Exhibit 99.2
PROSPECTUS SUPPLEMENT SUMMARY
Overview
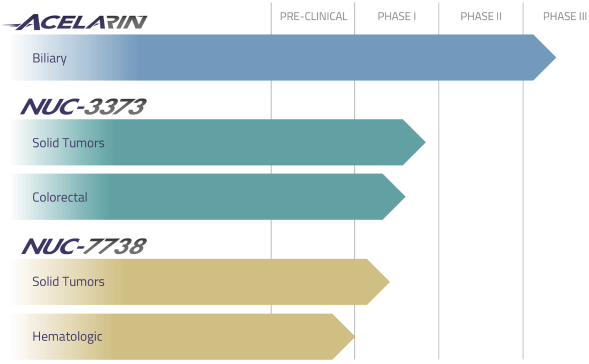
We are a clinical-stage biopharmaceutical company focused on significantly improving treatment outcomes for cancer patients by applying our ProTide™ technology to transform some of the most widely prescribed chemotherapy agents, nucleoside analogs, into more effective and safer medicines. While these conventional agents remain part of the standard of care for the treatment of many solid and hematological tumors, their efficacy is limited by cancer cell resistance mechanisms and they are often poorly tolerated. Utilizing our proprietary technology, we are developing new medicines, ProTides, designed to overcome key cancer resistance mechanisms and generate much higher concentrations of anti-cancer metabolites in cancer cells. Our most advanced ProTide candidates, Acelarin® and NUC-3373, are new chemical entities derived from the nucleoside analogs gemcitabine and 5-fluorouracil, respectively, two widely used chemotherapy agents. Acelarin is currently being evaluated in multiple clinical trials, including a Phase 3 clinical trial for patients with biliary tract cancer, a Phase 1b clinical trial for patients with biliary tract cancer, a Phase 2 clinical trial for patients with platinum-resistant ovarian cancer, and a Phase 3 clinical trial for patients with metastatic pancreatic cancer for which enrollment has been suspended. NUC-3373 is currently in a Phase 1 clinical trial in patients with advanced solid tumors and a Phase 1b clinical trial in patients with advanced colorectal cancer. Our third ProTide, NUC-7738, is a transformation of a novel nucleoside analog (3’-deoxyadenosine) that has never been successfully developed or approved as a chemotherapy but has shown potent anti-cancer activity in preclinical studies. We are evaluating NUC-7738 in a Phase 1 clinical trial for patients with advanced solid tumors. We have retained worldwide rights to these lead product candidates as well as our preclinical product candidates, all of which we refer to as ProTides.
Acelarin, our most advanced product candidate, is a first-in-class ProTide that has been evaluated in over 300 patients. Acelarin is a ProTide transformation of gemcitabine that we believe could replace gemcitabine in certain cancer indications and have utility across a range of other cancers. In a Phase 1 dose-ranging trial in 49 evaluable patients with advanced metastatic solid tumors, Acelarin was well tolerated, achieved a 78% disease control rate and was associated with intracellular levels of active anti-cancer metabolite over 200 times higher than those reported for gemcitabine. A subset of 14 evaluable patients with relapsed/refractory gynecological cancers achieved a 93% disease control rate. In a Phase 1b dose-ranging trial in 23 evaluable patients with recurrent ovarian cancer, Acelarin was combined with carboplatin and achieved a 96% disease control rate. Based on these disease control rates and the tolerability profile observed, a Phase 1b trial of Acelarin is being conducted in patients with locally advanced or metastatic biliary tract cancers to determine the optimal dose in combination with cisplatin. In October 2018, at the European Society for Medical Oncology (ESMO) 2018 Congress, we announced combined results from cohorts 1 and 2 of this trial, also known as the ABC-08 trial, in which Acelarin in combination with cisplatin was observed to continue to achieve approximately a doubling of the response rate expected with the standard of care, gemcitabine plus cisplatin. In addition, these results showed the combination was well-tolerated and several patients achieved significant reductions in their tumor volume as well as further tumor shrinkage over time. In June 2019, the FDA granted orphan drug designation for Acelarin for the treatment of advanced biliary tract cancer. In October 2019, the FDA cleared the IND for our Phase 3 clinical trial, also known as the NuTide:121 trial, of Acelarin in combination with cisplatin for patients with previously untreated locally advanced
or metastatic biliary tract cancer. We expect to complete recruitment for the first interim analysis in the second half of 2021. We believe Acelarin in combination with cisplatin has the potential to significantly improve the survival outcomes of patients with advanced biliary tract cancer. If approved, our goal is to establish Acelarin in combination with cisplatin as the global standard of care for the first-line treatment of patients with advanced biliary tract cancer.
NUC-3373, our second product candidate, is a ProTide transformation of the active anti-cancer metabolite of 5-fluorouracil, or 5-FU, which we believe has the potential to replace 5-FU as the standard of care in the treatment of a wide range of cancers. In preclinical studies, we observed that NUC-3373 overcame the key resistance mechanisms associated with 5-FU and generated intracellular levels of the active anti-cancer metabolite over 300 times higher than that of 5-FU. NUC-3373 is currently being evaluated in a Phase 1 clinical trial, also known as the NuTide:301 trial, of patients with advanced solid tumors. In this trial, NUC-3373 has generated high levels of the active anti-cancer metabolite inside the patients’ white blood cells resulting in complete inhibition of the target enzyme associated with cancer cell growth. The pharmacokinetic profile of NUC-3373 appears favorable, which supports our belief that NUC-3373 may enhance efficacy, improve safety and provide a more convenient dosing regimen compared with the standard of care 5-FU. In October 2018, we reported further interim data from this trial at ESMO 2018. These interim data showed that three patients had achieved stable disease after treatment, with progression-free survival, or PFS, lasting more than nine months at September 25, 2018, as well as a continued promising pharmacokinetic and pharmacodynamic, tolerability and dosage administration profile. Importantly, no patients developed hand-foot syndrome, as of data cut-off, which is a debilitating side effect occurring in 34% to 72% of patients treated with fluoropyrimidine therapy. The results of this trial suggest that NUC-3373 has the potential to overcome the key cancer resistance mechanisms associated with 5-FU and may be capable of achieving anti-cancer activity even in patients who have progressed on prior treatment with a fluoropyrimidine. We expect to report further data from the NuTide:301 trial in the first half of 2021.
In October 2018, we commenced a Phase 1b trial, also known as the NuTide:302 trial, in patients with advanced colorectal cancer in which NUC-3373 will be combined with agents typically combined with 5-FU, including leucovorin, irinotecan, oxaliplatin and monoclonal antibodies. In October 2019, we presented interim data from this trial at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics. These interim data supported the previously reported favorable pharmacokinetic profile of NUC-3373. The anti-cancer mechanism of action of NUC-3373 has been previously observed in preclinical studies, which we believe further supports the biological advantages of NUC-3373 over 5-FU. We believe NUC-3373 has significant commercial potential as approximately 500,000 patients in North America are estimated to receive intravenous 5-FU each year. The next stage of NUC-3373’s development has commenced in the NuTide:302 trial which is investigating NUC-3373 in patients with advanced colorectal cancer in combination with oxaliplatin or irinotecan. We then plan to open an expansion cohort in less heavily pre-treated patients. We expect to report data from the NuTide:302 trial in the second half of 2020 and in 2021. Contingent on regulatory guidance and other factors, we also plan to initiate a Phase 3 clinical trial in patients with advanced colorectal cancer in the second half of 2021.
NUC-7738, our third product candidate, is a ProTide transformation of 3’-deoxyadenosine, or 3’-dA, a novel nucleoside analog that has shown potent anti-cancer activity in preclinical studies. In March 2019, we opened a Phase 1 clinical trial, known as the NuTide:701 trial, with NUC-7738 in patients with advanced solid tumors. In October 2019, we announced preclinical data on NUC-7738, detailing multiple potential anti-cancer modes of action. In preclinical studies of NUC-7738, we have observed additional anti-cancer mechanisms of action to those previously reported for 3’-deoxyadenosine. Significantly higher levels of anti-cancer metabolites are generated inside cancer cells than with 3’-deoxyadenosine, causing increased cell injury. Preclinical data also suggest that NUC-7738 activates AMPK, which may inhibit the mTOR pathway, highlighting another potential anti-cancer mechanism of this candidate. We expect to report interim clinical data from the Phase 1 trial in the second half of 2020 and the first half of 2021. Contingent on regulatory guidance and other factors, we also plan to initiate a Phase 2 clinical trial in the second half of 2021.
2
Despite the widespread use of nucleoside analogs, their efficacy is severely limited by cancer cell resistance mechanisms and they are often poorly tolerated. Harnessing the power of phosphoramidate chemistry, we convert nucleoside analogs into activated nucleotide analogs with the addition of a phosphate group, which is protected by specific combinations of aryl, ester and amino acid groupings. By adding and protecting this phosphate group, we design our ProTides to avoid or overcome key cancer resistance mechanisms in the uptake, activation and breakdown of nucleoside analogs. As a result, we believe our ProTides have the potential to generate hundreds of times higher concentrations of the active anti-cancer metabolites inside tumor cells, potentially making our ProTides more effective than the current standards of care. Because our ProTides resist breakdown, and are thus more stable, we believe they are also able to reduce or eliminate the generation of toxic byproducts that can result from the breakdown of nucleoside analogs like gemcitabine, 5-FU and 3’-deoxyadenosine.
Our proprietary ProTide technology was invented in the Cardiff University laboratory of our late Chief Scientific Officer, Professor Christopher McGuigan, who conceived of and filed the original composition of matter patents for our initial ProTides. The unique feature of his discovery was the specific combination of aryl, ester and amino acid groupings that protect the activated, or phosphorylated, nucleoside analog. This phosphoramidate chemistry approach is the key to the ProTide technology. Every ProTide grouping is distinct, and Professor McGuigan and his team synthesized and tested thousands of compounds in order to identify the optimal ProTide grouping for each underlying nucleoside analog.
We have licensed what we believe to be the foundational patent estate for the application of phosphoramidate chemistry in oncology. We have granted patents in key markets, including the United States, Europe and Japan, protecting the composition of matter of Acelarin, NUC-3373 and other of our product candidates. Professor McGuigan’s work preceded and helped lead to the development of several FDA-approved anti-viral drugs containing nucleotide analogs, including: sofosbuvir, or Sovaldi®, which is also a key component of Harvoni®; and tenofovir alafenamide fumarate, or TAF, which is a key component of Genvoya®, Descovy® and Odefsey®.
We are led by Hugh S. Griffith, our founder and Chief Executive Officer, who brings over 25 years of experience in the biopharmaceutical industry, including at Abbott Laboratories (now AbbVie Inc.) and Parke-Davis Warner Lambert (now Pfizer Inc.). Before founding NuCana, he led the operations of Bioenvision, Inc. from start-up through its acquisition by Genzyme Corporation. While at Bioenvision, he was instrumental in developing and commercializing clofarabine, a nucleoside analog for the treatment of pediatric leukemia.
Recent Developments
Interim Data Presentation at ESMO Virtual Congress 2020
In August 2020, we announced that we had three posters accepted for presentation at the upcoming ESMO Virtual Congress 2020 to be held September 19, 2020 to September 21, 2020.
The first poster presents six patient case studies from the ongoing Phase 1 clinical trial of NUC-3373 in heavily pre-treated patients with metastatic colorectal cancer. These interim data from the trial showed that (i) some patients achieved stable disease for a longer period of time on NUC-3373 than the patient had achieved on their prior line of therapy, and (ii) some patients experienced tumor shrinkage, including one fluoropyrimidine-refractory patient. We believe these data support the potential of NUC-3373 to improve progression-free survival in patients who had relapsed or were refractory to prior 5-FU-containing regimens. We also believe these data show that NUC-3373’s pharmacokinetic and tolerability profile is favorable and unaffected by leucovorin.
The second poster presents two patient case studies from the ongoing Phase 1 clinical trial of NUC-7738 in patients with advanced solid tumors who have exhausted all standard therapies. These interim data observed significant reductions in tumor volume maintained over time in these patients. Additionally, we observed a positive change in character of a target lesion of one of the patients in the trial. We believe these data support the potential anti-cancer activity of NUC-7738 and indicate a favorable pharmacokinetic and tolerability profile.
The third poster provides an overview of the ongoing global Phase 3 clinical trial of Acelarin as a first-line treatment for patients with advanced biliary tract cancer currently being conducted at approximately 100 clinical sites across North America, Europe and Asia Pacific.
3
The patient case studies presented in the posters for NUC-3373 and NUC-7738 are preliminary and subject to change as further analyses are conducted. In addition, both of these clinical trials are ongoing and these patient cases studies represent only a subset of the patients expected to enroll in the trials. As a result, the interim data from both trials may change as further patient follow up occurs and more patient data become available.
Potential Appointment of a New Director
Following completion of this offering, we expect to appoint an individual designated by Abingworth LLP, an expected investor in this offering, to serve as a member of our board of directors.
Our Strategy
Our goal is to transform standards of care and improve survival for patients across a wide range of cancer indications. Our strategy includes the following key components:
| • | Rapidly develop Acelarin as a first-in-class nucleotide analog for the treatment of patients with cancer. We believe that Acelarin has the potential to replace the core chemotherapy component of treatment regimens for patients with various cancers, focusing initially on: |
| • | Biliary tract cancer. We reported interim data from a Phase 1b trial of Acelarin in combination with cisplatin in January 2018 and in October 2018. Following the FDA’s clearance of our IND in October 2019, we opened a global Phase 3 trial of Acelarin in combination with cisplatin as a first-line treatment for patients with biliary tract cancer. We expect to complete recruitment for the first interim analysis in the second half of 2021. |
| • | Rapidly develop NUC-3373 to replace 5-FU as the standard of care for the treatment of patients with various cancers. |
| • | Colorectal cancer. In October 2019, we presented interim data from NuTide:302, our Phase lb trial in patients with advanced, metastatic colorectal cancer who have already received 5-FU in combination with oxaliplatin and irinotecan. In this study NUC-3373 is being assessed for safety and a recommended Phase 2 dose when combined with many of the agents typically combined with 5-FU, including leucovorin, irinotecan, oxaliplatin and monoclonal antibodies. These interim data supported the previously reported favorable pharmacokinetic profile of NUC-3373. We plan to report further interim data from the NuTide:302 trial in the second half of 2020 and the first half of 2021. Contingent on regulatory guidance and other factors, we plan to initiate a Phase 3 clinical trial of NUC-3373 in combination with other agents for patients with colorectal cancer in the second half of 2021. |
| • | Advanced solid tumors. We plan to continue our Phase 1 monotherapy trial of NUC-3373 to establish the optimal dose and dosing schedule of single-agent NUC-3373 in patients with advanced solid tumors and report interim data in the first half of 2021. |
| • | Rapidly develop NUC-7738 as a treatment for patients with solid tumors. Our Phase 1 clinical trial with NUC-7738, a ProTide based on a novel nucleoside analog, for patients with advanced solid tumors is ongoing, and we expect to report interim data from the trial in the second half of 2020 and the first half of 2021. We expect to initiate a Phase 2 clinical trial in the second half of 2021. |
| • | Leverage our proprietary ProTide technology platform to develop additional product candidates. We are pursuing both the transformation of well-established and widely used nucleoside analogs as well as novel nucleoside analogs, which we believe have the potential to address additional areas of unmet medical need in oncology. |
| • | Continue to strengthen our intellectual property position. We own or have exclusive rights to the core technologies underlying our ProTide technology platform. We have been granted patents in key markets, including the United States, Europe and Japan, protecting the composition of matter of Acelarin, NUC-3373 and other of our product candidates. We intend to further expand and enhance our intellectual property position. We also have been granted or allowed patent protection in key markets for the proposed commercial formulation of Acelarin and for uses of Acelarin in targeting |
4
| cancer. Our patent portfolio has grown substantially in the past year and we are actively evaluating new intellectual property opportunities as they arise, with the intention of further expanding our intellectual property position. |
| • | Build a focused commercial organization. We have worldwide rights to all product candidates that we are developing. We believe that many of the cancers we are initially targeting with our ProTides can be addressed by a focused sales and marketing team. We plan to commercialize any product candidates for which we receive regulatory marketing approval using a specialized sales force in the United States and Europe. |
Our Pipeline
We take a scientifically driven approach to designing ProTides, which we believe have the potential to result in highly efficacious cancer therapies with improved tolerability. Our pipeline of product candidates is summarized below.
Intellectual Property
We actively seek to protect the intellectual property and proprietary technology that we believe is important to our business, including seeking, maintaining, enforcing and defending patent rights for our therapeutics and processes, whether developed internally or licensed from third parties. Our success will depend on our ability to obtain and maintain patent and other protection including data/market exclusivity for our product candidates and platform technology, preserve the confidentiality of our know-how and operate without infringing the valid and enforceable patents and proprietary rights of third parties. See “Risk Factors—Risks Related to Our Intellectual Property” included in this prospectus.
Our policy is to seek to protect our proprietary position, generally by filing an initial priority filing at the U.K. Intellectual Property Office. This is followed by the filing of a patent application under the Patent Co-operation Treaty claiming priority from the initial application(s) and then filing applications for patent grant in territories including, for example, the United States, Europe and Japan. In each case, we determine the strategy and territories required after discussion with our patent attorneys so that we obtain relevant coverage in territories that are commercially important to us and our product candidates. We additionally rely on data exclusivity,
5
market exclusivity and patent term extensions when available. We also rely on trade secrets and know-how relating to our underlying platform technology and product candidates. Prior to making any decision on filing any patent application, we consider with our patent attorneys whether patent protection is the most sensible strategy for protecting the invention concerned or whether the invention should be maintained as confidential.
As of September 7, 2020, we owned 610 granted patents (of which 14 are U.S.-issued patents) and 396 pending patent applications (of which 22 are U.S. pending patent applications). Commercially or strategically important non-U.S. jurisdictions in which we hold issued or pending patent applications include: Australia, Canada, China, Eurasia (in the form of a regional patent), Europe (in the form of a regional patent), Hong Kong, India, Israel, Japan, South Korea, Malaysia, Mexico, Philippines, Singapore and South Africa.
Acelarin
We own 91 granted patents covering the composition of matter of our Acelarin product candidate. The patent claims are directed to the Acelarin product candidate and to a genus around that candidate. Acelarin was originally formed as a mixture of two diastereoisomers, both of which are biologically active, and each of these composition of matter patents cover Acelarin both as a single diastereoisomer and as a mixture of diastereoisomers. The composition of matter patents for Acelarin have been granted in major territories, including United States, Europe and Japan. These granted patents are expected to expire in 2024, excluding any patent term adjustments and any patent term extensions.
Additionally, we own 85 granted patents, as well as 16 pending patent applications, directed towards Acelarin in single diastereoisomer form. The more soluble single diastereoisomer is being used for clinical development in our ongoing and planned upcoming clinical trials. A patent claiming the more soluble single diastereoisomer of Acelarin has been granted in the United States and Europe, and corresponding patent applications are pending in other major territories, including Japan. These granted patents and patents arising from the pending applications, if issued, are expected to expire in 2033 and 2035, excluding any patent term adjustments and any patent term extensions.
We own granted patents and patent applications covering formulations of Acelarin (including those used in the clinical trials), methods of making Acelarin (including as a single diastereoisomer), and specific uses of Acelarin, including the use of Acelarin in combination with carboplatin and Acelarin in combination with cisplatin. Patents claiming the clinical formulation of Acelarin have been granted in the United States and Europe. Patents arising from these pending applications have been filed in all major territories, including the United States, Europe and Japan and are expected to expire in 2035, 2036 and 2038, excluding any patent term adjustments and any patent term extensions.
NUC-3373
We own 60 granted patents and five pending applications covering the composition of matter of NUC-3373, a genus around NUC-3373 and specific uses of NUC-3373. Those patents were granted in major territories, including the United States, Europe and Japan. These granted patents and patents arising from the pending applications, if issued, are expected to expire in 2032, excluding any patent term adjustments and any patent term extensions.
We own patent applications covering formulations of NUC-3373 (including those used in the clinical trials), methods of making NUC-3373, and specific uses of NUC-3373. These patents and patents arising from these pending applications are expected to expire in 2036, 2037 and 2038 excluding any patent term adjustments and any patent term extensions.
NUC-7738
We own 47 granted patents and 25 pending applications covering the composition of matter of NUC-7738, a genus around NUC-7738 and specific uses of NUC-7738. This includes a granted composition of matter patent in the United States, Europe and Japan. There are patent applications pending in major territories,
6
including the United States, Europe and Japan. These granted patents and patents arising from these pending applications, if issued, are expected to expire in 2035 excluding any patent term adjustments and any patent term extensions.
We own patent applications covering formulations of NUC-7738, methods of making NUC-7738, and specific uses of NUC-7738. Patents arising from these pending applications are expected to expire in 2036, 2038 and 2040 excluding any patent term adjustments and any patent term extensions.
7