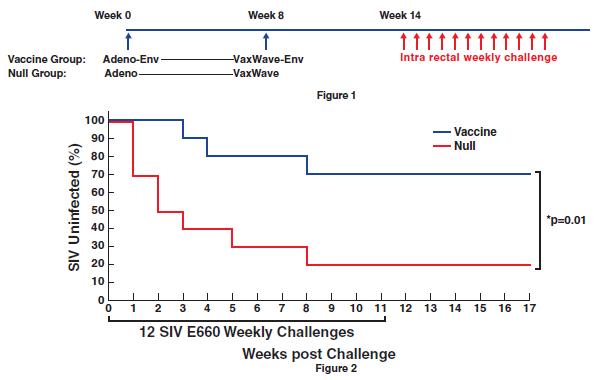
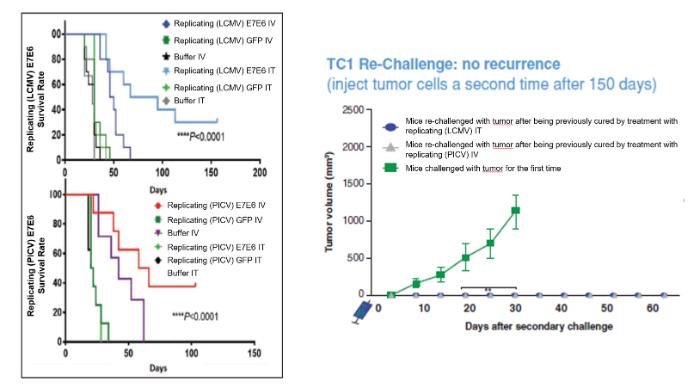
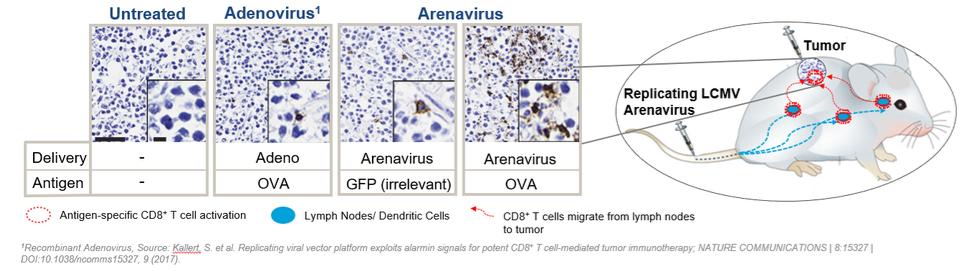
We are the owner or exclusive licensee to proprietary patent positions related to our replicating technology. Our patent portfolio related to our replicating technology includes a patent family exclusively licensed from the University of Geneva. This patent family includes a patent granted in the United States and patents granted in Europe (validated in Albania, Austria, Belgium, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Macedonia, Malta, Monaco, Norway, Poland, Portugal, Romania, Serbia, Slovakia, Slovenia, Spain, Sweden, Switzerland/LI, The Netherlands, Turkey and the United Kingdom), Hong Kong and Japan. The European patent in this family (European Patent No. 3218504) was opposed by a third party in April 2021. This patent family also includes pending applications in the United States, Europe, Canada, Australia, Japan, India, China and Hong Kong. The second patent family in our replicating platform portfolio is jointly owned by us and the University of Basel. The rights of the University of Basel under this patent family are exclusively licensed to us. This second patent family includes pending applications in various countries, including in the United States, Europe, Eurasia, Hong Kong, Korea, China, Canada, Australia, New Zealand, Mexico, Japan, Brazil, Singapore, India, and Israel. The granted United States patent from the first patent family is expected to expire in April 2037 due to patent term adjustment. The granted patents in Europe, Hong Kong and Japan, and the pending applications related to our replicating technology are expected to expire between 2035 and 2037, not giving effect to any potential patent term extensions or patent term adjustments and assuming payment of all appropriate maintenance, renewal, annuity or other