indications where there is a compelling scientific rationale, strong clinical tractability and significant unmet medical need.
Enhance our position as a leading metabolic disease company by developing, acquiring or in-licensing additional investigational product candidates. We are continually evaluating opportunities to build a robust pipeline of potential leading treatments for serious metabolic diseases. We may select additional assets for their potential as stand-alone monotherapies or for eventual use in combination with other products.
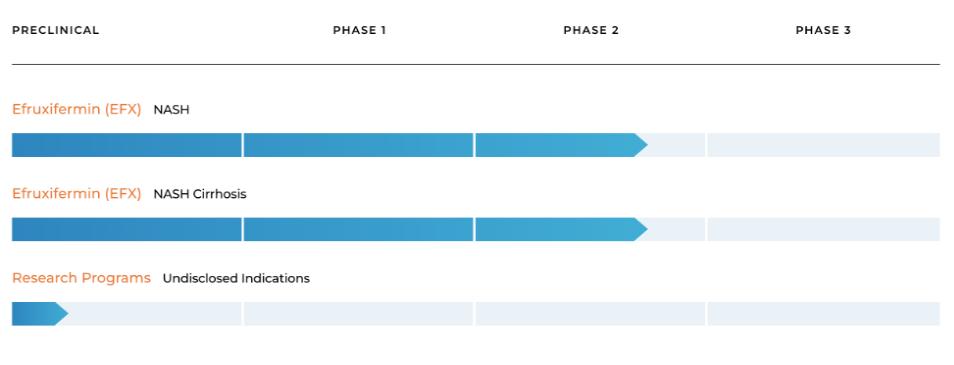
Our Pipeline
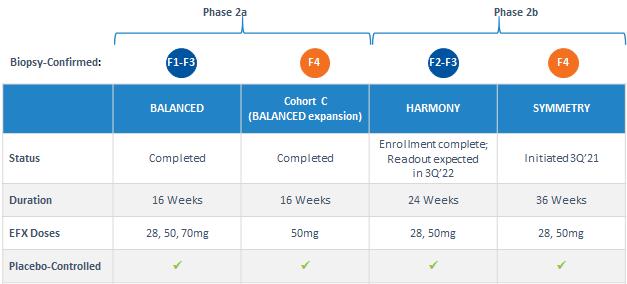
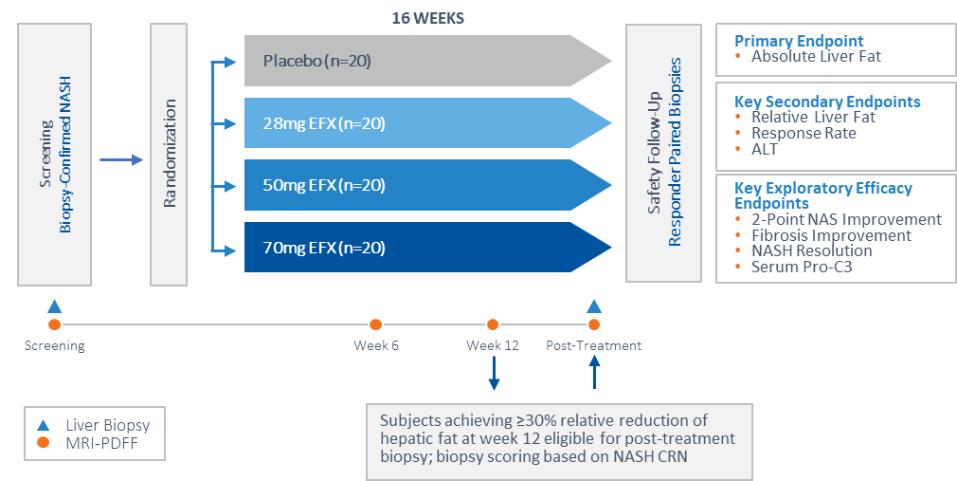
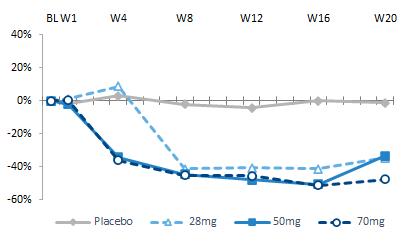
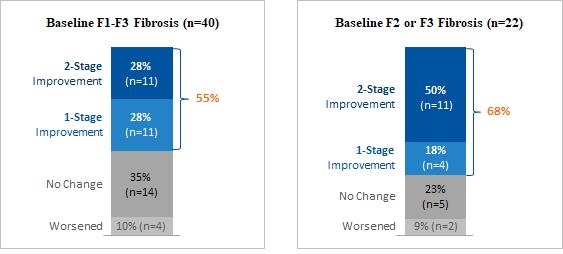
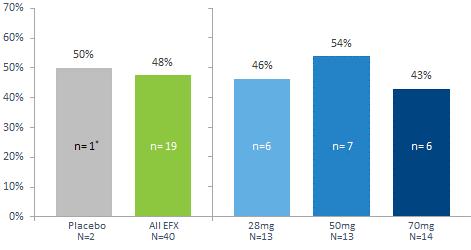
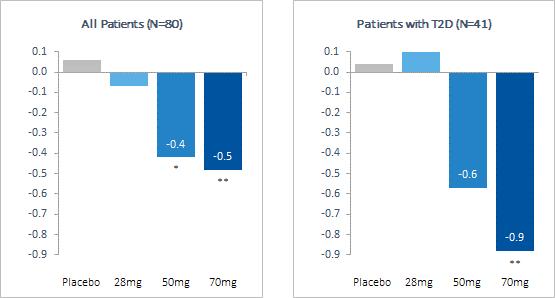
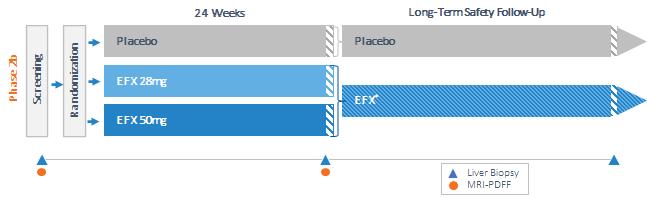
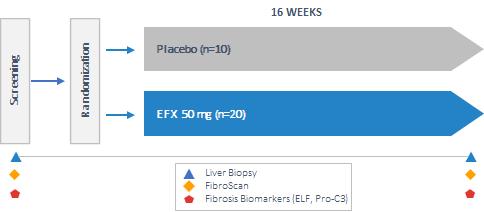
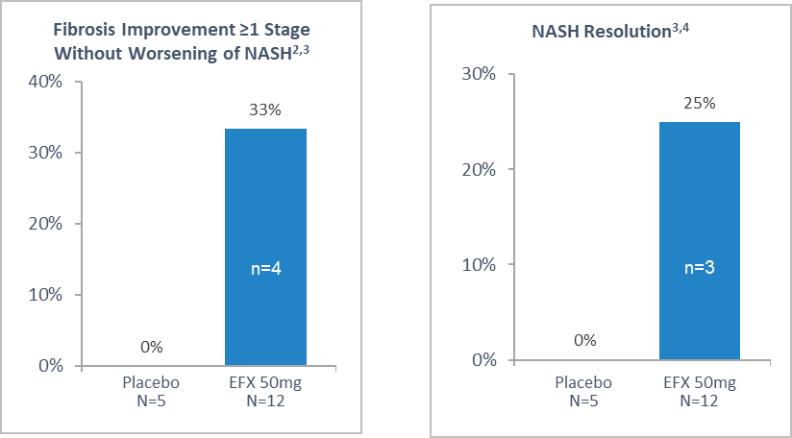
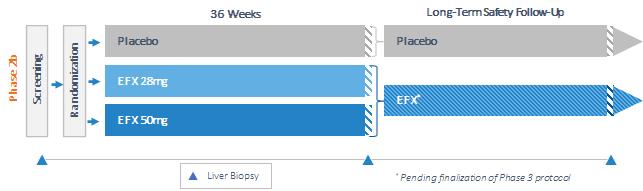
Our pipeline is anchored by EFX, a potential best-in-class FGF21 analog for treatment of NASH, if approved. We have one EFX program focused on patients with pre-cirrhotic NASH (F2-F3), which is supported by the HARMONY study, an ongoing Phase 2b clinical trial. We have a second EFX program focused on patients with cirrhotic NASH (F4, compensated), which is supported by the SYMMETRY study, an ongoing Phase 2b clinical trial. These two programs align with FDA guidance published in 2018 and 2019, which recommends different regulatory approval pathways for patients with pre-cirrhotic and cirrhotic NASH.
Akero’s Pipeline
NASH Overview
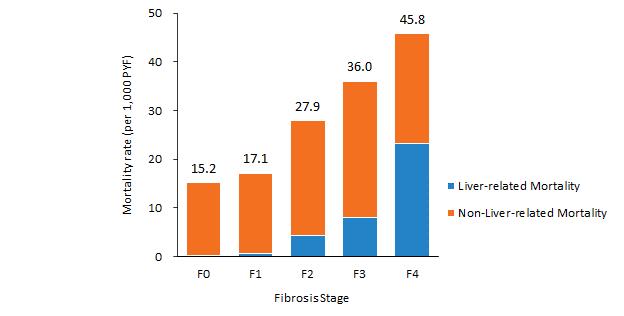
NASH is a severe form of NAFLD, which is driven by the global obesity epidemic. Patients with NAFLD have an excessive accumulation of fat in the liver resulting from an excess of caloric intake over energy needs. In patients with NASH, excessive liver fat leads to hepatocyte stress, which triggers localized inflammation and can cause extensive scarring, or fibrosis, of the liver, as the liver attemps to repair and replace damaged cells.
Patients with NASH are at increased risk of liver-related morbidity and mortality, including liver failure and hepatocellular carcinoma. As NASH progresses, cardiovascular-related morbidity and mortality also increase, with cardiovascular disease being the most frequent cause of death in patients with NASH. The prevalence of patients with advanced fibrosis (F2-F4) in the United States is projected to rise 14.1 million by 2030, representing a roughly 100% increase from an estimated 6.7 million in 2016.
Diagnosis and disease burden
NASH is currently diagnosed through liver biopsy and its severity is measured using scoring systems that assess the extent and severity of steatosis, lobular inflammation, hepatocellular ballooning and fibrosis. Some patients may be diagnosed with NASH after presenting with symptoms such as general fatigue and nondescript abdominal
8