UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
__________________________
FORM
__________________________
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Transition Period From to
Commission file number:
__________________________
(Exact name of Registrant as specified in its charter) |
__________________________
| ||
(State or other jurisdiction of incorporation or organization) |
| (I.R.S. Employer Identification No.) |
|
| |
| ||
(Address of principal executive offices) |
| (Zip Code) |
(
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
| Trading Symbol |
| Name of the exchange on which registered |
|
|
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer”, “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | ☐ | Accelerated filer | ☐ |
☒ | Smaller reporting company | ||
|
| Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes
The aggregate market value of the common stock held by non-affiliates of the registrant, based on the closing price of a share of common stock on June 30, 2023 as reported by the NYSE American on such date was approximately $
As of March 12, 2024, the registrant had
CATHETER PRECISION, INC.
TABLE OF CONTENTS
| 2 |
| Table of Contents |
CATHETER PRECISION, INC.
PART I
Forward-Looking Statements
This Annual Report on Form 10-K contains forward-looking statements that are based on our management’s beliefs and assumptions and on information currently available. This section should be read in conjunction with our audited financial statements and related notes included in Part II, Item 8 of this report. The statements contained in this Annual Report on Form 10-K that are not historical facts are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended.
Forward-looking statements can be identified by words such as “believe,” “anticipate,” “may,” “might,” “can,” “could,” “continue,” “depends,” “expect,” “expand,” “forecast,” “intend,” “predict,” “plan,” “rely,” “should,” “will,” “may,” “seek,” or the negative of these terms and other similar expressions, although not all forward-looking statements contain these words. You should read these statements carefully because they discuss future expectations, contain projections of future results of operations or financial condition, or state other “forward-looking” information. These statements relate to our future plans, objectives, expectations, intentions and financial performance and the assumptions that underlie these statements.
These forward-looking statements are subject to a number of risks, uncertainties, and assumptions, including, but not limited to, those described in “Risk Factors.” These forward-looking statements reflect our beliefs and views with respect to future events and are based on estimates and assumptions as of the date of this Annual Report on Form 10-K and are subject to risks and uncertainties. We discuss many of these risks in greater detail in the section entitled “Risk Factors” included in Part I, Item 1A and elsewhere in this report. Given these uncertainties, you should not place undue reliance on these forward-looking statements. We qualify all of the forward-looking statements in this Annual Report on Form 10-K by these cautionary statements. Except as required by law, we assume no obligation to update these forward-looking statements publicly, or to update the reasons actual results could differ materially from those anticipated in any forward-looking statements, whether as a result of new information, future events or otherwise.
This Annual Report on Form 10-K also contains estimates, projections and other information concerning our industry, our business, and the markets for certain diseases, including data regarding the estimated size of those markets. Information that is based on estimates, forecasts, projections, market research or similar methodologies is inherently subject to uncertainties and actual events or circumstances may differ materially from events and circumstances reflected in this information. Unless otherwise expressly stated, we obtained this industry, business, market, and other data from reports, research surveys, studies, and similar data prepared by market research firms and other third parties, industry, medical and general publications, government data, and similar sources.
| 3 |
| Table of Contents |
ITEM 1. BUSINESS
Overview
The registrant (together with our consolidated operating subsidiary, the “Company” or “Catheter”) was incorporated under the name “Ra Medical Systems, Inc.” as a Delaware corporation in July 2018. A predecessor had been incorporated in California in September of 2002, but was reincorporated in 2018 in connection with our initial public offering. The Company was initially formed to develop, commercialize and market an excimer laser-based platform for use in the treatment of vascular and dermatological immune-mediated inflammatory diseases, including the DABRA product line.
On January 9, 2023, the Company merged with Catheter Precision, Inc., or “Old Catheter”, a privately-held Delaware corporation (the “Merger”), and the business of Old Catheter became a wholly owned subsidiary of the Company, which today is our only operating subsidiary. Following the Merger, we discontinued the Company’s legacy lines of business and the use of any of its DABRA-related assets. For further information about these historical lines of business, see “Item 1. Business” of the Company’s Form 10-K for the fiscal year ended December 31, 2021. Since the Merger, we have shifted the focus of our operations to Old Catheter’s product lines. Accordingly, our current activities primarily relate to Old Catheter’s historical business which comprises the design, manufacture and sale of new and innovative medical technologies focused in the field of cardiac electrophysiology, or “EP.”
Our primary product is the VIVO System which is an anacronym for View into Ventricular Onset System(“VIVO” or “VIVO System”) which is a non-invasive imaging system that offers 3D cardiac mapping to help with localizing the sites of origin of idiopathic ventricular arrhythmias in patients with structurally normal hearts prior to EP procedures.
Our newest product, LockeT, is a suture retention device indicated for wound healing by distributing suture tension over a larger area in the patient in conjunction with a figure of eight suture closure. LockeT is intended to temporarily secure sutures and aid clinicians in locating and removing sutures efficiently.
Our product portfolio also includes the Amigo® Remote Catheter System, or Amigo, a robotic arm that serves as a catheter control device. Prior to 2018, Old Catheter marketed Amigo. We own the intellectual property related to Amigo, and this product is under consideration for future research and development of a generation 2 product.
Electrophysiology Market Overview
EP is one of healthcare’s largest sectors and rapidly growing. The EP market includes well known medical devices such as pacemakers, electrocardiogram, or ECG, systems and cardiac catheters, but also laboratory equipment such as intracardiac mapping systems and fluoroscopy systems (similar to x-ray in real time). The EP market includes large medical device companies such as Medtronic, Plc., Abbott Laboratories, Biosense-Webster (J&J) and Boston Scientific Corp. and is estimated to be $15.1 billion by 2028 (CAGR of 13.0%). Population growth, increasing rates of heart disease and the rising cost of healthcare are driving growth in the EP markets.
| 4 |
| Table of Contents |
Within the EP market, we focus our products on the catheter ablation market. The catheter ablation market was $3.2 billion in 2020 and estimated to grow to $6.4 billion in 2026. The catheter ablation market is growing at a faster rate (12.4% CAGR) than the EP market as a whole.
Within the last 10 years, ventricular ablation has become a fast-growing treatment option. Currently, there are about 80,000 ventricular ablations annually and VT ablations represent approximately 16% of ablations in the U.S. Currently, the market is underserved, and this number is expected to increase to over 250,000 procedures by 2026. The ventricular ablation market is expected to grow at a 21% CAGR through 2026, which is a faster rate than the global EP market and the catheter ablation market as a whole. The growth in the ventricular ablation market is driven by an aging population, advances in EP technology as well as updated physician guidelines. The Heart Rhythm Society, or HRS, Expert Consensus Statement on Catheter Ablation of Ventricular Arrhythmias, published in May 2019 recommends catheter ablation in preference to anti arrhythmic drugs or in the situation where anti arrhythmic therapy has failed or is not tolerated. The guidelines also recommend ablation for reducing recurrent VT and implantable cardioverter-defibrillator shocks.
Existing Treatments and Methods for Catheter Ablations
Traditionally, the first line of treatment for cardiac arrhythmias is medication. Unfortunately, this is not a permanent fix and most patients eventually need a catheter ablation.
Catheter Ablation Procedure Overview
An electrophysiologist stands next to the patient’s bed near the patient’s groin. A catheter or catheters are inserted into the femoral vein (located at the groin) and navigated into the right side of the heart. Depending on the type of arrhythmia, the catheter is inserted into the atrium or the ventricle. Once inserted, a diagnostic catheter is used in conjunction with an invasive (traditional) mapping system to create a map of the patient’s heart. This allows the physician to see the individual patient’s cardiac structures and size. Once the map is created, the physician begins to “pace map.” This process requires the physician to move the catheter from spot to spot to determine the electrical conduction at different areas to determine if the tissue in that area is responsible for the arrhythmia. Once the area is located, the physician will provide a form of energy (radiofrequency, cryo, etc.) to ablate the tissue in that spot.
Treatment Challenges for Ventricular Arrhythmias
Treatment of ventricular ablations with cardiac ablations is a relatively new treatment option. As a result, we believe that the patient population is underserved and is not as well understood, and the available techniques and technologies are limited when compared to the atrial ablation options.
| 5 |
| Table of Contents |
Ablation locations within the ventricle are very difficult to identify. Often, patients are highly symptomatic (dizzy, breathing difficulties, etc.) but the arrhythmia is infrequent. When this happens, it is hard to predict when the patient will be having an “active” arrhythmia. Because of this, the physician may not be able to identify the location even when using medication to induce the arrhythmia. Without confirmation during invasive mapping, the patient is removed from the electrophysiology lab without the ablation procedure being performed and the patient is required to return at a later date and try again for a successful outcome.
Even when a patient has frequent ventricular arrhythmias, the process of pace-mapping often takes 4 – 5 hours to identify the location for ablation, which can increase the likelihood of patient complications due to the extended time under anesthesia.
Lastly, many patients with untreated ventricular arrhythmias cannot tolerate anesthesia well, thus invasive mapping that takes a long time is not an option for them.
Treatment Challenges for Atrial Arrhythmias
Catheter ablation for atrial arrhythmias is more standardized and “advanced” than for ventricular ablations, thus less pace mapping is required. Instead, a procedure called Pulmonary Vein Isolation (“PVI”) is performed for atrial fibrillation, and a single line is ablated for atrial flutter. In pulmonary vein isolation, tiny scars are created in the left upper chamber of the heart in the area where the four lung (pulmonary) veins connect.
Despite steady improvement in the tools available to perform effective procedures, there is clear study evidence that catheter based atrial fibrillation treatment technology can become more effective. According to a study entitled “Long Term Outcomes of Catheter Ablation of Atrial Fibrillation: A Systematic Review and Meta- Analysis” published in the Journal of American Heart Association on March 18, 2013, which looked at multiple individual studies covering over 6,000 patients, “single procedure freedom from atrial fibrillation at long term follow up was 53.1%.” The same study found “with multiple procedures performed, the long-term success rate was 79.8%.” Ineffective treatment may result in patients undergoing two or more EP procedures to achieve relief from atrial fibrillation at an estimated cost in the range of $20,000 or more per procedure.
Specific reasons have not been proven for the lower success rate of initial ablation procedures. However, there is growing evidence that better results occur if the treating EP physician is able to make better lesions by maintaining stable contact force of the catheter against the heart wall, thereby reliably delivering the energy required to eliminate the abnormal rhythms. Variation in catheter contact force occurs as the physician attempts to manually position and hold the catheter tip in a stable position during cases lasting 2 to 3 hours in order to perform typically over 100 ablations of the cardiac anatomy.
Large multi-national medical device companies, such as Medtronic, Inc., Boston Scientific Corp., Abbott Laboratories, St. Jude Medical, Inc. and the Biosense Webster division of Johnson & Johnson, among others, continue to invest heavily to develop and introduce new devices and technologies to improve patient outcomes. Included among these are force-sensing catheters, including the Biosense SmartTouch TM catheter, which provide a continuous readout of the contact force between the catheter and the heart wall. Our Vivo System is focused on the controlled delivery of these catheter technologies to enhance both the performance of ablation procedures and the ease and safety for the physicians who perform them.
A recent peer-reviewed multicenter study sponsored by Biosense Webster, entitled “Paroxysmal AF Catheter Ablation with a Contact Force Sensing Catheter” published in 2014 found that catheter ablation success rates can be as high as 80% when the physician is able to maintain stable contact force within investigator selected working ranges. “When the CF (contact force) employed was between investigator selected working ranges > 80% of the time during therapy, outcomes were 4.25 times more likely to be successful.” Further, “stable CF during radiofrequency application increases the likelihood of twelve-month success.” However, it should be noted that, using manually controlled methods, the physicians in the study could only maintain optimal tissue contact in less than 30% of the patients studied.
In addition, another study, sponsored by St. Jude Medical, Inc. and published in 2015 showed similar findings using their recently FDA-approved contact-force sensing catheter, TOCCASTAR. In the TOCCASTAR study, 85.5% of ablation procedure patients were free of atrial fibrillation at one year after the procedure when optimal catheter tip contact force was maintained, versus only 67.7% when non-optimal contact force was achieved.
| 6 |
| Table of Contents |
VIVO Clinical Use and Studies
To date, VIVO has been used in more than 1,000 procedures, by more than 30 physicians in 10 countries. Initial clinical work was completed with the first-generation software, which resulted in FDA 510(k) Clearance in June 2019.
The U.S. multi-center study enrolled 51 patients from 5 centers. Of note, the Principal Investigator and center to have the highest enrollment was Johns Hopkins University in Baltimore, Maryland. This study was conducted to evaluate the accuracy of VIVO as compared to invasive mapping systems (current prevailing method for determining arrhythmia origins). VIVO met all study endpoints and correctly matched the predicted arrhythmia origin in 44/44 patients (100%; primary endpoint) and correctly matched paced sites in 225/226 locations (99.56%; secondary endpoint). In some instances, this study showed that VIVO has better predictability for arrhythmia origin than a physician’s manual review of a 12 lead ECG.
While conducting the initial clinical study for FDA submission, we developed generation 2 in parallel with a goal to have this version complete and ready to submit upon 510(k) clearance of generation 1. We successfully achieved this goal and received CE Mark and FDA 510(k) Clearance for generation 2 in 2020.
Additional clinical work has occurred with generation 2. Until recently, this data has been single center, physician-initiated research and has resulted in peer reviewed clinical science at electrophysiology conferences and in journals.
Three physicians, at different centers, in the UK conducted a feasibility study for Stereotactic Ablative Radiotherapy, or SABR, and published their data on nine patients. SABR is an ablation technique utilizing non-invasive methods akin to proton therapy for cancer treatment. To do a complete non-invasive ablation, accurately predicting the ablation location non-invasively is key to procedural success, and VIVO was utilized for this purpose. Non-invasive ablation is a new technique and requires additional data, but it is showing promise and has generated excitement within the EP community. If accepted for wide-spread treatment, this would allow for previously un-ablatable patients to receive lifesaving treatments.
In February 2023, a study from the Royal Brompton Hospital was published. This study enrolled 15 patients with 24 VTs (ventricular contractions) and PVCs (premature ventricular contractions). VIVO accurately identified VT and PVC origin in 23/24 (96%) and sub-localized in 100% of subjects. Acute success was achieved in 100% of cases. Standard ECG algorithms, conducted by 3 physicians in blind trials, only identified the correct chamber in 50-88% of the patients and sub-localized within the right ventricular outflow tract (septum v free wall) in 37 – 58% of subjects. Of note, six patients had previously attended for nine attempted ablations collectively, which were either unsuccessful or aborted owing to lack of spontaneously occurring clinical PVCs. One patient had previously reported for four separate attempts without PVCs and ablations were aborted, but collection of a single beat allowed VIVO to create an analysis map and provide the physician with information to complete the ablation for all these patients. In addition, this study showed a 27% reduction in procedure time when using VIVO as compared to a historical cohort. This study concluded that VIVO can accurately identify arrhythmia origin with an accuracy that is superior to that of established ECG algorithms.
In April 2022, one physician from the Netherlands presented an abstract at EHRA (European Heart Rhythm Association), focused on using VIVO as a way to screen patients prior to the ablation procedure. This study of 15 patients concludes that using VIVO pre-procedurally may enable the physician to determine procedure success rates and prevent unnecessary ablation procedures. This data will need to be further studied in larger numbers but determining success in advance of the procedure would improve ablation therapy, which has a high failure rate and thus requires additional ablation procedures.
In October 2021 the first patient was enrolled in the VIVO EU Registry. This registry aims to gather data about how VIVO is used in real-world settings, outside of a rigorous clinical study. The registry will enroll 125 patients across Europe and the UK and collect information about different workflows and applications for VIVO. Enrollment of 125 patients was completed in June 2023. The study requires 12 month follow-up and data collection is planned for completion in Q3 2024.This data serves multiple purposes including fulfilling European regulatory requirements for on-going data collection, publication of multi-center data, and future development of studies and improvements to the VIVO technology.
| 7 |
| Table of Contents |
Currently, there is an ongoing physician initiated study at Coventry hospital in the UK. This study will enroll 50 patients with Re-entrant Ventricular Tachycardia. These patients have hearts that are not structurally normal and scarred tissue is present in the ventricle. This data will be used for publication and to support an FDA submission to expand the current labeling of the existing product.
Our Products
VIVO™ System
Our lead product, VIVO, is an FDA-cleared and CE marked product that utilizes non-invasive inputs to locate the origin of ventricular arrhythmias. VIVO has been used in more than 1,000 procedures in leading U.S. and European hospitals under a limited commercial launch that commenced in the third quarter of 2021. A full scale commercial launch commenced in Q1 2023 in conjunction with the expansion of a direct sales force in the US.
VIVO is a non-invasive imaging system that offers 3D cardiac mapping to help with localizing the sites of origin of idiopathic ventricular arrhythmias in patients with structurally normal hearts prior to electrophysiology procedures. The VIVO system has achieved a CE Mark allowing it to be commercialized in the European Union and has been placed at several hospitals in Europe. FDA 510(k) Clearance in the United States was received in June 2019.
The VIVO software is provided on an off the shelf laptop, and the system includes a 3D camera. In addition, the system can only be used with a disposable component, the VIVO Positioning Patches, which are required for each procedure.
|
|
|
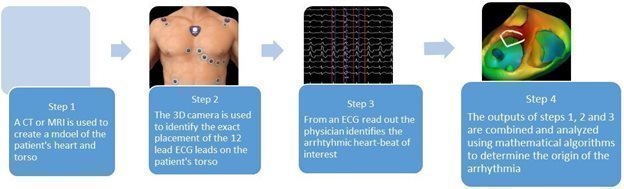
The VIVO software contains proprietary algorithms that are based on standard EP principles. However, the accuracy of the algorithms is improved because it does not use generalized assumptions and instead, uses patient specific information. VIVO uses standard clinical inputs such as a CT or MRI and a 12 lead ECG, both of which are routinely gathered for most EP procedures, allowing VIVO to seamlessly integrate into the workflow. A 3D photograph is obtained of the patient’s torso after the ECG leads are in place and all of these clinical inputs are combined to generate a 3D map of the patient’s heart with a location of the earliest onset of the ventricular arrhythmia.
| 8 |
| Table of Contents |
VIVO Workflow

LockeT
LockeT, a suture retention device, is a sterile, Class I product that was registered with the FDA in February 2023, at which time we began initial shipments to distributors. In May 2023, Catheter submitted LockeT for CE Mark approval. CE Mark approval is expected in the second half of 2024, at which time initial international shipments to distributors will begin. LockeT is indicated for wound healing by distributing suture tension over a larger area in the patient in conjunction with a figure of eight suture closure and is intended to temporarily secure sutures and aid clinicians in locating and removing sutures efficiently.
|
|
|
| 9 |
| Table of Contents |
Clinical studies for LockeT began during 2023. The three phases of the current studies are planned to show the product’s effectiveness and benefits, including faster wound closure, earlier ambulation, potentially leading to early hospital discharge, and lower costs for the healthcare provider and/or insurance payor. This data is intended to provide crucial data for marketing and to expand our indications for use with the FDA. See License and Other Agreements below.
The Phase I - First in Man Feasibility Study was completed in 2023 and showed the device works for its intended purpose, that there were no safety events and gathered initial data to support Phase II submission to Institutional Review Board (IRB). The results were submitted to the Journal of American Academy of Cardiologists in January 2024.
Phase II received IRB approval in late 2023 and is anticipated to be completed by June 2024. This phase will compare manual compression (standard of care) to LockeT in a one-to-one randomized study of 100 patients and assess improved time to hemostasis and ambulation when using LockeT versus manual compression.
Phase III IRB approval is in process and will compare LockeT to one or more competitive products in a one-to-one randomized study of 100 patients and will include cost comparisons and assess risk of hematoma when using LockeT versus those competitive products. We anticipate this phase to be completed in early 2025.
Our Previously Marketed DABRA Product
Prior to the Merger, we manufactured and marketed DABRA, a portable excimer laser console with proprietary, single-use catheters for the minimally invasive endovascular treatment of vascular blockages resulting from lower extremity vascular disease in both above and below the knee lesions.
The DABRA catheter transmitted energy from the laser to the vascular blockage. The laser energy traveled through the catheter and ablated the blockage, reducing it to chemicals that were found naturally in the bloodstream. The catheters were specifically designed for use with our excimer laser. The DABRA catheter used a liquid-filled plastic tubing allowing for the efficient and precise delivery of the laser energy.
Subsequent to the Merger, we no longer manufactured or marketed DABRA.
Our Solutions
Adoption of our VIVO System by electrophysiologists is expected to enhance their ability to diagnose and treat cardiac arrhythmias.
Non-invasive mapping prior to the ablation procedure provides a solution for patients that could not be ablated previously. First, many patients with VT do not tolerate anesthesia well. By providing a non-invasive solution to determine the ablation location, physicians are better able to understand where the arrhythmia originates and how easily one can access the ablation location, minimizing the amount of time that the patient may need to be anesthetized, and allowing many patients the ability to have an ablation that otherwise could not. Second, many patients are highly symptomatic, but do not have PVCs often. In these situations, the patients are often brought in for ablation procedures only to have no arrhythmia and sent home time and time again. In these instances, the physicians can monitor the patient prior to hospitalization and obtain information about the arrythmia. In this way, the patient can still proceed to an ablation procedure without having PVCs on the day of surgery.
Non-invasive mapping also enables planning prior to the start of the procedure. This enables the physicians to better understand where they are targeting, which enables them to make advanced decisions about where they are navigating the catheter and which catheter(s) they are using, reducing both procedure time and cost.
Surgery patients who are offered the LockeT device are expected to benefit from faster wound closure, more comfort than manual compression and earlier ambulation, potentially leading to early hospital discharge and lower costs for the healthcare provider and/or insurance payor.
| 10 |
| Table of Contents |
Our Strategy
Our goal is to become a leading medical imaging company in the field of cardiac electrophysiology, and we are dedicated to developing and delivering electrophysiology products to provide patients, hospitals, and physicians with novel technologies and solutions to improve the lives of patients with cardiac arrhythmias. We aim to establish VIVO as an integral tool used by cardiac electrophysiologists during ablation treatment of ventricular arrhythmias by reducing procedure time and patient complications and increasing procedural success.
Customers
Our primary customers are hospitals providing cardiac electrophysiology lab procedures. We believe there are 2,000 to 3,000 EP labs in the U.S. and a similar number of labs outside of the U.S. performing approximately 600,000 ablation procedures annually. During fiscal 2023, we had two individual customers that represented approximately 32% and 20% of our total revenues, respectively, and four customers (including the two just described) that in the aggregate represented approximately 72% of our total revenues.
Sales and Marketing
Today, we use a mix of distribution partners (Europe), independent sales agents (U.S.) specializing in EP products, and direct employees providing clinical support and product specialization. In the U.S., the VIVO System and patches are currently sold by direct employees and independent sales agents who call on electrophysiologists, lab staff and hospital administrators. This sales team qualifies appropriate prospective customers, and with support from our direct clinical specialists they conduct product demonstrations, and support customer training and case usage. In Europe, our products are sold through distributors, supported by three full time contracted employees.
In addition, in both the U.S. and Europe, we have entered into a co-marketing agreement with Stereotaxis, or STX. The goal is to leverage the compatibility of VIVO with their robotic system. STX customers are the same customers for VIVO, and VIVO provides their customers with an added tool to reduce procedure time. Pursuant to the agreement, STX can perform promotional activity at any hospital globally that has a Stereotaxis Robotic Magnetic Navigation System, referred to herein as a robotic hospital, and where VIVO has appropriate regulatory clearances. In addition, STX will act as a spot distributor for us at mutually agreed upon hospitals where the VIVO System is included as a line item within an STX quote. In exchange for its marketing, distribution and support activity, Stereotaxis receives a payment equal to 45% of the revenue generated from VIVO at robotic hospitals. After the initial sale of VIVO products to customers by Stereotaxis, we will be responsible for selling additional VIVO-related products to the customers but will continue to owe the 45% payment to Stereotaxis with respect to any such sales. The agreement has a term that runs through December 31, 2025, provided however, that the agreement will automatically extend for successive two-year terms unless either party provides the other written notice of termination at least one year prior to the next-scheduled termination date. Stereotaxis will continue to be entitled to receive the 45% payments described above for a period of six months following termination of the agreement.
We continue to hire additional clinical support and direct sales representation as we continue the full VIVO product launch that began in 2023. They are experienced in the electrophysiology field and will identify and target prospective customers to educate, and demonstrate our products, leading to adoption and purchase of our technology. We will continue to use direct clinical specialists to provide training and ongoing clinical support.
In the future, we intend to market our products in the U.S. and certain international markets using a combination of a direct sales force and independent distributors. This may require us to make a significant investment building our U.S. commercial infrastructure and sales force and in recruiting and training our sales representatives and clinical specialists for U.S. commercialization of VIVO. This is a lengthy process that requires recruiting appropriate sales representatives, establishing a commercial infrastructure in the United States, and training our sales representatives, and will require significant ongoing investment by us. Following initial training, our sales representatives typically require lead time in the field to grow their network of accounts, coordinate their sales efforts with each hospital’s capital budgeting and acquisition cycle and produce sales results. Successfully recruiting and training a sufficient number of productive sales representatives is required to achieve growth at the rate we desire.
| 11 |
| Table of Contents |
Marketing and market development activities will target increasing our product usage and expanding the applications of VIVO into the physician clinic and not just hospitals by employing a reimbursement specialist to provide reimbursement for VIVO in different settings.
Outside the U.S., we will continue to foster additional key partner relationships with distributors who will market, sell and support its products.
In addition, we believe there are opportunities to offer additional complementary products through our sales and marketing channels that would enhance the productivity of our sales force and provide additional scale to revenue, better covering fixed operating costs.
Manufacturing and Availability of Raw Materials
VIVO manufacturing, inventory and product fulfillment is housed in our approximate 2,000 square feet facility in Fort Mill, South Carolina. This facility currently has one full-time employee who oversees manufacturing, quality objectives, and order fulfillment. The VIVO system includes VIVO software, loaded onto an off-the-shelf laptop, which we equip with a 3D camera. We purchase laptops and cameras that have been manufactured by third parties. Disposable VIVO Positioning Patches are also required for use of the system, and the manufacture of the patches is outsourced. We also outsource updating and troubleshooting of the software, as needed, to a third party software engineering company from time to time. LockeT manufacturing, inventory and product fulfillment has been subcontracted to the company who is also providing research and development of the product.
Competition
The medical device industry is highly competitive, subject to rapid change and significantly affected by new product introductions and other activities of industry participants. We face potential competition from major medical device companies worldwide, many of which have longer, more established operating histories, and significantly greater financial, technical, marketing, sales, distribution, and other resources. Our overall competitive position is dependent upon a number of factors, including product performance and reliability, manufacturing cost, and customer support. Our primary competitors in the cardiac electrophysiology space include known medical devices such as pacemakers, electrocardiogram, or ECG, systems and cardiac catheters, but also laboratory equipment such as intracardiac mapping systems and fluoroscopy systems (similar to x-ray in real time). The EP market includes large medical device companies such as Medtronic, Plc., Abbott Laboratories, Biosense-Webster (J&J) and Boston Scientific Corp. LockeT’s direct competitors include Abbott’s Perclose device, Haemonetic’s VASCADE device and Inari Medical’s FlowStasis device.
Reimbursement
At this time, there is no reimbursement for VIVO. Ablation procedures are reimbursed using one current procedural technology, or CPT, code, which varies depending on the type and complexity of the procedure. The range of reimbursement for ablations varies within regions but can be as much as $20,000 or more.
We currently intend, in the future, to hire a reimbursement specialist to guide us through the process of obtaining a CPT code specifically for VIVO. Although a new Category III CPT codes is approved and available starting July 1, 2024, Category III codes, which are temporary, do not have a payment rate established, and payment is at the discretion of payors; further, payors generally require a high level of clinical data through long-term patient studies to demonstrate that a treatment produces favorable results in a cost-effective manner relative to other treatments, in order to be willing to provide reimbursement based on Category III codes. Successful execution of our current commercialization and build out for VIVO will be needed in order to move Category III codes to permanent Category I codes.
Research and Development
The major focus of our research and development team is to leverage our existing technology platform for new applications and improvements to our existing applications, including multiple engineering efforts to improve our current products. Future research and development efforts will involve continued enhancements to and cost reductions for VIVO and LockeT. We will also explore the development of other products that can be derived from our core technology platform and intellectual property. Our research and development team works together with our commercial team to set development priorities based on communicated customer needs. The feedback received from our customers is reviewed and evaluated for incorporation into new products.
| 12 |
| Table of Contents |
In the future we intend to develop a generation 3 of VIVO. This version would have expanded indications to include ischemic heart disease and improve usability by the hospital staff. It would also contain more automaticity, potentially reducing our need for clinical support.
Resources Material to Our Business
Patents and Proprietary Technology
Patents
We have a number of patents covering our intellectual property, both in the U.S., as well in a number of international countries. We consider the U.S. to be the most important market for our products, and hence, the most important country for the filing of patents. Any foreign filings are merely replicates of the U.S. filings. For the U.S., we have the following patent positions for the different product areas:
| · | VIVO – We have two U.S. patents granted on the original VIVO concept, which have been licensed from a third party. We consider the primary component to be the ideas around utilizing a 3D camera to identify the exact location of the body surface electrodes. These two patents expire in 2038. An additional two applications have been granted, which disclosed ideas around merging of the heart models to other heart images and expire in 2038 and 2040. An additional three applications were published, all filed in 2021, covering the idea of determining the thickness of the wall of the ventricle, covering the concept of the rendering of a heart model and likely outcomes of an EP procedure. An additional application was filed in September 2023 and is not yet published. |
|
|
|
| · | LockeT – Suture Retention Device - We have four published U.S. patent applications. These cover the basic concept, methods of use and the design of the conceived device. |
|
|
|
| · | AMIGO – We have twenty issued U.S. patents. The first patent, filed in 2006 and expiring in 2031, covers the basic idea, with a three way motor, a remote control, a sled device, and a docking station for a catheter. The more detailed ideas behind the original concept were covered in three patents filed between 2011 and 2013 and expiring in 2026. Additional concepts and methods were filed with six patents between 2010 and 2013, with expirations between 2029 and 2031. We consider the most relevant of the intellectual property to be the guiding track with opposing flexible guides to hold the catheter stable as it is advanced, the form and function of the controller handle, and the introducer interface of the arm to the introducer. An additional ten patents, filed between 2013 and 2017, and expiring in 2034 to 2037, are patents covering ideas not used in the original commercial device, but potential ideas for future embodiments. |
License and Other Agreements
PEACS, NV Software and Technology License Agreement
On May 1, 2016, we entered into a certain Software and Technology License Agreement with PEACS, NV, a Netherlands company, or the License Agreement, for the exclusive worldwide license of the underlying technology to its VIVO product, including intellectual property rights and patent applications pertaining thereto. The license was for use of the technology for the field of use defined as “the localization of the origin of cardiac activation for the electrophysiology treatment and/or detection of cardiac arrhythmias.” The License Agreement called for us to pay for the prosecution and maintenance of patents to protect the technology.
In May 2021, the License Agreement was modified to modify the field of use to specifically exclude the use of clinical applications for the implanting of atrial or ventricular pacemakers, including bi-ventricular pacemakers.
| 13 |
| Table of Contents |
LockeT Royalty Agreement
In February 2022, we agreed to an assignment and royalty agreement, or the Royalty Agreement, for the LockeT device. Pursuant to the Royalty Agreement, we agreed to pay a royalty fee of 5% on net sales up to $1 million. Thereafter, if a patent for the LockeT device is obtained from the U.S. Patent and Trademark Office, we will pay a royalty fee of 2% of net sales up to a total of $10 million in royalties. In addition, at the time of the Merger, additional royalty rights with respect to LockeT device were granted to certain holders, or the Noteholders, of Old Catheter’s outstanding convertible promissory notes in exchange for forgiveness of the interest that had accrued under those notes but remained unpaid, pursuant to the terms of certain Debt Settlement Agreements. The Debt Settlement Agreements provided for the Noteholders to receive, in the aggregate, approximately 12% of the net sales, if any, of the LockeT device, commencing upon the first commercial sale through December 31, 2035.
Trademarks
We own or have rights to trademarks that we use in connection with the operation of our business. We own or have rights to trademarks for Ra Medical Systems and Catheter Precision and their logos, as well as other trademarks such as AMIGO. In February 2024 we filed a trademark for LockeT.
Trade Secrets
We also have relied upon trade secrets, know-how and technological innovation, and may in the future rely upon licensing opportunities, to develop and maintain its competitive position. We have protected our proprietary rights through a variety of methods, including confidentiality agreements and proprietary information agreements with suppliers, employees, consultants and others who may have access to proprietary information.
Government Regulations
Governmental authorities in the U.S. (at the federal, state, and local levels) and abroad extensively regulate, among other things, the research and development, testing, manufacture, quality control, clinical research, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing, and export and import of products such as those we market and are developing. See Item 1.A. Risk Factors—Risks Related to Government Regulation.
United States Medical Device Regulation
In the U.S., medical devices are subject to extensive regulation by the Food and Drug Administration (“FDA”), pursuant to the Food, Drug and Cosmetic Act, or FDCA, and its implementing regulations, and certain other federal and state statutes and regulations. The laws and regulations govern, among other things, the design, manufacture, storage, recordkeeping, approval, labeling, promotion, post-approval monitoring and reporting, distribution and import and export of medical devices. Failure to comply with applicable requirements may subject a device and/or its manufacturer to a variety of administrative sanctions, such as FDA refusal to approve pending pre-market approval (“PMA”), applications or premarket notification submissions (commonly referred to as “510(k)s),” issuance of warning letters or untitled letters, product recalls, import detentions, civil monetary penalties, and/or judicial sanctions, such as product seizures, injunctions, and criminal prosecution.
The FDCA classifies medical devices into one of three categories based on the risks associated with the device and the level of control necessary to provide reasonable assurance of safety and effectiveness. Class I devices are deemed to be low risk and are subject to the fewest regulatory controls. Class II devices provide intermediate levels of risk. They are subject to general controls and must also comply with special controls. Class III devices are generally the highest risk devices and are subject to the highest level of regulatory control to provide reasonable assurance of the device’s safety and effectiveness. Class III devices must typically be approved by the FDA before they are marketed. LockeT is a sterile, Class I product and was registered with the FDA in February of 2023. It does not require FDA marketing authorization. VIVO is an FDA-cleared Class II product.
Establishments that manufacture devices are required to register their establishments with the FDA and provide the FDA a list of the devices that they handle at their facilities.
| 14 |
| Table of Contents |
The FDA conducts market surveillance and periodic visits, both announced and unannounced, to inspect or re-inspect equipment, facilities, laboratories and processes to confirm regulatory compliance. These inspections may include the manufacturing facilities of subcontractors. Following an inspection, the FDA may issue a report, known as a Form 483, listing instances where the manufacturer has failed to comply with applicable regulations and/or procedures or, if observed violations are severe and urgent, a warning letter. If the manufacturer does not adequately respond to a Form 483 or warning letter, the FDA (with assistance from the Justice Department in certain cases) make take enforcement action against the manufacturer or impose other sanctions or consequences, which may include:
| · | injunctions or consent decrees; |
|
|
|
| · | civil monetary penalties; |
|
|
|
| · | recall, detention or seizure of our products; |
|
|
|
| · | operating restrictions, partial or total shutdown of production facilities; |
|
|
|
| · | refusal of or delay in granting requests for 510(k) clearance, de novo classification, or premarket approval of new products or modified products; |
|
|
|
| · | withdrawing 510(k) clearances, de novo classifications, or premarket approvals that are already granted; |
|
|
|
| · | refusal to grant export approval or export certificates or devices; and |
|
|
|
| · | criminal prosecution. |
Pre-Market Authorization and Notification
While most Class I and some Class II devices can be marketed without prior FDA authorization, most medical devices can be legally sold within the U.S. only if the FDA has: (i) approved a PMA application prior to marketing, generally applicable to most Class III devices; (ii) cleared the device in response to a premarket notification, or 510(k) submission, generally applicable to Class I and II devices; or (iii) authorized the device to be marketed through the de novo process, generally applicable for novel Class I or II devices. Some devices that have been classified as Class III are regulated pursuant to the 510(k) requirements because the FDA has not yet called for PMAs for these devices.
510(k) Notification
Product marketing in the U.S. for most Class II and limited Class I devices typically follows a 510(k) pathway. To obtain 510(k) clearance, a manufacturer must submit a premarket notification demonstrating that the proposed device is substantially equivalent to a legally marketed device, referred to as the predicate device. A predicate device may be a previously 510(k) cleared device or a device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for submission of PMA applications, or a product previously granted de novo authorization. The manufacturer must show that the proposed device has the same intended use as the predicate device, and it either has the same technological characteristics, or it is shown to be equally safe and effective and does not raise different questions of safety and effectiveness as compared to the predicate device.
There are three types of 510(k)s: traditional; special, for certain device modifications; and abbreviated, for devices that conform to a recognized standard. The special and abbreviated 510(k)s are intended to streamline review. The FDA intends to process special 510(k)s within 30 days of receipt and abbreviated 510(k)s within 90 days of receipt. Though the FDA has a goal to clear a traditional 510(k) within 90 days of receipt, the clearance pathway for traditional 510(k)s can take substantially longer.
After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a new 510(k) clearance or could require a PMA. The FDA requires each manufacturer to make this determination in the first instance, but the FDA can review any such decision. If the FDA disagrees with a manufacturer’s decision not to seek a new 510(k) clearance for the modified device, the agency may retroactively require the manufacturer to seek 510(k) clearance or PMA. The FDA also can require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or PMA is obtained.
| 15 |
| Table of Contents |
VIVO was cleared by the FDA via a traditional 510(k) with supporting clinical data. This data was collected via a clinical study enrolling 51 subjects and took approximately 12 months to gather. In order to expand the indications for use of the current VIVO product with the FDA to include ischemic hearts, data collection via the Coventry study to support a new 510(k) submission will be required. It is expected that future generations of VIVO will require similar data collection and 510(k) submission to receive separate FDA clearance.
Because the LockeT device is a Class I product, it did not require clinical data or a formal submission process. After completing validation testing and compiling a Device History File, LockeT was added to our listing of registered devices. The regulatory pathway for future LockeT devices will depend on the intended use and desired labeling claims and the requirements for clinical data.
De Novo Classification
Devices of a new type that the FDA has not previously classified based on risk are automatically classified into Class III by operation of section 513(f) (1) of the FDCA, regardless of the level of risk they pose. To avoid requiring PMA review of low- to moderate-risk devices classified in Class III by operation of law, Congress enacted section 513(f)(2) of the FDCA. This provision allows the FDA to classify a low- to moderate-risk device not previously classified into Class I or II through the de novo classification pathway. The FDA evaluates the safety and effectiveness of devices submitted for review under the de novo classification pathway and devices determined to be Class II through this pathway can serve as predicate devices for future 510(k) applicants. The de novo classification pathway can require clinical data and is generally more burdensome than the 510(k) pathway and less burdensome than the PMA pathway.
Pre-Market Approval
A product not eligible for 510(k) clearance or de novo classification must follow the PMA pathway, which requires proof of the safety and effectiveness of the device to the FDA’s satisfaction.
Results from adequate and well-controlled clinical trials are required to establish the safety and effectiveness of a Class III PMA device for each indication for which FDA approval is sought. After completion of the required clinical testing, a PMA including the results of all preclinical, clinical, and other testing, and information relating to the product’s marketing history, design, labeling, manufacture, and controls, is prepared and submitted to the FDA.
The PMA process is generally more expensive, rigorous, lengthy, and uncertain than the 510(k) premarket notification process and de novo classification process and requires proof of the safety and effectiveness of the device to the FDA’s satisfaction. As part of the PMA review, the FDA will typically inspect the manufacturer’s facilities for compliance with Quality System Regulations, or QSR, requirements, which impose elaborate testing, control, documentation and other quality assurance procedures. The FDA’s review of a PMA application typically takes one to three years but may last longer. The FDA will often convene an independent advisory panel to review the submission. If the FDA’s evaluation of the PMA application is favorable, the FDA will issue a PMA for the approved indications, which can be more limited than those originally sought by the manufacturer. The PMA can include post-approval conditions that the FDA believes necessary to ensure the safety and effectiveness of the device including, among other things, restrictions on labeling, promotion, sale and distribution. Failure to comply with the conditions of approval can result in material adverse enforcement action, including the loss or withdrawal of the approval and/or placement of restrictions on the sale of the device until the conditions are satisfied.
Even after approval of a PMA, a new PMA or PMA supplement may be required in the event of a modification to the device, its labeling or its manufacturing process. Supplements to a PMA often require the submission of the same type of information required for an original PMA, except that the supplement is generally limited to that information needed to support the proposed change from the product covered by the original PMA.
Clinical Trials
A clinical trial is almost always required to support a PMA application and de novo classification and is sometimes required for a premarket notification. For significant risk devices, the FDA regulations require that human clinical investigations conducted in the U.S. be approved under an Investigational Device Exemption (“IDE”), which must become effective before clinical testing may commence. A nonsignificant risk device does not require FDA approval of an IDE. In some cases, one or more smaller IDE studies may precede a pivotal clinical trial intended to demonstrate the safety and efficacy of the investigational device. A 30-day waiting period after the submission of each IDE is required prior to the commencement of clinical testing in humans. If the FDA disapproves the IDE within this 30-day period, the clinical trial proposed in the IDE may not begin. In addition, even if the 30-day waiting period expires without objection by the FDA, the FDA can impose a clinical hold if safety issues arise.
| 16 |
| Table of Contents |
An IDE application must be supported by appropriate data, such as animal and laboratory test results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE application must also include a description of product manufacturing and controls, and a proposed clinical trial protocol. The FDA typically grants IDE approval for a specified number of patients to be treated at specified study centers. During the study, the sponsor must comply with the FDA’s IDE requirements for investigator selection, trial monitoring, reporting, and record keeping. The investigators must obtain patient informed consent, follow the investigational plan and study protocol, control the disposition of investigational devices, and comply with reporting and record keeping requirements. Prior to granting PMA, the FDA typically inspects the records relating to the conduct of the study and the clinical data supporting the PMA application for compliance with IDE requirements.
Clinical trials must be conducted: (i) in compliance with federal regulations; (ii) in compliance with good clinical practice, or GCP, an international standard intended to protect the rights and health of patients and to define the roles of clinical trial sponsors, investigators, and monitors; and (iii) under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. Pivotal clinical trials supporting premarket applications for devices are typically conducted at geographically diverse clinical trial sites and are designed to permit the FDA to evaluate the overall benefit-risk relationship of the device and to provide adequate information for the labeling of the device when considering whether a device satisfies the statutory standard for commercialization. Clinical trials, for significant and nonsignificant risk devices, must be approved by an institutional review board, or IRB—an appropriately constituted group that has been formally designated to review and monitor biomedical research involving human subjects and which has the authority to approve, require modifications in, or disapprove research to protect the rights, safety, and welfare of the human research subject.
The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with the FDA requirements or presents an unacceptable risk to the clinical trial patients. An IRB may also require the clinical trial it has approved to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions or sanctions.
Although the QSR does not fully apply to investigational devices, the requirement for controls on design and development does apply. The sponsor also must manufacture the investigational device in conformity with the quality controls described in the IDE application and any conditions of IDE approval that the FDA may impose with respect to manufacturing. Investigational devices may only be distributed for use in an investigation and must bear a label with the statement: “CAUTION-Investigational device. Limited by Federal law to investigational use.”
Post-Market Requirements
After a device is placed on the market, numerous regulatory requirements apply. These include: the QSR requirements, labeling regulations, the medical device reporting regulations (which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur), and reports of corrections and removals regulations (which require manufacturers to report recalls or removals and field corrections to the FDA if initiated to reduce a risk to health posed by the device or to remedy a violation of the FDCA). Failure to properly identify reportable events or to file timely reports, as well as failure to address observations to FDA’s satisfaction, can subject us to warning letters, recalls, or other sanctions and penalties.
Advertising, marketing and promotional activities for devices are also subject to FDA oversight and must comply with the statutory standards of the FDCA, and the FDA’s implementing regulations. The FDA’s oversight authority review of marketing and promotional activities encompasses, but is not limited to, direct-to-consumer advertising, healthcare provider-directed advertising and promotion, sales representative communications to healthcare professionals, promotional programming and promotional activities involving electronic media. The FDA also regulates industry-sponsored scientific and educational activities that make representations regarding product safety or efficacy in a promotional context.
| 17 |
| Table of Contents |
Manufacturers of medical devices are permitted to promote products solely for the uses and indications set forth in the approved or cleared product labeling. A number of enforcement actions have been taken against manufacturers that promote products for “off-label” uses (i.e., uses that are not described in the approved or cleared labeling), including actions alleging that claims submitted to government healthcare programs for reimbursement of products that were promoted for “off-label” uses are fraudulent in violation of the Federal False Claims Act or other federal and state statutes and that the submission of those claims was caused by off-label promotion. The failure to comply with prohibitions on “off-label” promotion can result in significant monetary penalties, revocation or suspension of a company’s business license, suspension of sales of certain products, product recalls, civil or criminal sanctions, exclusion from participating in federal healthcare programs, or other enforcement actions. In the United States, allegations of such wrongful conduct could also result in a corporate integrity agreement with the U.S. government that imposes significant administrative obligations and costs, as has occurred in the past with respect to our legacy products that we no longer market.
The Federal Trade Commission, or FTC, also oversees the advertising and promotion of our products (other than labeling) pursuant to its broad authority to police deceptive advertising for goods or services within the U.S. The FDA and FTC work together to regulate different aspects of activities by medical product manufacturers, consistent with the inter-agency Memorandum of Understanding. Under the Federal Trade Commission Act, or FTCA, the FTC is empowered, among other things, to (a) prevent unfair methods of competition and unfair or deceptive acts or practices in or affecting commerce; (b) seek monetary redress and other relief for conduct injurious to consumers; and (c) gather and compile information and conduct investigations relating to the organization, business, practices, and management of entities engaged in commerce. In the context of performance claims for products such as our devices and services, compliance with the FTC Act includes ensuring that there is scientific data to substantiate the claims being made, that the advertising is neither false nor misleading, and that any user testimonials or endorsements we or our agents disseminate related to the devices or services comply with disclosure and other regulatory requirements.
Violations of the FDCA or FTCA relating to the inappropriate promotion of approved products may lead to investigations alleging violations of federal and state healthcare fraud and abuse and other laws, including state consumer protection laws.
For a PMA or Class II 510(k) or de novo devices, the FDA also may require post-marketing testing, surveillance, or other measures to monitor the effects of an approved or cleared product. The FDA may place conditions on a PMA-approved device that could restrict the distribution or use of the product. In addition, quality-control, manufacture, packaging, and labeling procedures must continue to conform to QSRs and other applicable regulatory requirements after approval and clearance, and manufacturers are subject to periodic inspections by the FDA. Accordingly, manufacturers must continue to expend time, money, and effort in the areas of production and quality-control to maintain compliance with QSRs. If the FDA believes we or any of our contract manufacturers or regulated suppliers are not in compliance with these requirements and patients are being subjected to serious risks, the agency can shut down our manufacturing operations, require recalls of our medical device products, refuse to approve new marketing applications, initiate legal proceedings to detain or seize products, enjoin future violations, or assess civil and criminal penalties against us or our officers or other employees.
European Economic Area (EEA) Regulation
The EEA recognizes a single medical device approval (the CE Mark) which allows for distribution of an approved product throughout the EEA without additional general applications in each country. Individual EEA members, however, reserve the right to require additional labeling or information to address particular patient safety issues prior to allowing marketing. Third parties called “Notified Bodies” award the CE Mark. These Notified Bodies are approved and subject to review by the “Competent Authorities” of their respective countries. Our Notified Bodies perform periodic on-site inspections to independently review our compliance with systems and regulatory requirements. A number of countries outside of the EEA accept the CE Mark in lieu of marketing submissions as an addendum to that country’s application process. We have a CE Mark for the VIVO System. Beginning July 1, 2023, the United Kingdom requires its own medical device approval (UKCA). VIVO is currently registered with UK’s Medicines and Healthcare products Regulatory Agency to market the VIVO system in the UK. Since July 1, 2023, VIVO bears the UKCA symbol as required by the UK Medical Devices Regulations 2002 (SI 2002 No 618, as amended) (“MDR”) to continue UK distributions. MDR requirements now include on-going collection of clinical data to include in annual reports to ensure state of the art technology and safety requirements are met. We are currently collecting data via a multi-center (and country) European Registry. This registry concluded enrollment in June 2023 and is anticipated to complete data collection and study activities in September 2024.
| 18 |
| Table of Contents |
LockeT is currently undergoing MDR review and approval via the Notified Body. CE Mark is anticipated in August 2024.
Other Healthcare Laws
Our business operations and current and future arrangements with healthcare professionals, consultants, customers and patients, expose us to broadly applicable state, federal, and foreign fraud and abuse and other healthcare laws and regulations. These laws constrain the business and financial arrangements and relationships through which we conduct our operations, including how we research, market, sell and distribute our products. Such laws include, but are not limited to:
| · | the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under a U.S. healthcare program such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the U.S. federal Anti-Kickback Statute or specific intent to violate it in order to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act; |
|
|
|
| · | U.S. federal civil and criminal false claims laws and civil monetary penalties laws, including the federal civil False Claims Act, which, among other things, impose criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the U.S. government, claims for payment or approval that are false or fraudulent, knowingly making, using or causing to be made or used, a false record or statement material to a false or fraudulent claim, or from knowingly making a false statement to avoid, decrease or conceal an obligation to pay money to the U.S. government. Persons and entities can be held liable under these laws if they are deemed to “cause” the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers or promoting a product off-label; |
|
|
|
| · | the U.S. Health Insurance Portability and Accountability Act of 1996, or HIPAA, which imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement, in connection with the delivery of, or payment for, healthcare benefits, items or services. Similar to the U.S. federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the health care fraud statute implemented under HIPAA or specific intent to violate it in order to have committed a violation; |
|
|
|
| · | in addition, HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH Act, and its implementing regulations, imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information without appropriate authorization by covered entities subject to the rule, such as health plans, healthcare clearinghouses and certain healthcare providers as well as their business associates that perform certain services for or on their behalf involving the use or disclosure of individually identifiable health information; |
|
|
|
| · | the U.S. Physician Payments Sunshine Act, which requires applicable manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the government information related to payments or other “transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), non-physician healthcare professionals (defined to include physician assistants, nurse practitioners, clinical nurse specialists, certified registered nurse anesthetists and anesthesiologist assistants, and certified nurse-midwives) and teaching hospitals, as well as information regarding ownership and investment interests held by the physicians described above and their immediate family members; and |
|
|
|
| · | analogous state and non-U.S. laws and regulations, such as state anti-kickback and false claims laws, which may apply to our business practices, including, but not limited to, research, distribution, sales and marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers, or by the patients themselves; state laws that require pharmaceutical and device companies to comply with the industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the U.S. government, or otherwise report or restrict payments that may be made to healthcare providers and other potential referral sources; state laws and regulations that require manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures and pricing information; and state and non-U.S. laws governing the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
| 19 |
| Table of Contents |
In particular, activities and arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, waste and other abusive practices. These laws and regulations may restrict or prohibit a wide range of activities or other arrangements related to the development, marketing or promotion of products, including pricing and discounting of products, provision of customer incentives, provision of reimbursement support, other customer support services, provision of sales commissions or other incentives to employees and independent contractors and other interactions with healthcare practitioners, other healthcare providers and patients.
Because of the breadth of these laws and the narrow scope of the statutory or regulatory exceptions and safe harbors available, our business activities could be challenged under one or more of these laws. Relationships between medical product manufacturers and health care providers are an area of heightened scrutiny by the government.
Government expectations and industry best practices for compliance continue to evolve and past activities may not always be consistent with current industry best practices. Further, there is a lack of government guidance as to whether various industry practices comply with these laws, and government interpretations of these laws continue to evolve, all of which creates compliance uncertainties. Any non-compliance could result in regulatory sanctions, criminal or civil liability and serious harm to our reputation. It is not always possible to identify and deter misconduct, and the precautions we take to detect and prevent this activity may not be effective in preventing such conduct, mitigating risks, or reducing the chance of governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations.
If a government entity opens an investigation into possible violations of any of these laws (which may include the issuance of subpoenas), we would have to expend significant resources to defend ourselves against the allegations. Defending against any such actions can be costly, time-consuming and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired. If any of the above occur, it could adversely affect our ability to operate our business and our results of operations.
Allegations that we, our officers, or our employees violated any one of these laws can be made by individuals called “whistleblowers” who may be our employees, customers, competitors or other parties. Government policy is to encourage individuals to become whistleblowers and file a complaint in federal court alleging wrongful conduct. The government is required to investigate all of these complaints and decide whether to intervene. If the government intervenes and we are required to pay money back to the government as a result of a settlement or judgement, the whistleblower, as a reward, is awarded a percentage. If the government declines to intervene, the whistleblower may proceed on his or her own and, if successful, he or she will receive a percentage of any judgment or settlement amount the company is required to pay. The government may also initiate an investigation on its own. If any such actions are instituted against us, those actions could have a significant impact on our business, including the imposition of significant fines, and other sanctions that may materially impair our ability to run a profitable business. In particular, if our operations are found to be in violation of any of the laws described above or if we agree to settle with the government without admitting to any wrongful conduct or if we are found to be in violation of any other governmental regulations that apply to us, we, our officers and employees may be subject to sanctions, including civil and criminal penalties, damages, fines, exclusion from participation in government health care programs, such as Medicare and Medicaid, imprisonment, the curtailment or restructuring of our operations and the imposition of a corporate integrity agreement, any of which could adversely affect our business, results of operations and financial condition.
| 20 |
| Table of Contents |
Foreign Corrupt Practices Act
The Foreign Corrupt Practices Act, or FCPA, and similar anti-bribery laws in other jurisdictions, generally prohibit businesses and their representatives from offering to pay, paying, promising to pay or authorizing the payment of money or anything of value to a foreign official in order to influence any act or decision of the foreign official in his or her official capacity or to secure any other improper advantage in order to obtain or retain business. The FCPA also obligates companies whose securities are listed in the U.S. to comply with accounting provisions requiring us to maintain books and records, which in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the corporation, including international subsidiaries, if any, and to devise and maintain a system of internal accounting controls sufficient to provide reasonable assurances regarding the reliability of financial reporting and the preparation of financial statements. The scope of the FCPA includes interactions with certain healthcare professionals in many countries.
There is no assurance that our internal control policies and procedures will protect us from acts committed by our employees or agents. If we are found to be liable for FCPA or other violations (either due to our own acts or our inadvertence, or due to the acts or inadvertence of others), we could suffer from civil and criminal penalties or other sanctions, including contract cancellations or debarment, and loss of reputation, any of which could have a material adverse impact on our business, financial condition, and results of operations.
Privacy and Data Protection Laws
HIPAA, as amended by the HITECH Act, and the regulations that have been issued under it, impose certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of protected health information. In the U.S., certain states have additional privacy laws that also work towards the safeguarding of personal information.
HIPAA requirements and restrictions apply to “covered entities” (which include health care providers and insurers) as well as to their business associates that receive protected health information from them in order to provide services to or perform certain activities on their behalf. The statute and regulations also impose notification obligations on covered entities and their business associates in the event of a breach of the privacy or security of protected health information. We occasionally receive protected health information from our customers in the course of our business. As such, we believe that we are business associates and therefore subject to HIPAA’s requirements and restrictions with respect to handling such protected health information and have executed business associate agreements with certain customers.
It is possible the data protection laws may be interpreted and applied in a manner that is inconsistent with our practices. If so, this could result in government-imposed fines or orders requiring that we change our practices, which could adversely affect our business. In addition, these privacy regulations may differ from country to country and state to state and may vary based on whether testing is performed in the U.S. or in the local country. Complying with these various laws and regulations could cause us to incur substantial costs or require us to change our business practices and compliance procedures in a manner adverse to our business. Further, compliance with data protection laws and regulations could require us to take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, or in some cases, impact our ability to operate in certain jurisdictions. We can provide no assurance that we are or will remain in compliance with diverse privacy and security requirements in all of the jurisdictions in which we do business. If we fail to comply or are deemed to have failed to comply with applicable privacy protection laws and regulations such failure could result in government enforcement actions and create liability for us, which could include substantial civil and/or criminal penalties, as well as private litigation and/or adverse publicity that could negatively affect our operating results and business.
| 21 |
| Table of Contents |
Environmental Regulation
We are subject to federal, state and local regulations governing the storage, use and disposal of waste materials and products. Although we believe that our safety procedures for storing, handling and disposing of these materials and products comply with the standards prescribed by law and regulation, we cannot eliminate the risk of accidental contamination or injury from those hazardous materials. Federal regulations promulgated by the Occupational Safety and Health Administration impose additional requirements on us, including those protecting employees from exposure to elements such as blood-borne pathogens. We cannot predict the frequency of compliance, monitoring, or enforcement actions to which we may be subject as those regulations are being implemented, which could adversely affect our operations.
Segment Information
We operate our business as one segment which includes all activities related to the marketing, sales and development of medical technologies focused in the field of cardiac EP. The chief operating decision-maker reviews the operating results on an aggregate basis and manages the operations as a single operating segment.
Employees
As of March 12, 2024, we had a total of 14 employees, including 14 full-time employees, which includes finance and administrative, sales and marketing and clinical professionals. We also have retained a total of 5 persons as independent contractors. We are planning to increase our sales force in support of product launches but currently have no other plans to increase our staff.
Corporate Information
Our principal executive offices are located at 1670 Highway 160 West, Suite 205, Fort Mill, South Carolina 29708. Our telephone number is (973) 691-2000. Our corporate website address is www.catheterprecision.com. Information contained on, or that can be accessed through, our website is not incorporated by reference into this document, and you should not consider information on our website to be part of this document.
You may find on our website at www.catheterprecision.com electronic copies of our Annual Report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act of 1934, or Exchange Act. Such filings are placed on our website as soon as reasonably possible after they are filed with the Securities and Exchange Commission, or SEC.
| 22 |
| Table of Contents |
ITEM 1A. RISK FACTORS
Investing in our common stock involves a high degree of risk. You should carefully consider the risks and uncertainties described below, together with all of the other information in this Annual Report on Form 10-K, before making an investment decision. The risks and uncertainties described below may not be the only ones we face. If any of the risks actually occur, our business, financial condition, operating results, cash flows and prospects could be materially and adversely affected. In that event, the market price of our common stock could decline, and you could lose part or all of your investment.
Risk Factor Summary (This Summary is not intended to and does not describe all of the risk factors discussed below that may impact the Company. We urge investors to review the detailed descriptions of risk factors that follow.)
Risks Related to Our Financial Position and Need for Additional Capital
| · | We will be required to raise additional funds to finance our operations and continue as a going concern; We may not be able to do so when necessary, and/or the terms of any financings may not be advantageous to us. |
|
|
|
| · | Our business has a history of losses, will incur additional losses, and may never achieve profitability. |
Risks Related to Our Internal Controls
| · | We have identified material weaknesses in our internal control over financial reporting. These material weaknesses could adversely affect our ability to report our results of operations and financial condition accurately and in a timely manner. |
|
|
|
| · | Compliance with Sarbanes-Oxley Act Section 404 could have a material adverse impact on our business. |
Risks Related to Our Business and Products
| · | We will not be able to reach profitability unless we are able to achieve our product expansion and growth goals; our VIVO launch plans require significant investment in infrastructure and sales representatives. |
|
|
|
| · | Our research and development and commercialization efforts may depend on entering into agreements with corporate collaborators. |
|
|
|
| · | We have entered into joint marketing agreements with respect to our products, and may enter into additional join marketing agreements, that will reduce our revenues from product sales. |
|
|
|
| · | Royalty agreements with respect to LockeT, the surgical vessel closing pressure device, will reduce any future profits from this product. |
|
|
|
| · | If we experience significant disruptions in our information technology systems, our business may be adversely affected. |
|
|
|
| · | Litigation and other legal proceedings may adversely affect our business. |
|
|
|
| · | If we make acquisitions or divestitures, we could encounter difficulties that harm our business. |
|
|
|
| · | Failure to attract and retain sufficient qualified personnel could also impede our growth. |
|
|
|
| · | Our revenues may depend on our customers’ receipt of adequate reimbursement from private insurers and government sponsored healthcare programs. |
|
|
|
| · | We may be unable to compete successfully with companies in our highly competitive industry, many of whom have substantially greater resources than we do. |
|
|
|
| · | Our future operating results depend upon our ability to obtain components in sufficient quantities on commercially reasonable terms or according to schedules, prices, quality and volumes that are acceptable to us, and suppliers may fail to deliver components, or we may be unable to manage these components effectively or obtain these components on such terms. |
|
|
|
| · | If hospitals, physicians and patients do not accept our current and future products or if the market for indications for which any product candidate is approved is smaller than expected, we may be unable to generate significant revenue, if any. |
|
|
|
| · | The recent coronavirus outbreak (“COVID-19”) adversely affected our financial condition and results of operations and we cannot provide any certainty as to whether there will be future impacts from COVID-19 or another pandemic. |
|
|
|
| · | A variety of risks associated with marketing our products internationally could materially adversely affect our business. |
| 23 |
| Table of Contents |
| · | The impact of the military conflicts in Ukraine and Israel, and the actions that have been and could be taken by other countries, including new and stricter sanctions and actions taken in response to such sanctions, have affected, and may continue to affect, our business and results of operations, including our supply chain. |
|
|
|
| · | If the third parties on which we rely for the conduct of our clinical trials and results do not perform our clinical trial activities in accordance with good clinical practices and related regulatory requirements, we may be unable to obtain regulatory approval for or commercialize our product candidates. |
|
|
|
| · | We may be adversely affected by product liability claims, unfavorable court decisions or legal settlements. |
|
|
|
| · | Our ability to use our net operating loss carryforwards may be limited. |
|
|
|
| · | We may have to make milestone payments under the Settlement Agreement we entered into with the Department of Justice (“DOJ”). |
Risks Related to Government Regulation and our Industry
| · | We are subject to pervasive and continuing regulation by the FDA and other regulatory agencies. Our products may be subject to additional recalls, revocations or suspensions after receiving FDA or foreign approval or clearance, which could divert managerial and financial resources, harm our reputation, and adversely affect our business. |
|
|
|
| · | Changes in trade policies among the U.S. and other countries, in particular the imposition of new or higher tariffs, could place pressure on our average selling prices as our customers seek to offset the impact of increased tariffs on their own products. Increased tariffs or the imposition of other barriers to international trade could have a material adverse effect on our revenues and operating results. |
|
|
|
| · | Product clearances and approvals can often be denied or significantly delayed. |
|
|
|
| · | Although we have obtained regulatory clearance for our VIVO and LockeT products in the U.S. and certain non-U.S. jurisdictions, our business plans include expanding uses for our products, which will require additional clearances; and even after clearance is obtained, our products remain subject to extensive regulatory scrutiny. |
|
|
|
| · | If we or our suppliers fail to comply with the FDA’s Quality System Regulation, or QSR, or any applicable state equivalent, our operations could be interrupted, and our potential product sales and operating results could suffer. |
|
|
|
| · | Our products may be subject to additional recalls, revocations or suspensions after receiving FDA or foreign approval or clearance, which could divert managerial and financial resources, harm our reputation, and adversely affect our business. |
|
|
|
| · | If any of our products cause or contribute to a death or a serious injury, or malfunction in certain ways, we will be required to report under applicable medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions. |
|
|
|
| · | Healthcare reform initiatives and other administrative and legislative proposals may adversely affect our business, financial condition, results of operations and cash flows in our key markets. |
Risks Related to our Intellectual Property
| · | If we are unable to obtain and maintain patent protection for our products, our competitors could develop and commercialize products and technology similar or identical to ours, and our ability to successfully commercialize our existing products and any products we may develop, and our technology may be adversely affected. |
| 24 |
| Table of Contents |
Risks Related to Ownership of Our Common Stock Including Volatility and Highly Concentrated Ownership
Risks Related to Our Financial Position and Need for Additional Capital
We will be required to raise additional funds to finance our operations and continue as a going concern; We may not be able to do so when necessary, and/or the terms of any financings may not be advantageous to us.
Our operations to date have consumed substantial amounts of cash and our business, including the business of Old Catheter conducted prior to its being acquired by the Company, sustained negative cash flows from operations for the last several years. In addition, our auditors’ report on our financial statements included in this Form 10-K contains an explanatory paragraph about the substantial doubt to continue as a going concern. As of March 7, 2024, we have approximately $1.86 million in cash and cash equivalents, which, together with our anticipated cash from operations, is not adequate to meet our working capital needs through May of 2024, and our business is currently not profitable. During the first quarter of 2023 we raised approximately $9.3 million in proceeds from securities transactions, but Merger costs and other negative cash flows have substantially depleted our cash. As a result, we will require future additional capital infusions including public or private financing, strategic partnerships or other arrangements with organizations that have capabilities and/or products that are complementary to our own capabilities and/or products, in order to execute our strategic vision. However, there can be no assurances that we can complete any financings, strategic alliances or collaborative development agreements, and the terms of such arrangements may not be advantageous to us. In addition, any additional equity financing will be dilutive to our current stockholders, and debt financing, if available, may involve restrictive covenants. If we raise funds through collaborative or licensing arrangements, we may be required to relinquish, on terms that are not favorable to us, rights to some of our technologies or product candidates that we would otherwise seek to develop or commercialize. Our failure to raise capital when needed could materially harm our business, financial condition, and results of operations. See “—We have entered into joint marketing agreements with respect to our products, and may enter into additional join marketing agreements, that will reduce our revenues from product sales,” and “—Royalty agreements with respect to LockeT, the surgical vessel closing pressure device, will reduce any future revenues from this product.”
Our business has a history of losses and will incur additional losses, and we may never achieve profitability.
Our current business primarily derives revenues from the View into Ventricular Onset System or VIVO™ System (“VIVO” or “VIVO System”). VIVO is FDA cleared and CE marked, having received FDA 510(k) clearance in June 2019. Old Catheter began a limited commercial launch of VIVO in the third quarter of 2021, and we began a full-scale launch in 2023 in conjunction with the expansion of a direct sales force in the U.S. Our current business strategies include a plan to expand uses for VIVO, which will require additional clearances. While we do generate revenue, we are currently operating at a loss, and there is no guarantee that we will be able to grow revenues enough to offset our costs and realize profitability. To date, we have not been profitable, and our accumulated deficit was approximately $275.7 million at December 31, 2023. Historically, aside from Merger costs, our losses have resulted principally from costs incurred in research and development, and from general and administrative costs associated with our operations. During the first quarter 2023 we raised approximately $9.3 million in proceeds from securities transactions, but Merger costs and other negative cash flows have substantially depleted our cash. However, in order to continue the commercialization of our assets consistent with our vision, we will need to conduct substantial additional research, development and clinical trials. Our business strategy also includes expanding uses for our products which will require us to seek additional regulatory clearances in the United States, and we also must continue to expand our patents in order to obtain meaningful patent protection for and establish freedom to commercialize our product candidates. We must also complete further clinical trials and seek regulatory approvals for any new product candidates we discover, license or acquire. We cannot be sure whether and when we will obtain required regulatory approvals, or successfully research, develop, commercialize, manufacture and market any other product candidates. We expect that these activities, together with future general and administrative activities, will result in significant expenses for the foreseeable future. We may never achieve profitability.
| 25 |
| Table of Contents |
Risks Related to Our Internal Controls
We have identified material weaknesses in our internal control over financial reporting. These material weaknesses could adversely affect our ability to report our results of operations and financial condition accurately and in a timely manner.
Our management is responsible for establishing and maintaining adequate internal controls over financial reporting designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”). Our management is likewise required, on a quarterly basis, to evaluate the effectiveness of our disclosure controls and to disclose any material changes to our internal controls identified through such evaluation. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected and corrected on a timely basis. As described elsewhere in this Form 10-K and in our Quarterly Reports on Form 10-Q filed during 2023, we have identified material weaknesses in our internal control over financial reporting related to (1) the lack of segregation of duties, (2) the lack of designed and operating review controls with respect to oversight of the financial reporting process, (3) errors with respect to the review of work performed by service providers, (4) errors in connection with accounting for the royalty obligation acquired in the merger with Old Catheter, (5) use of an incorrect discount rate in calculating the fair value of the royalty obligation and (6) timing of revenue recognition. As a result of these material weaknesses, our management has concluded that our disclosure controls were not effective as of March 31, 2023, June 30, 2023, September 30, 2023 and December 31, 2023. For a discussion of management’s consideration of the material weaknesses described above, see below “Part II, Item 9A. Controls and Procedures: of this Annual Report on Form 10-K, and “Part I, Item 4. Controls and Procedures” included in our Quarterly Report on Form 10-Q for the quarter ended September 30, 2023.
As described below at “Part II, Item 9A. Controls and Procedures” of this Annual Report on Form 10-K and “Part I, Item 4. Controls and Procedures” included in our Quarterly Report on Form 10-Q for the quarter ended September 30, 2023, we have concluded that our disclosure controls were not effective as of March 31, 2023, June 30, 2023, September 30, 2023 and December 31, 2023 because material weaknesses existed in our internal control over financial reporting. We are in the process of formulating a plan to remediate the material weaknesses described therein; however, if we are unable to remediate our material weaknesses in a timely manner or we identify additional material weaknesses, we may be unable to provide required financial information in a timely or reliable manner and we may incorrectly report financial information. Likewise, if our financial statements are not filed on a timely basis, we could be subject to sanctions or investigations by the stock exchange on which our common stock is listed, the SEC or other regulatory authorities. In such a case, there could be a material adverse effect on our business. The existence of material weaknesses or significant deficiencies in internal control over financial reporting could adversely affect our reputation or investor perceptions of us, which could have a negative effect on the trading price of our stock. In addition, we may incur additional costs to remediate the material weaknesses in our internal control over financial reporting.
We can give no assurance that the measures we have taken and plan to take in the future will remediate the material weaknesses identified or that any additional material weaknesses or restatements of financial results will not arise in the future due to a failure to implement and maintain adequate internal control over financial reporting or circumvention of these controls or otherwise.
There may be additional undetected material weaknesses in our internal control over financial reporting, as a result of which we may not detect financial statement errors on a timely basis. Further, to the extent we identify additional material weaknesses, we will not be able to fully assess whether corrective measures will remediate the material weakness in our internal control over financial reporting until we have completed our implementation efforts and sufficient time passes in order to evaluate their effectiveness. In addition, if we identify additional errors that result in material weaknesses in our internal control over financial reporting, we may not detect errors on a timely basis and our financial statements may be materially misstated. Moreover, in the future we may engage in additional business transactions, such as acquisitions, reorganizations or implementation of new information systems, any of which could negatively affect our internal control over financial reporting and result in material weaknesses.
If we identify additional material weaknesses in our internal control over financial reporting or if we continue to be unable to assert that our internal control over financial reporting is effective, we may again be late with the filing of our periodic reports, investors may lose confidence in the accuracy and completeness of our financial reports, and the market price of our common stock could be negatively affected. As a result of any internal control failures, we could also become subject to investigations by the stock exchange on which our securities are listed, the SEC, or other regulatory authorities, and become subject to litigation from investors and stockholders, which could harm our reputation or divert financial and management resources from our core business, and which would have a material adverse effect on our business, financial condition and results of operations.
| 26 |
| Table of Contents |
Compliance with Section 404 of the Sarbanes-Oxley Act could have a material adverse impact on our business.
We are required, pursuant to Section 404 of the Sarbanes-Oxley Act, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. As a “smaller reporting company” that is a non-accelerated filer, we will avail ourselves of the exemption from the requirement that our independent registered public accounting firm attest to the effectiveness of our internal control over financial reporting under Section 404. However, we may no longer avail ourselves of this exemption when we cease to be a non-accelerated filer. When our independent registered public accounting firm is required to undertake an assessment of our internal control over financial reporting, the cost of our compliance with Section 404 will correspondingly increase. Our compliance with applicable provisions of Section 404 will require that we incur substantial accounting expense and expend significant management time on compliance-related issues as we implement additional corporate governance practices and comply with reporting requirements. Moreover, if we are not able to comply with the requirements of Section 404 to us in a timely manner, or if we or our independent registered public accounting firm identifies additional deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, the market price of our stock could decline and we could be subject to sanctions or investigations by the SEC or other regulatory authorities, which would require additional financial and management resources.
Risks Related to Our Business and Products
We will not be able to reach profitability unless we are able to achieve our product expansion and growth goals.
Our goal to achieve profitability is dependent upon establishing VIVO as an integral tool used by cardiac electrophysiologists during ablation treatment of ventricular arrhythmias, as well as upon developing and marketing new products, such as LockeT, the wound closure device, and the successful build out of our U.S. commercial infrastructure and sales force. During fiscal 2023, over 70% of our revenues were derived from four customers, two of whom represented over half of our revenues. In today’s healthcare environment, the process for new technologies to be adopted and penetrate market share has become more complex, with the need to win over multiple stakeholders within clinical, administrative and support teams in hospitals, and increasingly we must target the administrators in integrated delivery networks. To accomplish this, we will need to:
| · | Develop initial users that demonstrate clinical and economic benefits and support studies which provide evidence of tangible benefits to prospective customers, such as procedural success, no or minimal patient complications and reduced procedure times. |
|
|
|
| · | Collaborate with clinical thought leaders to establish clinical techniques, evolve our product features and demonstrate enhanced capabilities to broaden the appeal of VIVO and LockeT. |
|
|
|
| · | Acquire data and expand our FDA clearance to market our products for additional procedure types. In Europe, VIVO is cleared for pre-procedural planning in all types of hearts and procedures, including ischemic hearts. In the U.S., we will need to seek clearance for ischemic hearts to broaden the indications for use of our products, which can expand clinical demand. Data from the Coventry study focused on reentrant ventricular tachycardia will be used to support a clinical submission with existing version of VIVO. |
|
|
|
| · | Enhance the design, user utility and clinical capability of VIVO and LockeT through further product development and collaboration with clinical users. |
|
|
|
| · | Seek to engage collaboration with larger market participants and their larger sales force coverage to integrate the prospecting, sale and support of our products in conjunction with other products used in electrophysiology procedures. |
|
|
|
| · | Opportunistically identify acquisitions to enhance our enterprise scale, sales synergy and fixed cost coverage. |
|
|
|
| · | Seek to obtain permanent CPT codes for reimbursement from Medicare to broaden the appeal of using VIVO in the physician’s clinic. The process takes five to seven years to complete. To date, we have met with reimbursement specialists and are working to determine the best strategy. |
| 27 |
| Table of Contents |
In addition, our sales and marketing strategy for VIVO requires us to hire additional clinical support and sales representatives who are experienced in the EP field. In addition, we must make a significant investment building our U.S. commercial infrastructure and sales force, a lengthy process requiring ongoing investment and a certain amount of lead time to produce the growth rate we desire.
If we are unable to accomplish one or more of the foregoing, we may be unable to achieve our product expansion and growth goals, and may be unable to achieve profitability.
Our research and development and commercialization efforts may depend on entering into agreements with corporate collaborators.
We may need to seek out additional collaborations in order to commercialize our products. We will continue to seek research collaborations, co-development and marketing agreements, and licensing deals for our products in development; however, there is no guarantee that we will be successful in our efforts. Any collaborator with whom we may enter into such collaboration agreements may not support fully our research and commercial interests since our programs may compete for time, attention and resources with such collaborator’s internal programs. Therefore, these future collaborators may not commit sufficient resources to our programs to move them forward effectively, or the programs may not advance as rapidly as they might if we had retained complete control of all research, development, regulatory and commercialization decisions.
We have entered into joint marketing agreements with respect to our products, and may enter into additional joint marketing agreements, that will reduce our revenues from product sales.
Old Catheter entered into a Joint Marketing Agreement with Stereotaxis, Inc. in January 2021, as subsequently amended in January 2022 and May 2022, pursuant to which Stereotaxis agrees to promote our VIVO System to customers who may benefit from the use of VIVO in robotic or non-robotic electrophysiology procedures. Pursuant to the agreement, Stereotaxis can perform promotional activity at any hospital globally that has a Stereotaxis Robotic Magnetic Navigation System, referred to herein as a robotic hospital, and where VIVO has appropriate regulatory clearances. In addition, Stereotaxis will act as a spot distributor for us at mutually agreed upon hospitals where the VIVO System is included as a line item within a Stereotaxis quote. In exchange for its marketing, distribution and support activity, Stereotaxis receives a payment equal to 45% of the revenue generated from VIVO at robotic hospitals. After the initial sale of VIVO products to customers by Stereotaxis, Catheter will be responsible for selling additional VIVO-related products to the customers but will continue to owe the 45% payment to Stereotaxis with respect to any such sales. The agreement has a term that runs through December 31, 2025, provided however, that the agreement will automatically extend for successive two-year terms unless either party provides the other written notice of termination at least one year prior to the next-scheduled termination date. Stereotaxis will continue to be entitled to receive the 45% payments described above for a period of six months following termination of the agreement. Although we believe that this agreement is in the best interest of our business and our stockholders, it will materially reduce the revenues that we receive from VIVO products that are sold by Stereotaxis, and any similar agreements entered into in the future may have the same impact.
Royalty agreements with respect to LockeT, our surgical vessel closing pressure device, will reduce any future profits from this product.
In February 2022, Old Catheter agreed to an assignment and royalty agreement for the Surgical Vessel Closing Pressure Device (“LockeT”). Pursuant to the agreement, Old Catheter agreed to pay a royalty fee of 5% on net sales up to $1 million. Thereafter, if a patent for the Surgical Vessel Closing Pressure Device is obtained from the U.S. Patent and Trademark Office, Old Catheter will pay a royalty fee of 2% of net sales up to a total of $10 million in royalties. In addition, at the time of our merger with Old Catheter, additional royalty rights with respect to LockeT were granted to certain holders (the “Noteholders”) of Old Catheter’s outstanding convertible promissory notes in exchange for forgiveness of the interest that had accrued under those notes but remained unpaid, pursuant to the terms of certain Debt Settlement Agreements. The agreements provide for the Noteholders to receive, in the aggregate, approximately 12% of the net sales, if any, of the Surgical Vessel Closing Pressure Device, commencing upon the first commercial sale through December 31, 2035. As a result, even if the Surgical Vessel Closing Pressure Device is successfully developed and marketed, our revenues from this device will be reduced by the amount of these royalties.
| 28 |
| Table of Contents |
If we experience significant disruptions in our information technology systems, our business may be adversely affected.
We depend on our information technology systems for the efficient functioning of our business, as well as for accounting, financial reporting, data storage, compliance, purchasing and inventory management. We do not have redundant information technology systems at this time. Our information technology systems may be subject to computer viruses, ransomware or other malware, attacks by computer hackers, failures during the process of upgrading or replacing software, databases or components thereof, power outages, hardware failures, telecommunication failures and user errors, among other malfunctions. In addition, a variety of our software systems are cloud-based data management applications hosted by third-party service providers whose security and information technology systems are subject to similar risks. Technological interruptions could disrupt our operations, including our ability to timely ship and track product orders, project inventory requirements, manage our supply chain and otherwise adequately service our customers, or could disrupt our customers’ ability use our products for treatments. In the event we experience significant disruptions, we may be unable to repair our systems in an efficient and timely manner. Accordingly, such events may disrupt or reduce the efficiency of our entire operation and have a material adverse effect on our business, financial condition, and results of operations. Currently, we carry business interruption coverage to mitigate certain potential losses, but this insurance is limited in amount, subject to deductibles, and we cannot be certain that such potential losses will not exceed our policy limits. We are increasingly dependent on complex information technology to manage our infrastructure. Our information systems require an ongoing commitment of significant resources to maintain, protect and enhance our existing systems. Failure to maintain or protect our information systems and data integrity effectively could have a material adverse effect on our business, financial condition, and results of operations.
Litigation and other legal proceedings may adversely affect our business.
From time to time we are involved in and may become involved in legal proceedings relating to patent and other intellectual property matters, product liability claims, employee claims, tort or contract claims, federal regulatory investigations, securities class actions, and other legal proceedings or investigations, which could have an adverse impact on our reputation, business and financial condition and divert the attention of our management from the operation of our business. For example, we have previously been a party to securities class action and shareholder derivative litigation and other litigation. Litigation is inherently unpredictable and can result in excessive or unanticipated verdicts and/or injunctive relief that affect how we operate our business. We could incur judgments or enter into settlements of claims for monetary damages or for agreements to change the way we operate our business, or both. There may be an increase in the scope of these matters or there may be additional lawsuits, claims, proceedings or investigations in the future, which could have a material adverse effect on our business, financial condition, and results of operations. Adverse publicity about regulatory or legal action against us could damage our reputation and brand image, undermine our customers’ confidence and reduce long-term demand for our products, even if the regulatory or legal action is unfounded or not material to our operations.
We must indemnify or advance reasonable legal expenses for officers and directors, including, in certain circumstances, former employees and directors, in their defense against legal proceedings, unless certain conditions apply. A prolonged uninsured expense and indemnification obligation could have a material adverse effect on our business, financial condition, and results of operations.
If we make acquisitions or divestitures, we could encounter difficulties that harm our business.
We may acquire companies, products or technologies that we believe to be complementary to the present or future direction of our business, or that may be of a strategic nature with a focus on a new direction. If we engage in such acquisitions, we may have difficulty integrating the acquired personnel, financials, operations, products or technologies. Acquisitions may dilute our earnings per share, disrupt our ongoing business, distract our management and employees, increase our expenses, subject us to liabilities, and increase our risk of litigation, all of which could harm our business. If we use cash to acquire companies, products or technologies, it may divert resources otherwise available for other purposes. If we use our common stock to acquire companies, products or technologies, our stockholders may experience substantial dilution.
| 29 |
| Table of Contents |
Failure to attract and retain sufficient qualified personnel could also impede our growth.
Our current Interim Chief Financial Officer, Margrit Thomassen, is currently only working for us on an interim basis. As a result, we will need to hire a new, full-time Chief Financial Officer soon. We do not maintain “key man” insurance policies on the lives of any of our employees, including our Executive Chairman and Chief Executive Officer, David A. Jenkins. Mr. Jenkins is critical to our current business development, and we would not be able to easily replace him. Our success also depends on our ability to retain our other current key employees and continue to attract, retain and motivate highly skilled junior, mid-level and senior managers as well as junior, mid-level and senior scientific and medical personnel. If we are not successful in attracting and retaining highly qualified personnel, it would have a material adverse effect on our business, financial condition, and results of operations. We face intense competition for executive-level talent from a variety of sources, including from current and potential competitors in the medical device and healthcare industries, and there is no guarantee that we can locate suitable replacements when they are needed.
Our revenues may depend on our customers’ receipt of adequate reimbursement from private insurers and government sponsored healthcare programs.
Political, economic, and regulatory influences continue to change the healthcare industry in the United States. The ability of hospitals to pay fees for our products will partially depend on the extent to which reimbursement for the costs of such materials and related treatments will continue to be available from private health coverage insurers and other similar organizations. We may have difficulty gaining market acceptance for the products we sell if third-party payers do not provide adequate coverage and reimbursement to hospitals and/or other relevant healthcare providers.
Major third-party payers of hospitals, such as private healthcare insurers, periodically revise their payment methodologies based, in part, upon changes in government sponsored healthcare programs. We cannot predict these periodic revisions with certainty, and such revisions may result in stricter standards for reimbursement of hospital charges for certain specified products, potentially adversely impacting our business, results of operations, and financial condition when we start receiving reimbursement from third party payers.
The sales of our products and services will depend in part on the availability of reimbursement by third-party payers, such as government health administration authorities, private health insurers and other organizations. Third-party payers often challenge the price and cost-effectiveness of medical treatments and services. Governmental approval of health care products does not guarantee that these third-party payers will pay for the products. Even if third-party payers do accept our products and services, the amounts they pay may not be adequate to enable us to realize a profit. Legislation and regulations affecting the pricing of therapies may change before our products and services are approved for marketing, and any such changes could further limit reimbursement, if any.
We may be unable to compete successfully with companies in our highly competitive industry, many of whom have substantially greater resources than we do.
The healthcare industry is highly competitive. There are numerous approved products for treating the indications for which we have received clearance or approval and those that we may pursue in the future. Many of these cleared or approved products are well-established and are widely accepted by physicians, patients and third-party payors. Insurers and other third-party payors may encourage the use of competitors’ products. In addition, many companies are developing products, and we cannot predict what the standard of care will be in the future.
Our primary competitors in the cardiac electrophysiology, or EP, space include known medical devices such as pacemakers, electrocardiogram, or ECG, systems and cardiac catheters, but also laboratory equipment such as intracardiac mapping systems and fluoroscopy systems (similar to x-ray in real time). The EP market includes large medical device companies such as Medtronic, Plc., Abbott Laboratories, Biosense-Webster (J&J) and Boston Scientific Corp.
| 30 |
| Table of Contents |
Many of our competitors have substantially greater financial, manufacturing, commercial, and technical resources than we do. There has been consolidation in the industry, and we expect that to continue. Larger competitors may have substantially larger sales and marketing operations than we do. This may allow those competitors to spend more time with current and potential customers and to focus on a larger number of current and potential customers, which gives them a significant advantage over our sales and marketing team and our international distributors in making sales. In addition, we are often selling to customers who already utilize our competitors’ products and who have established relationships with our competitors’ sales representatives and familiarity with our competitors’ products.
Larger competitors may also have broader product lines, which enables them to offer customers bundled purchase contracts and quantity discounts. These competitors may have more experience than we have in research and development, marketing, manufacturing, preclinical testing, conducting clinical trials, obtaining FDA and non-U.S. regulatory clearances or approvals and marketing cleared or approved products. Our competitors may discover technologies and techniques, or enter into partnerships and collaborations, to develop competing products that are more effective or less costly than our products or the products we may develop. This may render our technology or products obsolete or noncompetitive. Our competitors may also be better equipped than we are to respond to competitive pressures. If we are unable to compete successfully in our industry, it would have a material adverse effect on our business, financial condition, and results of operations.
Our future operating results depend upon our ability to obtain components in sufficient quantities on commercially reasonable terms or according to schedules, prices, quality and volumes that are acceptable to us, and suppliers may fail to deliver components, or we may be unable to manage these components effectively or obtain these components on such terms.
We have historically obtained certain components globally, some of which were uniquely customized, from limited sources. This subjected us to significant supply and pricing risks and exposed us to multiple potential sources of component shortages. Many components, including those that are available from multiple sources, are at times subject to industry-wide shortages and significant commodity pricing fluctuations that could materially adversely affect our financial condition and operating results. We may source alternative parts to mitigate the challenges caused by these shortages, but there is no guarantee we may be able to continually do so as we scale production to meet our growth targets. The unavailability of any component or supplier could result in production delays, idle manufacturing facilities, product design changes and loss of access to important technology and tools for producing and supporting our products, as well as impact our capacity production. Our suppliers may not be willing or able to sustainably meet our timelines or our cost, quality and volume needs, or to do so may cost us more, which may require us to replace them with other sources. If our supply of components for a new or existing product continues to be delayed or constrained for any reason, including if an outsourcing partner delayed shipments of completed products to us or additional time is required to obtain sufficient quantities from the original source, or if we have to identify and obtain sufficient quantities from an alternative source, then our financial condition and operating results could be materially adversely affected. In addition, the continued availability of these components at acceptable prices, or at all, can be affected for any number of reasons, including if suppliers decide to concentrate on the production of common components or components for other customers instead of components customized to meet our requirements. While we have entered into agreements for the supply of certain components, there can be no assurance that we will be able to extend or renew these agreements on similar terms, or at all. Some of our components are purchased off the shelf and therefore we have no contractual certainties regarding their availability or pricing. Component suppliers may suffer from poor financial conditions, which can lead to business failure for the supplier or consolidation within a particular industry, further limiting our ability to obtain sufficient quantities of components on commercially reasonable terms. While we believe that we will be able to secure components as needed, and where necessary locate additional or alternate sources or develop our own replacements for relevant components, there is no assurance that we will be able to do so quickly or at all.
Additionally, we may be unable to obtain components on attractive terms and where applicable to achieve through negotiations with relevant suppliers cost reductions and/or avoid unfavorable changes to terms, source less expensive suppliers for certain parts and redesign certain parts to make them less expensive to produce. Any of these occurrences may harm our business, prospects, financial condition and operating results.
| 31 |
| Table of Contents |
If hospitals, physicians and patients do not accept our current and future products or if the market for indications for which any product candidate is approved is smaller than expected, we may be unable to generate significant revenue, if any.
Even when any of our product candidates obtain regulatory approval, they may not gain market acceptance among hospitals, physicians, patients, and third-party payers. Physicians may decide not to use our products for a variety of reasons including:
| · | timing of market introduction of competitive products; |
|
|
|
| · | demonstration of clinical safety and efficacy compared to other products; |
|
|
|
| · | cost-effectiveness; |
|
|
|
| · | limited or no coverage by third-party payers; |
|
|
|
| · | convenience and ease of administration; |
|
|
|
| · | prevalence and severity of adverse side effects; |
|
|
|
| · | restrictions in the label of the device; |
|
|
|
| · | other potential advantages of alternative treatment methods; and |
|
|
|
| · | ineffective marketing and distribution support of our products. |
If any of our product candidates are approved but fail to achieve market acceptance or such market is smaller than anticipated, we may not be able to generate significant revenue and our business would suffer.
The recent coronavirus, or COVID-19, outbreak adversely affected our financial condition and results of operations and we cannot provide any certainty as to whether there will be future impacts from COVID-19 or another pandemic.
The COVID-19 outbreak adversely affected our financial condition and results of operations. The impact of the outbreak of COVID-19 on the businesses and the economy in the United States and the rest of the world was significant. The extent to which the COVID-19 outbreak will continue to impact business and the economy is highly uncertain and cannot be predicted, and there can be no guarantee that a future pandemic will not have similar or worse impacts. Accordingly, we cannot predict the extent to which our financial condition and results of operation will be affected.
A variety of risks associated with marketing our products internationally could materially adversely affect our business.
In addition to selling our products in the U.S., we sell products outside of the U.S. We are subject to additional risks related to operating in foreign countries, including:
| · | differing regulatory requirements in foreign countries; |
|
|
|
| · | differing reimbursement regimes in foreign countries, including price controls and lower payment; |
|
|
|
| · | unexpected changes in tariffs, trade barriers, price and exchange controls and other regulatory requirements; |
|
|
|
| · | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
|
|
|
| · | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; |
|
|
|
| · | foreign taxes, including withholding of payroll taxes; |
|
|
|
| · | foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; |
|
|
|
| · | difficulties staffing and managing foreign operations; |
| 32 |
| Table of Contents |
| · | workforce uncertainty in countries where labor unrest is more common than in the U.S.; |
|
|
|
| · | potential liability under the FCPA or comparable foreign regulations; |
|
|
|
| · | challenges enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the U.S.; |
|
|
|
| · | product shortages resulting from any events affecting raw material or finished good supply or distribution or manufacturing capabilities abroad; |
|
|
|
| · | the impact of the current situation relating to trade with China and tariffs and other trade barriers that may be implemented by governmental authorities; |
|
|
|
| · | the impact of public health epidemics on the global economy; and |
|
|
|
| · | business interruptions resulting from geo-political actions, including war and terrorism. |
These and other risks associated with international operations may materially adversely affect our ability to attain or maintain profitable operations, which would have a material adverse effect on our business, financial condition, and results of operations.
The impact of the ongoing Russia-Ukraine and Israel-Gaza military conflicts, and other actions that have been and could be taken by other countries, including new and stricter sanctions and actions taken in response to such sanctions, have affected, and may continue to affect, our business and results of operations, including our supply chain.
On February 24, 2022, Russian forces launched significant military action against Ukraine, and sustained conflict and disruption in the region has occurred. The impact to Ukraine, as well as actions taken by other countries, including new and stricter sanctions imposed by Canada, the United Kingdom, the European Union, the U.S. and other countries and companies and organizations against officials, individuals, regions and industries in Russia and Ukraine, and actions taken by Russia in response to such sanctions, and each country’s potential response to such sanctions, tensions and military actions could have a material adverse effect on our operations. Any such material adverse effect from the conflict and enhanced sanctions activity may disrupt our supply chains and affect the delivery of our products and services or impair our ability to complete financial or banking transactions. We may suffer similar adverse effects from the Israel-Gaza conflict which has been ongoing since October 2023.
We also cannot predict the impact of any heightened geopolitical instability or the results that may follow, including reductions in consumer confidence, heightened inflation, cyber disruptions or attacks, higher natural gas costs, higher manufacturing costs and higher supply chain costs. The impact of armed conflicts such as those described above could cause our results to differ materially from the outlook presented in this Annual Report.
If the third parties on which we rely for the conduct of our clinical trials and results do not perform our clinical trial activities in accordance with good clinical practices and related regulatory requirements, we may be unable to obtain regulatory approval for or commercialize our product candidates.
We may use independent clinical investigators and other third-party service providers to conduct and/or oversee the clinical trials of our product candidates.
FDA requires us and our clinical investigators to comply with regulations and standards, commonly referred to as good clinical practices, for conducting, recording, and reporting the results of clinical trials to assure that data and reported results are credible and accurate, and that the trial participants are adequately protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. Third parties may not complete activities on schedule or may not conduct our clinical trials in accordance with regulatory requirements or the respective trial plans and protocols. The failure of these third parties to carry out their obligations could delay or prevent the development, approval, and commercialization of our product candidates or result in enforcement actions against us.
| 33 |
| Table of Contents |
We may be adversely affected by product liability claims, unfavorable court decisions or legal settlements.
We are exposed to potential product liability risks inherent in the design, manufacturing, and marketing of our products. These matters are subject to many uncertainties, and outcomes are not predictable. In addition, we may incur significant legal expenses regardless of whether we are found to be liable.
While we maintain product liability insurance, there can be no assurance that such coverage is sufficient to cover all product liabilities that we may incur. We are not currently subject to any product liability proceedings, and we have no reserves for product liability disbursements. However, we may incur material liabilities relating to product liability claims in the future, including product liability claims arising out of the usage and delivery of our products. Should we incur product-related liabilities exceeding our insurance coverage, we would be required to use available cash or raise additional cash to cover such liabilities.
Our ability to use our net operating loss carryforwards may be limited.
As of December 31, 2023, we had net operating loss carryforwards, or NOLs, available of approximately $147 million for federal income tax purposes and $111.7 million for state income tax purposes. Utilization of these NOLs depends on many factors, including our future income, which cannot be assured. These NOLs could expire unused and be unavailable to offset our future income tax liabilities. In addition, under Section 382 of the Internal Revenue Code of 1986, as amended, or IRC, and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50% change, by value, in its equity ownership by 5% stockholders over a three-year period, the corporation’s ability to use its pre-change NOLs and other pre-change tax attributes to offset its post-change income may be limited. We completed an IRC Section 382 analysis regarding the limitation of net operating losses through December 31, 2020 and determined that ownership changes occurred in May 2020. Management believes further ownership changes occurred during each of the years ended December 31, 2023, 2022 and 2021. Accordingly, utilization of our NOLs is subject to an annual limitation for federal tax purposes under IRC Section 382. Due to the changes in control, we estimated that $51.9 million of the $147 million federal NOLs are effectively eliminated, according to IRC Section 382. In addition, $40.8 million of our $111.7 million in state NOLs were also eliminated. As a result of these eliminations, our federal and state NOLs were reduced to $95.1 million and $70.9 million, respectively, before taking into consideration the valuation allowance.
We may have to make milestone payments under the Settlement Agreement we entered into with the DOJ.
We have entered into a Settlement Agreement with the Department of Justice, or DOJ, and agreements with the participating states, resolving a DOJ civil investigation concerning certain Covered Conduct (as defined in the Settlement Agreement), and the Office of Inspector General, or OIG, has agreed, in consideration of our full payment of amounts owed in the Settlement Agreement and our obligations under a Corporate Integrity Agreement, to release our permissive exclusion rights and refrain from instituting any administrative action seeking to exclude us from participating in Medicare, Medicaid, or other federal health care programs as a result of specified covered conduct. The Corporate Integrity Agreement has a five-year term expiring in December 2025 and imposes monitoring, reporting, certification, documentation, oversight, screening, and training obligations on us, including the hiring of a compliance officer and independent review organization; however, the OIG has agreed that we are not subject to the terms of the Corporate Integrity Agreement for so long as we do not carry on the legacy Ra Medical business or use the related business assets.
Pursuant to our Settlement Agreement with the DOJ, if during fiscal 2024 our revenues exceed $10 million, we have agreed to pay the United States and certain Medicaid participating states, $1.25 million. Payment must be made within 90 days after the end of the fiscal year.
| 34 |
| Table of Contents |
Risks Related to Governmental Regulation and our Industry
We are subject to pervasive and continuing regulation by the FDA and other regulatory agencies.
Our medical device operations are subject to pervasive and continuing FDA regulatory requirements.
Medical devices regulated by the FDA are subject to “general controls” which include:
| · | registration with the FDA; listing commercially distributed products with the FDA; |
|
|
|
| · | complying with applicable cGMPs under the Quality System Regulations, or QSR; |
|
|
|
| · | filing reports with the FDA of and keeping records relative to certain types of adverse events associated with devices under the medical device reporting regulation; |
|
|
|
| · | assuring that device labeling complies with device labeling requirements; |
|
|
|
| · | reporting recalls and certain device field removals and corrections to the FDA; and |
|
|
|
| · | obtaining premarket notification 510(k) clearance for devices prior to marketing. |
Some devices known as “510(k)-exempt” devices can be marketed without prior marketing clearance or approval from the FDA. In addition to the “general controls,” Class II medical devices are also subject to “special controls,” including, in many cases, adherence to a particular guidance document and compliance with the performance standard. As a Class II, 510(k)-cleared device, our VIVO product is subject to both general and special controls. Instead of obtaining 510(k) clearance, most Class III devices are subject to premarket approval, or PMA. We do not believe any of our current products are Class III devices, but future products could be, which would subject them to the PMA process.
Many medical devices are also regulated by the FDA as “electronic products.” In general, manufacturers and marketers of “electronic products” are subject to certain FDA regulatory requirements intended to ensure the radiological safety of the products. These requirements include, but are not limited to, filing certain reports with the FDA about the products and defects/safety issues related to the products as well as complying with radiological performance standards.
In addition, we may be required to conduct costly post-market testing and surveillance to monitor the safety or effectiveness of our products, and we must comply with medical device reporting, or MDR, requirements, including the reporting of adverse events and malfunctions related to our products. We are required to file MDRs if our products may have caused or contributed to a serious injury or death or malfunctioned in a way that could likely cause or contribute to a serious injury or death if it were to recur. Any such MDR that reports a significant adverse event could result in negative publicity, which could harm our reputation and future sales. Later discovery of previously unknown problems with our products, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements may result in changes to labeling, restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recalls, a requirement to repair, replace or refund the cost of any medical device we manufacture or distribute, fines, suspension of regulatory clearances or approvals, product seizures, injunctions or the imposition of civil or criminal penalties which may have a material adverse effect on our business, financial condition, and results of operations.
The medical device industry is now experiencing greater scrutiny and regulation by federal, state and foreign governmental authorities. Companies in our industry are subject to more frequent and more intensive reviews and investigations, often involving marketing, business practices, and product quality management. For example, as discussed above, on December 28, 2020, we entered into a Settlement Agreement with the DOJ to resolve a civil False Claims Act investigation and related civil action, and in connection with the Settlement Agreement, we also have reached agreements that resolve previously disclosed related investigations conducted by certain state attorneys general. Under the Settlement Agreement, and the agreements with the participating states, we were required to make an initial payment of $2.5 million, of which we paid $2.4 million in December 2020 and $0.1 million in April 2021. We also were required to make a payment of $5.0 million as a result of the January 2023 merger with Old Catheter in January 2023, which we made in February 2023. We may be required to make additional payments in the future upon the achievement of revenue targets.
| 35 |
| Table of Contents |
Additionally, federal, state and foreign governments and entities have enacted laws and issued regulations and other standards requiring increased visibility and transparency of our interactions with healthcare providers. For example, the U.S. Physician Payment Sunshine Act, now known as Open Payments, requires us to report to the Centers for Medicare & Medicaid Services, or CMS, payments and other transfers of value to all U.S. physicians and U.S. teaching hospitals, with the reported information made publicly available on a searchable website. On December 28, 2020, we entered into the Settlement Agreement with the DOJ relating to claims under the civil False Claims Act investigation concerning, among other things, whether we marketed and promoted DABRA devices, which we are no longer marketing, for unapproved uses that were not covered by federal healthcare programs, and whether we paid improper remuneration to physicians and other healthcare providers in violation of the Anti-Kickback Statute. Effective January 2022, we are also required to collect and report information on payments or transfers of value to physician assistants, nurse practitioners, clinical nurse specialists, certified registered nurse anesthetists, and certified nurse-midwives. Failure to comply with these legal and regulatory requirements could impact our business, and we have spent and will continue to spend substantial time and financial resources to develop and implement enhanced structures, policies, systems and processes to comply with these legal and regulatory requirements, which may also impact our business and which could have a material adverse effect on our business, financial condition, and results of operations for years after any resolution of these investigations and any resulting claims are resolved.
Product clearances and approvals can often be denied or significantly delayed.
Under FDA regulations, unless exempt, a new medical device may only be commercially distributed after it has received 510(k) clearance, is authorized through the de novo classification process, or is the subject of an approved PMA. The FDA will clear marketing of a medical device through the 510(k) process if it is demonstrated that the new product is substantially equivalent to another legally marketed product not subject to a PMA. Sometimes, a 510(k) clearance must be supported by preclinical and clinical data. Our ability to enroll patients in clinical trials could be impacted by a resurgence of the COVID-19 outbreak or another pandemic, as many patients would be likely to elect or would likely be asked to delay procedures at such a time.
The PMA process typically is more costly, lengthy and stringent than the 510(k) process. Unlike a 510(k) review which determines “substantial equivalence,” a PMA requires that the applicant demonstrate reasonable assurance that the device is safe and effective by producing valid scientific evidence, including data from preclinical studies and human clinical trials. Therefore, to obtain regulatory clearance or approvals, we typically must, among other requirements, provide the FDA and similar foreign regulatory authorities with preclinical and clinical data that demonstrate to their satisfaction that our products satisfy the criteria for approval. Preclinical testing and clinical trials must comply with the regulations of the FDA and other government authorities in the U.S. and similar agencies in other countries.
We may be required to obtain PMAs, PMA supplements or additional 510(k) premarket clearances to market modifications to our existing products, and our current business strategy contemplates expanding uses of our products to include uses that will require additional clearances. The FDA requires device manufacturers to make and document a determination of whether a device modification requires approval or clearance; however, the FDA can review a manufacturer’s decision. The FDA may not agree with our decisions not to seek approvals or clearances for particular device modifications. If the FDA requires us to obtain PMAs, PMA supplements or premarket clearances for any modification to a previously cleared or approved device, we may be required to cease manufacturing and marketing the modified device and perhaps also to recall such modified device until we obtain FDA clearance or approval. We may also be subject to significant regulatory fines or penalties.
The FDA may not approve future PMA applications or supplements or clear our 510(k) applications on a timely basis or at all. For example, the a new pandemic outbreak could affect the FDA’s ability to review applications or supplements. Such delays or refusals could have a material adverse effect on our business, financial condition, and results of operations.
The FDA may also change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions which may prevent or delay approval or clearance of our products under development or impact our ability to modify our currently approved or cleared products on a timely basis. Any of these actions could have a material adverse effect on our business, financial condition, and results of operations.
International regulatory approval processes may take more or less time than the FDA clearance or approval process. If we fail to comply with applicable FDA and comparable non-U.S. regulatory requirements, we may not receive regulatory clearances or approvals or may be subject to FDA or comparable non-U.S. enforcement actions. We may be unable to obtain future regulatory clearance or approval in a timely manner, or at all, especially if existing regulations are changed or new regulations are adopted. For example, the FDA clearance or approval process can take longer than anticipated due to requests for additional clinical data and changes in regulatory requirements. A failure or delay in obtaining necessary regulatory clearances or approvals would materially adversely affect our business, financial condition, and results of operations.
| 36 |
| Table of Contents |
Although we have obtained regulatory clearance for our VIVO and LockeT products in the U.S. and certain non-U.S. jurisdictions, our business plans include expanding uses for our products, which will require additional clearances; and even after clearance is obtained, our products will remain subject to extensive regulatory scrutiny.
Although our VIVO and LockeT products have received regulatory clearance in the U.S. and certain non-U.S. jurisdictions, they are subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping, conduct of post-marketing studies, and submission of safety, effectiveness, and other post-market information, including both federal and state requirements in the U.S. and requirements of comparable non-U.S. regulatory authorities. In addition, our business plans include expanding uses for our products, which will require additional clearances.
Any regulatory clearances or approvals that we have received for our products will be subject to limitations on the cleared or approved indicated uses for which the product may be marketed and promoted or to the conditions of approval or contain requirements for potentially costly post-marketing testing. We are required to report certain adverse events and production problems, if any, to the FDA and comparable foreign regulatory authorities. Any new legislation addressing product safety issues could result in increased costs to assure compliance. The FDA and other agencies, including the DOJ, closely regulate and monitor the post-clearance or post-approval marketing and promotion of products to ensure that they are marketed and distributed only for the cleared or approved indications and in accordance with the provisions of the cleared or approved labeling.
Promotional communications with respect to devices are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s cleared or approved labeling. As such, we may not promote our products for indications or uses for which they do not have clearance or approval. However, physicians can use their independent and professional judgment and use our products for off-label purposes, as FDA regulations do not restrict a physician’s choice of treatment with the practice of medicine. Prior to making certain changes to a cleared product, including certain changes to product labeling, the holder of a cleared 510(k) application may be required to submit a new premarket application and obtain clearance or approval.
If a regulatory agency discovers previously unknown problems with our products, such as adverse events of unanticipated severity or frequency, or problems with our facility where the product is manufactured, or disagrees with the promotion, marketing or labeling of our products, such regulatory agency or enforcement authority may impose restrictions on that product or us, including requiring withdrawal of the product from the market. In addition to this type of penalty for failing to comply with applicable regulatory requirements, a regulatory agency or enforcement authority may, among other things:
| · | subject us to an adverse inspectional finding or Form 483, or other compliance or enforcement notice, communication, or correspondence; |
|
|
|
| · | issue warning or untitled letters that would result in adverse publicity or may require corrective advertising; |
|
|
|
| · | impose civil or criminal penalties; |
|
|
|
| · | suspend or withdraw regulatory clearances or approvals; |
|
|
|
| · | refuse to clear or approve pending applications or supplements to approved applications submitted by us; |
|
|
|
| · | impose restrictions on our operations, including closing our sub-assembly suppliers’ facilities; |
|
|
|
| · | seize or detain products; or |
|
|
|
| · | require a product recall. |
In addition, violations of the Federal Food, Drug, and Cosmetic Act, or FDCA, relating to the promotion of approved products may lead to investigations alleging violations of federal and state healthcare fraud and abuse and other laws, as well as state consumer protection laws. As disclosed previously, we settled a DOJ civil False Claims Act investigation concerning, among other things, whether we marketed and promoted our DABRA devices for unapproved uses that were not covered by federal healthcare programs. We are no longer marketing DABRA devices.
| 37 |
| Table of Contents |
Any government adverse finding, regulatory sanction or investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. Any failure to comply with ongoing regulatory requirements may significantly and adversely affect our ability to commercialize and generate revenue from our products. If regulatory sanctions are applied or if regulatory clearance or approval is withdrawn, it would have a material adverse effect on our business, financial condition, and results of operations.
Our products may be subject to additional recalls, revocations or suspensions after receiving FDA or foreign approval or clearance, which could divert managerial and financial resources, harm our reputation, and adversely affect our business.
The FDA and similar foreign governmental authorities have the authority to order the recall of our products because of any failure to comply with applicable laws and regulations, or defects in design or manufacture. A government mandated or voluntary product recall by us could occur because of, for example, component failures, device malfunctions, or other adverse events, such as serious injuries or deaths, or quality-related issues such as manufacturing errors or design or labeling defects.
For example, prior to our acquisition of Old Catheter and switch in focus to developing and marketing its products, we conducted four recalls related to our previously marketed DABRA product. We no longer market DABRA, but any government-mandated recall or additional voluntary recall by us of VIVO, LockeT or another product we market in the future could occur as a result of component failures, manufacturing errors, design or labeling defects or other issues. These voluntary recalls and any future recalls of our products could divert managerial and financial resources, harm our reputation and adversely affect our business.
In addition, the FDA conducted an unannounced facility inspection in December 2019 in connection with our previously marketed DABRA product. The FDA issued to us a Form 483 that included observations, related to our previously marketed DABRA product, that schedules for the adjustment, cleaning, and other maintenance of equipment have not been adequately established, a device master record index was not current, and document control procedures have not been fully established. We responded to the FDA with the corrective measures we are taking and to address the issues identified in the Form 483 and based on this information, the FDA issued to us an Establishment Inspection Report, or EIR, closing out the inspection. All actions were completed, and the final Form 483 report was sent to the FDA on September 25, 2020. We are no longer operating this facility, but the FDA could conduct inspections of our current facilities.
Depending on the corrective action we take to address a product’s deficiencies or defects, the FDA may require, or we may voluntarily decide, that we will need to seek and obtain new approvals or clearances for a device before we may market or distribute the corrected device. Seeking such approvals or clearances may delay our ability to replace the recalled devices in a timely manner. Moreover, if we do not adequately address problems associated with our devices, we may face additional regulatory enforcement action, including adverse inspection findings, FDA warning letters, product seizure, injunctions, administrative penalties, or civil or criminal fines. We may also be required to bear other costs or take other actions that may have a negative impact on our sales as well as face significant adverse publicity or regulatory consequences, which could harm our business, including our ability to market our products in the future.
In addition, we are subject to medical device reporting regulations that require us to report to the FDA or similar foreign governmental authorities if one of our products may have caused or contributed to a death or serious injury or if we become aware that it has malfunctioned in a way that would be likely to cause or contribute to a death or serious injury if the malfunction recurred. After a May 2018 inspection related to our previously marketed DABRA product, the FDA issued to us a Form 483 that included observations for failure to properly evaluate whether certain complaints related to our previously marketed DABRA product that we received rose to a level required to be reported to the FDA. At that time, in response, we informed the FDA that we had modified our complaint review procedures and we completed a retrospective evaluation and did not find any complaints which required a submission to the FDA. We have not requested, and the FDA has not issued, an EIR related to this inspection. We no longer market DABRA.
| 38 |
| Table of Contents |
The failure by us to properly identify reportable events or to file timely reports with the FDA can subject us to sanctions and penalties, including warning letters and recalls. Physicians, hospitals and other healthcare providers may make similar reports to regulatory authorities. Any such reports may trigger an investigation by the FDA or similar foreign regulatory bodies, which could divert managerial and financial resources, harm our reputation and have a material adverse effect on our business, financial condition, and results of operations.
If we or our suppliers fail to comply with the FDA’s Quality System Regulation, or QSR, or any applicable state equivalent, our operations could be interrupted, and our potential product sales and operating results could suffer.
We and our suppliers are required to comply with the FDA’s QSR, which delineates, among other things, the design controls, document controls, purchasing controls, identification and traceability, production and process controls, acceptance activities, nonconforming product requirements, corrective and preventive action requirements, labeling and packaging controls, handling, storage, distribution and installation requirements, complaint handling, records requirements, servicing requirements, and statistical techniques potentially applicable to the production of our medical devices. We and our suppliers are also subject to the regulations of foreign jurisdictions regarding the manufacturing process if we market products overseas. The FDA enforces the QSR through periodic and announced or unannounced inspections of manufacturing facilities. We anticipate that we and certain of our third-party component suppliers will be subject to future inspections. If our facility or manufacturing processes or our suppliers’ facilities or manufacturing processes are found to be in non-compliance or fail to take satisfactory corrective action in response to adverse QSR inspectional findings, the FDA could take legal or regulatory enforcement actions against us and/or our products, including but not limited to the cessation of sales or the initiation of a recall of distributed products, which could impair our ability to produce our products in a cost-effective and timely manner in order to meet our customers’ demands. We may also be required to bear other costs or take other actions that may have a negative impact on our future sales and our ability to generate profits.
Current regulations depend heavily on administrative interpretation. If the FDA does not believe that we are in compliance with applicable FDA regulations, the agency could take legal or regulatory enforcement actions against us and/or our products. We are also subject to periodic inspections by the FDA, other governmental regulatory agencies, as well as certain third-party regulatory groups. Future interpretations made by the FDA or other regulatory bodies made during the course of these inspections may vary from current interpretations and may adversely affect our business and prospects. The FDA’s and other comparable non-U.S. regulatory agencies’ statutes, regulations, or policies may change, and additional government regulation or statutes may be enacted, which could increase post-approval regulatory requirements, or delay, suspend, or prevent marketing of any cleared or approved products or necessitate the recall of distributed products. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or abroad.
The medical device industry has been under heightened FDA scrutiny as the subject of government investigations and enforcement actions. If our operations and activities are found to be in violation of any FDA laws or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines and other legal and/or agency enforcement actions. Any penalties, damages, fines, or curtailment or restructuring of our operations or activities could adversely affect our ability to operate our business and our financial results. The risk of us being found in violation of FDA laws is increased by the fact that many of these laws are broad and their provisions are open to a variety of interpretations. Any action against us for violation of these laws, even if we successfully defend ourselves against that action and its underlying allegations, could cause us to incur significant legal expenses and divert management’s attention from the operation of our business. Where there is a dispute with a federal or state governmental agency that cannot be resolved to the mutual satisfaction of all relevant parties, we may determine that the costs, both real and contingent, are not justified by the commercial returns to us from maintaining the dispute or the product.
Various claims, design features or performance characteristics of our medical devices, that we regarded as permitted by the FDA without new marketing clearance or approval, may be challenged by the FDA or state or foreign regulators. The FDA or state or foreign regulatory authorities may find that certain claims, design features or performance characteristics, in order to be made or included in the products, are required to be supported by further clinical studies and marketing clearances or approvals, which could be lengthy, costly and possibly unobtainable.
| 39 |
| Table of Contents |
If any of our products cause or contribute to a death or a serious injury, or malfunction in certain ways, we will be required to report under applicable medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions.
Under the FDA medical device reporting regulations, or MDR regulations, medical device manufacturers are required to report to the FDA information that a device has or may have caused or contributed to a death or serious injury or has malfunctioned in a way that would likely cause or contribute to death or serious injury if the malfunction of the device or one of our similar devices were to recur.
If we fail to report events required to be reported to the FDA within the required timeframes, or at all, the FDA could take enforcement action against us. Any such adverse event involving our products also could result in future voluntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection or enforcement action. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require our time and capital, distract management from operating our business, and may harm our reputation and have a material adverse effect on our business, financial condition, and results of operations.
Healthcare reform initiatives and other administrative and legislative proposals may adversely affect our business, financial condition, results of operations and cash flows in our key markets.
There have been and continue to be proposals by the federal government, state governments, regulators and third-party payors to control or manage the increasing costs of healthcare and, more generally, to reform the U.S. healthcare system.
Certain of these proposals could limit the prices we are able to charge for our products or the coverage and reimbursement available for our products and could limit the acceptance and availability of our products on the market. The adoption of proposals to control costs could have a material adverse effect on our business, financial condition, and results of operations.
For example, in the U.S., in March 2010, the Patient Protection and Affordable Care Act, or PPACA, was passed. The PPACA was intended to make significant changes to the way healthcare is financed by both federal and state governments and private insurers, with direct impacts to the medical device industry. Among other provisions, the PPACA imposed, with limited exceptions, a deductible excise tax of 2.3% on sales of medical devices by entities that manufacture or import certain medical devices offered for sale in the U.S. The Consolidated Appropriations Act, 2016 (Pub. L. 114-113), signed into law in December 2015, included a two-year moratorium on the medical device excise tax. A second two-year moratorium on the medical device excise tax was signed into law in January 2018 as part of the Extension of Continuing Appropriations Act, 2018 (Pub. L. 115-120), extending the moratorium through December 31, 2019. On December 20, 2019, President Trump signed into law a permanent repeal of the medical device tax under the PPACA, but there is no guarantee that Congress or the President will not reverse course in the future. If such an excise tax on sales of any of our products in the U.S. is enacted, it could have a material adverse effect on our business, financial condition, and results of operations.
In addition, the PPACA and the Medicare Access and CHIP Reauthorization Act of 2015 substantially changed the way healthcare is delivered and financed by both governmental and private insurers. These changes included the creation of demonstration programs and other value-based purchasing initiatives that provide financial incentives for physicians and hospitals to reduce costs. Under the Trump Administration, there were ongoing efforts to modify or repeal all or part of PPACA or take executive action that affects its implementation. Tax reform legislation was passed that includes provisions that impact healthcare insurance coverage and payment such as the elimination of the tax penalty for individuals who do not maintain health insurance coverage (the so-called “individual mandate”). Such actions or similar actions could have a negative effect on the utilization of our products.
On December 18, 2019, the U.S. Court of Appeals for the Fifth Circuit upheld a lower court’s determination in Texas v. Azar, 4:18-cv-00167, that the individual mandate was unconstitutional and remanded the case to the lower court for further analysis as to whether PPACA as a whole is unconstitutional because the individual mandate is not severable from other provisions of the law. In June 2021, the U.S. Supreme Court held that Texas and other challengers had no legal standing to challenge the PPACA, dismissing the case on procedural grounds without specifically ruling on the constitutionality of the PPACA. Thus, the PPACA remains in effect in its current form. Further, legislative and regulatory changes under the PPACA remain possible, although the federal administration under President Biden has signaled that it plans to build on the PPACA and expand the number of people who are eligible for health insurance under it. It is unclear how future litigation and healthcare measures promulgated by the Biden administration or future administrations will impact the implementation of the PPACA and our business, financial condition and results of operations. Complying with any new legislation or reversing changes implemented under the PPACA could be time-intensive and expensive, resulting in a material adverse effect on our business.
| 40 |
| Table of Contents |
Other healthcare reform legislative changes have also been proposed and adopted in the U.S. since the PPACA was enacted. In August 2011, the Budget Control Act of 2011, among other things, led to aggregate reductions of Medicare payments to providers of 2% per fiscal year. These reductions went into effect in April 2013, which, due to subsequent legislative amendments, will stay in effect through 2031, with the exception of a temporary suspension implemented under various COVID-19 relief legislation from May 1, 2020 through March 31, 2022, unless additional congressional action is taken. Under current legislation, the actual reduction in Medicare payments will vary from 1% in 2022 to up to 4% in the final fiscal year of the sequester. In January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Further, recently there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several U.S. Congressional inquiries and proposed, and enacted, federal legislation designed to bring transparency to product pricing and reduce the cost of products and services under government healthcare programs. As a result of reform of the U.S. healthcare system, changes in reimbursement policies or healthcare cost containment initiatives may limit or restrict coverage and reimbursement for procedures using our products and cause our revenue to decline. Additionally, individual states in the U.S. have also become increasingly active in passing legislation and implementing regulations designed to control product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures. Moreover, Medicare, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what products to purchase, and which suppliers will be included in their healthcare programs. Adoption of price controls and other cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures may prevent or limit our ability to generate revenue, attain profitability.
Various new healthcare reform proposals are emerging at the federal and state level. Any new federal and state healthcare initiatives that may be adopted could limit the amounts that federal and state governments will pay for healthcare products and services and could have a material adverse effect on our business, financial condition, and results of operations.
Healthcare cost containment pressures and legislative or administrative reforms resulting in restrictive coverage and reimbursement practices of third-party payors could decrease the demand for our products and the number of procedures performed using our devices, which could have an adverse effect on our business.
The ability of our customers to obtain reimbursement for procedures that are performed using our products from government and private third-party payors is critical to our success. The availability of coverage and reimbursement for procedures performed using our products affects which products customers purchase and the prices they are able to pay to us.
Reimbursement can vary based on geographical location, type of provider/customer, and third-party payor and can significantly influence the acceptance of new products and services. Third-party payors may view some procedures performed using our products as experimental and may not provide coverage. Third-party payors may not cover and reimburse our customers for certain procedures performed using our products in whole or in part in the future, or payment rates may decline and not be adequate, or both. Further, coverage and reimbursement by third-party payors to our customers is also related to billing codes to describe procedures performed using our products. Hospitals and physicians use several billing codes to bill for such procedures. Obtaining Category I CPT codes for VIVO will be important to our future success. However, even if these codes are obtained, third-party payors may not recognize CPT codes available for use by our customers. Further, CPT codes may change over time, undermining our customer’s ability to continue to use those codes, and reimbursement may be interrupted. Furthermore, some payors may not accept these new or revised codes for payment.
| 41 |
| Table of Contents |
Reimbursement rates are unpredictable, and we cannot project how our business may be affected by future legislative and regulatory developments. Future legislation or regulation, or changing payment methodologies, may have a material adverse effect on our business, financial condition, and results of operations, and reimbursement may not be adequate for all customers. From time to time, typically on an annual basis, payment amounts are updated and revised by third-party payors. To be successful, our business model requires it to be possible for the cost of our products to generally be recovered by the healthcare provider as part of the payment for performing a procedure and not separately reimbursed. For that reason, even after we achieve CPT codes for our products, these annual updates, especially lower payments, could continue to directly impact the demand for our products. For example, in July 2013, the Centers for Medicare and Medicaid Services, or CMS, proposed reimbursement changes that would have decreased reimbursement for procedures in an outpatient-based facility, such as a catheterization lab. Although CMS chose not to implement those changes in 2013, we cannot assure you that CMS will not take similar actions in the future.
After we develop new products or seek to market our products for new approved or cleared indications, there tends to be limited demand for the product unless government and private third-party payors provide adequate coverage and reimbursement to our customers. However, obtaining codes and reimbursement for new products requires an extended, multi-year effort.
Even after reimbursement approval and coverage by government and private payors is obtained, providers submitting reimbursement claims for new products or existing products with new approved or cleared indications may face delay in payment if there is confusion by providers or payors regarding the appropriate codes to use in seeking reimbursement. Such delays may create an unfavorable impression within the marketplace regarding the level of reimbursement or coverage available for our products.
Demand for our products or new approved indications for our existing products may fluctuate over time if federal or state legislative or administrative policy changes affect coverage or reimbursement levels for our products or the services related to our products. In the U.S., there have been, and we expect there will continue to be legislative and regulatory proposals to change the healthcare system, such as the potential repeal of the PPACA, some of which could significantly affect our business. It is uncertain what impact the current U.S. presidential administration or future administrations will have on healthcare spending. If enacted and implemented, any measures to restrict healthcare spending could result in decreased revenue from the sale of our products and decreased potential returns from our research and development initiatives. Other legislative or administrative reforms to the U.S. or international reimbursement systems in a manner that significantly reduces reimbursement for procedures performed using our products or denies coverage for those procedures could have a material adverse effect on our business, financial condition, and results of operations.
We are regulated by federal Anti-Kickback Statutes.
The Federal Anti-Kickback Statute is a provision of the Social Security Act of 1972 that prohibits as a felony offense the knowing and willful offer, payment, solicitation or receipt of any form of remuneration in return for, or to induce, (1) the referral of a patient for items or services for which payment may be made in whole or part under Medicare, Medicaid, or other federal healthcare programs, (2) the furnishing or arranging for the furnishing of items or services reimbursable under Medicare, Medicaid, or other federal healthcare programs or (3) the purchase, lease, or order or arranging or recommending the purchasing, leasing or ordering of any item or service reimbursable under Medicare, Medicaid or other federal healthcare programs. The Patient Protection and Affordable Care Act, or PPACA, amended section 1128B of the Social Security Act to make it clear that a person need not have actual knowledge of the statute, or specific intent to violate the statute, as a predicate for a violation. The OIG, which has the authority to impose administrative sanctions for violation of the statute, has adopted as its standard for review a judicial interpretation which concludes that the statute prohibits any arrangement where even one purpose of the remuneration is to induce or reward referrals. A violation of the Anti-Kickback Statute is a felony punishable by imprisonment, criminal fines of up to $25,000, civil fines of up to $50,000 per violation, and three times the amount of the unlawful remuneration. A violation also can result in exclusion from Medicare, Medicaid or other federal healthcare programs. In addition, pursuant to the changes to the PPACA, a claim that includes items or services resulting from a violation of the Anti-Kickback Statute is a false claim for purposes of the False Claims Act. We cannot assure that the applicable regulatory authorities will not determine that some of our arrangements with hospitals or physicians violate the federal Anti-Kickback Statute or other applicable laws. An adverse determination could subject us to different liabilities, including criminal penalties, civil monetary penalties and exclusion from participation in Medicare, Medicaid or other health care programs, any of which could have a material adverse effect on our business, financial condition or results of operations.
| 42 |
| Table of Contents |
We are regulated by the federal Stark Law.
The federal Stark Law, 42 U.S.C. 1395nn, also known as the physician self-referral law, generally prohibits a physician from referring Medicare and Medicaid patients to an entity (including hospitals) providing ‘designated health services,’ if the physician or a member of the physician’s immediate family has a ‘financial relationship’ with the entity, unless a specific exception applies. Designated health services include, among other services, inpatient hospital services, outpatient prescription drug services, clinical laboratory services, certain imaging services (e.g., MRI, CT, ultrasound), and other services that our affiliated hospitals may order for their patients. The prohibition applies regardless of the reasons for the financial relationship and the referral. Like the Anti-Kickback Statute, the Stark Law contains statutory and regulatory exceptions intended to protect certain types of transactions and arrangements. Unlike safe harbors under the Anti-Kickback Statute with which compliance is voluntary, an arrangement must comply with every requirement of a Stark Law exception or the arrangement is in violation of the Stark Law.
Because the Stark Law and implementing regulations continue to evolve and are detailed and complex, while we attempt to structure our relationships to meet an exception to the Stark Law, there can be no assurance that the arrangements entered into by us with affiliated hospitals will be found to be in compliance with the Stark Law, as it ultimately may be implemented or interpreted. The penalties for violating the Stark Law can include the denial of payment for services ordered in violation of the statute, mandatory refunds of any sums paid for such services, and civil penalties of up to $15,000 for each violation, double damages, and possible exclusion from future participation in the governmental healthcare programs. A person who engages in a scheme to circumvent the Stark Law’s prohibitions may be fined up to $100,000 for each applicable arrangement or scheme.
Some states have enacted statutes and regulations against self-referral arrangements similar to the federal Stark Law, but which may be applicable to the referral of patients regardless of their payer source and which may apply to different types of services. These state laws may contain statutory and regulatory exceptions that are different from those of the federal law and that may vary from state to state. An adverse determination under these state laws and/or the federal Stark Law could subject us to different liabilities, including criminal penalties, civil monetary penalties and exclusion from participation in Medicare, Medicaid or other health care programs, any of which could have a material adverse effect on our business, financial condition or results of operations.
We must comply with Health Information Privacy and Security Standards.
HIPAA and regulations thereunder contain detailed requirements concerning the use and disclosure of individually identifiable patient health information by various healthcare providers, such as medical groups. HIPAA covered entities must implement certain administrative, physical, and technical security standards to protect the integrity, confidentiality and availability of certain electronic health information received, maintained, or transmitted. HIPAA also implemented standard transaction code sets and standard identifiers that covered entities must use when submitting or receiving certain electronic healthcare transactions, including billing and claim collection activities. Violations of the HIPAA privacy and security rules may result in civil and criminal penalties, including a tiered system of civil money penalties that range from $100 to $50,000 per violation, with a cap of $1.5 million per year for identical violations. A HIPAA covered entity must also promptly notify affected individuals where a breach affects more than 500 individuals and report breaches affecting fewer than 500 individuals annually. State attorneys general may bring civil actions on behalf of state residents for violations of the HIPAA privacy and security rules, obtain damages on behalf of state residents, and enjoin further violations.
Many states also have laws that protect the privacy and security of confidential, personal information, which may be similar to or even more stringent than HIPAA. Some of these state laws may impose fines and penalties on violators and may afford private rights of action to individuals who believe their personal information has been misused. We expect increased federal and state privacy and security enforcement efforts.
| 43 |
| Table of Contents |
If a breach of our measures protecting personal data covered by HIPAA, as amended by the HITECH Act, occurs, we may incur significant liabilities.
HIPAA, as amended by the HITECH Act, and the regulations that have been issued under it, imposes certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of protected health information. The requirements and restrictions apply to “covered entities” (which include health care providers and insurers) as well as to their business associates that receive protected health information from them in order to provide services to or perform certain activities on their behalf. The statute and regulations also impose notification obligations on covered entities and their business associates in the event of a breach of the privacy or security of protected health information. We occasionally receive protected health information from our customers in the course of our business. As such, we believe that we are business associates and therefore subject to HIPAA’s requirements and restrictions with respect to handling such protected health information and have executed business associate agreements with certain customers.
It is possible the data protection laws may be interpreted and applied in a manner that is inconsistent with our practices. If so, this could result in government-imposed fines or orders requiring that we change our practices, which could adversely affect our business. In addition, these privacy regulations may differ from country to country and state to state and may vary based on whether testing is performed in the U.S. or in the local country. Complying with these various laws and regulations could cause us to incur substantial costs or require us to change our business practices and compliance procedures in a manner adverse to our business. Further, compliance with data protection laws and regulations could require us to take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, or in some cases, impact our ability to operate in certain jurisdictions. We can provide no assurance that we are or will remain in compliance with diverse privacy and security requirements in all of the jurisdictions in which we do business. If we fail to comply or are deemed to have failed to comply with applicable privacy protection laws and regulations such failure could result in government enforcement actions and create liability for us, which could include substantial civil and/or criminal penalties, as well as private litigation and/or adverse publicity that could negatively affect our operating results and business.
A cyber security incident could cause a violation of HIPAA and/or state consumer privacy laws, breach of customer and patient privacy, or other negative impacts.
We rely extensively on our information technology (or IT) systems to manage scheduling and financial data, communicate with hospitals and their patients, vendors, and other third parties, and summarize and analyze operating results. In addition, we have made significant investments in technology, including the engagement of a third-party IT provider. A cyber-attack that bypasses our IT security systems could cause an IT security breach, a loss of protected health information, or other data subject to privacy laws, a loss of proprietary business information, or a material disruption of our IT business systems. This in turn could have a material adverse impact on our business and result of operations. In addition, our future results of operations, as well as our reputation, could be adversely impacted by theft, destruction, loss, or misappropriation of public health information, other confidential data, or proprietary business information.
Computer malware, viruses, and hacking and phishing attacks by third parties have become more prevalent in our industry and may occur on our systems in the future. Because techniques used to obtain unauthorized access to or sabotage systems change frequently and generally are not recognized until successfully launched against a target, we may be unable to anticipate these techniques or to implement adequate preventative measures. As cyber-security threats develop and grow, it may be necessary to make significant further investments to protect data and infrastructure. If an actual or perceived breach of our security occurs, (i) we could suffer severe reputational damage adversely affecting customer or investor confidence, (ii) the market perception of the effectiveness of our security measures could be harmed, (iii) we could lose potential sales and existing customers, our ability to deliver our services or operate our business may be impaired, (iv) we may be subject to litigation or regulatory investigations or orders, and (v) we may incur significant liabilities. Our insurance coverage may not be adequate to cover the potentially significant losses that may result from security breaches.
| 44 |
| Table of Contents |
We must comply with environmental and Occupational Safety and Health Administration Regulations.
We are subject to federal, state and local regulations governing the storage, use and disposal of waste materials and products. Using hazardous substances in our operations exposes us to the risk of accidental injury, contamination or other liability from the use, storage, importation, handling, or disposal of hazardous materials. If our or our suppliers’ operations result in the contamination of the environment or expose individuals to hazardous substances, we could be liable for damages and fines, and any liability could significantly exceed our insurance coverage and have a material adverse effect on our business, financial condition, and results of operations. Future changes to environmental and health and safety laws could cause us to incur additional expenses or restrict our operations, which could have a material adverse effect on our business, financial condition, and results of operations. Although we believe that our safety procedures for storing, handling and disposing of these materials and products comply with the standards prescribed by law and regulation, we cannot eliminate the risk of accidental contamination or injury from those hazardous materials. In the event of an accident, we could be held liable for any damages that result and any liability could exceed the limits or fall outside the coverage of our insurance coverage, which we may not be able to maintain on acceptable terms, or at all. We could incur significant costs and attention of our management could be diverted to comply with current or future environmental laws and regulations. Federal regulations promulgated by the Occupational Safety and Health Administration impose additional requirements on us, including those protecting employees from exposure to elements such as blood-borne pathogens. We cannot predict the frequency of compliance, monitoring, or enforcement actions to which we may be subject as those regulations are being implemented, which could adversely affect our operations.
We must comply with a range of other Federal and State Healthcare Laws.
We are also subject to other federal and state healthcare laws that could have a material adverse effect on our business, financial condition or results of operations. The Health Care Fraud Statute prohibits any person from knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, which can be either a government or private payer plan. Violation of this statute, even in the absence of actual knowledge of or specific intent to violate the statute, may be charged as a felony offense and may result in fines, imprisonment, or both. The Health Care False Statement Statute prohibits, in any matter involving a federal health care program, anyone from knowingly and willfully falsifying, concealing or covering up, by any trick, scheme or device, a material fact, or making any materially false, fictitious or fraudulent statement or representation, or making or using any materially false writing or document knowing that it contains a materially false or fraudulent statement. A violation of this statute may be charged as a felony offense and may result in fines, imprisonment or both. Under the Civil Monetary Penalties Law of the Social Security Act, a person (including an organization) is prohibited from knowingly presenting or causing to be presented to any United States officer, employee, agent, or department, or any state agency, a claim for payment for medical or other items or services where the person knows or should know (a) the items or services were not provided as described in the coding of the claim, (b) the claim is a false or fraudulent claim, (c) the claim is for a service furnished by an unlicensed physician, (d) the claim is for medical or other items or service furnished by a person or an entity that is in a period of exclusion from the program, or (e) the items or services are medically unnecessary items or services. Violations of the law may result in penalties of up to $10,000 per claim, treble damages, and exclusion from federal healthcare programs.
In addition, the OIG may impose civil monetary penalties against any physician who knowingly accepts payment from a hospital (as well as against the hospital making the payment) as an inducement to reduce or limit medically necessary services provided to Medicare or Medicaid program beneficiaries. Further, except as permitted under the Civil Monetary Penalties Law, a person who offers or transfers to a Medicare or Medicaid beneficiary any remuneration that the person knows or should know is likely to influence the beneficiary’s selection of a particular provider of Medicare or Medicaid payable items or services may be liable for civil monetary penalties of up to $10,000 for each wrongful act.
In addition to the state laws previously described, we may also be subject to other state fraud and abuse statutes and regulations if we expand our operations nationally. Many states have adopted a form of anti-kickback law, self-referral prohibition, and false claims and insurance fraud prohibition. The scope of these laws and the interpretations of them vary from state to state and are enforced by state courts and regulatory authorities, each with broad discretion. Generally, state laws reach to all healthcare services and not just those covered under a governmental healthcare program. A determination of liability under any of these laws could result in fines and penalties and restrictions on our ability to operate in these states. We cannot assure you that our arrangements or business practices will not be subject to government scrutiny or be found to violate applicable fraud and abuse laws.
| 45 |
| Table of Contents |
Governmental export or import controls could limit our ability to compete in foreign markets and subject us to liability if we violate them.
Our products may be subject to U.S. export controls. Governmental regulation of the import or export of our products, or our failure to obtain any required import or export authorization for our products, when applicable, could harm our international sales and adversely affect our revenue. Compliance with applicable regulatory requirements regarding the export of our products may create delays in the introduction of our products in international markets or, in some cases, prevent the export of our products to some countries altogether. Furthermore, U.S. export control laws and economic sanctions prohibit the shipment of certain products and services to countries, governments, and persons targeted by U.S. sanctions. If we fail to comply with export and import regulations and such economic sanctions, we may be fined or other penalties could be imposed, including a denial of certain export privileges. Moreover, any new export or import restrictions, new legislation or shifting approaches in the enforcement or scope of existing regulations, or in the countries, persons, or technologies targeted by such regulations, could result in decreased use of our products by, or in our decreased ability to export our products to existing or potential customers with international operations. Any decreased use of our products or limitation on our ability to export or sell access to our products would likely materially and adversely affect our business, financial condition, and results of operations.
Changes in trade policies among the U.S. and other countries, in particular the imposition of new or higher tariffs, could place pressure on our average selling prices as our customers seek to offset the impact of increased tariffs on their own products. Increased tariffs or the imposition of other barriers to international trade could have a material adverse effect on our revenues and operating results.
In addition to current and proposed economic sanctions on Russia, which may increase or continue for an indefinite period of time as a result of Russia’s invasion of Ukraine, the U.S. has imposed or proposed new or higher tariffs on certain products exported by a number of U.S. trading partners, including China, Europe, Canada, and Mexico. In response, many of those trading partners, including China, have imposed or proposed new or higher tariffs on American products. Continuing changes in government trade policies create a heightened risk of further increased tariffs that impose barriers to international trade.
Tariffs on our customers’ products may adversely affect our gross profit margins in the future due to the potential for increased pressure on our selling prices by customers seeking to offset the impact of tariffs on their own products. We believe that increases in tariffs on imported goods or the failure to resolve current international trade disputes could have a material adverse effect on our business and operating results.
Risks Related to our Intellectual Property
If we are unable to obtain and maintain patent protection for our products, our competitors could develop and commercialize products and technology similar or identical to ours, and our ability to successfully commercialize our existing products and any products we may develop, and our technology may be adversely affected.
As with other medical device companies, our ability to maintain and solidify a proprietary position for our products will depend upon our success in obtaining effective patent claims that cover such products, their manufacturing processes and their intended methods of use, and enforcing those claims once granted. Furthermore, in some cases, we may not be able to obtain issued claims covering VIVO or LockeT, as well as other technologies that are important to our business, which are sufficient to prevent third parties, such as our competitors, from utilizing our technology. Any failure to obtain or maintain patent protection with respect to VIVO, LockeT or any new devices that we market could have a material adverse effect on our business, financial condition, and results of operations.
Changes in either the patent laws or their interpretation in the U.S. and other countries may diminish our ability to protect our inventions, obtain, maintain, and enforce our intellectual property rights and, more generally, could affect the value of our intellectual property or narrow the scope of our issued patents. Additionally, we cannot predict whether the patent applications we are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient protection from competitors or other third parties.
| 46 |
| Table of Contents |
The patent prosecution process is expensive, time-consuming, and complex, and we may not be able to file, prosecute, maintain, enforce, or license all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output in time to obtain patent protection. Although we enter into non-disclosure and confidentiality agreements with parties who have access to confidential or patentable aspects of our research and development output, such as our employees, corporate collaborators, outside scientific collaborators, suppliers, consultants, advisors and other third parties, any of these parties may breach the agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek patent protection. In addition, our ability to obtain and maintain valid and enforceable patents depends on whether the differences between our inventions and the prior art allow our inventions to be patentable over the prior art. Furthermore, publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the U.S. and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot be certain that we were the first to make the inventions claimed in any of our pending patent applications, or that we were the first to file for patent protection of such inventions. Moreover, in some circumstances, we may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the patents, covering technology that we license from or license to third parties and are therefore reliant on our licensors or licensees. Therefore, these and any of our patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business. Defects of form in the preparation or filing of our patents or patent applications may exist, or may arise in the future, for example, with respect to proper priority claims, inventorship, and the like, although we are unaware of any such defects that we believe are of material importance. If we or any future licensors or licensees fail to establish, maintain or protect such patents and other intellectual property rights, such rights may be reduced or eliminated. If any future licensors or licensees are not fully cooperative or disagree with us as to the prosecution, maintenance or enforcement of any patent rights, such patent rights could be compromised. If there are material defects in the form, preparation or prosecution of our patents or patent applications, such patents or applications may be invalid and unenforceable. Any of these outcomes could impair our ability to prevent competition from third parties, which may have an adverse impact on our business.
In addition, if any patents are issued in the future, they may not provide us with any competitive advantages, or may be successfully challenged by third parties. Agreement terms that address non-competition are difficult to enforce in many jurisdictions and may not be enforceable in any particular case. To the extent that our intellectual property and other proprietary rights are not adequately protected, third parties might gain access to our proprietary information, develop and market products or services similar to ours, or use trademarks similar to ours, each of which could materially harm our business. Existing U.S. federal and state intellectual property laws offer only limited protection. Moreover, the laws of other countries in which we now, or may in the future, conduct operations or contract for services may afford little or no effective protection of our intellectual property. The failure to adequately protect our intellectual property and other proprietary rights could materially harm our business.
The strength of patent rights involves complex legal and scientific questions and can be uncertain. This uncertainty includes changes to the patent laws through either legislative action to change statutory patent law or court action that may reinterpret existing law or rules in ways affecting the scope or validity of issued patents. The patent applications that we own may fail to result in issued patents in the U.S. or foreign countries with claims that cover our products or services. Even if patents do successfully issue from the patent applications that we own, third parties may challenge the validity, enforceability or scope of such patents, which may result in such patents being narrowed, invalidated or held unenforceable. Any successful challenge to our patents could deprive us of exclusive rights necessary for the successful commercialization of our products and services. Furthermore, even if they are unchallenged, our patents may not adequately protect our products and services, provide exclusivity for our products and services, or prevent others from designing around our claims. If the breadth or strength of protection provided by the patents we hold or pursue with respect to our products and services is challenged, it could dissuade companies from collaborating with us to develop, or threaten our ability to commercialize, our products and services.
Patents have a limited lifespan. In the U.S., the natural expiration of a utility patent is generally 20 years after its effective filing date and the natural expiration of a design patent is generally 14 years after its issue date, unless the filing date occurred on or after May 13, 2015, in which case the natural expiration of a design patent is generally 15 years after its issue date. Various extensions may be available; however, the life of a patent, and the protection it affords, is limited. Without patent protection for our products and services, we may be open to competition. Further, if we encounter delays in our development efforts, the period of time during which we could market our products and services under patent protection would be reduced.
| 47 |
| Table of Contents |
In addition to the protection afforded by patents, we also rely on trade secret protection to protect proprietary know-how that may not be patentable or that we elect not to patent, processes for which patents may be difficult to obtain or enforce, and any other elements of our products and services that involve proprietary know-how, information or technology that is not covered by patents. However, trade secrets can be difficult to protect. If the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating any trade secrets. Misappropriation or unauthorized disclosure of our trade secrets could significantly affect our competitive position and may have a material adverse effect on our business. Furthermore, trade secret protection does not prevent competitors from independently developing substantially equivalent products.
We may not be able to protect our intellectual property and proprietary rights throughout the world.
Third parties may attempt to commercialize competitive products or services in foreign countries where we do not have any patents or patent applications where legal recourse may be limited. This may have a significant commercial impact on our foreign business operations.
Filing, prosecuting, and defending patents on our products in all countries throughout the world would be prohibitively expensive, and the laws of foreign countries may not protect our rights to the same extent as the laws of the U.S. Consequently, we may not be able to prevent third parties from utilizing our inventions in all countries outside the U.S., or from selling or importing products made using our inventions in and into the U.S. or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection but where enforcement is not as strong as that in the U.S. These products may compete with our products, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets, and other intellectual property protection, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our intellectual property and proprietary rights generally. Proceedings to enforce our intellectual property and proprietary rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property and proprietary rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Many countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we are forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be impaired, and our business, financial condition, results of operations and prospects may be adversely affected.
| 48 |
| Table of Contents |
Risks Related to Ownership of Our Common Stock
The price of our stock has been and may continue to be volatile, which could result in substantial losses for investors. Further, an active, liquid and orderly trading market for our common stock may not be sustained and we do not know what the market price of our common stock will be, and as a result it may be difficult for you to sell your shares of our common stock.
Prior to our listing on the New York Stock Exchange in September 2018, there was no public market for shares of our common stock. Although our common stock is now listed on the NYSE American, the market for our shares has demonstrated varying levels of trading activity. Furthermore, an active trading market for our shares may not be sustained in the future. You may not be able to sell your shares quickly or at the market price if trading in shares of our common stock is not active. Further, an inactive market may also impair our ability to raise capital by selling shares of our common stock and may impair our ability to enter into strategic partnerships or acquire companies or products by using shares of our common stock as consideration, which could have a material adverse effect on our business, financial condition, and results of operations. In addition, the trading price of our common stock is likely to be highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control, including limited trading volume. In addition to the factors discussed in this Risk Factors section and elsewhere in this Annual Report, these factors include:
| · | our failure to increase the sales of our products; |
|
|
|
| · | the failure by our customers to obtain adequate reimbursements or reimbursement levels that would be sufficient to support product sales to our customers and pricing of our products to support revenue projections; |
|
|
|
| · | unanticipated serious safety concerns related to the use of our products; |
|
|
|
| · | changes in our organization; |
|
|
|
| · | introduction of new products or services offered by us or our competitors; |
|
|
|
| · | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors; |
|
|
|
| · | our ability to effectively manage our future growth; |
|
|
|
| · | the size and growth of our target markets; |
|
|
|
| · | actual or anticipated variations in quarterly operating results; |
|
|
|
| · | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
|
|
|
| · | significant lawsuits, including shareholder litigation, government actions or litigation related to intellectual property; |
|
|
|
| · | our cash position; |
|
|
|
| · | our failure to meet the estimates and projections of the investment community or that we may otherwise provide to the public; |
|
|
|
| · | publication of research reports about us or our industry, or positive or negative recommendations or withdrawal of research coverage, by securities analysts; |
|
|
|
| · | any delay in any regulatory filings for our future products and any adverse development or perceived adverse development with respect to the applicable regulatory authority’s review of such products; |
|
|
|
| · | adverse regulatory decisions, including failure to receive regulatory approval of our future products, failure to maintain regulatory approval for our existing products or failure to obtain regulatory approval for additional indications for our existing products; |
|
|
|
| · | changes in laws or regulations applicable to our products; |
|
|
|
| · | adverse developments concerning our suppliers or distributors; |
|
|
|
| · | our inability to obtain adequate supplies and components for our products or inability to do so at acceptable prices; |
|
|
|
| · | our inability to establish and maintain collaborations if needed; |
|
|
|
| · | changes in the market valuations of similar companies; |
|
|
|
| · | overall performance of the equity markets; |
|
|
|
| · | sales of large blocks of our common stock including sales by our executive officers and directors; |
|
|
|
| · | trading volume of our common stock; |
| 49 |
| Table of Contents |
| · | limited “public float” in the hands of a small number of persons whose sales or lack of sales of our common stock could result in positive or negative pricing pressure on the market price for our common stock; |
|
|
|
| · | additions or departures of key scientific or management personnel; |
|
|
|
| · | changes in accounting practices; |
|
|
|
| · | ineffectiveness of our internal controls; |
|
|
|
| · | general political and economic conditions; and |
|
|
|
| · | other events or factors, many of which are beyond our control. |
In addition, the stock market in general, and medical device companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance. In the past, securities class action litigation has often been instituted against companies following periods of volatility in the market price of a company’s securities. This type of litigation, if instituted, could result in substantial costs and a diversion of management’s attention and resources, which could have a material adverse effect on our business, financial condition, and results of operations.
The ownership of our common stock is highly concentrated, and may become more so in the near future, which may prevent you and other stockholders from influencing significant corporate decisions and may result in conflicts of interest that could cause the company stock price to decline.
David A. Jenkins, our Executive Chairman of the Board, and his affiliates and family members, beneficially own or control, in the aggregate, approximately 16.9% of our outstanding shares of common stock. In addition, if the outstanding shares of our Series X convertible preferred stock, or Series X Preferred Stock, qualify to convert into common stock on or after July 9, 2024, which will occur if we satisfy the initial listing standards of the New York American or another securities exchange or are delisted from the NYSE American, it is possible that David A. Jenkins and affiliates and family members will beneficially own more than 50% of our outstanding common stock. Accordingly, these persons have a substantial influence, and in the future may have de facto control, over the election of directors, any merger, consolidation or sale of all or substantially all of our assets or any other significant corporate transactions. These stockholders may also delay or prevent a change of control, even if such a change of control would benefit the other stockholders. This significant concentration of stock ownership may adversely affect the trading price of our common stock due to investors’ perception that conflicts of interest may exist or arise, and may adversely affect the liquidity of our common stock. In addition, it is possible that after July 9, 2024, we will satisfy the controlled company provisions of the NYSE American, in which case the combined company would not be required to satisfy all of the corporate governance requirements of the NYSE American, including without limitation, requirements that a majority of the Board be independent and that the combined company have independent compensation and nominating committees. See “—In the near future, we may be a “controlled company” within the meaning of NYSE American rules and, as a result, we may qualify for, and may choose to rely on, exemptions from certain corporate governance requirements”.
In the future, we may be a “controlled company” within the meaning of NYSE American rules and, as a result, we may qualify for, and may choose to rely on, exemptions from certain corporate governance requirements.
If the outstanding shares of our Series X Preferred Stock qualify to convert into common stock on or after July 9, 2024, which will occur if we satisfy the initial listing standards of the New York American or another securities exchange or are delisted from the NYSE American, it is possible that David A. Jenkins and affiliates will beneficially own more than 50% of our outstanding common stock. In that case, the Company will be a “controlled company” as defined in Section 801 of the NYSE American Company Guide. Under the NYSE American rules, a company of which more than 50% of the voting power is held by an individual, group or another company is a “controlled company” and may elect not to comply with certain NYSE American corporate governance requirements, including:
| · | the requirement that a majority of the Company’s board of directors consists of independent directors; |
|
|
|
| · | the requirement that the Company’s directors must be nominated by a Nominating Committee composed by a majority of independent directors; and |
|
|
|
| · | the requirement that executive compensation must be determined or recommended to the Company’s board of directors for determination, by a Compensation Committee comprised of independent directors or by a majority of the independent directors on the Company’s board. |
| 50 |
| Table of Contents |
Accordingly, if we qualify as a controlled company, we will likely elect to be treated as such and our stockholders will not be afforded the same protections generally as stockholders of other NYSE American-listed companies.
We are a smaller reporting company, and we cannot be certain if the reduced reporting requirements applicable to smaller reporting companies will make our common stock less attractive to investors.
We are a smaller reporting company, as defined by SEC rules. For as long as we continue to be a smaller reporting company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not smaller reporting companies, including reduced financial statement requirements, and reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock, and our stock price may be more volatile.
Future sales and issuances of a substantial number of shares of our common stock or rights to purchase common stock by our stockholders in the public market could result in additional dilution of the percentage ownership of our stockholders and cause our stock price to fall.
If our stockholders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market, the trading price of our common stock could decline. As of March 12, 2024, we had 7,573,403 outstanding shares of our common stock and outstanding options to purchase up to 614,593 shares of our common stock. At our special meeting of stockholders held on March 21, 2023, our stockholders approved the conversion of 1,993.581 shares of our Series X Preferred Stock into 1,993,581 shares of our common stock. The remaining 12,656.011 shares of Series X Preferred Stock may be convertible into 12,656,011 shares of our common stock on or after July 9, 2024, in the event that we meet the initial listing standards of the NYSE American or another securities exchange or have been delisted from the NYSE American. Also at the special meeting, our stockholders authorized the issuance of 497,908 shares of our common stock and 7,203 shares of our convertible Series A preferred stock, which are convertible into up to 4,501,060 shares of our common stock, as well as the issuance of warrants described below. Since the issuance of the Series A stock on March 21, 2023, 3,500 shares have been converted into 2,187,104 shares of our common stock. There are 3,703 shares of our convertible Series A preferred stock currently outstanding, which are convertible into up to 2,313,956 shares of our common stock.
In connection with our February 2022 equity offering, July 2022 warrant repricing and 2020 equity offerings, we issued warrants to investors and our placement agents and, in connection with the sale of the Dermatology Business in 2021, we issued a warrant to the broker. In connection with our January 2023 warrant repricing, we issued a warrant to purchase up to 331,608 shares of common stock at $4.00 per share. Pursuant to a private placement in January 2023, as approved by the stockholders at our March 21, 2023 special meeting of stockholders, we also issued warrants to purchase up to 9,998,186 shares of common stock at a purchase price of $3.00 per share. We had an aggregate of 11,042,137 warrants outstanding as of March 12, 2024. During the first quarter of 2020, we adopted the 2020 Inducement Equity Incentive Plan, or the 2020 Plan, for the purpose of attracting, retaining and incentivizing employees in furtherance of our success. As of December 31, 2023, 540 shares were available for issuance under the 2020 Plan. In July 2023, we adopted the 2023 Equity Incentive Plan, or the 2023 Plan. As of March 12, 2024, 146,545 shares were available for issuance under the 2023 Plan, and options to purchase 410,000 shares were outstanding. The 2023 Equity Incentive Plan provides for quarterly increases in the number of shares authorized for issuance under the Plan based on a percentage of the increase in the number of shares outstanding during the quarter. We assumed options to purchase 753,699 shares in connection with the merger with Old Catheter, and as of March 12, 2024, 204,520 of these options remained outstanding. In addition, in the first quarter of 2024, we issued employee and director stock options to purchase an aggregate of 410,000 shares. If these additional shares of common stock are issued and sold, or if it is perceived that they will be sold, in the public market, this could result in additional dilution and the trading price of our common stock could decline.
| 51 |
| Table of Contents |
Further, additional capital may be needed in the future to continue our planned operations, including commercialization efforts, expanded research and development activities and costs associated with operating a public company. To raise capital, we may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner, we determine from time to time. If we sell common stock, convertible securities or other equity securities, investors may be materially diluted by subsequent sales. Such sales may also result in material dilution to our existing stockholders, and new investors could gain rights, preferences and privileges senior to the holders of our common stock.
Anti-takeover provisions under our charter documents and Delaware law could delay or prevent a change of control which could limit the market price of our common stock and may prevent or frustrate attempts by our stockholders to replace or remove members of our board of directors or our current management and may adversely affect the market price of our common stock.
Our certificate of incorporation and bylaws contain provisions that could delay or prevent a change of control of our company or changes in our board of directors that our stockholders might consider favorable. Some of these provisions include:
| · | that our board of directors is divided into three classes serving staggered three-year terms, such that not all members of the board is elected at one time, which could delay the ability of stockholders to change the membership of a majority of our board of directors; |
|
|
|
| · | the ability of our board of directors to issue shares of preferred stock and to determine the price and other terms of those shares, including preferences and voting rights, without stockholder approval, which could be used to significantly dilute the ownership of a hostile acquirer; |
|
|
|
| · | the exclusive right of our board of directors to elect a director to fill a vacancy created by the expansion of our board of directors or the resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our board of directors; |
|
|
|
| · | a prohibition on stockholder action through written consent, which requires that all stockholder actions be taken at an annual or special meeting of our stockholders; |
|
|
|
| · | a requirement that special meetings of stockholders be called only by the chairperson of the board of directors, the chief executive officer or president (in the absence of a chief executive officer) or a majority vote of our board of directors, which could delay the ability of our stockholders to force consideration of a proposal or to take action, including the removal of directors; |
|
|
|
| · | the requirement for the affirmative vote of holders of at least 66 2/3% of the voting power of all of the then outstanding shares of the voting stock, voting together as a single class, to amend the provisions of our certificate of incorporation relating to the issuance of preferred stock and management of our business or our bylaws, which may inhibit the ability of an acquirer to affect such amendments to facilitate an unsolicited takeover attempt; |
|
|
|
| · | the ability of our board of directors, by majority vote, to amend our bylaws, which may allow our board of directors to take additional actions to prevent an unsolicited takeover and inhibit the ability of an acquirer to amend our bylaws to facilitate an unsolicited takeover attempt; and |
|
|
|
| · | advance notice procedures with which stockholders must comply to nominate candidates to our board of directors or to propose matters to be acted upon at a stockholders’ meeting, which may discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain control of us. |
In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporate Law, which may prohibit certain business combinations with stockholders owning 15% or more of our outstanding voting stock. These anti-takeover provisions and other provisions in our certificate of incorporation and bylaws could make it more difficult for stockholders or potential acquirers to obtain control of our board of directors or initiate actions that are opposed by the then-current board of directors and could also delay or impede a merger, tender offer or proxy contest involving our company. These provisions could also discourage proxy contests and make it more difficult for you and other stockholders to elect directors of your choosing or cause us to take other corporate actions you desire. Any delay or prevention of a change of control transaction or changes in our board of directors could cause the market price of our common stock to decline.
| 52 |
| Table of Contents |
Our certificate of incorporation provides that the Court of Chancery of the State of Delaware and the federal district courts of the U.S. are the exclusive forums for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our certificate of incorporation provides that the Court of Chancery of the State of Delaware is the exclusive forum for any derivative action or proceeding brought on our behalf; any action asserting a breach of fiduciary duty; any action asserting a claim against us arising under the Delaware General Corporation Law, our certificate of incorporation or our bylaws; any action to interpret, apply, enforce or determine the validity of our certificate of incorporation or our bylaws; and any action asserting a claim against us that is governed by the internal affairs doctrine. This provision would not apply to suits brought to enforce a duty or liability created by the Exchange Act or any other claim for which the U.S. federal courts have exclusive jurisdiction.
Our certificate of incorporation further provides that the federal district courts of the U.S. are the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act. The enforceability of similar exclusive federal forum provisions in other companies’ organizational documents has been challenged in legal proceedings, and while the Delaware Supreme Court has ruled that this type of exclusive federal forum provision is facially valid under Delaware law, there is uncertainty as to whether other courts would enforce such provisions.
These exclusive forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees. Alternatively, if a court were to find either exclusive forum provision in our certificate of incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could have a material adverse effect on our business, financial condition, and results of operations.
We are subject to the continued listing requirements of the NYSE American. If we are unable to comply with such requirements, our common stock would be delisted from the NYSE American, which would limit investors’ ability to effect transactions in our common stock and subject us to additional trading restrictions.
Shares of our common stock are currently listed on the NYSE American. In order to maintain our listing, we must maintain certain share prices, financial and share distribution targets, including maintaining a minimum amount of stockholders’ equity, minimum public float, and a minimum number of public stockholders. In addition to these objective standards, the NYSE American may delist the securities of any issuer if, in its opinion, the issuer’s financial condition and/or operating results appear unsatisfactory; if it appears that the extent of public distribution or the aggregate market value of the security has become so reduced as to make continued listing on the NYSE American inadvisable; if the issuer sells or disposes of principal operating assets or ceases to be an operating company; if an issuer fails to comply with the NYSE American’s listing requirements; if an issuer’s common stock sells at what the NYSE American considers a “low selling price” (generally trading below $0.20 per share for an extended period of time); or if any other event occurs or any condition exists which makes continued listing on the NYSE American, in its opinion, inadvisable. On August 31, 2022, we received a deficiency letter from the NYSE American indicating that we were not in compliance with Section 1003(f)(v) of the NYSE American Company Guide, because shares of our common stock have been selling for a low price per share for a substantial period time. We have since regained compliance with this Section, but there can be no guarantee that our stock price will not fall below the required levels again. We also received similar letters related to our late Form 10-Q filings during 2023, but we have since filed all late Forms and have remedied those deficiencies.
| 53 |
| Table of Contents |
If the NYSE American delists our shares of common stock from trading on its exchange and we are not able to list our securities on another national securities exchange, we expect our common stock would qualify to be quoted on an over-the-counter market. If this were to occur, we could face significant material adverse consequences, including:
| · | a limited availability of market quotations for our securities; |
|
|
|
| · | reduced liquidity for our securities; |
|
|
|
| · | a determination that our common stock is a “penny stock” which will require brokers trading in our common stock to adhere to more stringent rules and possibly result in a reduced level of trading activity in the secondary trading market for our securities; |
|
|
|
| · | a limited amount of news and analyst coverage; |
|
|
|
| · | a decreased ability to issue additional securities or obtain additional financing in the future. |
The National Securities Markets Improvement Act of 1996, which is a federal statute, prevents or preempts the states from regulating the sale of certain securities, which are referred to as “covered securities.” Because our shares of common stock are listed on the NYSE American, our shares of common stock qualify as covered securities under such statute. Although the states are preempted from regulating the sale of our securities, the federal statute does allow the states to investigate companies if there is a suspicion of fraud, and, if there is a finding of fraudulent activity, then the states can regulate or bar the sale of covered securities in a particular case. If we were no longer listed on the NYSE American, our securities would not be covered securities and we would be subject to regulation in each state in which we offer our securities.
We have not paid dividends in the past and have no immediate plans to pay dividends.
We plan to reinvest all of our earnings, to the extent we have earnings, in order to market our products and to cover operating costs and to otherwise become and remain competitive. We do not plan to pay any cash dividends with respect to our securities in the foreseeable future. We cannot assure you that we would, at any time, generate sufficient surplus cash that would be available for distribution to the holders of our common stock as a dividend.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 1C. CYBERSECURITY
The Company approaches cybersecurity as an enterprise-wide risk and uses external resources to assess risk and manage its IT and 24x7 cybersecurity operations, including managed service providers who assist in the support of key business systems. The Company may also periodically engage external consultants to assist with cybersecurity incident management, particularly where advanced or specialized expertise may be required. If a material cybersecurity breach occurs, the incident will be reviewed by management to determine whether further escalation is appropriate. The Executive Chairman of the Board and Chief Executive Officer and the Interim Chief Financial Officer have primary responsibility for this review and assessment. Any incident assessed as potentially being or becoming material will immediately be escalated for further assessment and reported to designated members of our executive team, and if deemed necessary or appropriate, the Board of Directors. The Board Audit Committee is responsible for reviewing and discussing the adequacy and effectiveness of the Company’s information security policies and the procedures and controls regarding information systems. In addition, we plan to consult with outside counsel as appropriate, including on materiality analyses and disclosure matters, and to assist in making the final materiality determination regarding disclosure and other compliance decisions. We also plan to keep our independent public accounting firm informed of such incidents as appropriate.
The Company maintains a cyber liability insurance policy that is designed to cover certain expenses, business losses, business interruption, and fines and penalties associated with a data breach or other similar incident. Cyber liability insurance also provides coverage in the event of a ransomware attack.
To date, the Company has not had a cybersecurity event that materially impacted or is reasonably likely to materially affect its business strategy, results of operations, financial condition, or the security of its proprietary data.
| 54 |
| Table of Contents |
ITEM 2. PROPERTIES
Our manufacturing, inventory and order fulfillment activities are performed in our approximate 2,000 square foot headquarters facility in Fort Mill, South Carolina under a lease that expires in November 2025. We conduct administrative and accounting activities in an approximate 1,100 square foot facility in Augusta, New Jersey under a lease that expires in December 2024. Administrative activities are also conducted in an approximate 1,200 square foot facility in Park City, Utah under a lease that expires in April 2026.
We believe that our existing facilities are sufficient to meet our current needs and that suitable additional space will be available as and when needed.
ITEM 3. LEGAL PROCEEDINGS
In the normal course of business, we are at times subject to pending and threatened legal actions. In management’s opinion, any potential loss resulting from the resolution of these matters will not have a material effect on our results of operations, financial position or cash flows.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
| 55 |
| Table of Contents |
PART II — FINANCIAL INFORMATION
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information and Holders
Our common stock is traded on the NYSE American under the symbol “VTAK”. Prior to August 17, 2023 the Company’s common stock was traded on the NYSE American under the symbol “RMED”.
On March 12, 2024, the last reported sales price of our common stock was $0.58 and, according to our transfer agent, as of March 12, 2024, there were 112 record holders of our common stock. The actual number of stockholders is greater than the number of record holders and includes stockholders who are beneficial owners but whose shares are held in street name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust or by other entities.
Dividend Policy
We have never declared or paid cash dividends on our common stock. We currently intend to retain all available funds and any future earnings for use in the operation of our business and do not anticipate paying any dividends on our common stock in the foreseeable future. Any future determination to declare dividends will be made at the discretion of our board of directors and will depend on, among other factors, our financial condition, operating results, capital requirements, general business conditions, the terms of any future credit agreements and other factors that our board of directors may deem relevant.
ITEM 6. [Reserved]
| 56 |
| Table of Contents |
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Investing in our common stock involves a high degree of risk. You should carefully consider the risks and uncertainties described below, together with all of the other information in this Annual Report on Form 10-K, including the Risk Factors contained in Item 1A, before making an investment decision. The risks and uncertainties described in this Annual Report on Form 10-K may not be the only ones we face. If any of the risks actually occur, our business, financial condition, operating results, cash flows and prospects could be materially and adversely affected. In that event, the market price of our common stock could decline, and you could lose part or all of your investment. Refer to the Current Report on Form 8-K filed on March 28, 2023 for management’s discussion and analysis of financial condition and results of operations for Catheter Precision, Inc.’s historical financial results.
Overview
The registrant (together with our consolidated operating subsidiary, the “Company” or “Catheter”) was incorporated under the name “Ra Medical Systems, Inc.” as a Delaware corporation in July 2018. A predecessor had been incorporated in California in September of 2002, but was reincorporated in 2018 in connection with our initial public offering. The Company was initially formed to develop, commercialize and market an excimer laser-based platform for use in the treatment of vascular and dermatological immune-mediated inflammatory diseases, including the DABRA product line.
On January 9, 2023, the Company merged with the former Catheter Precision, Inc., or “Old Catheter”, a privately-held Delaware corporation (the “Merger”), and the business of Old Catheter became a wholly owned subsidiary of the Company, which today is our only operating subsidiary. Following the Merger, we discontinued the Company’s legacy lines of business and the use of any of its DABRA-related assets. For further information about these historical lines of business, see “Item 1. Business” of the Company’s Form 10-K for the fiscal year ended December 31, 2021. Since the Merger, we have shifted the focus of our operations to Old Catheter’s product lines. Accordingly, our current activities primarily relate to Old Catheter’s historical business which comprises the design, manufacture and sale of new and innovative medical technologies focused in the field of cardiac electrophysiology, or “EP.”
Our primary product is the View into Ventricular Onset System or VIVO System (“VIVO” or “VIVO System”) which is a non-invasive imaging system that offers 3D cardiac mapping to help with localizing the sites of origin of idiopathic ventricular arrhythmias in patients with structurally normal hearts prior to EP procedures.
Our newest product, LockeT, is a suture retention device indicated for wound healing by distributing suture tension over a larger area in the patient in conjunction with a figure of eight suture closure. LockeT is intended to temporarily secure sutures and aid clinicians in locating and removing sutures efficiently.
Our product portfolio also includes the Amigo® Remote Catheter System, or Amigo, a robotic arm that serves as a catheter control device. Prior to 2018, Old Catheter marketed Amigo. We own the intellectual property related to Amigo, and this product is under consideration for future research and development of a generation 2 product.
Pre-Merger Operations
The Company owns intellectual property related to an advanced excimer laser-based platform for use in the treatment of vascular immune-mediated inflammatory diseases. The Destruction of Arteriosclerotic Blockages by laser Radiation Ablation, laser and single-use catheter, together referred to as the DABRA Excimer Laser System or DABRA, was developed as a tool in the treatment of Peripheral Artery Disease which commonly occurs in the legs. The Company also previously marketed the Pharos laser which was used to treat proliferative skin conditions. The Company completed the sale of its Pharos laser business, or Dermatology Business, to STRATA Skin Sciences, Inc. on August 16, 2021.
| 57 |
| Table of Contents |
The board of directors approved a reduction in force ("RIF") effective June 6, 2022, under which approximately 65% of Ra Medical's full-time employees were immediately terminated and provided one-time severance payments totaling approximately $0.6 million. In August and September 2022, an additional 20% of Legacy Ra Medical’s employees were terminated and provided one-time severance payments totaling approximately $0.3 million. The purpose of the RIF was to preserve capital with the goal of maximizing the opportunities available to Legacy Ra Medical during the board of directors’ review of strategic alternatives.
As a result of the RIF and the board of directors’ review of strategic alternatives, the Company paused all engineering activities in June 2022. The Company has ceased marketing the DABRA Excimer Laser System and does not currently intend to commercialize the DABRA 2.0 catheter.
Post-Merger Operations
Looking forward, we do not expect to use our legacy DABRA-related assets or continue the Company's legacy lines of business, but instead have shifted the focus of our operations to Old Catheter’s product lines. Accordingly, our current activities primarily relate to Old Catheter’s historical business, which comprises the design, manufacture and sale of new and innovative medical technologies focused in the field of cardiac electrophysiology, or EP.
Our primary product is the VIVO System. We are focused on the design, market development and usage adoption of our VIVO System by cardiac electrophysiologists to enhance their ability to diagnose and treat cardiac arrhythmias. We have completed development, received regulatory clearance, and initiated sales of the VIVO System in the U.S. and Europe.
Our business strategy is to become a leading medical imaging company in the field of cardiac electrophysiology, and we are dedicated to developing and delivering electrophysiology products to provide patients, hospitals, and physicians with novel technologies and solutions to improve the lives of patients with cardiac arrhythmias. We aim to establish VIVO as an integral tool used by cardiac electrophysiologists during ablation treatment of ventricular arrhythmias by reducing procedure time and patient complications and increasing procedural success.
We have received FDA clearance to market and promote the VIVO System in the United States as a pre-procedure planning tool for patients with structurally normal hearts undergoing ablation treatment for idiopathic ventricular arrhythmias. VIVO allows for the acquisition, analysis, display and storage of cardiac electrophysiological data and maps for analysis by a physician. We began a limited commercial launch of VIVO in 2021 and to date, VIVO has been utilized in more than 1,000 procedures in the U.S. and EU by over 30 physicians, with no reported device-related complications.
We have been cleared to label the VIVO System with the CE Mark in the EU and certain other countries. The CE Mark designation, which affirms the product’s conformity with European health, safety, and environmental protection standards, allows us to market that product in countries that are members of the EU and the European Free Trade Association. Catheter has commenced limited sales of the VIVO System in Europe and the UK through independent distributors. Catheter’s international distributors are supported by two EU based full time consultants.
In addition, LockeT, a suture retention device, is a sterile, Class I product that was registered with the FDA in February 2023, at which time we began initial shipments to distributors. In May 2023, Catheter submitted LockeT for CE Mark approval. CE Mark approval is expected in the second half of 2024, at which time initial international shipments to distributors will begin. LockeT is indicated for wound healing by distributing suture tension over a larger area in the patient in conjunction with a figure of eight suture closure, and it is intended to temporarily secure sutures and aid clinicians in locating and removing sutures efficiently.
| 58 |
| Table of Contents |
Clinical studies for LockeT began during the year ended December 31, 2023. The three phases of the current studies are planned to show the product’s effectiveness and benefits, including faster wound closure, earlier ambulation, potentially leading to early hospital discharge, and lower costs for the healthcare provider and/or insurance payor. This data is intended to provide crucial data for marketing and to expand our indications for use with the FDA.
Recent Developments
Settlement Agreements with the Department of Justice and Participating States
On December 28, 2020, the Company entered into a settlement agreement with the U.S., acting through the Department of Justice ("DOJ") and on behalf of the Office of Inspector General, and other settlement agreements with certain state attorneys general (collectively the "Settlement Agreements"), to resolve investigations and a related civil action concerning its marketing of the DABRA laser system and DABRA-related remuneration to certain physicians. Pursuant to the terms of the Settlement Agreements, if the Company was acquired or was otherwise involved in a change in control transaction (as defined in the Settlement Agreements) before the end of 2024, the Company was required to pay a settlement amount of $5.0 million. As a result of the Merger, the Company made payments of $4.7 million and $0.3 million to the DOJ and participating states, respectively, in February 2023. Such amounts were included in accrued expenses in the balance sheet at December 31, 2022.
Warrant Inducement Offer
On January 9, 2023, we reduced the exercise price of certain existing warrants, or the Existing Warrants, exercisable for 331,608 shares of the Company's common stock held by a certain investor (the “Investor”), with exercise prices ranging from $14.00 to $526.50 per share to $4.00 per share, or the Warrant Repricing. In connection with the Warrant Repricing, we entered into a warrant inducement offer letter, or the Inducement Letter, with the Investor pursuant to which it would exercise up to all of the 331,608 Existing Warrants, or the Inducement Offer. In consideration for exercising the Existing Warrants pursuant to the terms of the Inducement Letter, we received approximately $1.3 million in gross proceeds. We paid the placement agent aggregate cash fees of approximately $0.2 million related to the Inducement Offer which represented 8.0% of the gross proceeds received from the Inducement Offer plus other offering costs. In consideration for exercising the Existing Warrants pursuant to the terms of the Inducement Letter, we issued the Investor a new Series E common stock purchase warrant, or Series E Warrant, to purchase 331,608 shares of common stock at an exercise price of $4.00 per share. The Series E Warrant is exercisable for five years from the date of stockholder approval. Exercise of the Series E Warrant in full was approved by the Company's stockholders at the special Stockholders’ Meeting held on March 21, 2023. The incremental fair value of the repriced warrants amounted to $0.3 million and the fair value of Series E warrant totaled $1.9 million. The relative fair values of such amounts were recorded to additional paid-in capital concurrent with the exercise of the Existing Warrants. The Company registered the shares of common stock underlying the Series E Warrant for resale in February 2023.
Securities Purchase Agreement
On January 9, 2023, we entered into a Securities Purchase Agreement (the "Securities Purchase Agreement"), for a private placement (the "Private Placement"), with the Investor. Pursuant to the Securities Purchase Agreement, on March 23, 2023, the Investor purchased, for an aggregate purchase price of approximately $8.0 million, (a) 497,908 Class A Units at a price of $1.60029 per Class A Unit, each consisting of one share of common stock, one Series F Common Stock Purchase Warrant, or Series F Warrant, and one Series G Common Stock Purchase Warrant, or Series G Warrant, and together with the Series F Warrant, the PIPE Warrants, and (b) 4,501,060 Class B Units at a price of $1,000 per unit, each consisting of one share of a new series of the Company’s preferred stock, designated as Series A Convertible Preferred Stock, par value $0.0001, or the PIPE Preferred Stock, and one Series F Warrant and one Series G Warrant for each share of the Company’s common stock underlying the PIPE Preferred Stock, each share of which is convertible into approximately 625 shares of the Company’s common stock, or the Preferred Conversion Rate. The closing under the Securities Purchase Agreement and the sale and issuance of the Class A Units and Class B Units (and the issuance of any underlying common stock) was approved at the special Stockholders’ Meeting held March 21, 2023.
| 59 |
| Table of Contents |
The PIPE Warrants are exercisable at an exercise price of $3.00 per share, subject to adjustments as provided under the terms of the PIPE Warrants. The PIPE Warrants are exercisable at any time until the expiration thereof, except that the PIPE Warrants cannot be exercised if, after giving effect thereto, the purchaser would beneficially own more than 4.99%, or the Maximum Percentage, of the outstanding shares of common stock of the Company, which Maximum Percentage may be increased or decreased by the purchaser with written notice to the Company to any other percentage specified not in excess of 9.99%. The Series F Warrants have a term of two years from the date of stockholder approval, and the Series G Warrants have a term of six years from the date of stockholder approval. Stockholder approval of the Series F Warrants and Series G Warrants was obtained at the special Stockholders’ Meeting held on March 21, 2023.
Shares of PIPE Preferred Stock, the conversion of which was approved at the special Stockholders’ Meeting held on March 21, 2023, convert into common stock at the option of the holder at the Preferred Conversion Rate, subject to certain ownership limitations as described below. The conversion price is subject to adjustment in the case of stock splits, stock dividends, combinations of shares and similar recapitalization transactions.
Subject to limited exceptions, holders of shares of PIPE Preferred Stock do not have the right to convert any portion of their Preferred Stock if the holder, together with its affiliates, would beneficially own in excess of 9.99% of the number of shares of the Company’s common stock outstanding immediately after giving effect to its conversion.
Holders of PIPE Preferred Stock are entitled to receive dividends on shares of PIPE Preferred Stock equal, on an as-if-converted-to-common stock basis, and in the same form as dividends actually paid on shares of the common stock. Except as otherwise required by law, the PIPE Preferred Stock does not have voting rights. However, as long as any shares of PIPE Preferred Stock are outstanding, the Company will not, without the affirmative vote of the holders of a majority of the then outstanding shares of the PIPE Preferred Stock, (a) alter or change adversely the powers, preferences or rights given to the PIPE Preferred Stock, (b) alter or amend the Certificate of Designation for the PIPE Preferred Stock, (c) amend its certificate of incorporation or other charter documents in any manner that adversely affects any rights of the holders of PIPE Preferred Stock, (d) increase the number of authorized shares of PIPE Preferred Stock, or (e) enter into any agreement with respect to any of the foregoing. The PIPE Preferred Stock does not have a preference upon any liquidation, dissolution or winding-up of the Company. The holders of PIPE Preferred Stock shall be entitled to receive out of the assets, whether capital or surplus, of the Company the same amount that a holder of the Company’s common stock would receive if the PIPE Preferred Stock were fully converted (disregarding for such purposes any conversion limitations) to the Company’s common stock, which amounts will be paid pari passu with all holders of the Company’s common stock.
The Company also entered into a registration rights agreement with the purchasers requiring the Company to register the resale of the shares of its common stock, the shares issuable upon exercise of the PIPE Warrants and the shares issuable upon the conversion of the PIPE Preferred Stock. These registration statements were declared effective in April 2023.
The net proceeds from the Private Placement and the Warrant Repricing have been used to advance the development and commercialization of our novel electrophysiology technologies and solutions and to support general corporate purposes.
| 60 |
| Table of Contents |
Conversion of Series X Convertible Preferred Stock
On March 21, 2023, the Company held a special meeting of stockholders (the “Stockholders’ Meeting”), at which the stockholders approved, among other things, the issuance of 1,993,581 shares of common stock upon conversion of 1,993.581 of Series X Convertible Preferred Stock which were issued upon the closing of the Merger (see Note 3, Business Combination of our accompanying audited consolidated financial statements). On March 23, 2023, the Company issued 1,974,905 shares of common stock upon the conversion of 1,974.905 shares of Series X Convertible Preferred Stock. On October 24, 2023, the remaining 18,676 shares of common stock were issued upon the conversion of 18.676 shares of Series X Convertible Preferred Stock. The remaining 12,656.011 shares of Series X Convertible Preferred Stock are expected to remain outstanding until at least July 9, 2024, and will convert thereafter up to 12,656,011 shares of common stock, only if the Company meets the initial listing standards of the NYSE American or another national securities exchange or is delisted from the NYSE American.
Issuance of Securities upon Conversion of Series A Preferred
On July 5, 2023 the Company issued 1,093,552 shares of its common stock in connection with the conversion of 1,750 shares of its outstanding Series A Convertible Preferred Stock. The shares were issued in connection with two separate conversions of 875 shares of Series A Convertible Preferred Stock into 546,776 shares of common stock that occurred on July 3, 2023. Each share of Series A Convertible Preferred Stock is convertible into approximately 625 shares of common stock.
On July 24, 2023, the Company issued 546,776 shares of its common stock in connection with the conversion of 875 shares of its outstanding Series A Convertible Preferred Stock.
On January 24, 2024, the Company issued 546,776 shares of its common stock in connection with the conversion of 875 shares of its outstanding Series A Convertible Preferred Stock.
Adoption of 2023 Equity Incentive Plan
On July 11, 2023, we held an Annual Meeting where our stockholders approved the 2023 Equity Incentive Plan (“2023 Plan”) that authorizes us to grant options, restricted stock and other equity-based awards. No issuance of options were granted under the 2023 Plan during the year ended December 31, 2023. Options to purchase an aggregate of 435,000 shares were granted in January and February of 2024 and 121,545 shares currently remain available for grant under the 2023 Plan, subject to adjustment as provided therein.
Components of our Results of Operations for the Years Ended December 31, 2023 and 2022
Revenues
Product sales revenues prior to the Merger consisted of sales of catheters for use with the DABRA laser in our atherectomy clinical trials.
After the Merger, our legacy DABRA laser is no longer in use and we have shifted the focus of our operations to Old Catheter’s product lines. Accordingly, our current activities primarily relate to Old Catheter’s historical business which comprises the design, manufacture and sale of new and innovative medical technologies focused in the field of cardiac electrophysiology, or EP.
Our revenues post-Merger primarily consist of VIVO, which is a non-invasive imaging system that offers 3D cardiac mapping to help with localizing the sites of origin of idiopathic ventricular arrhythmias in patients with structurally normal hearts prior to EP procedures. In addition to the VIVO System, customers are provided with VIVO Positioning Patch Sets, which are custom patches, that are used in conjunction with the VIVO System to complete the intended output of the VIVO System. The delivery of the VIVO System, including the VIVO Positioning Patch Sets represents the Company’s primary performance obligation. The Company recognizes revenue upon the delivery of the VIVO system. The Company also provides customers with the option to pay for software upgrades in advance at the time of the contract's inception. Software upgrades are stand-ready services, whereby the Company will provide software upgrade services to the customer when and as upgrades are available. Terms of the period covered by the payment of software upgrades in advance can range from one year to multiple years. Customers have the option to renew terms covered by software upgrades at the end of each term. The stand-ready software upgrades represent the Company's second separate performance obligation and revenue is recognized over the term of the period.
| 61 |
| Table of Contents |
The Company is a business that has operations within multiple countries. During 2023, approximately 25% of the Company’s sales were derived from customers outside the United States.
Cost of Revenues
Cost of revenues for product sales consisted primarily of costs of components for use in our products, the labor used to produce our products, and the manufacturing overhead that supports production.
Selling, General and Administrative Expenses
Selling, general and administrative ("SG&A"), expenses consist of employee-related costs, including salaries, benefits and stock-based compensation expenses. Other SG&A expenses include amortization of intangible assets and accretion of royalties payable acquired in the Merger, professional services fees, including legal, audit and tax fees, insurance costs, general corporate expenses and facility-related expenses.
Research and Development Expenses
Research and development ("R&D"), expenses are expensed as incurred and include the following: product development, certain employee-related expenses, including salaries, benefits and an allocated portion of stock-based compensation expense; cost of clinical studies to support new products and product enhancements, including expanded indications; supplies used for internal R&D and clinical activities; and cost of outside consultants who assist with technology development and clinical affairs.
Results of Operations for the Years Ended December 31, 2023 and 2022
The following table sets forth the results of the Company's operations for the periods presented ($ in thousands):
|
| For the Year Ended December 31, |
|
|
| |||||||
|
| 2023 |
|
| 2022 |
|
| Change |
| |||
Revenue |
| $ | 442 |
|
| $ | 14 |
|
| $ | 428 |
|
Cost of revenues |
|
| 30 |
|
|
| 161 |
|
|
| (131 | ) |
Selling, general and administrative expenses |
|
| 17,122 |
|
|
| 16,250 |
|
|
| 872 |
|
Research and development expenses |
|
| 475 |
|
|
| 6,392 |
|
|
| (5,917 | ) |
Restructuring/impairment charges |
|
| 60,934 |
|
|
| 4,172 |
|
|
| 56,762 |
|
Change in fair value of royalties payable |
|
| 7,208 |
|
|
| — |
|
|
| 7,208 |
|
Other income, net |
|
| 339 |
|
|
| 99 |
|
|
| 240 |
|
Revenues
The increase in revenues of approximately $428 thousand for the year ended December 31, 2023 as compared to the corresponding period in the prior year was due to product sales of the VIVO system, as a result of the merger that took place in January 2023.
| 62 |
| Table of Contents |
Cost of Revenues
The decrease in cost of revenues of approximately $131 thousand for the year ended December 31, 2023 as compared to the corresponding period in the prior year was due to the cost of sales of the VIVO System, as a result of the Merger that took place in January 2023, which were substantially lower than the cost of revenues for the Ra Medical legacy products during the comparable prior year periods, which legacy products have been discontinued.
Selling, General and Administrative Expenses
The increase in SG&A of approximately $0.9 million for the year ended December 31, 2023 as compared to the corresponding period in the prior year was due primarily to the increase in depreciation and amortization of approximately $2.0 million that resulted from intangible assets acquired in the Merger, an increase in salaries and benefits of $2.0 million related to the Company's former Chief Executive Officer, an increase in stock based compensation of approximately $0.8 million, which was related to the one time stock compensation for Old Catheter stock options assumed in the Merger, an increase in consulting expenses of $0.7 million and an increase in other selling, general and administrative expenses of $0.4 million. The increase was partially offset by a decrease in professional fees of approximately $3.6 million, which were incurred in connection with the Merger, a decrease in insurance expense of approximately $0.8 million, and a decrease of investor relations and SEC fees of approximately $0.6 million.
Research and Development Expenses
The decrease in R&D expenses of approximately $5.9 million for the year ended December 31, 2023 as compared to the corresponding period in the prior year was due primarily to a decrease in R&D salaries and benefits expenses of $3.5 million, a decrease of parts and materials of $0.7 million, a decrease in clinical study costs of $0.6 million, a decrease in R&D professional fees of $0.6 million, a decrease in R&D facilities allocation expenses of $0.3 million, and a decrease in other expenses of $0.4 million. These decreases were primarily the result of the discontinuation of the historical products of Ra Medical that were under development. The decrease was partially offset by an increase in suture retention device development of $0.2 million.
| 63 |
| Table of Contents |
Restructuring and Impairment Charges
We test for goodwill impairment at the reporting level annually in the fourth quarter or more frequently if a change in circumstances or the occurrence of events indicates that potential impairment exists. As a result of the Merger with Old Catheter, the Company recognized $60.9 million of goodwill. Due to a sustained decrease in our share price during the quarters ended March 31, 2023 and June 30, 2023, we concluded that in accordance with ASC 350 a triggering event occurred indicating that potential impairment exists that required us to assess if impairment existed as of March 31, 2023 and June 30, 2023. In accordance with ASC 350 we performed a quantitative goodwill impairment test, which resulted in the carrying amount of the reporting unit exceeding its fair value, indicating that the goodwill of the reporting unit was impaired. We utilized a combination of an income and market approach to assess the fair value of the reporting unit as of March 31, 2023 and June 30, 2023. The income approach considered the discounted cash flow model, considering projected future cash flows (including timing and profitability), discount rate reflecting the risk inherent in future cash flows, perpetual growth rate, and projected future economic and market conditions while the guideline public company market approach considered marketplace earnings multiples from within a peer public company group. We recorded the impairment charge of $60.9 million within loss on impairment of goodwill in the consolidated statement of operations. As of December 31, 2023, cumulative goodwill impairment charges of $60.9 million were incurred related to our single reporting unit.
Restructuring costs of $4.2 million were incurred during the year ended December 31, 2022, due to the RIF and the board of directors’ decisions to discontinue manufacturing activities and enrollment in the clinical trial of the legacy DABRA products. There were no restructuring costs incurred during the year ended December 31, 2023.
Change in fair value of royalties payable
As of the date of the Merger, the royalties payable was calculated using a discounted cash flow method utilizing a discount rate of 24.1%. At each reporting period, the fair value of the royalties payable is calculated using the discounted cash flow method. At December 31, 2023, the discount rate was 28.0%. The change in fair value of the royalties payable from the date of the Merger to December 31, 2023, was a decrease of $7.2 million. There were no royalties payable for the year ended December 31, 2022 and therefore no fair value measurement.
Other income, Net
The increase in other income (expense), net of approximately $0.2 million for the year ended December 31, 2023 as compared to the corresponding periods in the prior year was primarily due to an increase in interest income.
Liquidity and Capital Resources
As of December 31, 2023, we had cash and cash equivalents of approximately $3.6 million and an accumulated deficit of approximately $275.7 million. For the year ended December 31, 2023, net cash used from operating activities was approximately $20.6 million. We have incurred recurring net losses from operations and negative cash flows from operating activities since inception.
| 64 |
| Table of Contents |
In January 2023, we raised gross proceeds of $1.3 million from a Warrant Repricing and, in March 2023, we completed a Private Placement and raised gross proceeds of $8.0 million (see Note 13, Equity Offerings, of our accompanying audited consolidated financial statements). Despite the additional financing, we expect operating losses and negative cash flows to continue for the foreseeable future as we invest in our commercial capabilities. These negative cash flows and additional costs associated with the Merger paid during the year ended December 31, 2022 and during the year ended December 31, 2023 have substantially depleted our cash. Following the Merger with Old Catheter, we further reduced staff and other costs while assuming the operating costs of Old Catheter. Of the Company’s cash flows used in operating activities of $20.6 million, a portion of them are cash outflows related to the Merger and are non-recurring in nature. Specifically, we paid approximately $5.0 million in settlement costs that had been accrued as of December 31, 2022 (see Note 8, Accrued Expenses, of our accompanying audited consolidated financial statements) and $1.75 million in severance to our former Chief Executive Officer. We will continue to monitor our operating costs and seek to reduce our current liabilities. Such actions may impair our ability to proceed with certain strategic activities. As of March 7, 2024 we had $1.86 million of cash and cash equivalents. We believe that this amount will not be sufficient to fund our operations through May 2024. Because expected revenues are not adequate to fund our planned expenditures and anticipated operating costs beyond such point, we are currently evaluating potential means of raising cash through future capital transactions. If we are unable to do so, we will be required to reduce our spending rate to align with expected revenue levels and cash reserves, although there can be no guarantee that we will be successful in doing so. Accordingly, we will likely be required to raise additional cash through debt or equity transactions to continue our operations, and if we are unable to do so, we will be required to suspend a portion or all of our operations. We may not be able to secure financing in a timely manner or on favorable terms, if at all.
As a result of these factors, management has concluded that there is substantial doubt about the Company’s ability to continue as a going concern for a period of one year after the date the consolidated financial statements are issued. The Company’s consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Cash Flows for the Years Ended December 31, 2023 and 2022 ($ in thousands)
|
| For the Year Ended December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Net cash provided by (used in): |
|
|
|
|
|
| ||
Operating activities |
| $ | (20,619 | ) |
| $ | (22,568 | ) |
Investing activities |
|
| (61 | ) |
|
| 21 |
|
Financing activities |
|
| 8,386 |
|
|
| 23,361 |
|
Net change in cash and cash equivalents |
| $ | (12,294 | ) |
| $ | 814 |
|
Net Cash Used in Operating Activities
During the year ended December 31, 2023, net cash used in operating activities of $20.6 million consisted of a net loss of $70.6 million, a decrease in operating assets and liabilities of $7.1 million, partially offset by non-cash expenses of $57.0 million, consisting primarily of a loss on impairment of goodwill of $60.9 million, non-cash stock-based compensation of $1.2 million, depreciation and amortization of $2.1 million, and a change in fair value of royalties payable of $7.2 million.
During the year ended December 31, 2022, net cash used in operating activities of $22.6 million consisted of a net loss of $26.9 million, partially offset by non-cash expenses of $3.8 million, consisting primarily of non-cash restructuring costs of $2.9 million and stock-based compensation and depreciation and amortization each of $0.4 million, partially offset by a non-cash gain of $0.1 million related to the write-off of our right-of-use asset and liability due to the termination of the lease for our manufacturing and office space. In addition, there was a net change in operating assets and liabilities of $0.5 million.
| 65 |
| Table of Contents |
Net Cash (Used in)/Provided by Investing Activities
During the year ended December 31, 2023, net cash used in investing activities of $61 thousand consisted of purchases of property and equipment of approximately $76 thousand, offset by proceeds from cash acquired as part of business combination of approximately $15 thousand.
During the year ended December 31, 2022, net cash provided by investing activities of $21 thousand consisted of proceeds from sales of property and equipment of approximately $38 thousand, partially offset by purchases of property and equipment of approximately $17 thousand.
Net Cash Provided by Financing Activities
During the year ended December 31, 2023, net cash provided by financing activities of $8.4 million- primarily consisted of net cash proceeds from the private placement of $8.0 million, proceeds from the exercise of warrants of $1.3 million, and proceeds from issuance of common stock and warrants of $0.2 million, partially offset by the payment of offering costs of $0.6 million, payments of convertible promissory notes and accrued interest of $0.3 million, payments of costs related to the warrant repricing of $0.2 million, and payments on note payable of $0.1 million.
During the year ended December 31, 2022, net cash provided by financing activities of $23.4 million consisted primarily of net proceeds of $11.5 million from the issuance of common stock and warrants in the February 2022 offering, $7.4 million under our ATM offerings and $5.7 million from the exercises of warrants.
Off-Balance Sheet Arrangements
We do not engage in transactions that generate relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, as a part of our ongoing business. Accordingly, we did not have any off-balance sheet arrangements during any of the periods presented.
The Company’s Critical Accounting Estimates
The information set forth below relates to the Company’s critical accounting policies and estimates. The discussion and analysis of our financial position and results of operations is based on our audited consolidated financial statements included elsewhere in this Annual Report, which have been prepared in accordance with U.S. GAAP. We believe certain of our accounting policies are critical to understanding our financial position and results of operations.
Management’s discussion and analysis of the Company's financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of these consolidated financial statements requires us to make estimates and assumptions for the reported amounts of assets, liabilities, revenue, expenses and related disclosures. We regularly evaluate estimates and assumptions related to business combinations, including the determination of the purchase price and related allocations to the fair value of assets acquired and liabilities assumed, provisions for legal contingencies, income taxes, deferred income tax, asset valuation allowances, valuation of warrant liabilities, share based compensation and revenues. Our estimates are based on current facts, historical experience and various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions and any such differences may be material.
| 66 |
| Table of Contents |
We believe the following discussion addresses our most critical accounting policies, which are those that are most important to our financial condition and results of operations and require our most difficult, subjective and complex judgments.
Business Combinations
We account for business combinations under the provisions of ASC Topic 805-10, Business Combinations (“ASC 805-10”), which requires that the purchase method of accounting be used for all business combinations. Assets acquired and liabilities assumed, including non-controlling interests, are recorded at the date of acquisition at their respective fair values. ASC 805-10 also specifies criteria that intangible assets acquired in a business combination must meet to be recognized and reported apart from goodwill. Goodwill represents the excess purchase price over the fair value of the tangible net assets and intangible assets acquired in a business combination. Acquisition-related expenses are recognized separately from the business combinations and are expensed as incurred.
Accounting for Long-Lived Assets-Useful Lives
Intangible assets acquired from business combinations are initially measured at their estimated fair values and are then amortized on a straight-line basis over their estimated useful lives. Management evaluates whether events or circumstances have occurred that indicate the remaining useful life or carrying value of the amortizing intangible should be revised and adjusted, if necessary. Should the sum of the undiscounted expected future net cash flows be less than the carrying value, the Company would recognize an impairment loss at that date.
Goodwill
Goodwill, which represents the excess of purchase price of Old Catheter over the fair value of net assets acquired, is carried at cost. Goodwill is not amortized; rather, it is subject to a periodic assessment for impairment by applying a fair value-based test.
To determine whether goodwill is impaired, annually or more frequently if needed, the Company performs a multi-step impairment test. The Company first has the option to assess qualitative factors to determine if it is more likely than not that the carrying value of a reporting unit exceeds its estimated fair value. The Company may also elect to skip the qualitative testing and proceed directly to the quantitative testing. When performing quantitative testing, the Company first estimates the fair values of its reporting units using a combination of an income and market approach. To determine fair values, the Company is required to make assumptions about a wide variety of internal and external factors. Significant assumptions used in the impairment analysis include financial projections of free cash flow (including significant assumptions about operations including the rate of future revenue growth, capital requirements, and income taxes), long-term growth rates for determining terminal value and discount rates. Comparative market multiples are used to corroborate the results of the discounted cash flow test. These assumptions require significant judgment. Pursuant to ASU 2017-04, Simplifying the Test for Goodwill Impairment, the single step is to determine the estimated fair value of the reporting unit and compare it to the carrying value of the reporting unit, including goodwill. To the extent the carrying amount of goodwill exceeds the implied goodwill, the difference is the amount of the goodwill impairment. The Company also completes a reconciliation between the implied equity valuation prepared and the Company’s market capitalization. The majority of the inputs used in the discounted cash flow model are unobservable and thus are considered to be Level 3 inputs. The inputs for the market capitalization calculation are considered Level 1 inputs.
| 67 |
| Table of Contents |
Stock-Based Compensation
We calculate the cost of awards of equity instruments based on the grant date fair value of the awards issued to employees, members of our board of directors and nonemployee consultants using the Black-Scholes option pricing valuation model, or Black-Scholes model, which incorporates various assumptions including volatility, expected term and risk-free interest rate. The expected term of the options is the estimated period of time until exercise and was determined using the SEC’s safe harbor rules, using an average of vesting and contractual terms, as we did not have sufficient historical experience of similar awards. Expected stock price volatility is based on historical volatilities of certain “guideline” companies, as the Company does not have sufficient historical stock price data. The risk-free interest rate is based on the implied yield available on U.S. Treasury zero-coupon issues with an equivalent term. The estimated fair value of stock-based compensation awards is amortized on a straight-line basis over the relevant vesting period, adjusted for actual forfeitures at the time they occur.
Royalties Payable
We are obligated to pay royalties under various royalty agreements Old Cather had entered into. On January 9, 2023, prior to the consummation of the Merger, Old Catheter entered in an agreement with its Convertible Promissory Noteholders, which substantially consisted of amounts due to David A. Jenkins, previously Old Catheter's Chairman of the Board of Directors prior to the Merger, and, currently, the Company’s Executive Chairman of the Board of Directors and Chief Executive Officer, to forgive all accrued interest and future interest expense in exchange for a future royalty right. We will pay to the Noteholders a total royalty equal to approximately 12% of net sales of LockeT, commencing upon the first commercial sale, through December 31, 2035.
In addition, Old Catheter had entered into an agreement with the inventor of LockeT in exchange for the assignment and all rights to LockeT. Pursuant to the agreement, we will pay a 5% royalty on net sales up to $1 million in royalties. After $1 million has been paid, and if, and only if, a U.S. patent is granted by the United States Patent and Trademark Office, then we will continue to pay a royalty at a rate of 2% of LockeT net sales, until total cumulative royalties of $10 million have been paid.
During 2006 and 2007, Old Catheter entered into two investment grant agreements with a non-profit foundation for the purpose of funding the initial development of Old Catheter's AMIGO System. The agreement calls for the payment of sales-based royalties to the foundation, upon successful commercialization of the AMIGO System. We are not currently selling the AMIGO System.
New Accounting Pronouncements
In June 2022, the FASB issued ASU No. 2022-03, Fair Value Measurement (Topic 820): Fair Value Measurement of Equity Securities Subject to Contractual Sale Restrictions (“ASU 2022-03”) which clarifies guidance for fair value measurement of an equity security subject to a contractual sale restriction and establishes new disclosure requirements for such equity securities. ASU 2022-03 is effective for fiscal years beginning after December 15, 2023 and for interim periods within those fiscal years, with early adoption permitted. The Company is currently evaluating the impact of ASU 2022-03 on its consolidated financial statements.
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures, which requires public entities to disclose consistent categories and greater disaggregation of information in the rate reconciliation and for income taxes paid. It also includes certain other amendments to improve the effectiveness of income tax disclosures. The guidance is effective for financial statements issued for annual periods beginning after December 15, 2024, with early adoption permitted. The Company is required to adopt this standard prospectively in fiscal year 2025 for the annual reporting period ending December 31, 2025. The accounting pronouncement is not expected to have a material impact on the Company's related disclosures.
Effective January 1, 2023, repurchases are subject to a nondeductible excise tax under the Inflation Reduction Act of 2022 equal to 1.0% of the fair market value of the shares repurchased, subject to certain limitations. There was no impact to our financial condition or results of operations in 2023 as a result of the excise tax.
| 68 |
| Table of Contents |
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Our financial statements and supplementary data required by this item are set forth at the pages indicated in Item 15.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Previously reported.
ITEM 9A. CONTROLSAND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Executive Chairman of the Board and Chief Executive Officer and Interim Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as defined in Rule 13a-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act, as of December 31, 2023, including the disclosure controls and procedures of Old Catheter. Our objective in designing our disclosure controls and procedures is that they provide reasonable assurance of achieving their objectives of ensuring that information we are required to disclose in the reports we file or submit under the Exchange Act is accumulated and communicated to our management, including our Chief Executive Officer and Interim Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosures, and is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their desired control objectives, and management necessarily is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based upon this evaluation, due to the existence of the material weaknesses found in our internal controls over financial reporting described below, our Chief Executive Officer and Interim Chief Financial Officer concluded that, as of December 31, 2023, our disclosure controls and procedures were not effective at the reasonable assurance level. As disclosed in our Form 10-Qs for the quarters ended June 30, 2023 and September 30, 2023, for the reasons set forth therein, our Chief Executive Officer and then-Chief Financial Officer concluded that, as of March 31, 2023, June 30, 2023, and September 30, 2023, our disclosure controls and procedures were not effective at the reasonable assurance level, excluding at that time the disclosure controls and procedures of Old Catheter. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected and corrected on a timely basis. In preparation of our financial statements for the period covered by this report, we identified material weaknesses in internal control over financial reporting related to our control environment that existed as of December 31, 2023, as described below. Specifically, we identified material weaknesses with respect to (1) the lack of segregation of duties, (2) the lack of designed and operating review controls with respect to oversight of the financial reporting process, (3) errors with respect to the review of work performed by service providers, (4) errors in connection with accounting for the royalty obligation acquired in the merger with Old Catheter, (5) use of an incorrect discount rate in calculating the fair value of the royalty obligation, and (6) timing of revenue recognition. Notwithstanding the identified material weaknesses, management believes that the Financial Statements and related financial information included in this Annual Report for the year ended December 31, 2023 fairly present, in all material respects, our balance sheets, statements of operations, shareholders’ equity and cash flows as of and for the periods presented.
Remediation Plan
Management is in the process of developing a remediation plan. The material weaknesses will not be considered remediated until management designs and implements effective controls that operate for a sufficient period of time and management has concluded, through testing, that these controls are effective. The Company will monitor the effectiveness of its remediation plans and will make changes management determines to be appropriate. Anticipated remediation measures include continuing assessment of the need to expand the Company’s current accounting and financial reporting teams to include individuals with requisite experience to meet the requirements associated with the increasing operations of a publicly traded company, establishment of policies and procedures to ensure full review and sign offs with respect to the inputs sent to third-party service providers as well as the reports and documentation upon the completion of their work prior to any adjustments being made to the financial statements, establishment of policies and procedures related to the review of all contracts the Company enters into to ensure any terms or conditions are evaluated for any accounting required or accounting treatment or disclosure, and establishment of policies and procedures to review the inputs to royalty liability and other fair value calculations as well as the outputs impacting the balance at each reporting period. We have taken the following steps associated with material weaknesses related to Old Catheter: (1) for segregation of duties, we had hired additional employees, including a Chief Financial Officer; however, we are now seeking a new, permanent Chief Financial Officer, (2) we have begun recording revenue when the product is received by the customer, and (3) as a result of the Merger, Old Catheter no longer has derivative liabilities, so a previously identified material weakness related to this is no longer applicable to our current business.
| 69 |
| Table of Contents |
Changes in Internal Control over Financial Reporting
There have been no changes in the Company’s internal control over financial reporting during the quarter ended December 31, 2023 that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting other than those related to Old Catheter and the Merger with Old Catheter, which include changes necessitated by the change in the Company’s line of business and the remediation of the material weaknesses with respect to Old Catheter as described above.
Management’s Annual Report on Internal Control Over Financial Reporting and Attestation Report of the Registered Public Accounting Firm
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act). Management conducted an assessment of the effectiveness of our internal control over financial reporting, including Old Catheter’s internal control over financial reporting, based on the criteria set forth in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework). Based on the assessment, management has concluded that its internal control over financial reporting was not effective as of December 31, 2023 to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with GAAP, due to the material weaknesses discussed above at “Evaluation of Disclosure Controls and Procedures.” Our independent registered public accounting firm, WithumSmith+Brown, PC ("Withum"), is not required to and has not issued an attestation report as of December 31, 2023 because we are not an “accelerated filer” or a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act.
Inherent Limitations on Effectiveness of Controls
Management recognizes that a control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud or error, if any, have been detected. These inherent limitations include the realities that judgments in decision making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by management override of the controls. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions; over time, controls may become inadequate because of changes in conditions, or the degree of compliance with policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
| 70 |
| Table of Contents |
ITEM 9B. OTHER INFORMATION
No director or officer (as defined in Rule 16a–1(f) under the Exchange Act) of the Company adopted or terminated (i) any contract, instruction or written plan for the purchase or sale of securities of the registrant intended to satisfy the affirmative defense conditions of Rule 10b5–1(c) under the Exchange Act; and/or (ii) any “non-Rule 10b5–1 trading arrangement” as defined in paragraph (c) of Item 408 of Regulation S-K, during the quarter ended December 31, 2023.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
None.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Composition of the Board
Our business and affairs are managed under the direction of our board of directors, or the Board, which currently consists of four members, three of whom are “independent” under NYSE American listing standards. Our bylaws provide that the number of directors will be fixed from time to time by resolution of the Board. All directors hold office until their successors have been elected and qualified or until their earlier death, resignation, disqualification or removal. We have divided the terms of office of the directors into three classes with staggered three year terms: Class I, whose term expires at the 2025 Annual Meeting of Stockholders; Class II, whose term expires at the 2026 Annual Meeting of Stockholders; and Class III, whose term expires at the 2024 Annual Meeting of Stockholders.
Information about the Board of Directors
The following table sets forth the names, ages as of March 12, 2024, and certain other information regarding each member of the Board. The following information has been furnished to us by the directors.
Name |
| Class |
| Age |
| Position |
| Director Since |
| Current Term Expires |
David A. Jenkins |
| II |
| 66 |
| Executive Chairman of the Board of Directors and Chief Executive Officer |
| 2023 |
| 2026 |
Martin Colombatto |
| I |
| 65 |
| Director |
| 2017 |
| 2025 |
James Caruso |
| III |
| 63 |
| Director |
| 2023 |
| 2024 |
John P. Francis |
| III |
| 58 |
| Director |
| 2024 |
| 2024 |
David A. Jenkins became Executive Chairman of the Board in January 2023. He became Interim Chief Executive Officer in April 2023 and was named Chief Executive Officer in January 2024. He has spent most of his career as an entrepreneur in the medical device industry, and has established numerous companies including Old Catheter, where he served as the CEO and as Chairman of Old Catheter’s Board. He has been Chairman of the Board of Old Catheter since Catheter’s inception in 2006 and has served as CEO of Old Catheter since December 2020. His prior experience includes having served as Chairman and CEO of Arrhythmia Research and overseeing the introduction to the market of Cardiolab, the first dual monitor, 32 channel electrophysiology recording system. This technology was later acquired by General Electric and continues to be sold into the market place today. Another of Mr. Jenkins’ companies, EP MedSystems, Inc., was sold to St. Jude Medical, Inc., now part of Abbott, for approximately $93 million in 2008. Mr. Jenkins also founded and served as the CEO of Transneuronix, Inc., a maker of implantable stimulators for the treatment of weight loss, which was later sold to Medtronic for $267 million in 2005. Mr. Jenkins holds a degree in accounting from the University of Kansas, and a master’s degree in business from the University of Texas, Austin. He began his career in public accounting with Coopers and Lybrand. We believe that Mr. Jenkins is qualified to serve as a director because of his extensive experience in the medical device industry.
| 71 |
| Table of Contents |
Martin Colombatto has served as a director of the Company since January 2017. Mr. Colombatto has served as a Venture and Industry Partner of Seven Peaks Ventures LLP, a venture capital fund based in Bend, OR, since January 2016. From December 2013 to August 2014, Mr. Colombatto served as a director of PLX Technology, Inc., a technology company. Mr. Colombatto has also served as the Chief Executive Officer and President of Staccato Communications, Inc., an Ultra-Wideband semiconductor company, from January 2006 to March 2009 and as Executive Chairman of Staccato Communications, Inc., from January 2006 to September 2010. Prior to joining Staccato, Mr. Colombatto served as Vice President and General Manager of the Networking Business unit of Broadcom Corp., a broadband communication semiconductor company, from July 1996 to July 2002. Mr. Colombatto was also previously employed by LSI Logic, an application specific semiconductor company, from August 1987 to July 1996. Mr. Colombatto also previously held engineering positions at Reliance Electric, a production automation and control company, from August 1985 to June 1987 and Texas Instruments, an electronics company, from June 1982 to April 1985. Mr. Colombatto holds a Bachelor of Science degree in Electronic Engineering Technology from California State Polytechnic University, Pomona. We believe that Mr. Colombatto is qualified to serve as a member of our board of directors due to his extensive management experience and familiarity with our business and strategy.
James Caruso has held senior level financial positions in both public and private companies for more than 40 years, including serving as Chief Financial Officer at several publicly traded and privately held medical device companies. He has managed all financial aspects of businesses and is proficient in SEC reporting and compliance requirements. Mr. Caruso also has extensive operational experience and has led post-acquisition business integration activities on several occasions. Mr. Caruso served as Chief Financial Officer of Catheter Precision from 2010 through 2016. Mr. Caruso also served as Chief Financial Officer of EP MedSystems, Inc. (NASDAQ:EPMD), a company focused on cardiac electrophysiology that was acquired by St Jude Medical in 2008; Hi-Tronics Designs, Inc., a privately held medical device design and manufacturing company that was acquired by Advanced Neuromodulation Systems, Inc. in 2001; and Micron Products, Inc., a publicly traded medical device manufacturing company that was acquired by Arrhythmia Research Technology in 1991. Mr. Caruso spent five years in the audit practice at Deloitte (formerly Deloitte & Touche). Mr. Caruso received his Bachelor of Science in Business Administration from Rutgers University and an MBA from Fordham University and is a Certified Public Accountant. We believe that Mr. Caruso is qualified to serve as a director because of his senior level financial experience with public and private companies.
John P. Francis has served as Managing Member of Francis Capital Management, LLC, an investment management firm specializing in small capitalization equities, since 2000. Mr. Francis has more than 20 years of experience in investment management, finance and accounting. Mr. Francis has extensive experience investing in small cap medical device companies. Mr. Francis earned his bachelor's degree in economics from UCLA and MBA from the UCLA Anderson School of Management. Mr. Francis' qualifications to serve as a director include his financial, business and accounting experience. Mr. Francis is a Chartered Financial Analyst and a Certified Public Accountant (inactive).
Executive Officers
David A Jenkins became Executive Chairman of the Board in January 2023. He became Interim Chief Executive Officer in April 2023 and was named Chief Executive Officer in January 2024. His biographical information is set forth above at “Information About the Board of Directors.”
Margrit Thomassen, age 54, became interim Chief Financial Officer and Secretary as of January 2, 2024. Ms. Thomassen has served as Controller of the Company since the merger with Old Catheter on January 9, 2023. From 2005 until 2023 she worked as Chief Financial Officer of SeaCap Management LLC, an affiliate of Mr. Jenkins, on various investment opportunities handling accounting, tax, management and human resources tasks. In 2021, she assumed the role of Controller at Old Catheter.
| 72 |
| Table of Contents |
Delinquent Section 16(a) Reports
Section 16(a) of the Exchange Act requires our executive officers and directors, and persons who own more than 10% of a registered class of our equity securities, to file reports of ownership and changes of ownership on Forms 3, 4 and 5 with the SEC. Such directors, executive officers and 10% stockholders are required by SEC regulations to furnish us with copies of all Section 16(a) forms they file.
Based solely on our review of the copies of such forms, and written representations that we have received from certain reporting persons that they filed all required reports, we believe that all of our officers, directors and greater than 10% stockholders complied with all Section 16(a) filing requirements applicable to them with respect to transactions during 2023, other than one late Form 4 filed by Susanne Meline, who served as a director during 2023, with respect to one transaction.
Audit Committee
The members of our Audit Committee are John Francis and James Caruso. Mr. Caruso serves as the chairperson of our Audit Committee. The Board has determined that each member of the Audit Committee is an independent director under the NYSE American listing rules, satisfies the additional independence criteria for audit committee members and satisfies the requirements for financial literacy under the NYSE American listing rules and Rule 10A-3 of the Exchange Act, as applicable. The Board has also determined that Mr. Caruso qualifies as an audit committee financial expert within the meaning of the applicable rules and regulations of the SEC and satisfies the financial sophistication requirements of the NYSE American listing rules.
Corporate Governance Principles and Code of Ethics and Conduct
The Board has adopted corporate governance principles. These principles address items such as the qualifications and responsibilities of our directors and director candidates and corporate governance policies and standards applicable to us in general. In addition, the Board has adopted a written code of ethics and conduct that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. A copy of our corporate governance principles and code of ethics and conduct are available on our website, www.catheterprecision.com, under the Investor Relations tab under “Governance”, then “Governance Documents.” If the Board makes any substantive amendments to, or grants any waivers from, the code of ethics and conduct for any officer or director, it will disclose the nature of such amendment or waiver on the Company’s website.
ITEM 11. EXECUTIVE COMPENSATION
Director Compensation
With respect to 2023, the compensation committee and the Board did not retain a compensation consultant in connection with determining compensation of non-employee directors. Following the merger with Old Catheter in January 2023, the Board set 2023 compensation at an annual cash retainer of $50,000. In January 2024, the compensation committee recommended, and the Board approved, 2024 compensation to all non-employee directors consisting of a cash retainer of $50,000 and an award of non-qualified stock options to purchase 25,000 shares of Company common stock to each non-employee director. Options were granted on January 8, 2024, have a purchase price of $0.40 per share, a 10-year term, and vest quarterly over three years. Retainer cash payments will be paid in cash on or about the last day of each fiscal quarter of the Company in arrears to each non-employee director.
We also reimburse our non-employee directors for reasonable, customary and documented travel expenses to attend meetings of our board of directors and committees of our board of directors.
Our non-employee directors remain eligible to receive equity awards and cash or other compensation outside of the compensation described above, as may be provided from time to time at the discretion of our Board of Directors. No such awards or payments were made in 2023.
| 73 |
| Table of Contents |
2023 Director Compensation Table
The following table sets forth information regarding compensation earned or paid to our non-employee directors during the year ended December 31, 2023:
|
| Fees Earned or Paid in Cash ($) |
|
| Option Awards ($)(1) |
|
| Total ($) |
| |||
Martin Colombatto (2) |
|
| 50,000 |
|
|
| — |
|
|
| 50,000 |
|
James Caruso |
|
| 50,000 |
|
|
| — |
|
|
| 50,000 |
|
Susanne Meline |
|
| 50,000 |
|
|
| — |
|
|
| 50,000 |
|
(1) | No option awards were granted to the directors during the year ended December 31, 2023. |
|
|
(2) | Mr. Colombatto held vested options to purchase 73 shares of Company common stock as of December 31, 2023. |
See Executive Compensation for information about the compensation of Mr. David Jenkins, a director who is also an executive officer, and Mr. Will McGuire, a former director who was also an executive officer during a portion of 2023.
Processes and Procedures for Executive Compensation
The Compensation Committee assists the Board in discharging the Board’s responsibilities relating to oversight of the compensation of the chief executive officer and other executive officers, including reviewing and making recommendations to the Board with respect to the compensation, plans, policies and programs for the chief executive officer and other executive officers and administering the equity compensation plans for executive officers and employees.
The Compensation Committee annually reviews the compensation, plans, policies and programs for the chief executive officer and other executive officers. In connection therewith, the Compensation Committee considers, among other things, each executive officer’s performance in light of established individual and corporate goals and objectives and the recommendations of our chief executive officer. In particular, the Compensation Committee considers the recommendations of the chief executive officer when reviewing base salary and incentive performance compensation levels of the executive officers and when setting specific individual and corporate performance targets under the annual incentive bonus plan for the executive officers. While the chief executive officer provides input on his compensation, he does not participate in compensation committee or Board deliberations regarding his own compensation. The Compensation Committee may delegate its authority to a subcommittee, but it may not delegate any power or authority required by agreement, law, regulation or listing standard to be exercised by the Compensation Committee as a whole.
Named Executive Officers
The named executive officers for 2023 (“NEOs”), which consist of our principal executive officer and our former Chief Financial Officer, who were our only executive officers as of December 31, 2023, as well as our former Chief Executive Officer, who would have been one of our next two most highly compensated executive officers but for the fact that he was not serving as an executive officer as of December 31, 2023, were as follows:
| · | David A. Jenkins, Executive Chairman and Interim Chief Executive Officer; |
|
|
|
| · | Steve Passey, former Chief Financial Officer; and |
|
|
|
| · | Jonathan Will McGuire, former Chief Executive Officer and Secretary. |
Mr. Jenkins was appointed Executive Chairman upon effectiveness of the Merger on January 9, 2023. Mr. McGuire served as Chief Executive Officer until April 28, 2023. Mr. Jenkins was appointed interim Chief Executive Officer beginning April 28, 2023, and Chief Executive Officer beginning January 2, 2024. Mr. Passey served as Chief Financial Officer from April 1, 2023, through December 31, 2023.
| 74 |
| Table of Contents |
Summary Compensation Table
The following table provides information regarding the compensation of the NEOs for 2023 and 2022, as applicable:
Name and Principal Position |
| Year |
| Salary ($) |
|
| Bonus ($) |
|
| Stock Awards ($) |
|
| Option Awards ($) |
|
| Non-Equity Incentive Plan Compensation ($) |
|
| All Other Compensation ($) |
|
| Total ($) |
| |||||||
David A. Jenkins |
| 2023 |
|
| 300,000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 300,000 |
| |||||
Executive Chairman and Interim Chief Executive Officer |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Jonathan Will McGuire |
| 2023 |
|
| 173,077 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1,750,000 | (1) |
|
| 1,923,077 |
| ||||
Former Chief Executive Officer and Secretary |
| 2022 |
|
| 500,000 |
|
|
| — |
|
|
| — |
|
|
| — |
|
|
| — |
|
|
| 43,953 | (2) |
|
| 543,953 |
|
Steve Passey |
| 2023 |
|
| 189,664 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 189,664 |
|
Former Chief Financial Officer |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) | represents severance pay that Mr. McGuire became entitled to upon his resignation, which was effective April 28, 2023, and which was paid in May 2023. |
|
|
(2) | Amounts include Company matching contributions to 401(k) plan; taxable amounts from vested stock awards; and amounts paid for a supplemental health insurance plan. |
Executive Employment Agreements and Arrangements
David A. Jenkins
In January 2023, we entered into an oral employment agreement with David A. Jenkins, Chairman of the Board. In accordance with the terms of Mr. Jenkins’ employment agreement, he is entitled to annual compensation of $300,000.
Jonathan Will McGuire
The Company entered into an offer letter with Mr. McGuire dated March 9, 2020 which provided for at-will employment. The offer letter provided for an initial base salary of $500,000 and eligibility annually for a target cash bonus of 100% of his annual base salary, based on achieving performance objectives established by our board of directors or a committee of our board of directors. Mr. McGuire was also entitled to certain severance benefits and change in control payments, as more fully described in Mr. McGuire’s Change in Control and Severance Agreement below.
Steve Passey
During his employment with the Company, Mr. Passey received an annual salary of $250,000. He was also eligible to receive bonuses and equity awards at the discretion of the Compensation Committee. No such bonuses or awards were awarded.
Mr. McGuire’s Change in Control and Severance Agreement
The Company entered into a change in control and severance agreement with Mr. McGuire on March 30, 2020. The agreement provided for certain severance benefits, including the change in control payments described below, if the termination was by the Company without cause, or by Mr. McGuire for good reason, as such terms are defined in the agreement. If such termination occurred within a certain Change in Control Period, the change in control payments described below became payable. In January 2023, the agreement was amended to provide that the Change in Control Period would begin three months before a Change in Control, including the Merger, and end 24 months following such event.
| 75 |
| Table of Contents |
Prior to an April 2023 amendment, if the termination described above occurred within the Change in Control Period, Mr. McGuire’s became entitled to the following:
| · | a lump-sum payment equal to 24 months of the executive officer’s annual base salary as in effect immediately prior to such termination (or if such termination is due to a resignation for good reason based on a material reduction in base salary, then as in effect immediately prior to the reduction) or if greater, at the level in effect immediately prior to the change in control); |
|
|
|
| · | a lump-sum payment equal to 150% of the executive officer’s target annual bonus as in effect for the fiscal year in which such termination occurs; |
|
|
|
| · | payment of premiums for coverage under COBRA for the executive officer and the named executive officer’s eligible dependents, if any, for up to 24 months, or taxable monthly payments for the equivalent period in the event payment of the COBRA premiums would violate or be subject to an excise tax under applicable law; and |
|
|
|
| · | 100% accelerated vesting and exercisability of all outstanding equity awards and, in the case of an equity award with performance-based vesting, all performance goals and other vesting criteria generally will be deemed achieved at target. |
The agreement provided that if any of the amounts provided for above or otherwise payable to Mr. McGuire would constitute “parachute payments” within the meaning of Section 280G of the Internal Revenue Code and could be subject to the related excise tax, he would be entitled to receive either full payment of benefits under the change in control or severance agreement or such lesser amount which would result in no portion of the benefits being subject to the excise tax, whichever resulted in the greater amount of after-tax benefits to the executive officer. The change in control and severance agreement did not require us to provide any tax gross-up payments.
The agreement was amended in April 2023 to clarify that Mr. McGuire’s severance payment would be based on his 2022 salary and bonus target, and that he would not be entitled to COBRA benefits. Mr. McGuire resigned for “good reason” as defined in the agreement, effective April 28, 2023. Upon his resignation, he became entitled to receive a payment of approximately $1.75 million under the agreement, which amount was paid in May 2023.
Outstanding Equity Awards at 2023 Fiscal Year-End
As of December 31, 2023, none of the NEOs named above held unexercised stock options, unvested stock awards, or any unearned and unvested shares, units or other rights awarded under equity incentive plans. All of Mr. McGuire’s unvested awards vested upon his resignation in April 2023, and his stock options became no longer exercisable three months following his resignation. Ms. Thomassen held options to purchase Company stock on December 31, 2023, which were received pursuant to the Merger, as described in Item 13 below.
Perquisites, Health, Welfare and Retirement Benefits
Our named executive officers are eligible to participate in our employee benefit plans, including our medical, dental, vision, group life, disability and accidental death and dismemberment insurance plans, in each case on the same basis as all of our other employees.
We generally do not provide perquisites or personal benefits to our named executive officers, except in limited circumstances. Our board of directors may elect to adopt qualified or non-qualified benefit plans in the future if it determines that doing so is in our best interests.
401(k) Savings Plan
Prior to the Merger, the Company maintained a tax-qualified retirement plan that provided eligible employees, including named executive officers, with an opportunity to save for retirement on a tax advantaged basis. All participants’ interests in their deferrals were 100% vested when contributed. Pre-tax and after-tax contributions were allocated to each participant’s individual account and were then invested in selected investment alternatives according to the participant’s directions. The Company, in its sole discretion, could make certain contributions to the plan. The 401(k) plan was intended to qualify under Sections 401(a) and 501(a) of the Internal Revenue Code. As a tax-qualified retirement plan, contributions to the 401(k) plan and earnings on those contributions were not taxable to the employees until distributed from the 401(k) plan, and all contributions, if any, were deductible by the Company when made. As a result of the Merger, the Company terminated the 401(k) Savings Plan and liquidated all assets in March 2023.
| 76 |
| Table of Contents |
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth certain information with respect to the beneficial ownership of our common stock as of March 12, 2024 by:
| · | each person, or group of affiliated persons, who we know to beneficially own more than 5% of our common stock; |
|
|
|
| · | each of our named executive officers; |
|
|
|
| · | each of our directors; and |
|
|
|
| · | all of our executive officers and directors as a group. |
The percentage ownership information shown in the table is based on an aggregate of 7,573,403 shares of our common stock outstanding as of March 12, 2024.
We have determined beneficial ownership in accordance with the rules of the Securities and Exchange Commission. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities. In addition, the rules include shares of common stock issuable pursuant to: (i) the exercise of stock options that are either immediately exercisable or exercisable on or before May 11,2024, which is 60 days after March 12, 2024 and (ii) outstanding warrants to purchase common stock held by that person that are either immediately exercisable or exercisable on or before May 11, 2024, which is 60 days after March 12, 2024. These shares are deemed to be outstanding and beneficially owned by the person holding those options and warrants for the purpose of computing the percentage ownership of that person, but they are not treated as outstanding for the purpose of computing the percentage ownership of any other person.
Unless otherwise noted below, the address of each of the individuals and entities named in the table below is c/o Catheter Precision, Inc., 1670 Highway 160 West, Suite 205, Fort Mill, South Carolina 29708. Beneficial ownership representing less than 1% is denoted with an asterisk (*).
Unless otherwise indicated, the persons or entities identified in this table have sole voting and investment power with respect to all shares shown as beneficially owned by them, subject to applicable community property laws.
|
| Number of Shares of Common Stock Beneficially Owned |
|
| Percentage of Common Stock Beneficially Owned |
| ||
5% Stockholders: |
|
|
|
|
|
| ||
Armistice Capital Master Fund Ltd. (1) |
|
| 752,874 |
|
|
| 9.99 | % |
Directors and Named Executive Officers: |
|
|
|
|
|
|
|
|
Jonathan Will McGuire (2) |
|
| 504 |
|
| * |
| |
David A. Jenkins (3) |
|
| 1,044,087 |
|
|
| 13.79 | % |
Steven K. Passey |
|
| — |
|
|
| — |
|
James J. Caruso (4) |
|
| 3,862 |
|
| * |
| |
Martin Colombatto (5) |
|
| 2,773 |
|
| * |
| |
John P. Francis (6) |
|
| 6,242 |
|
| * |
| |
All directors and executive officers as a group (5 persons) (2)(3)(4)(5)(6)(7) |
|
| 1,074,232 |
|
| 14.14 | % | |
(1) | The number of shares presented as beneficially owned by shareholder was obtained from Schedule 13G filed by the shareholder on February 14, 2024 and represents shares issuable upon conversion of Series A Preferred Stock. The precise number of shares beneficially owned by the shareholder depends upon the operation of certain beneficial ownership blockers and the number of shares outstanding, and therefore may be greater or less than the number presented from time to time. The table does not include other derivative securities that are not currently exercisable due to beneficial ownership blockers. Address of stockholder is 510 Madison Avenue, 7th Floor, New York, NY 10022. |
| 77 |
| Table of Contents |
(2) | Mr. McGuire resigned as an officer, director and employee of the Company effective April 28, 2023. |
(3) | Includes (i) 2,264 shares held by a family charitable trust of which Mr. Jenkins is the trustee; (ii) 2,264 shares held by a charitable remainder unitrust of which Mr. Jenkins’ wife is the trustee; and (iii) 709,703 shares held by a partnership of which Mr. Jenkins is the manager member of the managing partner. Excludes 235,320 shares held by certain adult immediate family members of Mr. Jenkins. Does not include 8,190.261 shares of Series X Preferred Stock held by Mr. Jenkins and his affiliates which are convertible into 8,190,261 shares of common stock but which are subject to certain beneficial ownership blockers and which may not be converted, at the earliest, until July 9, 2024. Also does not include 1,049.024 shares of Series X Preferred Stock held, in the aggregate, by certain adult immediate family members of Mr. Jenkins and which are convertible into 1,049,024 shares of common stock, but which are subject to certain beneficial ownership blockers and which may not be converted, at the earliest, until July 9, 2024. Also does not include exercisable options to purchase 144,169 shares of common stock and unvested options to purchase 25,000 shares of common stock held by Missiaen Huck, the non-executive chief operating officer of Catheter and Mr. Jenkins’s adult daughter. |
(4) | Does not include 7.932 shares of Series X Preferred Stock held by Mr. Caruso which are convertible into 7,932 shares of common stock but which are subject to certain beneficial ownership blockers and which may not be converted, at the earliest, until July 9, 2024. Includes currently exercisable options to purchase 2,083 shares of common stock. Does not include unvested options to purchase 22,917 shares of common stock. |
(5) | Includes (i) 73 shares of common stock subject to options exercisable within 60 days of March 12, 2024, and (ii) 30 shares held of record by M. Colombatto Trust, of which Mr. Colombatto serves as trustee, and (iii) exercisable options to purchase 2,083 shares of common stock. Does not include unvested options to purchase 22,917 shares of common stock. |
(6) | Includes (i) 1,548 shares of common stock subject to warrants exercisable within 60 days of March 12, 2024 held of record by Catalysis Partners (CP), (ii) exercisable options to purchase 2,083 shares of common stock, and (iii) 36 shares held by his spouse. Does not include unvested options to purchase 22,917 shares of common stock. Mr. Francis has an investment interest in CP and, together with his spouse, owns a controlling interest in Francis Capital Management LLC, which also has an investment interest in CP and serves as both its Managing Member and Investment Manager. Mr. Francis disclaims beneficial interest of these securities except to the extent of his pecuniary interest therein. Does not include 11.481 shares of Series X Preferred Stock held by a retirement fund for the benefit of Mr. Francis which are convertible into 11,481 shares of common stock but which are subject to certain beneficial ownership blockers and which may not be converted, at the earliest, until July 9, 2024. |
(7) | Includes 16,764 shares of common stock underlying vested stock options held by Margrit Thomassen, the Company’s interim Chief Financial Officer and Secretary; excludes unvested options to purchase 25,000 shares of common stock held by Ms. Thomassen. |
| 78 |
| Table of Contents |
EQUITY COMPENSATION PLAN INFORMATION
Information as of December 31, 2023, regarding the Company’s equity compensation plans is summarized in the following table:
|
| Number of Securities to be Issued Upon Exercise of Outstanding Options and Restricted Stock Units |
|
| Weighted-Average Exercise Price of Outstanding Options (1) |
|
| Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans (Excluding Securities Reflected in Column (a)) |
| |||
Plan Category |
| (a) |
|
| (b) |
|
| (c) |
| |||
Equity compensation plans approved by security holders (2) |
|
| 73 |
|
| $ | 17,161.30 |
|
|
| 502,408 |
|
Equity compensation plans not approved by security holders (3) |
|
| 214,579 |
|
| $ | .63 |
|
|
| 0 |
|
Total |
|
| 214,652 |
|
| $ | 6.47 |
|
|
| 502,408 |
|
(1) | The weighted average exercise price is based solely on outstanding options. |
(2) | Outstanding options were issued under the Company’s 2018 Equity Incentive Plan (as amended, the “2018 Plan”). The number of securities remaining available includes both the 2018 Plan and the 2023 Equity Incentive Plan (the “2023 Plan”). Excludes 54,678 shares which become available on March 1, 2024, and additional shares which will become available in future quarters, pursuant to an adjustment feature under the 2023 Plan. Under the adjustment features, the number of shares available for issuance under the 2023 Plan increases on the first day of each fiscal quarter (each, an “Adjustment Date”) by an amount equal to the lesser of: (i) 10% of the number equal to the number of shares of common stock outstanding on the applicable Adjustment Date less the number of shares of Common Stock outstanding at the beginning of the fiscal quarter immediately preceding the Adjustment Date, but if such number is a negative number, then the increase will be zero; or (ii) such lesser number of Shares as may be determined by the Board. (3) Represents Old Catheter options assumed in connection with the January 9, 2023 acquisition of Old Catheter. |
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Related Person Transactions
Pursuant to SEC rules, a “transaction” with a related party includes any transaction, arrangement or relationship (or any series of similar transactions, arrangements or relationships) in which the Company was or is a participant and the related person had or will have a direct or indirect material interest where the amount involved exceeds the lesser of $120,000 or one percent of the average of the Company’s total assets at year-end for the last two completed fiscal years. Accordingly, the applicable threshold for us is $120,000.
Since January 1, 2022, we have engaged in the following transactions in which our executive officers, directors, promoters or beneficial owners of more than 5% of our common stock had or will have a direct or indirect material interest, other than compensation arrangements which are described under “Executive Compensation.” We believe that all of these transactions were on terms as favorable as could have been obtained from unrelated third parties.
Employment and Related Agreements
We currently do not have written employment agreements with our executive officers. For information about our employment agreements with our Named Executive Officers, who are former employees, refer to “Executive Compensation—Executive Employment Agreements and Arrangements.”
In January 2023, we entered into an oral employment agreement with Missiaen Huck, Mr. David Jenkins’ adult daughter. Ms. Huck serves as the non-executive chief operating officer of Catheter and receives annual compensation of $165,000. In January 2024, Ms. Huck received a grant of options to purchase 25,000 shares of Company common stock which have an exercise price of $0.40 per share, vest ratably over five years and have a term of 10 years. As noted below, Ms. Huck also holds options to purchase an additional 144,169 shares of Company common stock which were originally issued by Old Catheter and assumed in the Merger.
| 79 |
| Table of Contents |
Margrit Thomassen became interim Chief Financial Officer and Secretary in January 2024. She joined the Company as controller in January 2023, in connection with the Merger with Old Catheter. Ms. Thomassen’s annual salary for both 2023 and 2024 is $144,000. In January 2024, Ms. Thomassen received a grant of options to purchase 25,000 shares of Company common stock which have an exercise price of $0.40 per share, vest ratably over five years and have a term of 10 years. As noted below, Ms. Thomassen also holds options to purchase an additional 16,764 shares of Company common stock which were originally issued by Old Catheter and assumed in the Merger.
Merger-Related Transactions
Mr. Jenkins and his affiliates held approximately $25.1 million of Old Catheter’s Convertible Promissory Notes, or the Notes, that were converted in the Old Catheter merger into 7,856.251 shares of Series X Preferred Stock. Upon consummation of the merger, each such Noteholder received, in exchange for discharge of the principal of his or its Notes, a number of shares of our Series X Preferred Stock representing a potential right to convert into our common stock in an amount equal to one common share for each $3.20 of principal amount. In consideration for forgiving the interest accrued but remaining unpaid under the Notes in an aggregate amount of approximately $13.9 million, Mr. Jenkins and his affiliates also received royalties equal to 11.77% of the net sales, if any, of the LockeT device, commencing upon the first commercial sale and through December 31, 2035.
In addition to the shares described above that were issued in connection with the Notes, Mr. Jenkins and his affiliates received 1,325.838 shares of Series X Preferred Stock in the merger, and Mr. Jenkins’ adult children received 1,284.344 shares of Series X Preferred Stock in the merger, all in exchange for their equity interests in Old Catheter in accordance with the merger exchange ratio. Additional, noninterest-bearing demand loans totaling $1,075,000 from David Jenkins to Old Catheter were repaid by the Company at or shortly after the closing of the merger.
Mr. Jenkins’ daughter, Missiaen Huck, received options to purchase 144,169 shares of the Company’s common stock upon the closing of the Merger in exchange for her options to purchase shares of Catheter common stock, converted based on the exchange ratio in the merger. Of the total options to purchase 144,169 shares of the Company’s common stock, 140,816 options have an exercise price of $0.59 per share, and the remaining 3,353 options have an exercise price of $2.02 per share.
Ms. Thomassen received options to purchase 16,764 shares of the Company’s common stock upon the closing of the Merger in exchange for her options to purchase shares of Catheter common stock, converted based on the exchange ratio in the Merger. The options have an exercise price of $0.59 per share.
Following stockholder approval on March 21, 2023, we issued 991,828 shares of common stock to Mr. Jenkins and affiliates upon conversion of 991.828 shares of Series X Preferred Stock, and 235,320 shares of common stock to his adult children upon conversion of 235.320 shares of Series X Preferred Stock.
Warrant Inducement Offer
On January 9, 2023, the Company reduced the exercise price of certain existing warrants (the "Existing Warrants"), exercisable for 331,608 shares of the Company’s common stock held by Armistice Capital Master Fund Ltd. (“Armistice”) with exercise prices ranging from $14.00 to $526.50 per share to $4.00 per share (the "2023 Warrant Repricing"). In connection with the 2023 Warrant Repricing, the Company entered into a warrant inducement offer letter (the "2023 Inducement Letter") with Armistice pursuant to which it would exercise up to all of the 331,608 Existing Warrants (the "Inducement Offer"). In consideration for exercising the Existing Warrants pursuant to the terms of the 2023 Inducement Letter, the Company received approximately $1.3 million in gross proceeds. The Company paid the placement agent aggregate cash fees of approximately $0.2 million related to the Inducement Offer which represented 8.0% of the gross proceeds received from the Inducement Offer plus other offering costs resulting in net proceeds to the Company of $1.1 million. In consideration for exercising the Existing Warrants pursuant to the terms of the 2023 Inducement Letter, the Company issued Armistice a new Series E common stock purchase warrant, or Series E Warrant (the "Series E Warrant"), to purchase 331,608 shares of common stock at an exercise price of $4.00 per share. The Series E Warrant is exercisable for five years from the date of stockholder approval. Exercise of the Series E Warrant in full was subject to approval of the Company's stockholders other than Armistice which was obtained at the stockholders’ meeting held on March 21, 2023 (the ”Stockholders’ Meeting”).
Private Placement
On January 9, 2023, the Company entered into a Securities Purchase Agreement (“Securities Purchase Agreement”) for a private placement (“Private Placement”), with Armistice. Pursuant to the Securities Purchase Agreement, Armistice agreed to purchase, for an aggregate purchase price of approximately $8.0 million, (a) Class A units at a price that was the lower of $3.00 per unit and 90% of the 5 day volume weighted average price of the Company’s common stock immediately prior to obtainment of the approval of the Company’s stockholders of conversion of the PIPE Preferred Stock and PIPE Warrants (as each are defined below), each consisting of one share of common stock, one Series F common stock purchase warrant, or Series F Warrant, and one Series G common stock purchase warrant, or Series G Warrant, and together with the Series F Warrants (the “PIPE Warrants”) and (b) Class B units at a price of $1,000 per unit, each consisting of one share of a new series of the Company’s preferred stock, designated as Series A Convertible Preferred Stock (the “PIPE Preferred Stock”), par value $0.0001, and one Series F Warrant and one Series G Warrant for each share of the Company’s common stock underlying the PIPE Preferred Stock (each share of which is convertible into a number of shares of the Company’s common stock equal to $1,000 divided by the lower of $3.00 and 90% of the 5 day volume weighted average closing price of the Company’s common stock immediately prior to the obtainment of the approval of the Company’s stockholders of conversion of the PIPE Preferred Stock and PIPE Warrants, or the Preferred Conversion Rate). The closing under the Securities Purchase Agreement and the sale and issuance of the Class A units and Class B units (and the issuance of any underlying common stock) were approved at the Stockholders’ Meeting. At the closing of the Private Placement, the Company issued 497,908 Class A units for proceeds of approximately $0.9 million and 7,203 Class B units for proceeds of approximately $7.1 million, the preferred stock underlying which is convertible into up to 4,501,060 shares of common stock, as well as the issuance of warrants described below.
The PIPE Warrants, including Series F warrants and Series G warrants, are exercisable at an exercise price of $3.00 per share, subject to adjustments as provided under the terms of the PIPE Warrants. The PIPE Warrants are exercisable at any time on or after the closing date of the Private Placement until the expiration thereof, except that the PIPE Warrants cannot be exercised if, after giving effect thereto, the purchaser would beneficially own more than 4.99%, or the Maximum Percentage, of the outstanding shares of common stock of the Company, which Maximum Percentage may be increased or decreased by the purchaser with written notice to the Company to any other percentage specified not in excess of 9.99%. The Series F Warrants have a term of two years from the date of stockholder approval, and the Series G Warrants have a term of six years from the date of stockholder approval. The Series F Warrants and Series G Warrants were approved at the Stockholders’ Meeting.
Issuance of Securities upon Conversion of Series A Preferred
On July 5, 2023 the Company issued 1,093,552 shares of its common stock in connection with the conversion of 1,750 shares of its outstanding Series A Convertible Preferred Stock held by Armistice Capital Master Fund Ltd. (“Armistice”). The shares were issued in connection with two separate conversions of 875 shares of Series A Convertible Preferred Stock into 546,776 shares of common stock that occurred on July 3, 2023. Each share of Series A Convertible Preferred Stock is convertible into approximately 625 shares of common stock.
On July 24, 2023, the Company issued 546,776 shares of its common stock in connection with the conversion of 875 shares of its outstanding Series A Convertible Preferred Stock held by Armistice.
On January 24, 2024, the Company issued 546,776 shares of its common stock in connection with the conversion of 875 shares of its outstanding Series A Convertible Preferred Stock held by Armistice.
Indemnification of Officers and Directors
We have historically entered into indemnification agreements with directors and executive officers, in addition to the indemnification provided for in our amended and restated certificate of incorporation and amended and restated bylaws, and we may also do so in the future. The indemnification agreements and our amended and restated certificate of incorporation and amended and restated bylaws require us to indemnify our directors and officers to the fullest extent permitted by Delaware law.
| 80 |
| Table of Contents |
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
Our independent registered public accounting firm is Withum (auditor ID: 100) since June 21, 2023. Withum was also the independent registered public accounting firm that audited the financial statements of Old Catheter for the fiscal year ended December 31, 2022.
Fees Paid to the Independent Registered Public Accounting Firms
The following table represents aggregate fees for services provided to us in the fiscal year ended December 31, 2023 by Withum. It does not include fees billed to us for services rendered by our previous auditor, Haskell & White LLP, during 2023, or fees billed to us by Withum for pre-Merger services rendered to Old Catheter.:
|
| 2023 (Withum) |
| |
Audit Fees (1) |
| $ | 660,872 |
|
Audit-Related Fees (2) |
|
| — |
|
Tax Fees (3) |
|
| — |
|
All Other Fees (4) |
|
| — |
|
Total Fees |
| $ | 660,872 |
|
(1) | “Audit Fees” consist of fees billed for professional services rendered during the respective fiscal year in connection with the audit of our annual financial statements, review of our quarterly financial statements, and services that are normally provided in connection with statutory and regulatory filings or engagements for those fiscal years. This includes consents and other services related to SEC matters and registration statements. |
(2) | “Audit-Related Fees” generally include fees incurred for assurance and related services that are reasonably related to the performance of the audit or review of the Company’s financial statements but are not otherwise included as Audit Fees. For fiscal 2023, the fees presented consist of fees billed for professional services rendered in connection with the purchase price allocation and valuation of the business combination with regards to the Merger. |
(3) | “Tax Fees” consist of permissible tax compliance and tax advisory service fees. Withum did not bill us for any tax fees for the year ended December 31, 2023. |
(4) | “All Other Fees” consist of fees billed for services other than the services reported in Audit Fees, Audit-Related Fees, and Tax Fees. |
Auditor Independence
During the year ended December 31, 2023, there were no other professional services provided by Withum that would have required our audit committee to consider their compatibility with maintaining Withum’s independence.
Pre-Approval Policy
Our audit committee’s policy is to pre-approve all audit and permissible non-audit services provided by the independent accountants and the related estimated fees. These services may include audit services, audit-related services, tax services and other services. Our audit committee generally pre-approves particular services or categories of services on a case-by-case basis. The independent registered public accounting firm and management are required to periodically report to our audit committee regarding the extent of services provided by the independent registered public accounting firm in accordance with these pre-approvals, and the fees for the services performed to date. All of Withum’s services to the Company for fiscal year 2023 described above were pre-approved by our audit committee.
| 81 |
| Table of Contents |
PART IV — FINANCIAL INFORMATION
ITEM 15. EXHIBIT AND FINANCIAL STATEMENT SCHEDULES.
(a) We have filed the following documents as part of this Annual Report:
1. Financial Statements.
| Page |
F-1 | |
F-7 | |
F-8 | |
F-9 | |
F-10 | |
F-11 |
2. Financial Statement Schedules.
There are no financial statement schedules provided because the information called for is either not required or is shown either in the financial statements or the notes thereto.
| 82 |
| Table of Contents |
3. Exhibits.
Exhibit Number |
| Description |
| Incorporated by Reference | ||||||
|
|
|
| Form |
| File No. |
| Exhibit |
| Filing Date |
|
| 8-K |
| 001-38677 |
| 2.1 |
| 1/13/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
| Amended and Restated Certificate of Incorporation of the Registrant. |
| 8-K |
| 001-38677 |
| 3.1 |
| 10/1/2018 | |
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 3.1 |
| 11/17/2020 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 3.1 |
| 9/20/2022 | ||
|
|
|
|
|
|
|
|
|
|
|
| Certificate of Designation of Series X Convertible Preferred Stock. |
| 8-K |
| 001-38677 |
| 3.1 |
| 1/13/2023 | |
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 3.2 |
| 1/13/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 3.1 |
| 8/4/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 3.2 |
| 10/1/2018 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 3.1 |
| 8/17/2022 | ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 4.1 |
| 5/22/2020 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 4.2 |
| 5/22/2020 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 4.3 |
| 5/22/2020 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| S-1 |
| 333-239887 |
| 4.3 |
| 7/16/2020 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| S-1 |
| 333-239887 |
| 4.4 |
| 7/16/2020 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| S-1 |
| 333-239887 |
| 4.5 |
| 7/16/2020 | ||
|
|
|
|
|
|
|
|
|
|
|
4.9 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| S-1/A |
| 333-262195 |
| 4.9 |
| 2/3/2022 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 4.1 |
| 7/22/2022 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 4.4 |
| 2/9/2022 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 10-Q |
| 001-38677 |
| 4.7 |
| 8/15/2022 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 4.1 |
| 1/13/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 4.2 |
| 1/13/2023 | ||
|
| 83 |
| Table of Contents |
Exhibit Number |
| Description |
| Incorporated by Reference | ||||||
|
|
|
| Form |
| File No. |
| Exhibit |
| Filing Date |
|
| 8-K |
| 001-38677 |
| 4.3 |
| 1/13/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
10.1 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 2018 Form of Indemnification Agreement between the Registrant and directors and executive officers. |
| S-1 |
| 333-226191 |
| 10.2 |
| 8/24/2018 | |
|
|
|
|
|
|
|
|
|
|
|
| Ra Medical Systems, Inc. 2018 Stock Compensation Plan and Forms of Award Agreement thereunder. |
| S-1 |
| 333-226191 |
| 10.3 |
| 7/16/2018 | |
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 99.1 |
| 10/13/2020 | ||
10.5 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.6 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.7 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.8 |
| [omitted] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.9 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 10.11 |
| 4/16/2020 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 10.6 |
| 1/13/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 10.1 |
| 1/19/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| S-1 |
| 333-237701 |
| 10.15 |
| 4/16/2020 | ||
|
|
|
|
|
|
|
|
|
|
|
10.12 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.13 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.14 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 10-K |
| 001-38677 |
| 10.19 |
| 3/17/2021 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 10-K |
| 001-38677 |
| 10.20 |
| 3/17/2021 | ||
|
|
|
|
|
|
|
|
|
|
|
| Notice of Suspension of Corporate Integrity Agreement, dated January 11, 2023. |
| 10-K |
| 001-38677 |
| 10.16.1 |
| 3/28/2023 | |
|
|
|
|
|
|
|
|
|
|
|
10.17 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.18 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.19 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.20 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.21 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.22 |
| [omitted.] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 10.1 |
| 7/22/2022 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 10.4 |
| 1/13/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 3.2 |
| 1/13/2023 |
|
| 84 |
| Table of Contents |
Exhibit Number |
| Description |
| Incorporated by Reference | ||||||
|
|
|
| Form |
| File No. |
| Exhibit |
| Filing Date |
|
| - Ex. B to January 2023 SPA (form of Registration Rights Agreement). |
| 8-K |
| 001-38677 |
| 10.5 |
| 1/13/2023 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 4.2 |
| 1/13/2023 | |
|
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 4.3 |
| 1/13/2023 | |
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 10.5 |
| 1/13/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 8-K |
| 001-38677 |
| 10.3 |
| 1/13/2023 | ||
|
| 10-K |
| 001-38677 |
| 10.27.1 |
| 3/28/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 10-K |
| 001-38677 |
| 10.27.2 |
| 3/28/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 10-K |
| 001-38677 |
| 10.27.3 |
| 3/28/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 10-K |
| 001-38677 |
| 10.28 |
| 3/28/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 10-K |
| 001-38677 |
| 10.29 |
| 3/28/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
| Extension Agreement dated January 11, 2022 to the Stereotaxis Marketing Agreement. |
| 10-K |
| 001-38677 |
| 10.29.1 |
| 3/28/2023 | |
|
|
|
|
|
|
|
|
|
|
|
| Addendum One dated May 27, 2022 to the Stereotaxis Marketing Agreement. |
| 10-K |
| 001-38677 |
| 10.29.2 |
| 3/28/2023 | |
|
|
|
|
|
|
|
|
|
|
|
|
| 10-K |
| 001-38677 |
| 10.30.1 |
| 3/28/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
| Consulting Agreement dated February 1, 2018, with Patricia Kennedy. |
| 10-K |
| 001-38677 |
| 10.31 |
| 3/28/2023 | |
|
|
|
|
|
|
|
|
|
|
|
|
| 10-K |
| 001-38677 |
| 10.31.1 |
| 3/28/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| DEF 14A |
| 001-38677 |
| Annex C |
| 05/25/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
| 2023 Form of Nonstatutory Stock Option Agreement Under 2023 Equity Incentive Plan |
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
| 2023 Form of Incentive Stock Option Agreement Under 2023 Equity Incentive Plan |
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
| Software and Technology License Agreement dated May 1, 2016, with Peacs BV. |
| 10-K |
| 001-38677 |
| 10.32 |
| 3/28/2023 | |
|
|
|
|
|
|
|
|
|
|
|
| Settlement and Amendment Agreement dated May 24, 2021 with Peacs BV. |
| 10-K |
| 001-38677 |
| 10.32.1 |
| 3/28/2023 |
|
| 85 |
| Table of Contents |
Exhibit Number |
| Description |
| Incorporated by Reference | ||||||
|
|
|
| Form |
| File No. |
| Exhibit |
| Filing Date |
|
| 8-K |
| 001-38677 |
| 16.1 |
| 6/26/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
|
| 10-K |
| 001-38677 |
| 21.1 |
| 3/28/2023 | ||
|
|
|
|
|
|
|
|
|
|
|
| Consent of WithumSmith+Brown, PC, Independent Registered Public Accounting Firm. |
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
| Consent of Haskell & White LLP, Independent Registered Public Accounting Firm. |
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
| Policy Relating to the Recovery of Erroneously Awarded Compensation |
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
101.INS* |
| Inline XBRL Instance Document. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.SCH* |
| Inline XBRL Taxonomy Extension Schema Document. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.CAL* |
| Inline XBRL Taxonomy Extension Calculation Linkbase Document. |
|
|
|
|
|
|
|
|
101.DEF* |
| Inline XBRL Taxonomy Extension Definition Linkbase Document. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.LAB* |
| Inline XBRL Taxonomy Extension Label Linkbase Document. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.PRE* |
| Inline XBRL Taxonomy Extension Presentation Linkbase Document |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
104* |
| Cover Page Interactive Data File (formatted as Inline XBRL) |
|
|
|
|
|
|
|
|
* | Filed herewith. |
| The information in this exhibit is furnished and deemed not filed with the Securities and Exchange Commission for purposes of section 18 of the Exchange Act of 1934, as amended (Exchange Act), and is not to be incorporated by reference into any filing of Ra Medical Systems, Inc. under the Securities Act of 1933, as amended (Securities Act), or the Exchange Act, whether made before or after the date hereof, regardless of any general incorporation language in such filing. |
+ | Indicates a management contract or compensatory plan. |
ITEM 16. FORM 10–K SUMMARY.
None.
| 86 |
| Table of Contents |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| CATHETER PRECISION, INC. |
| |
|
|
| |
|
|
|
|
Date: March 29, 2024 | By: | /s/ David A. Jenkins |
|
|
| David A. Jenkins |
|
|
| Executive Chairman and Chief Executive Officer |
|
| 87 |
| Table of Contents |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints David A. Jenkins and Margrit Thomassen, and each of them, his true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, to sign any and all amendments (including post-effective amendments) to this Annual Report on Form 10-K and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto each of said attorneys-in-fact and agents, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that each of said attorneys-in-facts and agents, or his substitute or substitutes, or any of them, shall do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
Signature |
| Title |
| Date |
|
|
|
|
|
/s/ David A. Jenkins |
| Executive Chairman of the Board and Chief Executive Officer |
| March 29, 2024 |
David A. Jenkins |
| (Principal Executive Officer) |
|
|
|
|
|
|
|
/s/ Margrit Thomassen |
| Interim Chief Financial Officer |
| March 29, 2024 |
Margrit Thomassen |
| (Principal Financial and Accounting Officer) |
|
|
|
|
|
|
|
/s/ James Caruso |
| Director |
| March 29, 2024 |
James Caruso |
|
|
|
|
|
|
|
|
|
/s/ Martin Colombatto |
| Director |
| March 29, 2024 |
Martin Colombatto |
|
|
|
|
|
|
|
|
|
/s/ John P. Francis |
| Director |
| March 29, 2024 |
John P. Francis |
|
|
|
|
| 88 |
| Table of Contents |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Catheter Precision, Inc.:
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheet of Catheter Precision, Inc., (the “Company”) as of December 31, 2023, the related consolidated statements of operations, stockholders’ equity, and cash flows, for the year ended December 31, 2023, and the related notes (collectively referred to as the "consolidated financial statements"). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023, and the results of its operations and its cash flows for the year ended December 31, 2023, in conformity with accounting principles generally accepted in the United States of America.
Substantial Doubt Regarding Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the entity has incurred recurring losses from operations and expects to continue to incur operating losses that raise substantial doubt about its ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty. Our opinion is not modified with respect to this matter.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's consolidated financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audit provides a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
| F-1 |
| Table of Contents |
Business Combination
Description of the Matter
On January 9, 2023, the Company completed the acquisition of Catheter Precision Inc. a privately held Delaware Corporation (“Old Catheter”) (the “Merger”). As further described in Note 3 to the consolidated financial statements, the Company accounted for the purchase consideration and related valuation and allocation in accordance with Accounting Standards Codification (“ASC”) 805, Business Combinations (ASC 805).
Management evaluated all criteria in ASC 805 including the makeup of management and governance before and after the transaction as well as the percentage of voting interests in the Company. Based on the evaluation, the Company concluded that they were the accounting acquirer. Total consideration for the Merger was $72.5 million which represents the sum of the (i) estimated fair value of $69.1 million for the 14,649.592 shares of newly designated Series X Convertible Preferred Stock issued and (ii) the estimated fair value of $3.4 million associated with stock options issued as replacements of Old Catheter share-based payment awards. The Company engaged valuation specialists to fair value the Series X Convertible Preferred Stock that was issued as well as assist in the allocation of purchase price for the acquired assets and liabilities assumed.
We identified the evaluation of the Company’s determination of the accounting acquirer and the determination of the purchase price associated with the Merger to be a critical audit matter. The business combination was deemed to be a critical audit matter due to the complexity and subjectivity involved in (1) the evaluation of identifying the accounting acquirer and (2) the determination of the purchase consideration and related valuation and allocation of such consideration.
| · | A high degree of auditor judgment was required in evaluating the relative importance of the indicative factors, individually and in the aggregate, including the post combination voting rights, composition of the board of directors and management, the terms of the newly created Series X Convertible Preferred Stock issued in the Merger, and the entity initiating the business combination. |
| · | The determination of the purchase price and the related allocation of the purchase price to the underlying assets acquired and liabilities assumed is a complex process that requires involvement from valuation specialists and significant professional judgment regarding the various inputs into an array of valuations to support the consideration given in the transaction and the underlying allocations. |
A different conclusion would result in a material difference in the accounting for the Merger.
How the Critical Audit Matter was Addressed in the Audit
The following are the primary procedures we performed to address this critical audit matter.
| · | We tested the Company’s conclusions that the Company was the accounting acquirer by: |
| · | reviewing management’s evaluation process which involved documenting an understanding of the terms in the Amended and Restated Agreement and Plan of Merger and related exhibits as follows: |
| · | evaluated management’s assessment of the post combination voting rights, |
| · | reviewed the composition of the board of directors and management, |
| F-2 |
| Table of Contents |
| · | reviewed the public filings associated with and leading up to and subsequent to the Merger, |
| · | reviewed the terms of the newly created Series X Convertible Preferred Stock, |
| · | examined the documents associated with the Merger including the Merger Agreement, corporate documents including the articles of incorporation and bylaws of the Company, investor presentations, and board minutes of both the Company and Old Catheter pre- and post-merger, |
| · | corroborated our understanding the structure and form of the Merger with external legal counsel. |
| · | We evaluated management’s valuations including obtaining audit evidence that supports management’s inputs in generating the forecasts used by the valuation specialists as follows: |
| · | audited projections and other significant inputs to each of the calculations to assess reasonableness of the final purchase price allocation, |
| · | assessed the professional competence, experience, and objectivity of the Company's external valuation specialist. |
| · | utilized our internal valuation specialists to assess the fair value of the Series X Convertible Preferred Stock. |
| · | utilized our internal valuation specialist to evaluate the reasonableness of the methodology and valuations of acquired intangible assets. |
Fair Value of Royalties Payable
Description of the Matter
As described in Note 10 to the consolidated financial statements, the Company had $6.97 million of royalties payable as of December 31, 2023, based on the fair value of the royalties payable related to the Amigo and LockeT royalty agreements acquired in connection with the Merger. We identified the fair value of royalties payable as a critical audit matter due to its highly sensitive inputs. In determining the fair value management must generate revenue projections through the expiration of the royalty agreements. They also must calculate a revenue-adjusted discount rate which is then applied to calculate the present value of the royalties payable. There is significant uncertainty associated with the projections due to the fact that management must forecast sales for a new product which, while there is interest in the market place and it is being evaluated for use in the hospital environment, it has had no sales as of the date of the valuation. In addition, the calculation of the discount rate requires the involvement of management's valuation specialists.
How the Critical Matter was Addressed in the Audit
To determine the reasonableness of the fair value of the royalties payable, we:
| · | audited the projections through detailed review of management’s memo and forecast schedules along with review of supporting evidence including: |
| o | industry standards, |
| o | external data sources, |
| o | regulatory factors, |
| o | Company press releases and related SEC filings, |
| o | copies of presentations given by the Company, |
| · | considered any potentially contradicting information, |
| · | reviewed the present value calculation of the royalties payable, |
| · | assessed the professional competence, experience, and objectivity of the Company's external valuation specialist, |
| · | utilized our internal valuation specialist to evaluate the reasonableness of the methodology of the calculation of the revenue-adjusted discount rate. |
| F-3 |
| Table of Contents |
Assessment of ASC 350 and ASC 360 impairment analysis
Description of the Matter
As described in Note 2 to the consolidated financial statements, in accordance with ASC 350, Intangibles – Goodwill and Other (ASC 350) and ASC 360, Impairment and Disposal of Long-Lived Assets (ASC 360), the Company, at least annually or more frequently if certain events or changes in circumstances indicate the carrying value may not be recoverable, management performs an impairment analysis. As a result of the sustained decline of the Company’s stock price from the date of the Merger to the date of each reporting period, the Company assessed their goodwill, intangible assets, and long-lived assets for impairment.
The Company performed a quantitative goodwill impairment test, which resulted in the carrying amount of the reporting unit exceeding the estimated fair value of the reporting unit, indicating that the goodwill of the reporting unit was impaired. The Company utilized a combination of an income and market approach to assess the fair value of the reporting unit. The income approach considered the discounted cash flow model, considering projected future cash flows (including timing and profitability), discount rate reflecting the risk inherent in future cash flows, perpetual growth rate, and projected future economic and market conditions. The guideline public company market approach considered marketplace earnings multiples from within a peer public company group. As of December 31, 2023, cumulative goodwill impairment charges of $60.9 million were incurred related to the Company’s single reporting unit.
With respect to its long-lived assets, to determine whether the carrying amount of the long-lived asset group is recoverable, the Company determined the estimated future cash flows of the group for a period consistent with that of the primary assets of the group. The sum of the undiscounted cash flows was then compared to the carry amount of the long-lived assets, as of December 31, 2023, to conclude whether the asset group carrying value is recoverable.
We identified the assessment of ASC 350 and ASC 360 impairment analysis as a critical audit matter due to the estimation and subjectivity needed to identify impairment triggers and perform an impairment test. The inputs to test are subjective as they are based on management’s forecasts. Additionally, there is complexity that requires the Company to involve valuation specialists in performing the quantitative test.
How the Critical Matter was Addressed in the Audit
To determine the reasonableness of the goodwill impairment and conclusion the long-lived assets were not impaired we:
| · | reviewed the impairment analyses performed as of each interim reporting period as well as at year-end, |
| · | audited the projections utilized by the valuation specialist in the impairment assessment through detailed review of management’s memo and forecast schedules along with review of supporting evidence including: |
| o | industry standards, |
| o | external data sources, |
| o | regulatory factors, |
| o | Company press releases and related SEC filings, |
| o | copies of presentations given by the Company, |
| F-4 |
| Table of Contents |
| · | assessed the professional competence, experience, and objectivity of the Company's external valuation specialist, |
| · | utilized our internal valuation specialists to review the methodologies for each quarter’s quantitative impairment analysis. |
Equity Offerings - Private Placement
Description of the Matter
As discussed in Note 13, Private Placement, on January 9, 2023, the Company entered into a Securities Purchase Agreement for a private placement for which the Company would issue (a) Class A units consisting of one share of common stock, one Series F common stock purchase warrant, and one Series G common stock purchase warrant and (b) Class B units consisting of one share of newly designated Series A Convertible Preferred Stock, one Series F common stock purchase warrant and one Series G common stock purchase warrant. The Company engaged valuation specialists to fair value the underlying instruments and allocate proceeds accordingly. Management evaluated all criteria under ASC 480, Distinguishing Liabilities from Equity, to determine the proper accounting treatment for each instrument issued. We identified accounting for this equity-based transaction as a critical audit matter due to the complexity of the offering consisting of preferred stock, common stock and warrants in addition to the related valuation and classification of the instruments.
How the Critical Matter was Addressed in the Audit
The following are the primary procedures we performed to address this critical audit matter:
| · | reviewed the underlying agreements with the investor, |
| · | reviewed management’s accounting treatment analysis memorandum, |
| · | due to the complexity of the underlying instrument, we evaluated whether the various instruments issued were free-standing, could be classified as debt or equity, and whether they contained any derivatives, |
| · | vouched the proceeds that were raised, |
| · | audited management’s allocation of proceeds analysis by |
| o | footing/cross footing the schedule for mathematical accuracy, |
| o | agreeing amounts to the Company’s accounting treatment analysis memorandum, |
| o | agreeing the fair value to the valuation reports prepared by the Company’s valuation specialist, |
| o | confirming the price of the Company’s common stock, |
| o | agreed share issuances to the Company’s transfer agent report. |
| · | utilized our internal valuation specialists to evaluate volatility inputs of the Black-Scholes models that were utilized in allocating the value of each instrument based on the net proceeds raised. |
/s/
We have served as the Company’s auditor since 2023.
March 29, 2024
PCAOB ID Number
| F-5 |
| Table of Contents |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of Catheter Precision, Inc.
(formerly, Ra Medical Systems, Inc.)
Opinion on the Financial Statements
We have audited the accompanying balance sheet of Catheter Precision, Inc. (formerly, Ra Medical Systems, Inc.) (the “Company”) as of December 31, 2022, the related statements of operations, stockholders' equity, and cash flows for the year ended December 31, 2022 and the related notes (collectively referred to as the “financial statements“). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2022, and the results of its operations and its cash flows for the year ended December 31, 2022, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company's financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
| /s/ HASKELL & WHITE LLP |
|
| HASKELL & WHITE LLP |
|
We have served as the Company's auditor from 2021 to 2023.
Irvine, California
March 28, 2023
| F-6 |
| Table of Contents |
CATHETER PRECISION, INC. (formerly known as RA MEDICAL SYSTEMS, INC.)
Consolidated Balance Sheets
(in thousands, except par value data)
|
| December 31, 2023 |
|
| December 31, 2022 |
| ||
ASSETS |
|
|
|
|
|
| ||
Current Assets |
|
|
|
|
|
| ||
Cash and cash equivalents |
| $ |
|
| $ |
| ||
Accounts receivable, net |
|
|
|
|
|
| ||
Inventories |
|
|
|
|
|
| ||
Prepaid expenses and other current assets |
|
|
|
|
|
| ||
Total current assets |
|
|
|
|
|
| ||
Property and equipment, net |
|
|
|
|
|
| ||
Lease right-of-use assets |
|
|
|
|
|
| ||
Intangible assets, net |
|
|
|
|
|
| ||
Other non-current assets |
|
|
|
|
|
| ||
TOTAL ASSETS |
| $ |
|
| $ |
| ||
LIABILITIES AND STOCKHOLDERS' EQUITY |
|
|
|
|
|
|
|
|
Current Liabilities |
|
|
|
|
|
|
|
|
Accounts payable |
| $ |
|
| $ |
| ||
Accrued expenses |
|
|
|
|
|
| ||
Notes payable |
|
|
|
|
|
| ||
Current portion of operating lease liabilities |
|
|
|
|
|
| ||
Total current liabilities |
|
|
|
|
|
| ||
Royalties payable |
|
|
|
|
|
| ||
Operating lease liabilities |
|
|
|
|
|
| ||
Total liabilities |
|
|
|
|
|
| ||
Commitments and contingencies (see Note 17) |
|
|
|
|
|
|
|
|
Stockholders' Equity |
|
|
|
|
|
|
|
|
Preferred Stock, $ |
|
|
|
|
|
|
|
|
Series A Convertible Preferred Stock, $ |
|
|
|
|
|
| ||
Series X Convertible Preferred Stock, $ |
|
|
|
|
|
| ||
Common stock, $ |
|
|
|
|
|
| ||
Additional paid-in capital |
|
|
|
|
|
| ||
Accumulated deficit |
|
| ( | ) |
|
| ( | ) |
Total stockholders' equity |
|
|
|
|
|
| ||
TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY |
| $ |
|
| $ |
| ||
See accompanying notes to consolidated financial statements.
| F-7 |
| Table of Contents |
CATHETER PRECISION, INC. (formerly known as RA MEDICAL SYSTEMS, INC.)
Consolidated Statements of Operations
(in thousands, except per share data)
|
| Year Ended December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Revenues |
|
|
|
|
|
| ||
Product sales |
| $ |
|
| $ |
| ||
Cost of revenues |
|
|
|
|
|
|
|
|
Product sales |
|
|
|
|
|
| ||
Service and other |
|
|
|
|
|
| ||
Total cost of revenues |
|
|
|
|
|
| ||
Gross profit (loss) |
|
|
|
|
| ( | ) | |
Operating expenses |
|
|
|
|
|
|
|
|
Loss on impairment of goodwill |
|
|
|
|
|
| ||
Selling, general and administrative |
|
|
|
|
|
| ||
Research and development |
|
|
|
|
|
| ||
Restructuring costs |
|
|
|
|
|
| ||
Total operating expenses |
|
|
|
|
|
| ||
Operating loss |
|
| ( | ) |
|
| ( | ) |
Other income, net |
|
|
|
|
|
|
|
|
Interest income |
|
|
|
|
|
| ||
Other income (expense), net |
|
| ( | ) |
|
|
| |
Change in fair value of royalties payable |
|
|
|
|
|
| ||
Total other income, net |
|
|
|
|
|
| ||
Loss from operations before income taxes |
|
| ( | ) |
|
| ( | ) |
Income taxes |
|
|
|
|
|
| ||
Net loss |
| $ | ( | ) |
| $ | ( | ) |
Deemed dividend - warrant inducement offer |
|
| ( | ) |
|
|
| |
Net loss attributable to common stockholders |
| $ | ( | ) |
| $ | ( | ) |
Net loss per share attributable to common stockholders, basic and diluted |
| $ | ( | ) |
| $ | ( | ) |
Weighted average common shares used in computing net loss per share, basic and diluted |
|
|
|
|
|
| ||
See accompanying notes to consolidated financial statements.
| F-8 |
| Table of Contents |
CATHETER PRECISION, INC. (formerly known as RA MEDICAL SYSTEMS, INC.)
Consolidated Statements of Stockholders' Equity
(in thousands, except share data)
|
| Series A Convertible Preferred Stock |
|
| Series X Convertible Preferred Stock |
|
| Common Stock |
|
| Additional |
|
| Accumulated |
|
| Total Stockholders' |
| ||||||||||||||||||
|
| Shares |
|
| Amount |
|
| Shares |
|
| Amount |
|
| Shares |
|
| Amount |
|
| Paid-In Capital |
|
| Deficit |
|
| Equity |
| |||||||||
Balance at December 31, 2021 |
|
| — |
|
| $ |
|
|
| — |
|
| $ |
|
|
|
|
| $ |
|
| $ |
|
| $ | ( | ) |
| $ |
| ||||||
Common stock issued, net |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Warrants issued, net |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Warrants exercised |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Restricted stock awards cancelled or vested |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Common stock issued pursuant to the vesting of restricted stock units and purchases under employee stock purchase plan |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Stock-based compensation |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Net loss |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | ||||
Balance at December 31, 2022 |
|
| — |
|
| $ |
|
|
| — |
|
| $ |
|
|
|
|
| $ |
|
| $ |
|
| $ | ( | ) |
| $ |
| ||||||
Common stock issued upon exercise of options |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Restricted stock awards cancelled or vested |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Stock-based compensation |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Issuance of Series X Convertible Preferred Stock in merger |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Conversion of Series X Convertible Preferred Stock |
|
| — |
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Issuance of Series A Convertible Preferred Stock in connection with private placement, net |
|
|
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||
Warrants exercised net |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Conversion of Series A Convertible Preferred Stock |
|
| ( | ) |
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Deemed dividend - warrant inducement offer |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Net loss |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | ||||
Balance at December 31, 2023 |
|
|
|
| $ |
|
|
|
|
| $ |
|
|
|
|
| $ |
|
| $ |
|
| $ | ( | ) |
| $ |
| ||||||||
See accompanying notes to consolidated financial statements.
| F-9 |
| Table of Contents |
CATHETER PRECISION, INC. (formerly known as RA MEDICAL SYSTEMS, INC.)
Consolidated Statements of Cash Flows
(in thousands, except share data)
|
| For the Years Ended December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
CASH FLOWS FROM OPERATING ACTIVITIES: |
|
|
|
|
|
| ||
Net loss |
| $ | ( | ) |
| $ | ( | ) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
Restructuring charges |
|
|
|
|
|
| ||
Loss on impairment of goodwill |
|
|
|
|
|
| ||
Depreciation and amortization |
|
|
|
|
|
| ||
Stock-based compensation |
|
|
|
|
|
| ||
Change in fair value of royalties payable |
|
| ( | ) |
|
|
| |
Gain on write-off of right-of-use asset and liability |
|
|
|
|
| ( | ) | |
Loss on sales and disposals of property and equipment |
|
|
|
|
|
| ||
Provision for credit losses |
|
|
|
|
|
| ||
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
Accounts receivable |
|
| ( | ) |
|
|
| |
Inventories |
|
|
|
|
| ( | ) | |
Prepaid expenses and other assets |
|
|
|
|
| ( | ) | |
Lease right-of-use assets and lease liabilities |
|
|
|
|
|
| ||
Accounts payable |
|
| ( | ) |
|
| ( | ) |
Accrued expenses |
|
| ( | ) |
|
|
| |
Accrued interest - related parties |
|
| ( | ) |
|
|
| |
Other liabilities |
|
|
|
|
| ( | ) | |
Net cash used in operating activities |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
CASH FLOWS FROM INVESTING ACTIVITIES: |
|
|
|
|
|
|
|
|
Purchases of property and equipment |
|
| ( | ) |
|
| ( | ) |
Proceeds from sales of property and equipment |
|
|
|
|
|
| ||
Cash acquired as part of business combination |
|
|
|
|
|
| ||
Net cash (used in) provided by investing activities |
|
| ( | ) |
|
|
| |
|
|
|
|
|
|
|
|
|
CASH FLOWS FROM FINANCING ACTIVITIES: |
|
|
|
|
|
|
|
|
Proceeds from issuance of common stock and warrants |
|
|
|
|
|
| ||
Payments of offering costs related to the issuance of common stock and warrants |
|
|
|
|
| ( | ) | |
Payments on note payable |
|
| ( | ) |
|
|
| |
Proceeds from exercise of warrants |
|
|
|
|
|
| ||
Proceeds from issuance of common stock in connection with the employee stock purchase plan |
|
|
|
|
|
| ||
Payments of costs related to exercise of warrants |
|
| ( | ) |
|
| ( | ) |
Payments of convertible promissory notes |
|
| ( | ) |
|
|
| |
Proceeds from the private placement of securities |
|
|
|
|
|
| ||
Payments of offering costs related to the private placement of securities |
|
| ( | ) |
|
|
| |
Net cash provided by financing activities |
|
|
|
|
|
| ||
NET CHANGE IN CASH AND CASH EQUIVALENTS |
|
| ( | ) |
|
|
| |
CASH AND CASH EQUIVALENTS, beginning of year |
|
|
|
|
|
| ||
CASH AND CASH EQUIVALENTS, end of year |
| $ |
|
| $ |
| ||
SUPPLEMENTAL CASH FLOW INFORMATION |
|
|
|
|
|
|
|
|
Unpaid offering costs |
| $ |
|
| $ |
| ||
Cash payments for income taxes |
| $ |
|
| $ |
| ||
Non-cash consideration for Catheter acquisition |
| $ |
|
| $ |
| ||
Conversion of Series A Convertible Preferred Stock for common stock |
| $ |
|
| $ | |
| |
Cash payments for interest |
| $ |
|
| $ |
| ||
See accompanying notes to consolidated financial statements.
| F-10 |
| Table of Contents |
CATHETER PRECISION, INC. (formerly known as RA MEDICAL SYSTEMS, INC.)
Notes to Consolidated Financial Statements
(in thousands, except share data)
Note 1. Organization and Nature of Operations
The Company
Catheter Precision Inc. (formerly known as Ra Medical Systems, Inc.) ("Catheter" or the "Company or "Legacy RA Medical"), was incorporated in Delaware in July 2018. Catheter was initially formed to develop, commercialize and market its advanced excimer laser-based platform for use in the treatment of vascular and dermatological immune-mediated inflammatory diseases.
On January 9, 2023, Catheter entered into the Amended and Restated Agreement and Plan of Merger, or the "Merger Agreement", with Catheter Precision, Inc., or “Old Catheter”, a privately-held Delaware corporation. Under the terms of the Merger Agreement, Old Catheter became a wholly owned subsidiary of Catheter, together referred to as the Company, in a stock-for-stock merger transaction, or the "Merger".
After the Merger and looking forward, the legacy Destruction of Arteriosclerotic Blockages by laser Radiation Ablation laser and single-use catheter, together referred to as "DABRA," related assets were no longer used and Catheter’s legacy lines of business were discontinued, but instead the Company has shifted the focus of its operations to Old Catheter’s product lines. Accordingly, the Company’s current activities primarily relate to Old Catheter’s historical business which comprises the design, manufacture and sale of new and innovative medical technologies primarily focused in the field of cardiac electrophysiology, or EP.
The Company’s primary product is the View into Ventricular Onset System (“VIVO” or “VIVO System”) which is a non-invasive imaging system that offers 3D cardiac mapping to help with localizing the sites of origin of idiopathic ventricular arrhythmias in patients with structurally normal hearts prior to EP procedures. The VIVO system has achieved a CE Mark allowing it to be commercialized in the European Union and has been placed at several hospitals in Europe. United States Food and Drug Administration ("FDA") 510(K) clearance in the United States was received and the Company began a limited commercial release of VIVO in 2021.
The Company’s newest product, Surgical Vessel Closing Pressure Device ("LockeT"), is a suture retention device indicated for wound healing by distributing suture tension over a larger area in the patient in conjunction with a figure of eight suture closure and is intended to temporarily secure sutures and aid clinicians in locating and removing sutures efficiently. In addition, LockeT is a sterile, Class I product that was registered with the FDA in February 2023, at which time initial shipments began to distributors. Clinical studies for LockeT began during the quarter ended September 30, 2023. These studies are planned to show the product’s effectiveness and benefits, including faster wound closure, earlier ambulation, potentially leading to early hospital discharge, and cost benefits. This information is intended to provide crucial data for marketing and to expand the Company's indications for use with the FDA.
The Company’s product portfolio also includes the Amigo® Remote Catheter System (the "AMIGO" or "AMIGO System"), a robotic arm that serves as a catheter control device. Prior to 2018, Old Catheter marketed Amigo. The Company owns the intellectual property related to Amigo, and this product is under consideration for future research and development of a generation 2 product.
Prior to the Merger, Catheter developed an advanced excimer laser-based platform for use in the treatment of vascular immune-mediated inflammatory diseases. DABRA was developed as a tool in the treatment of Peripheral Artery Disease which commonly occurs in the legs. The Company has ceased marketing DABRA.
Effective June 6, 2022, the Company’s board of directors approved a staggered reduction in force (“RIF”). On September 2, 2022, the Company completed the RIF. The purpose of the RIF was to preserve capital with the goal of maximizing the opportunities available to the Company in furtherance of the board of directors’ review of strategic alternatives. As a result of the RIF, the Company paused all engineering and manufacturing activities during the third quarter of 2022 for its legacy DABRA Products.
| F-11 |
| Table of Contents |
Going Concern
As of December 31, 2023, the Company had cash and cash equivalents of approximately $
Management expects operating losses and negative cash flows to continue for the foreseeable future as the Company invests in its commercial capabilities. Additional costs associated with the Merger paid during the years ended December 31, 2023 and 2022, respectively, have substantially depleted the Company’s cash. Following the Merger with Old Catheter, management further reduced staff and other costs while assuming the operating costs of Old Catheter. Of the Company’s cash flows used in operating activities of $
As a result of these factors, management has concluded that there is substantial doubt about the Company’s ability to continue as a going concern for a period of one year after the date the consolidated financial statements are issued. The Company’s consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Note 2. Summary of Significant Accounting Policies
Principles of Consolidation
The consolidated financial statements of the Company include the accounts of the Company and Old Catheter. All intercompany transactions have been eliminated in consolidation. The financial results of Old Catheter are included in the consolidated financial statements from the date of completion of the Merger to December 31, 2023.
Basis of Presentation
The consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”). The Financial Accounting Standards Board (“FASB”) establishes these principles to ensure financial condition, results of operations, and cash flows are consistently reported. Any reference in these notes to applicable accounting guidance is meant to refer to the authoritative nongovernmental GAAP as found in the FASB Accounting Standards Codification ("ASC").
Use of Estimates
The preparation of the consolidated financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting periods. Actual results could differ from those estimates. The Company’s consolidated financial statements are based upon a number of estimates including, but not limited to, the accounting for the Old Catheter business combination (see Note 3, Business Combination), allowance for credit losses, evaluation of impairment of long-lived assets and goodwill, valuation of long-lived assets and their associated estimated useful lives, reserves for warranty costs, fair value of royalties payable, evaluation of probable loss contingencies, fair value of preferred stock and warrants issued, and the fair value of equity awards granted.
| F-12 |
| Table of Contents |
Reclassifications
Certain reclassifications have been made to the financial statements for the nine months ended September 30, 2023 to conform to the current period financial statement presentation. Certain regulatory costs of $
Concentrations of Credit Risk
The Company's financial instruments that are exposed to concentrations of credit risk consist primarily of cash and cash equivalents and accounts receivable. Cash equivalents represent highly liquid investments with maturities of 90 days or less at the date of purchase. Credit risk related to cash and cash equivalents is based on the creditworthiness of the financial institutions at which these funds are held. The Company has cash balances at financial institutions which throughout the year may exceed the federally insured limit of $250,000. Any loss incurred or a lack of access to such funds could have a significant adverse impact on the Company's financial condition, results of operations, and cash flows. To reduce its risk associated with the failure of any such financial institution, the Company evaluates the rating of the financial institution in which it holds deposits. Any material loss that the Company may experience in the future could have an adverse effect on its ability to pay its operational expenses or make other payments and may require the Company to move its cash to other high quality financial institutions. Currently, the Company is reviewing its bank relationships in order to mitigate its risk to ensure that its exposure is limited or reduced to the Federal Deposit Insurance Corporation protection limits.
The Company extends credit to customers in the normal course of business. Concentrations of credit risk with respect to accounts receivable exist to the full extent of amounts presented in the consolidated financial statements. The Company does not require collateral from its customers to secure accounts receivable.
The Company had three customers that represented more than 10% of the Company’s consolidated revenue as of December 31, 2023.
The Company has no significant off-balance sheet risk such as foreign exchange contracts, option contracts, or other hedging arrangements.
Segment Reporting
The Company operates in one business segment, which is the marketing, sales and development of medical technologies focused in the field of cardiac electrophysiology.
Cash and Cash Equivalents
The Company considers all short-term, highly liquid investments with original maturities of 90 days or less to be cash equivalents. Cash equivalents primarily represent funds invested in readily available checking and money market accounts. The Company maintains deposits in financial institutions in excess of federally insured limits of $250,000, in the amount of $
Fair Value Measurements
Fair value represents the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants and is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or liability. A three-tier fair value hierarchy is used to identify inputs used in measuring fair value as follows:
Level 1 - Observable inputs that reflect quoted market prices (unadjusted) for identical assets or liabilities in active markets;
Level 2 - Inputs other than the quoted prices in active markets that are observable either directly or indirectly in the marketplace for identical or similar assets and liabilities; and
Level 3 - Unobservable inputs that are supported by little or no market data, which require the Company to develop its own assumptions.
| F-13 |
| Table of Contents |
Cash equivalents, prepaid expenses, trade accounts receivable, accounts payable, and accrued expenses are reported on the consolidated balance sheets at carrying value which approximates fair value due to the short-term maturities of these instruments.
The following table details the fair value measurements within the fair value hierarchy of the Company’s financial instruments:
|
|
|
| Fair value at December 31, 2023 |
|
|
| |||||||||
|
| Total |
|
| Level 1 |
|
| Level 2 |
|
| Level 3 |
| ||||
Assets: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Cash Equivalents |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Mutual Fund |
| $ |
|
| $ |
|
| $ |
|
| $ |
| ||||
Money Market fund |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Total assets |
| $ |
|
| $ |
|
| $ |
|
| $ |
| ||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Liabilities |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Royalties payable |
| $ |
|
| $ |
|
| $ |
|
| $ |
| ||||
Total liabilities |
| $ |
|
| $ |
|
| $ |
|
| $ |
| ||||
The royalties payable have unobservable inputs that are not supported by any market data. As such the Company developed its own assumptions and identified the inputs as level 3. The revenue adjusted discount rate (“RADR”) was calculated using a weighted average cost of capital (“WACC”) approach for the level 3 measurement. The RADR considers the WACC from the Company’s impairment analysis and adjusts certain inputs to represent the risk profile of the revenue. Under the cost of equity section, the risk-free rate has changed to be commensurate with the royalties payable term. Additionally, the Beta and Company Specific Risk Premium have been adjusted to Revenue Beta and Revenue Specific Risk Premium, respectively. This adjustment was calculated by multiplying the respective metric by the quotient of equity volatility over revenue volatility. The remaining inputs from the Impairment WACC have remained unchanged.
|
|
|
| Fair value at December 31, 2022 |
|
|
| |||||||||
|
| Total |
|
| Level 1 |
|
| Level 2 |
|
| Level 3 |
| ||||
Assets: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Cash Equivalents |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Certificate of Deposit |
| $ |
|
| $ |
|
| $ |
|
| $ |
| ||||
Money Market fund |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Total assets |
| $ |
|
| $ |
|
| $ |
|
| $ |
| ||||
Financial Instruments — Credit Losses (ASU 2016-13)
In June 2016, the FASB issued Accounting Standards Update ("ASU") 2016-13, Financial Instruments - Credit Losses (“CECL”). The amendments in this update introduce a new accounting model to measure credit losses for financial assets measured at amortized cost. The FASB has also issued additional ASUs to clarify the scope and provide additional guidance for ASU 2016-13. Credit losses for financial assets measured at amortized cost should be determined based on the total current expected credit losses over the life of the financial asset or group of financial assets. In effect, the financial asset or group of financial assets should be presented at the net amount expected to be collected. Credit losses will no longer be recorded under the current incurred loss model for financial assets measured at amortized cost. The amendments also modify the accounting for available-for-sale debt securities whereby credit losses will be recorded through an allowance for credit losses rather than a write-down to the security’s cost basis, which allows for reversals of credit losses when estimated credit losses decline. Credit losses for available-for-sale debt securities should be measured in a manner similar to current GAAP.
| F-14 |
| Table of Contents |
There was no impact of applying the CECL methodology upon adoption effective on January 1, 2020.
Under the CECL impairment model, the Company develops and documents its allowance for credit losses on its trade receivables based on three portfolio segments: Hospitals – United States, Hospitals – Europe, and Distributors. The determination of portfolio segments is based primarily on the customers’ industry and geographical location.
Our quantitative allowance for credit loss estimates under CECL was determined using the method that uses an aging schedule. The Company also considers qualitative adjustments that may relate to unique risks, changes in current economic conditions that may not be reflected in quantitatively derived results, or other relevant factors to further inform our estimate of the allowance for credit losses.
Accounts Receivable and Allowances for Doubtful Accounts
Trade accounts receivable are recorded at invoiced amounts, net of allowance for credit losses, if applicable, and are unsecured and do not bear interest.
The allowance for doubtful accounts is based on the probability of future collection under the current expected credited loss impairment model under CECL, which was adopted by the Company on January 1, 2020. Under the CECL impairment model, the Company determines its allowance by applying the method based on an aging schedule. The Company also considers reasonable and supportable current information in determining its estimated loss rates, such as external forecasts, macroeconomic trends or other factors including customers’ credit risk and historical loss experience. The adequacy of the allowance is evaluated on a regular basis. Account balances are written off after all means of collection are exhausted and the balance is deemed uncollectible. Subsequent recoveries are credited to the allowance. Changes in the allowance are recorded as adjustments to bad debt expense in the period incurred.
Accounts receivable consists of the following:
|
| December 31, 2023 |
|
| December 31, 2022 |
| ||
Trade accounts receivable |
| $ |
|
| $ |
| ||
Less: Reserve for expected credit losses |
|
|
|
|
| ( | ) | |
Accounts receivable, net - balance at end of period |
| $ |
|
| $ |
| ||
Inventories
Inventories are stated at the lower of cost (first-in, first-out method) or net realizable value. Cost includes materials, labor and manufacturing overhead related to the purchase and production of inventories. The Company reduced the carrying value of inventories for those items that were potentially excess, obsolete or slow-moving based on changes in customer demand, technological developments or other economic factors.
Property and Equipment
Property and equipment are recorded at cost and depreciated on a straight-line basis over their estimated useful lives as follows:
Machinery and equipment |
| |
Computer hardware and software |
| |
VIVO DEMO/Clinical Systems |
| |
Furniture and fixtures |
|
Leasehold improvements are depreciated over the shorter of the useful life of the leasehold improvement or the term of the underlying property’s lease.
| F-15 |
| Table of Contents |
The Company periodically reviews the residual values and estimated useful lives of each class of its property and equipment for ongoing reasonableness, considering long-term views on its intended use of each class of property and equipment and the planned level of improvements to maintain and enhance assets within those classes.
When assets are retired or otherwise disposed of, the cost and related accumulated depreciation are removed from the account balances and any resulting gain or loss is recognized in income for the period. The cost of repairs and maintenance is expensed as incurred, whereas significant betterments are capitalized.
Impairment of Long-Lived Assets
In accordance with ASC 360, Impairment and Disposals of Long-lived Assets, the Company periodically reviews its long-lived assets for impairment when certain events or changes in circumstances indicate that the carrying value of the long-lived assets may not be recoverable. Should the sum of the undiscounted expected future net cash flows be less than the carrying value, the Company would recognize an impairment loss at that date.
As a result of the sustained decline of the Company's stock price from the date of the Merger, the Company assesses its long-lived assets for impairment. To determine whether the carrying amount of the long-lived asset group is recoverable, the Company determined the estimated future cash flows of the group for a period consistent with that of the primary assets of the group. The sum of the undiscounted cash flows was then compared to the carrying amount of the long-lived assets, as of December 31, 2023. The Company concluded there was no impairment as of December 31, 2023.
Due to the Company’s RIF and the decision to discontinue enrollment of patients in its DABRA related clinical trial, the Company ceased manufacturing activities of DABRA. The Company’s property and equipment was determined to be impaired as of June 30, 2022, resulting in an impairment charge of $
Goodwill
In accordance with ASC 350, Intangibles – Goodwill and Other, goodwill is calculated as the difference between the acquisition date fair value of the consideration transferred and the fair value of net assets acquired. Goodwill, which represents the excess of purchase price of Old Catheter over the fair value of net assets acquired, is carried at cost. Goodwill is not amortized; rather, it is subject to a periodic assessment for impairment by applying a fair value-based test. The Company reviews goodwill for possible impairment annually during the fourth quarter, or whenever events or circumstances indicate that the carrying amount may not be recoverable.
To determine whether goodwill is impaired, annually or more frequently if needed, the Company performs a multi-step impairment test. The Company first has the option to assess qualitative factors to determine if it is more likely than not that the carrying value of a reporting unit exceeds its estimated fair value. The Company may also elect to skip the qualitative testing and proceed directly to the quantitative testing. When performing quantitative testing, the Company first estimates the fair values of its reporting units using a combination of an income and market approach. To determine fair values, the Company is required to make assumptions about a wide variety of internal and external factors. Significant assumptions used in the impairment analysis include financial projections of free cash flow (including significant assumptions about operations including the rate of future revenue growth, capital requirements, and income taxes), long-term growth rates for determining terminal value and discount rates. Comparative market multiples are used to corroborate the results of the discounted cash flow test. These assumptions require significant judgment. Pursuant to ASU 2017-04, Simplifying the Test for Goodwill Impairment, the single step is to determine the estimated fair value of the reporting unit and compare it to the carrying value of the reporting unit, including goodwill. To the extent the carrying amount of goodwill exceeds the implied goodwill, the difference is the amount of the goodwill impairment. The Company also completes a reconciliation between the implied equity valuation prepared and the Company’s market capitalization. The majority of the inputs used in the discounted cash flow model are unobservable and thus are considered to be Level 3 inputs. The inputs for the market capitalization calculation are considered Level 1 inputs. There were impairment charges of $60.9 million recognized during the year ended December 31, 2023, see Note 3, Business Combination and Note 7, Goodwill, for additional details.
Royalties Payable
The Company is obligated to pay royalties under various royalty agreements Old Cather had entered into. On January 9, 2023, prior to the consummation of the Merger, Old Catheter entered in an agreement with its Convertible Promissory Noteholders (“Noteholders”), which substantially consisted of amounts due to David A. Jenkins, previously Old Catheter's Chairman of the Board of Directors prior to the Merger, and, currently, the Company’s Executive Chairman of the Board of Directors and Chief Executive Officer, to forgive all accrued interest and future interest expense in exchange for a future royalty right.
| F-16 |
| Table of Contents |
In addition,
During 2006 and 2007, Old Catheter entered into two investment grant agreements with a non-profit foundation for the purpose of funding the initial development of Old Catheter's AMIGO System. The agreement calls for the payment to the foundation, upon successful commercialization of the AMIGO System (see Note 10, Royalties Payable).
As of the date of the Merger, the royalties payable had an estimated fair value of approximately $
Product Warranty
The Company’s current products are warrantied against defects in material and workmanship when properly used for their intended purpose and properly maintained.
Similarly, the DABRA products were warrantied against defects in material and workmanship when properly used for their intended purpose and appropriately maintained. Accordingly, the Company generally replaced catheters that kinked or failed to calibrate. The product warranty liability was determined based on historical information such as past experience, product failure rates or number of units repaired, estimated cost of material and labor. The product warranty liability also includes the estimated costs of a product recall.
The warranty accrual is included in accrued expenses in the accompanying consolidated balance sheets. Warranty expenses are included in cost of revenues in the accompanying consolidated statements of operations. Changes in estimates to previously established warranty accruals resulted from current period updates to assumptions regarding repair and product recall costs and are included in current period warranty expense.
Distinguishing Liabilities from Equity
The Company relies on the guidance provided by ASC Topic 480, Distinguishing Liabilities from Equity, to classify certain redeemable and/or convertible instruments. The Company first determines whether a financial instrument should be classified as a liability. The Company will determine the liability classification if the financial instrument is mandatorily redeemable, or if the financial instrument, other than outstanding shares, embodies a conditional obligation that the Company must or may settle by issuing a variable number of its equity shares.
Once the Company determines that a financial instrument should not be classified as a liability, the Company determines whether the financial instrument should be presented between the liability section and the equity section of the balance sheet. The Company will determine temporary equity classification if the redemption of the financial instrument is outside the control of the Company (i.e. at the option of the holder). Otherwise, the Company accounts for the financial instrument as permanent equity.
Revenue Recognition
The Company applies the provisions of FASB ASC Topic 606, Revenue from Contracts with Customers (“ASC 606”), and all related appropriate guidance. The core principle of this standard is that a company should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services.
| F-17 |
| Table of Contents |
The Company measures revenue based upon the consideration specified in the client arrangement, and revenue is recognized when the performance obligations in the client arrangement are satisfied. A performance obligation is a promise in a contract to transfer a distinct service to the customer. The transaction price of a contract is allocated to each distinct performance obligation. Under ASC 606, revenue is recognized when a customer obtains control of promised goods. To achieve this core principal, the Company applies the following five steps:
Step 1: Identify the contract with the customer
Step 2: Identify the performance obligations in the contract
Step 3: Determine the transaction price
Step 4: Allocate the transaction price to the performance obligations in the contract
Step 5: Recognize revenue when the Company satisfies a performance obligation
The Company’s primary product in 2023 was the VIVO System. The VIVO System offers 3D cardiac mapping to help with localizing the sites of origin of idiopathic ventricular arrhythmias in patients with structurally normal hearts prior to electrophysiology studies. In addition to the VIVO System, customers are provided with VIVO Positioning Patch Sets, which are custom patches, that are used in conjunction with the VIVO System to complete the intended output of the VIVO System. The delivery of the VIVO System, including the VIVO Positioning Patch Sets represents the Company’s primary performance obligation. The Company recognizes revenue upon the delivery of the VIVO system. The Company also provides customers with the option to pay for software upgrades in advance at the time of the contract's inception. Software upgrades are stand-ready services, whereby the Company will provide software upgrade services to the customer when and as upgrades are available. Terms of the period covered by the payment of software upgrades in advance can range from one year to multiple years. Customers have the option to renew terms covered by software upgrades at the end of each term. The stand-ready software upgrades represent the Company's second separate performance obligation and revenue is recognized over the term of the period.
The Company invoices the customers after physical possession and control of the VIVO System is transferred to the customer and recognizes revenue upon delivery. The timing of payment for the corresponding invoices is dependent upon the credit terms identified in each contract. The Company invoices customers who pay for software upgrades in advance in conjunction with the invoice for the delivery of the VIVO System, and subsequent renewals of software upgrades are invoiced at the inception of the term. Revenue for these stand-ready services is recognized evenly over the term of the upgrade period, consistently with similar stand-ready services under ASC 606. Similar to the delivery of the VIVO System, the timing of payment for the corresponding invoices is dependent upon the credit terms identified in each contract. The Company has elected the practical expedient to expense costs to obtain a contract, as incurred, as opposed to recognizing the cost as an asset upon occurrence.
Disaggregation of Revenue
The following table summarizes disaggregated product sales by geographic area ($ in thousands):
|
| Year Ended December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Product Sales |
|
|
|
|
|
| ||
US |
| $ |
|
| $ |
| ||
Europe |
|
|
|
|
|
| ||
|
| $ |
|
| $ |
| ||
| F-18 |
| Table of Contents |
Shipping and Handling Costs
Shipping and handling costs charged to customers are included in net product sales, while all other shipping and handling costs are included in selling, general and administrative expenses in the accompanying consolidated statements of operations.
Advertising and Marketing
Advertising costs are expensed as incurred and included in selling, general and administrative expenses. Advertising costs were $
Patents
The Company expenses patent costs, including related legal costs, as incurred and records such costs as selling, general and administrative expenses in the accompanying consolidated statements of operations.
Research and Development
Major components of research and development costs include personnel expenses, consulting, supplies and clinical trial expenses. Research and development expenses are charged to operations in the period incurred.
Stock-Based Compensation
The Company records stock-based compensation expense associated with stock options, restricted stock awards (“RSAs”) and restricted stock units (“RSUs”) issued to employees, members of the Company’s board of directors and consultants in accordance with the authoritative guidance for stock-based compensation. The Company evaluates whether an award should be classified and accounted for as a liability award or equity award for all stock-based compensation awards granted. The cost of an award of an equity instrument is measured at the grant date, based on the estimated fair value of the award using the Black-Scholes option pricing valuation model (“Black-Scholes model”) which incorporates various assumptions including expected term, volatility and risk-free interest rate, and is recognized as expense on a straight-line basis over the requisite service period of the award, which is generally the vesting period of the respective award. Share-based compensation for an award with a performance condition is recognized when the achievement of such performance condition is determined to be probable. If the outcome of such performance condition is not determined to be probable or is not met, no compensation expense is recognized, and any previously recognized compensation expense is reversed. Forfeitures are recognized as a reduction of stock-based compensation expense as they occur.
As a result of the Merger, all unvested Old Catheter stock options were subject to accelerated vesting and therefore became fully vested, as of the closing date of the business combination. The Company recognized the fair value of the replacement options as included in consideration transferred to the extent they do not exceed the fair value of the equivalent Old Catheter options. Any incremental fair value was recognized in compensation expense in the post-combination period, with this recognized as a Day 1 expense due to the Old Catheter options becoming fully vested concurrent with the closing of the business combination.
Income Taxes
The Company accounts for income taxes using the asset and liability method. Under this method, deferred tax assets and liabilities are determined based on differences between the financial reporting and tax basis of assets and liabilities and are measured using enacted tax rates and laws that are expected to be in effect when the differences reverse. Any resulting net deferred tax assets are evaluated for recoverability and, accordingly, a valuation allowance is provided when it is more likely than not that all or some portion of the deferred tax asset will not be realized.
The Company accounts for uncertainty in income taxes using a two-step approach to recognizing and measuring uncertain tax positions. The first step is to evaluate the tax position for recognition by determining whether it is more likely than not that the position will be sustained on audit, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit as the largest amount that is more than 50% likely of being realized upon ultimate settlement. An uncertain tax position is considered effectively settled on completion of an examination by a taxing authority if certain other conditions are satisfied. Should the Company incur interest and penalties relating to tax uncertainties, such amounts would be classified as a component of interest expense and other expense, respectively.
| F-19 |
| Table of Contents |
Basic and Diluted Net Loss per Share of Common Stock
The Company calculates basic net loss per share by dividing net loss by the weighted average number of common shares outstanding during the reporting period. A net loss cannot be diluted so when the Company is in a net loss position, basic and diluted loss per common share are the same. If in the future the Company achieves profitability, the denominator of a diluted earnings per common share calculation will include both the weighted average number of shares outstanding and the number of common stock equivalents, if the inclusion of such common stock equivalents would be dilutive. Anti-dilutive common stock equivalents excluded from the computation of diluted net loss per share include warrants, stock options, non-vested restricted stock awards, restricted stock units, Series A Convertible Preferred Stock, and Series X Convertible Preferred (see Note 12, Net loss per Share).
Net loss attributable to common stockholders consists of net income or loss, as adjusted for actual and deemed dividends declared. The Company recorded a deemed dividend for the modification of existing warrants and issuance of new warrants during the year ended December 31, 2023 of $
Recently Announced Accounting Pronouncements
In June 2022, the FASB issued ASU No. 2022-03, Fair Value Measurement (Topic 820): Fair Value Measurement of Equity Securities Subject to Contractual Sale Restrictions (“ASU 2022-03”) which clarifies guidance for fair value measurement of an equity security subject to a contractual sale restriction and establishes new disclosure requirements for such equity securities. ASU 2022-03 is effective for fiscal years beginning after December 15, 2023 and for interim periods within those fiscal years, with early adoption permitted. The Company is currently evaluating the impact of ASU 2022-03 on its consolidated financial statements.
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures, which requires public entities to disclose consistent categories and greater disaggregation of information in the rate reconciliation and for income taxes paid. It also includes certain other amendments to improve the effectiveness of income tax disclosures. The guidance is effective for financial statements issued for annual periods beginning after December 15, 2024, with early adoption permitted. The Company is required to adopt this standard prospectively in fiscal year 2025 for the annual reporting period ending December 31, 2025. The accounting pronouncement is not expected to have a material impact on the Company's related disclosures.
Note 3. Business Combination
On January 9, 2023, the Company completed the acquisition of Old Catheter for the purpose of acquiring Old Catheter’s existing and developing product lines based on unique electrophysiology technology.
Pursuant to the Merger Agreement, all Old Catheter common stock shares issued and outstanding and convertible promissory notes, representing an aggregate principal of $
The total purchase consideration for the Merger was $
The fair value of the Series X Convertible Preferred Stock includes certain discounts applied to the closing stock price of the Company, on January 9, 2023, of $
| F-20 |
| Table of Contents |
The following table summarizes the fair value of the consideration associated with the Merger ($ in thousands):
Description |
| Fair Value as of January 9, 2023 |
| |
Fair value of 14,649.592 Series X convertible preferred stock issued |
| $ |
| |
Fair value of Old Catheter’s fully vested stock options |
|
|
| |
Total Purchase Price |
| $ |
| |
The Merger is being accounted for as a business combination in accordance with Topic 805 and the Company has been determined to be the accounting acquirer. The Company allocated the purchase price to the assets acquired and liabilities assumed at fair value. The preliminary purchase price allocation reflects various preliminary fair value estimates and analyses, including certain tangible assets acquired and liabilities assumed, the valuation of intangible assets acquired, liabilities assumed, and goodwill, which were subject to change within the measurement period as preliminary valuations were being finalized (generally one year from the acquisition date). Measurement period adjustments were recorded in the reporting period in which the estimates are finalized, and adjustment amounts were determined. During the three months ended June 30, 2023, the Company recorded measurement period adjustments based on changes to certain estimates and assumptions and their related impact to the purchase price allocation. Developed technology was revised from $
The following table summarizes the final purchase price allocations relating to the Merger ($ in thousands):
Description |
| Fair Value |
| |
Assets acquired: |
|
|
| |
Cash and cash equivalents |
| $ |
| |
Accounts receivable |
|
|
| |
Inventories |
|
|
| |
Prepaid expenses and other current assets |
|
|
| |
Property and equipment, net |
|
|
| |
Lease right-of-use assets |
|
|
| |
Other assets |
|
|
| |
Developed technology |
|
|
| |
Customer relationships |
|
|
| |
Trademarks |
|
|
| |
Goodwill |
|
|
| |
Total assets acquired |
| $ |
| |
|
|
|
|
|
Liabilities assumed: |
|
|
|
|
Accounts payable |
| $ |
| |
Accrued expenses |
|
|
| |
Lease liability |
|
|
| |
Interest payable |
|
|
| |
Convertible promissory notes |
|
|
| |
Royalties payable |
|
|
| |
Total liabilities assumed |
|
|
| |
Total purchase price |
| $ |
| |
| F-21 |
| Table of Contents |
All intangible assets acquired are subject to amortization and their associated estimated acquisition date fair values and estimated useful lives are as follows:
Intangible Assets |
| Estimated Fair Value |
|
| Estimated Useful Life |
| ||
Developed technology- VIVO |
| $ |
|
|
|
| ||
Developed technology- LockeT |
|
|
|
|
|
| ||
Customer relationships |
|
|
|
|
|
| ||
Trademark- VIVO |
|
|
|
|
|
| ||
Trademark- LockeT |
|
|
|
|
|
| ||
|
| $ |
|
|
|
|
| |
Notwithstanding the above, as described in Note 7, management determined that there were indicators of asset impairment during the year ended December 31, 2023, and assessed the carrying values of the Company’s intangible assets and goodwill. As a result, the Company recorded an impairment charge relating to goodwill of $
Transaction costs incurred in connection with this business combination amounted to approximately $
Pro Forma Financial Information
The following table represents the revenue, net loss and net loss per share effect of the acquired company, as reported on a pro forma basis as if the acquisition occurred on January 1, 2022. These pro forma results are not necessarily indicative of the results that would have occurred if the acquisition had occurred on the first day of the period presented, nor does the pro forma financial information purport to represent the results of operations for future periods. The following information for the years ended December 31, 2023 and 2022 is presented in thousands except for the per share data ($ in thousands, except per share data):
|
| For the Years Ended December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Revenues |
| $ |
|
| $ |
| ||
Net loss |
| $ | ( | ) |
| $ | ( | ) |
Net loss attributable to common stockholders |
| $ | ( | ) |
| $ | ( | ) |
Basic and diluted net loss per share – on a pro forma basis |
| $ | ( | ) |
| $ | ( | ) |
Note 4. Inventories
Inventories consisted of the following ($ in thousands):
|
| December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Raw materials |
| $ |
|
| $ |
| ||
Finished goods |
|
|
|
|
|
| ||
Inventories |
| $ |
|
| $ |
| ||
There were no inventory obsolescence charges for the year ended December 31, 2023. Due to the Company's RIF and decision to discontinue enrollment of patients in its DABRA clinical trial, the Company suspended manufacturing activities of DABRA products in June 2022 and disposed of substantially all DABRA related inventories in July 2022, resulting in a write-down of $1.0 million in its inventories to net realizable value. Such expense is included in restructuring and impairment charges in the consolidated statements of operations for the year ended December 31, 2022.
| F-22 |
| Table of Contents |
Note 5. Property and Equipment
Property and equipment, net consisted of the following ($ in thousands):
|
| December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Machinery and equipment |
| $ |
|
| $ |
| ||
Computer hardware and software |
|
|
|
|
|
| ||
VIVO DEMO/Clinical Systems |
|
|
|
|
|
| ||
Property and equipment, gross |
|
|
|
|
|
| ||
Accumulated depreciation |
|
| ( | ) |
|
|
| |
Property and equipment, net |
| $ |
|
| $ |
| ||
Depreciation expense was $
Due to the Company’s decision to discontinue enrollment of patients in its DABRA clinical trial and the RIF, the Company suspended manufacturing activities of DABRA products in June 2022. The Company’s property and equipment was determined to be impaired as of June 30, 2022, resulting in an impairment charge of $
Note 6. Intangible Assets
The following table summarizes the Company’s intangible assets as of December 31, 2023 ($ in thousands):
|
| Estimated Useful Life in Years |
|
| Gross Carrying Amount at January 9, 2023 |
|
| Accumulated Amortization |
|
| Net Book Value at December 31, 2023 |
| ||||
Developed technology ‐ VIVO |
|
|
|
| $ |
|
| $ | ( | ) |
| $ |
| |||
Developed technology ‐ LockeT |
|
|
|
|
|
|
|
| ( | ) |
|
|
| |||
Customer relationships |
|
|
|
|
|
|
|
| ( | ) |
|
|
| |||
Trademarks/trade names ‐ VIVO |
|
|
|
|
|
|
|
| ( | ) |
|
|
| |||
Trademarks/trade names ‐ LockeT |
|
|
|
|
|
|
|
| ( | ) |
|
|
| |||
|
|
|
|
|
| $ |
|
| $ | ( | ) |
| $ |
| ||
As of December 31, 2022 the Company did not have any intangible assets.
The estimated future amortization expense for the next five years and thereafter is as follows ($ in thousands):
Years ending December 31, |
| Future Amortization Expense |
| |
2024 |
| $ |
| |
2025 |
|
|
| |
2026 |
|
|
| |
2027 |
|
|
| |
2028 |
|
|
| |
Thereafter |
|
|
| |
Total |
| $ |
| |
| F-23 |
| Table of Contents |
The Company uses the straight-line method to determine the amortization expense for its definite lived intangible assets. Amortization expense, included within selling, general and administrative expenses, relating to the purchased intangible assets was $
The weighted average remaining amortization period for the Company’s intangible assets as of December 31, 2023, is
Note 7. Goodwill
In connection with the Merger, the excess of the purchase price over the estimated fair value of the net assets assumed of $
The Company tests Goodwill for impairment at the reporting unit level annually in the fourth quarter or more frequently if a change in circumstances or the occurrence of events indicates that potential impairment exists. Due to a sustained decrease in the Company’s share price during the quarters ended March 31, 2023 and June 30, 2023, the Company concluded that, in accordance with ASC 350, a triggering event occurred indicating that potential impairment exists and required the Company to assess if impairment exists as of March 31, 2023 and June 30, 2023. In accordance with ASC 350, the Company performed a quantitative goodwill impairment test, which resulted in the carrying amount of the reporting unit exceeding the estimated fair value of the reporting unit, indicating that the goodwill of the reporting unit was impaired. The Company utilized a combination of an income and market approach to assess the fair value of the reporting unit. The income approach considered the discounted cash flow model, considering projected future cash flows (including timing and profitability), discount rate reflecting the risk inherent in future cash flows, perpetual growth rate, and projected future economic and market conditions. The guideline public company market approach considered marketplace earnings multiples from within a peer public company group. As of December 31, 2023, cumulative goodwill impairment charges of $
The following is a roll forward of goodwill as of December 31, 2023 ($ in thousands):
Balance at beginning of year |
| $ |
| |
Goodwill recognized in connection with the Merger (Note 3) |
|
|
| |
Impairment charge |
|
| ( | ) |
Balance at end of year |
| $ |
|
Note 8. Accrued Expenses
Accrued expenses consisted of the following ($ in thousands):
|
| December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Legal expenses |
| $ |
|
| $ |
| ||
DOJ settlement |
|
|
|
|
|
| ||
Offering costs |
|
|
|
|
|
| ||
Compensation and related benefits |
|
|
|
|
|
| ||
Warranty expenses |
|
|
|
|
|
| ||
Other accrued expenses |
|
|
|
|
|
| ||
Accrued expenses |
| $ |
|
| $ |
| ||
| F-24 |
| Table of Contents |
Activity in the product warranty accrual is included in accrued expenses in the consolidated balance sheets and consisted of the following ($ in thousands):
|
| Year Ended December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Balance at beginning of year |
| $ |
|
| $ |
| ||
Claims satisfied |
|
|
|
|
| ( | ) | |
Removal of accrued warranty |
|
| ( | ) |
|
|
| |
Balance at end of year |
| $ |
|
| $ |
| ||
The warranty relates to the voluntary recall of DABRA catheters, which was initiated in September 2019. The recall was closed by the FDA in July 2023 and no claims have been submitted in approximately 2 years. As such, the Company derecognized the warranty liability as of December 31, 2023.
Note 9. Notes Payable
The Company purchased director and officer liability insurance coverage on October 16, 2023 for $
Note 10. Royalties Payable
LockeT Royalty
On January 9, 2023 Old Catheter entered into an agreement with the Noteholders to forgive all accrued interest and future interest expense in exchange for a future royalty right. Under these agreements, the Company is obligated to pay the Noteholders a total royalty equal to approximately
An additional royalty will be paid to the
AMIGO System Royalty
During 2006 and 2007, Old Catheter entered into two investment grant agreements with a non-profit foundation for the purpose of funding the initial development of Old Catheter's AMIGO System, receiving a total of $
The agreement calls for the payment of the following sales-based royalties, by Old Catheter, to the foundation, upon successful commercialization of the AMIGO System:
Royalty Percentage |
|
| Until Royalty Payment Reaches a Total of |
| ||
|
| $ |
| |||
|
| $ |
| |||
|
|
| ||||
| F-25 |
| Table of Contents |
The Company is not actively marketing and selling the AMIGO System. There was no royalty expense recorded for the years ended December 31, 2023 and 2022 in relation to the AMIGO System. The AMIGO System royalty has been earned and payment has been deferred to a future date.
The table below represents the change in fair value of level 3 royalties payable for the year ended December 31, 2023. See Note 2, Summary of Significant Accounting Policies, for valuation techniques.
Balance at beginning of year |
| $ |
| |
AMIGO royalty payable recognized in connection with the Merger |
|
|
| |
LockeT royalty payable recognized in connection with the Merger |
|
|
| |
Change in fair value of royalties payable |
|
| ( | ) |
Balance at end of year |
| $ |
|
Note 11. Leases
For the years ended December 31, 2023 and 2022, operating lease expense was $
The Company's lease agreements generally do not provide an implicit borrowing rate. Therefore, the Company used a benchmark approach to derive an appropriate imputed discount rate. The Company benchmarked itself against other companies with similar credit ratings and of comparable quality and derived an imputed rate, which was used in a portfolio approach to discount its real estate lease liabilities. Management used an estimated incremental borrowing rate as detailed below for each lease.
Lease Terms and Discount Rate
The table below presents certain information related to the weighted average remaining lease term and the weighted average discount rate for the Company’s operating leases, as of December 31, 2023:
Weighted average remaining lease term (in years) - operating leases |
|
|
| |
Weighted average discount rate - operating leases |
|
| % |
California Operating Lease
The Company had an operating lease for office and manufacturing space which required it to pay base rent and certain utilities.
On October 24, 2022, the Company entered into a lease termination agreement (the “Lease Termination Agreement”) with the landlord, pursuant to which it terminated the lease agreement for its office and manufacturing space in Carlsbad, California, effective October 28, 2022. In accordance with the terms of the Lease Termination Agreement, the Company agreed to (i) release its right to the security deposit of approximately $
| F-26 |
| Table of Contents |
South Carolina Office Lease Agreement
On September 27, 2022, Old Catheter entered into a lease agreement for office space located in Fort Mill, South Carolina. The space is used for office and general use. The term of the lease began on October 1, 2022, is 38 months, and includes two months of free rental from the commencement date of the lease. The lease contains two separate
New Jersey Office Lease Agreement
On December 7, 2022, Old Catheter entered into a lease agreement for office space located in Augusta, New Jersey. The space is used for office and general use. The term of the lease is
Park City Office Lease Agreement
On March 19, 2023, the Company entered into a lease agreement for office space located in Park City, Utah. The space is used for office and general use. The term of the lease is for
Future lease payments for all lease obligations for the following five fiscal years and thereafter are as follows ($ in thousands):
Years ending December 31: |
| Operating Lease |
| |
2024 |
| $ |
| |
2025 |
|
|
| |
2026 |
|
|
| |
Total minimum lease payments |
|
|
| |
Less effects of discounting |
|
| ( | ) |
Present value of future minimum lease payments |
| $ |
| |
| F-27 |
| Table of Contents |
Lease right-of-use lease assets and lease liabilities for the Company's operating leases were recorded in the consolidated balance sheets as follows ($ in thousands):
|
| December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Assets |
|
|
|
|
|
| ||
Lease right-of-use assets |
| $ |
|
| $ |
| ||
Total lease assets |
| $ |
|
| $ |
| ||
|
|
|
|
|
|
|
|
|
Liabilities |
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
Lease liabilities - current portion |
| $ |
|
| $ |
| ||
Non-current liabilities: |
|
|
|
|
|
|
|
|
Lease liabilities - net of current portion |
|
|
|
|
|
| ||
Total lease liabilities |
| $ |
|
| $ |
| ||
Note 12. Net Loss per Share
The Company’s outstanding warrants to purchase common stock have participation rights to any dividends that may be declared in the future and are therefore considered to be participating securities. Participating securities have the effect of diluting both basic and diluted earnings per share during periods of income. During periods of loss, no loss is allocated to the participating securities since the holders have no contractual obligation to share in the losses of the Company.
Anti-dilutive common share equivalents excluded from the computation of diluted net loss per share at December 31, 2023 consisted of Series A convertible preferred stock of
Anti-dilutive common share equivalents excluded from the computation of diluted net loss per share at December 31, 2022 consisted of warrants of
Net loss attributable to common stockholders consists of net loss, as adjusted for deemed dividends. The Company recorded a deemed dividend for the modification of existing warrants and issuance of the Series E warrants (see Note 13, Equity Offerings) of $
Note 13. Equity Offerings
Public Offering
On February 8, 2022, the Company completed an offering (the "2022 Offering") in which it issued and sold (i)
| F-28 |
| Table of Contents |
The Series A warrants and Series B warrants were valued at approximately $4.2 million and $7.4 million, respectively, for a total of $
|
| Series A |
|
| Series B |
| ||
Risk-free interest rate |
|
| % |
|
| % | ||
Volatility |
|
| % |
|
| % | ||
Expected dividend yield |
|
| % |
|
| % | ||
Expected life (in years) |
|
|
|
|
|
| ||
Pursuant to the exercise of the Overallotment Option in February 2022, the Company issued
Net proceeds received from the 2022 Offering were approximately $
The Company entered into an agreement with a former placement agent that, subject to satisfaction of the requirements contained therein, called for a cash tail fee payable based on capital raised from certain investors for a definitive time following the expiration of the agreement. The accrued cash tail fee of approximately $
2022 Warrant Repricing
On July 22, 2022, the Company reduced the exercise price of all outstanding warrants, consisting of Series A warrants and Series B warrants, that were issued in the public offering on February 8, 2022 (the "2022 Offering") from $25.00 per share to $
The 2022 Warrant Repricing resulted in an immediate and incremental increase of approximately $
| F-29 |
| Table of Contents |
The Series A warrants and Series B warrants were valued on the date of the 2022 Warrant Repricing using the Black-Scholes model based on the following assumptions:
|
| Series A |
|
| Series B |
| ||
Risk-free interest rate |
|
| % |
|
| % | ||
Volatility |
|
| % |
|
| % | ||
Expected dividend yield |
|
| % |
|
| % | ||
Expected life (in years) |
|
|
|
|
|
| ||
The Series C warrants were valued on the date of the 2022 Warrant Repricing at approximately $
Risk-free interest rate |
|
| % | |
Volatility |
|
| % | |
Expected dividend yield |
|
| % | |
Expected life (in years) |
|
|
|
The Company entered into an agreement with a former placement agent that, subject to satisfaction of the requirements contained therein, called for a cash tail fee payable based on capital raised from certain investors for a definitive time following the expiration of the agreement. The accrued cash tail fee of approximately $
At-The-Market Sales Agreement
On September 2, 2022, the Company entered into the At-The-Market Sales Agreement (the “ATM Agreement”) under which the Company could sell its common stock from time to time having an aggregate offering price of up to $
Warrant Inducement Offer
On January 9, 2023, the Company reduced the exercise price of certain existing warrants (the "Existing Warrants"), exercisable for
| F-30 |
| Table of Contents |
As a result of the 2023 Warrant Repricing and Inducement Offer, the Company presents a deemed dividend for the modification of Existing Warrants and issuance of the Series E Warrants of $
The warrants, other than the Series E Warrants which are presented in a separate table below, were valued on the date of the 2023 Warrant Repricing using the Black-Scholes model based on the following assumptions:
|
| 5/22/2020 Raise |
|
| 8/3/20 Raise |
|
| Series B |
|
| Series C |
| ||||
Risk-free interest rate |
|
| % |
|
| % |
|
| % |
|
| % | ||||
Volatility |
|
| % |
|
| % |
|
| % |
|
| % | ||||
Expected dividend yield |
|
| % |
|
| % |
|
| % |
|
| % | ||||
Expected life (in years) |
|
|
|
|
|
|
|
|
|
|
|
| ||||
The Series E warrants were also valued on the date of the 2023 Warrant Repricing at approximately $
Risk-free interest rate |
|
| % | |
Volatility |
|
| % | |
Expected dividend yield |
|
| % | |
Expected life (in years) |
|
|
|
Private Placement
On January 9, 2023, the Company entered into a Securities Purchase Agreement (“Securities Purchase Agreement”) for a private placement (“Private Placement”), with the Investor. Pursuant to the Securities Purchase Agreement, the Investor agreed to purchase, for an aggregate purchase price of approximately $8.0 million, (a) Class A units at a price that was the lower of $
The PIPE Warrants, including Series F warrants and Series G warrants, are exercisable at an exercise price of $3.00 per share, subject to adjustments as provided under the terms of the PIPE Warrants. The PIPE Warrants are exercisable at any time on or after the closing date of the Private Placement until the expiration thereof, except that the PIPE Warrants cannot be exercised if, after giving effect thereto, the purchaser would beneficially own more than 4.99%, or the Maximum Percentage, of the outstanding shares of common stock of the Company, which Maximum Percentage may be increased or decreased by the purchaser with written notice to the Company to any other percentage specified not in excess of 9.99%. The Series F Warrants have a term of two years from the date of stockholder approval, and the Series G Warrants have a term of six years from the date of stockholder approval. The Series F Warrants and Series G Warrants were approved at the Stockholders’ Meeting.
| F-31 |
| Table of Contents |
The Series F warrants and Series G warrants were valued, in aggregate, at approximately $
|
| Series F |
|
| Series G |
| ||
Risk-free interest rate |
|
| % |
|
| % | ||
Volatility |
|
| % |
|
| % | ||
Expected dividend yield |
|
| % |
|
| % | ||
Expected life (in years) |
|
|
|
|
|
| ||
The proceeds from the Securities Purchase Agreement were allocated to the equity instruments issued based on their relative fair values and recorded in additional paid-in capital.
Shares of PIPE Preferred Stock, the conversion of which was approved at the Stockholders’ Meeting, convert into common stock at the option of the holder at the Preferred Conversion Rate, subject to certain ownership limitations as described below. The conversion price is subject to adjustment in the case of stock splits, stock dividends, combinations of shares and similar recapitalization transactions.
Subject to limited exceptions, holders of shares of PIPE Preferred Stock will not have the right to convert any portion of their Preferred Stock if the holder, together with its affiliates, would beneficially own in excess of 9.99% of the number of shares of the Company’s common stock outstanding immediately after giving effect to its conversion.
Holders of PIPE Preferred Stock will be entitled to receive dividends on shares of PIPE Preferred Stock equal, on an as-if-converted-to-common stock basis, and in the same form as dividends actually paid on shares of the common stock. Except as otherwise required by law, the PIPE Preferred Stock does not have voting rights.
The Company also entered into a registration rights agreement with the purchasers requiring the Company to register the resale of the shares of common stock, the shares issuable upon exercise of the Warrants and the shares issuable upon the conversion of the PIPE Preferred Stock.
Conversion of Preferred Stock Issued in Private Placement
On July 5, 2023 the Company issued
On July 24, 2023, the Company issued 546,776 shares of its common stock in connection with the conversion of
See Note 20, Subsequent Events, for additional conversion.
| F-32 |
| Table of Contents |
Warrants
The following table presents the number of common stock warrants outstanding:
Warrants outstanding, December 31, 2021 |
|
|
| |
Issued |
|
|
| |
Exercised |
|
| ( | ) |
Expired |
|
|
| |
Warrants outstanding, December 31, 2022 |
|
|
| |
Issued |
|
|
| |
Exercised |
|
| ( | ) |
Expired |
|
| ( | ) |
Warrants outstanding, December 31, 2023 |
|
|
|
The following table presents the number and type of common stock warrants outstanding, their exercise price, and expiration dates as of December 31, 2023:
Warrant Type |
| Warrants Outstanding |
|
| Exercise Price |
|
| Expiration Date | |||
May 2020 Warrants |
|
|
|
| $ |
|
| ||||
May 2020 Placement Agent Warrants |
|
|
|
| $ |
|
| ||||
August 2020 Warrants |
|
|
|
| $ |
|
| ||||
August 2020 Placement Agent Warrants |
|
|
|
| $ |
|
| ||||
August 2021 Pharos Banker Warrants |
|
|
|
| $ |
|
| ||||
February 2022 Series B Warrants |
|
|
|
| $ |
|
| ||||
July 2022 Series C Warrants |
|
|
|
| $ |
|
| ||||
January 2023 Series E Warrants |
|
|
|
| $ |
|
| ||||
March 2023 Series F Warrants |
|
|
|
| $ |
|
| ||||
March 2023 Series G Warrants |
|
|
|
| $ |
|
| ||||
|
|
|
|
|
|
|
|
|
| ||
As of December 31, 2023, the warrants issued by the Company had a weighted average exercise price of $
Note 14. Preferred Stock
Series X Convertible Preferred Stock
As described in Note 3, above, pursuant to the Merger Agreement, all Old Catheter common stock shares issued and outstanding and convertible promissory notes, representing an aggregate principal of $
Series X Convertible Preferred Stock has no voting rights prior to the conversion into common stock. While there are generally no voting rights of the Series X Convertible Preferred Stock, there are protective rights regarding the sales of the company, change of control, etc. No currently outstanding share of Series X Preferred may convert into common stock until on or after July 9, 2024, and then, only if the Company’s common stock has been delisted from the NYSE American or has been approved for initial listing on the NYSE American or another stock exchange, at a rate of
| F-33 |
| Table of Contents |
Upon consummation of the merger, each holder of Old Catheter convertible promissory notes received, in exchange for discharge of the principal of his or its Notes, a number of shares of the
On March 21, 2023, the Company held the Stockholders’ Meeting, at which the stockholders approved, among other things, the issuance of
Series A Convertible Preferred Stock
As described in Note 13, on January 9, 2023, the Company entered into a Securities Purchase Agreement for a Private Placement, with the Investor. Pursuant to the Securities Purchase Agreement, shares of Series A Convertible Preferred Stock were issued, the conversion of which was approved at the Stockholders’ Meeting. The Series A Convertible Preferred Stock converts into common stock at the option of the holder at the Preferred Conversion Rate, subject to certain ownership limitations as described below. The conversion price is subject to adjustment in the case of stock splits, stock dividends, combinations of shares and similar recapitalization transactions.
Subject to limited exceptions, holders of shares of
Holders of Series A Convertible Preferred Stock will be entitled to receive dividends on shares of Series A Convertible Preferred Stock equal, on an as-if-converted-to-common stock basis, and in the same form as dividends actually paid on shares of the common stock. Except as otherwise required by law, the Series A Convertible Preferred Stock does not have voting rights.
The Company also entered into a registration rights agreement with the purchasers requiring the Company to register the shares of common stock, issuable upon the conversion of the Series A Convertible Preferred Stock. The shares have been registered for resale on an effective registration statement on Form S-1.
Note 15. Stock-Based Compensation
2018 Equity Incentive Plan
In September 2018, the Company’s board of directors adopted, and the Company’s stockholders approved, the 2018 Equity Incentive Plan (the “2018 Plan”) which provided for the grant of incentive stock options, non-statutory stock options, restricted stock awards, restricted stock units, performance-based stock awards and other forms of equity compensation to the Company’s employees, directors and consultants. Stock options granted under the 2018 Plan generally vest one-fourth on the first anniversary of the vesting commencement date with the balance vesting monthly over the remaining three years. Restricted stock units granted under the 2018 Plan generally vest one third on the first anniversary of the vesting commencement date and one sixth every six months thereafter such that the award will be fully vested on the third anniversary of the vesting commencement date. As of December 31, 2023 and December 31, 2022,
| F-34 |
| Table of Contents |
2020 Inducement Equity Incentive Plan
In March 2020, the Company adopted the 2020 Inducement Equity Incentive Plan (the “2020 Plan”) for the purpose of attracting, retaining and incentivizing employees in furtherance of the Company’s success. The 2020 Plan was adopted without stockholder approval pursuant to Rule 303A.08 of the New York Stock Exchange. The 2020 Plan is used to offer equity awards as material inducements for new employees to join the Company. Upon adoption of the 2020 Plan,
2023 Equity Incentive Plan
In July 2023, the Company’s stockholders approved, the 2023 Equity Incentive Plan (the “2023 Plan”) which provided for the grant of incentive stock options, non-statutory stock options, restricted stock awards, restricted stock units, performance-based stock awards and other forms of equity compensation to the Company’s employees, directors and consultants. Stock options granted under the 2023 Plan to employees and consultants generally will vest annually over a five-year period or as determined by the Board’s Compensation Committee, while grants to non-employee directors generally vest quarterly over a three-year period. As of December 31, 2023,
Stock Options Assumed in Merger (See Note 3, Business Combination)
At the closing of the Merger, each outstanding option to purchase Old Catheter common stock that had not previously been exercised prior to the closing of the Merger was assumed and converted into options to purchase
The following is a summary of stock option activity for the year ended December 31, 2023:
|
| Stock Options |
|
| Weighted Average Exercise Price |
|
| Weighted Average Remaining Life (in years) |
|
| Aggregate Intrinsic Value (in thousands) |
| ||||
Outstanding at December 31, 2022 |
|
|
|
| $ |
|
|
|
|
|
|
| ||||
Options assumed in Old Catheter Merger |
|
|
|
| $ |
|
|
| — |
|
|
| — |
| ||
Options exercised |
|
| ( | ) |
| $ |
|
|
| — |
|
|
| — |
| |
Canceled/forfeited |
|
| ( | ) |
| $ |
|
|
| — |
|
|
| — |
| |
Outstanding at December 31, 2023 |
|
|
|
| $ |
|
|
|
|
| $ | — |
| |||
Vested and expected to vest at December 31, 2023 |
|
|
|
| $ |
|
|
|
|
| $ | — |
| |||
Exercisable at December 31, 2023 |
|
|
|
| $ |
|
|
|
|
| $ | — |
| |||
| F-35 |
| Table of Contents |
The Company did not grant any stock options during the year ended December 31, 2023.
Restricted Stock Units
The following is a summary of the restricted stock unit activity for the 2018 Plan for the year ended December 31, 2023:
|
| Restricted Stock Units |
|
| Weighted Average Grant Date Fair Value |
| ||
Outstanding at December 31, 2022 |
|
|
|
| $ |
| ||
Vested |
|
| ( | ) |
| $ |
| |
Forfeited |
|
| ( | ) |
| $ |
| |
Outstanding at December 31, 2023 |
|
|
|
| $ |
| ||
Restricted Stock Awards
A summary of the restricted stock award activity for the year ended December 31, 2023 is presented below:
|
| Restricted Stock Awards |
|
| Weighted Average Grant Date Fair Value |
| ||
Outstanding at December 31, 2022 |
|
|
|
| $ |
| ||
Vested |
|
| ( | ) |
| $ |
| |
Forfeited |
|
| ( | ) |
| $ |
| |
Outstanding at December 31, 2023 |
|
|
|
| $ |
| ||
Employee Stock Purchase Plan
In September 2018, the Company’s board of directors adopted the 2018 Employee Stock Purchase Plan (the “ESPP”) which permitted eligible employees to purchase the Company’s common stock at a discount through payroll deductions during defined offering periods. Eligible employees could elect to withhold up to
The Company paused the ESPP in May 2022. For the year ended December 31, 2022, cash received from the exercise of purchase rights under the ESPP was approximately $
As of December 31, 2023, the Company had issued
Stock-based compensation expense recorded in operating expenses was as follows ($ in thousands):
|
| For the Year Ended December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Selling, general and administrative |
| $ |
|
| $ |
| ||
Research and development |
|
|
|
|
|
| ||
Stock-based compensation in operating expenses |
| $ |
|
| $ |
| ||
Stock-based compensation of approximately $
| F-36 |
| Table of Contents |
There was no unrecognized estimated stock-based compensation expense for stock options, restricted stock awards or restricted stock units at December 31, 2023.
Note 16. Income Taxes
A reconciliation of the differences between the U.S. statutory federal income tax rate and the effective tax rate as provided in the consolidated statements of operations is as follows:
|
| For the Year Ended December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Tax computed at the federal statutory rate |
|
| % |
|
| % | ||
Section 382 NOL limitation |
|
| — |
|
|
| ( | )% |
Nondeductible expenses |
|
| ( | )% |
|
| ( | )% |
State income taxes, net of federal benefits |
|
| ( | )% |
|
| % | |
Stock-based compensation |
|
| ( | )% |
|
| — |
|
Other |
|
| — |
|
|
| % | |
Change in valuation allowance |
|
| ( | )% |
|
| % | |
Purchase accounting |
|
| % |
|
| — |
| |
Goodwill impairment |
|
| ( | )% |
|
| — |
|
Royalty mark to market |
|
| % |
|
| — |
| |
|
|
| — |
|
|
| — |
|
The federal and state income tax provision is summarized as follows (in thousands):
|
| For the Year Ended December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Current |
|
|
|
|
|
| ||
Federal |
| $ |
|
| $ |
| ||
State |
|
|
|
|
|
| ||
|
|
|
|
|
|
| ||
Deferred |
|
|
|
|
|
|
|
|
Federal |
|
|
|
|
|
| ||
State |
|
|
|
|
|
| ||
|
|
|
|
|
|
| ||
Income tax expense |
| $ |
|
| $ |
| ||
Deferred income taxes reflect the net tax effects of (a) temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for tax purposes, and (b) operating losses and tax credit carryforwards.
| F-37 |
| Table of Contents |
The tax effects of significant components of the Company’s deferred tax assets (liabilities) are as follows (in thousands):
|
| December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Deferred tax assets: |
|
|
|
|
|
| ||
Net operating loss carryforwards |
| $ |
|
| $ |
| ||
Stock-based compensation |
|
|
|
|
|
| ||
Capitalized research and development |
|
|
|
|
|
| ||
Reserves |
|
|
|
|
|
| ||
Intangible assets |
|
|
|
|
|
| ||
Accrued legal settlement |
|
|
|
|
|
| ||
Operating lease liabilities |
|
|
|
|
|
| ||
Accrued compensation |
|
|
|
|
|
| ||
Other accruals |
|
|
|
|
|
| ||
R&D credits |
|
|
|
|
|
| ||
Total gross deferred tax assets |
|
|
|
|
|
| ||
Deferred tax liabilities: |
|
|
|
|
|
|
|
|
Fixed asset basis |
|
| ( | ) |
|
|
| |
Operating lease right-of-use assets |
|
| ( | ) |
|
|
| |
Intangible assets |
|
| ( | ) |
|
|
| |
Total gross deferred tax liabilities |
|
| ( | ) |
|
|
| |
Valuation allowance |
|
| ( | ) |
|
| ( | ) |
Total deferred taxes |
| $ |
|
| $ |
| ||
At December 31, 2023, and December 31, 2022 the Company had available Federal Net Operating Loss (NOL) carryforwards of $
The valuation allowance relates to deferred tax assets for certain items that will be deductible for income tax purposes under very limited circumstances and for which the Company believes it is not more likely than not that it will realize the associated tax benefit. However, in the event that the Company determines that it would be able to realize more or less than the recorded amount of net deferred tax assets, an adjustment to the deferred tax asset valuation allowance would be recorded in the period such a determination is made. In assessing the realizability of deferred tax assets, management considers whether it is more-likely-than-not that some portion of all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Management considers the scheduled reversal of deferred tax liabilities (including the impact of available carryback and carryforward periods), projected future taxable income, and tax planning strategies in making this assessment. Based upon the levels of historical taxable income, projections of future taxable income and the reversal of deferred tax liabilities over the periods in which the deferred tax assets are deductible, management believes it is more-likely-than-not that the Company will not realize the benefits of these deductible differences, net of the existing valuation allowance. The amount of deferred tax asset considered realizable, however, could change in the near term if estimates which require significant judgment of future taxable income during the carryforward period are increased or decreased.
| F-38 |
| Table of Contents |
The Company recognizes interest and penalties relating to uncertain tax positions in income tax expense. No amounts were recorded in 2023 and 2022.
Effective January 1, 2023, repurchases of Company stock are subject to a nondeductible excise tax under the Inflation Reduction Act of 2022 equal to 1.0% of the fair market value of the shares repurchased, subject to certain limitations. There was no impact to the Company’s financial condition or results of operations in 2023 as a result of the excise tax.
The Company files income tax returns as prescribed by tax laws of the jurisdictions in which it operates. In the normal course of business, the Company is subject to examination by federal, state and local jurisdictions where applicable based on the statute of limitations that apply in each jurisdiction. The Company has no open income tax audits with any taxing authority as of December 31, 2023. The Company is still subject to income tax examinations by U.S. federal and state tax authorities for the years 2018 through 2022. However, to the extent allowed by law, the tax authorities may have the right to examine prior periods where net operating losses were generated and carried forward, and make adjustments up to the amount of the net operating loss carryforward amount.
Note 17. Commitments and Contingencies
In the normal course of business, the Company is at times subject to pending and threatened legal actions. In management’s opinion, any potential loss resulting from the resolution of these matters will not have a material effect on the results of operations, financial position or cash flows of the Company.
As of December 31, 2023, the Company had no outstanding litigation.
Note 18. Employee Benefit Plan
In January 2019, the Company established a defined contribution plan under Section 401(k) of the Internal Revenue Code (“401(k) Plan”). Under the terms of the 401(k) Plan, all full-time employees were eligible to make voluntary contributions as a percentage or defined amount of compensation. The Company made matching contributions based on
Note 19. Related Parties
Prior to the Merger, David A. Jenkins, the Company’s current Executive Chairman of the Board and Chief Executive Officer, and Old Catheter’s then Chairman of the Board of Directors, and his affiliates held approximately $
In addition to the shares described above that were issued in connection with the Notes,
In connection with the Merger (see Note 3, Business Combination), the Company assumed $
| F-39 |
| Table of Contents |
Mr. Jenkins’ daughter, the Company’s non-executive Chief Operating Officer, received options to purchase
Following stockholder approval on March 21, 2023, the Company issued
Note 20. Subsequent Events
Issuance of Securities in Private Placement
On January 24, 2024, Catheter Precision, Inc. issued
Options Issued Under 2023 Equity Incentive Plan
On January 8, 2024, the Board approved the issuance of a total of
On February 26, 2024, the Board approved the issuance of a total of
| F-40 |