UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM
(Mark One)
For the Fiscal Year Ended
OR
Commission File Number:
(Exact Name of Registrant as Specified in Its Charter)
(State or Other Jurisdiction of |
(I.R.S. Employer |
Incorporation or Organization) |
Identification No.) |
(Address of Principal Executive Offices, Including Zip Code)
(
(Registrant’s Telephone Number, Including Area Code)
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
|
|
Securities registered pursuant to Section 12(g) of the Act: None.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer |
☐ |
☒ |
|
Non-accelerated filer |
☐ |
Smaller reporting company |
|
|
|
Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 USC. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No
The aggregate market value of the voting common shares held by non-affiliates of the registrant was approximately $
The number of shares of the registrant’s Class A common stock outstanding as of February 15, 2023 was
DOCUMENTS INCORPORATED BY REFERENCE
Certain information required to be provided in Part III of this Annual Report on Form 10-K will be provided by a Definitive Proxy Statement for our 2023 Annual Meeting of Stockholders (the “Proxy Statement”) to be filed with the Securities and Exchange Commission on or before May 1, 2023.
Auditor Firm Id: |
Auditor Name: |
Auditor Location: |
ORGANOGENESIS HOLDINGS INC.
ANNUAL REPORT ON FORM 10-K
FOR FISCAL YEAR ENDED DECEMBER 31, 2022
TABLE OF CONTENTS
|
|
Page |
|
|
|
|
|
|
Item 1. |
2 |
|
Item 1A. |
34 |
|
Item 1B. |
70 |
|
Item 2. |
70 |
|
Item 3. |
70 |
|
Item 4. |
70 |
|
|
|
|
|
|
|
|
|
|
Item 5. |
71 |
|
Item 6. |
72 |
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
73 |
Item 7A. |
86 |
|
Item 8. |
87 |
|
Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
87 |
Item 9A. |
87 |
|
Item 9B. |
88 |
|
Item 9C. |
Disclosure Regarding Foreign Jurisdictions that Prevent Inspections |
88 |
|
|
|
|
|
|
|
|
|
Item 10. |
89 |
|
Item 11. |
89 |
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
89 |
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
89 |
Item 14. |
89 |
|
|
|
|
|
|
|
|
|
|
Item 15. |
90 |
|
Item 16. |
92 |
|
|
|
|
93 |
||
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K, including the sections entitled “Business,” “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” contains forward-looking statements. These statements may relate to, but are not limited to, expectations of our future results of operations, business strategies and operations, financing plans, potential growth opportunities, potential market opportunities and the effects of competition, as well as assumptions relating to the foregoing. Forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified. These risks and other factors include, but are not limited to, those listed under “Risk Factors.” In some cases, you can identify forward-looking statements by terminology such as “may,” “will,” “should,” “could,” “expect,” “plan,” “anticipate,” “believe,” “estimate,” “predict,” “intend,” “potential,” “might,” “would,” “continue” or the negative of these terms or other comparable terminology. These statements are only predictions. Actual events or results may differ materially.
As used herein, except as otherwise indicated by context, references to “we,” “us,” “our,” “the Company,” “Organogenesis” and “ORGO” will refer to Organogenesis Holdings Inc. and its subsidiaries.
TRADEMARKS AND SERVICE MARKS
All trademarks, trade names, product names, graphics and logos of Organogenesis contained herein are trademarks or registered trademarks of Organogenesis Holdings Inc. or its subsidiaries, as applicable, in the United States and/or other countries. All other party trademarks, trade names, product names, graphics and logos contained herein are the property of their respective owners. The use or display of other parties’ trademarks, trade names, product names, graphics or logos is not intended to imply, and should not be construed to imply, a relationship with, or endorsement or sponsorship of Organogenesis by such other party.
Solely for convenience, the trademarks, service marks and trade names referred to in this annual report are listed without the ®, (sm) and (TM) symbols, but we will assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensors to these trademarks, service marks and trade names.
1
PART I
ITEM 1. BUSINESS
Overview
Organogenesis is a leading regenerative medicine company focused on the development, manufacture and commercialization of solutions for the Advanced Wound Care and Surgical & Sports Medicine markets. Our products have been shown through clinical and scientific studies to support and in some cases accelerate tissue healing and improve patient outcomes. We are advancing the standard of care in each phase of the healing process through multiple breakthroughs in tissue engineering and cell therapy. Our solutions address large and growing markets driven by aging demographics and increases in comorbidities such as diabetes, obesity, cardiovascular and peripheral vascular disease and smoking. We offer our differentiated products and in-house customer support to a wide range of health care customers including hospitals, wound care centers, government facilities, ambulatory service centers (“ASCs”) and physician offices. Our mission is to provide integrated healing solutions that substantially improve medical outcomes and the lives of patients while lowering the overall cost of care.
We offer a comprehensive portfolio of products in the markets we serve that address patient needs across the continuum of care. We have and intend to continue to generate data from clinical trials, real-world outcomes and health economics research that validate the clinical efficacy and value proposition offered by our products. Several of our existing and pipeline products in our portfolio have Premarket Application (“PMA”) approval, or 510(k) clearance from the United States Food and Drug Administration (“FDA”). Given the extensive time and cost required to conduct clinical trials and receive FDA approvals, we believe that our data and regulatory approvals provide us a strong competitive advantage. Our product development expertise and multiple technology platforms provide a robust product pipeline, which we believe will drive future growth.
In the Advanced Wound Care market, we focus on the development and commercialization of advanced wound care products for the treatment of chronic and acute wounds in various treatment settings. We have a comprehensive portfolio of regenerative medicine products, capable of supporting patients from early in the wound healing process through wound closure regardless of wound type. Our advanced wound care products include Apligraf for the treatment of venous leg ulcers (“VLUs”) and diabetic foot ulcers (“DFUs”); Dermagraft for the treatment of DFUs (manufacturing currently suspended pending transition to a new manufacturing facility or engagement of a third-party manufacturer); PuraPly AM as an antimicrobial barrier for a broad variety of wound types; and the Affinity, Novachor, and NuShield wound coverings to address a variety of wound sizes and types. We have a highly trained and specialized direct wound care sales force paired with comprehensive customer support services.
In the Surgical & Sports Medicine market, we are leveraging our broad regenerative medicine capabilities to address chronic and acute surgical wounds and tendon and ligament injuries. Our Sports Medicine products include NuShield for surgical applications in targeted soft tissue repairs; and Affinity, Novachor, PuraPly MZ, and PuraPly AM for management of open wounds in the surgical setting. We currently sell these products through independent agencies and our direct sales force.
As of December 31, 2022, we had approximately 1,030 full-time employees worldwide. For the year ended December 31, 2022, we generated revenue of $450.9 million and we incurred operating expenses of $323.6 million.
Competitive Strengths
We believe we have several unique strengths that have been instrumental to our success and position us well for future growth:
2
Our Business Strategy
We believe the following strategies will play a critical role in our future growth:
3
Industry Overview
We focus our efforts on medical conditions that involve difficult-to-heal wounds and musculoskeletal injuries. Healing difficulties may arise from a variety of causes and in various types of tissue and anatomic areas. Impaired healing is commonly associated with an inability to move beyond the inflammatory stages of healing, resulting in a chronic wound or injury, an ongoing inflammatory cycle, and an inability to achieve normal tissue healing. Biofilm and other infectious conditions also play a key role in disrupting wound healing processes. Regenerative medicine is a collection of technologies aimed at generating tissue as close as possible to native or natural tissue, to replace damaged tissue, and to fill or replace defects. Demand for these technologies is increasing as physician understanding of the underlying wound healing processes grows and as demographic and population health trends result in the increased prevalence of systemic comorbidities that contribute to healing problems throughout the body.
Our products use regenerative medicine technologies to provide solutions in the Advanced Wound Care (Chronic Wound) and Surgical (Acute Wound) & Sports Medicine markets. Based on industry reports and management estimates, we believe that our addressable Advanced Wound Care and Surgical & Sports Medicine markets totaled approximately $24 billion in 2021, which included an estimated $10 billion addressable market for Advanced Wound Care and an estimated $14 billion for Surgical & Sports Medicine. Within the Advanced Wound Care market in 2021, 49% of treatments used advanced wound dressings, 18% used biologics, 21% used external wound healing devices, and 13% consisted of more traditional wound care dressings. The skin substitute market, within biologics, is expected to grow from $1.1 billion in 2021 to $2 billion in 2026. Within the Surgical & Sports Medicine market, the surgical/acute wound sub-market accounts for $9.1 billion, the chronic inflammatory and degenerative condition sub-market accounts for approximately $3.8 billion, and the tendon and ligament injuries sub-market accounts for approximately $1.0 billion in 2015.
Key drivers of growth in these two markets include:
4
Advanced Wound Care Market
Wounds represent a large and growing burden on the public health as well as a significant cost to the health care system. Wounds are divided into two primary types, chronic and acute. It is estimated that approximately 80 million patients suffer from chronic and acute wounds globally each year, excluding surgical incisions. Chronic wounds account for most of the expenses due to their complexity and length of treatment.
Chronic Wounds
Chronic wounds are wounds that have not appropriately closed after four weeks of treatment with traditional treatment such as dressings. Chronic wounds include:
While the underlying etiology of these chronic wounds is different, at a cellular level many of the problems that result in failed healing are the same. These include uncontrolled inflammatory processes, shortages of cell types, and growth factors secreted by cells that are critical to healing, and that result in disrupted cell signaling pathways.
Relative Prevalence of Wounds
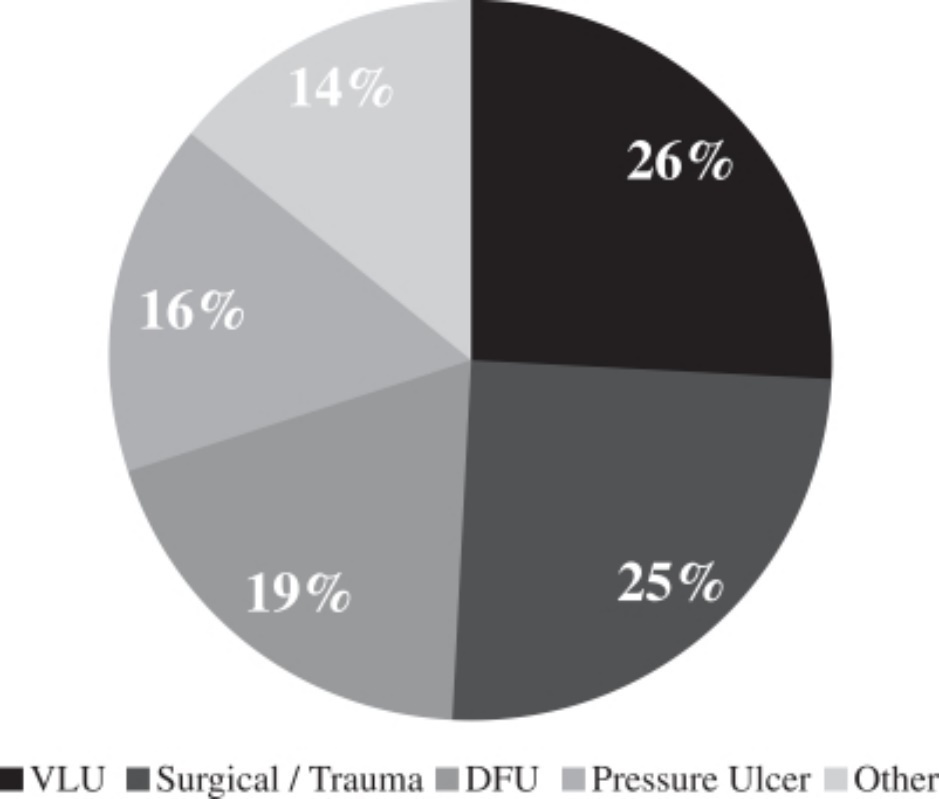
Our customers in outpatient wound care facilities are faced with a wide variety of types of wounds with different anatomical locations and underlying causes. Based on a retrospective cohort study of data from wound care centers from June 2008 and June 2012, the distribution of wound types in hospital outpatient wound care centers is detailed below:
Distribution of Wound Types*
* Based on a September 2013 JAMA Dermatology published retrospective cohort study.
Due to the breadth of our wound care portfolio, our products are able to address both chronic and acute wounds across all of these wound types.
5
Our Solution
The wound care market includes traditional dressings such as bandages, gauzes, and ointments and advanced wound care products such as mechanical devices, advanced dressings, and biologics. These advanced wound care products target chronic and acute wounds not adequately addressed by traditional therapies. Our products are primarily classified as skin substitutes, which fall within the biologics category of the Advanced Wound Care market.
According to Grand View Research, the Global Advanced Wound Care market was estimated to be approximately $10 billion in 2021 and is expected to grow at a compound annual growth rate, or CAGR, of 4% through 2028. This market consists of several product categories including advanced wound dressings, external wound healing devices such as negative pressure wound therapy, or NPWT, biologics such as skin substitute and growth factors and other traditional wound dressings. The approximate breakdown for these product categories in 2021 is set forth below.
Advanced Wound Care Market
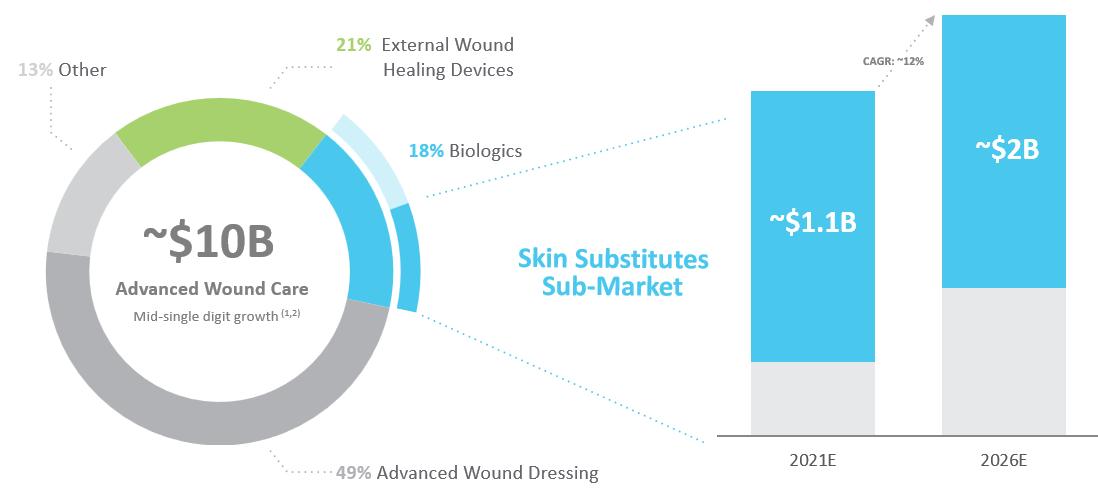
Wound biologics represents one of the smallest segments of the Advanced Wound Care market but is the fastest growing and has seen the highest level of innovation. According to BCC Research, the worldwide wound biologics market, which includes skin substitutes and growth factors, was estimated to be approximately $1.7 billion in 2021, of which skin substitute products are estimated to represent approximately 62%. Skin substitutes, bioengineered or biologic grafts that cover skin defects and support healing, are one of the fastest-growing categories of the Advanced Wound Care market. The skin substitute market, within biologics, is expected to grow from $1.1 billion in 2021 to $2 billion in 2026. Going forward, the skin substitute market is projected to continue to grow as patients with hard-to-heal wounds transition from other therapies to skin substitute treatment.
We expect this market to continue to grow at a rapid rate as physicians are educated about the use of these products and understand the benefits as compared to other currently marketed products, payers incentivize doctors to use more cost-effective treatments, patients demand more effective treatment solutions and advanced wound care becomes more common outside of the United States. We also believe that adoption of these products will increase as clinical evidence supporting the benefits of skin substitutes over traditional therapies continues to grow. Skin substitutes have demonstrated improved chronic and acute wound healing rates at a lower overall cost than the current standard of care. In a matched cohort study we commissioned, Medicare treatment costs for DFUs treated with Apligraf were $5,253 (p=0.49) lower per patient than the standard of care and for DFUs treated with Dermagraft, these costs were $6,991 (p=0.84) lower per patient than the standard of care. See Rice et al. “Economic outcomes among Medicare patients receiving bioengineered cellular technologies for treatment of diabetic foot ulcers.” J Med Econ. 2015;18(8):586-95.
Our products compete with other skin substitutes as well as other advanced wound care products such as NPWT and growth factors. Due to its market position as a skin substitute with antimicrobial properties appropriate for the treatment of wounds with biofilm or otherwise at high risk of infection, our PuraPly AM product also competes with antimicrobial dressings. Antimicrobial wound products have historically represented a more than $1 billion annual market. We are a market leader in the antimicrobial skin substitute market and have supported the expansion of that market with our comprehensive marketing and educational campaigns.
6
Finally, the skin substitute market remains substantially underpenetrated. According to BioMed GPS, over 7.9 million wounds globally, 3.7 million in the United States, require medical care and are classified as difficult-to-heal wounds where traditional therapies are unlikely to succeed. Market growth will be propelled by the aging population and rise of diabetes, obesity, and cardiovascular disease — all associated with poor vascularity increasing the susceptibility of chronic, hard-to-heal wounds. Despite the vast need and proven benefits of advanced wound care products in general including skin substitutes, market penetration remains low in relation to the size of the total addressable market. Our internal estimates indicate that if the potentially addressable market were completely penetrated today, annual skin substitute revenue in the United States alone could exceed $9 billion.
We believe that we are well positioned in the skin substitute market as adoption continues to increase. According to BioMed GPS, we are a leading skin substitute company in the United States, and we have an experienced and established sales force with deep relationships with clinicians, wound care centers, and hospitals. We also have a diverse array of products to address the different varieties of wounds throughout the wound healing process.
Surgical & Sports Medicine Market
An estimated 313 million surgical procedures are performed worldwide annually. An analysis of Medicare beneficiaries reveals that surgical wound care is associated with the highest wound care expenses, followed by DFUs. Trauma wounds, including burns, are included in the surgical/acute wound area. It is estimated that traumatic injury is responsible for more than 5 million deaths worldwide per year. Sports Medicine has displayed considerable growth as compared to other healthcare fields as a result of the rise in incidence of sports-associated injuries along with increase in awareness among people regarding physical fitness. We estimate the immediate addressable Surgical & Sports Medicine market for our products to be approximately $14 billion with a CAGR of approximately 6% through 2028.

Surgical/Acute Wounds
A surgical and/or acute wound is an injury that causes a rapid break in the skin and sometimes the underlying tissue. Acute wounds can be traumatic wounds, such as abrasions, lacerations, penetrating injuries and burns, or surgical wounds (grafts, dehiscences, necrotizing soft tissue infections) from surgical incisions. In contrast to chronic wounds, which would normally heal but stall due to biologic factors, acute wounds can be so severe that they overwhelm the body’s normal healing capacity. Biofilm and other infectious conditions, particularly in acute wounds with a high risk of infection such as open fractures, may also pose challenges to the healing of acute wounds. According to BCC Research, in the United States alone more than 150,000 deaths stem from traumatic injuries and there are more than 3 million nonfatal injuries per year. An estimated 180,000 deaths every year are caused by burns, and nonfatal burn injuries are a leading cause of morbidity. According to the American Burn Association, approximately 450,000 Americans sustain serious burn injuries every year, and more than 40,000 require hospitalization and advanced medical care.
7
Tendon and Ligament Injuries
Tendon and ligament injuries are common orthopedic conditions in an active and aging population. There are approximately 250,000 rotator cuff repairs performed in the United States annually. Additionally, in 2015, there were approximately 40,000 outpatient Achilles tendon repairs in the United States. Re-rupture and reoperation continue to be a significant source of concern with non-operative management, occurring in 4.8% of Achilles tendon repair cases and as many as 25% or more rotator cuff repair cases. Comorbidities such as diabetes and obesity, as well as age, are correlated with a higher risk of failed healing and re-rupture. Regenerative tissue scaffolds may be used to support the healing of tendons, ligaments, and other soft tissues. According to Technavio, the annual regenerative tissue scaffold market is estimated to exceed $1 billion.
Sports Medicine--Orthobiologics
While our current portfolio of products has applicability across a wide variety of clinical specialties and wound types in the advanced wound care and surgical wound care market, our goal is to re-enter the regenerative orthobiologics market in the future (if BLA approval for ReNu is obtained). Orthobiologics are biologic substances that are used to address injuries of the musculoskeletal system. Orthobiologic products are used to treat people with long-term disabling musculoskeletal disorders and injuries. The majority of musculoskeletal injuries occur due to recreational and sports activities. The patient demographics include both younger populations and those involved in professional sports, as well as the elderly population, usually requiring treatment for degenerative disorders and chronic diseases. The market has seen an increase in surgical volumes in part due to a higher incidence of comorbidities and chronic inflammatory and degenerative conditions, such as osteoarthritis (“OA”) and tendonitis. The growing and aging population affected with OA that is still looking to remain active will continue to seek non-surgical or minimally invasive alternatives. The prevalence of knee OA has been increasing over the past several decades in the U.S., mirroring the aging population and the growing obesity epidemic.
Chronic Inflammatory and Degenerative Conditions (Future Pipeline Opportunity)
Chronic inflammatory and degenerative orthopedic conditions are increasingly prevalent, driven in part by an aging demographic and higher levels of comorbidities such as diabetes and obesity. OA is the most common chronic condition of the joints, affecting approximately 27 million individuals in the United States. OA can affect multiple joints in the body, with arthritis of the knee being the most commonly treated. One in two adults will develop symptoms of knee OA during their lives. Other chronic inflammatory conditions such as Achilles and rotator cuff tendinosis and plantar fasciitis are also increasingly common. Similar to many of the other conditions that we seek to address, chronic inflammatory and degenerative orthopedic conditions are often correlated with smoking, obesity, and diabetes, among other factors. Collectively, these and other related conditions were treated with an estimated 9 million injections in 2016, including steroids and hyaluronic acid, or HA. According to Grand View Research, the global chronic inflammatory and degenerative orthopedic market (Viscosupplementation Market) exceeded $3.8 billion in 2020.
Our Solution
We believe our multiple regenerative technology platforms will allow us to build a broad portfolio covering the full range of needs in the Surgical & Sports Medicine market. In the short term, our focus will be on providing clinicians with wound covering and solutions to support soft tissue healing solutions with our placental-based technologies for open acute wounds and tendon and ligament surgical repair procedures. In the long-term, we plan to deepen our focus and provide solutions for chronic inflammatory and degenerative conditions, and in particular, OA as illustrated by our current Phase III Clinical Trial for ReNu. We intend to address patient needs with our portfolio in the inpatient hospital, ASC, and clinic settings. We estimate the immediate addressable Surgical & Sports Medicine market for our products to be approximately $14 billion respectively with a CAGR of approximately 6% through 2028.
For surgical/acute wounds, as skin substitutes continue to gain market adoption based on their demonstrated efficacy in improving healing rates with lower overall costs for these comprised healing situations, we believe we are well positioned with our comprehensive portfolio of technologies. Our placental-based technologies (Affinity, Novachor, NuShield) and skin substitute with antimicrobial properties (PuraPly AM) are highly differentiated both in composition along with their level of clinical utility. These product attributes coupled with our current market-leading position and high level of organizational competency give us the confidence that we have the ability to capture a high percent share of this growing market.
In tendon and ligament repair, conventional surgical approaches rely on mechanical fixation to temporarily approximate damaged tissues, assuming that the natural healing process will then result in a permanent repair. Patients with impaired healing may be unable to generate the necessary tissue structures, resulting in unacceptable failure rates over time. As additional clinical evidence and technology adoption is gained with our placental-based technologies, we believe we are well positioned with our current offering (NuShield as a surgical barrier) and our native collagen surgical matrix (PuraForce for soft tissue reinforcement).
OA and other degenerative conditions, as well as soft tissue injuries such as tendinosis and fasciitis, are currently treated by injection with steroids or HA. However, steroids offer pain relief for only a limited period and have been shown to further degrade
8
some types of tissues over time, worsening the underlying condition. The evidence of HA’s efficacy has been questioned, and it is clear that a significant percentage of patients do not respond to HA treatment. Patients who fail these less invasive therapies have limited options and may require surgical intervention, including total joint replacement.
Orthobiologics have been shown to be an effective alternative to traditional treatments. Due to their anti-inflammatory and pro-healing effects, they go beyond mechanical intervention to support the healing process in the damaged tissue and often result in faster healing times and shorter hospital stays. The orthobiologics market includes bone morphogenetic protein, viscosupplementation with HA, synthetic bone graft substitutes, and stem cell therapy, in addition to DBM and allograft. Our current product pipeline includes Sports Medicine solutions based on placental-based technologies (ReNu). There is a rapidly growing body of clinical and scientific evidence indicating the potential of these products, particularly orthobiologics, in surgical applications, resulting in increased adoption of these products.
Our Products
Advanced Wound Care
In the Advanced Wound Care (Chronic Wound) market, we focus on the development and commercialization of a broad portfolio of cellular and acellular wound care offerings that treat patients from the earliest indication of impaired healing to wound closure. Our suite of products helps treat a wide range of chronic wounds such as VLUs, DFUs, and pressure ulcers.
The breadth and depth of our portfolio allow physicians to tailor solutions to meet the needs of individual wound care patients. Wounds of all types normally progress through predictable phases of healing, starting with inflammation, progressing to cell proliferation, and finally remodeling to form normal skin. Wounds may stall during this process, typically in the inflammatory phase, for a variety of reasons. These reasons include biofilm or infection, uncontrolled inflammatory processes, shortages of cell types and growth factors secreted by cells that are critical to healing and disrupted cell signaling pathways.
It is increasingly recognized that addressing biofilm is an important step in healing any wound. Biofilm is generated by densely packed microbial communities that are attached to the wound surface and enclosed in a matrix of self-produced extracellular polymeric substance, or EPS. Biofilm is present in at least 78% of chronic wounds and can inhibit the healing of all wound types. We engage with the physician at the earliest indication of impaired healing with our PuraPly AM product, which helps control biofilm as an antimicrobial barrier via the broad-spectrum antimicrobial PHMB. If reduction of biofilm and control of the excessive inflammatory response is sufficient to result in healing, as is many times the case, PuraPly AM may be the only product required to achieve wound closure. If underlying healing issues persist, we offer an array of bioactive products and placental-based wound coverings tailored for a wide variety of wound sizes and types.
Our advanced wound care products are used in wound clinics that are located in an outpatient hospital setting as well as in physician offices and ASCs. The table below summarizes our comprehensive advanced wound care product suite:
Product (Launch Year)
|
Description
|
Regulatory
|
Clinical Application
|
|
Affinity (2014)†
|
Fresh amniotic membrane wound covering in which viable cells, growth factors/cytokines, and ECM proteins in the native tissue are preserved. |
361 HCT/P |
Chronic and acute wounds |
|
|
|
|
|
|
Novachor (2021)
|
Fresh chorion membrane wound covering in which viable cells, growth factors/cytokines, and ECM proteins in the native tissue are preserved. |
361 HCT/P |
Chronic and acute wounds |
|
|
|
|
|
|
Apligraf (1998)
|
Bioengineered living cell therapy that contains two living cell types, keratinocytes, and fibroblasts, that produce a broad spectrum of cytokines and growth factors |
PMA |
VLUs; DFUs |
|
|
|
|
|
|
9
Product (Launch Year)
|
Description
|
Regulatory
|
Clinical Application
|
|
|
|
|
Dermagraft (2001)*
|
Bioengineered product with living human fibroblasts seeded on a bioabsorbable scaffold, that produces human collagen, ECM, proteins, cytokines, and growth factors |
PMA |
DFUs |
|
|
|
|
|
|
|
|
NuShield (2010)†
|
Dehydrated placental tissue wound covering preserved to retain all layers of the native tissue including both the amnion and chorion membranes, with the epithelial layer and the spongy/intermediate layer intact |
361 HCT/P |
Chronic and acute wounds |
|
|
|
|
|
|
|
|
PuraPly AM (2016)
|
Antimicrobial barrier comprised of purified native collagen matrix with broad-spectrum polyhexamethylene biguanide, or PHMB, antimicrobial agent. Line extensions include PuraPly XT, which contains additional layers of collagen matrix and a higher level of PHMB. Extra-fenestrated (EF) versions of the products allow for added conformability and fluid drainage. |
510(k) |
Chronic and acute wounds (except 3rd degree burns) |
† Launched by NuTech Medical; acquired by Organogenesis in 2017.
* Launched by Smith & Nephew; acquired by Organogenesis in 2014.
Affinity & Novachor
Affinity & Novachor are fresh, amnion & chorion allograft wound coverings for application in the care of chronic and acute wounds. We believe both products are one of only a few placental tissue products containing viable amniotic cells, and are unique in that they undergo our proprietary AlloFresh process that hypothermically stores the products in their fresh state, never dried or frozen, which retains their native benefits and structure. Regulated as human cells, tissues, and cellular and tissue-based product, or HCT/P, under Section 361 of the PHSA, these products are referred to as Section 361 HCT/Ps, or simply 361 HCT/Ps. Affinity was launched in 2014 by NuTech Medical and acquired by us in 2017. Novachor was launched in December 2021.
Apligraf
Apligraf is a bioengineered bi-layered skin substitute that is the only product that has, to date, received PMA approval for the treatment of both VLUs and DFUs. Launched in 1998, Apligraf drives faster healing and more complete wound closure through its tissue-engineered structure, which includes an outer layer of protective skin cells (human epidermal keratinocytes), and an inner layer of cells (human dermal fibroblasts) contained within a collagen matrix. Apligraf is the leading skin substitute product for the treatment of VLUs, and its effectiveness has been established based on an extensive clinical history with over one million units shipped. We believe Apligraf is also the first and only wound-healing therapy to demonstrate in a randomized controlled trial, or RCT, a significant change in patients’ VLU wound tissue, showing a shift from a non-healing gene profile to a healing profile. Apligraf plays an active role in healing by providing the wound with living human skin cells, growth factors and other proteins produced by the cells, and a collagen matrix.
Dermagraft
Dermagraft is a dermal substitute grown from human dermal fibroblasts and has received PMA approval for the treatment of DFUs. Launched in 2001 by Smith & Nephew and acquired by us in 2014, this product helps to restore the compromised wound bed to facilitate healing. The living cells in Dermagraft produce many of the same proteins and growth factors that support the healing response in healthy skin. In addition to an FDA-monitored RCT demonstrating its superiority to conventional therapy in the healing of
10
DFUs, studies based on real-world electronic health records and Medicare data have demonstrated its superior clinical efficacy and value as compared to competitive wound care products and conventional therapy. Dermagraft can be applied weekly (up to eight times) over a twelve-week period and does not need to be removed from the wound during this period because it contains a temporary mesh fabric that is dissolvable and becomes part of the body’s own healing processes. Manufacturing of Dermagraft was suspended in the fourth quarter of 2021 and sales of Dermagraft were suspended in the second quarter of 2022 as part of our plan to transition our Dermagraft manufacturing to a new manufacturing facility or engage a third-party manufacturer, which we expect will result in substantial long-term cost savings. In the period when Dermagraft is not available, we expect that customers will be willing to substitute Apligraf for Dermagraft and that the suspension of Dermagraft sales will not have a material impact on our net revenue.
NuShield
NuShield is a dehydrated placental tissue wound covering and surgical barrier that is topically or surgically applied to the target tissue to support native healing. Regulated as a 361 HCT/P, NuShield is processed using our proprietary LayerLoc process, which preserves the native structure of the amnion and chorion membranes, including the intermediate or spongy layer, and their native structural and regulatory proteins. NuShield is available in multiple sizes, can be used as a wound covering to help support native healing of chronic and acute wounds of many sizes, and can be stored at room temperature with a five-year shelf life. NuShield was launched in 2010 by NuTech Medical and acquired by us in 2017.
PuraPly Antimicrobial
PuraPly Antimicrobial, or PuraPly AM, was developed to address the challenges posed by bioburden and excessive inflammation in the wound. Functioning as an antimicrobial barrier skin substitute, PuraPly AM is a purified native porcine type I collagen matrix embedded with polyhexamethylene biguanide, or PHMB, a localized broad-spectrum antimicrobial. PuraPly AM was launched in 2016 and has received 510(k) clearance for the management of multiple wound types, including partial and full-thickness wounds, pressure ulcers, venous ulcers, diabetic ulcers, chronic vascular ulcers, tunneled/undermined wounds, surgical wounds, trauma wounds, draining wounds, and first- and second-degree burns. The combination of PHMB with a native collagen matrix helps manage bioburden while supporting healing across a wide variety of wound types, regardless of severity or duration. Line extensions include PuraPly XT, which contains additional layers of collagen matrix and a higher level of PHMB. Extra-fenestrated (EF) versions of the products allow for added conformability and fluid drainage. We also developed and received 510(k) clearance for PuraPly without PHMB, which we refer to as “PuraPly,” including PuraPly MZ, for those patients who do not require an antimicrobial agent.
Surgical & Sports Medicine
In the Surgical & Sports Medicine market, we focus on the development and commercialization of products that support the healing of surgical/acute wounds, and musculoskeletal injuries including tendon repair and chronic degenerative conditions such as
11
OA. Our products in this market are used predominantly in the inpatient and outpatient hospital and ASC settings. The table below summarizes the principal products in our Surgical & Sports Medicine product suite:
Product (Launch Year)
|
Description
|
Regulatory
|
Clinical Application
|
|
|
|
|
|
|
|
|
|
|
|
|
NuShield (2010)
|
Dehydrated placental tissue barrier membrane preserved to retain all layers of the native tissue including both the amnion and chorion membranes, with the epithelial layer and the spongy/intermediate layer intact |
361 HCT/P |
Barrier membrane to support repair of tendon, ligament, and other soft tissue injuries |
|
|
|
|
Affinity (2014)
|
Fresh amniotic membrane wound covering in which viable cells, growth factors/cytokines, and ECM proteins in the native tissue are preserved |
361 HCT/P |
Wound covering for acute surgical wounds |
|
|
|
|
Novachor (2021)
|
Fresh chorion membrane wound covering in which viable cells, growth factors/cytokines, and ECM proteins in the native tissue are preserved. |
361 HCT/P |
Wound covering for acute surgical wounds |
|
|
|
|
PuraPly AM (2016)
|
Purified native collagen matrix with broad-spectrum PHMB antimicrobial agent. Line extensions include PuraPly XT, which contains additional layers of collagen matrix and a higher level of PHMB. Extra-fenestrated (EF) versions of the products allow for added conformability and fluid drainage. |
510(k) |
Antimicrobial barrier for management of open wounds in the surgical setting |
|
|
|
|
PuraForce (2019)
|
PuraForce is a bioengineered porcine collagen surgical matrix for use in soft tissue reinforcement applications that is intended for 510(k) indications for the reinforcement of all tendons in the body. PuraForce has high biomechanical strength per unit thickness, making it ideal for extremities applications. We commercially launched this product in 2019 |
510(k) |
Indicated for the reinforcement of soft tissues repaired by sutures or suture anchors during tendon repair surgery |
|
|
|
|
PuraPly MZ (2022)
|
PuraPly MZ is a micronized particulate version of PuraPly that allows application in powder or gel form to deep and tunneling wounds. PuraPly MZ is intended for indications for the management of open wounds in the surgical setting. |
510(k) |
Chronic and acute wounds (except 3rd degree burns) |
12
NuShield, Affinity, Novachor, PuraPly AM, PuraForce, and PuraPly MZ
We market our NuShield product for surgical and orthopedic applications. NuShield may be used as a surgical barrier or as an on-lay or wrap barrier to support soft tissue repairs. When used as a barrier membrane, the native biological characteristics of this placental tissue may help support the healing of soft tissue defects, particularly in difficult-to-heal locations or challenging patient populations. We market our Affinity and Novachor products as wound coverings for acute surgical wounds and our PuraPly AM product as an antimicrobial barrier for the management of open wounds in the surgical setting. PuraForce is a bioengineered porcine collagen surgical matrix for use in soft tissue reinforcement applications. PuraPly MZ is a micronized particulate version of PuraPly that allows application in powder or gel form for the management of open wounds in the surgical setting.
Bone Allograft Products
Our bone allograft products, which are derived from donated human cadaveric bone, include FiberOS and OCMP. Each of these products is used as a bone void filler, primarily in orthopedic and neurosurgical applications requiring bony fusion, such as spinal fusions and foot and ankle fusions. FiberOS is a blend of demineralized cortical fibers, mineralized cortical powder, and demineralized cortical powder and OCMP is a freeze-dried allograft cancellous (spongy or mesh-like) and demineralized cortical mixture.
Product Pipeline
We have a robust pipeline of products under development for both the Advanced Wound Care and Surgical & Sports Medicine markets. We believe our pipeline efforts will deepen our comprehensive portfolio of offerings as well as allow us to address additional clinical applications. The following table summarizes our pipeline products and potential timeline for their commercial launch:
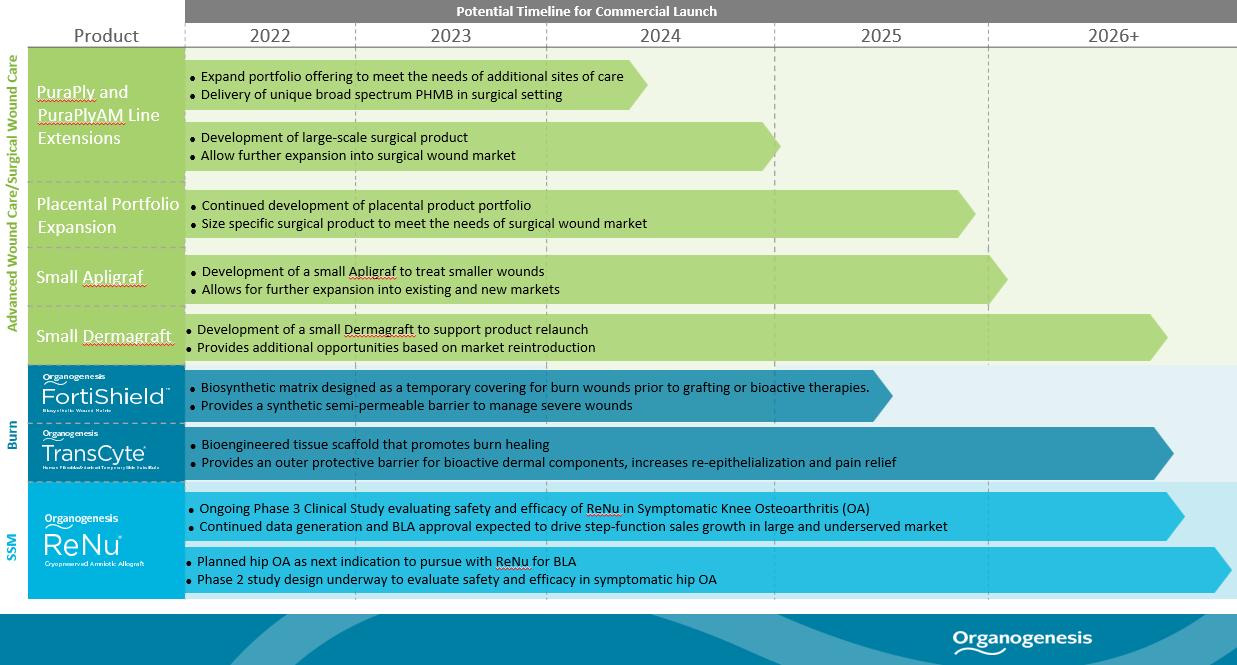
PuraPly and PuraPlyAM Line Extensions
The PuraPly portfolio is comprised of a purified native collagen matrix. PuraPly AM is an antimicrobial barrier leveraging the purified native collagen matrix with broad-spectrum PHMB antimicrobial agent. The design objective of line extensions in development is to leverage our knowledge and expertise to develop products to specifically meet the needs of additional sites of care.
Placental Portfolio Expansion
13
We have placental products under development. The design objective is to develop a larger graft with a long shelf life stored at room temperature to meet the needs of the surgical wound market.
Our R&D team continues to research and develop additional product concepts from our placental technology platform, as well as to collaborate with our Business Development team to assess additional product in-licensing or acquisition opportunities.
Apligraf and Dermagraft Line Extensions
We have two development projects underway to develop additional sizes of Apligraf and Dermagraft. The objective is to develop at least one additional smaller size of each product to optimize clinical utilization for smaller wounds such as DFUs. These types of changes to living cell-based products require significant development and validation work, and will require FDA PMA Supplement approval for the changes. Therefore, we expect the duration of the development projects to be several years before commercial products will be available. Manufacturing of Dermagraft line extensions is dependent on the completion of manufacturing and supply capabilities for the product.
FortiShield
FortiShield is a biosynthetic wound matrix made from a semi-permeable silicone membrane bonded to a kitted nylon fabric and coated with collagen, to provide a flexible dressing that is designed to adhere to the application site, provide a barrier to the external environment, and allow for excess exudate drainage. FortiShield is intended for use as a temporary wound covering, and to provide a moist wound healing environment on cleanly debrided wounds after hemostasis has been established. The primary indication for the product is as a transitional wound matrix for second degree burns. There are additional chronic and acute wound applications. A 510(k) application has been filed, and FDA has requested additional testing which is under review by the agency. If the product receives 510(k) clearance, we plan to commercially launch it for acute and chronic wound applications. This is also dependent on the completion of manufacturing and supply capabilities for the product.
TransCyte
TransCyte is a bioengineered tissue scaffold that promotes burn healing, and has received PMA approval for the treatment of deep second- and third-degree burns. We acquired the product from Shire, and it was previously marketed by Smith & Nephew. TransCyte complements our portfolio to address all severities of burn wounds. TransCyte is a flexible, durable product that provides bioactive dermal components, an outer protective barrier, increased re-epithelialization and pain relief for patients suffering from burns. We believe TransCyte will address a sizable market opportunity with limited competition, with only two other PMA approved products that would be directly competitive to TransCyte currently on the market, and only one competitor product containing a biosynthetic barrier to protect wounds. We conducted a clinical experience program with burn surgeons in 2022 with a limited supply of product manufactured at the closed La Jolla facility. Full launch is dependent on the completion of manufacturing capabilities.
ReNu
ReNu is a cryopreserved suspension derived from human amniotic tissue and cells derived from amniotic fluid, formulated for office use. It has been used to support healing of soft tissues, particularly in degenerative conditions such as OA and joint and tendon injuries such as tendinosis and fasciitis. The initial target indication for ReNu is for the management of symptoms associated with knee OA. A clinical study of ReNu for knee OA has been published, which we believe may indicate signs of its safety and suggest potential efficacy for a period of more than a year. On May 31, 2021, we suspended commercial distribution of ReNu in connection with the end of the FDA’s enforcement grace period for certain products that previously were marketed as 361 HCT/Ps. We are continuing to conduct clinical studies of ReNu to support BLA approval for the management of symptoms associated with knee OA, and are in the planning stages to conduct clinical studies of ReNu to support the management of symptoms associated with Hip OA. We believe ReNu may have potential as a treatment for additional OA and tissue regeneration applications, which would need to be clinically evaluated further before any such approved uses. ReNu was launched in 2015 by NuTech Medical and acquired by us in 2017.
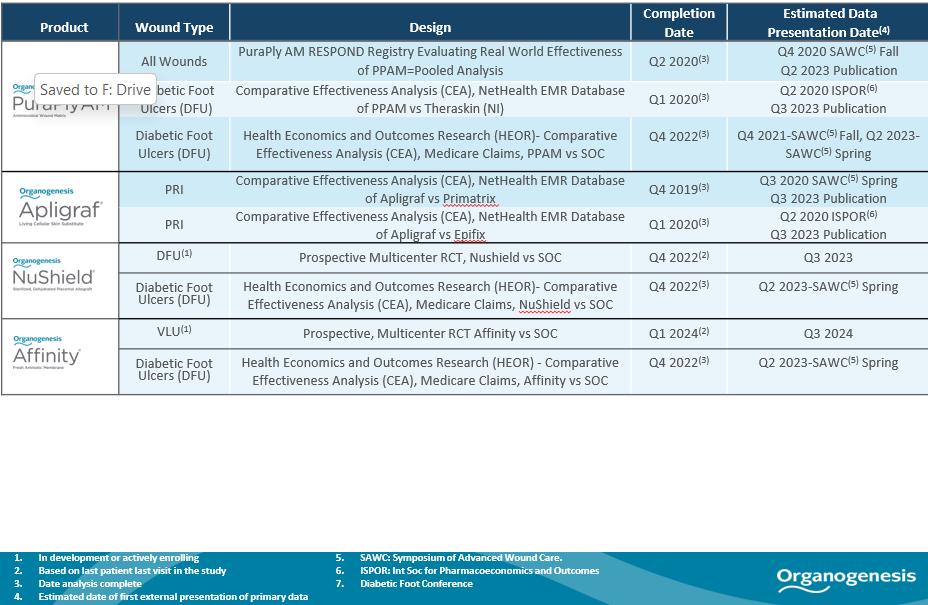
Ongoing Clinical Studies
We believe gathering robust and comprehensive clinical and real-world outcomes data is an essential component of developing a competitive product portfolio and driving further penetration in the markets where we compete. We have three ongoing prospective trials and six comparative effectiveness studies. We continue to invest in generating clinical data for our Advanced Wound Care and Surgical & Sports Medicine products, and believe such data enhance sales efforts with physicians and reimbursement dynamics with payers over time. The tables below summarize the status of our recent clinical studies for our Advanced Wound Care and Surgical & Sports Medicine products. As used herein, p value is a measure of statistical significance. The lower the p value, the more likely it is that the results of a clinical trial or study are statistically significant rather than an experimental anomaly. Generally, to be considered statistically significant, such results must have a p value <0.05.
14
Advanced Wound Care
15
Sports Medicine

Selected Published Clinical Studies
PuraPly AM
In a published prospective, multicenter, cohort study of 307 patients on the use of PuraPly AM in cutaneous wounds including acute and chronic wounds, 52, 62, and 73% of all wounds achieved closure at week 20, 26, and 32 respectively, with a median time to wound closure of 17 weeks. The wounds studied included 67 (22%) venous leg ulcers, 62 (20%) diabetic foot ulcers, 45 (15%) pressure ulcers, 54 (18%) post-surgical wounds, and 79 (26%) other wounds. For all 307 wounds, the incidence of achieving greater than a 60% reduction in baseline area and depth was 81 and 71% respectively. In addition, the incidence of wounds demonstrating greater than a 75% reduction in baseline volume was 85%.
Two subgroup analyses from the PuraPly AM multicenter, cohort study of 307 patients were published. In the venous leg ulcer (n=67) cohort, wound closure frequencies were 33%, 42%, 45%, 53%, and 73% at weeks 8, 12, 16, 24, and 32, respectively. The median time to closure was 22 weeks. Incidences of achieving a greater than 60% reduction in baseline area and depth were 78% and 70%, respectively, with 87% showing a reduction of greater than 75% in volume.
In the pressure injury (n=45) cohort, wound closure frequencies were 5%, 39%, 49%, and 62% at weeks 4, 16, 24, and 32 weeks, respectively. The median time to wound closure for all wounds was 32 weeks. Incidences of achieving a greater than 60% reduction in baseline area and depth were 78% and 64%, respectively, with approximately 82% of wounds showing a reduction in volume greater than 75%.
Affinity
In a published randomized controlled clinical trial of Affinity for use in diabetic foot ulcers comparing the use of Affinity and the standard of care (n=38) to the use of the standard of care alone (n=38), 60% of wounds in the Affinity and standard of care group achieved wound closure at 12 weeks compared to 38% of wounds in the standard of care group (p=0.04) and 63% of wounds in the Affinity and standard of care group achieved wound closure at 16 weeks compared to 38% of wounds in the standard of care group (p=0.04). In addition: 82% of wounds in the Affinity and standard of care group achieved a greater than 60% reduction in wound area as compared to 58% of wounds in the standard of care group (p=0.02); 65% of wounds in the Affinity and standard of care group achieved a greater than 60% reduction in wound depth as compared to 39% in the standard of care group (p=0.04); and 81% of wounds in the Affinity and standard of care group achieved a greater than 75% reduction in wound volume as compared to 58% in the standard of care group.
16
NuShield
In a published clinical study of clinical experience using NuShield for the management of 50 wounds (VLUs (n=14), DFUs (n=24) and other wounds (n=12)), 45 (90%) of the wounds had wound closure percentages between 60% to 100%. The median time to complete wound closure (or healing) for all wounds was 102 days (14.6 weeks), and the percent healing rate of all wounds healed at 16 and 24 weeks was 56% and 73%, respectively. For DFUs treated with NuShield, the median time to healing was 120 days (17.1 weeks) and the percent healing rates at 16 and 24 weeks were 43% and 59%, respectively. For VLUs treated with NuShield, the median time to healing was 90 days (12.9 weeks), with percent healing rates of 56% and 85% at 16 and 24 weeks, respectively. For all other wounds treated with NuShield (including pressure ulcers, nonhealing surgical, ischemic, mixed etiology, and nonhealing amputation), the median time to healing was 48 days (6.9 weeks), with percent healing rates of 57% and 100% at 16 and 24 weeks, respectively.
ReNu
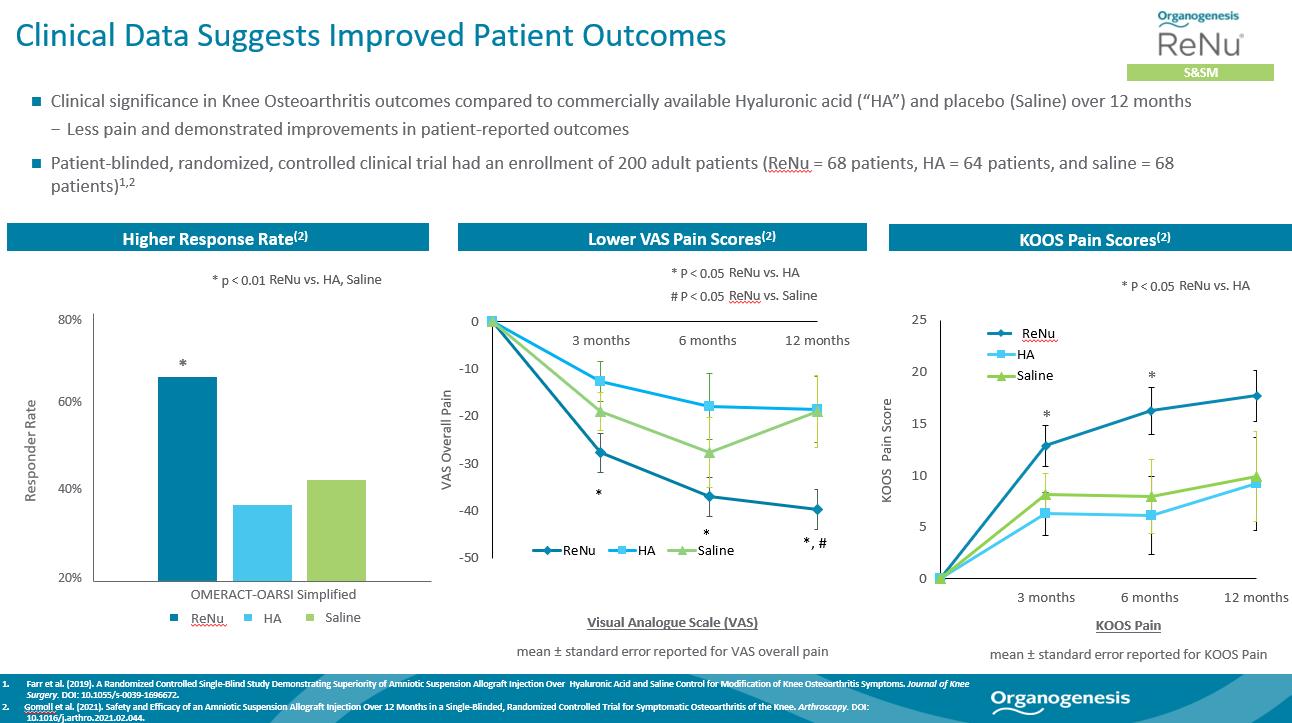
In a 200-patient randomized controlled multicenter single-blind study comparing the treatment of knee OA symptoms with ReNu (n=68), a commercially available hyaluronic acid, or HA (n=64), and saline (n=68), patients treated with ReNu reported a clinically meaningful and statistically significant reduction in Visual Analogue Scale (“VAS”) pain and higher OMERACT-OARSI responder rate at 12 months follow-up compared to patients treated with HA or saline. Pain was also evaluated using the Knee Injury and Osteoarthritis Outcome Score (“KOOS”) Pain score, and ReNu resulted in a statistically greater improvement in pain compared to HA at both 3 and 6 months.
A 474-patient Phase 3 prospective, multicenter, double-blind, placebo-controlled study is underway to evaluate the efficacy of ReNu (Amniotic Suspension Allograft, “ASA”) for the treatment of symptomatic knee OA (NCT04636229). Patients will be randomly assigned in a 1:1 ratio to receive a single intra-articular (IA) injection of 2 mL of ASA (plus 2 mL of normal saline) or 4 mL of normal saline. The primary efficacy endpoint has been defined as the difference in change from baseline in WOMAC Pain scale at 6 months between ASA- and placebo-treated patients. The design and statistical methodology of the current Phase III multi-center trial were informed and optimized based on the results of the 200 patient study. In March 2023, we reported the positive outcome of a pre-specified interim analysis of the data from 50% of the 474 required patients in our Phase 3 clinical trial for management of symptoms associated with knee OA that focused on the 6-month primary endpoint for sample size re-estimation. Based on the interim analysis, the independent data monitoring committee (“DMC”) recommended that the trial proceed without modification and continue without change to sample size. The DMC also found the safety data to be consistent with the known safety profile for ReNu.
17
TransCyte
In a published study of the safety and efficacy of TransCyte for the treatment of partial thickness burns, the mean timing to achieve greater than 90% wound epithelialization was 11 days for patients treated with TransCyte as compared to 18 days for patients treated with silver sulfadiazine cream (p=0.002).
Previously Published Clinical Studies for FDA-Approved Products
We also have accumulated a significant body of clinical evidence demonstrating the efficacy of our FDA-approved products, Apligraf and Dermagraft. We continue to invest in generating similar data for other Advanced Wound Care and Surgical & Sports Medicine products, and believe such data enhance sales efforts with physicians and reimbursement dynamics with payers over time. Our product Apligraf is the only product that has obtained FDA approval for the treatment of both VLUs and DFUs. Our product Dermagraft has also received FDA approval for DFUs. Below is a summary of the primary data supporting each product, and a description of the clinical studies that are currently in progress. As used herein, p value is a measure of statistical significance. The lower the p value, the more likely it is that the results of a clinical trial or study are statistically significant rather than an experimental anomaly. Generally, to be considered statistically significant, such results must have a p value <0.05.
Apligraf
Two pivotal studies were initially conducted with Apligraf demonstrating the safety and efficacy of the product in the treatment of full- and partial-thickness VLUs and DLUs. As a result, Apligraf obtained FDA approval for these indications. We have conducted a number of additional studies that provide further clinical evidence of the safety and efficacy of the product, including recent comparative effectiveness, cost effectiveness, and mechanism of action studies.
Pivotal FDA Registration Trials
For the DFU indication, a multi-center prospective RCT of Apligraf for the treatment of DFUs versus standard of care was conducted. Two hundred eight patients with Type 1 and 2 diabetes were enrolled, who had a plantar DFU of full- or partial-thickness. Patients with a chronic wound that exhibited less than 30% healing prior to treatment were eligible for the clinical trial. All patients’ ulcers were off-loaded using either crutches or a wheelchair for the first six weeks, followed by customized pressure-relieving footwear for at least four weeks post closure. Mean ulcer size was 2.97 cm2 and 2.83 cm2 in the Apligraf and the control group, respectively. The mean duration of the ulcer was 12 months in the Apligraf group and 11 months in the control group.
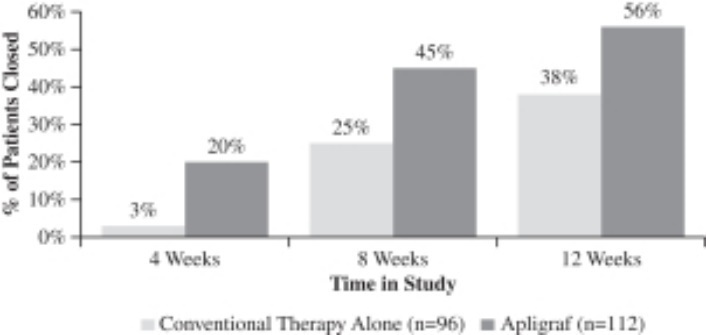
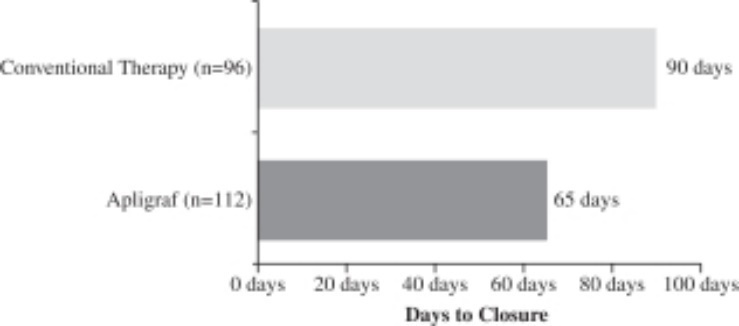
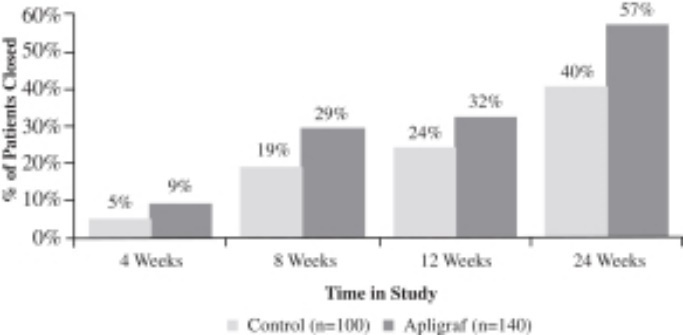
Apligraf was significantly more effective than conventional therapy for the incidence of complete wound closure over time. By 12 weeks of treatment, 56% (63 of 112 patients) of DFUs treated with Apligraf plus conventional therapy (debridement, saline dressings, total off-loading) were 100% closed, compared to 38% (36 of 96 subjects) of ulcers treated with conventional therapy alone (p=.0042). The median time to 100% wound closure was 65 days for DFUs treated with Apligraf plus conventional therapy versus 90 days for ulcers treated with conventional therapy alone (p=.0026).
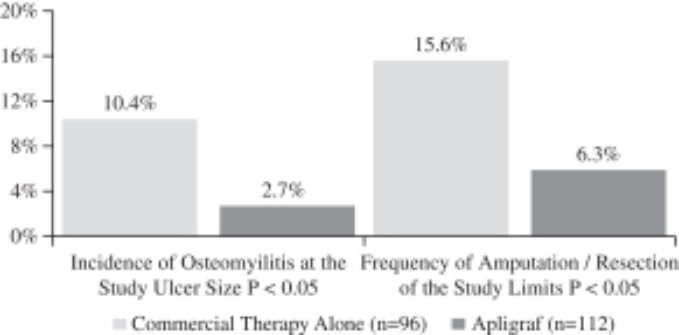
Recurrence is an important measure of healing durability, and in the study, 96% of ulcers treated with Apligraf remained closed at six months versus 87% in the control group. An important outcome of the study was an observed reduction in the incidence of reported adverse events of osteomyelitis and amputations/resections. Patients receiving Apligraf had a statistically significant (p<.05) lower incidence of osteomyelitis at the study ulcer site (2.7% vs. 10.4%) compared to patients treated with conventional therapy at six months. Apligraf-treated patients required significantly fewer amputations or resections of the study limb (6.3% vs. 15.6%) (p <.05) compared to patients treated with conventional therapy at six months. The primary results of the study are presented in the figures below.
Incidence of 100% Wound Closure |
Median Time to 100% Wound Closure |
|
|
18
Reduction in Osteomyelitis and Amputation / Resection
For the VLU pivotal trial, the efficacy of Apligraf was evaluated in a prospective, parallel-group, randomized, controlled, multi-center study involving 240 patients with VLUs. Subjects receiving Apligraf in combination with compression therapy were compared with an active treatment concurrent control of zinc paste gauze and compression therapy. Apligraf plus compression therapy was more effective in achieving complete wound closure by week 24 (57% vs 40%, p=.022). In patients with long-standing VLUs with greater than one year’s duration (n=120), Apligraf plus compression therapy was more than twice as effective in achieving complete wound closure by week 24 (47% vs 19%, p=.002). The primary results of the study are presented in the figures below.
All Patients Achieving 100% Closure
Comparative Effectiveness and Economic Studies
We conducted four comparative effectiveness studies with Apligraf utilizing our proprietary access to data collected in Net Health’s Wound Expert® Electronic Medical Record, or EMR, database. Net Health’s wound care software is utilized by more than 1,000 wound care centers across the United States. In collaboration with statistical experts and leading clinicians, we analyzed outcomes of treatment with Apligraf versus other skin substitutes including EpiFix (owned by MiMedx), Theraskin (owned by Bioventus, Inc.), Oasis (owned by Smith & Nephew), and Primatrix (Owned by Integra). All four studies showed that Apligraf improved overall healing rates as well as time to healing. For example, patients treated with Apligraf showed a 53% relative improvement in healing over patients treated with EpiFix at 24 weeks. All four studies have been published in peer-reviewed journals.
The Analysis Group, a private economics consulting firm, conducted a study to evaluate the economic outcomes of Medicare patients receiving Apligraf and Dermagraft, assessing the real-world medical services utilization and associated costs compared to patients receiving conventional care. Data for 502 matched Apligraf and conventional care patient pairs and 222 matched Dermagraft and conventional care patient pairs were analyzed. Increased costs associated with outpatient service utilization relative to matched conventional care patients were offset by lower amputation rates, fewer days hospitalized and fewer emergency department visits among Apligraf and Dermagraft patients. Consequently, Apligraf and Dermagraft patients with DFUs had per-patient average healthcare costs during the 18-month follow-up period that were lower than their respective matched conventional care counterparts (Apligraf was $5,253 (p=0.49), lower per patient, while Dermagraft was $6,991 (p=0.84) lower). These findings suggest that use of Apligraf and Dermagraft for treatment of DFU may lower overall medical costs through reduced utilization of costly healthcare services.
Mechanism of Action Clinical Study
To elucidate the mechanisms through which Apligraf promotes healing of chronic VLUs, the University of Miami Miller School of Medicine Department of Dermatology & Cutaneous Surgery conducted an RCT in which 24 patients with non-healing VLUs were treated with either standard of care (compression therapy) or Apligraf together with standard of care. Tissue biopsies were collected
19
from the VLU edge before and one week after treatment, and the samples underwent a comprehensive analysis of gene expression and protein analyses. The analyses conducted suggest that Apligraf induced a shift from a non-healing to a healing tissue response, involving modulation of inflammatory and growth factor signaling, keratinocyte activation, and attenuation of signaling involved in the chronic ulcer impaired state. In these ways, Apligraf application orchestrated a shift from the chronic non-healing ulcer microenvironment to a distinctive healing milieu resembling that of an acute, healing wound.
Dermagraft
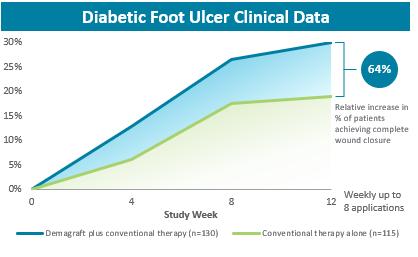
Dermagraft was approved as a Class III medical device for the treatment of DFUs based on the results of a large pivotal clinical trial. Three hundred fourteen patients were enrolled in a prospective RCT to evaluate the safety and efficacy of Dermagraft in conjunction with conventional therapy compared to a control arm of conventional therapy alone. Conventional therapy involved the sharp debridement and cleaning of the ulcer, application of a wet-to-dry gauze, and the use of therapeutic, pressure-reducing footwear. Patients were eligible to be screened for the trial if they had a plantar DFU on the heel or forefoot that was greater than 1cm2 and less than 20cm2. At the screening visit, the patients began receiving conventional therapy. If the DFU had not decreased in size by more than 50% during the next two weeks and the patient met all other inclusion and exclusion criteria, the patient was randomized into one of two treatment groups: Dermagraft plus conventional therapy or conventional therapy alone. Patients in the Dermagraft group received a weekly application of Dermagraft and conventional therapy for up to eight weeks. The primary endpoint for the trial was superiority in complete DFU closure by 12 weeks.
Pivotal FDA Registration Trial
In the pivotal clinical trial, the weekly application of Dermagraft and conventional therapy for up to eight weeks increased the proportion of DFUs that achieved 100% closure at 12 weeks by 64%, when compared to the use of conventional therapy alone. Patients treated in the Dermagraft group were 1.7 times more likely to achieve 100% closure than patients receiving conventional therapy alone. These results demonstrated statistically significant improvements. The incidence of adverse events among the Dermagraft and control groups was generally consistent across both groups, with the most common adverse events being infection at the DFU site, infection not at the DFU site, accidental injury and skin dysfunction/blister. However, the percentage of patients who developed an infection at the DFU site was significantly lower in the Dermagraft treatment group as compared with the control group, 10.4% versus 17.9%, respectively. No adverse laboratory findings were associated with the use of Dermagraft and no adverse device effects were reported in the trial. In addition, no immunological responses or rejections from patients that received Dermagraft were reported in this trial or in patients treated to date. The primary healing data for the trial is presented in the figure below.
Percent of Patients with Complete Healing by 12 Weeks
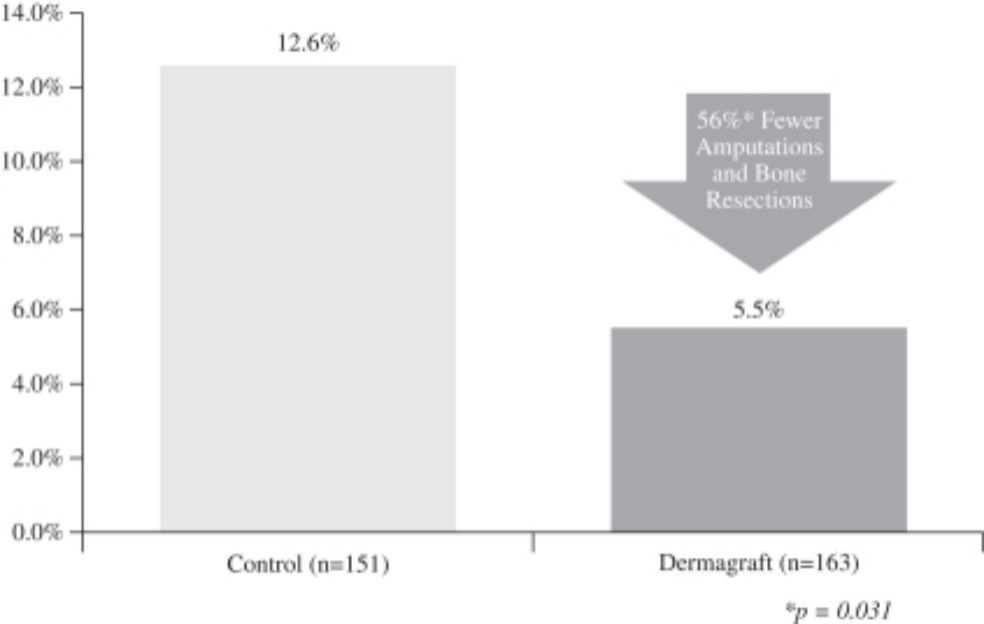
In a post-hoc analysis, it was determined that in patients treated with Dermagraft, there was a significant reduction in incidence of amputations or bone resections, as compared to the control group (12.6% versus 5.5%, respectively, p=0.031). No adverse laboratory findings were associated with the use of Dermagraft and no adverse device effects were reported in the trial. In addition, no
20
immunological responses or rejections from patients that received Dermagraft were reported in this trial or in patients treated to date. The amputation or bone resection data is presented in the figure below.
Frequency of Patients Experiencing a Study Ulcer-Related Amputation or
Bone Resection at 12 Weeks
Comparative Effectiveness and Economic Studies
We have conducted three comparative effectiveness studies with Dermagraft, which utilizes our proprietary access to data collected in the EMR database. In collaboration with statistical experts and leading clinicians, we analyzed outcomes of treatment with Dermagraft versus other skin substitutes including EpiFix (owned by MiMedx), Primatrix (owned by Integra), and Grafix (owned by Smith & Nephew). All three studies showed that Dermagraft improved overall healing rates as well as time to healing. In one study, patients treated with Dermagraft showed a 52% relative improvement in healing over EpiFix by week 24.
The economic study of Dermagraft in a Medicare population conducted by the Analysis Group is described under the heading “—Our Products— Previously Published Clinical Studies for FDA-Approved Products—Apligraf—Comparative Effectiveness and Economic Studies” above.
Platform Technologies
Our proven research and development capabilities and established technology platforms support a robust and adaptable product pipeline for future applications. The platform technologies in which we have deep experience include:
21
Commercial Infrastructure
Sales and Marketing
We have dedicated substantial resources to establish a multi-faceted sales capability in the United States. Our current Advanced Wound Care portfolio is sold throughout the United States via an experienced direct sales force, which focuses its efforts on wound care in various sites of care. We use a mix of direct sales representatives and independent agencies to service the Surgical & Sports Medicine market. As of December 31, 2022, we had approximately 360 direct sales representatives and approximately 150 independent agencies who have substantial medical device sales experience in our target end markets. These sales representatives are supported by teams of professionals focused on sales management, sales operations and effectiveness, ongoing training, analytics, and marketing.
We have historically focused our market development and commercial activities in the United States, but we have obtained marketing registrations, developed commercial and distribution capabilities, and are currently selling products in several countries outside of the United States. Our Apligraf product is currently distributed by our direct sales force in Switzerland, and through independent sales agents in Saudi Arabia and Kuwait. Our NuShield product is also distributed by our direct sales force in Switzerland, and through independent sales agents in Kuwait. We have obtained marketing registration for our Dermagraft product in Mexico, but we are not currently distributing it. Additionally, we are evaluating the regulatory pathways and market potential for our products in other major markets, including the European Union. Sales generated by our direct sales forces in the United States have represented, and we anticipate will continue to represent, a majority of our revenues.
Customer Support Services
We offer our customers in-house customer support services, including services provided by our experienced reimbursement support team, our medical and technical support team, and our field-based medical science liaison team. We believe that we have a competitive advantage by providing these essential support services in-house in that we are able to align the support services closely with our sales efforts as appropriate and improve the customer’s overall experience.
Research and Development
Our research and development team has extensive experience in developing regenerative medicine products, and works to design products that are intended to improve patient outcomes, simplify techniques, shorten procedures, reduce hospitalization and rehabilitation times, and, as a result, reduce costs. We conduct research and development activities at our laboratory facilities in Canton, MA, Birmingham, AL, and San Diego, CA. We have recruited and retained staff with significant experience and skills, gained through both industry experience and training at leading colleges and universities with regenerative medicine graduate programs. In addition to our internal staff, our external network of development labs, testing labs, and expert clinicians aid us in our research and development process. We continue to build our clinical operations capabilities to effectively run multiple concurrent multicenter clinical trials, including trials intended for FDA regulatory submissions (e.g. BLA). We have significant regulatory affairs capabilities to prepare and manage our regulatory submissions for product approvals.
The majority of our product portfolio, including Apligraf, our PuraPly product family, our collagen biomaterial technology platform product family, and all of our placental-based products, was developed by our research and development team at our three facilities. We have proven competencies to bring products to market via a broad range of regulatory classifications, as evidenced by FDA approval or clearance of our products via PMA approval of a Class III medical device; BLA approval of a biologics product; and 510(k) clearance of a Class II medical device, in addition to our 361 HCT/P allograft products and several products for which we have obtained international registrations.
Manufacturing and Suppliers
We manufacture internally our primary non-placental-based products and use third-party manufacturers for our placental-based products. We have significant expansion capabilities in our in-house manufacturing facilities and we believe that our contract manufacturers are well positioned to support future expansion.
We have robust internal compliance processes to maintain the high quality and reliability of our products. We use annual internal audits, combined with external audits by regulatory agencies to monitor our quality control practices. We are registered with the FDA as a medical device manufacturing establishment and a HCT/P registered establishment. We are also accredited by the
22
American Association of Tissue Banks (“AATB”) and licensed with several states per their tissue banks regulations. All of our contract manufacturers are registered with the FDA as HCT/P establishments and are AATB accredited.
We utilize third-party raw material suppliers to support our internal manufacturing processes. We select all of our suppliers through a rigorous process to ensure high quality and reliability with the capacity to support our expanding production levels. Only raw material from approved suppliers is used in the manufacture of our products. To confirm quality and identify any risks, our approved suppliers are audited at pre-determined intervals. Historically, we have not experienced any significant difficulty locating and obtaining the suppliers or materials necessary to fulfill our production requirements. In the first quarter of 2019, however, we suspended production of our product Affinity due to production issues at one of our suppliers. As this was our sole supplier of Affinity, it resulted in a disruption of our production capabilities. We identified an alternate supplier and were able to resume commercial-scale production in the second quarter of 2020. Subsequently, we have added a second source to provide additional capacity and redundancy in supply.
The manufacture of our products is dependent on the availability of sufficient quantities of source tissue, which is the primary component of our products. Source tissue includes donated human tissue, porcine tissue, and bovine tissue. We acquire donated human tissue directly through institutional review board-approved protocols at multiple hospitals, as well as through tissue procurement firms engaged by us or by our contract manufacturers. We have two qualified porcine tissue suppliers, and currently one source of bovine tissue. Our processing of these tissues is, and our supplier sources are required to be, compliant with applicable FDA current Good Tissue Practice, or cGTP, regulations, AATB standards, and U.S. Department of Agriculture, or USDA, requirements.
Reimbursement
Overview
Our customers primarily consist of hospitals, wound care centers, government facilities, ASCs, and physician offices, all of which rely on coverage and reimbursement for our products by Medicare, Medicaid, and other third-party payers. Governmental healthcare programs, such as Medicare and Medicaid, typically have published and defined coverage criteria and published reimbursement rates for medical products, services, and procedures that are established by law or regulation. Non-government payers have their own coverage criteria and often negotiate payment rates for medical products, services, and procedures. Many also require prior authorization as a prerequisite to coverage. In addition, in the United States, an increasing percentage of insured individuals are receiving their medical care through managed care programs, which monitor utilization and also may require prior authorization for the products and services that a member receives. Coverage and reimbursement from government and commercial payers are not assured and are subject to change.
Medicare, the federally funded program that provides healthcare coverage for senior citizens and people with disabilities, is the largest third-party payer in the United States. The Centers for Medicare and Medicaid Services (“CMS”) administers the Medicare program and uses Medicare Administrative Contractors (“MACs”) to process claims, develop coverage policies and make payments within designated geographic jurisdictions. CMS does not have a national coverage determination related to skin substitutes. Coverage for our products falls under the jurisdiction of the Part A/B MACs. Medicare coverage for our products is determined by each MAC for its specific jurisdiction. Currently, all the MACs, even those without published local coverage determinations (“LCDs”), cover our products in the outpatient hospital, physician office, and ASC settings.
Private payers often, but not always, follow the lead of Medicare or other governmental payers in making coverage and reimbursement determinations. Therefore, achieving favorable Medicare coverage and reimbursement can sometimes be a significant factor in obtaining favorable coverage and reimbursement for products by private payers. While most private payers currently cover Apligraf and Dermagraft, and some cover Affinity, most of those payers do not cover many of our other products, such as PuraPly, PuraPly AM, and NuShield.
Currently, Medicare makes a separate payment for our products when used in the physician office at a payment rate based on average sales price (“ASP”) methodology, including ASP plus 6% for some products. In the outpatient hospital and ASC settings, Medicare payment for all our products is bundled into the payment for the application procedure.
23
All Medicare payment amounts, including separate payment for our products, are affected by sequestration. In 2020, legislation was enacted that temporarily discontinued the sequestration rate of 2% of the government portion, which was imposed under the Budget Control Act of 2011 (“BCA”); under 2% sequestration, the final payment rate for products paid based on ASP is ASP+4.3%. The sequestration began again on April 1, 2022 at a rate of 1%. Starting on July 1, 2022, the sequestration rate returned to 2%. Sequestration may also be ordered under the Statutory Pay-As-You-Go Act of 2010 (“Statutory PAYGO”), which requires deficit neutrality in most laws passed by Congress. The $1.9 trillion American Rescue Plan Act of 2021 was expected to trigger Statutory PAYGO at the end of the 2021 Congressional session, but Congress has delayed a Statutory PAYGO sequestration order until after 2024.
The proposed update to the Medicare Physician Fee Schedule (“MPFS”) for calendar year 2023 included a proposal to stop making separate payments for all skin substitutes, including all of our products, in 2024 or 2025. Instead of making separate payment for skin substitutes, Medicare would bundle the payment for skin substitutes into the payment made for the application procedure. As part of this proposal, Medicare would consider all skin substitutes to be supplies instead of biologicals and would require manufacturers of skin substitutes, including us, to apply for new HCPCS codes that would be effective starting in 2024. In the 2023 MPFS final rule, published on November 1, 2022, CMS did not finalize this bundling proposal and will consider more public input in the future; however, they may propose the same policy again or make other proposals in the future that could affect our business and our revenue.
All skin substitute products administered in the hospital outpatient department and ASC settings are bundled. No skin substitute products currently have pass-through status. Pursuant to the Appropriations Act, PuraPly AM and PuraPly had pass-through status from October 1, 2018 through September 30, 2020, at which time the pass-through status expired. As of October 1, 2020, payment for PuraPly and PuraPly AM is bundled into the payment rate for the application procedure.
Skin Substitutes Used for Wound Care
All of our Advanced Wound Care products are classified as “skin substitutes” for Medicare reimbursement purposes. In 2014, CMS instituted “bundled” payments in the hospital outpatient and ASC setting for skin substitutes using a two-tier payment system. The Medicare payment system bundles payment for our products (and all skin substitutes) into the payment for the application of the skin substitute, resulting in a single payment to the provider that includes both the application of the product and the product itself. There is one bundled payment amount for procedures that involve high-cost products, i.e., products whose cost exceeds a threshold amount, and another bundled payment amount for procedures that involve low-cost products that do not meet the threshold. The bundled payment rate is updated annually and is also geographically adjusted. Currently, all of our wound care products are assigned to the high-cost bundle; it is not possible to predict, however, whether those products will continue to be assigned to the high-cost bundle or the rates that will be paid for each bundle. Further, under the bundling policy, there is an inherent incentive to use the cheapest products available, even if those products are less effective.
The bundled payment rates are also geographically adjusted. This geographic adjustment may result in significant payment variations among regions; sixty percent of the hospital payment rate and fifty percent of the ASC payment is adjusted to take into account the region’s wage-index, which can vary widely from one region to another. The wage-index adjustment can increase or decrease the unadjusted payment amount and may result in reimbursement being insufficient to account for the cost of skin substitute products and sizes in one geographic area that are fully reimbursed in other geographic areas.
Medicare has signaled that it may revise its two-tiered bundled payment policy for skin substitutes. Medicare solicited comments in the calendar year 2019 proposed rule related to proposed updates and policy changes under the Medicare Hospital Outpatient Prospective Payment System (OPPS) and ASC Payment System. Medicare specifically solicited comments on whether it should eliminate the two-tiered bundle policy and establish a single bundle for all products. However, CMS has not implemented any changes to its two-tiered payment structure for skin substitutes in response to those comments. In the calendar year 2023 proposed rule, CMS did not solicit comments on changes to its two-tiered payment structure. However, if CMS finalizes any revisions to its two-tiered payment policy, those changes could result in decreased reimbursement for our products which could decrease utilization and reduce our revenues. Moreover, any new policy could result in a financial incentive for hospitals and ASCs to use our competitor’s products, thereby reducing our market share and revenue.
In the physician office setting, payment for skin substitutes is not bundled into the payment for the administration of the product. Skin substitutes are paid separately from the application procedure and the Medicare payment rate for all biological skin substitutes (including ours) is calculated based on the ASP methodology on a per square centimeter basis with the total payment for the product being the per square centimeter ASP-based payment rate multiplied by the total number of centimeters. In the physician office setting the Medicare payment rates for all biological skin substitutes (including ours) are updated quarterly based on the ASP methodology and are not geographically adjusted. All Medicare payment amounts, including separate payment for our products, are affected by sequestration. Under the BCA, sequestration reduces by two percent the federal portion of the Medicare payment amount; the
24
beneficiary coinsurance amount of 20 percent is unaffected by sequestration. Congress had suspended sequestration during the COVID-19 public health emergency until April 1, 2022, at which time the reduction was one percent. Starting on July 1, 2022, the sequestration rate returned to two percent.
The ASP-based payment methodology applies only to physician offices. However, in the future, it is possible, through legislation or regulation, that Medicare will institute bundled payment for skin substitutes in the physician office setting. In fact, the proposed updates to the MPFS for calendar year 2023 included a proposal to stop making separate payment for all skin substitutes, including all of our products, in 2024 or 2025. Instead of making separate payment for skin substitutes, Medicare would bundle the payment for skin substitutes into the payment made for the application procedure. In the 2023 MPFS final rule, published on November 1, 2022, CMS did not finalize this bundling proposal and will consider more public input in the future; however, they may propose the same policy again or make other proposals in the future that could affect our business and our revenue.
Before calendar year 2022, Medicare did not require us to report ASP for some of our products because they are regulated by the FDA as medical devices; we voluntarily reported ASP data for most products. However, starting on April 30, 2022, we were required to report ASP for all our products because of a provision enacted in the Consolidated Appropriations Act of 2020, signed into law on December 27, 2020. CMS does not necessarily include all products that report ASP data in the quarterly ASP file. The local Part A/B MACs establish local payment for drugs and biologics whose ASP does not appear in the quarterly ASP file. MACs have the discretion to pay for such products based on invoices submitted by providers, Wholesale Acquisition Cost (“WAC”) + 3%, or they may contact CMS to determine if there are unpublished ASP data.
Section 90004 of the Infrastructure Investment and Jobs Act, enacted in November 2021, requires manufacturers to pay a refund to the federal government if more than a certain applicable percentage of their single-use product is not administered to a patient and is discarded (“wasted”) by providers. Because there is a lack of consistency and uniformity in wound sizes, it is likely that some skin substitute product is discarded with every treatment. Providers are only required to report discarded product when the product is paid separately (not part of a bundled payment rate.) The rebate obligation took effect on January 1, 2023, and CMS proposed a methodology to implement the rebate in the MPFS rulemaking. The applicable percentage is required to be at least 10 percent of total allowed charges for the drug in a given calendar quarter. CMS has the authority to increase the applicable percentage that applies to refunds for discarded product if there are “unique circumstances”. We submitted comments on the proposal noting the unique circumstances related to skin substitutes and asking CMS to apply a higher percentage. In the 2023 MPFS final rule, published on November 1, 2022, CMS did not apply a higher applicable percentage to any products other than the hydrogel example they used in the proposed rule and stated that they plan to collect additional information about products that may have unique circumstances such that an increased applicable percentage (higher than 10 percent) would apply. CMS estimated the wastage percentage for three of our products - Apligraf, Dermagraft, and PuraPly - based on 2020 data. We do not know if the refund amounts calculated in 2023 will be similar to these estimates but if they are, we may owe rebates, which could be material, on these products and possibly other products. The total amount of any discarded product rebate liability is not known at this time.
In the calendar year 2022 Final Rule for the MPFS, CMS established ten healthcare common procedure coding system (HCPCS), codes that describe synthetic skin substitutes, and more of these codes for synthetic skin substitutes have been established since. CMS has directed MACs to make separate payments for these codes when they are reported with the CPT codes for the application of skin substitutes. Because manufacturers of these products are not required to establish a WAC, or submit an ASP (because they are not treated as drugs or biologics by Medicare), it is likely the Part A/B MACs will pay for these products based on invoices. We do not know what effect this will have on our business or revenue.
Commercial insurers contract with participating providers such as hospitals, wound care centers, government facilities, ASCs, and physician offices to establish agreed-upon payment rates for items and services, including skin substitutes. Usually, these rates are in the form of a fee-schedule but sometimes there is a bundled payment rate. In many cases, the fee schedules are based on Medicare payment rates, which are bundled in hospitals and ASCs, but not in physician offices. These rates may vary by insurer, by provider and by region.
Medicaid coverage and payment rates and policies as to the types of providers (e.g., podiatrists) who are allowed to apply our products are determined by each state’s Medicaid program. Some states may bundle Medicaid payment for skin substitutes into the payment for the application procedure, like Medicare, while other states may pay separately. State Medicaid programs may reach different conclusions regarding the medical necessity of products used in treating Medicaid patients.
Currently, three MACs (Novitas, FCSO, and CGS) are in the process of issuing LCDs for skin substitutes for the treatment of DFUs and VSUs. Each of the proposed LCDs has the PuraPly products listed as non-covered. We have commented on the proposed policies and are working to reverse this in the final LCDs, but if the noncoverage of the PuraPly products stands in the final LCDs, then this noncoverage could adversely impact utilization of the PuraPly products and our revenue. In addition, these proposed policies would (1) limit the number of skin substitutes that can be applied to a wound, (2) prohibit switching skin substitutes during a course of
25
treatment, and (3) would require us to get certification from the FDA that our amniotic products are solely regulated under Section 361 of the Public Health Services Act. These LCDs have only been released in draft form and we do not know if or when they might be finalized or what policies will be included in any final LCD. If any of these proposals are finalized, our business and revenue could be adversely affected.
Surgical & Sports Medicine Products
Surgical & Sports Medicine products administered on an inpatient basis in a hospital are reimbursed by Medicare as part of a bundled payment based on the Medicare Severity Diagnosis Related Group (“MS-DRG”), to which a patient is assigned upon discharge from the hospital. MS-DRG assignment is determined according to the patient’s primary diagnosis, but can also be affected by other secondary diagnoses and the provision of certain surgical procedures. Certain MS-DRGs account for complications and comorbidities, which may increase the reimbursement amount.
The MS-DRG payment rate is a consolidated prospective payment for all services provided by the hospital during the patient’s hospitalization, based on the average cost of care calculated from Medicare claims data. With extremely few exceptions, the MS-DRG payment is inclusive of all services, products, and resources. Products administered during surgical procedures are not typically coded or paid separately when provided to a hospital inpatient. MS-DRG payments are case rates and hospitals profit when their costs for a particular patient are below the case rate and they are at risk of a loss if their costs are above the case rate.
Some private payers use the MS-DRG based system to reimburse facilities for inpatient services.
Competition
We operate in highly competitive markets that are subject to rapid technological change. Success in these markets depends primarily on product efficacy, ease of product use, product price, availability of coverage and adequate third-party reimbursement, customer support services for technical, clinical, and reimbursement support, and customer preference for, and loyalty to, the products.
We believe that the demonstrated clinical efficacy of our products, the breadth of our product portfolio, our in-house customer support services, our customer relationships and reputation offer us advantages over our competitors. In addition, we believe we are one of the few regenerative medicine companies offering PMA approved and 510(k) cleared products in addition to our 361 HCT/Ps.
Our products compete primarily with skin substitute products, placental-based technology products, orthobiologics products, other advanced wound care and traditional wound care products, among others. Our competitors include Amniox Medical, Inc., Arthrex, Inc., Bioventus Inc., Convatec Group Plc., Integra LifeSciences Holdings Corporation, MiMedx Group, Inc., Smith & Nephew plc and 3M, Incorporated.
We also compete in the marketplace to recruit and retain qualified scientific, management and sales personnel, as well as to acquire technologies and technology licenses complementary to our products or advantageous to our business.
We are aware of several companies that compete, or are developing technologies, in our current and future product areas. As a result, we expect competition to remain intense. Our ability to compete successfully will depend on our ability to develop proprietary products that reach the market in a timely manner, receive adequate coverage and reimbursement, are cost effective, and are safe and effective.
Intellectual Property
Our success depends in part on our ability to protect our proprietary technology and intellectual property and operate without infringing the patents and other proprietary rights of third parties. We rely on a combination of trademark, trade secret, patents, copyright, and other intellectual property rights and measures to protect the intellectual property rights that we consider important to our business. We also rely on know-how and continuing technological innovation to develop and maintain our competitive position. Other than a license from Novartis Pharma AG for trademark and domain name rights to Apligraf and an exclusive license from RESORBA Medical GmbH, or Resorba, to a U.S. patent for a collagen-based wound dressing containing PHMB, we do not have any additional material licenses to any technology or intellectual property rights. Under the terms of the exclusive license from Resorba, we were obligated to make minimum royalty payments of $1.0 million in each of 2018 and 2019, and were subject to a $2.5 million minimum royalty payment in 2017, as part of an ongoing low single-digit royalty payment on net sales of PuraPly AM; the term of the license shall continue for the life of the patent, which expires in October 2026. We may also terminate the license upon written notice to Resorba in the event that (i) the patent is invalidated or (ii) we stop all activities that would require a license to the patent, and either party may terminate the license in the event of a material breach by the other party, subject to notice and an ability to cure. In addition,
26
we were obligated to make upfront and maintenance payments totaling $0.6 million at specified periods prior to April 1, 2019, including a payment of $0.2 million that was made on July 1, 2018. The license is assignable but not sub-licensable.
As of December 31, 2022, we owned 36 issued patents globally, of which 15 were U.S. patents. As of December 31, 2022, we owned 13 pending patent applications, of which 9 were patent applications pending in the United States. Subject to payment of required maintenance fees, annuities, and other charges, many of our issued patents are currently expected to expire between 2027 and 2042. The expiration of these patents is not expected to have a material impact on our business. In addition, many of our products, including our Apligraf, Dermagraft, and NuShield products, are not covered by our issued patents or pending patent applications. Our issued patents are drawn to the following main areas: methods of making and using cultured tissue constructs, methods for preparing multi-layer stacks of living tissue, methods for treating recessed oral gingiva using cultured tissue constructs, methods of making and using osteogenic implants comprising a placental membrane sheet, wound treatment methods using amniotic stem cell solutions and placental membrane sheets, methods of generating cartilage in a skeletal joint using placental membrane preparations, hepatocyte growth factor- and hyaluronic acid-containing compositions and methods of using such compositions, methods making placental membrane preparations comprising hyaluronic acid, methods of harvesting or proliferating human prenatal stem cells, hypothermic morselized placental membrane storage methods, uses of human amniotic fluid for treating chronic wounds and joint diseases, and adjustable debridement curette apparatuses. Our pending patent applications encompass additional areas, including wound treating methods using morselized amnion tissue and amniotic-derived cells, methods of assessing native stem cell populations using cultured isolated stem cells and reference cell sources, visco-supplement compositions and musculoskeletal inflammatory treatment methods using same, wound care treatment and methods of making and using such treatment, model systems and methods to characterize anti-inflammatory activity, and porcine collagen compositions and methods of using such compositions. Our pending patent applications may not result in issued patents and we can give no assurance that any patents that have been issued or might be issued in the future will protect our current or future products or provide us with any competitive advantage. See the section titled “Risk Factors—Risks Related to Our Intellectual Property” for additional information.
Additionally, we own or have rights to trademarks or trade names that are used in our business and in conjunction with the sale of our products, including 10 U.S. trademark registrations and 11 foreign trademark registrations, as of December 31, 2022.
We also seek to protect our proprietary rights through a variety of methods, including confidentiality agreements and proprietary information agreements with suppliers, employees, consultants, and others who may have access to our proprietary information.
Government Regulation
FDA Regulation of Product Registration, Manufacture, and Promotion
We market medical products in the United States that have either been approved or cleared by the FDA prior to marketing, or do not require FDA premarket review. Our marketed products that have received marketing authorization from the FDA have done so under one of the following agency pathways: 510(k) clearance for a Class II medical device or approval of a PMA for a Class III medical device. These medical products are regulated by the FDA under the PHSA or the FDCA along with the FDA’s implementing regulations. These federal statutes and regulations govern, among other things, the following activities that we perform or are performed on our behalf and will continue to perform or have performed on our behalf: the production, research, development, testing, manufacture, quality control, packaging, labeling, storage, approval, advertising, and promotion, distribution of our products into interstate commerce, record keeping, service and surveillance, complaint handling, repair or recall of products, adverse event reporting and other field safety corrective actions.
FDA Regulatory Review and Approval Process
Unless an exemption applies or the product is a Class I device, each medical device that we market must first receive either 510(k) clearance or PMA approval from the FDA. In addition, certain modifications made to marketed devices also may require 510(k) clearance or approval of a PMA supplement. We maintain necessary clearances and approvals for products derived from porcine, bovine, and human tissues that are regulated by the FDA. PuraPly, PuraPly AM, PuraPly XT, PuraPly MZ, and PuraForce are medical devices that have been cleared for marketing under a number of 510(k)s for uses such as wound dressing, intraoral barrier, and surgical mesh. We also maintain medical device approvals for the Apligraf (P950032) and Dermagraft (P000036) devices, both approved by the FDA as chronic wound treatments.
27
With respect to the manufacture of medical devices and biologics, the FDA regulates and inspects equipment, facilities, laboratories, and processes used in the manufacturing and testing of products prior to providing approval to market products. After receiving approval from the FDA, additional regulatory review or inspection may be required if we make a material change in manufacturing equipment, location or process. Our manufacturing processes must comply with the FDA’s Quality System Regulation, or QSR, for our medical device products. The QSR requires that each device manufacturer establish and implement a quality system under which the manufacturer monitors the manufacturing process and maintains records that show compliance with FDA regulations and the manufacturer’s written specifications and procedures relating to the devices. Among other things, these regulations require that manufacturers establish performance requirements before production and follow requirements applicable to design controls, testing, record keeping, documentation, manufacturing standards, labeling, complaint handling, and management review.
Manufacturers of biologics must comply with applicable cGMP regulations, including quality control and quality assurance and maintenance of records and documentation. Manufacturers and others involved in the manufacture and distribution of such products also must register their establishments with the FDA and certain state agencies. Both domestic and foreign manufacturing establishments must register and provide additional information to the FDA upon their initial participation in the manufacturing process. Concurrent with clinical trials, companies usually complete additional preclinical studies and must also develop additional information about the physical characteristics of the biologic product candidate, as well as finalize a process for manufacturing the product candidate in commercial quantities in accordance with cGMP requirements. To help reduce the risk of the introduction of adventitious agents or of causing other adverse events with the use of biologic products, the PHSA emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other requirements, the sponsor must develop methods for testing the identity, strength, quality, potency, and purity of the final biologic product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the biologic product candidate does not undergo unacceptable deterioration over its shelf life.
The FDA conducts periodic visits, both announced and unannounced, to re-inspect our equipment, facilities, laboratories, and processes to confirm regulatory compliance. These inspections may include the manufacturing facilities of subcontractors. Following an inspection, the FDA may issue a report, known as a 483, listing instances where the manufacturer has failed to comply with applicable regulations and/or procedures or, if observed violations are severe and urgent, a warning letter. If the manufacturer does not adequately respond to a 483 or warning letter, the FDA may take enforcement action against the manufacturer or impose other sanctions or consequences, which may include:
In addition, we must comply with medical device reporting regulations and corrections and removal reporting regulations. Medical device reporting regulations require that manufacturers report to the FDA if their devices may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur. Corrections and removal reporting regulations require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health. The FDA may also order a mandatory recall if there is a reasonable probability that the device would cause serious adverse health consequences or death.
Certain human cells, tissues, and cellular and tissue-based products, or HCT/Ps, are regulated under Section 361 of the PHSA and are referred to as “Section 361 HCT/Ps” or simply “361 HCT/Ps,” while other HCT/Ps are subject to the FDA’s regulatory requirements for medical devices and/or biologics. A product that is regulated as a 361 HCT/P may be commercially distributed without prior FDA clearance or approval. Pursuant to 21 CFR 1271.10, in order to be regulated as a 361 HCT/P, and hence exempt from premarket review, an HCT/P must be minimally manipulated, intended for homologous use, and manufactured without being combined with another article (except for water, crystalloids, or sterilizing, preserving, or storage agents). The HCT/P must also either
28
have no systemic effect and not be dependent upon the metabolic activity of living cells for its primary function or, if it has a systemic effect, be intended for autologous use, for allogeneic use in a first-degree or second-degree blood relative or for reproductive use. We believe that Affinity and NuShield generally fulfill the relevant criteria under 21 CFR 1271.10. In light of the 361 HCT/P Guidance, our labeling and marketing claims for Affinity and NuShield clarify that they are intended for use as wound coverings, and thus qualify as Section 361 HCT/Ps. However, the FDA could disagree with our conclusion and require premarket approval or clearance for Affinity, NuShield, or any placental-based sheet product we presently have or may have in the future market, which would disrupt the marketing of these products, potentially expose us to regulatory sanctions, and have a material adverse effect on our business, financial condition and results of operations. Section 361 HCT/Ps are subject to specific FDA regulations that include cGTPs, donor eligibility determination requirements, adverse event reporting, and advertising and labeling requirements. cGTP regulations govern the methods used in, and the facilities and controls used for, the manufacture of HCT/Ps, including but not limited to all steps in recovery, donor screening, donor testing, processing, storage, labeling, packaging, and distribution.
Before testing any biologic product candidate in humans, the product candidate must undergo preclinical testing. Preclinical tests, also referred to as nonclinical studies, include laboratory evaluations of product chemistry, potency, toxicity, and formulation, as well as in vivo studies to assess the potential safety and activity of the product candidate and to establish a rationale for therapeutic use. The conduct of the preclinical tests must comply with federal regulations and requirements including GLPs. Concurrent with clinical trials, companies usually must complete some long-term preclinical testing, such as animal tests of reproductive adverse events and carcinogenicity, and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the drug in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality, and purity of the final drug product.
The clinical trial sponsor must submit the results of the preclinical studies, together with manufacturing information, analytical data, any available clinical data or literature, and a proposed clinical protocol, to the FDA as part of the Investigational New Drug Application (“IND”). The FDA may impose clinical holds on a biologic product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, clinical trials may not recommence without FDA authorization and then only under terms authorized by the FDA. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to commence, or that, once begun, issues will not arise that suspend or terminate such trials.
Clinical trials involve the administration of the biologic product candidate to volunteers or patients under the supervision of qualified investigators who generally are physicians not employed by, or under, the control of the trial sponsor. Clinical trials are conducted under written study protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical trial will be stopped if certain adverse events should occur. Each protocol and certain amendments to the protocol must be submitted to the FDA as part of the IND. Submission of an IND may or may not result in the FDA allowing clinical trials to commence. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations comprising the GCP requirements, including the requirement that all research subjects provide informed consent. Further, each clinical trial must be reviewed and approved by an Institutional Review Board (“IRB”) at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers items such as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical trial subject, or their legal representative, reviews and approves the trial protocol, and must monitor the clinical trial until completed.
29
Human clinical trials are typically conducted in three sequential phases that may overlap, be combined, or be bifurcated into two parts:
Post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up. Sometimes approval for a product is conditional upon the completion of post-marketing clinical studies.
During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the FDA. Written IND safety reports must be promptly submitted to the FDA, the IRB, and the investigators for: serious and unexpected suspected adverse reactions; any findings from other trials; findings from animal or in vivo laboratory tests or in vitro testing that suggest a significant risk for human subjects; or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report as soon as possible, but in no case later than 15 calendar days after the sponsor determines that the information qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction as soon as possible but no later than seven calendar days after the sponsor’s initial receipt of the information.
The FDA or the sponsor or its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the biologic product candidate has been associated with unexpected serious harm to patients.
Expedited Development and Review Programs
The FDA is authorized to expedite the review of BLAs in several ways. Under the Fast Track program, the sponsor of a biologic product candidate may request the FDA to designate the product for a specific indication as a Fast Track product concurrent with or after the filing of the IND. Biologic products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product candidate and the specific indication for which it is being studied. In addition to other benefits, such as the ability to have greater interactions with the FDA, the FDA may initiate review of sections of a Fast Track BLA before the application is complete, a process known as rolling review.
Any product submitted to the FDA for marketing, including under a Fast Track program, may be eligible for other types of FDA programs intended to expedite development and review, such as breakthrough therapy designation, regenerative medicine advance therapy designation, priority review and accelerated approval.
30
Fast Track designation, breakthrough therapy designation, RMAT designation and accelerated approval do not change the standards for approval but may expedite the development or approval process.
Post-approval Requirements
FDA regulation of biologic products continues after approval, particularly with respect to cGMP requirements, including quality control and quality assurance and maintenance of records and documentation. Other post-approval requirements applicable to biologic products include reporting of cGMP deviations that may affect the identity, potency, purity and overall safety of a distributed product, record-keeping requirements, reporting of adverse effects, reporting updated safety and efficacy information and complying with electronic record and signature requirements. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant or manufacturer to administrative or judicial civil or criminal actions and adverse publicity. These actions could include refusal to approve pending applications or supplemental applications, withdrawal of an approval, clinical hold, suspension or termination of a clinical trial by an IRB, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines or other monetary penalties, refusals of government contracts, mandated corrective advertising or communications with healthcare providers, debarment, restitution, disgorgement of profits or other civil or criminal penalties.
Medical Product Marketing and Promotion
Advertising, marketing and promotional activities for devices and biologics are also subject to FDA oversight and must comply with the statutory standards of the FDCA, and the FDA’s implementing regulations. The FDA’s oversight authority review of marketing and promotional activities encompasses, but is not limited to, direct-to-consumer advertising, healthcare provider-directed advertising and promotion, sales representative communications to healthcare professionals, promotional programming and promotional activities involving electronic media. The FDA also regulates industry-sponsored scientific and educational activities that make representations regarding product safety or efficacy in a promotional context. A sponsor also must comply with the FDA’s advertising and promotion requirements, such as the prohibition on promoting products for uses or in patient populations that are not described in the product’s approved labeling (known as “off-label use”). The FDA may take enforcement action against a company for promoting unapproved uses of a product or for other violations of its advertising and labeling laws and regulations. Enforcement actions may include product seizures, injunctions, civil or criminal penalties or regulatory letters, which may require corrective advertising or other corrective communications to healthcare professionals.
31
Government Advocacy
We engage in public policy advocacy with policymakers and continue to work to demonstrate that our therapeutic products provide value to patients and to those who pay for health care. We advocate with government policymakers to encourage a long-term approach to sustainable health care financing that ensures access to innovative medicines and does not disproportionately target FDA-regulated medical devices and biologics as a source of budget savings. In markets with historically low rates of health care spending, we encourage those governments to increase their investments and adopt market reforms in order to improve their citizens’ access to appropriate health care.
Regulations Governing Reimbursement/Fraud and Abuse
Within the United States, our products and our customers are subject to extensive regulation by a wide range of federal and state agencies. These agencies regulate the coverage and reimbursement of our products, and prohibit activities that might result in health care fraud and abuse against patients and insurance programs. Internationally, other governments also impose regulations in connection with their health care reimbursement programs and the delivery of health care items and services.
U.S. federal health care fraud and abuse laws generally apply to our activities because our products are covered under federal healthcare programs such as Medicare and Medicaid. The principal U.S. federal health care fraud and abuse laws applicable to us and our activities include: (1) the Anti-Kickback Statute, which prohibits the knowing and willful offer, solicitation, payment or receipt of anything of value in order to generate business reimbursable by a federal health care program; (2) the False Claims Act, which prohibits the submission of false or otherwise improper claims for payment to a federally funded health care program, including claims resulting from a violation of the Anti-Kickback Statute; and (3) health care fraud statutes that prohibit false statements and fraudulent and abusive claims made to any third-party payer.
The Anti-Kickback Statute is particularly relevant because of its broad applicability. Specifically, the Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving, or providing remuneration, directly or indirectly, in exchange for, or to induce, either the referral of an individual, or the furnishing, arranging for or recommending a good or service for which payment may be made in whole or part under federal health care programs, such as the Medicare and Medicaid programs. Depending on the circumstances, almost any financial interaction with a healthcare provider, patient or customer could implicate the Anti-Kickback Statute. Statutory exceptions and regulatory safe harbors protect certain interactions from prosecution if all specified requirements are met. However, most safe harbors or exceptions require, among other things, fair market value exchanges. The government can exercise enforcement discretion in taking action against unprotected activities. Many types of interactions in which we commonly engage, such as customer support services, could implicate the Anti-Kickback Statute, are not protected by a safe harbor or exception and have been the subject of government scrutiny and enforcement action when not structured appropriately. If the government determines that these activities are abusive, we could be subject to enforcement action. Other companies that manufacture wound care products have been subject to government scrutiny and enforcement action. For example, in early 2017, Shire Pharmaceuticals LLC and other subsidiaries of Shire plc agreed to pay $350 million to settle federal and state False Claims Act allegations that Shire and the company that Shire acquired in 2011, Advanced BioHealing, employed kickbacks and other unlawful methods to induce clinics and physicians to use or overuse its product Dermagraft (a product we subsequently acquired). Penalties for Anti-Kickback Statute violations may include both criminal penalties such as imprisonment and civil sanctions such as fines and possible exclusion from Medicare, Medicaid, and other federal health care programs. Exclusion would mean that our products would no longer be eligible for reimbursement under federal healthcare programs.
There are similar state false claims, anti-kickback, and insurance laws that apply to state-funded Medicaid and other health care programs as well as to commercial third-party payers. Insurance companies may also bring a private cause of action for treble damages against a manufacturer for a pattern of causing false claims to be filed under the federal Racketeer Influenced and Corrupt Organizations Act, or RICO. In addition, the Foreign Corrupt Practices Act, or FCPA, may be used to prosecute companies in the United States for arrangements with physicians, or other parties outside the United States if the physician or party is a government official of another country and the arrangement violates the laws of that country.
In addition to receiving scrutiny and providing potential grounds for action under the Anti-Kickback Statute, pricing, sales and marketing practices of medical device and pharmaceutical manufacturers are also subject to tightly focused regulation at the federal and state levels. Federal law and regulation, for example, establish pricing methodologies for government health insurance programs and require regular reporting of sales information to CMS in support of manufacturer price calculations. In recent years, the federal government and a growing number of states have introduced new drug price transparency requirements that can require extensive information disclosures to agencies or potential purchasers relating to drug price increases. Health care laws and regulations generally limit financial interactions between manufacturers and health care providers; require pharmaceutical and medical device companies to comply with voluntary compliance standards issued by industry associations and the relevant compliance guidance promulgated by the U.S. federal government; and/or require disclosure to the government and/or public of financial interactions (so-called “sunshine laws”). Many of these laws and regulations contain ambiguous requirements or require administrative guidance for implementation. Manufacturers must adopt reasonable interpretations of requirements if there is ambiguity and those interpretations could be
32
challenged. Given the lack of clarity in laws and their implementation, our activities could be subject to the penalty provisions of the pertinent federal and state laws and regulations.
The healthcare laws and regulations applicable to us, including those described above, are subject to evolving interpretations and enforcement discretion. If a governmental authority were to conclude that we are not in compliance with applicable laws and regulations, we and our officers and employees could be subject to severe criminal and civil financial penalties, including, for example, exclusion from participation as a supplier of product to beneficiaries covered by Medicare or Medicaid. Any failure to comply with laws and regulations relating to reimbursement and health care goods and services could adversely affect our reputation, business, financial condition and cash flows. To help ensure compliance with the laws and regulations governing the provision of health care goods and services, we have implemented a comprehensive compliance program based on the HHS Office of Inspector General’s Seven Elements of an Effective Compliance Program. Despite our compliance program, we cannot be certain that we have always operated in full compliance with all applicable healthcare laws.
Our profitability and operations are subject to risks relating to changes in legislative, regulatory, and reimbursement policies and decisions as well as changes to private payer reimbursement coverage and payment decisions and policies. Implementation of further legislative or administrative reforms to reimbursement systems, or adverse decisions relating to our products by administrators of these systems in coverage or reimbursement, could significantly reduce reimbursement or result in the denial of coverage, which could have an impact on the acceptance of and demand for our products and the prices that our customers are willing to pay for them.
Seasonality
Revenues during our fourth quarter tend to be stronger than other quarters because many hospitals increase their purchases of our products during the fourth quarter to coincide with the end of their budget cycles in the United States. Satisfaction of patient deductibles through the course of the year also results in increased revenues later in the year. In general, our first quarter usually has lower revenues than the preceding fourth quarter, the second and third quarters have higher revenues than the first quarter, and the fourth quarter revenues are the highest in the year.
Human Capital Resources
As of December 31, 2022, we had approximately 1,030 employees worldwide. None of our employees are represented by a collective bargaining agreement. We have never experienced a work stoppage. We believe our employee relations are good.
In managing our business, we focus on a number of measures and objectives with respect to the attraction, development, and retention of our employees that we believe are important to our business, including diversity, communication, compensation, tenure, professional development, and health, well-being and safety:
33
Available Information
Our Internet website address is http://www.organogenesis.com. Through our website, we make available, free of charge, our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and any amendments to those reports, as well as proxy statements, and, from time to time, other documents as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities and Exchange Commission, or SEC. These SEC reports can be accessed through the “Investors” section of our website. The information found on our website is not part of this or any other report we file with or furnish to the SEC.
Item 1A. Risk factors
Summary of Risk Factors
Below is a summary of the principal factors that make an investment in our Class A common stock speculative or risky. This summary does not address all of the risks that we face. Additional discussion of the risks summarized in this risk factor summary, and other risks that we face, can be found below under the heading “Risk Factors” and should be carefully considered, together with other information in this Form 10-K and our other filings with the SEC before making an investment decision regarding our Class A common stock.
34
35
Risk Factors
You should carefully consider the risks and uncertainties described below, together with the information included elsewhere in this Annual Report on Form 10-K and other documents we file with the SEC. The risks and uncertainties described below are those that we have identified as material, but are not the only risks and uncertainties facing us. Our business is also subject to general risks and uncertainties that affect many other companies, such as overall U.S. and non-U.S. economic and industry conditions including a global economic slowdown, geopolitical events, changes in laws or accounting rules, fluctuations in interest and exchange rates, terrorism, international conflicts, major health concerns, natural disasters or other disruptions of expected economic and business conditions. Additional risks and uncertainties not currently known to us or that we currently believe are immaterial also may impair our business operations and liquidity.
Risks Related to Organogenesis and its business
Our operating results may fluctuate significantly as a result of a variety of factors, many of which are outside of our control.
We are subject to the following factors, among others, that may negatively affect our operating results:
36
We have based our current and future expense levels largely on our investment plans and estimates of future events, although certain of our expense levels are, to a large extent, fixed. We may be unable to adjust spending in a timely manner to compensate for any unexpected revenue shortfall. Accordingly, any significant shortfall in revenue relative to our planned expenditures would have an immediate adverse effect on our business, results of operations, and financial condition. Further, as a strategic response to changes in the competitive environment or to changes in laws and regulations, we may from time to time make certain pricing, service, or marketing decisions (e.g., reduce prices) that could have a material and adverse effect on our business, results of operations, and financial condition. Due to the foregoing factors, our revenue and operating results are and will remain difficult to forecast.
We have incurred significant losses in past years and, notwithstanding our reported net income for the 2020, 2021, and 2022 fiscal years, we may incur losses in the future.
To date, we have financed our operations primarily through debt and equity financings, and, with the exception of the fiscal years ended December 31, 2022, 2021, and 2020, in which we reported net income of $15.5 million, $94.2 million and $17.2 million, respectively, we have incurred losses from operations in many years since our inception. As of December 31, 2022, we had an accumulated deficit of $45.3 million. We expect to incur significant sales and marketing costs to support the sale of our products. Our prior losses, combined with any potential future losses, may have an adverse effect on our business, results of operations, and financial condition.
We have identified material weaknesses in our internal control over financial reporting, and our management has concluded that our disclosure controls and procedures are not effective.
A “material weakness” is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our financial statements will not be prevented or detected on a timely basis. We did not design and maintain effective controls (i) to properly identify and assess significant non-routine transactions and (ii) over information technology general controls and proper segregation of duties to support the proper initiation and recording of transactions and the resulting impact on business process controls and applications that rely on such data.
While we have successfully addressed certain of the internal control deficiencies that were included in the aggregation of the previously reported material weakness, we are continuing to work on remediating the remaining internal control deficiencies, as well as other internal control deficiencies identified during the current period, that collectively are aggregating to form the material weaknesses in our internal controls over financial reporting that exists as of December 31, 2022. However, we cannot assure you that additional material weaknesses or significant deficiencies will not occur in the future. If our internal control over financial reporting or our disclosure controls and procedures are not effective, we may not be able to accurately report our financial results or prevent fraud, which may cause investors to lose confidence in our reported financial information and may lead to a decline in our stock price.
Although we have made certain progress in remediating these material weaknesses, we concluded that the material weaknesses described above continued to exist as of December 31, 2022. We have taken actions to remediate the deficiencies in our internal controls over financial reporting and implemented additional processes and controls designed to address the underlying causes of the above-mentioned material weaknesses. If we do not successfully remediate the material weaknesses described above, or if other material weaknesses or other deficiencies arise in the future, we may be unable to accurately report our financial results, which could cause our financial results to be materially misstated and require restatement.
Rapid technological change could cause our products to become obsolete, and if we do not enhance our product offerings through our research and development efforts, we may be unable to effectively compete.
The technologies underlying our products are subject to rapid and profound technological change. Competition intensifies as technical advances in each field are made and become more widely known. We can give no assurance that others will not develop services, products, or processes with significant advantages over the products, services, and processes that we offer or are seeking to develop. Any such occurrence could have a material and adverse effect on our business, results of operations, and financial condition.
We plan to enhance and broaden our product offerings in response to changing customer demands and competitive pressure and technologies, but we may not be successful. The success of any new product offering or enhancement to an existing product will depend on numerous factors, including our ability to:
37
If we do not develop and, when necessary, obtain regulatory clearance or approval for new products or product enhancements in time to meet market demand, or if there is insufficient demand for these products or enhancements, our results of operations will suffer. Our research and development efforts may require a substantial investment of time and resources before we are adequately able to determine the commercial viability of a new product, technology, material or other innovation. In addition, even if we are able to successfully develop enhancements or new generations of our products, these enhancements or new generations of products may not be covered or reimbursed by government healthcare programs such as Medicare or private health plans, may not produce sales in excess of the costs of development and/or may be quickly rendered obsolete by changing customer preferences or the introduction by our competitors of products embodying new technologies or features.
To be commercially successful, we must convince physicians that our products are safe and effective alternatives to existing treatments and that our products should be used in their procedures.
We believe physicians will only adopt our products if they determine, based on experience, clinical data and published peer-reviewed journal articles, that the use of our products in a particular procedure is a favorable alternative to conventional methods. Physicians also are more interested in using cost-effective products and may practice in settings like Accountable Care Organizations, or ACOs, or Medical Homes, where they face considerable cost-containment pressure. In general, physicians may be slow to change their medical treatment practices and use of our products for the following reasons, among others:
The degree of market acceptance of our products will continue to depend on a number of factors, including:
38
In addition, we are currently conducting clinical studies for some of our products that were brought to market as 361 HCT/Ps to generate efficacy data in various clinical applications. Unfavorable results from these 361 HCT/P clinical trials such as lack of clinical efficacy or serious treatment-related side effects could negatively affect the use and adoption of our products by physicians and hospitals, thereby compromising our market acceptance.
We believe recommendations for, and support of our products by, influential physicians are essential for market acceptance and adoption. If we do not receive this support (e.g., because we are unable to demonstrate favorable long-term clinical data), physicians and hospitals may not use our products, which would significantly reduce our ability to achieve expected revenue and would prevent us from sustaining profitability.
In the course of conducting our business, we must comply with regulatory quality requirements, and adequately address quality issues that may arise with our products, as well as defects in third-party components included in our products. Although we have established internal procedures to minimize risks that may arise from quality issues, we may not be able to eliminate or mitigate these risks and quality issues may arise in which case we would be subject to liability. If the quality of our products does not meet the expectations of regulators, physicians, or patients, then we could be subject to regulatory sanctions and our brand and reputation could suffer and our business, results of operations, and financial condition could be adversely impacted.
We face the risk of product liability claims and may not be able to obtain or maintain adequate product liability insurance.
Our business exposes us to the risk of product liability claims that are inherent in the manufacturing, processing, investigating, and marketing of medical devices and human tissue products. We are, and may in the future be, subject to product liability claims and lawsuits, including potential class actions or mass tort claims, alleging that our products have resulted or could result in an unsafe condition or injury. Product liability claims may be made by patients and their families, healthcare providers, or others selling our products. Defending a lawsuit, regardless of merit, could be costly, divert management attention, and result in adverse publicity, which could result in the withdrawal of, or reduced acceptance of, our products in the market. If we cannot successfully defend against product liability claims, we could incur substantial liability and costs. In addition, regardless of merit or eventual outcome, product liability claims may result in:
Although we have product liability insurance that we believe is adequate, this insurance is subject to deductibles and coverage limitations and we may not be able to maintain this insurance. Also, it is possible that claims could exceed the limits of our coverage or be excluded from coverage under our policy. If we are unable to maintain product liability insurance at an acceptable cost or on acceptable terms with adequate coverage or otherwise protect ourselves against potential product liability claims or we underestimate the amount of insurance we need, we could be exposed to significant liabilities, which may harm our business. One or more product liability claims could cause our stock price to decline and, if our liability exceeds our insurance coverage, could adversely affect our business, results of operations, and financial condition.
Interruptions in the supply of our products or inventory loss may adversely affect our business, results of operations, and financial condition.
Our products are manufactured using technically complex processes requiring specialized facilities, highly specific raw materials, and other production constraints. The complexity of these processes, as well as strict company and government standards
39
for the manufacture and storage of our products, subjects us to production risks. In addition to ongoing production risks, process deviations or unanticipated effects of approved process changes may result in non-compliance with regulatory requirements including stability requirements or specifications. Most of our products must be stored and transported within a specified temperature range. For example, if environmental conditions deviate from that range, our products’ remaining shelf-lives could be impaired or their safety and efficacy could be adversely affected, making them unsuitable for use. These deviations may go undetected. The occurrence of actual or suspected production and distribution problems can lead to lost inventories, and in some cases recalls, with consequential reputational damage and the risk of product liability. The investigation and remediation of any identified problems can cause production delays and result in substantial additional expenses. Production of our Affinity product, for example, was suspended in the first quarter of 2019 due to production issues at one of our suppliers. As a result, we identified an alternate supplier, and were only able to resume commercial-scale production in the second quarter of 2020. Subsequently, we have added a second source to provide additional capacity and redundancy in supply. This disruption in supply resulted in reduced Affinity revenue. Although we were able to partially offset the lost Affinity revenue by increasing production of our other products, there can be no assurance that we will be able to do so in the event of any future suspensions or failures in the storage or manufacturing of Affinity, Dermagraft or our other products. Any future failure in the storage or manufacture of our products or loss in supply could result in a loss of our market share and negatively affect our revenues and operations.
As noted above, manufacturing of Dermagraft was suspended in the fourth quarter of 2021, and sales of Dermagraft were suspended in the second quarter of 2022. We plan to transition our Dermagraft manufacturing to a new manufacturing facility or engage a third-party manufacturer, which we expect will result in substantial long-term cost savings. In the period when Dermagraft is not available, we expect that customers will be willing to substitute Apligraf for Dermagraft and that the suspension of Dermagraft sales will not have a material impact on our net revenue. However, if we do not realize the expected substantial long-term cost savings or if customers are unwilling to substitute Apligraf for Dermagraft during the period in which Dermagraft is unavailable, it could have an adverse effect on our net revenue and results of operations.
Because we depend upon a limited group of suppliers and manufacturers for our products, including our NuShield, Affinity, Apligraf and PuraPly Antimicrobial products, we may incur significant product development costs and experience material delivery delays if we lose any significant supplier, which could materially impact sales of our products.
We obtain some of the components for our products from a limited group of suppliers. For us to be successful, our suppliers must be able to provide us with these components in substantial quantities, in compliance with regulatory requirements, in accordance with agreed-upon specifications, at acceptable costs, and on a timely basis. Our efforts to maintain a continuity of supply and high quality and reliability may not be successful. Manufacturing disruptions experienced by our suppliers may jeopardize our supply of these components. Due to the stringent regulations and requirements of the FDA regarding the manufacture of our products, we may not be able to quickly establish additional or replacement sources for certain components or materials. A change in suppliers could require significant effort or investment in circumstances where the items supplied are integral to product performance or incorporate unique technology. A reduction or interruption in manufacturing (including the current suspension of Dermagraft manufacturing pending its transition to a new manufacturing facility or engagement of a third-party manufacturer), or an inability to secure alternative sources of raw materials or components, could have a material effect on our business, results of operations, and financial condition. In addition, one or more of our suppliers may refuse to extend us credit with respect to our purchasing or leasing equipment, supplies, products, or components, or may only agree to extend us credit on significantly less favorable terms or subject to more onerous conditions. This could significantly disrupt our ability to purchase or lease required equipment, supplies, products and components in a cost-effective and timely manner and could have a material adverse effect on our business, results of operations, and financial condition. Any casualty, natural disaster, other disruption of any of our sole-source suppliers’ operations, or any unexpected loss of any existing exclusive supply contract, could have a material adverse effect on our business, results of operations, and financial condition.
Our products are dependent on the availability of tissue from human donors, and any disruption in supply could adversely affect our business, results of operations, and financial condition.
Many of the products that we manufacture require that we obtain human tissue. The success of our business depends upon, among other factors, the availability of tissue from human donors. Any failure to obtain tissue from our sources will interfere with our ability to effectively meet the demand for our products incorporating human tissue. The processing of human tissue for our products is very labor-intensive and it is therefore difficult to maintain a steady supply stream. The availability of donated tissue could also be adversely impacted by regulatory changes, public opinion of the donor process as well as our own reputation in the industry. The challenges we may face in obtaining adequate supplies of human tissue involve several risks, including limited control over the availability, quality, and delivery schedules. In addition, any interruption in the supply of any human tissue component could materially harm our ability to manufacture our products until a new source of supply, if any, could be found. We may be unable to find a sufficient alternative supply channel in a reasonable time period or on commercially reasonable terms, if at all, which would have a material adverse effect on our business, results of operations, and financial condition.
40
Increased prices for, or unavailability of, raw materials used in our products could adversely affect our business, results of operations, and financial condition.
Our profitability is affected by the prices of the raw materials used in the manufacture of our products. These prices may fluctuate based on a number of factors beyond our control, including changes in supply and demand, general economic conditions, labor costs, fuel-related delivery costs, competition, import duties, excises and other indirect taxes, currency exchange rates, and government regulation. Due to the highly competitive nature of the healthcare industry and the cost containment efforts of our customers and third-party payers, we may be unable to pass along cost increases for key components or raw materials through higher prices to our customers. If the cost of key components or raw materials increases, and we are unable fully to recover these increased costs through price increases or offset these increases through other cost reductions, we could experience lower margins and profitability. Significant increases in the prices of raw materials, due to inflation or otherwise, that cannot be recovered through productivity gains, price increases or other methods could adversely affect our business, results of operations, and financial condition.
We continue to invest significant capital to maximize our sales and marketing infrastructure, and there can be no assurance that these efforts will result in significant increases in sales.
We are committed to maximizing our internal sales and marketing capabilities, including by optimizing our sales force to further support the marketing and sales of the products acquired in connection with our 2017 acquisition of NuTech Medical and our 2020 acquisition of CPN Biosciences. As a result, we continue to invest in sales and marketing resources for our products to allow us to reach new customers and potentially increase sales. These expenses impact our operating results, and there can be no assurance that we will continue to be successful in significantly increasing the sales of our products.
The impairment or termination of our relationships with independent sales agencies, whom we do not control, could materially and adversely affect our ability to generate revenues and profits. We intend to develop additional relationships with independent sales agencies in order to increase revenue from certain of our products; our inability to do so may prevent us from increasing sales.
We derive a portion of our revenues through our relationships with independent sales agencies. The impairment or termination of these relationships for any reason could materially and adversely affect our ability to generate revenues and profits. Because the independent sales agency often controls the customer relationships within its territory, there is a risk that if our relationship with the independent sales agency ends, our relationship with the customer will be lost. Also, because we do not control an independent sales agency’s field sales agents, there is a risk we will be unable to ensure that our sales processes, regulatory compliance, and other priorities will be consistently communicated and executed by the distributor. If we fail to maintain relationships with our key independent sales agencies, or fail to ensure that our independent sales agencies adhere to our sales processes, regulatory compliance, and other priorities, this could have an adverse effect on our business, results of operations, and financial condition. We may have liability for the actions of independent sales agencies in marketing our products and our lack of control over their activities impedes our ability to prevent, detect or address such non-compliance.
We intend to develop relationships and arrangements with additional independent sales agencies in order to increase our sales with respect to certain of our products. However, we may fail to develop such relationships, in which case we may not be able to increase our sales. Our success is partially dependent upon our ability to retain and motivate our independent sales agencies and their representatives to sell our products in certain territories. They may not be successful in implementing our marketing plans. Some of our independent sales agencies may not sell our products exclusively and may offer similar products from other companies. Our independent sales agencies may terminate their contracts with us, may devote insufficient sales efforts to our products, or may focus their sales efforts on other products that produce greater commissions for them, which could have an adverse effect on our business, results of operations, and financial condition. We also may not be able to find additional independent sales agencies who will agree to market and/or distribute those products on commercially reasonable terms, if at all. If we are unable to establish new independent sales agency relationships or renew current sales agency agreements on commercially acceptable terms, our business, results of operations, and financial condition could be materially and adversely affected. In addition, because we do not control these independent sales agencies as closely as our employees, while we may take steps to mitigate the risks associated with noncompliance by independent sales agencies, there remains a risk they do not comply with regulatory requirements or our requirements or our policies which could also adversely affect our business.
We will need to continue to expand our organization, and managing growth may be more difficult than expected.
Managing our growth may be more difficult than we expect. We anticipate that a period of significant expansion will be required to penetrate and service the markets for our existing and anticipated future products and to continue to develop new products. This expansion will place a significant strain on management, operational and financial resources. To manage the expected growth of our operations, we must both modify our existing operational and financial systems, procedures and controls and implement new systems, procedures and controls. We must also expand our finance, administrative, and operations staff. Management may be unable
41
to hire, train, retain, motivate, and manage necessary personnel or to identify, manage, and exploit existing and potential strategic relationships and market opportunities.
In addition to expanding our organization, we are expanding our manufacturing capabilities, which requires significant capital expenditures. If these capital expenditures are higher than expected, it may adversely affect our financial condition and capital resources. In addition, if the expansion of our manufacturing facilities is delayed, for regulatory or other reasons, it may limit our ability to expand the size of our organization and to meet our corporate goals. Even if we are able to expand our manufacturing facilities as we plan, we may not realize the full expected benefit of our investment.
We may expand our business through acquisitions, similar to our acquisitions of NuTech Medical and CPN Biosciences, licenses, investments, and other commercial arrangements in other companies or technologies. Such acquisitions or commercial arrangements may entail significant risks.
We periodically evaluate strategic opportunities to acquire companies, divisions, technologies, products, and rights through licenses, distribution agreements, investments, and outright acquisitions to grow our business, such as our acquisitions of NuTech Medical and CPN Biosciences. In connection with one or more of those transactions, we may:
Any of these items could materially and adversely affect our revenues, financial condition, and profitability. Business acquisitions also involve the risk of unknown liabilities associated with the acquired business, which could be material. Our acquisition of NuTech Medical and CPN Biosciences expanded our wound care portfolio and our acquisition of NuTech Medical broadened our addressable market to include the Surgical & Sports Medicine market. We may not realize the increased revenues, cost savings, and synergies that we anticipate from this acquisition in the near term or at all due to many factors, including delays in the integration process, an inability to successfully penetrate the amniotic category of the wound care market or an inability to obtain necessary regulatory approvals. Additional liabilities related to acquisitions could include a lack of compliance with government regulations that could subject us to investigation and civil and criminal sanctions. For example, we may acquire a company that was not compliant with FDA quality requirements or was making payments or other forms of remuneration to physicians to induce them to use their products. Incurring unknown liabilities or the failure to realize the anticipated benefits of an acquisition could materially and adversely affect our business and we may lose our entire investment or be unable to recover our initial investment, which could include the cost of acquiring licenses or distribution rights, acquiring products, purchasing initial inventory, or investments in early-stage companies. Inability to recover our investment, or any write off of such investment, associated goodwill, or assets, could have a material and adverse effect on our business, results of operations, and financial condition.
New lines of business or new products and services may subject us to additional risks.
From time to time, we may implement or may acquire new lines of business, such as our Surgical & Sports Medicine products that were acquired in connection with our acquisition of NuTech Medical, or we may offer new products and services within existing lines of business. There are risks and uncertainties associated with these efforts, particularly in instances where the markets are not fully developed or are evolving. In developing and marketing new lines of business and new products and services, we may invest significant time and resources. External factors, such as regulatory compliance obligations, competitive alternatives, lack of market acceptance, and shifting market preferences, may also affect the successful implementation of a new line of business or a new product
42
or service. Failure to successfully manage these risks in the development and implementation of new lines of business or new products or services could have a material adverse effect on our business, results of operations, and financial condition.
Significant disruptions of information technology systems or breaches of information security could adversely affect our business, results of operations, and financial condition.
We rely to a large extent upon sophisticated information technology systems to operate our business. In the ordinary course of business, we collect, store, and transmit large amounts of confidential information (including, but not limited to, personal information and intellectual property). We also have outsourced significant elements of our operations to third parties, including significant elements of our information technology infrastructure and, as a result, we are managing many independent vendor relationships with third parties who may or could have access to our confidential information. The size and complexity of our information technology and information security systems, and those of our third-party vendors with whom we contract (and the large amounts of confidential information that is present on them), make such systems potentially vulnerable to service interruptions or to security breaches from inadvertent or intentional actions by our employees or vendors, or from malicious attacks by third parties. Such attacks are of ever-increasing levels of sophistication and are made by groups and individuals with a wide range of motives (including, but not limited to, industrial espionage and market manipulation) and expertise. While we have invested significantly in the protection of data and information technology, there can be no assurance that our efforts will prevent service interruptions or security breaches. For example, in August 2020, our information technology (“IT”) systems were exposed to a ransomware attack, which partially impaired certain IT systems for a short period of time. We finished investigating the incident, together with legal counsel and other incident response professionals. We did not experience any material losses related to the ransomware attack and were able to recover all data quickly, with only a minimal and temporary interruption to our business. While we have implemented measures to protect our data security and information technology systems, such measures may not prevent these events. Although we have cyber-insurance coverage that may cover certain events described above, this insurance is subject to deductibles and coverage limitations and we may not be able to maintain this insurance. Also, it is possible that claims could exceed the limits of our coverage. Any interruption or breach in our systems could adversely affect our business operations and/or result in the loss of critical or sensitive confidential information or intellectual property, and could result in financial, legal, business, and reputational harm to us or allow third parties to gain material, inside information that they use to trade in our securities.
If a breach of our measures protecting personal data covered by HIPAA, the HITECH Act, or the CCPA occurs, we may incur significant liabilities.
The Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the HITECH Act, and the regulations that have been issued under it, impose certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of protected health information. The requirements and restrictions apply to “covered entities” (which include health care providers and insurers) as well as to their business associates that receive protected health information from them in order to provide services to or perform certain activities on their behalf. The statute and regulations also impose notification obligations on covered entities and their business associates in the event of a breach of the privacy or security of protected health information. We occasionally receive protected health information from our customers in the course of our business. As such, we believe that we are business associates and therefore subject to HIPAA’s requirements and restrictions with respect to handling such protected health information, and have executed business associate agreements with certain customers.
In addition, California has enacted the California Consumer Privacy Act (“CCPA”), which came into effect on January 1, 2020. Pursuant to the CCPA, certain businesses are required, among other things, to make certain enhanced disclosures related to California residents regarding the use or disclosure of their personal information, allow California residents to opt-out of certain uses and disclosures of their personal information without penalty, provide Californians with other choices related to personal data in our possession, and obtain opt-in consent before engaging in certain uses of personal information relating to Californians under the age of 16. The California Attorney General may seek substantial monetary penalties and injunctive relief in the event of our non-compliance with the CCPA. The CCPA also allows for private lawsuits from Californians in the event of certain data breaches. Aspects of the CCPA remain uncertain, and we may be required to make modifications to our policies or practices in order to comply. Aside from California, Texas and several other major states impose rigorous local medical privacy requirements.
It is possible the data protection laws may be interpreted and applied in a manner that is inconsistent with our practices. If so, this could result in government-imposed fines or orders requiring that we change our practices, which could adversely affect our business. In addition, these privacy regulations may differ from country to country and state to state, and may vary based on whether testing is performed in the United States or in the local country. Complying with these various laws and regulations could cause us to incur substantial costs or require us to change our business practices and compliance procedures in a manner adverse to our business. Further, compliance with data protection laws and regulations could require us to take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, or in some cases, impact our ability to operate in certain jurisdictions. We can provide no assurance that we are or will remain in compliance with diverse privacy and security requirements in all of the jurisdictions
43
in which we do business. If we fail to comply or are deemed to have failed to comply with applicable privacy protection laws and regulations such failure could result in government enforcement actions and create liability for us, which could include substantial civil and/or criminal penalties, as well as private litigation and/or adverse publicity that could negatively affect our operating results and business.
We engage in transactions with related parties and such transactions present possible conflicts of interest that could have an adverse effect on our business, results of operations, and financial condition.
We have entered into a significant number of transactions with related parties. Related party transactions create the possibility of conflicts of interest with regard to our management, including that:
Such conflicts could cause an executive officer or a director to seek to advance his or her economic interests or the economic interests of certain related parties above ours. Conversely, we may not be able to enter into transactions with third parties on terms as favorable as the terms of existing transactions with related parties. Further, the appearance of conflicts of interest created by related party transactions could impair the confidence of our investors. It is possible that a conflict of interest could have a material adverse effect on our business, results of operations, and financial condition.
Our financial performance may be adversely affected by medical device tax provisions in healthcare reform laws.
The Patient Protection and Affordable Care Act (the “PPACA”) imposed, among other things, an excise tax of 2.3% on any entity that manufactures or imports medical devices offered for sale in the United States. Under these provisions, the Congressional Research Service predicted that the total cost to the medical device industry may be up to $20 billion over a decade. The Internal Revenue Service issued final regulations implementing the tax in December 2012, which required, among other things, bi-monthly payments and quarterly reporting. The Consolidated Appropriations Act, 2016 (Pub. L. 114-113), signed into law in December 2015, included a two-year moratorium on the medical device excise tax. A second two-year moratorium on the medical device excise tax was signed into law in January 2018 as part of the Extension of Continuing Appropriations Act, 2018 (Pub. L. 115-120), extending the moratorium through December 31, 2019. On December 20, 2019, President Trump signed into law a permanent repeal of the medical device tax under the PPACA, but there is no guarantee that Congress will not reverse course in the future. If such an excise tax on sales of our products in the United States is enacted, it could have a material adverse effect on our business, results of operations, and financial condition.
We could incur asset impairment charges related to certain leasehold improvements, which could adversely affect our business, results of operations, and financial condition.
Our long-term assets include property, plant and equipment of $102.5 million and $79.2 million as of December 31, 2022 and 2021, respectively. We review our long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. If an asset is determined to be impaired, the asset is written down to fair value, which is determined based on appraised value. Any such impairment could result in a non-cash charge equal to the full value of these improvements. During the years ended December 31, 2022, 2021, and 2020, we did not recognize an impairment charge with respect to our long-lived assets. Changes in our assumptions with respect to our expected use of these assets may result in an impairment charge in the future, which could adversely affect our business, results of operations, and financial condition.
We may be required to record a significant charge to earnings if our goodwill and other amortizable intangible assets, or other investments become impaired.
We are required under generally accepted accounting principles to test goodwill for impairment at least annually and to review our goodwill, amortizable intangible assets, and other assets acquired through merger and acquisition activity, for impairment when events or changes in circumstance indicate the carrying value may not be recoverable. Factors that could lead to impairment of goodwill, amortizable intangible assets, and other assets acquired via acquisitions include significant adverse changes in the business climate and actual or projected operating results (affecting our company as a whole or affecting any particular segment) and declines
44
in the financial condition of our business. We may be required in the future to record additional charges to earnings if our goodwill, amortizable intangible assets, or other investments become impaired. Any such charge would adversely impact our financial results.
Our ability to use our net operating loss carryforwards may be subject to certain limitations.
As of December 31, 2022, we had approximately $44.4 million of federal net operating loss carry-forwards available for the reduction of future years’ federal taxable income, all of which can be carried forward indefinitely. Under the Internal Revenue Code of 1986, as amended, or the Code, the deductibility of the net operating loss-carry-forward as of December 31, 2022 and all future net operating loss-carry-forwards is limited to 80% of taxable income, limiting or delaying in part the use of net operating loss-carry-forwards. As of December 31, 2022, we also had state net operating loss carry-forwards of approximately $14.3 million expiring from the year ended December 31, 2031 through 2038. It is uncertain whether and to what extent applicable state tax laws will conform to the federal rule, though we are already subject to limitations in net operating loss utilization in certain states.
In addition, our ability to utilize our federal net operating loss carryforwards may be limited under Section 382 of the Code. In the event of an “ownership change”, Section 382 imposes an annual limitation on the amount of post-ownership change taxable income that may be offset with pre-ownership change net operating losses of the loss corporation experiencing the ownership change. An “ownership change” is defined by Section 382 as a cumulative change in ownership of our company of more than 50% within a three-year period. As of December 31, 2021, we performed a study and determined that there is no limitation on our federal net operating losses. Current or future changes in our stock ownership may trigger an “ownership change,” some of which may be outside our control. Accordingly, our ability to utilize our net operating loss carryforwards to offset federal taxable income, if any, could be limited by Section 382, which could potentially result in increased future tax liability to us.
We are dependent on the proper functioning of our and third-party manufacturing facilities, our supply chain, and our sales force, all of which could be negatively impacted by public health emergencies, including the COVID-19 pandemic, or other factors, in a manner that could materially adversely affect our business, financial condition or results of operations.
We manufacture our non-placental-based products and use third-party manufacturers for our placental-based products and we use third-party raw material suppliers to support our internal manufacturing processes. If our manufacturing capabilities or the manufacturing capabilities of our suppliers are impacted as a result of a public health emergency, including a resurgence of the COVID-19 pandemic, it may not be possible for us to timely manufacture relevant products at the required levels or at all. While the COVID-19 pandemic has not had a material adverse effect on our business to date, a reduction or interruption in any of our manufacturing processes as a result of a public health emergency in the future (including a resurgence of the COVID-19 pandemic) could have a material adverse effect on our business, results of operations, financial condition and cash flows.
We also may be unable to obtain the raw materials necessary to support our internal manufacturing processes due to the additional constraints on suppliers. The manufacture of our products is dependent on the availability of sufficient quantities of source tissue, which is the primary component of our products. Source tissue includes donated human tissue, porcine tissue, and bovine tissue. We acquire donated human tissue directly through institutional review board-approved protocols at multiple hospitals, as well as through tissue procurement firms engaged by us or by our contract manufacturers. Any failure to obtain tissue from our sources, including any failures related to public health emergencies, like the COVID-19 pandemic, will interfere with our ability to effectively meet the demand for our products. Any interruption in the supply of source tissue could materially harm our ability to manufacture our products until a new source of supply, if any, could be found. We may be unable to find a sufficient alternative supply channel in a reasonable time period or on commercially reasonable terms, if at all, which would have a material adverse effect on our business, results of operations, and financial condition.
Our current Advanced Wound Care portfolio is sold throughout the United States via an experienced direct sales force, which focuses its efforts on wound care in various sites of care. We use a mix of direct sales representatives and independent agencies to service the Surgical & Sports Medicine market. These sales representatives are supported by teams of professionals focused on sales management, sales operations and effectiveness, ongoing training, analytics and marketing. Our direct sales force functions by meeting in person with physicians and health care providers to discuss our products. Public health emergencies, like COVID-19, may negatively affect demand for our products by limiting the ability of our sales personnel to maintain their customary contacts with physicians and health care providers. In such case, we cannot assure you that our direct sales representatives or independent agencies will increase or maintain our current sales levels, which could have a material adverse effect on our business, results of operations, financial condition and cash flows. We may also experience significant and unpredictable reductions in demand for certain of our
45
products if patients are unable to access certain advanced therapies due to stay-at-home orders or other governmental actions taken to address a public health emergency.
Our ability to comply with financial covenants under our credit agreement and raise capital may be materially adversely impacted by COVID-19 or other factors.
We have funded our operations and capital spending, in part, through third-party debt and proceeds from the sale of our Class A common stock. Our 2021 Credit Agreement requires that we comply with certain financial covenants that include Consolidated Fixed Charge Coverage Ratio and Consolidated Total Net Leverage Ratio, tested quarterly. If we are unable to meet these financial covenants due to the economic impact of COVID-19 or otherwise, the borrowings under the 2021 Credit Agreement may become due and payable immediately unless we obtain an amendment from our lenders and we would be prohibited from making any borrowings under the Revolving Facility. There can be no assurance that our lenders would agree to any such amendment on acceptable terms, or at all. In addition, any sustained disruption in the capital markets from the COVID-19 pandemic could negatively impact our ability to raise capital from the offering of equity or debt securities.
Risks Related to Regulation of Our Products and Other Government Regulations
Our products are subject to the Infrastructure Investment and Jobs Act and rebate obligations that took effect on January 1, 2023, and we may owe rebates, which could be material, on our Apligraf, Dermagraft, and PuraPly products and possibly other products.
Section 90004 of the Infrastructure Investment and Jobs Act, enacted in November 2021, requires manufacturers to pay a refund to the federal government if more than a certain applicable percentage of their single-use product is not administered to a patient and is discarded (“wasted”) by providers. Because there is a lack of consistency and uniformity in wound sizes, it is likely that some skin substitute product is discarded with every treatment. The rebate obligation took effect January 1, 2023, and CMS proposed a methodology to implement the rebate in the MPFS rulemaking. The applicable percentage is required to be at least 10 percent of total allowed charges for the drug in a given calendar quarter. CMS has the authority to increase the applicable percentage that applies to refunds for discarded product if there are “unique circumstances.” We submitted comments on the proposal noting the unique circumstances related to skin substitutes and asking CMS to apply a higher percentage. In the 2023 MPFS final rule, published on November 1, 2022, CMS did not apply a higher applicable percentage to any products other than the hydrogel example they used in the proposed rule and stated that they plan to collect additional information about products that may have unique circumstances such that an increased applicable percentage (higher than 10 percent) would apply. CMS estimated the wastage percentage for three of our products - Apligraf, Dermagraft, and PuraPly - based on 2020 data. We do not know if the refund amounts calculated in 2023 will be similar to these estimates but if they are then we may owe rebates, which could be material, on these products and possibly other products. The total amount of any discarded product rebate liability is not known at this time.
We may encounter substantial delays or difficulties in our clinical trials.
Before obtaining marketing approval from regulatory authorities for the sale of our product candidates, we must conduct extensive clinical trials to demonstrate the safety and efficacy of the product candidates. Clinical testing is expensive, time-consuming and uncertain as to the outcome. We have limited experience with clinical trials. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, if at all. A failure of one or more clinical trials can occur at any stage of testing. Events that may prevent successful or timely completion of clinical development include:
46
ReNu is in Phase 3 clinical development for the management of symptoms associated with knee OA. Our anticipated timeline for these and other trials and studies on our clinical trial candidates may be subject to delays due to factors such as those discussed above.
Any inability to successfully complete preclinical and clinical development could result in additional costs to us or impair our ability to generate revenues from product sales, regulatory, development and commercialization milestones and royalties. In addition, if we make manufacturing or formulation changes to our product candidates, we may need to conduct additional studies to bridge our modified product candidates to earlier versions. Clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates and may harm our business, financial condition, results of operations and prospects.
Success in research and preclinical studies or early clinical trial results may not be indicative of results obtained in later trials. Likewise, preliminary, initial or interim data from clinical trials should be considered carefully and with caution since the final data may be materially different from the preliminary, initial or interim data, particularly as more patient data become available.
Results from preclinical studies or early clinical trials, including feasibility studies, or earlier conducted clinical trials are not necessarily predictive of future clinical trial results, and interim results of a clinical trial are not necessarily indicative of final results. Our clinical trial candidates, including ReNu, may fail to show the desired safety and efficacy in clinical development despite demonstrating positive results in preclinical studies or having successfully advanced through initial or earlier clinical trials or preliminary stages of clinical trials. From time to time, we have and may in the future publish or report preliminary, initial or interim data. Preliminary, initial or interim data from our clinical trials and those of our partners may not be indicative of the final results of the trial and are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and/or more patient data become available. In this regard, such data may show initial evidence of clinical benefit, but as patients continue to be followed and more patient data becomes available, there is a risk that any therapeutic effects will not be durable in patients and/or will decrease over time, or cease entirely. Preliminary, initial or interim data also remain subject to audit and
47
verification procedures that may result in the final data being materially different from such preliminary, initial or interim data. As a result, preliminary, initial or interim data should be considered carefully and with caution until the final data are available.
There is no guarantee that any of our clinical trials will be successful. In addition, there is a high failure rate for drugs, biologic products and cell therapies proceeding through clinical trials. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in preclinical testing and earlier-stage clinical trials. Data obtained from preclinical and clinical activities are subject to varying interpretations, which may delay, limit or prevent regulatory approval. Any such setbacks could adversely affect our business, financial condition, results of operations and prospects.
Obtaining the necessary regulatory approvals or clearances for certain of our products will be expensive and time-consuming and may impede our ability to fully exploit our technologies or otherwise limit our ability to meet other business objectives.
As biological products and medical devices, many of the products that we market require regulatory approvals or clearances from the FDA, or from similar regulatory authorities outside of the United States, before they may legally be distributed in commerce. In particular, such products may require FDA approval of Biologics License Applications, or BLAs, under Section 351 of the Public Health Service Act (the “PHSA”), Premarket Approval, or PMA, submissions under Section 515 of the Federal Food, Drug, and Cosmetic Act, or FDCA, or may require clearance under Section 510(k) of the FDCA. Although we believe that we have all necessary regulatory approvals or clearances legally required for the products that we currently market, the introduction of new or modified products may require us to secure new approvals or clearances. Additionally, the FDA may take the position that some of the products that we currently market without premarket approval or clearance in fact require such approval or clearance. The process of obtaining an approved BLA or PMA requires the expenditure of substantial time, effort and financial resources and may take years to complete. Although obtaining clearance under section 510(k) is somewhat less burdensome, it is also associated with significant costs and resource commitments. The fee for filing a BLA, PMA or 510(k) notification, and the annual user fees for any establishment that manufactures biologics or medical devices, as well as product fees applicable to each approved product are substantial.
In January 2021, we announced that the first patient was enrolled in the pivotal Phase 3 clinical trial evaluating the safety and efficacy of ReNu for the management of symptoms associated with knee OA. There are significant costs associated with conducting clinical trials to support approvals that cannot necessarily be estimated with any accuracy until investigational plans have been developed. Moreover, data obtained from clinical activities may show a lack of safety or efficacy or may be inconclusive or susceptible to varying interpretations, any of which could delay, limit or prevent regulatory approval. Failure or delay can occur at any time during the clinical trial process. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful. Even product candidates in later stages of clinical trials may fail to show the required safety profile or meet the efficacy endpoints despite having progressed through preclinical studies and initial clinical trials. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials. We cannot be certain that we will not face similar setbacks. Even with positive clinical trial results, there may be other barriers to approval or clearance, and the FDA may not grant approval or clearance on a timely basis, or at all. Even if the FDA clears or approves our products, the clinical data submitted to the FDA may not be sufficient for payers to cover and/or adequately reimburse our customers for use of our products. Additionally, the FDA may limit the indications for use in an approval or clearance, or place other conditions on an approval, that could restrict the commercial application of the products.
Regenerative medicine advanced therapy, or RMAT, designation for our product candidates may not lead to faster development or regulatory processes nor does it increase the likelihood that such product candidates will receive marketing approval.
RMAT was introduced as a new designation under the 21st Century Cures Act for the development and review of certain regenerative medicine therapies. To receive RMAT designation, a regenerative medicine product candidate must be intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition with preliminary clinical evidence indicating that the drug has the potential to address the unmet medical needs. RMAT designation does not require evidence to indicate that the drug may offer a substantial improvement over available therapies, as breakthrough designation requires.
An RMAT product candidate receives intensive guidance on an efficient product development program; involvement of senior managers and experienced staff on a proactive, collaborative and cross-disciplinary review; and a rolling review. Regenerative medicine therapies that qualify for RMAT designation may also qualify for other FDA expedited programs, including fast track designation, breakthrough therapy designation, accelerated approval and priority review designation, if they meet the criteria for such programs. However, RMAT designation does not assure that marketing approval will be granted and, if granted, that the approval process would be any faster than it would have otherwise been.
48
In January 2021, we announced RMAT designation for ReNu for the management of symptoms associated with knee OA. However, there is no guarantee that the receipt of RMAT designation will result in a faster development process, review or approval for ReNu for the management of symptoms associated with knee OA or increase the likelihood that ReNu will be granted marketing approval for the management of symptoms associated with knee OA. Likewise, any future RMAT designation or other expedited review status such as breakthrough therapy designation for any of our other product candidates neither guarantees a faster development process, review or approval nor improves the likelihood of the grant of marketing approval by FDA for any such product candidate compared to drugs considered for approval under conventional FDA procedures. In addition, the FDA may withdraw any RMAT or other expedited review status at any time. We may seek RMAT or breakthrough therapy designation for our other product candidates, but the FDA may not grant this status to any such product candidates.
We may seek fast track designation by the FDA for one or more of our product candidates, but we might not receive such designation, and even if we do, such designation may not actually lead to a faster development or regulatory review or approval process.
If a product is intended for the treatment of a serious or life-threatening condition and the product demonstrates the potential to address unmet needs for this condition, the treatment sponsor may apply for FDA fast track designation. Even if we receive fast track designation, fast track designation does not ensure that we will receive marketing approval or that approval will be granted within any particular time frame. We may not experience a faster development, regulatory review or approval process with fast track designation compared to conventional FDA procedures. Additionally, the FDA may withdraw fast track designation if it believes that the designation is no longer supported by data from our clinical development program. Fast track designation alone does not guarantee qualification for the FDA’s priority review procedures.
A breakthrough therapy designation by the FDA for a product candidate may not lead to a faster development or regulatory review or approval process, and it would not increase the likelihood that the product candidate will receive marketing approval.
We may seek a breakthrough therapy designation for one or more product candidates. A breakthrough therapy is defined as a product candidate that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the product candidate may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. For product candidates that have been designated as breakthrough therapies, interaction and communication between the FDA and the sponsor of the trial can help to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens. Product candidates designated as breakthrough therapies by the FDA are also eligible for priority review if supported by clinical data at the time of the submission of the new drug application.
Designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if we believe that one of our product candidates meets the criteria for designation as a breakthrough therapy, the FDA may disagree and instead determine not to make such designation. In any event, the receipt of a breakthrough therapy designation for a product candidate may not result in a faster development process, review or approval compared to product candidates considered for approval under conventional FDA procedures and it would not assure ultimate approval by the FDA. In addition, even if one or more of our product candidates qualify as breakthrough therapies, the FDA may later decide that the product candidate no longer meets the conditions for qualification or it may decide that the time period for FDA review or approval will not be shortened.
We must comply with applicable post-marketing regulatory obligations, which could include obtaining new regulatory approvals or clearances.
Following approval or clearance, some types of changes to the approved or cleared product, such as adding new indications or additional labeling claims or introducing manufacturing changes, are subject to FDA review and approval, which may require further nonclinical or clinical testing. The costs and other resource burdens associated with obtaining new regulatory approvals or clearances for existing or future products may limit the resources available to us to fully exploit our technologies or may otherwise limit our ability to carry out other business activities. Depending on the nature of the change, we may determine that the change may be carried out without obtaining premarket approval or clearance. The FDA or another regulatory body could disagree with our conclusion and require such premarket approval or clearance, which would disrupt the marketing of these products, potentially expose us to regulatory sanctions, and have a material adverse effect on our business, financial condition and results of operations.
49
The FDA may determine that certain of our products that are, or are derived from, human cells or tissues, such as Affinity, Novachor, and NuShield, do not qualify for regulation solely under Section 361 of the Public Health Services Act, or PHSA, and may require that we revise our labeling and marketing claims for these products or that we suspend sales of such products until FDA approval is obtained, which could adversely affect our business, results of operations, and financial condition.
Certain of the products that we manufacture, process and distribute are, or are derived from, human cells or tissues, including amniotic tissue. The FDA has specific regulations governing human cells, tissues and cellular and tissue-based products, or HCT/Ps. In particular, HCT/Ps that meet certain criteria set forth in the FDA’s regulations at 21 C.F.R. § 1271.10 are regulated solely under Section 361 of the PHSA, so-called “Section 361 HCT/Ps”, and are not subject to any premarket clearance or approval requirements. They are also subject to less stringent post-market regulatory requirements than products regulated under Section 351 of the PHSA and/or under Sections 505, 510 or 515 of the FDCA. The Company has believed that certain of our HCT/Ps, including our products derived from amniotic membrane, qualify for regulation as Section 361 HCT/Ps. However, the regulatory classification of an HCT/P as a Section 361 HCT/P depends in part on the purposes for which the product is intended and in part on the processing to which an HCT/P is subject. On November 16, 2017, the FDA issued a final guidance document entitled, “Regulatory Considerations for Human Cells, Tissues, and Cellular and Tissue-Based Products: Minimal Manipulation and Homologous Use”, or 361 HCT/P Guidance, which provides FDA’s current thinking on how to apply the existing regulatory criteria for regulation as a Section 361 HCT/P. These include, in addition to other requirements, requirements that an HCT/P be both minimally manipulated and intended for homologous use. In general, “minimal manipulation” is a standard referring to the degree to which the original characteristics of an HCT/P have been altered by processing and “homologous use” refers to the requirement that an HCT/P perform the same basic function in the donor as in the recipient. Any action by the FDA to apply the principles set forth in the 361 HCT/P Guidance to the HCT/Ps that we distribute could have adverse consequences for us and make it more difficult or expensive for us to conduct our business.
In light of the 361 HCT/P Guidance, our labeling and marketing claims for our placental-based membrane products, including our Affinity, NuShield, and Novachor products, clarify that they are intended as wound coverings, and thus meet the homologous use requirement to qualify as Section 361 HCT/Ps. However, the FDA could disagree with our conclusion and require premarket approval or clearance for Affinity, NuShield, or any placental-based sheet product we market, which would disrupt the marketing of these products, potentially expose us to regulatory sanctions, and have a material adverse effect on our business, financial condition and results of operations. Further, we believe it is necessary to obtain FDA approval of a BLA for NuCel and ReNu because those products may be deemed to be more than minimally manipulated, not for homologous use, or otherwise not regulated as Section 361 HCT/Ps. We continue to conduct clinical studies of ReNu to support FDA approval of a BLA for the management of symptoms associated with knee OA and, based on favorable feasibility studies that are subject to further evaluation, we believe ReNu has potential as a treatment for additional OA and tissue regeneration applications. We have discontinued clinical development of NuCel. If we obtain BLA approval for ReNu or NuCel, compliance with applicable post-market regulatory requirements will involve significant time and substantial costs. Even for those products that remain regulated as Section 361 HCT/Ps, increasing regulatory scrutiny within the industry in which we operate could lead to heightened requirements, compliance with which could be costly. The costs and other resource burdens associated with any of these regulatory outcomes may limit the resources available to us to fully exploit our technologies or may otherwise limit our ability to carry out other business activities.
The 361 HCT/P Guidance originally indicated that the FDA was providing a 36-month enforcement grace period to allow time for distributors of HCT/Ps to make any regulatory submissions and obtain any premarket approvals necessary to comply with the guidance. In July 2020, the FDA announced that the enforcement grace period would be extended until May 31, 2021 as a result of the challenges presented by the COVID-19 public health emergency. On April 21, 2021, the FDA reaffirmed that the enforcement grace period would end on May 31, 2021, at which time we ceased commercial distribution of ReNu and NuCel. Although we believe our suspension of ReNu and NuCel commercialization was timely and proper, the FDA and other regulators may disagree with how or when such commercialization practices were conducted, which could expose us to regulatory sanctions, and have a material adverse effect on our business, financial condition and results of operations.
To the extent that the FDA may determine that certain of our products that are, or are derived from, human cells or tissues do not qualify for regulation solely under Section 361 of the PHSA, the introduction of new tissue products would become more expensive, expansion of our tissue product offerings could be significantly delayed, and we could be subject to additional post-market regulatory requirements or suspension of product sales until FDA approval is obtained.
As stated above, in light of the 361 HCT/P Guidance, the FDA may determine that the types of cell- and tissue-based products that we distribute—and in particular, products derived from allografts consisting of human skin or amniotic tissue—are subject to premarket clearance or approval requirements. Should the FDA make such a determination, products of this type, including future products that we seek to introduce, will be much more costly to commercialize, as we will likely have to carry out preclinical work in animals and/or clinical trials in humans to support approval. Such preclinical work and clinical trials are expensive and time-consuming with no guarantee of success. In addition, these products will be subject to more stringent post-market regulatory requirements than those that currently apply, including but not limited to more stringent restrictions on advertising and promotion of these products, as well as more extensive adverse event reporting. In the future, we may also wish to market our existing HCT/P
50
products for new intended uses that may render them ineligible for regulation as Section 361 HCT/Ps and cause them to require premarket clearance or approval and comply with post-market regulations under the medical device or biological product provisions of the FDCA and/or PHSA instead. Compliance with these requirements will involve significant time and substantial costs and could limit the resources available to us to fully exploit our technologies, including limiting our ability to introduce new allograft-derived products.
We conduct a range of nonclinical, as well as clinical trials, comparative effectiveness, economic and other studies of our products. Unfavorable results from these trials or studies or from similar trials or studies conducted by others may negatively affect the use or adoption of our products by physicians, hospitals, and payers, which could have a negative impact on the market acceptance of these products and their profitability.
We conduct a variety of nonclinical and clinical trials, comparative effectiveness studies and economic and other studies of our products, including our ongoing clinical trial for ReNu, in an effort to generate comprehensive clinical and real-world outcomes data and cost-effectiveness data in order to obtain product approval and drive further penetration in the markets we serve. In the event that these trials and studies, or similar trials and studies conducted by others, yield unfavorable results, those results could negatively affect the use or adoption of our products by physicians, hospitals, and payers, thereby compromising market acceptance and profitability.
Our business is subject to continuing significant regulatory obligations by the FDA and other authorities, compliance with which is expensive and time-consuming and may impede our ability to fully exploit our technologies or otherwise limit our ability to meet other business objectives.
Aside from the obligation to obtain regulatory approvals or clearances, companies such as ours have ongoing regulatory obligations that are expensive and time-consuming to meet. In particular, the production and marketing of our products are subject to extensive regulation and review by the FDA and numerous other governmental authorities both in the United States and abroad. As noted above, some of the products that we distribute are considered Section 361 HCT/Ps. The FDA’s regulation of HCT/Ps includes requirements for registration and listing of products; donor screening and testing; processing and distribution, known as “Current Good Tissue Practices,” or cGTP; labeling; record keeping and adverse-reaction reporting; and inspection and enforcement. Moreover, it is likely that the FDA’s regulation of HCT/Ps will continue to evolve in the future. Complying with any such new regulatory requirements may entail significant time delays and expense, which could have a material adverse effect on our business, results of operations, and financial condition. Our other products are regulated as biologics and medical devices, which are subject to even more stringent regulation by the FDA. As noted above, these products are subject to rigorous premarket review processes, and an approval or clearance may place substantial restrictions on the indications for which the product may be marketed or the population for whom it may be marketed, may require warnings to accompany the product or may impose other restrictions on the sale and/or use of the product. In addition, approved and cleared products are subject to continuing obligations to comply with other substantial regulatory requirements, including the FDA’s cGTP regulations, the FDA’s QSR and/or the FDA’s Current Good Manufacturing Practices, or cGMP regulations, adverse event reporting, and FDA inspections. The costs and other resource burdens associated with maintaining regulatory approvals or clearances for our products and otherwise meeting our regulatory obligations may limit the resources available to us to fully exploit our technologies or may otherwise limit our ability to carry out other business activities.
In some states, the manufacture, storage, or distribution of HCT/Ps requires a license or permit to operate as a tissue bank or tissue distributor. We believe that we have all required state licenses or permits applicable to the distribution of HCT/Ps, but there is a risk that there may be state or local license or permit requirements of which we are unaware or with which we have not complied. In the event that such noncompliance exists in a given jurisdiction, we could be precluded from distributing HCT/Ps in that jurisdiction and also could be subject to fines or other penalties. If any such actions were to be instituted against us, it could adversely affect our business and/or financial condition.
The American Association of Tissue Banks, or AATB, has issued operating standards for tissue banking. Compliance with these standards is a requirement in order to become an accredited tissue bank. In addition, some states have their own tissue banking regulations. In addition, procurement of certain human organs and tissue for transplantation is subject to the restrictions of the National Organ Transplant Act, or NOTA, which prohibits the transfer of certain human organs, including skin and related tissue for valuable consideration, but permits the reasonable payment associated with the removal, transportation, implantation, processing, preservation, quality control and storage of human tissue and skin. We reimburse tissue banks, hospitals, and physicians for their services associated with the recovery, storage, and transportation of donated human tissue. Although we have independent third-party appraisals that confirm the reasonableness of the service fees we pay, if we were to be found to have violated NOTA’s prohibition on the sale or transfer of human tissue for valuable consideration, we, our officers, or employees, would potentially be subject to criminal enforcement sanctions, which could materially and adversely affect our business, results of operations, and financial condition.
51
Many of the products we manufacture and process are derived from human tissue and therefore have the potential for disease transmission.
The utilization of human tissue creates the potential for transmission of communicable diseases, including, but not limited to, human immunodeficiency virus, or HIV, viral hepatitis, syphilis and other viral, fungal or bacterial pathogens. We are required to comply with federal and state regulations intended to prevent communicable disease transmission.
Although we maintain strict quality controls over the procurement and processing of our tissue, there is no assurance that these quality controls will be adequate. In addition, negative publicity concerning disease transmission from other companies’ improperly processed donated tissue could have a negative impact on the demand for our products. If any of our products are implicated in the transmission of any communicable disease, our officers, employees and we could be subject to government sanctions including but not limited to recalls, and civil and criminal liability, with sanctions that include exclusion from doing business with the federal government. We could also be exposed to product liability claims from those who used or received our products as well as loss of our reputation.
Defects, failures, or quality issues associated with our products could lead to product recalls or safety alerts, adverse regulatory actions, litigation, including product liability claims, and negative publicity that could erode our competitive advantage and market share and materially adversely affect our reputation, business, results of operations, and financial condition.
Quality is extremely important to us and our customers due to the serious and costly consequences of product failure. Quality and safety issues may occur with respect to any of our products, and our future operating results will depend on our ability to maintain an effective quality control system and effectively train and manage our workforce with respect to our quality system. The development, manufacture, and control of our products are subject to extensive and rigorous regulation by numerous government agencies, including the FDA and similar foreign agencies. Compliance with these regulatory requirements, including but not limited to the FDA’s QSR, GMPs, and adverse events/recall reporting requirements in the United States and other applicable regulations worldwide, is subject to continual review and is monitored rigorously through periodic inspections by the FDA and foreign regulatory authorities. The FDA and foreign regulatory authorities may also require post-market testing and surveillance to monitor the performance of approved products. Our manufacturing facilities and those of our suppliers and independent sales agencies are also subject to periodic regulatory inspections. If the FDA or a foreign authority were to conclude that we have failed to comply with any of these requirements, it could institute a wide variety of enforcement actions, ranging from a public warning letter to more severe sanctions, such as product recalls or seizures, withdrawals, monetary penalties, consent decrees, injunctive actions to halt the manufacture or distribution of products, import detentions of products made outside the United States, export restrictions, restrictions on operations or other civil or criminal sanctions. Civil or criminal sanctions could be assessed against our officers, employees, or us. Any adverse regulatory action, depending on its magnitude, may restrict us from effectively manufacturing, marketing, and selling our products.
In addition, we cannot predict the results of future legislative activity or future court decisions, any of which could increase regulatory requirements, subject us to government investigations or expose us to unexpected litigation. Any regulatory action or litigation, regardless of the merits, may result in substantial costs, divert management’s attention from other business concerns, and place additional restrictions on our sales or the use of our products. In addition, negative publicity, including regarding a quality or safety issue, could damage our reputation, reduce market acceptance of our products, cause us to lose customers, and decrease demand for our products. Any actual or perceived quality issues may also result in issuances of physician’s advisories against our products or cause us to conduct voluntary recalls. Any product defects or problems, regulatory action, litigation, negative publicity or recalls could disrupt our business and have a material adverse effect on our business, results of operations, and financial condition.
We may implement a product recall or voluntary market withdrawal, which could significantly increase our costs, damage our reputation and disrupt our business.
The manufacturing, marketing, and processing of our products involve an inherent risk that our products or processes may not meet manufacturing specifications, applicable regulatory requirements or quality standards. In that event, we may voluntarily implement a recall or market withdrawal or may be required to do so by a regulatory authority. A recall or market withdrawal of one of our products would be costly and would divert management resources. A recall or withdrawal of one of our products, or a similar product processed by another entity, also could impair sales of our products as a result of confusion concerning the scope of the recall or withdrawal, or as a result of the damage to our reputation for quality and safety.
We are subject to various governmental regulations relating to the labeling, marketing, and sale of our products.
Both before and after a product is commercially released, we have ongoing responsibilities under regulations promulgated by the FDA, the Federal Trade Commission, and similar U.S. and foreign regulations governing product labeling and advertising, distribution, sale, and marketing of our products.
52
Manufacturers of medical devices and biological products are permitted to promote products solely for the uses and indications set forth in the approved or cleared product labeling. A number of enforcement actions have been taken against manufacturers that promote products for “off-label” uses (i.e., uses that are not described in the approved or cleared labeling), including actions alleging that claims submitted to government healthcare programs for reimbursement of products that were promoted for “off-label” uses are fraudulent in violation of the Federal False Claims Act or other federal and state statutes and that the submission of those claims was caused by off-label promotion. The failure to comply with prohibitions on “off-label” promotion can result in significant monetary penalties, revocation or suspension of a company’s business license, suspension of sales of certain products, product recalls, civil or criminal sanctions, exclusion from participating in federal healthcare programs, or other enforcement actions. In the United States, allegations of such wrongful conduct could also result in a corporate integrity agreement with the U.S. government that imposes significant administrative obligations and costs.
We and our employees and contractors are subject, directly or indirectly, to federal, state and foreign healthcare fraud and abuse laws, including false claims laws. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
Our operations are subject to various federal, state, and foreign fraud and abuse laws. These laws may constrain our operations, including the financial arrangements and relationships through which we market, sell, and distribute our products.
U.S. federal and state laws that affect our ability to operate include, but are not limited to:
Activities and arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, waste, and other abusive practices. These laws and regulations may restrict or prohibit a wide range of activities or other arrangements related to the development, marketing, or promotion of products, including pricing and discounting of products, provision of customer incentives, provision of reimbursement support, other customer support services, provision of sales commissions or other incentives to employees and independent contractors and other interactions with healthcare practitioners, other healthcare providers and patients.
Because of the breadth of these laws and the narrow scope of the statutory or regulatory exceptions and safe harbors available, our business activities could be challenged under one or more of these laws. Relationships between medical product manufacturers and health care providers are an area of heightened scrutiny by the government. We engage in various types of activities, including the conduct of speaker programs to educate physicians, the provision of reimbursement advice and support to customers, and the
53
provision of customer and patient support services, that have been the subject of government scrutiny and enforcement action within the medical device industry.
Government expectations and industry best practices for compliance continue to evolve and our past activities may not always be consistent with current industry best practices. Further, there is a lack of government guidance as to whether many varied industry practices comply with these laws, and government interpretations of these laws continue to evolve, all of which create compliance uncertainties. Any non-compliance could result in regulatory sanctions, criminal or civil liability, and serious harm to our reputation. Although we have a comprehensive compliance program designed to ensure that our employees’ and commercial partners’ activities and interactions with healthcare professionals and patients are appropriate, ethical, and consistent with all applicable laws, regulations, guidelines, policies, and standards, it is not always possible to identify and deter misconduct, and the precautions we take to detect and prevent this activity may not be effective in preventing such conduct, mitigating risks, or reducing the chance of governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations.
If a government entity opens an investigation into possible violations of any of these laws (which may include the issuance of subpoenas or civil investigative demands), we would have to expend significant resources to defend ourselves against the allegations. Allegations that we, our officers, or our employees violated any one of these laws can be made by individuals called “whistleblowers” who may be our employees, customers, competitors, or other parties. Government policy is to encourage individuals to become whistleblowers and file a complaint in federal court alleging wrongful conduct. The government is required to investigate all of these complaints and decide whether to intervene. If the government intervenes and we are required to pay money back to the government, the whistleblower, as a reward, is awarded a percentage of the collection. If the government declines to intervene, the whistleblower may proceed on their own and, if they are successful, they will receive a percentage of any judgment or settlement amount the company is required to pay. The government may also initiate an investigation on its own. Such actions could have a significant impact on our business, including the imposition of significant fines, and other sanctions that may materially impair our ability to run a profitable business. In particular, if our operations are found to be in violation of any of the laws described above or if we agree to settle with the government without admitting to any wrongful conduct or if we are found to be in violation of any other governmental regulations that apply to us, we, our officers and employees may be subject to sanctions, including civil and criminal penalties, damages, fines, exclusion from participation in government health care programs, such as Medicare and Medicaid, imprisonment, the curtailment or restructuring of our operations and the imposition of a corporate integrity agreement, any of which could adversely affect our business, results of operations, and financial condition.
We could be subject to legal exposure if we do not report the average sales prices, or ASP, to government agencies or if our reporting is not accurate and complete.
Our products are reimbursed by Medicare in physician office settings at a rate of ASP plus 6%. All Medicare payments, including payments based on the ASP methodology, are subject to sequestration. Congress previously suspended sequestration imposed under the BCA, and there was no sequestration through March 31, 2022. On April 1, 2022, there was a 1% sequestration and beginning on July 1, 2022, the sequestration returned to 2%. Sequestration applies to the government’s payment portion, which is 80% of the total payment amount. Additionally, in future years, it is possible that an up-to 4% Medicare sequestration could be ordered under Statutory PAYGO, which requires deficit neutrality in most laws passed by Congress. Until January 2022, we were not required to report ASP for all our skin substitute products that are paid separately as biologics because they are regulated as medical devices by the FDA, although we chose to report ASP for some of our products. Starting with the reporting deadline for the first quarter of 2022, we were required to report ASP for all our skin substitute products that are paid separately as biologics as a result of provisions included in the Consolidated Appropriations Act of 2020. As of January 1, 2022, we began reporting ASP for all our skin substitute products that are paid separately as biologics. The first such ASP report was made on April 30, 2022 for Q1 2022. Government price reporting requirements are complex. If we do not report ASP correctly, we may have to restate ASP for prior quarters and we could be subject to civil monetary penalties and/or, if the violation is knowing or reckless, be subject to False Claims Act liability. In the case of very serious or repeated violations, we could be excluded from doing business with the Medicare program and other federal healthcare programs.
We face significant uncertainty in the industry due to government healthcare reform and other legislative action.
There have been and continue to be laws enacted by the federal government, state governments, regulators, and third-party payers to control healthcare costs, and generally, to reform the healthcare system in the United States. For example, the Patient Protection and Affordable Care Act of 2010 (“PPACA”) and the Medicare Access and CHIP Reauthorization Act of 2015 substantially changed the way healthcare is delivered and financed by both governmental and private insurers. These changes included the creation of demonstration programs and other value-based purchasing initiatives that provide financial incentives for physicians and hospitals to reduce costs, including incentives for furnishing low-cost therapies for chronic wounds even if those therapies are less effective than our products. There were extensive efforts recently to modify or repeal all or part of PPACA. Tax reform legislation was passed that includes provisions that impact healthcare insurance coverage and payment such as the elimination of the tax penalty for individuals who do not maintain health insurance coverage (the so-called “individual mandate”). Such actions or similar actions
54
could have a negative effect on the utilization of our products. We expect such efforts to continue and that there may be additional reform proposals at federal and state levels. On December 18, 2019, the United States Court of Appeals for the Fifth Circuit upheld a lower court’s determination in California v. Texas (orig. Texas v. Azar, 4:18-cv-00167), that the individual mandate was unconstitutional and remanded the case to the lower court for further analysis as to whether PPACA as a whole is unconstitutional because the individual mandate is not severable from other provisions of the law. The United States Supreme Court agreed to review the case and on June 17, 2021, ordered that the Fifth Circuit’s decision be reversed and that the case be dismissed.
Additionally, on August 16, 2022, Congress passed legislation to limit the price of drugs and biological under the Medicare program. The Inflation Reduction Act (IRA) establishes a Drug Price Negotiation Program that requires the Secretary of Health and Human Services to negotiate the price of certain high expenditure Medicare drugs that do not have generic or biosimilar competition. The law also establishes Medicare Part B inflationary rebates, effective Q1 2023. Generally, manufacturers of Part B drugs with an ASP+6% that exceeds the inflation-adjusted payment amount from Q3 2021 will be required to pay a rebate to the Medicare program. These and similar drug pricing reforms could increase pricing pressure on our products.
General legislative action may also affect our business. For example, the Budget Control Act of 2011 included provisions to reduce the federal deficit. The Budget Control Act, as amended, resulted in the imposition of reductions of up to 2% in Medicare payments to providers which began in April 2013 and are scheduled to remain in effect through the first six months of 2032. The Coronavirus Aid, Relief, and Economic Security (CARES) Act and subsequent legislation suspended the payment adjustment from May 1, 2020 through March 31, 2022. There was 1% Medicare sequestration from April 1 to June 30, 2022, and the 2% Medicare sequester was reinstated on July 1, 2022. Additionally, under Statutory PAYGO, a 4% Medicare sequester could be ordered at the end of the 2024 Congressional session. These or other similar reductions in government healthcare spending could result in reduced demand for our products or additional pricing pressure.
Bills currently before the United States Congress may also affect our business, if enacted. For example, during the 117th Congressional session, the Cures 2.0 Act, H.R. 6000, 117th Cong. (2021) was introduced into the United States House of Representatives. If reintroduced in a similar form, it may contain provisions that could result in legal and regulatory changes that affect our business. These changes may include a new payment pathway for breakthrough medical devices that are FDA approved or cleared on or after a certain date. The enactment of Cures 2.0 (or similar legislation) may also accelerate FDA timelines for designation of breakthrough and RMAT therapies and also result in new requirements for the use of patient experience data and real-world evidence in regulating certain FDA products. If enacted, these changes could make it easier for our competitors to bring comparable or more advanced products to market quickly, resulting in reduced demand for our products.
Our sales into foreign markets expose us to risks associated with international sales and operations.
We are currently selling into foreign markets and plan to expand such sales. Managing a global organization is difficult, time-consuming, and expensive. Conducting international operations subjects us to risks that could be different from those faced by us in the United States. The sale and shipment of our products across international borders, as well as the purchase of components and products from international sources, subject us to extensive U.S. and foreign governmental trade, import and export and customs regulations and laws, including but not limited to, the Export Administration Regulations and trade sanctions against embargoed countries, which are administered by the Office of Foreign Assets Control within the Department of the Treasury, as well as the laws and regulations administered by the Department of Commerce. These regulations limit our ability to market, sell, distribute, or otherwise transfer our products or technology to prohibited countries or persons.
Compliance with these regulations and laws is costly, and failure to comply with applicable legal and regulatory obligations could adversely affect us in a variety of ways that include, but are not limited to, significant criminal, civil, and administrative penalties, including imprisonment of individuals, fines and penalties, denial of export privileges, seizure of shipments and restrictions on certain business activities. Also, the failure to comply with applicable legal and regulatory obligations could result in the disruption of our distribution and sales activities.
These risks may limit or disrupt our expansion, restrict the movement of funds, or result in the deprivation of contractual rights or the taking of property by nationalization or expropriation without fair compensation. Operating in international markets also requires significant management attention and financial resources.
We could be adversely affected by violations of the U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws.
The U.S. Foreign Corrupt Practices Act, or FCPA, the U.K. Bribery Act of 2010, and similar anti-bribery laws in other jurisdictions generally prohibit companies and their intermediaries from making improper payments for the purpose of obtaining or retaining business. Our policies mandate compliance with these anti-bribery laws, including the requirements to maintain accurate information and internal controls. We operate in many parts of the world that have experienced governmental corruption to some degree and in certain circumstances, strict compliance with anti-bribery laws may conflict with local customs and practices. There is no assurance that our internal control policies and procedures will protect us from acts committed by our employees or agents. If we
55
are found to be liable for FCPA or other violations (either due to our own acts or our inadvertence, or due to the acts or inadvertence of others), we could suffer from civil and criminal penalties or other sanctions, including contract cancellations or debarment, and loss of reputation, any of which could have a material adverse impact on our business, financial condition, and results of operations.
Risks Related to Reimbursement for our Products
The rate of reimbursement and coverage for the purchase of our products by government and private insurance is subject to change.
Sales of almost all of our products depend partly on the ability of our customers to obtain reimbursement for the cost of our products under government healthcare programs such as Medicare and Medicaid and from other global government authorities. Government healthcare programs and private health plans continuously seek to reduce healthcare costs. For example, in 2014, Medicare established a policy to stop making separate payment for our products in certain clinical settings. This policy required us to reduce prices for our products which caused significant reduction in our revenue.
Our success will depend in part on the extent to which coverage and adequate reimbursement for the costs of such products and related treatments will be available from government health administration authorities, private health insurers, and other third-party payers and we do not know whether such reimbursement will be available. For example, currently most private payers provide limited coverage for our PuraPly AM, PuraPly, Novachor, and NuShield products and as a result, there is limited use of these products for patients covered by private payers.
The continuing efforts of government agencies, private health plans, and other payers of healthcare services to contain or reduce costs of healthcare may adversely affect:
The proposed updates to the MPFS for calendar year 2023 included a proposal to stop making separate payments for all skin substitutes, including all of our products, in 2024 or 2025. Instead of making separate payment for skin substitutes, Medicare would bundle the payment for skin substitutes into the payment made for the application procedure. As part of this proposal, Medicare would consider all skin substitutes to be supplies instead of biologicals and would require manufacturers of skin substitutes, including us, to apply for new HCPCS codes that would be effective starting in 2024. In the 2023 MPFS final rule, published on November 1, 2022, CMS did not finalize this bundling proposal and will consider more public input in the future; however, they may propose the same policy again or make other proposals in the future that could affect our business and our revenue. If Medicare reproposes and finalizes a policy to stop making separate payment for skin substitutes in calendar year 2024 or calendar year 2025, reimbursement for our products may not be adequate and our business, results of operations, and financial condition may be negatively affected.
Payers are increasingly attempting to contain healthcare costs by limiting both the breadth of coverage and the level of reimbursement, particularly for new therapeutic products generally or specifically for new therapeutic products that target an indication that is perceived to be well served by existing treatments. Specifically, the Patient Protection and Affordable Care Act, or PPACA, enacted in 2010, contains provisions for Medicare demonstration programs that create financial incentives to treat patients with chronic wounds conservatively and not use our products. Furthermore, all our products are not paid separately in the outpatient hospital setting which is our largest customer base. This payment policy has created incentives to use our competitors’ products. Accordingly, even if coverage and reimbursement are provided, market acceptance of our products has been and will be adversely affected if access to coverage is administratively burdensome to obtain and/or use of our products is administratively burdensome or unprofitable for healthcare providers or less profitable than alternative treatments. In addition, Medicare, which is the major source of revenue for most of our customers, reimburses the same amounts for most of our products and the products of our competitors targeting the same indications in the hospital outpatient setting. Because in some sites of care, the reimbursement amount is not based on the cost we charge our customers for our products or the cost our competitors charge for products targeting the same indication, our customers may elect to use products cheaper than ours in order to increase their margins, which could have a material adverse effect on our business, results of operations, and financial condition.
Reimbursement from Medicare, Medicaid, and other third-party payers is usually adjusted yearly as a result of legislative, regulatory, and policy changes as well as budgetary pressures. In fact, Medicare has signaled that it may discontinue its two-tier bundling policy when it solicited comments on alternatives in its calendar year 2019 rulemaking. Changes in the policy could occur as early as calendar year 2023 and could include the establishment of a single bundle for all products which could place our products at a
56
significant competitive disadvantage. Possible reductions in, or eliminations of, coverage or reimbursement by third-party payers, or the denial of, or provision of uneconomical reimbursement for new products, as a result of these changes may affect our customers’ revenue and ability to purchase our products. Any changes in the healthcare regulatory, payment, or enforcement landscape relative to our customers’ healthcare services also have the potential to significantly affect our operations and revenue. In addition, Medicare uses regional contractors called Medicare Administrative Contractors, or MACs, to process claims, develop coverage policies and make payments within designated geographic jurisdictions. While our products are currently covered by most MACs, we cannot be certain they will be in the future.
Wound care supplies, such as our product line acquired from CPN Biosciences, are subject to coding verification from CMS’s Pricing, Data Analysis and Coding contractor (the “PDAC”). The PDAC is responsible for verifying the HCPCS Level II DMEPOS Codes for all wound care supplies. Our current wound care supplies sold through CPN have received coding verification from the PDAC and all products have HCPCS Level II codes. Additional wound care supplies that we develop or acquire will also be subject to the PDAC coding verification process. We cannot guarantee the outcome of the PDAC coding verification process. If we are unsuccessful in receiving verification of the applicable HCPCS codes for our products, our wound care supplies could be ineligible for reimbursement or reimbursed at a lower rate than appropriate for our supplies.
While we cannot predict the outcome of current or future legislation, we anticipate, particularly given the recent focus on healthcare reform legislation, that governmental authorities will continue to introduce initiatives directed at lowering the total cost of healthcare and restricting coverage and reimbursement for our products. If we are not successful in obtaining adequate reimbursement for our products from third-party payers, the market’s acceptance of our products could be adversely affected. Inadequate reimbursement levels also likely would create downward price pressure on our products. Even if we do succeed in obtaining widespread reimbursement for our products, future changes in reimbursement policies could have a negative impact on our business, financial condition and results of operations.
The rate of reimbursement and coverage for the purchase of our products by government and private insurance (including by Medicare Administrative Contractors) is subject to uncertainty.
Our products are subject to varying forms of governmental and private payor reimbursement, and fluctuations in these forms of payment may adversely affect our business. For example, in sites of service where payment for skin substitutes is based on the ASP methodology, Medicare pays for skin substitutes separately from the application procedure. In this case, the Medicare payment rate for all skin substitutes (including ours) is calculated on a per square centimeter basis. These rates are adjusted quarterly based on manufacturer ASP reporting, and the payment amount is ASP plus 6%, WAC plus 3%, or invoice pricing. All Medicare payment amounts, including separate payments under the ASP methodology, are subject to sequestration. The Medicare sequestration of 2%, under the BCA, was temporarily suspended and that suspension continued through March 31, 2022. On April 1, 2022, the sequestration became 1% and it returned to 2% as of July 1, 2022. Additionally, under Statutory PAYGO, a 4% Medicare sequester could be ordered at the end of the 2024 Congressional session. Before January 2022, the Medicare statute did not require us to report ASP for our products because they are regulated by the FDA as medical devices. However, starting with the reporting deadline for the first quarter of 2022, we were required to report ASP for our products based on a provision within the Consolidated Appropriations Act of 2020, signed into law on December 27, 2020.
When ASP data are not available in the quarterly ASP file published by CMS (for instance our Affinity product in the fourth quarter of 2021), the Part A/B MACs establish payment for drugs and biologics in their jurisdiction(s). In these situations, MACs can update their reimbursement methodology as frequently as quarterly, without notice. MACs also have the discretion to establish coverage policies for all skin substitute products (including ours). Accordingly, even if coverage and reimbursement are provided, market acceptance of our products has been and will be adversely affected if access to coverage is administratively burdensome to obtain, use of our products is administratively burdensome, or is unprofitable for healthcare providers or less profitable than alternative treatments.
Furthermore, Medicare has signaled that it may revise its two-tiered bundled payment policy for skin substitutes. Medicare solicited comments in rulemaking for calendar year 2019 related to proposed updates and policy changes under the Medicare Hospital Outpatient Prospective Payment System (OPPS) and Ambulatory Surgical Center (ASC) Payment System. Medicare specifically solicited comments on whether it should eliminate the two-tiered bundle policy and establish a single bundle for all products. However, Medicare did not make any changes to its two-tiered payment policy in response to those comments. If CMS proposes and finalizes any revisions to its two-tiered payment policy, those changes could result in decreased reimbursement for our products which could decrease utilization and reduce our revenues. Moreover, any new policy could result in a financial incentive for hospitals and ASCs to use our competitor’s products, thereby reducing our market share and revenue.
57
Cost-containment efforts of our customers, purchasing groups, third-party payers, and governmental organizations could adversely affect our business, results of operations, and financial condition.
Many existing and potential customers for our products within the United States are members of GPOs and/or IDNs, including accountable care organizations or public-based purchasing organizations, and our business is partly dependent on major contracts with these organizations. Our products can be contracted under national tenders or with larger hospital GPOs. GPOs and IDNs negotiate pricing arrangements with healthcare product manufacturers and distributors and offer the negotiated prices to affiliated hospitals and other members. GPOs and IDNs typically award contracts on a category-by-category basis through a competitive bidding process. At any given time, we are typically at various stages of responding to bids and negotiating and renewing GPO and IDN agreements, including agreements that would otherwise expire. Bids are generally solicited from multiple manufacturers or service providers with the intention of obtaining lower pricing. Due to the highly competitive nature of the bidding process and the GPO and IDN contracting processes in the United States, we may not be able to obtain or maintain contract positions with major GPOs and IDNs across our product portfolio. Failure to be included in certain of these agreements could have a material adverse effect on our business, financial condition and results of operations. In addition, while having a contract with a major purchaser, such as a GPO or IDN, for a given product category can facilitate sales, sales volumes of those products may not be maintained. For example, GPOs and IDNs are increasingly awarding contracts to multiple suppliers for the same product category. Even when we are the sole contracted supplier of a GPO or IDN for a certain product category, members of the GPO or IDN generally are free to purchase from other suppliers. Furthermore, GPO and IDN contracts typically are terminable without cause upon 60 to 90 days’ notice. The healthcare industry has been consolidating, and the consolidation among third-party payers into larger purchasing groups will increase their negotiating and purchasing power. Such consolidation may result in greater pricing pressure on us due to pricing concessions and may further exacerbate the risks described above.
Risks Related to Our Intellectual Property
Our patents and other intellectual property rights may not adequately protect our products.
Our ability to compete effectively will depend, in part, on our ability to maintain the proprietary nature of our technology and manufacturing processes. We rely on manufacturing and other know-how, patents, trade secrets, trademarks, license agreements, and contractual provisions to establish our intellectual property rights and protect our products. These legal means, however, afford only limited protection and may not adequately protect our rights. The failure to obtain, maintain, enforce, or defend such intellectual property rights, for any reason, could allow third parties to make competing products or impact our ability to develop, manufacture and market our own products on a commercially viable basis, or at all, which could have a material adverse effect on our revenues, financial condition or results of operations.
In particular, we rely primarily on trade secrets, know-how, and other unpatented technology, which are difficult to protect. Although we seek such protection in part by entering into confidentiality agreements with our vendors, employees, consultants, and others who may have access to proprietary information, we cannot be certain that these agreements will not be breached, adequate remedies for any breach would be available or our trade secrets, know-how, and other unpatented proprietary technology will not otherwise become known to or be independently developed by our competitors. If we are unsuccessful in protecting our intellectual property rights, sales of our products may suffer and our ability to generate revenue could be severely impacted.
We have filed applications to register various trademarks for use in connection with our products in various countries and also, with respect to certain products, rely on the trademarks of third parties. These trademarks may not afford adequate protection. We or these third parties also may not have the financial resources to enforce the rights under these trademarks which may enable others to use the trademarks and dilute their value. Additionally, our marks may be found to conflict with the trademarks of third parties. In such a case, we may not be able to derive any value from such trademarks or, even, may be required to cease using the conflicting mark. The value of our trademarks may also be diminished by our own actions, such as failing to impose appropriate quality control when licensing our trademarks. Any of the foregoing could impair the value of, or ability to use, our trademarks and have an adverse effect on our business.
Most of the key patents related to our marketed products are expired. We have no patent protection covering, for example, our Apligraf, Dermagraft, or NuShield products. However, in addition to trade secrets, trademarks, know-how, and other unpatented technology, we have pursued and plan to continue to pursue patent protection where we believe that doing so offers potential commercial benefits. However, we may be incorrect in our assessments of whether or when to pursue patent protection. Moreover, patents may not issue from any of our pending patent applications. Even if we obtain or in-license issued patents, such patent rights may not provide valid patent protection sufficiently broad to prevent any third party from developing, using, or commercializing products that are similar or functionally equivalent to our products or technologies, or otherwise provide any competitive advantage. In addition, these patent rights may be challenged, revoked, invalidated, infringed, or circumvented by third parties. Laws relating to such rights may in the future be changed or withdrawn in a manner adverse to us.
58
Additionally, our products or the technologies or processes used to formulate or manufacture our products may now, or in the future, infringe the patent rights of third parties. It is also possible that third parties will obtain patent or other proprietary rights that might be necessary or useful for the development, manufacture, or sale of our products. In such cases, we may need or choose to obtain licenses for intellectual property rights from others and it is possible that we may not be able to obtain these licenses on commercially reasonable terms, if at all.
Pending and future intellectual property litigation could be costly and disruptive and may have an adverse effect on our business, results of operations, and financial condition.
We operate in an industry characterized by extensive intellectual property litigation. Defending intellectual property litigation is expensive and complex, takes significant time and diverts management’s attention from other business concerns, and the outcomes are difficult to predict. We have in the past been subject to claims that our products or technology violate a third party’s intellectual property rights, and we may be subject to such assertions in the future. Any pending or future intellectual property litigation may result in significant damage awards, including treble damages under certain circumstances, and injunctions that could prevent the manufacture and sale of affected products or could force us to seek a license and/or make significant royalty or other payments in order to continue selling the affected products. Such licenses may not be available on commercially reasonable terms, if at all. We have in the past and may in the future choose to settle disputes involving third-party intellectual property by taking a license. Such licenses or other settlements may involve, for example, upfront payments, yearly maintenance fees and royalties. At any given time, we may be involved as either a plaintiff or a defendant in a number of intellectual property actions, the outcomes of which may not be known for prolonged periods of time. A successful claim of patent or other intellectual property infringement or misappropriation against us could materially adversely affect our business, results of operations, and financial condition.
We may be subject to damages resulting from claims that we, our employees, or our independent contractors have wrongfully used or disclosed alleged trade secrets, proprietary or confidential information of our competitors or are in breach of non-competition or non-solicitation agreements with our competitors.
Some of our employees were previously employed at other medical device, pharmaceutical, or biotechnology companies. We may also hire additional employees who are currently employed at other medical device, pharmaceutical, or biotechnology companies, including our competitors. Additionally, consultants or other independent agents with whom we may contract may be or have been in a contractual arrangement with one or more of our competitors. Although no claims are currently pending, we may be subject to claims that we, our employees, or our independent contractors have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of these former employers or competitors. In addition, we have been and may in the future be subject to claims that we caused an employee to breach the terms of his or her non-competition or non-solicitation agreement. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. If we fail to defend such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. There can be no assurance that this type of litigation will not occur, and any future litigation or the threat thereof may adversely affect our ability to hire additional direct sales representatives, or other personnel. A loss of key personnel or their work product could hamper or prevent our ability to market existing or new products, which could severely harm our business.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time-consuming, and ultimately unsuccessful.
Competitors may infringe or misappropriate the patents or other intellectual property that we own or license. In response, we may be required to file infringement claims, which can be expensive and time-consuming. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us, such as alleging that we infringe their patents. In addition, in a patent infringement proceeding, a court may decide that a patent that we own or license is invalid or unenforceable, in whole or in part, construe the patent’s claims narrowly or conclude that there is no infringement. An adverse result in any litigation or defense proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Interference proceedings provoked by third parties or brought by us may be necessary to determine the priority of inventions with respect to the patents or patent applications that we own or license. An unfavorable outcome could require us to cease using the invention or attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our defense of litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
59
If we are unable to protect the confidentiality of our trade secrets and know-how, our business and competitive position would be harmed.
We seek to protect our proprietary technology and processes, in part, by entering into confidentiality and assignment of inventions agreements with our employees, consultants, scientific advisors, and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. Despite our efforts, agreements may be breached and security measures may fail, and we may not have adequate remedies for any breach or failure. In addition, our trade secrets and know-how may otherwise become known or be independently discovered by competitors. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive, and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. Moreover, if any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they communicate it, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
We may be subject to claims challenging the inventorship or ownership of the patents and other intellectual property that we own or license.
We may be subject to claims that former employees, collaborators, or other third parties have an ownership interest in the patents and intellectual property that we own or license. While it is our policy to require our employees and contractors who may be involved in the development of intellectual property to execute agreements obligating them to assign such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own; our licensors may face similar obstacles. We could be subject to ownership disputes arising, for example, from conflicting obligations of consultants or others who are involved in developing our product candidates. Litigation may be necessary to defend against any claims challenging inventorship or ownership. If we fail in defending any such claims, we may have to pay monetary damages and may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, intellectual property, which could adversely impact our business, results of operations, and financial condition.
Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees, and other fees on patents and patent applications will be due to be paid to the U.S. Patent and Trademark Office and similar foreign agencies in several stages over the lifetime of the patents and patent applications. We rely on our outside counsel to pay these fees due to foreign patent agencies. The U.S. Patent and Trademark Office and various foreign patent agencies require compliance with a number of procedural, documentary, fee payment, and other provisions during the patent application process. We employ law firms and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, potential competitors might be able to enter the market, which could have a material adverse effect on our business, results of operations, and financial condition.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
Success in the biopharmaceutical industry is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the pharmaceutical industry involve both technological and legal complexity, and therefore obtaining and enforcing pharmaceutical patents is costly, time-consuming, and inherently uncertain.
Recent patent reform legislation could increase the uncertainties and costs of prosecuting patent applications and enforcing and defending patents. Enacted in 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, made significant changes to U.S. patent law, including provisions that affect the prosecution of patent applications and also affect patent litigation. The U.S. Patent and Trademark Office developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, including the first to file provisions, only became effective in March 2013. The full impact of the Leahy-Smith Act on our business is not yet clear, but it could result in increased costs and more limited patent protection, either of which could adversely affect our business, results of operations, and financial condition.
60
Moreover, recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. In addition to increasing uncertainty regarding our ability to obtain patents in the future, this combination of events has created uncertainty regarding the value of any patents we do obtain. Depending on decisions by the U.S. Congress, the federal courts, and the U.S. Patent and Trademark Office, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce any current or future patents that we may own or license.
Risks Related to Our Indebtedness
Our indebtedness could have a material adverse effect on our business, results of operations, and financial condition.
As of December 31, 2022, we had approximately $71.3 million of aggregate principal amount of indebtedness outstanding under our 2021 Credit Agreement. Our indebtedness increases the risk that we may be unable to generate cash sufficient to pay amounts due in respect of our indebtedness and could have other important consequences to our debt holders and significant effects on our business. For example, it could:
In addition, the credit agreements governing our senior secured credit facilities collateralize substantially all of our personal property and assets, including our intellectual property, and contain restrictive covenants that limit our ability to engage in activities that may be in our long-term best interests. Our failure to comply with those covenants could result in an event of default that, if not cured or waived, could result in the acceleration of all of our indebtedness.
Despite our current level of indebtedness, we may incur substantially more debt. This could further exacerbate the risks associated with our substantial leverage.
We may incur significant additional indebtedness in the future. Although the credit agreements governing our senior secured and subordinated credit facilities limit our ability and the ability of our present and future subsidiaries to incur additional indebtedness, the terms of the senior secured and subordinated credit facilities permit us to incur significant additional indebtedness under certain circumstances. In addition, the credit agreements governing our senior secured and subordinated credit facilities do not prohibit us from incurring obligations that do not constitute indebtedness as defined therein. To the extent that we incur additional indebtedness or such other obligations, the risk associated with our substantial indebtedness described above, including our potential inability to service our debt, will increase.
We will require a significant amount of cash to service our debt, and our ability to generate cash depends on many factors beyond our control, and any failure to meet our debt service obligations could materially adversely affect our business, results of operations, and financial condition.
Our ability to make payments on and to refinance our indebtedness and to fund working capital needs and planned capital expenditures will depend on our ability to generate cash in the future. This, to a certain extent, is subject to general economic, financial, competitive, business, legislative, regulatory, and other factors that are beyond our control.
If our business does not generate sufficient cash flow from operations or if future borrowings are not available to us in an amount sufficient to enable us to pay our indebtedness or to fund our other liquidity needs, we may need to refinance all or a portion of our indebtedness on or before the maturity thereof, sell assets, reduce or delay capital investments or seek to raise additional capital, any of which could have a material adverse effect on our business, results of operations, and financial condition. In addition, we may
61
not be able to effect any of these actions, if necessary, on commercially reasonable terms or at all. Our ability to restructure or refinance our indebtedness will depend on the condition of the capital markets and our financial condition at such time. Any refinancing of our debt could be at higher interest rates and may require us to comply with more onerous covenants, which could further restrict our business operations. The terms of existing or future debt instruments, including the credit agreements governing our senior and subordinated secured credit facilities, may limit or prevent us from taking any of these actions. In addition, any failure to make scheduled payments of interest and principal on our outstanding indebtedness would likely result in a reduction of our credit rating, which could harm our ability to incur additional indebtedness on commercially reasonable terms or at all. Our inability to generate sufficient cash flow to satisfy our debt service obligations, or to refinance or restructure our obligations on commercially reasonable terms or at all, would have an adverse effect, which could be material, on our business, results of operations, and financial condition, as well as on our ability to satisfy our obligations in respect of the senior and subordinated secured credit facilities and our other indebtedness.
Our failure to comply with the agreements relating to our outstanding indebtedness, including as a result of events beyond our control, could result in an event of default that could materially adversely affect our business, results of operations, and financial condition.
If there were an event of default under any of the agreements relating to our outstanding indebtedness, the holders of the defaulted debt could cause all amounts outstanding with respect to that debt to be due and payable immediately. We cannot guarantee that our assets or cash flow would be sufficient to fully repay borrowings under our outstanding debt instruments if accelerated upon an event of default. Further, if we are unable to repay, refinance or restructure our indebtedness under our secured debt, the holders of such debt could proceed against the collateral securing that indebtedness. In addition, any event of default or declaration of acceleration under one debt instrument could also result in an event of default under one or more of our other debt instruments. As a result, any default by us on our indebtedness could have a material adverse effect on our business, results of operations, and financial condition.
The credit agreements governing our senior secured credit facility and our subordinated credit facility restrict our current and future operations, particularly our ability to respond to changes or to take certain actions.
The credit agreements governing our senior secured credit facility and our subordinated credit facility are collateralized by substantially all of our assets, including our intellectual property, and impose significant operating and financial restrictions and limit our ability and our other restricted subsidiaries’ ability to, among other things:
As a result of these covenants and restrictions, we are and will be limited in how we conduct our business, and we may be unable to raise additional debt or equity financing to compete effectively or to take advantage of new business opportunities. In addition, our senior secured credit facility requires us to comply with a minimum consolidated revenue covenant (measured on a trailing twelve-month basis) and a minimum monthly liquidity ratio (measured as of the last day of each month). The operating and financial restrictions and covenants in the senior secured credit facility, as well as any future financing agreements that we may enter into, may restrict our ability to finance our operations, engage in business activities or expand or fully pursue our business strategies. Our ability to comply with these covenants may be affected by events beyond our control, and we may not be able to meet those covenants. For example, in the past, we have not been in compliance with certain financial covenants in our debt agreements, which may occur again in the future. We cannot guarantee that we will be able to maintain compliance with these covenants in the future and, if we fail to do so, that we will be able to obtain waivers from the lenders and/or amend the covenants.
62
Our failure to comply with the restrictive covenants described above as well as others contained in our future debt instruments from time to time could result in an event of default, which, if not cured or waived, could result in our being required to repay these borrowings before their due date. If we are forced to refinance these borrowings on less favorable terms, our business, results of operations, and financial condition could be adversely affected.
Risks Related to Our Class A Common Stock
The Significant Stockholder Group exercises significant control over us, and their interests may conflict with yours in the future.
Alan A. Ades, Albert Erani, Glenn H. Nussdorf, Dennis Erani, Starr Wisdom, and certain of their respective affiliates, including Organo PFG LLC, Organo Investors LLC, Dennis Erani 2012 Issue Trust, Alan Ades as Trustee of the Alan Ades 2014 GRAT, Albert Erani Family Trust dated 12/29/2012, GN 2016 Family Trust u/a/d August 12, 2016, GN 2016 Organo 10-Year GRAT u/a/d September 30, 2016 and RED Holdings, LLC, who we refer to collectively as the Significant Stockholder Group, control a significant amount of the voting power of the outstanding Class A common stock. As of February 15, 2023, the Significant Stockholder Group collectively beneficially owns approximately 45% of the Company’s Class A common stock. As a result of this voting control, the Significant Stockholder Group collectively can effectively determine the outcome of all matters requiring stockholder approval, including, but not limited to, the election and removal of the Company’s directors (including the right to designate four of our directors pursuant to the terms of an agreement between the Company and the Significant Stockholder Group), as well as other matters of corporate or management policy (such as potential mergers or acquisitions, payment of dividends, asset sales, and amendments to the Company’s certificate of incorporation and bylaws). This concentration of ownership may delay or deter possible changes in control and limit the liquidity of the trading market for the Company’s Class A common stock, which may reduce the value of an investment in its Class A common stock. This voting control could also deprive stockholders of an opportunity to receive a premium for their shares of Class A common stock as part of a potential sale of the Company. So long as the Significant Stockholder Group and their affiliates continue to own a significant amount of the Company’s combined voting power, they may continue to be able to strongly influence or effectively control its decisions. The interests of the Significant Stockholder Group and their affiliates may not coincide with the interests of other holders of the Company Class A common stock.
In the ordinary course of their business activities, the Significant Stockholder Group and their affiliates may engage in activities where their interests conflict with our interests or those of our other stockholders. In addition, the Significant Stockholder Group may have an interest in pursuing acquisitions, divestitures, and other transactions that, in their judgment, could enhance their investment, even though such transactions might involve risks to you.
Our stock price has been, and is likely to continue to be, volatile. Fluctuations in revenue or results of operations could cause additional volatility in our stock price and thus our stockholders could incur substantial losses.
Our stock price has been volatile and could be subject to wide fluctuations in response to various factors, many of which are beyond our control. The stock market in general and the market for biotechnology companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies, which in some cases has been exacerbated by the COVID-19 pandemic. Any unanticipated shortfall in our revenue in any fiscal quarter could have an adverse effect on our results of operations in that quarter. The effect on our net income of such a shortfall could be exacerbated by the relatively fixed nature of most of our costs, which primarily include personnel costs as well as facilities costs. These fluctuations could cause the trading price of our stock to be negatively affected. Our quarterly operating results have varied substantially in the past and may vary substantially in the future.
Some companies that have experienced volatility in the trading price of their shares have been the subject of securities class action litigation, as we are and as disclosed in Item 3, “Legal Proceedings”. Any lawsuit to which we are a party, with or without merit, may result in an unfavorable judgment. We also may decide to settle lawsuits on unfavorable terms.
Any such negative outcome could result in payments of substantial damages or fines, damage to our reputation or adverse changes to our business practices. Defending against litigation is costly and time-consuming, and could divert our management’s attention and our resources. Furthermore, during the course of litigation, there could be negative public announcements of the results of hearings, motions or other interim proceedings or developments, which could have a negative effect on the market price of our Class A common stock.
63
The Company bylaws designate the Court of Chancery of the State of Delaware, to the fullest extent permitted by law, as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by the Company stockholders, which could limit the ability of the Company stockholders to obtain a favorable judicial forum for disputes with the Company or with directors, officers or employees of the Company and may discourage stockholders from bringing such claims.
Under the Company bylaws, unless the Company consents in writing to the selection of an alternative forum, the sole and exclusive forum will be the Court of Chancery of the State of Delaware for:
These provisions of the Company’s certificate of incorporation and bylaws could limit the ability of the Company stockholders to obtain a favorable judicial forum for certain disputes with the Company or with its directors, officers or other employees, which may discourage such lawsuits against the Company and its directors, officers, and employees. Alternatively, if a court were to find these provisions of the Company’s certificate of incorporation or bylaws inapplicable to, or unenforceable in respect of, one or more of the types of actions or proceedings listed above including, without limitation, any actions asserted under the Securities Act of 1933, as amended, the Company may incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect its business, financial condition and results of operations. In addition, there is uncertainty as to whether a court would enforce the Company’s forum selection provision with respect to any actions asserted under the Securities Act of 1933, as amended, as investors cannot waive compliance with the federal securities laws and the rules and regulations thereunder.
Provisions in the Company’s charter may inhibit a takeover of the Company, which could limit the price investors might be willing to pay in the future for the Company's Class A common stock and could entrench management.
The Company’s certificate of incorporation contains provisions that may discourage unsolicited takeover proposals that shareholders may consider to be in their best interests. These provisions include the ability of the Board of Directors to designate the terms of and issue new series of preferred shares, which may make more difficult the removal of management and may discourage transactions that otherwise could involve payment of a premium over prevailing market prices for the Company’s securities.
General Risk Factors
We are currently and in the future may be, subject to securities class action litigation or other litigation that could cause us to incur significant legal expenses, divert management’s attention, and result in harm to our business.
We are exposed to potential liabilities and reputational risk associated with securities class action litigation. We are party to a securities class action lawsuit as disclosed in Item 3, “Legal Proceedings”. We may be subject to additional lawsuits, including class action or securities derivative lawsuits as well as incur additional legal fees and may face negative impacts to our stock price and reputation. In addition, we are obligated to indemnify and advance expenses to certain individuals involved in certain of these proceedings.
Any adverse judgment in or settlement of any pending or any future litigation could result in significant payments, fines and penalties that could have a material adverse effect on our business, results of operations, financial condition and reputation. Such payments, damages or settlement costs, if any, related to these matters could be in excess of our insurance coverage. The amount of time that is required to resolve these lawsuits is unpredictable and any litigation or claims against us, even those without merit, may cause us to incur substantial costs, divert management’s attention from the day-to-day operation of our business, and materially harm our reputation.
We face significant and continuing competition, which could adversely affect our business, results of operations, and financial condition.
64
We face significant and continuing competition in our business, which is characterized by rapid technological change and significant price competition. Market share can shift as a result of technological innovation and other business factors. Our customers consider many factors when selecting a product, including product reliability, clinical outcomes, economic outcomes, price, and services provided by the manufacturer. Our ability to compete depends in large part on our ability to provide compelling clinical and economic benefits to our customers and payers, develop and commercialize new products and technologies and anticipate technological advances. Product introductions or enhancements by competitors which may have advanced technology, better features, or lower pricing may make our products obsolete or less competitive. In addition, consolidation in the healthcare industry continues to lead the demand for price concessions or to the exclusion of some suppliers from certain of our markets, which could have an adverse effect on our business, results of operations or financial condition. The presence of this competition in our market may lead to pricing pressure, which would make it more difficult to sell our products at a price that will make us profitable or prevent us from selling our products at all. As a result, we will be required to devote continued efforts and financial resources to bring our products under development to market, deliver cost-effective clinical outcomes, expand our geographic reach, enhance our existing products, and develop new products for the advanced wound care and soft tissue repair markets. Even if we develop cost effective and/or new products, they may not be covered or reimbursed due to cost-containment and other financial pressures from payers.
Our future capital needs are uncertain and we may need to raise funds in the future, and such funds may not be available on acceptable terms or at all.
Continued expansion of our business will be expensive and we may seek funds from stock offerings, borrowings under our existing or future credit facilities or other sources. Our capital requirements will depend on many factors, including:
Our operating plan may change as a result of many factors currently unknown to us and we may need additional funds sooner than planned. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. Furthermore, if we issue equity or convertible debt securities to raise capital, you may experience dilution, and the new equity or convertible debt securities may have rights, preferences, and privileges that are senior to or otherwise adversely affect your rights as a stockholder. In addition, if we raise capital through collaboration, licensing or other similar arrangements, it may be necessary to relinquish valuable rights to our products, potential products or proprietary technologies, or grant licenses on terms that are not favorable to us. If we cannot raise capital on acceptable terms, we may not be able to develop our product candidates, enhance our existing products, execute our business plan, take advantage of future opportunities, or respond to competitive pressure, changes in our supplier relationships, or unanticipated customer requirements. Any of these events could adversely affect our ability to achieve our development and commercialization goals, which could have a material adverse effect on our business, results of operations, and financial condition.
Our future success depends on our ability to retain key employees, consultants and advisors, and to attract, retain and motivate qualified personnel.
We are highly dependent on our executive officers, the loss of whose services may adversely impact the achievement of our objectives. In particular, we depend on Gary Gillheeney, our President and Chief Executive Officer. Recruiting and retaining other qualified employees, consultants and advisors for our business, including scientific and technical personnel, will also be critical to our success. There is currently a shortage of skilled executives and scientific personnel in our industry, which is likely to continue. As a result, competition for skilled personnel is intense and the turnover rate can be high. We may not be able to attract and retain personnel on acceptable terms given the competition among numerous medical device companies for individuals with similar skill sets. The inability to recruit or loss of the services of any executive, key employee, consultant or advisor may impede the progress of our research, development, and sales growth objectives.
65
Our ability to recruit, retain and motivate our employees and consultants will depend in part on our ability to offer attractive compensation. We may also need to increase the level of cash compensation that we pay to them, which may reduce funds available for research and development and support of our sales growth objectives. There can be no assurance that we will have sufficient cash available to offer our employees and consultants attractive compensation.
Despite our efforts to retain valuable employees, members of our management, scientific and development teams may terminate their employment with us. The loss of the services of any of our executive officers or other key employees and our inability to find suitable replacements could potentially harm our business, prospects, financial condition or results of operations. We do not maintain “key person” insurance policies on the lives of these individuals or any of our other employees.
Many of the companies that we compete against for qualified personnel have substantially greater financial and other resources and different risk profiles than we do. They may also provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high-quality candidates than what we can offer. If we are unable to continue to attract and retain high-quality personnel, the rate and success at which we can discover, develop and commercialize product candidates will be limited.
Business or economic disruptions or global health concerns could seriously harm our business.
Broad-based business or economic disruptions could adversely affect our business and the sale of our products. For example, in December 2019 an outbreak of a novel strain of coronavirus (COVID-19) originated in Wuhan, China, and spread to a number of other countries, including the United States. This outbreak resulted in extended shutdowns of certain businesses throughout the world. While the COVID-19 pandemic has not materially adversely affected our financial results and business operations through December 31, 2022, COVID-19 continues to present risks to the Company and we continue to closely monitor the impact of the pandemic on all aspects of our business. Global health concerns, such as the COVID-19 pandemic, often disproportionately impact the hospitals, clinics, and healthcare providers to whom we sell our products, which could have a material adverse effect on our business and our results of operation and financial condition.
Uncertainty and adverse changes in the general economic conditions may negatively affect our business.
If general economic conditions in the United States decline, or if consumers fear that economic conditions will decline, sales of our products may decline. Adverse changes may occur as a result of adverse economic conditions, fluctuating oil prices, supply chain problems, inflation, political instability, declining consumer confidence, a continuation or worsening of the COVID-19 pandemic or another pandemic, unemployment, fluctuations in stock markets, contraction of credit availability, or other factors affecting economic conditions generally. These changes may negatively affect the sales of our existing or development of future products, increase the cost, and decrease the availability of financing, or increase costs associated with producing and distributing our products and potential product candidates.
Changes in accounting standards and subjective assumptions, estimates and judgments by management related to complex accounting matters could significantly affect our business, results of operations, and financial condition.
United States generally accepted accounting principles (“GAAP”) and related accounting pronouncements, implementation guidelines and interpretations with regard to a wide range of matters that are relevant to our business are highly complex. These matters include, but are not limited to, revenue recognition, leases, income taxes, impairment of goodwill and long-lived assets and equity-based compensation. Changes in these rules, guidelines or interpretations could significantly change our reported or expected financial performance or financial condition.
In addition, the preparation of financial statements in conformity with GAAP requires management to make assumptions, estimates and judgments that affect the amounts reported in the consolidated financial statements and accompanying notes. We base our estimates and judgments on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. The results of these estimates form the basis for making judgments about the carrying values of assets, liabilities and equity, and the amount of net revenues and expenses that are not readily apparent from other sources. Our operating results may be adversely affected if our assumptions change or if actual circumstances differ from those in our assumptions, which could cause our operating results to fall below the expectations of securities analysts and investors, resulting in a decline in our stock price.
Our failure to comply with regulatory obligations could result in negative effects on our business.
The failure by us or one of our suppliers to comply with applicable regulatory requirements could result in, among other things, the FDA or other governmental authorities:
66
Failure to comply with applicable regulatory requirements could also result in civil actions against us by private parties (e.g., under the federal Lanham Act and/or state unfair competition laws), and other unanticipated negative consequences. If any of these actions were to occur it could harm our reputation and cause our product sales to suffer and may prevent us from generating revenue.
Our officers, employees, independent contractors, principal investigators, consultants and commercial partners may engage in misconduct or activities that are improper under other laws and regulations, which would create liability for us.
We are exposed to the risk that our officers, employees, independent contractors (including contract research organizations, or CROs), principal investigators, consultants and commercial partners may engage in fraudulent conduct or other illegal activity and/or may fail to disclose unauthorized activities to us. Misconduct by these parties could include, but is not limited to, intentional, reckless and/or negligent failures to comply with:
In particular, companies involved in the manufacture of medical products are subject to laws and regulations intended to ensure that medical products that will be used in patients are safe and effective, and specifically that they are not adulterated or contaminated, that they are properly labeled, and have the identity, strength, quality and purity that which they are represented to possess. Further, companies involved in the research and development of medical products are subject to extensive laws and regulations intended to protect research subjects and ensure the integrity of data generated from clinical trials and of the regulatory review process. Any misconduct in any of these areas — whether by our own employees or by contractors, vendors, business associates, consultants, or other entities acting as our agents — could result in regulatory sanctions, criminal or civil liability and serious harm to our reputation. Although we have a comprehensive compliance program designed to ensure that our employees’, CRO partners’, principal investigators’, consultants’, and commercial partners’ activities and interactions with healthcare professionals and patients are appropriate, ethical, and consistent with all applicable laws, regulations, guidelines, policies and standards, it is not always possible to identify and deter misconduct, and the precautions we take to detect and prevent this activity may not be effective in preventing such conduct, mitigating risks, or reducing the chance of governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, those actions could have a significant impact on our business, including the imposition of significant fines, and other sanctions that may materially impair our ability to run a profitable business.
67
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on our business, results of operations, and financial condition.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment, manufacture and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials or other work-related injuries, this insurance may not provide adequate coverage against potential liabilities. In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
Unanticipated changes in effective tax rates or adverse outcomes resulting from examination of the Company’s income or other tax returns could adversely affect the Company’s financial condition and results of operations.
The Company is subject to income tax in the United States and Switzerland, and the Company’s domestic tax liabilities will be subject to the allocation of expenses in differing jurisdictions. The Company’s future effective tax rates could be subject to volatility or adversely affected by a number of factors, including:
In addition, the Company may be subject to audits of the Company’s income, sales and other taxes by U.S. federal, state, local and non-U.S. taxing authorities. Outcomes from these audits could have an adverse effect on the Company’s financial condition and results of operations.
A market for the Company’s securities may not continue, which would adversely affect the liquidity and price of the Company’s securities.
The price of the Company’s securities may fluctuate significantly due to general market and economic conditions. An active trading market for the Company’s securities may never develop or, if developed, it may not be sustained. In addition, the price of the Company’s securities can vary due to general economic conditions and forecasts, the Company’s general business condition and the release of the Company’s financial reports. Additionally, if the Company’s securities are not listed on, or become delisted from, Nasdaq for any reason, and are quoted on the OTC Bulletin Board, an inter-dealer automated quotation system for equity securities that is not a national securities exchange, the liquidity and price of the Company’s securities may be more limited than if the Company was quoted or listed on Nasdaq or another national securities exchange. You may be unable to sell your securities unless a market can be established or sustained.
The Company’s quarterly operating results may fluctuate significantly and could fall below the expectations of securities analysts and investors due to seasonality and other factors, some of which are beyond the Company’s control, resulting in a decline in the Company’s stock price.
The Company’s quarterly operating results may fluctuate significantly because of several factors, including:
68
If securities or industry analysts do not publish or cease publishing research or reports about the Company, its business, or its market, or if they change their recommendations regarding the Company Class A common stock adversely, then the price and trading volume of the Company Class A common stock could decline.
The trading market for the Company Class A common stock will be influenced by the research and reports that industry or securities analysts may publish about us, the Company’s business, the Company’s market, or the Company’s competitors. Securities and industry analysts may stop publishing research on the Company. If any analyst who covers the Company were to cease coverage of the Company or fail to regularly publish reports on it, we could lose visibility in the financial markets, which could cause the Company’s stock price or trading volume to decline. If any of the analysts who cover the Company change their recommendation regarding the Company’s stock adversely, or provide more favorable relative recommendations about the Company’s competitors, the price of the Company Class A common stock would likely decline.
Changes in laws, regulations or rules, or a failure to comply with any laws, regulations or rules, may adversely affect the Company’s business, investments and results of operations.
The Company is subject to laws, regulations and rules enacted by national, regional and local governments and Nasdaq. In particular, the Company is required to comply with certain SEC, Nasdaq and other legal or regulatory requirements. Compliance with, and monitoring of, applicable laws, regulations and rules is difficult, time-consuming and costly. Those laws, regulations or rules and their interpretation and application may also change from time to time and those changes could have a material adverse effect on the Company’s business, investments and results of operations. In addition, a failure to comply with applicable laws, regulations or rules, as interpreted and applied, could have a material adverse effect on the Company’s business and results of operations.
Our failure to meet the continued listing requirements of Nasdaq could result in a delisting of our securities.
If we fail to satisfy the continued listing requirements of Nasdaq such as the corporate governance requirements or the minimum closing bid price requirement, Nasdaq may take steps to delist our securities. Such a delisting would likely have a negative effect on the price of the securities and would impair your ability to sell or purchase the securities when you wish to do so. In the event of a delisting, we can provide no assurance that any action taken by us to restore compliance with listing requirements would allow our securities to become listed again, stabilize the market price or improve the liquidity of our securities, prevent our securities from dropping below the Nasdaq minimum bid price requirement or prevent future non-compliance with Nasdaq’s listing requirements. Additionally, if our securities are not listed on, or become delisted from, Nasdaq for any reason, trading our common stock could be conducted only in the over-the-counter (“OTC”) market or on an electronic bulletin board established for unlisted securities such as the OTC Bulletin Board, an inter-dealer automated quotation system for equity securities that is not a national securities exchange, the liquidity and price of our securities may be more limited than if we were quoted or listed on Nasdaq or another national securities exchange. You may be unable to sell your securities unless a market can be established or sustained.
69
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
Our corporate headquarters is located on our four-building campus in Canton, Massachusetts, comprising approximately 300,000 square feet of leased and purchased space devoted to manufacturing, shipping, operations, and research and development. Three of the buildings are leased. The leases were initially set to expire on December 31, 2022, and were subsequently extended to December 31, 2027 when we exercised an option to renew these leases for an additional five-year term in December 2021. We lease the buildings in Canton from entities that are controlled by Alan A. Ades, Albert Erani, Dennis Erani and Glenn H. Nussdorf, who are also our stockholders. In addition, Messrs. Ades and Nussdorf are members of our Board of Directors.
In Norwood, Massachusetts, we have a leased facility of approximately 43,850 square feet for office and laboratory use. The lease commenced on March 13, 2019. The rent commencement date was February 1, 2020. The initial lease term is ten years from the rent commencement date and was extended for additional five years in December 2021. We have an option to extend the term for another ten years if exercised within 16-24 months from the end of the lease term.
With the expiration of the La Jolla facility leases on December 31, 2021, we entered into a lease in August 2020 for approximately 23,000 square feet in San Diego, California for office and laboratory use. The lease commenced on April 1, 2021. The initial lease term is ten years from the lease commencement date, with an option to extend the term for a period of five years.
In San Diego, California, we have leased a warehouse of approximately 19,000 square feet to store the manufacturing equipment from the La Jolla facilities. The lease expires on June 30, 2024.
In Birmingham, Alabama, we had a leased facility of approximately 25,000 square feet to support our amniotic products. The lease expired on December 31, 2022. We moved the operation from Birmingham, Alabama to our Canton, Massachusetts campus. We continue to maintain our R&D operations in Birmingham in a small leased facility.
ITEM 3. LEGAL PROCEEDINGS
On December 10, 2021, a class action complaint captioned Somogyi v. Organogenesis Holdings Inc., et al. was filed on behalf of a putative class of all purchasers of our securities against us and our Chief Executive Officer and Chief Financial Officer in the United States District Court for the Eastern District of New York. The court appointed Donald Martin as lead plaintiff. Mr. Martin filed an amended complaint on October 24, 2022 that brings claims on behalf of a purported class of all purchasers of our securities from August 10, 2020 through August 9, 2022 and alleges violations of federal securities law in connection with alleged false and misleading statements with respect to, among other matters, revenue, sales growth and ability to compete in connection with our Affinity and PuraPly XT products. The complaint alleges violations of the Securities Exchange Act of 1934 and Rule 10b-5 promulgated thereunder, and seeks unquantified damages as well as attorneys’ fees, expert fees and other costs. The action is in the early stages of litigation. We believe the claims are without merit and intend to vigorously contest them. We are currently preparing our papers in support of our motion to dismiss the litigation for failure to state a claim upon which relief can be granted.
We are not a party to any other material legal proceedings. From time to time, we may become involved in litigation or other legal proceedings relating to claims arising from the ordinary course of business. These matters may include intellectual property, employment and other general claims. With respect to our outstanding legal matters, based on our current knowledge, we believe that the amount or range of reasonably possible loss will not, either individually or in the aggregate, have a material adverse effect on our business, consolidated financial position, results of operations, or cash flows. However, the outcome of such legal matters is inherently unpredictable and subject to significant uncertainties.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
70
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our Class A common stock is listed on the Nasdaq Capital Market under the symbol “ORGO”. As of February 15, 2023, a total of 131,176,200 shares of our Class A common stock were outstanding and we had 583 holders of record of our Class A common stock. This number does not include shareholders for whom shares are held in “nominee” or “street” name.
Dividend policy
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain all available funds and future earnings, if any, to finance the growth and development of our business. We do not expect to pay any cash dividends on our Class A common stock in the foreseeable future. In addition, the terms of our 2021 Credit Agreement restrict our ability to pay cash dividends on our capital stock without the bank’s consent.
Stock Performance Graph(1)
The following graph shows a comparison from December 29, 2017 through December 31, 2022 of cumulative total return on assumed investments of $100.00 in cash in each of our Class A common stock, the NASDAQ Composite Index and the NASDAQ Biotechnology Index. Such returns are based on historical results and are not intended to suggest future performance. Data for the NASDAQ Composite Index and the NASDAQ Biotechnology Index assume reinvestment of dividends. For the period beginning on December 29, 2017 through December 10, 2018, the graph reflects cumulative total return based on the price per share of the Class A common stock of our predecessor company, Avista Healthcare Public Acquisition Corp., prior to the closing of our business combination.
COMPARISON OF FIVE YEARS CUMULATIVE TOTAL RETURN
Among Organogenesis Holdings Inc., the NASDAQ Composite Index,
and the NASDAQ Biotechnology Index
71
ITEM 6. RESERVED
72
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of financial condition and results of operations together with our consolidated financial statements and related notes included elsewhere in this Annual Report on Form 10-K. This discussion and other parts of this Annual Report on Form 10-K contain forward-looking statements that involve risks and uncertainties, such as statements regarding our plans, objectives, expectations, intentions and projections. Our actual results could differ materially from those discussed in these forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those discussed in the “Risk Factors” section of this Annual Report on Form 10-K.
Unless the context otherwise requires, for purposes of this section, the terms “we,” “us,” “the Company,” “Organogenesis” or “our company” refer to Organogenesis Holdings Inc. and its subsidiaries as they currently exist.
Overview
Organogenesis is a leading regenerative medicine company focused on the development, manufacture, and commercialization of solutions for the Advanced Wound Care and Surgical & Sports Medicine markets. Our products have been shown through clinical and scientific studies to support and in some cases accelerate tissue healing and improve patient outcomes. We are advancing the standard of care in each phase of the healing process through multiple breakthroughs in tissue engineering and cell therapy. Our solutions address large and growing markets driven by aging demographics and increases in comorbidities such as diabetes, obesity, cardiovascular and peripheral vascular disease and smoking. We offer our differentiated products and in-house customer support to a wide range of health care customers including hospitals, wound care centers, government facilities, ambulatory service centers (“ASCs”) and physician offices. Our mission is to provide integrated healing solutions that substantially improve medical outcomes and the lives of patients while lowering the overall cost of care.
We offer a comprehensive portfolio of products in the markets we serve that address patient needs across the continuum of care. We have and intend to continue to generate data from clinical trials, real-world outcomes and health economics research that validate the clinical efficacy and value proposition offered by our products. Several of our existing and pipeline products in our portfolio have premarket approval (“PMA”), or 510(k) clearance from the FDA. Given the extensive time and cost required to conduct clinical trials and receive FDA approvals, we believe that our data and regulatory approvals provide us with a strong competitive advantage. Our product development expertise and multiple technology platforms provide a robust product pipeline, which we believe will drive future growth.
In the Advanced Wound Care market, we focus on the development and commercialization of advanced wound care products for the treatment of chronic and acute wounds in various treatment settings. We have a comprehensive portfolio of regenerative medicine products capable of supporting patients from early in the wound healing process through wound closure regardless of wound type. Our Advanced Wound Care products include Apligraf for the treatment of venous leg ulcers (“VLUs”) and diabetic foot ulcers (“DFUs”); Dermagraft for the treatment of DFUs (manufacturing currently suspended pending transition to a new manufacturing facility or engagement of a third-party manufacturer); PuraPly AM as an antimicrobial barrier for a broad variety of wound types; and the Affinity, Novachor and NuShield wound coverings to address a variety of wound sizes and types. We have a highly trained and specialized direct wound care sales force paired with comprehensive customer support services.
In the Surgical & Sports Medicine market, we are leveraging our broad regenerative medicine capabilities to address chronic and acute surgical wounds and tendon and ligament injuries. Our Sports Medicine products include NuShield for surgical applications in targeted soft tissue repairs; and Affinity, Novachor, PuraPly MZ, and PuraPly AM for management of open wounds in the surgical setting. We currently sell these products through independent agencies and our direct sales force.
We generated net revenue of $450.9 million, $467.4 million, and $338.3 million for the years ended December 31, 2022, 2021, and 2020, respectively. We reported net income of $15.5 million, $94.2 (which includes a $48.3 million benefit from release of a tax valuation allowance), and $17.2 million for the years ended December 31, 2022, 2021, and 2020, respectively. While we reported net income for the most recent three years, we have incurred significant losses since inception and we may incur operating losses in the future as we expend resources as part of our efforts to grow our organization to support the planned expansion of our business. As of December 31, 2022, we had an accumulated deficit of $45.3 million. Our primary sources of capital to date have been from sales of our products, borrowings from related parties and institutional lenders and proceeds from the sale of our Class A common stock. We operate as one segment of regenerative medicine.
73
COVID-19 pandemic
On January 30, 2023, the Biden Administration announced it will end the public health emergency (and national emergency) declarations related to COVID-19 on May 11, 2023. While the COVID-19 pandemic has not materially adversely affected our financial results and business operations through December 31, 2022, COVID-19 continues to present risks to the Company and we continue to closely monitor the impact of the pandemic on all aspects of our business. We are unable to predict the impact that COVID-19 (including the emergence of new variants) will have on our financial position and operating results in the future.
CPN Acquisition
On September 17, 2020, we acquired certain assets and assumed certain liabilities of CPN Biosciences, LLC (“CPN”) pursuant to an asset purchase agreement dated July 24, 2020. This transaction was accounted for as a business combination using the acquisition method of accounting in accordance with ASC Topic 805, Business Combinations. The aggregated consideration amounted to $19.0 million as of the acquisition date which consisted of $6.4 million in cash, 2,151,438 shares of our common stock with a fair value of $8.8 million, and a contingent consideration (the “Earnout”) with a fair value at such time of $3.8 million. At the closing, we paid $5.8 million in cash and issued 1,947,953 shares of our Class A common stock. The remaining consideration of $1.4 million was held back and was released in April 2022 by the Company paying $0.6 million in cash and issuing 203,485 shares of the Company’s Class A common stock to the former equity holders of CPN. As of the conclusion of the Earnout period on June 30, 2022, the Company calculated the Earnout liability to be $0.
The results of operations of CPN have been included in our consolidated financial statements beginning on the acquisition date.
Dermagraft
As previously disclosed, manufacturing of Dermagraft was suspended in the fourth quarter of 2021 and sales of Dermagraft were suspended in the second quarter of 2022. We currently plan to transition our Dermagraft manufacturing to a new manufacturing facility or engage a third-party manufacturer, which we expect will result in substantial long-term cost savings. In the period when Dermagraft is not available, we expect that customers will be willing to substitute Apligraf for Dermagraft and that the suspension of Dermagraft sales will not have a material impact on our net revenue. However, if we do not realize the expected substantial long-term cost savings or if customers are unwilling to substitute Apligraf for Dermagraft during the period in which Dermagraft is unavailable, it could have an adverse effect on our net revenue and results of operations.
Management’s Use of Non-GAAP Measures
Our management uses financial measures that are not in accordance with generally accepted accounting principles in the United States, or GAAP, in addition to financial measures in accordance with GAAP to evaluate our operating results. These non-GAAP financial measures should be considered supplemental to, and not a substitute for, our reported financial results prepared in accordance with GAAP. Our management uses Adjusted EBITDA to evaluate our operating performance and trends and make planning decisions. Our management believes Adjusted EBITDA helps identify underlying trends in our business that could otherwise be masked by the effect of the items that we exclude. Accordingly, we believe that Adjusted EBITDA provides useful information to investors and others in understanding and evaluating our operating results, enhancing the overall understanding of our past performance and future prospects, and allowing for greater transparency with respect to key financial metrics used by our management in its financial and operational decision-making.
We define EBITDA as net income (loss) before depreciation and amortization, interest expense and income taxes. We define Adjusted EBITDA as EBITDA, further adjusted for the impact of certain items that we do not consider indicative of our core operating performance. These items include non-cash equity compensation, restructuring charges, loss on the extinguishment of debt, mark-to-market adjustments on our Earnout liability, transaction costs related to CPN acquisition, gain on settlement of deferred acquisition consideration, recovery of certain notes receivable from related parties, write-off of the capitalized costs related to certain unfinished construction work, the cancellation fee for terminating certain agreements or pausing a certain construction project, and settlement fee for a certain dispute. We have presented Adjusted EBITDA in this Annual Report on Form 10-K because it is a key measure used by our management and Board of Directors to understand and evaluate our operating performance, generate future operating plans and make strategic decisions regarding the allocation of capital. In particular, we believe that the exclusion of certain items in calculating Adjusted EBITDA can produce a useful measure for period-to-period comparisons of our business.
Our Adjusted EBITDA is not prepared in accordance with GAAP, and should not be considered in isolation of, or as an alternative to, measures prepared in accordance with GAAP. There are a number of limitations related to the use of Adjusted EBITDA
74
rather than net income (loss), which is the most directly comparable financial measure calculated and presented in accordance with GAAP. Some of these limitations are:
Because of these limitations, we consider, and you should consider, Adjusted EBITDA together with other operating and financial performance measures presented in accordance with GAAP. A reconciliation of Adjusted EBITDA from net income (loss), the most directly comparable financial measure calculated in accordance with GAAP, has been included herein.
Components of Our Consolidated Results of Operations
In assessing the performance of our business, we consider a variety of performance and financial measures. We believe the items discussed below provide insight into the factors that affect these key measures.
Revenue
We derive our net revenue from our portfolio of Advanced Wound Care and Surgical & Sports Medicine products. We primarily sell our Advanced Wound Care products through direct sales representatives who manage and maintain the sales relationships with hospitals, wound care centers, government facilities, ASCs, and physician offices. We primarily sell our Surgical & Sports Medicine products through third-party agencies.
We recognize revenue from sales of our Advanced Wound Care and Surgical & Sports Medicine products when the customer obtains control of our product, which occurs at a point in time and may be upon procedure date, shipment, or delivery, based on the contractual terms of a contract. We record revenue net of a reserve for returns, discounts and GPO rebates, which represent a direct reduction to the revenue we recognize.
Several factors affect our reported revenue in any period, including product, payer and geographic sales mix, operational effectiveness, pricing realization, marketing and promotional efforts, the timing of orders and shipments, regulatory actions including healthcare reimbursement scenarios, competition and business acquisitions.
Cost of goods sold and gross profit
Cost of goods sold includes personnel costs, product testing costs, quality assurance costs, raw materials and product costs, manufacturing costs, and the costs associated with our manufacturing and warehouse facilities. The changes in our cost of goods sold correspond with the changes in sales units and are also affected by product mix.
Gross profit is calculated as net revenue less cost of goods sold and generally increases as revenue increases. Our gross profit is affected by product and geographic sales mix, realized pricing of our products, the efficiency of our manufacturing operations and the costs of materials used and fees charged by third-party manufacturers to produce our products. Regulatory actions, including healthcare reimbursement scenarios, which may require costly expenditures or result in pricing pressures, may decrease our gross profit.
Selling, general and administrative expenses
Selling, general and administrative expenses generally include personnel costs for sales, marketing, sales support, customer support, and general and administrative personnel, sales commissions, incentive compensation, insurance, professional fees, depreciation, amortization, bad debt expense, royalties, information systems costs, gain or loss on disposal of long-lived assets, and costs associated with our administrative facilities. We generally expect our selling, general and administrative expenses to continue to
75
increase due to increased investments in market development and the geographic expansion of our sales forces as we drive for continued revenue growth.
Research and development expenses
Research and development expenses include personnel costs for our research and development personnel, expenses related to improvements in our manufacturing processes, enhancements to our currently available products, and additional investments in our product and platform development pipeline. Our research and development expenses also include expenses for clinical trials. We expense research and development costs as incurred. We generally expect that research and development expenses will increase as we continue to conduct clinical trials on new and existing products, move products through the regulatory pathway (e.g., seek BLA approval), bring new products to market, and enhance our manufacturing process and procedures.
Other expense, net
Interest expense—Interest expense consists of interest on our outstanding indebtedness, including amortization of debt discount and debt issuance costs, net of interest income recognized.
Loss on the extinguishment of debt— In August 2021, upon entering into the 2021 Credit Agreement, we paid an aggregate amount of $70.6 million associated with the termination of the 2019 Credit Agreement, including unpaid principal, accrued interest, the Final Payment and a prepayment fee. We recognized $1.9 million as loss on the extinguishment of the loan for the year ended December 31, 2021.
Gain on settlement of deferred acquisition consideration—In February 2020, we settled a dispute on the $5.0 million deferred acquisition consideration with the sellers of NuTech Medical for $4.0 million and assumed from the sellers of NuTech Medical the responsibilities related to a legacy lawsuit of NuTech Medical which was settled in October 2020. In connection with the settlement of this dispute and the legacy lawsuit, we recorded a gain of $2.2 million for the year ended December 31, 2020.
Income taxes
We account for income taxes using an asset and liability approach. Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Valuation allowances are provided when necessary to reduce net deferred tax assets to an amount that is more likely than not to be realized.
In determining whether a valuation allowance for deferred tax assets is necessary, we analyze both positive and negative evidence related to the realization of deferred tax assets including projected future taxable income, recent financial results and estimates of future reversals of deferred tax assets and liabilities. In addition, we consider whether it is more likely than not that the tax position will be sustained on examination by taxing authorities based on the technical merits of the position. In consideration of the factors discussed above, in the fourth quarter of 2021, we determined it was more likely than not that our deferred tax assets would be realized in the future and released the valuation allowance on our net U.S. deferred tax assets as of December 31, 2021, resulting in a benefit of $48.3 million in income taxes. We maintained the same position that our net U.S. deferred tax assets did not require a valuation allowance as of December 31, 2022.
Our U.S. provision for income taxes relates to current tax expense associated with taxable income that could not be offset by state net operating losses. We have utilized net operating losses to offset all of the 2022 federal taxable income but have exhausted net operating losses and are subject to limitations in the net operating loss utilization in certain states. We have also recorded a foreign provision for income taxes related to our wholly-owned subsidiary in Switzerland.
We account for uncertainty in income taxes recognized in the consolidated financial statements by applying a two-step process to determine the amount of tax benefit to be recognized. First, the tax position must be evaluated to determine the likelihood that it will be sustained upon external examination by the taxing authorities. If the tax position is deemed more-likely-than-not to be sustained, the tax position is then assessed to determine the amount of benefit to recognize in the consolidated financial statements. The amount of the benefit that may be recognized is the largest amount that has a greater than 50% likelihood of being realized upon ultimate settlement. The provision for income taxes includes the effects of any resulting tax reserves, or unrecognized tax benefits, that are considered appropriate as well as the related net interest and penalties.
Results of Operations
The following table sets forth, for the periods indicated, our results of operations:
76
|
|
Year Ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
Net revenue |
|
$ |
450,893 |
|
|
$ |
467,359 |
|
|
$ |
338,298 |
|
Cost of goods sold |
|
|
105,019 |
|
|
|
114,199 |
|
|
|
87,319 |
|
Gross profit |
|
|
345,874 |
|
|
|
353,160 |
|
|
|
250,979 |
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|||
Selling, general and administrative |
|
|
283,808 |
|
|
|
250,200 |
|
|
|
204,193 |
|
Research and development |
|
|
39,762 |
|
|
|
30,742 |
|
|
|
20,086 |
|
Total operating expenses |
|
|
323,570 |
|
|
|
280,942 |
|
|
|
224,279 |
|
Income from operations |
|
|
22,304 |
|
|
|
72,218 |
|
|
|
26,700 |
|
Other expense, net: |
|
|
|
|
|
|
|
|
|
|||
Interest expense |
|
|
(2,009 |
) |
|
|
(7,236 |
) |
|
|
(11,279 |
) |
Gain on settlement of deferred acquisition consideration |
|
|
- |
|
|
|
- |
|
|
|
2,246 |
|
Loss on the extinguishment of debt |
|
|
- |
|
|
|
(1,883 |
) |
|
|
- |
|
Other income (loss), net |
|
|
(13 |
) |
|
|
(13 |
) |
|
|
97 |
|
Total other expense, net |
|
|
(2,022 |
) |
|
|
(9,132 |
) |
|
|
(8,936 |
) |
Net income before income taxes |
|
|
20,282 |
|
|
|
63,086 |
|
|
|
17,764 |
|
Income tax (expense) benefit |
|
|
(4,750 |
) |
|
|
31,116 |
|
|
|
(530 |
) |
Net income |
|
$ |
15,532 |
|
|
$ |
94,202 |
|
|
$ |
17,234 |
|
EBITDA and Adjusted EBITDA
The following table presents a reconciliation of GAAP net income to non-GAAP EBITDA and non-GAAP Adjusted EBITDA, for each of the periods presented:
|
|
Year Ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(in thousands) |
|
|||||||||
Net income |
|
$ |
15,532 |
|
|
$ |
94,202 |
|
|
$ |
17,234 |
|
Interest expense |
|
|
2,009 |
|
|
|
7,236 |
|
|
|
11,279 |
|
Income tax expense (benefit) |
|
|
4,750 |
|
|
|
(31,116 |
) |
|
|
530 |
|
Depreciation |
|
|
5,845 |
|
|
|
5,781 |
|
|
|
4,438 |
|
Amortization |
|
|
4,883 |
|
|
|
4,949 |
|
|
|
3,745 |
|
EBITDA |
|
|
33,019 |
|
|
|
81,052 |
|
|
|
37,226 |
|
Stock-based compensation expense |
|
|
6,552 |
|
|
|
3,864 |
|
|
|
1,661 |
|
Restructuring charge (1) |
|
|
2,268 |
|
|
|
4,704 |
|
|
|
618 |
|
Gain on settlement of deferred acquisition consideration (2) |
|
|
- |
|
|
|
- |
|
|
|
(2,246 |
) |
Recovery of certain notes receivable from related parties (3) |
|
|
- |
|
|
|
(179 |
) |
|
|
(1,516 |
) |
Cancellation fee (4) |
|
|
- |
|
|
|
- |
|
|
|
1,950 |
|
Write-off of certain assets (5) |
|
|
4,200 |
|
|
|
1,104 |
|
|
|
- |
|
Change in fair value of Earnout (6) |
|
|
- |
|
|
|
(3,985 |
) |
|
|
203 |
|
Settlement fee (7) |
|
|
2,600 |
|
|
|
700 |
|
|
|
- |
|
Loss on extinguishment of debt (8) |
|
|
- |
|
|
|
1,883 |
|
|
|
- |
|
Facility construction project pause (9) |
|
|
632 |
|
|
|
- |
|
|
|
- |
|
CPN transaction costs (10) |
|
|
- |
|
|
|
- |
|
|
|
929 |
|
Adjusted EBITDA |
|
$ |
49,271 |
|
|
$ |
89,143 |
|
|
$ |
38,825 |
|
77
Comparison of the Years Ended December 31, 2022, 2021, and 2020
Revenue
|
|
Years Ended December 31, |
|
|
Change |
|
||||||||||||||||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|
2022 to 2021 |
|
|
2021 to 2020 |
|
|||||||||||||
|
|
(in thousands, except for percentages) |
|
|||||||||||||||||||||||||
Advanced Wound Care |
|
$ |
422,231 |
|
|
$ |
430,237 |
|
|
$ |
294,624 |
|
|
$ |
(8,006 |
) |
|
|
(2 |
%) |
|
$ |
135,613 |
|
|
|
46 |
% |
Surgical & Sports Medicine |
|
|
28,662 |
|
|
|
37,122 |
|
|
|
43,674 |
|
|
|
(8,460 |
) |
|
|
(23 |
%) |
|
|
(6,552 |
) |
|
|
(15 |
%) |
Net revenue |
|
$ |
450,893 |
|
|
$ |
467,359 |
|
|
$ |
338,298 |
|
|
$ |
(16,466 |
) |
|
|
(4 |
%) |
|
$ |
129,061 |
|
|
|
38 |
% |
For the year ended December 31, 2022, net revenue from our Advanced Wound Care products decreased by $8.0 million, or 2%, as compared to the year ended December 31, 2021. The decrease in Advanced Wound Care net revenue was primarily attributable to a decrease in sales of certain of our non-PuraPly products and the settlement fee with a GPO recorded as a direct reduction of revenue in the year ended December 31, 2022.
For the year ended December 31, 2022, net revenue from our Surgical & Sports Medicine products decreased by $8.5 million, or 23%, as compared to the year ended December 31, 2021. The decrease in Surgical & Sports Medicine net revenue was primarily due to the continued impact of the suspension of marketing of our ReNu and NuCel products in connection with the expiration of the FDA’s enforcement grace period on May 31, 2021.
For the year ended December 31, 2021, net revenue from our Advanced Wound Care products increased by $135.6 million, or 46%, as compared to the year ended December 31, 2020. The increase in Advanced Wound Care net revenue was primarily attributable to the expanded sales force, increased sales to existing and new customers, increased adoption of our placental-based product portfolio, including our Affinity product, as well as increased adoption of our PuraPly line extensions launched in the second half of 2020.
For the year ended December 31, 2021, net revenue from our Surgical & Sports Medicine products decreased by $6.6 million, or 15%, as compared to the year ended December 31, 2020. The decrease in Surgical & Sports Medicine net revenue was primarily due to the impact of the suspension of marketing of our ReNu and NuCel products in connection with the expiration of the FDA’s enforcement grace period on May 31, 2021.
Included within net revenue is PuraPly revenue of $242.7 million, $198.1 million, and $147.3 million for the years ended December 31, 2022, 2021, and 2020, respectively. The continued increase in PuraPly revenue in the years ended December 31, 2022 and 2021 was due to the expanded sales forces, expanded sites of care, and increased adoption, by existing and new customers, of our PuraPly line extensions.
Cost of Goods Sold and Gross Profit
78
|
|
Years Ended December 31, |
|
|
Change |
|
||||||||||||||||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|
2022 to 2021 |
|
|
2021 to 2020 |
|
|||||||||||||
|
|
(in thousands, except for percentages) |
|
|||||||||||||||||||||||||
Cost of goods sold |
|
$ |
105,019 |
|
|
$ |
114,199 |
|
|
$ |
87,319 |
|
|
$ |
(9,180 |
) |
|
|
(8 |
%) |
|
$ |
26,880 |
|
|
|
31 |
% |
Gross profit |
|
$ |
345,874 |
|
|
$ |
353,160 |
|
|
$ |
250,979 |
|
|
$ |
(7,286 |
) |
|
|
(2 |
%) |
|
$ |
102,181 |
|
|
|
41 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
For the year ended December 31, 2022, cost of goods sold decreased by $9.2 million, or 8%, as compared to the year ended December 31, 2021. The decrease in cost of goods sold was primarily due to decreased sales volume in our Advanced Wound Care and Surgical & Sports Medicine products.
For the year ended December 31, 2022, gross profit decreased by $7.3 million, or 2%, as compared to the year ended December 31, 2021. The decrease in gross profit resulted primarily from decreased sales volume and increased manufacturing-related costs, partially offset by a shift in product mix to our higher gross margin products.
For the year ended December 31, 2021, cost of goods sold increased by $26.9 million, or 31%, as compared to the year ended December 31, 2020. The increase in cost of goods sold was primarily due to increased unit volumes, and additional manufacturing and quality control headcount.
For the year ended December 31, 2021, gross profit increased by $102.2 million, or 41%, as compared to the year ended December 31, 2020. The increase in gross profit resulted primarily from increased sales volume due to the strength in our Advanced Wound Care products as well as a shift in product mix to our higher gross margin products.
Selling, General and Administrative Expenses
|
|
Years Ended December 31, |
|
|
Change |
|
||||||||||||||||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|
2022 to 2021 |
|
|
2021 to 2020 |
|
|||||||||||||
|
|
(in thousands, except for percentages) |
|
|||||||||||||||||||||||||
Selling, general and administrative |
|
$ |
283,808 |
|
|
$ |
250,200 |
|
|
$ |
204,193 |
|
|
$ |
33,608 |
|
|
|
13 |
% |
|
$ |
46,007 |
|
|
|
23 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
For the year ended December 31, 2022, selling, general and administrative expenses increased by $33.6 million, or 13%, as compared to the year ended December 31, 2021. The increase in selling, general and administrative expenses was primarily due to a $12.2 million increase related to additional headcount, primarily in our direct sales force, a $10.0 million increase related to increased travel and marketing programs amid the relaxed COVID-19 travel restrictions, a $5.6 million increase in legal, royalty and consulting costs associated with the ongoing operations of our business and the ERP system implementation, and a $4.2 million charge for disposal of certain equipment related to the construction in progress at one of the Company's Canton Massachusetts facilities and $0.6 million cancellation fees incurred in connection with the Company’s decision to pause the construction project for the same facility. In addition, in the year ended December 31, 2021, the Company recorded a $4.0 million reduction to the selling, general and administrative expenses related to the CPN Earnout fair value adjustments. These increases were partially offset by a $1.7 million miscellaneous decrease and a $1.3 million decrease in restructuring costs due to the smaller scale of the restructuring activities associated with closing the Birmingham office in 2022 as compared to the restructuring activities associated with closing the La Jolla office in 2021.
For the year ended December 31, 2021, selling, general and administrative expenses increased by $46.0 million, or 23%, as compared to the year ended December 31, 2020. The increase in selling, general and administrative expenses was primarily due to a $33.0 million increase related to additional headcount, primarily in our direct sales force and increased sales commissions due to increased sales, a $10.9 million increase in various administrative costs resulting from increased revenue and increased in legal, consulting fees and other costs associated with the ongoing operations of our business, a $4.3 million increase related to increased travel and marketing programs amid the relaxed COVID-19 travel restrictions, a $2.9 million increase in restructuring cost associated with closing the La Jolla office, and a $1.1 million write-off of certain design and consulting fees previously capitalized related to the unfinished construction work at one of the Company’s Canton, Massachusetts facilities. These increases were partially offset by a $4.2 million decrease resulting from the CPN Earnout fair value adjustment and a $2.0 million decrease in the cancellation fee incurred in the three months ended March 31, 2020 to cancel certain product development and consulting agreements.
79
Research and Development Expenses
|
|
Years Ended December 31, |
|
|
Change |
|
||||||||||||||||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|
2022 to 2021 |
|
|
2021 to 2020 |
|
|||||||||||||
|
|
(in thousands, except for percentages) |
|
|||||||||||||||||||||||||
Research and development |
|
$ |
39,762 |
|
|
$ |
30,742 |
|
|
$ |
20,086 |
|
|
$ |
9,020 |
|
|
|
29 |
% |
|
$ |
10,656 |
|
|
|
53 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
For the year ended December 31, 2022, research and development expenses increased by $9.0 million, or 29%, as compared to the year ended December 31, 2021. The increase in research and development expenses was primarily due to increased headcount associated with our existing Advanced Wound Care and Surgical & Sports Medicine products, an increase in product costs associated with our pipeline products not yet commercialized and an increase in the clinical study and related costs necessary to seek regulatory approvals for certain of our products.
For the year ended December 31, 2021, research and development expenses increased by $10.7 million, or 53%, as compared to the year ended December 31, 2020. The increase in research and development expenses was primarily due to increased headcount associated with our existing Advanced Wound Care and Surgical & Sports Medicine products, an increase in product costs associated with our pipeline products not yet commercialized and an increase in the clinical study and related costs necessary to seek regulatory approvals for certain of our products.
Other Expense, Net
|
|
Years Ended December 31, |
|
|
Change |
|
||||||||||||||||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|
2022 to 2021 |
|
|
2021 to 2020 |
|
|||||||||||||
|
|
(in thousands, except for percentages) |
|
|||||||||||||||||||||||||
Interest expense |
|
$ |
(2,009 |
) |
|
$ |
(7,236 |
) |
|
$ |
(11,279 |
) |
|
$ |
5,227 |
|
|
|
(72 |
%) |
|
$ |
4,043 |
|
|
|
(36 |
%) |
Gain on settlement of deferred acquisition consideration |
|
|
- |
|
|
|
- |
|
|
|
2,246 |
|
|
|
- |
|
|
|
0 |
% |
|
|
(2,246 |
) |
|
|
(100 |
%) |
Loss on the extinguishment of debt |
|
|
- |
|
|
|
(1,883 |
) |
|
|
- |
|
|
|
1,883 |
|
|
|
(100 |
%) |
|
|
(1,883 |
) |
|
** |
|
|
Other income (expense), net |
|
|
(13 |
) |
|
|
(13 |
) |
|
|
97 |
|
|
|
- |
|
|
|
0 |
% |
|
|
(110 |
) |
|
|
(113 |
%) |
Total other expense, net |
|
$ |
(2,022 |
) |
|
$ |
(9,132 |
) |
|
$ |
(8,936 |
) |
|
$ |
7,110 |
|
|
|
(78 |
%) |
|
$ |
(196 |
) |
|
|
2 |
% |
** not meaningful
For the year ended December 31, 2022, total other expense, net, decreased by $7.1 million, or 78%, as compared to the year ended December 31, 2021. The decrease in interest expense in 2022 resulted from the lower interest rate for the borrowings under the 2021 Credit Agreement. Loss on extinguishment of debt of $1.9 million in 2021 was related to loss recognized on the extinguishment of the 2019 Credit Agreement upon repayment in August 2021.
For the year ended December 31, 2021, total other expense, net, increased by $0.2 million, or 2%, as compared to the year ended December 31, 2020. Interest expense decreased by $4.0 million, or 36%, primarily due to the reduced interest rate for borrowings under the 2021 Credit Agreement. Loss on extinguishment of debt of $1.9 million in 2021 was related to loss recognized on the extinguishment of the 2019 Credit Agreement upon repayment in August 2021. The gain of $2.2 million in 2020 on the settlement of deferred acquisition consideration was related to the settlement of the deferred acquisition consideration dispute with the sellers of NuTech Medical in February 2020 as well as the decrease in legal accruals related to the settlement in October 2020 of a legacy lawsuit which we assumed from the sellers of NuTech Medical as part of the resolution of the aforementioned dispute.
Income Tax (Expense) Benefit
|
|
Years Ended December 31, |
|
|
Change |
|||||||||||||||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|
2022 to 2021 |
|
|
2021 to 2020 |
||||||||||||
|
|
(in thousands, except for percentages) |
||||||||||||||||||||||||
Income tax (expense) benefit |
|
$ |
(4,750 |
) |
|
$ |
31,116 |
|
|
$ |
(530 |
) |
|
$ |
(35,866 |
) |
|
|
(115 |
%) |
|
$ |
31,646 |
|
|
** |
** not meaningful
For the year ended December 31, 2022, income tax expense of $4.8 million included $2.8 million of current income taxes and $2.0 million of deferred income taxes. The effective tax rate for 2022 was 23.4% and was computed based on the statutory rate of 21%
80
adjusted primarily for state and local income taxes, nondeductible officer compensation and an out-of-period adjustment for an error included in the beginning balance of the deferred tax asset. For the year ended December 31, 2021, income tax benefit was $31.1 million, which primarily resulted from the release of the valuation allowance previously recorded against the full amount of our net U.S. deferred tax assets as of December 31, 2021. See footnote “15. Income Taxes” to our audited financial statements included in this Annual Report on Form 10-K .
For the year ended December 31, 2021, income tax benefit was $31.1 million, as compared to income tax expense of $0.5 million for the year ended December 31, 2020. The change is due to the deferred tax benefit of $32.0 million recognized for the year ended December 31, 2021, which primarily resulted from the release of the valuation allowance previously recorded against the full amount of our net U.S. deferred tax assets as of December 31, 2021. See footnote “15. Income Taxes” to our audited financial statements included in this Annual Report on Form 10-K.
Liquidity and Capital Resources
Since our inception, we have funded our operations and capital expenditures through cash flows from product sales, loans from affiliates and entities controlled by certain of our affiliates, third-party debt and proceeds from the sale of our capital stock. As of December 31, 2022, we had an accumulated deficit of $45.3 million and working capital of $147.6 million which included $102.5 million in cash and cash equivalents. We also have $125.0 million available for future revolving borrowings under our Revolving Facility (see footnote “12. Long-Term Debt Obligations” to our audited financial statements included in this Annual Report on Form 10-K). For the year ended December 31, 2022, we reported $450.9 million in net revenue, $15.5 million in net income and $24.9 million of cash inflows from operating activities. We expect that our cash on hand and other components of working capital as of December 31, 2022, availability under the 2021 Credit Agreement, plus net cash flows from product sales, will be sufficient to fund our operating expenses, capital expenditure requirements and debt service payments for at least 12 months beyond the filing date of this Annual Report on Form 10-K.
We continue to closely monitor ongoing developments in connection with the COVID-19 pandemic, which may negatively affect our commercial prospects, cash position and access to capital in fiscal 2023 or beyond. We will continue to assess our cash and other sources of liquidity and, if circumstances warrant, we will make appropriate adjustments to our operating plan. Please see “Item 1A. Risk Factors” in this Annual Report on Form 10-K for an additional discussion of risks.
Our primary uses of cash are working capital requirements, capital expenditure and debt service payments. Additionally, from time to time, we may use capital for acquisitions and other investing and financing activities. Working capital is used principally for our personnel as well as manufacturing costs related to the production of our products. Our working capital requirements vary from period to period depending on manufacturing volumes, the timing of shipments and the payment cycles of our customers and payers. Our capital expenditures consist primarily of building improvements, manufacturing equipment, and computer hardware and software.
To the extent additional funds are necessary to meet our long-term liquidity needs as we continue to execute on our business strategy, we anticipate that they will be obtained through additional equity or debt financings, other strategic transactions or a combination of these potential sources of funds. There can be no assurance that we will be able to obtain additional funds on terms acceptable to us, on a timely basis or at all.
The following table presents our cash and outstanding debt as of the dates indicated:
|
|
December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(in thousands) |
|
|||||||||
Cash and cash equivalents |
|
$ |
102,478 |
|
|
$ |
113,929 |
|
|
$ |
84,394 |
|
|
|
|
|
|
|
|
|
|
|
|||
Line of credit |
|
$ |
- |
|
|
$ |
- |
|
|
$ |
10,000 |
|
Term loan net of debt discount and issuance cost |
|
|
70,769 |
|
|
|
73,425 |
|
|
|
59,710 |
|
Finance lease obligations |
|
|
- |
|
|
|
200 |
|
|
|
15,061 |
|
Total debt |
|
$ |
70,769 |
|
|
$ |
73,625 |
|
|
$ |
84,771 |
|
Under the line of credit or the Revolving Facility, we have up to $125.0 million available for future revolving borrowings, subject to maintaining compliance with financial and non-financial covenants.
81
Cash Flows
The following table summarizes our cash flows for each of the periods presented:
|
|
Year Ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(in thousands) |
|
|||||||||
Net cash provided by operating activities |
|
$ |
24,859 |
|
|
$ |
61,978 |
|
|
$ |
5,466 |
|
Net cash used in investing activities |
|
|
(33,898 |
) |
|
|
(31,220 |
) |
|
|
(23,498 |
) |
Net cash provided by (used in) financing activities |
|
|
(2,199 |
) |
|
|
(1,036 |
) |
|
|
42,468 |
|
Net increase (decrease) in cash and restricted cash |
|
$ |
(11,238 |
) |
|
$ |
29,722 |
|
|
$ |
24,436 |
|
Operating Activities
During the year ended December 31, 2022, net cash provided by operating activities was $24.9 million, resulting from our net income of $15.5 million, non-cash charges of $43.4 million, partially offset by net cash used in connection with changes in our operating assets and liabilities of $34.1 million. Net cash used in changes in our operating assets and liabilities included an increase in accounts receivable of $8.8 million, an increase in inventory and prepaid expenses of $9.8 million, a decrease in operating lease liability of $7.0 million and a decrease of accrued expenses of $11.9 million, all of which were partially offset by an increase in accounts payable and other liabilities of $3.3 million.
During the year ended December 31, 2021, net cash provided by operating activities was $62.0 million, resulting from our net income of $94.2 million, non-cash charges of $4.8 million, partially offset by net cash used in connection with changes in our operating assets and liabilities of $37.0 million. The non-cash charges of $4.8 million consisted of $36.8 million standard non-cash items primarily related to depreciation and amortization, stock-based compensation expense, and inventory reserve, partially offset by the deferred tax benefit of $32.0 million primarily resulting from the release of the valuation allowance previously recorded against the full amount of our net U.S. deferred tax assets as of December 31, 2021. Net cash used in changes in our operating assets and liabilities included an increase in accounts receivable of $28.7 million, an increase in inventory of $9.3 million, and a decrease in operating leases and other liabilities of $12.2 million, all of which were partially offset by an increase in accounts payable, accrued expenses and other current liabilities of $13.2 million.
During the year ended December 31, 2020, net cash provided by operating activities was $5.5 million, resulting from our net income of $17.2 million and non-cash charges of $16.0 million, partially offset by net cash used in connection with changes in our operating assets and liabilities of $27.8 million. Net cash used in changes in our operating assets and liabilities included an increase in accounts receivable and other current assets of $17.9 million, an increase in inventory of $6.7 million, and a decrease in accounts payable and other liabilities of $4.6 million, all of which were partially offset by an increase in accrued expenses and other current liabilities of $1.4 million.
Investing Activities
During the year ended December 31, 2022, we used $33.9 million of cash in investing activities solely consisting of capital expenditures.
During the year ended December 31, 2021, we used $31.2 million of cash in investing activities solely consisting of capital expenditures.
During the year ended December 31, 2020, we used $23.5 million of cash in investing activities consisting of capital expenditures of $17.7 million and payment of $5.8 million related to the acquisition of CPN.
Financing Activities
During the year ended December 31, 2022, net cash used in financing activities was $2.2 million. This consisted primarily of the payment of term loan and finance lease obligations of $3.0 million and the payment of $0.6 million related to the CPN deferred acquisition consideration, partially offset by the net receipts of $1.4 million in connection with stock awards activities.
During the year ended December 31, 2021, net cash used in financing activities was $1.0 million. This consisted primarily of the repayment of borrowings of $70.0 million under the 2019 Credit Agreement, the payment of $1.6 million to extinguish this debt facility, the payment of finance lease obligations of $2.6 million, and the payment of $2.2 million related to other financing activities.
82
The net cash used in financing activities was principally offset by $73.2 million in net proceeds from the 2021 Credit Agreement and $2.2 million in proceeds from the exercise of common stock options.
During the year ended December 31, 2020, net cash provided by financing activities was $42.5 million. This consisted primarily of $59.1 million in net proceeds from the issuance of Class A common stock and $2.8 million in proceeds from the exercise of options. The net cash provided by financing activities was partially offset by the payment of finance lease obligations of $2.4 million, the payment of $3.5 million related to the NuTech Medical deferred acquisition consideration and the net debt repayment of $13.5 million under our 2019 Credit Agreement.
Indebtedness
2021 Credit Agreement
In August 2021, we and our subsidiaries entered into a credit agreement with SVB and several other lenders, which we refer to as the 2021 Credit Agreement. The 2021 Credit Agreement, as amended, provides for a term loan facility not to exceed $75.0 million (the “Term Loan Facility”) and a revolving credit facility not to exceed $125.0 million (the “Revolving Facility”).
Advances made under the 2021 Credit Agreement may be either SOFR Loans or ABR Loans, at our option. For SOFR Loans, the interest rate is a per annum interest rate equal to the Adjusted Term SOFR plus an Applicable Margin between 2.00% to 3.25% based on the Total Net Leverage Ratio. For ABR Loans, the interest rate is equal to (1) the highest of (a) the Wall Street Journal Prime Rate, (b) the Federal Funds Rate plus 0.50% and (c) the Adjusted Term SOFR rate plus 1.0%, plus (2) an Applicable Margin between 1.00% to 2.25% based on the Total Net Leverage Ratio.
The 2021 Credit Agreement requires us to make consecutive quarterly installment payments equal to the following: (a) from September 30, 2021 through and including June 30, 2022, $0.5 million; (b) from September 30, 2022 through and including June 30, 2023, $0.9 million; (c) from September 30, 2023 through and including June 30, 2025, $1.4 million and (d) from September 30, 2025 and the last day of each quarter thereafter until August 6, 2026 (the “Term Loan Maturity Date”), $1.9 million. We may prepay the Term Loan Facility. Once repaid, amounts borrowed under the Term Loan Facility may not be re-borrowed.
We must pay in arrears, on the first day of each quarter prior to August 6, 2026 (the “Revolving Termination Date”) and on the Revolving Termination Date, a fee for our non-use of available funds (the “Commitment Fee”). The Commitment Fee rate is between 0.25% to 0.45% based on the Total Net Leverage Ratio. We may elect to reduce or terminate the Revolving Facility in its entirety at any time by repaying all outstanding principal and unpaid accrued interest.
Under the 2021 Credit Agreement, we are required to comply with certain financial covenants including the Consolidated Fixed Charge Coverage Ratio and Consolidated Total Net Leverage Ratio, tested quarterly. In addition, we are also required to make representations and warranties and comply with certain non-financial covenants that are customary in loan agreements of this type, including restrictions on the payment of dividends, repurchase of stock, incurrence of indebtedness, dispositions and acquisitions.
As of December 31, 2022, we were in compliance with the covenants under the 2021 Credit Agreement. We had outstanding borrowings of $71.3 million under our Term Loan Facility and no borrowings outstanding under our Revolving Facility with $125 million available for future revolving borrowings, respectively.
2019 Credit Agreement
In March 2019, we, our subsidiaries and SVB, and the several other lenders thereto entered into a credit agreement, as amended (the “2019 Credit Agreement”), providing for a term loan facility of $40.0 million and a revolving credit facility of up to $60.0 million. Both facilities were set to mature in 2024. The interest rate for the term loan facility was a floating per annum interest rate equal to the greater of 3.75% above the Wall Street Journal Prime Rate and 9.25%. The interest rate for advances under the revolving facility was a floating per annum interest rate equal to the greater of the Wall Street Journal Prime Rate and 5.50%. If we elected to prepay the loan or terminate the facilities, we were required to pay a certain percentage of the outstanding principal as a prepayment fee. A final payment fee (the “Final Payment”) of 6.5% multiplied by the original aggregate principal amount of term loan facility was due upon the earlier to occur, the maturity date of the term loan or prepayment of all outstanding principal.
In August 2021, upon entering into the 2021 Credit Agreement, we paid an aggregate amount of $70.6 million due under the 2019 Credit Agreement, including unpaid principal, accrued interest, the Final Payment and a prepayment fee, with proceeds from the
83
2021 Credit Agreement, and the 2019 Credit Agreement was terminated. Upon termination of the 2019 Credit Agreement, the Company recognized $1.9 million as loss on the extinguishment of the loan for the year ended December 31, 2021.
Critical Accounting Policies and Significant Judgments and Estimates
Our consolidated financial statements have been prepared in accordance with GAAP. The preparation of our consolidated financial statements requires us to make estimates, assumptions and judgments that affect the reported amounts of assets, liabilities, and the disclosure at the date of the financial statements, as well as revenue and expenses recorded during the reporting periods. Management bases its estimates, assumptions and judgments on historical experience and on various other factors that it believes to be reasonable under the circumstances. Different assumptions and judgments would change the estimates used in the preparation of our consolidated financial statements, which, in turn, could materially change our results from those reported. Management evaluates its estimates, assumptions and judgments on an ongoing basis. Historically, our critical accounting estimates have not differed materially from actual results. However, if our assumptions change, we may need to revise our estimates or take other corrective actions, either of which may also have a material adverse effect on our consolidated statements of operations, liquidity and financial condition.
We believe the following critical accounting policies involve significant areas where management applies judgments and estimates in the preparation of our consolidated financial statements.
Revenue Recognition
We generate revenue through the sale of Advanced Wound Care and Surgical & Sports Medicine products. There is a single performance obligation in all of our contracts, which is our promise to transfer our product to customers based on specific payment and shipping terms in the arrangement. The entire transaction price is allocated to this single performance obligation. Product revenue is recognized when a customer obtains control of our product which occurs at a point in time and may be upon shipment, procedure date, or delivery, based on the terms of the contract. Revenue is recorded net of a reserve for returns, discounts and GPO rebates, which represent a direct reduction to the revenue we recognize. These reductions are accrued at the time revenue is recognized, based upon historical experience and specific circumstances.
Accounts Receivable, Net
Accounts receivables are stated at invoice value less estimated allowances for doubtful accounts. We continually monitor customer payments and maintain a reserve for estimated losses resulting from our customers’ inability to make required payments. We consider factors such as historical experience, credit quality, age of the accounts receivable balances, geography-related risks and economic conditions that may affect a customer’s ability to pay. In cases where there are circumstances that may impair a specific customer’s ability to meet its financial obligations, a specific allowance is recorded against amounts due, and thereby reduces the net recognized receivable to the amount reasonably believed to be collectible. Accounts receivables are written off when deemed uncollectible. Recoveries of accounts receivables previously written off are recorded when received.
Inventory
Inventory is stated at the lower of cost (determined under the first-in first-out method) or net realizable value. Inventory includes raw materials, work in process and finished goods. It also includes cell banks and the cost of tests mandated by regulatory agencies, of the materials to qualify them for production.
We regularly review inventory quantities on hand and record a provision to write down excess and obsolete inventory to its estimated net realizable value based upon management’s assumptions of future material usage, yields and obsolescence, which are a result of future demand and market conditions and the effective life of certain inventory items. Our excess and obsolete inventory review process includes analysis of sales forecasts and historical sales as compared to inventory on hand and working with operations to maximize recovery of excess inventory. The estimate of excess quantities is subjective and primarily dependent on our estimate of future demand for a particular product. If the estimate of future demand is inaccurate based on actual sales, we may increase the write-down for excess inventory for that component.
Goodwill
Goodwill represents the excess of the purchase price of an acquired business over the fair value of the identifiable assets acquired and liabilities assumed. Goodwill is not amortized but is tested for impairment at least annually (as of December 31), or more frequently if events or circumstances indicate the carrying value may no longer be recoverable and that an impairment loss may have occurred. Circumstances that could trigger an impairment test include, but are not limited to, a significant adverse change in the
84
business climate or legal factors, an adverse action or assessment by a regulator, or unanticipated competition. We operate as one segment, which is considered to be the sole reporting unit, and therefore goodwill is tested for impairment at the consolidated level.
In accordance with ASC Topic 350, Intangibles - Goodwill and Other, we may first assess qualitative factors to determine whether it is necessary to perform the quantitative goodwill impairment test. If after assessing the totality of events or circumstances, we determine that it is more likely than not (i.e. greater than 50% likelihood) that the fair value of the reporting unit is less than its carrying amount, then the quantitative test is required. Otherwise, no further testing is required. Alternatively, we can bypass the qualitative assessment and proceed directly to the quantitative test. The quantitative goodwill impairment test requires us to estimate and compare the fair value of the reporting unit with its carrying value. If the fair value of the reporting unit exceeds the carrying value of the net assets, goodwill is not impaired. If the fair value of the reporting unit is less than the carrying value, the difference is recorded as an impairment loss up to the amount of goodwill. At December 31, 2022, we elected to perform a quantitative analysis directly. We used the Company’s market capitalization to approximate the fair value of the reporting unit. The fair value exceeded the carrying value and no impairment was recorded.
Application of the goodwill impairment test requires judgments, including a qualitative assessment to determine whether there are any impairment indicators, and determining the fair value of the reporting unit. We used the Company’s market capitalization to determine the fair value of the reporting unit. There is no assurance that the Company’s market capitalization will not decline significantly from the level used in the impairment analysis. Goodwill impairment charges may be recognized in future periods to the extent changes in factors or circumstances occur, including deterioration in the macroeconomic environment and industry, deterioration in the Company’s performance or its future projections, or changes in plans for its reporting unit.
There were no impairments of goodwill recorded during 2022, 2021, and 2020.
Impairment of Long-Lived Assets
We review other long-lived assets (including identifiable definite lived intangible assets) for impairment whenever events or changes in circumstances indicate that the useful life is shorter than originally estimated or the carrying amount of an asset or asset group may not be recoverable. If such facts and circumstances exist, we assess the recoverability of the identified assets by comparing the projected undiscounted net cash flows associated with the related asset or group of assets over their remaining lives to their respective carrying amounts. Impairments, if any, are based on the excess of the carrying amount over the fair value of those assets and occur in the period in which the impairment determination is made.
There were no impairments of long-lived assets recorded during 2022, 2021, and 2020.
Income Taxes
We account for income taxes using an asset and liability approach. Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Valuation allowances are provided when necessary to reduce net deferred tax assets to an amount that is more likely than not to be realized.
In determining whether a valuation allowance for deferred tax assets is necessary, we analyze both positive and negative evidence related to the realization of deferred tax assets including projected future taxable income, recent financial results and estimates of future reversals of deferred tax assets and liabilities. In addition, we consider whether it is more likely than not that the tax position will be sustained on examination by taxing authorities based on the technical merits of the position. In consideration of the factors discussed above, in the fourth quarter of 2021, we determined it was more likely than not that our deferred tax assets would be realized in the future and released the valuation allowance on our net U.S. deferred tax assets as of December 31, 2021, resulting in a benefit of $48.3 million in income taxes. We maintained the same position that our net U.S. deferred tax assets did not require a valuation allowance as of December 31, 2022.
We account for uncertainty in income taxes recognized in the consolidated financial statements by applying a two-step process to determine the amount of tax benefit to be recognized. First, the tax position must be evaluated to determine the likelihood that it will be sustained upon external examination by the taxing authorities. If the tax position is deemed more-likely-than-not to be sustained, the tax position is then assessed to determine the amount of benefit to recognize in the consolidated financial statements. The amount of the benefit that may be recognized is the largest amount that has a greater than 50% likelihood of being realized upon ultimate
85
settlement. The provision for income taxes includes the effects of any resulting tax reserves, or unrecognized tax benefits, that are considered appropriate as well as the related net interest and penalties.
Valuation of Contingent Consideration
In connection with our acquisition of CPN, we recognized a non-current liability, at the time of the acquisition in 2020, for the fair value of the contingent consideration (the “Earnout”). The Earnout liability was classified as a Level 3 measurement for which fair value was derived from inputs that were unobservable and significant to the overall fair value measurement. The fair value of such Earnout liability was estimated using a Monte Carlo simulation model that utilized key assumptions including forecasted revenues and volatilities of the underlying financial metrics during the Earnout period. We assessed the fair value of the Earnout liability at each reporting period. Any subsequent changes in the estimated fair value of the liability were reflected in selling, general and administrative expenses until the liability was settled.
Stock-Based Compensation
We measure stock-based awards granted based on the fair value of the awards on the date of grant and recognize compensation expense for those awards over the requisite service period, which is generally the vesting period of the respective award. Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Generally, we issue stock-based awards with only service-based vesting conditions and record the expense for these awards using the straight-line method. We have not issued any stock-based awards with performance-based vesting conditions.
We recognize stock-based compensation expense within selling, general and administrative expenses in the consolidated statement of operations for all share-based payments based upon the estimated grant-date fair value for the awards expected to ultimately vest.
The fair value of each restricted stock unit is based on the fair market value of our Class A common stock on the date of grant. The fair value of each stock option grant is estimated on the date of grant using the Black-Scholes option pricing model. We have been a public company for a short period of time, have limited public float and lack company-specific historical and implied volatility information for our Class A common stock. Therefore, we estimate our expected stock price volatility based on the historical volatility of publicly traded peer companies and expect to continue to do so until such time as we have adequate historical data regarding the volatility of our own traded stock price. The expected term of our stock options has been determined utilizing the “simplified” method for awards that qualify as “plain-vanilla” options. The risk-free interest rate is determined by reference to the U.S. Treasury yield curve in effect at the time of grant of the award for time periods approximately equal to the expected term of the award. Expected dividend yield is based on the fact that we have never paid cash dividends on Class A common stock and do not expect to pay any cash dividends in the foreseeable future.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined in the rules and regulations of the SEC.
Recently Issued Accounting Pronouncements
For a description of recently issued accounting pronouncements, including the expected dates of adoption and the estimated effects, if any, on our consolidated financial statements, see footnote “2. Significant Accounting Policies” to our consolidated financial statements appearing at the end of this Annual Report on Form 10-K.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are exposed to various market risks, including fluctuations in interest rates and variability in currency exchange rates. We have established policies, procedures and internal processes governing our management of market risk.
Interest Rate Risk
As of December 31, 2022, we had $71.3 million in borrowings outstanding under our Term Loan Facility and no borrowings outstanding under our Revolving Facility, respectively. Borrowings under our 2021 Credit Agreement bear interest at variable rates. Based on the principal amount outstanding as of December 31, 2022, an immediate 10% change in the interest rate would not have a material impact on our financial position, results of operations or cash flows.
86
Foreign Currency and Market Risk
The majority of our employees and our major operations are currently located in the United States. The functional currency of our foreign subsidiary in Switzerland is the U.S. dollar. We have, in the normal course of business, engaged in contracts with contractors or other vendors in a currency other than the U.S. dollar. To date, we have had minimal exposure to fluctuations in foreign currency exchange rates as the time period from the date that transactions are initiated and the date of payment or receipt of payment is generally of short duration. Accordingly, we believe we do not have a material exposure to foreign currency risk.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Our consolidated financial statements, together with the report of our independent registered public accounting firm, appear on pages F-1 through F-30 of this Annual Report on Form 10-K.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of the Company’s Disclosure Controls
The Company’s management, with the participation of its principal executive officer and principal financial officer, evaluated the effectiveness of its disclosure controls and procedures as of December 31, 2022. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the rules and forms promulgated by the SEC. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on that evaluation, our management, including our principal executive officer and principal financial officer, concluded that, as of December 31, 2022, our disclosure controls and procedures were ineffective because our internal control over financial reporting was not designed properly to ensure proper identification of non-routine transactions and ensure appropriate segregation of duties.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, the Company’s principal executive officer and principal financial officer and effected by the Company’s board of directors, management and other personnel to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America.
Management conducted the assessment of the effectiveness of the Company’s internal control over financial reporting based on criteria established in Internal Control— Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission in 2013. As a result of this assessment, management concluded that, as of December 31, 2022, our internal control over financial reporting was ineffective due to the material weaknesses described below. As a result, the disclosure controls and procedures were ineffective in ensuring that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms and is accumulated and communicated to our management, including the chief executive officer and chief financial officer, as appropriate to allow timely decisions regarding disclosure.
We did not design and maintain effective controls (i) to properly identify and assess significant non-routine transactions and (ii) over information technology general controls and proper segregation of duties to support the proper initiation and recording of transactions and the resulting impact on business process controls and applications that rely on such data.
Although management has made certain progress in remediating these material weaknesses, management concluded that the material weaknesses described above continued to exist as of December 31, 2022. Management has taken actions to remediate the deficiencies in its internal controls over financial reporting and implemented additional processes and controls designed to address the
87
underlying causes of the above-mentioned material weaknesses. Management is committed to finalizing the remediation of the material weaknesses. Management’s internal control remediation efforts include the following:
As management continues to evaluate and work to improve our internal control over financial reporting, management may determine it is necessary to take additional measures to address the material weaknesses. However, we believe the above actions will be effective in remediating the material weaknesses and we will continue to devote significant time and attention to these remediation efforts. Until the controls have been operating for a sufficient period of time and management has concluded, through testing, that these controls are executed consistently and operating effectively, the material weaknesses described above will continue to exist.
The effectiveness of the Company’s internal control over financial reporting as of December 31, 2022, has been audited by RSM US LLP, an independent registered public accounting firm, as stated in their attestation report, which appears in Item 8 above.
Changes in Internal Control Over Financial Reporting
There have been no changes in our internal control over financial reporting that occurred during the quarter ended December 31, 2022 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting other than those described above related to remediation efforts. However, as the implementation of the new ERP system continues and as the material weaknesses are remediated, we will change our processes and procedures, which in turn, could result in changes to our internal control over financial reporting. As such changes occur, we will evaluate quarterly whether such changes materially affect our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
None.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
88
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item is incorporated by reference to our Definitive Proxy Statement for our 2023 Annual Meeting of Stockholders which will be filed with the Securities and Exchange Commission no later than 120 days after the end of our fiscal year (the “Proxy Statement”).
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item is incorporated by reference to our Proxy Statement.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item is incorporated by reference to our Proxy Statement.
The information required by this item is incorporated by reference to our Proxy Statement.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by this item is incorporated by reference to our Proxy Statement.
89
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a) Documents filed as a part of this Report:
(1) Financial Statements —See Index to Consolidated Financial Statements and Item 8 of this Annual Report on Form 10-K.
(2) Financial Statement Schedules —Schedules are omitted because they are not applicable, or are not required, or because the information is included in the Consolidated Financial Statements and notes thereto.
(3) Index to Exhibits.
Exhibit Index
Exhibit No. |
|
Exhibit |
|
|
|
|
|
|
3.1 |
|
|
|
|
|
3.2 |
|
|
|
|
|
3.3 |
|
|
|
|
|
4.1 |
|
|
|
|
|
10.1 |
|
|
|
|
|
10.2 |
|
|
|
|
|
10.3 |
|
|
|
|
|
10.4 |
|
|
|
|
|
10.5 |
|
|
|
|
|
10.6 |
|
|
|
|
|
10.7 |
|
|
|
|
|
10.8‡ |
|
|
|
|
|
90
Exhibit No. |
|
Exhibit |
10.9‡ |
|
|
|
|
|
10.10‡ |
|
|
|
|
|
10.11‡ |
|
|
|
|
|
10.12‡ |
|
|
|
|
|
10.13‡ |
|
|
|
|
|
10.14‡ |
|
|
|
|
|
10.15‡ |
|
|
|
|
|
10.16‡ |
|
|
|
|
|
10.17‡ |
|
|
|
|
|
10.18‡ |
|
|
|
|
|
10.19‡ |
|
|
|
|
|
10.20† |
|
|
|
|
|
10.21 |
|
|
|
|
|
10.22 |
|
|
|
|
|
10.23 |
|
|
|
|
|
10.24 |
|
|
|
|
|
10.25 |
|
|
|
|
|
10.26 |
|
|
91
Exhibit No. |
|
Exhibit |
10.27‡ |
|
|
10.28‡
|
|
|
10.29‡ |
|
|
10.30‡ |
|
|
10.31‡ |
|
|
10.32 |
|
|
10.33* |
|
|
10.34 |
|
|
21.1* |
|
|
|
|
|
23.1* |
|
|
|
|
|
31.1* |
|
|
|
|
|
31.2* |
|
|
|
|
|
32.1* |
|
|
|
|
|
101* |
|
The following materials from the Annual Report of Organogenesis Holdings Inc. on Form 10-K for the year ended December 31, 2022, formatted in XBRL (eXtensible Business Reporting Language): (i) Consolidated Balance Sheets as of December 31, 2022 and December 31, 2021 of Organogenesis Holdings Inc., (ii) Consolidated Statements of Operations for the years ended December 31, 2022, 2021, and 2020 of Organogenesis Holdings Inc., (iii) Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2022, 2021, and 2020 of Organogenesis Holdings Inc., (iv) Consolidated Statements of Cash Flows for the years ended December 31, 2022, 2021, and 2020 of Organogenesis Holdings Inc., and (v) Notes to Consolidated Financial Statements of Organogenesis Holdings Inc. |
104 |
|
Cover Page Interactive Data File (embedded within the Inline XBRL document) |
* Filed herewith.
† Confidential treatment granted as to portions of this Exhibit. The confidential portions of this Exhibit have been omitted and are marked by asterisks.
‡ Management contract or compensatory plan or arrangement.
ITEM 16. FORM 10-K SUMMARY
None.
92
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
ORGANOGENESIS HOLDINGS INC. |
|
|
|
By: |
/s/ Gary S. Gillheeney, Sr. |
|
Gary S. Gillheeney, Sr. President and Chief Executive Officer |
Date: |
March 1, 2023 |
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report has been signed below by the following persons on behalf of the Company and in the capacities and on the dates indicated.
Signature |
|
Title |
|
Date |
|
|
|
|
|
/s/ Gary S. Gillheeney, Sr.
Gary S. Gillheeney, Sr. |
|
Chief Executive Officer, President and Director (Principal Executive Officer) |
|
March 1, 2023 |
|
|
|
|
|
/s/ David Francisco
David Francisco |
|
Chief Financial Officer (Principal Financial and Accounting Officer) |
|
March 1, 2023 |
|
|
|
|
|
/s/ Alan A. Ades
Alan A. Ades |
|
Director |
|
March 1, 2023 |
|
|
|
|
|
/s/ Robert Ades
Robert Ades |
|
Director |
|
March 1, 2023 |
|
|
|
|
|
/s/ Michael J. Driscoll
Michael J. Driscoll |
|
Director |
|
March 1, 2023 |
|
|
|
|
|
/s/ Prathyusha Duraibabu
Prathyusha Duraibabu |
|
Director |
|
March 1, 2023 |
|
|
|
|
|
/s/ David Erani
David Erani |
|
Director |
|
March 1, 2023 |
|
|
|
|
|
/s/ Jon Giacomin
|
|
Director |
|
March 1, 2023 |
Jon Giacomin |
|
|
|
|
|
|
|
|
|
/s/ Michele Korfin Michele Korfin |
|
Director |
|
March 1, 2023 |
|
|
|
|
|
/s/ Arthur S. Leibowitz
Arthur S. Leibowitz |
|
Director |
|
March 1, 2023 |
|
|
|
|
|
/s/ Glenn H. Nussdorf
Glenn H. Nussdorf |
|
Director |
|
March 1, 2023 |
|
|
|
|
|
/s/ Gilberto Quintero Gilberto Quintero |
|
Director |
|
March 1, 2023 |
|
|
|
|
|
93
ORGANOGENESIS HOLDINGS INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
F-1
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Organogenesis Holdings Inc.
Opinions on the Financial Statements and Internal Control Over Financial Reporting
We have audited the accompanying consolidated balance sheets of Organogenesis Holdings Inc. and subsidiaries (the Company) as of December 31, 2022 and 2021, and the related consolidated statements of operations, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2022, and the related notes (collectively, the financial statements). We also have audited the Company’s internal control over financial reporting as of December 31, 2022, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission in 2013.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2022 and 2021, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2022, in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, because of the effect of the material weaknesses described below on the achievement of the objectives of the control criteria, the Company has not maintained effective internal control over financial reporting as of December 31, 2022, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission in 2013.
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis. The following material weaknesses have been identified and included in management's assessment. The Company did not design and maintain effective controls (i) to properly identify and assess significant non-routine transactions and (ii) over information technology general controls and proper segregation of duties to support the proper initiation and recording of transactions and the resulting impact on business process controls and applications that rely on such data. These material weaknesses were considered in determining the nature, timing and extent of audit tests applied in our audit of the 2022 financial statements, and this report does not affect our report dated March 1, 2023 on those financial statements.
Basis for Opinions
The Company's management is responsible for these financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company's financial statements and an opinion on the Company's internal control over financial reporting based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud, and whether effective internal control over financial reporting was maintained in all material respects.
Our audits of the financial statements included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
Definition and Limitations of Internal Control Over Financial Reporting
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial
F-2
statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing separate opinions on the critical audit matter or on the accounts or disclosures to which it relates.
Identification of significant non-routine transactions
Non-routine transactions represent activities that occur only periodically and are generally not part of the routine flow of transactions. During the year ended December 31, 2022, the Company entered into certain non-routine transactions that required management to apply judgement in determining the appropriate accounting treatment, including the agreement discussed in Note 2 to the financial statements with a Group Purchasing Organization (GPO) to settle previously disputed GPO fees for $3,300, which included $2,600 recorded as charge to earnings during the year ended December 31, 2022, and certain indicators of impairment from the disposal of equipment and a pause in construction during the year ended December 31, 2022 discussed in Note 8 to the financial statements, which resulted in the Company testing certain assets of its OI East asset group for impairment. Management must exercise significant judgment in the identification of significant non-routine transactions given that they are unique and not part of the routine flow of the Company’s transactions.
We identified management’s identification and assessment of significant non-routine transactions as a critical audit matter due to the subjectivity in determining the sufficiency of the results of the actions taken by management to identify all significant non-routine transactions.
Our audit procedures related to the Company’s identification of significant non-routine transactions included the following, among others:
/s/ RSM US LLP
We have served as the Company's auditor since 2004.
Boston, Massachusetts
March 1, 2023
F-3
ORGANOGENESIS HOLDINGS INC.
CONSOLIDATED BALANCE SHEETS
(in thousands, except share and per share amounts)
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Assets |
|
|
|
|
|
|
||
Current assets: |
|
|
|
|
|
|
||
Cash and cash equivalents |
|
$ |
|
|
$ |
|
||
Restricted cash |
|
|
|
|
|
|
||
Accounts receivable, net |
|
|
|
|
|
|
||
Inventory |
|
|
|
|
|
|
||
Prepaid expenses and other current assets |
|
|
|
|
|
|
||
Total current assets |
|
|
|
|
|
|
||
Property and equipment, net |
|
|
|
|
|
|
||
Intangible assets, net |
|
|
|
|
|
|
||
Goodwill |
|
|
|
|
|
|
||
Operating lease right-of-use assets, net |
|
|
|
|
|
|
||
Deferred tax asset, net |
|
|
|
|
|
|
||
Other assets |
|
|
|
|
|
|
||
Total assets |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
Liabilities and Stockholders’ Equity |
|
|
|
|
|
|
||
Current liabilities: |
|
|
|
|
|
|
||
Current portion of deferred acquisition consideration |
|
$ |
- |
|
|
$ |
|
|
Current portion of term loan |
|
|
|
|
|
|
||
Current portion of finance lease obligations |
|
|
- |
|
|
|
|
|
Current portion of operating lease obligations |
|
|
|
|
|
|
||
Accounts payable |
|
|
|
|
|
|
||
Accrued expenses and other current liabilities |
|
|
|
|
|
|
||
Total current liabilities |
|
|
|
|
|
|
||
Term loan, net of current portion |
|
|
|
|
|
|
||
Operating lease obligations, net of current portion |
|
|
|
|
|
|
||
Other liabilities |
|
|
|
|
|
|
||
Total liabilities |
|
|
|
|
|
|
||
(Note 18) |
|
|
|
|
|
|
||
Stockholders’ equity: |
|
|
|
|
|
|
||
Preferred stock, $ |
|
|
|
|
|
|
||
Common stock, $ |
|
|
|
|
|
|
||
Additional paid-in capital |
|
|
|
|
|
|
||
Accumulated deficit |
|
|
( |
) |
|
|
( |
) |
Total stockholders' equity |
|
|
|
|
|
|
||
Total liabilities and stockholders' equity |
|
$ |
|
|
$ |
|
||
The accompanying notes are an integral part of these consolidated financial statements
F-4
ORGANOGENESIS HOLDINGS INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except share and per share amounts)
|
|
Year Ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
Net revenue |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Cost of goods sold |
|
|
|
|
|
|
|
|
|
|||
Gross profit |
|
|
|
|
|
|
|
|
|
|||
Operating expenses: |
|
|
|
|
|
|
|
|
|
|||
Selling, general and administrative |
|
|
|
|
|
|
|
|
|
|||
Research and development |
|
|
|
|
|
|
|
|
|
|||
Total operating expenses |
|
|
|
|
|
|
|
|
|
|||
Income from operations |
|
|
|
|
|
|
|
|
|
|||
Other expense, net: |
|
|
|
|
|
|
|
|
|
|||
Interest expense |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Gain on settlement of deferred acquisition consideration |
|
|
- |
|
|
|
- |
|
|
|
|
|
Loss on the extinguishment of debt |
|
|
- |
|
|
|
( |
) |
|
|
- |
|
Other income (loss), net |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
Total other expense, net |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Net income before income taxes |
|
|
|
|
|
|
|
|
|
|||
Income tax (expense) benefit |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
Net income |
|
$ |
|
|
$ |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
Net income, per share: |
|
|
|
|
|
|
|
|
|
|||
Basic |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Diluted |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Weighted-average common shares outstanding |
|
|
|
|
|
|
|
|
|
|||
Basic |
|
|
|
|
|
|
|
|
|
|||
Diluted |
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|||
The accompanying notes are an integral part of these consolidated financial statements
F-5
ORGANOGENESIS HOLDINGS INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(in thousands, except share amounts)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
Additional |
|
|
|
|
|
Total |
|
|||||
|
|
|
|
Common Stock |
|
|
Paid-in |
|
|
Accumulated |
|
|
Stockholders’ |
|
||||||||
|
|
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Equity |
|
|||||
Balance as of December 31, 2019 |
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
Exercise of stock options |
|
|
|
|
|
|
|
|
|
|
|
|
|
- |
|
|
|
|
||||
Issuance of common stock associated with CPN acquisition |
|
|
|
|
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
|
|||
Stock-based compensation expense |
|
|
|
|
- |
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
|
||
Stock issued in the 2020 Underwritten Public Offering, net of issuance costs of $ |
|
|
|
|
|
|
|
|
|
|
|
|
|
- |
|
|
|
|
||||
Net income |
|
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
|
|
|
||
Balance as of December 31, 2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||
Exercise of stock options |
|
|
|
|
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
|
|||
Vesting of RSUs, net of shares surrendered to pay taxes |
|
|
|
|
|
|
|
- |
|
|
|
( |
) |
|
|
- |
|
|
|
( |
) |
|
Stock-based compensation expense |
|
|
|
|
- |
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
|
||
Net income |
|
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
|
|
|
||
Balance as of December 31, 2021 (as reported) |
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||
Adjustment due to settlement of GPO fee dispute |
|
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
( |
) |
|
|
( |
) |
Balance as of December 31, 2021 (as restated) |
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||
Exercise of stock options |
|
|
|
|
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
|
|||
Vesting of RSUs, net of shares surrendered to pay taxes |
|
|
|
|
|
|
|
- |
|
|
|
( |
) |
|
|
- |
|
|
|
( |
) |
|
Issuance of common stock associated with CPN acquisition |
|
|
|
|
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
|
|||
Stock-based compensation expense |
|
|
|
|
- |
|
|
|
- |
|
|
|
|
|
|
- |
|
|
|
|
||
Net income |
|
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
|
|
|
|
||
Balance as of December 31, 2022 |
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
The accompanying notes are an integral part of these consolidated financial statements
F-6
ORGANOGENESIS HOLDINGS INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
|
|
Year Ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|||
Net income |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Adjustments to reconcile net income to net cash provided by operating activities: |
|
|
|
|
|
|
|
|
|
|||
Depreciation |
|
|
|
|
|
|
|
|
|
|||
Amortization of intangible assets |
|
|
|
|
|
|
|
|
|
|||
Amortization of operating lease right-of-use assets |
|
|
|
|
|
|
|
|
- |
|
||
Non-cash interest expense |
|
|
|
|
|
|
|
|
|
|||
Deferred interest expense |
|
|
|
|
|
|
|
|
|
|||
Deferred rent expense |
|
|
- |
|
|
|
- |
|
|
|
|
|
Gain on settlement of deferred acquisition consideration |
|
|
- |
|
|
|
- |
|
|
|
( |
) |
Deferred tax expense (benefit) |
|
|
|
|
|
( |
) |
|
|
|
||
Loss on disposal of property and equipment |
|
|
|
|
|
|
|
|
|
|||
Provision recorded for doubtful accounts |
|
|
|
|
|
|
|
|
|
|||
Adjustment for excess and obsolete inventories |
|
|
|
|
|
|
|
|
|
|||
Stock-based compensation |
|
|
|
|
|
|
|
|
|
|||
Loss on extinguishment of debt |
|
|
- |
|
|
|
|
|
|
- |
|
|
Change in fair value of Earnout liability |
|
|
- |
|
|
|
( |
) |
|
|
|
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
|||
Accounts receivable |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Inventory |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Prepaid expenses and other current assets |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Operating leases |
|
|
( |
) |
|
|
( |
) |
|
|
- |
|
Accounts payable |
|
|
|
|
|
|
|
|
( |
) |
||
Accrued expenses and other current liabilities |
|
|
( |
) |
|
|
|
|
|
|
||
Other liabilities |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
Net cash provided by operating activities |
|
|
|
|
|
|
|
|
|
|||
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|||
Purchases of property and equipment |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Cash paid for business acquisition |
|
|
- |
|
|
|
- |
|
|
|
( |
) |
Net cash used in investing activities |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|||
Line of credit repayments under the 2019 Credit Agreement |
|
|
- |
|
|
|
( |
) |
|
|
( |
) |
Term loan borrowings (repayments) under the 2019 Credit Agreement |
|
|
- |
|
|
|
( |
) |
|
|
|
|
Proceeds from term loan under the 2021 Credit Agreement, net of debt discount and issuance cost |
|
|
- |
|
|
|
|
|
|
- |
|
|
Term loan repayments under the 2021 Credit Agreement |
|
|
( |
) |
|
|
( |
) |
|
|
- |
|
Proceeds from equity financing |
|
|
- |
|
|
|
- |
|
|
|
|
|
Payment of equity issuance costs |
|
|
- |
|
|
|
- |
|
|
|
( |
) |
Principal repayments of finance lease obligations |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Proceeds from the exercise of stock options |
|
|
|
|
|
|
|
|
|
|||
Payments of withholding taxes in connection with RSUs vesting |
|
|
( |
) |
|
|
( |
) |
|
|
- |
|
Payments of deferred acquisition consideration |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Payment to extinguish debt |
|
|
- |
|
|
|
( |
) |
|
|
- |
|
Net cash provided by (used in) financing activities |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
Change in cash, cash equivalents and restricted cash |
|
|
( |
) |
|
|
|
|
|
|
||
Cash, cash equivalents, and restricted cash, beginning of year |
|
|
|
|
|
|
|
|
|
|||
Cash, cash equivalents, and restricted cash, end of year |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Supplemental disclosure of cash flow information: |
|
|
|
|
|
|
|
|
|
|||
Cash paid for interest |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Cash paid for income taxes |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Supplemental disclosure of non-cash investing and financing activities: |
|
|
|
|
|
|
|
|
|
|||
Reimbursement of offering expenses included in prepaid expenses and other current assets |
|
$ |
- |
|
|
$ |
- |
|
|
$ |
|
|
Fair value of shares issued for business acquisition |
|
$ |
- |
|
|
$ |
- |
|
|
$ |
|
|
Deferred acquisition consideration and earnout liability recorded for business acquisition |
|
$ |
|
|
$ |
- |
|
|
$ |
|
||
Purchases of property and equipment in accounts payable and accrued expenses |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Right-of-use assets obtained through lease obligations |
|
$ |
|
|
$ |
|
|
$ |
- |
|
||
The accompanying notes are an integral part of these consolidated financial statements
F-7
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Amounts in thousands, except share and per share amounts)
1. Nature of Business and Basis of Presentation
Organogenesis Holdings Inc. (“ORGO” or the “Company”) is a leading regenerative medicine company focused on the development, manufacture, and commercialization of solutions for the Advanced Wound Care and Surgical & Sports Medicine markets. Several of the existing and pipeline products in the Company’s portfolio have Premarket Application (“PMA”) approval, or Premarket Notification 510(k) clearance from the United States Food and Drug Administration (“FDA”). The Company’s customers include hospitals, wound care centers, government facilities, ambulatory service centers (“ASCs”) and physician offices. The Company has
COVID-19 pandemic
On January 30, 2023, the Biden Administration announced it will end the public health emergency (and national emergency) declarations related to coronavirus (COVID-19) on May 11, 2023. While the COVID-19 pandemic has not materially adversely affected the Company’s financial results and business operations through the year ended December 31, 2022, the COVID-19 pandemic continues to present risks to the Company and the Company is unable to predict the impact that COVID-19 will have on its financial position and operating results in future periods.
2. Significant Accounting Policies
Restatement to Previously Issued Financial Statements
In August 2022, the Company reached an agreement with a Group Purchasing Organization (“GPO”) to settle previously disputed GPO fees for $
|
|
|
March 31, 2022 |
|
|
|
December 31, 2021 |
|
||||||||||||||||||
CONSOLIDATED BALANCE SHEETS |
|
|
As Previously Reported |
|
|
Adjustments |
|
|
As Restated |
|
|
|
As Previously Reported |
|
|
Adjustments |
|
|
As Restated |
|
||||||
Accrued expenses and other current liabilities |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
|
$ |
|
|
$ |
|
|
$ |
|
||||||
Total current liabilities |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
|
$ |
|
|
$ |
|
|
$ |
|
||||||
Total liabilities |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
|
$ |
|
|
$ |
|
|
$ |
|
||||||
Accumulated deficit |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Total stockholders’ equity |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
Three Months Ended March 31, 2022 |
|
|
|
Year Ended December 31, 2021 |
|
||||||||||||||||||
CONSOLIDATED STATEMENTS OF OPERATIONS |
|
|
As Previously Reported |
|
|
Adjustments |
|
|
As Restated |
|
|
|
As Previously Reported |
|
|
Adjustments |
|
|
As Restated |
|
||||||
Net revenue |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
Gross profit |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
Income from operations |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
Net income before income taxes |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
Net income |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
F-8
|
|
|
Three Months Ended March 31, 2022 |
|
|
|
Year Ended December 31, 2021 |
|
||||||||||||||||||
CONSOLIDATED STATEMENTS OF CASH FLOWS |
|
|
As Previously Reported |
|
|
Adjustments |
|
|
As Restated |
|
|
|
As Previously Reported |
|
|
Adjustments |
|
|
As Restated |
|
||||||
Net income / (loss) |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
Accrued expenses and other current liabilities |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
|
|
|
Three Months Ended March 31, 2022 |
|
|
|
Year Ended December 31, 2021 |
|
||||||||||||||||||
Revenue by Product Category: |
|
|
As Previously Reported |
|
|
Adjustments |
|
|
As Restated |
|
|
|
As Previously Reported |
|
|
Adjustments |
|
|
As Restated |
|
||||||
Advanced Wound Care |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
Surgical & Sports Medicine |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
Net revenue |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
Three Months Ended March 31, 2022 |
|
|
|
Year Ended December 31, 2021 |
|
||||||||||||||||||
Miscellaneous Items |
|
|
As Previously Reported |
|
|
Adjustments |
|
|
As Restated |
|
|
|
As Previously Reported |
|
|
Adjustments |
|
|
As Restated |
|
||||||
GPO fees |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
|
$ |
|
|
$ |
|
|
$ |
|
||||||
PuraPly revenue |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||||
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements and the reported results of operations during the reporting periods. In preparing the consolidated financial statements, the estimates and assumptions that management considers to be significant and that present the greatest amount of uncertainty include revenue recognition; sales returns and credit losses; inventory reserve; recognition and measurement of current and deferred income tax assets and liabilities; the assessment of recoverability of long-lived assets, assessing impairment of goodwill; valuation of assets and liabilities that use unobservable inputs, and the valuation and recognition of stock-based compensation. Actual results and outcomes may differ significantly from those estimates and assumptions.
Principles of Consolidation
The consolidated financial statements include the accounts and results of operations of Organogenesis Holdings Inc., and its wholly-owned subsidiaries. All intercompany balances and transactions have been eliminated in consolidation.
Segment Reporting
Operating segments are defined as components of an enterprise about which discrete financial information is available that is evaluated regularly by the chief operating decision maker, or decision-making group, in making decisions on how to allocate resources and assess performance for the organization. The Company’s chief operating decision maker is the Chief Executive Officer. The Company’s chief operating decision maker reviews consolidated operating results to make decisions about allocating resources and assessing performance for the entire Company. Accordingly, the Company has determined that it has a single operating segment—regenerative medicine.
The Company manages its operations as a single operating segment for the purposes of assessing performance and making operating decisions. The Company’s portfolio includes regenerative medicine products in various stages, ranging from preclinical to late stage development, and commercialized advanced wound care and surgical and sports medicine products which support healing across a wide variety of wound types at many different types of facilities.
Cash and Cash Equivalents
The Company primarily maintains its cash in bank deposit accounts in the United States which, at times, may exceed the federally insured limits. The Company has not experienced losses in such accounts and believes it is not exposed to significant credit risk on cash. The Company considers all highly liquid investments purchased with an original maturity of three months or less to be cash equivalents.
F-9
Restricted Cash
The Company had restricted cash of $
Accounts Receivable, Net
Accounts receivable are stated at invoice value less estimated allowances for doubtful accounts. The Company continually monitors customer payments and maintains a reserve for estimated losses resulting from its customers’ inability to make required payments. The Company considers factors when estimating the allowance for doubtful accounts such as historical experience, credit quality, age of the accounts receivable balances, geography-related risks and economic conditions that may affect a customer’s ability to pay. In cases where there are circumstances that may impair a specific customer’s ability to meet its financial obligations, a specific allowance is recorded against amounts due, thereby reducing the net recognized receivable to the amount reasonably believed to be collectible. Accounts receivables are written off when deemed uncollectible. Recoveries of accounts receivables previously written off are recorded when received.
Inventories
Inventories are stated at the lower of cost (determined under the first-in first-out method) or net realizable value. Work in process and finished goods include materials, labor and allocated overhead. Inventories also include cell banks and the cost of tests mandated by regulatory agencies of the materials to qualify them for production.
The Company regularly reviews inventory quantities on hand and records a provision to write down excess and obsolete inventory to its estimated net realizable value based upon management’s assumptions of future material usage, yields and obsolescence, which are a result of future demand and market conditions and the effective life of certain inventory items.
The Company also tests other components of its inventory for future growth projections. The Company determines the average yield of the component and compares it to projected revenue to ensure it is properly reserved.
Property and Equipment, Net
Property and equipment are recorded at cost and depreciated over the estimated useful lives of the respective assets on a straight-line basis. As of December 31, 2022 and 2021, the Company’s property and equipment consisted of leasehold improvements, building, furniture and computers, and equipment. Property and equipment’s estimated useful lives are as follows:
Leasehold improvements |
|
|
Building |
|
|
Furniture and computers |
|
|
Equipment |
|
Upon retirement or sale, the cost of assets disposed of, and the related accumulated depreciation are removed from the accounts and any resulting gain or loss is included in the consolidated statement of operations. Expenditures for repairs and maintenance are charged to expense as incurred. Expenditures for major improvements that extend the useful lives of the related asset are capitalized and depreciated over their remaining estimated useful lives. Construction in progress costs are capitalized when incurred until the assets are placed in service, at which time the costs will be transferred to the related property and equipment, and depreciated over their respective useful lives.
Goodwill
Goodwill represents the excess of the purchase price of an acquired business over the fair value of the identifiable assets acquired and liabilities assumed. Goodwill is not amortized, but is tested for impairment at least annually (as of December 31), or more frequently if events or circumstances indicate the carrying value may no longer be recoverable and that an impairment loss may have occurred. Circumstances that could trigger an impairment test include, but are not limited to, a significant adverse change in the business climate or legal factors, an adverse action or assessment by a regulator, or unanticipated competition. The Company operates as one segment, which is considered to be the sole reporting unit, and therefore goodwill is tested for impairment at the consolidated level.
In accordance with ASC Topic 350, Intangibles - Goodwill and Other, the Company may first assess qualitative factors to determine whether it is necessary to perform the quantitative goodwill impairment test. If after assessing the totality of events or
F-10
circumstances, the Company determines that it is more likely than not (i.e. greater than 50% likelihood) that the fair value of the reporting unit is less than its carrying amount, then the quantitative test is required. Otherwise, no further testing is required. Alternatively, the Company can bypass the qualitative assessment and proceed directly to the quantitative test. The quantitative goodwill impairment test requires the Company to estimate and compare the fair value of the reporting unit with its carrying value. If the fair value of the reporting unit exceeds the carrying value of the net assets, goodwill is not impaired. If the fair value of the reporting unit is less than the carrying value, the difference is recorded as an impairment loss up to the amount of goodwill. At December 31, 2022, we elected to perform a quantitative analysis directly. We used the Company’s market capitalization to approximate the fair value of the reporting unit. The fair value exceeded the carrying value and
There was
Intangible Assets Subject to Amortization
Intangible assets include intellectual property either owned by the Company or for which the Company has a license. Intangible assets acquired in a business combination are recognized at fair value using generally accepted valuation methods deemed appropriate for the type of intangible asset acquired. Intangible assets are reported net of accumulated amortization, separately from goodwill. Intangible assets with finite lives are amortized over their estimated useful lives. Intangible assets include developed technology and patents, trade names, trademarks, customer relationships and non-compete agreements obtained through business acquisitions.
Trade names and trademarks |
|
|
Developed technology |
|
|
Customer relationships |
|
|
Non-compete agreements |
|
Impairment of Long-Lived Assets
Long-lived assets consist primarily of property and equipment. The Company reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset or asset group may not be recoverable. Factors that the Company considers in deciding when to perform an impairment review include, but not limited to, significant underperformance of the business in relation to expectations, significant negative industry or economic trends and significant changes or planned changes in the use of the assets. When such an event occurs, the Company determines whether there has been impairment by comparing the anticipated undiscounted future net cash flows to the related asset group’s carrying value. If an asset is determined to be impaired, the asset is written down to fair value, which is determined based either on discounted cash flows or appraised value, depending on the nature of the asset. The Company did not record any impairment of long-lived assets during the years ended December 31, 2022, 2021, or 2020.
Revenue Recognition
Product Revenue
The Company generates revenue through the sale of Advanced Wound Care and Surgical & Sports Medicine products. There is a single performance obligation in all of the Company’s contracts, which is the Company’s promise to transfer the Company’s product to customers based on specific payment and shipping terms in the arrangement. The entire transaction price is allocated to this single performance obligation. Product revenue is recognized when a customer obtains control of the Company’s product which occurs at a point in time and may be upon shipment, procedure date, or delivery, based on the terms of the contract.
Reserves for Variable Consideration
Revenues from product sales are recorded net of reserves for variable consideration which includes but is not limited to product return, discounts, rebates and GPO fees that are offered within contracts between the Company and its customers relating to the Company’s sales of its products. These reserves are based on the amounts earned or to be claimed by its customers on the related sales and are recorded as a reduction of accounts receivable or an establishment of a liability. Where appropriate, these estimates take into consideration a range of possible outcomes which are probability-weighted for relevant factors such as the Company’s historical experience, current contractual and statutory requirements, specific known market events and trends, industry data and forecasted customer buying and payment patterns. Overall, these reserves reflect the Company’s best estimates of the amount of consideration to which it is entitled based on the terms of the contract and is included in the net sales price to the extent that it is probable that a significant reversal in the amount of the cumulative revenue recognized will not occur in a future period. Actual amounts of
F-11
consideration ultimately paid may differ from the Company’s estimates. If actual results vary from the Company’s estimates, the Company adjusts these estimates, which would affect net product revenue and earnings in the period such variances become known.
Product Returns
Consistent with industry practice, the Company generally offers customers a limited right of return for product purchased. The Company estimates the amount of its product sales that may be returned by its customers and records this estimate as a reduction of revenue in the period the related product revenue is recognized. The Company currently estimates product return reserves using its historical return rates as well as factors that it becomes aware of that it believes could significantly impact its expected returns, including product recalls, pricing changes, or changes in reimbursement rates. The Company does not record an asset for the returned product as the product is discarded upon receipt.
Rebates and Allowances
The Company provides certain customers with rebates and allowances that are explicitly stated in the Company’s contracts, resulting in a reduction of revenue and the establishment of a liability that is included in accrued expenses in the accompanying consolidated balance sheets in the period the related product revenue is recognized.
GPO Fees
The Company pays fees to GPOs for administrative services that the GPOs perform in connection with the purchases of the product by the GPO members. These fees are based on a contractually-determined percentage of the Company’s applicable sales. The Company classifies these GPO fees as a reduction of revenue based on the substance of the relationship of all parties involved in the transaction. For the years ended December 31, 2022, 2021, and 2020, the Company recorded GPO fees of $
Other Revenue Policies
Sales, value add, and other taxes collected on behalf of third parties are excluded from revenue.
Applying the practical expedient in paragraph ASC 606-10-32-18, the Company does not assess whether a contract has a significant financing component if the expectation at contract inception is such that the period between payment by the customer and the transfer of the promised products to the customer will be one year or less, which is the case with substantially all customers.
Applying the practical expedient in ASC 340-40-25-4, the Company recognizes the incremental costs of obtaining contracts as an expense when incurred if the amortization period of the assets that the Company otherwise would have recognized is one year or less. These costs are included in selling, general, and administrative expenses.
Applying the practical expedient in ASC 606-10-25-18B, the Company accounts for shipping and handling activities related to contracts with customers as costs to fulfill the promise to transfer the associated products. The Company records the related costs as part of the cost of goods sold.
Disaggregation of Revenue
The following table sets forth revenue by product category:
|
|
Year Ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
Advanced Wound Care revenue |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Surgical and Sports Medicine revenue |
|
|
|
|
|
|
|
|
|
|||
Total revenue |
|
$ |
|
|
$ |
|
|
$ |
|
|||
For all periods presented, net revenue generated outside the United States represented less than 1% of total net revenue.
Stock-Based Compensation
The Company measures stock-based awards granted based on the fair value of the awards on the date of grant and recognizes compensation expense for those awards over the requisite service period, which is generally the vesting period of the respective award. Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from
F-12
those estimates. Generally, the Company issues stock-based awards with only service-based vesting conditions and records the expense for these awards using the straight-line method. The Company has not issued any stock-based awards with performance-based vesting conditions.
The Company recognizes stock-based compensation expense within selling, general and administrative expenses in the consolidated statement of operations for all share-based payments based upon the estimated grant-date fair value for the awards expected to ultimately vest.
The fair value of each restricted stock unit grant is based on the fair market value of the Company’s Class A common stock on the date of grant. The fair value of each stock option grant is estimated on the date of grant using the Black-Scholes option-pricing model. The Company has been a public company for a short period of time, has limited public float and lacks company-specific historical and implied volatility information for its Class A common stock. Therefore, it estimates its expected stock price volatility based on the historical volatility of publicly traded peer companies and expects to continue to do so until such time as it has adequate historical data regarding the volatility of its own traded stock price. The expected term of the Company’s stock options has been determined utilizing the “simplified” method for awards that qualify as “plain-vanilla” options. The risk-free interest rate is determined by reference to the U.S. Treasury yield curve in effect at the time of grant of the award for time periods approximately equal to the expected term of the award. Expected dividend yield is based on the fact that the Company has never paid cash dividends on its Class A common stock and does not expect to pay any cash dividends in the foreseeable future.
Advertising
Advertising costs are expensed as incurred and are included in selling, general and administrative expenses in the consolidated statements of operations. Advertising costs were approximately $
Research and Development Costs
Research and development expenses include personnel costs for the Company’s research and development personnel, expenses related to improvements in manufacturing processes, enhancements to the Company’s currently available products, and additional investments in the product and platform development pipeline. Research and development expenses also include expenses for clinical trials. The Company expenses research and development costs as incurred.
Foreign Currency
The Company’s functional currency, including the Company’s Swiss subsidiary, Organogenesis GmbH, is the U.S. dollar. Foreign currency gains and losses resulting from re-measurement of assets and liabilities held in foreign currencies and transactions settled in a currency other than the functional currency are included separately as non-operating income or expense in the consolidated statements of operations as a component of other expense, net. The foreign currency amounts recorded for all periods presented were insignificant.
Valuation of Contingent Purchase Earnout
In connection with the acquisition of CPN, the Company recognized a non-current liability for the fair value of the contingent consideration (the “Earnout”) at the time of the acquisition in 2020. The Earnout liability was classified as a Level 3 measurement for which fair value was derived from inputs that were unobservable and significant to the overall fair value measurement. The fair value of such Earnout liability was estimated using a Monte Carlo simulation model that utilized key assumptions including forecasted revenues and volatilities of the underlying financial metrics during the Earnout period. The Company assessed the fair value of the Earnout liability at each reporting period. Any subsequent changes in the estimated fair value of the liability were reflected in selling, general and administrative expenses until the liability was settled.
Income Taxes
The Company accounts for income taxes using the asset and liability method which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in the consolidated financial statements or in the Company’s tax returns. Deferred tax assets and liabilities are determined on the basis of the differences between the consolidated financial statement and the tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. Changes in deferred tax assets and liabilities are recorded in the provision for income taxes. The Company quarterly assesses the likelihood that its deferred tax assets will be recovered from future taxable income and, to the extent it believes, based upon the weight of available evidence, that it is more likely than not that all or a portion of the deferred tax assets will not be realized, a valuation allowance is established through a charge to income tax expense. In determining whether a valuation
F-13
allowance for deferred tax assets is necessary, the Company analyzes both positive and negative evidence related to the realization of deferred tax assets, including projected future taxable income, recent financial results and estimates of future reversals of deferred tax assets and liabilities. In addition, the Company considers whether it is more likely than not that the tax position will be sustained on examination by taxing authorities based on the technical merits of the position. In consideration of the factors discussed above, in the fourth quarter of 2021, the Company determined it was more likely than not that its deferred tax assets would be realized in the future and released the valuation allowance on the net U.S. deferred tax assets as of December 31, 2021, resulting in a benefit of $
The Company accounts for uncertain income tax positions recognized in the consolidated financial statements by applying a two-step process to determine the amount of tax benefit to be recognized. First, the tax position must be evaluated to determine the likelihood that it will be sustained upon external examination by the taxing authorities. If the tax position is deemed more-likely-than-not to be sustained, the tax position is then assessed to determine the amount of benefit to recognize in the consolidated financial statements. The amount of the benefit that may be recognized is the largest amount that has a greater than 50% likelihood of being realized upon ultimate settlement. The provision for income taxes includes the effects of any resulting tax reserves, or unrecognized tax benefits, that are considered appropriate as well as the related net interest and penalties.
Fair Value of Financial Instruments
Certain assets and liabilities of the Company are carried at fair value under GAAP. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. Financial assets and liabilities carried at fair value are to be classified and disclosed in one of the following three levels of the fair value hierarchy, of which the first two are considered observable and the last is considered unobservable:
The carrying values of accounts receivable, inventory, prepaid expenses and other current assets, accounts payable and accrued expenses approximate their fair values due to the short-term nature of these assets and liabilities. The fair value of the Earnout liability was determined according to Level 3 inputs in the fair value hierarchy described above (see footnote “4. Fair Value Measurement of Financial Instruments”). The carrying values of outstanding borrowings under the Company’s debt arrangements (see footnote “12. Long-Term Debt Obligations”) approximate their fair values as determined based on a discounted cash flow model, which represents a Level 3 measurement.
Earnings (Loss) per Share (EPS)
The Company determines earnings (loss) per share in accordance with the authoritative guidance in ASC Topic 260, Earnings Per Share. The Company has one class of common stock (Class A common stock) for purposes of the EPS calculation and therefore computes basic EPS by dividing net income (loss) by the weighted average number of common shares outstanding for the applicable period. Diluted EPS is computed in the same manner as basic EPS, except that the number of shares is computed by giving effect to all potential dilutive common shares. For purpose of this calculation, outstanding stock options, and unvested restricted stock are considered potential dilutive common shares.
Emerging Growth Company
Before December 31, 2021, the Company was an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. Section 107 of the JOBS Act provides that an emerging growth company can take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards. The Company elected to use the extended transition period for complying with new or revised accounting standards (such as ASU 2016-02, Leases (Topic 842)) and, as a result of this election, the Company’s financial statements prior to 2021 may not be comparable to companies that comply with public company effective dates. Effective December 31, 2021, the Company is no longer an emerging growth company.
F-14
Recently Adopted Accounting Pronouncements
In March 2020, the FASB issued ASU No. 2020-04, Reference Rate Reform (Topic 848): Facilitation of the Effects of Reference Rate Reform on Financial Reporting (“ASU 2020-04”). ASU 2020-04 provides temporary optional expedients and exceptions to the US GAAP guidance on contract modifications and hedge accounting to ease the financial reporting burdens related to the expected market transition from the London Interbank Offered Rate (LIBOR) and other interbank offered rates to alternative reference rates. In January 2021, the FASB issued ASU No. 2021-01, Reference Rate Reform (Topic 848): Scope (“ASU 2021-01”), to clarify certain optional expedients and exceptions in Topic 848 for contract modifications and hedge accounting to apply to derivatives that are affected by the discounting transition. Both ASU 2020-04 and ASU 2021-01 are effective upon issuance through December 31, 2022.
In December 2022, the Company executed an amendment to its debt agreement, replacing LIBOR with the secured overnight financing rate (“SOFR”). The Company utilized the relief provided in these ASUs. As the amendment did not affect the amount or timing of the contractual cash flows and the contemporaneous modifications to the other terms were related to the reference rate reform, the amendment was not substantial in accordance with ASC 848-20-35-8. Therefore, the amendment did not impact the Company’s consolidated financial statements.
Recently Issued Accounting Pronouncements Not Yet Adopted
In June 2016, the FASB issued ASU 2016-13, Financial Instruments—Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments (“ASU 2016-13”). Subsequent to the issuance of ASU 2016-13, the FASB has issued the following updates: ASU 2018-19, Codification Improvements to Topic 326, Financial Instruments- Credit Losses, ASU 2019-04, Codification Improvements to Topic 326, Financial Instruments—Credit Losses, Topic 815, Derivatives and Hedging, and Topic 825, Financial Instruments, ASU 2019-05, Financial Instruments—Credit Losses (Topic 326)—Targeted Transition Relief and ASU 2019-11, Codification Improvements to Topic 326, Financial Instruments—Credit Losses. The objective of ASU 2016-13 and all the related updates is to provide financial statement users with more decision-useful information about the expected credit losses on financial instruments and other commitments to extend credit held by a reporting entity at each reporting date. The amendments in this ASU replace the incurred loss impairment methodology in current GAAP with a methodology that reflects expected credit losses and requires consideration of a broader range of reasonable and supportable information to inform credit loss estimates. ASU 2016-13 and the related updates are effective for fiscal years, and interim periods within those years, beginning after December 15, 2019 for public business entities excluding entities eligible to be smaller reporting companies and for fiscal years, and interim periods within those years, beginning after December 15, 2022 for all other entities. Early adoption is permitted. As the Company was a smaller reporting company when the standard was issued, the Company took advantage of the extended transition period and will adopt this standard and the related improvements on January 1, 2023 by recognizing a cumulative-effect adjustment to retained earnings. The Company has finished evaluating the effects of adopting ASU 2016-13 and the related improvements and has determined that there will be an immaterial impact on the Company’s consolidated financial statements.
3. Acquisition
On September 17, 2020 (the “Acquisition Date”), the Company acquired certain assets and assumed certain liabilities of CPN Biosciences, LLC (“CPN”) pursuant to an asset purchase agreement dated July 24, 2020. CPN offered a physician office management solution and advanced wound care products.
The aggregate consideration amounted to $
The Company was obligated to pay the Earnout to CPN’s former equity holders if CPN’s legacy product revenue in the Earnout Period (July 1, 2021 to June 30, 2022), exceeded CPN’s 2019 revenue. The amount of the Earnout, if any, would be equal to
F-15
4. Fair Value Measurement of Financial Instruments
Earnout Liability
In connection with accounting for the CPN acquisition on September 17, 2020, the Company recorded an Earnout liability of $
The following table provides a roll-forward of the fair value of the Company’s Earnout liability, for which fair value was determined using Level 3 inputs until the end of the Earnout Period on June 30, 2022.
|
|
|
|
Earnout liability |
|
|
Balance as of December 31, 2019 |
|
|
|
$ |
|
|
Acquisition Date fair value |
|
|
|
|
|
|
Change in fair value |
|
|
|
|
|
|
Balance as of December 31, 2020 |
|
|
|
|
|
|
Change in fair value |
|
|
|
|
( |
) |
Balance as of December 31, 2021 |
|
|
|
|
|
|
Change in fair value |
|
|
|
|
|
|
Balance as of June 30, 2022 |
|
|
|
$ |
|
|
The Company did not have any financial assets and liabilities measured at fair value on a non-recurring basis as of December 31, 2022 and 2021.
5. Accounts receivable, net
Accounts receivable consisted of the following:
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Accounts receivable |
|
$ |
|
|
$ |
|
||
Less - allowance for doubtful accounts |
|
|
( |
) |
|
|
( |
) |
|
|
$ |
|
|
$ |
|
||
The Company’s allowance for doubtful accounts was comprised of the following:
Balance as of December 31, 2020 |
|
$ |
|
|
Additions |
|
|
|
|
Write-offs |
|
|
( |
) |
Balance as of December 31, 2021 |
|
$ |
|
|
Additions |
|
|
|
|
Write-offs |
|
|
( |
) |
Balance as of December 31, 2022 |
|
$ |
|
F-16
6. Inventories
Inventories, net of related reserves for excess and obsolescence, consisted of the following:
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Raw materials |
|
$ |
|
|
$ |
|
||
Work in process |
|
|
|
|
|
|
||
Finished goods |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
Raw materials include various components used in the Company’s manufacturing process. The Company’s excess and obsolete inventory review process includes analysis of sales forecasts and historical sales as compared to inventory levels and working with operations to maximize recovery of excess inventory. During the years ended December 31, 2022, 2021, and 2020, the Company charged $
7. Prepaid Expenses and Other Current Assets
Prepaid expenses and other current assets consisted of the following:
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Subscriptions |
|
$ |
|
|
$ |
|
||
Conferences and marketing expenses |
|
|
|
|
|
|
||
Deposits |
|
|
|
|
|
|
||
Insurance |
|
|
|
|
|
|
||
Other |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
Deposits are funds held by vendors which are expected to be released within twelve months and therefore they are recorded as current assets.
8. Property and Equipment, Net
Property and equipment consisted of the following:
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Leasehold improvements |
|
$ |
|
|
$ |
|
||
Building |
|
|
|
|
|
|
||
Furniture, computers and equipment |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Accumulated depreciation |
|
|
( |
) |
|
|
( |
) |
Construction in progress |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
Depreciation expense was $
During the year ended December 31, 2022, the Company recorded a charge of $
F-17
In connection with this decision, the Company recorded a charge of $
This facility was part of the primary assets in one of the Company's two asset groups. The Company considered the equipment disposal and construction pause, among other things, to be triggering events under ASC 360. The triggering events indicated that the Company’s long-lived assets might be impaired. The Company performed a recoverability test during 2022, on the affected asset group in accordance with ASC 360, Property, Plant and Equipment. The estimated undiscounted cash flow directly attributable to the asset group exceeded the carrying value of the asset group. Therefore, no impairment was identified.
9. Goodwill and Intangible Assets
Goodwill was $
Identifiable intangible assets consisted of the following as of December 31, 2022:
|
|
Original |
|
|
Accumulated |
|
|
Net Book |
|
|||
|
|
Cost |
|
|
Amortization |
|
|
Value |
|
|||
Developed technology |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||
Trade names and trademarks |
|
|
|
|
|
( |
) |
|
|
|
||
Customer relationship |
|
|
|
|
|
( |
) |
|
|
|
||
Independent sales agency network |
|
|
|
|
|
( |
) |
|
|
- |
|
|
Patent |
|
|
|
|
|
( |
) |
|
|
- |
|
|
Non-compete agreements |
|
|
|
|
|
( |
) |
|
|
|
||
Total |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||
Identifiable intangible assets consisted of the following as of December 31, 2021:
|
|
Original |
|
|
Accumulated |
|
|
Net Book |
|
|||
|
|
Cost |
|
|
Amortization |
|
|
Value |
|
|||
Developed technology |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||
Trade names and trademarks |
|
|
|
|
|
( |
) |
|
|
|
||
Customer relationship |
|
|
|
|
|
( |
) |
|
|
|
||
Independent sales agency network |
|
|
|
|
|
( |
) |
|
|
- |
|
|
Patent |
|
|
|
|
|
( |
) |
|
|
- |
|
|
Non-compete agreements |
|
|
|
|
|
( |
) |
|
|
|
||
Total |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||
Amortization of intangible assets, calculated on a straight-line basis or using an accelerated method, which reflects the pattern in which the economic benefits of the intangible assets are consumed, was $
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
Thereafter |
|
|
|
|
Total |
|
$ |
|
F-18
10. Accrued Expenses and Other Current Liabilities
Accrued expenses and other current liabilities consisted of the following:
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Personnel costs |
|
$ |
|
|
$ |
|
||
Royalties |
|
|
|
|
|
|
||
Accrued but unpaid lease obligations and interest |
|
|
|
|
|
|
||
Accrued settlement fee |
|
|
- |
|
|
|
|
|
Accrued taxes |
|
|
|
|
|
|
||
Other |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
The accrued but unpaid lease obligations and the interest accrual on these obligations are related to the buildings in Canton, Massachusetts. See footnote “17. Leases”. See footnote “2. Significant Accounting Policies” for accrued settlement fee.
11. Restructuring
In order to reduce the Company’s cost structure and improve operating efficiency, the Company consolidates its manufacturing operations in various locations into Massachusetts facilities.
On October 21, 2020, the Company committed to a plan to restructure the workforce and operations in its La Jolla, California facilities. The restructuring involved
On March 9, 2022, the Company committed to a plan to restructure the workforce and operations in its Birmingham, Alabama facilities. The restructuring involved approximately
As a result of the restructuring activities, the Company incurred pre-tax charges of $
|
|
Employee |
|
|
Other |
|
|
Total |
|
|||
Liability balance as of December 31, 2019 |
|
$ |
- |
|
|
$ |
- |
|
|
$ |
- |
|
Expenses |
|
|
|
|
|
- |
|
|
|
|
||
Cash distributions |
|
|
- |
|
|
|
- |
|
|
|
- |
|
Liability balance as of December 31, 2020 |
|
|
|
|
|
- |
|
|
|
|
||
Expenses |
|
|
|
|
|
|
|
|
|
|||
Cash distributions |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Liability balance as of December 31, 2021 |
|
|
|
|
|
|
|
|
|
|||
Expenses |
|
|
|
|
|
|
|
|
|
|||
Cash distributions |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Liability balance as of December 31, 2022 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
F-19
12. Long-Term Debt Obligations
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Line of credit |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
Term loan |
|
|
|
|
|
|
||
Less debt discount and debt issuance cost |
|
|
( |
) |
|
|
( |
) |
Term loan, net of debt discount and debt issuance cost |
|
$ |
|
|
$ |
|
||
2021 Credit Agreement
In August 2021, the Company, as borrower, its subsidiaries, as guarantors, and Silicon Valley Bank (“SVB”), and the several other lenders thereto (collectively, the “Lenders”) entered into a credit agreement, as amended (the “2021 Credit Agreement”), providing for a term loan facility not to exceed $
Advances made under the 2021 Credit Agreement may be either SOFR Loans or ABR Loans, at the Company’s option.
The 2021 Credit Agreement requires the Company to make consecutive quarterly installment payments equal to the following: (a) from September 30, 2021 through and including June 30, 2022, $
The Company must pay in arrears, on the first day of each quarter prior to August 6, 2026 (the “Revolving Termination Date”) and on the Revolving Termination Date, a fee for the Company’s non-use of available funds (the “Commitment Fee”). The Commitment Fee rate is between
The Company recorded debt issuance costs and related fees of $
As of December 31, 2022 and 2021, the Company had outstanding borrowings of $
Future payments of the 2021 Credit Agreement, as of December 31, 2022, are as follows for the calendar years ending December 31:
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
Total |
|
$ |
|
F-20
2019 Credit Agreement
In March 2019, the Company, its subsidiaries and SVB, and the several other lenders thereto entered into a credit agreement, as amended (the “2019 Credit Agreement”), providing for a term loan facility of $
In August 2021, upon entering into the 2021 Credit Agreement, the Company paid an aggregate amount of $
13. Stockholders’ Equity
As of December 31, 2022, the issued shares of Class A common stock include
Each share of Class A common stock entitles the holder to
At December 31, 2022 and 2021, the Company reserved the following shares of Class A common stock for future issuance:
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Shares reserved for issuance for outstanding options |
|
|
|
|
|
|
||
Shares reserved for issuance for outstanding restricted stock units |
|
|
|
|
|
|
||
Shares reserved for issuance for future grants |
|
|
|
|
|
|
||
Total shares of authorized common stock reserved for future issuance |
|
|
|
|
|
|
||
2020 Underwritten Public Offering
In November 2020, the Company closed a public offering (the “2020 Underwritten Public Offering”) of
14. Share-Based Compensation
Stock Incentive Plans-the 2018 Plan
On November 28, 2018, the Board of Directors of the Company adopted, and on December 10, 2018, the Company’s stockholders approved, the Organogenesis 2018 Equity and Incentive Plan (the “2018 Plan”). The purposes of the 2018 Plan are to provide long-term incentives and rewards to the Company’s employees, officers, directors and other key persons (including consultants), to attract and retain persons with the requisite experience and ability, and to more closely align the interests of such employees, officers, directors and other key persons with the interests of the Company’s stockholders.
F-21
The 2018 Plan authorizes the Company’s Board of Directors or a committee of not less than two independent directors (in either case, the “Administrator”) to grant the following types of awards: non-statutory stock options; incentive stock options; restricted stock awards; restricted stock units; stock appreciation rights; unrestricted stock awards; performance share awards; and dividend equivalent rights. The 2018 Plan is administered by the Company’s Board of Directors.
At the adoption of the 2018 Plan, a total of
Stock Incentive Plans-the 2003 Plan
The Organogenesis 2003 Stock Incentive Plan (the “2003 Plan”), provides for the Company to issue restricted stock awards, or to grant incentive stock options or non-statutory stock options. Incentive stock options may be granted only to the Company’s employees. Restricted stock awards and non-statutory stock options may be granted to employees, members of the Board of Directors, outside advisors and consultants of the Company.
Effective December 10, 2018, no additional awards may be made under the 2003 Plan and as a result (i) any shares in respect of stock options that are expired or terminated under the 2003 Plan without having been fully exercised will not be available for future awards; (ii) any shares in respect of restricted stock that are forfeited to, or otherwise repurchased by the Company, will not be available for future awards; and (iii) any shares of Class A common stock that are tendered to the Company by a participant to exercise an award will not be available for future awards.
Stock-Based Compensation Expense
Stock options awarded under the stock incentive plans expire
During the years ended December 31, 2022, 2021, and 2020, the Company recorded stock-based compensation expenses of $
Restricted Stock Units (RSUs)
During the year ended December 31, 2022, the Company granted
The activity of restricted stock units is set forth below:
|
|
|
|
|
Weighted |
|
||
|
|
|
|
|
Average |
|
||
|
|
Number |
|
|
Grant Date |
|
||
|
|
of Shares |
|
|
Fair Value |
|
||
Unvested at December 31, 2021 |
|
|
|
|
$ |
|
||
Granted |
|
|
|
|
|
|
||
Vested |
|
|
( |
) |
|
|
|
|
Canceled/Forfeited |
|
|
( |
) |
|
|
|
|
Unvested at December 31, 2022 |
|
|
|
|
$ |
|
||
As of December 31, 2022, the total unrecognized compensation cost related to unvested restricted stock units expected to vest was $
F-22
Stock Options
The stock options granted during the years ended December 31, 2022, and 2021 were
|
|
|
|
|
|
Year Ended |
|
|||||
|
|
|
|
|
|
December 31, |
|
|||||
|
|
|
|
|
|
2022 |
|
|
2021 |
|
||
|
|
|
|
|
|
|
|
|
|
|
||
Risk-free interest rate |
|
|
|
|
|
|
% |
|
|
% |
||
Expected term (in years) |
|
|
|
|
|
|
|
|
|
|
||
Expected volatility |
|
|
|
|
|
|
% |
|
|
% |
||
Expected dividend yield |
|
|
|
|
|
|
% |
|
|
% |
||
Exercise price |
|
|
|
|
|
$ |
|
|
$ |
|
||
Underlying stock price |
|
|
|
|
|
$ |
|
|
$ |
|
||
These assumptions resulted in an estimated weighted-average grant-date fair value per share of stock options granted during the years ended December 31, 2022 and 2021 of $
The following table summarizes the Company’s stock option activity since December 31, 2021:
|
|
|
|
|
|
|
|
Weighted |
|
|
|
|
||||
|
|
|
|
|
Weighted |
|
|
Average |
|
|
|
|
||||
|
|
|
|
|
Average |
|
|
Remaining |
|
|
Aggregate |
|
||||
|
|
Number of |
|
|
Exercise |
|
|
Contractual |
|
|
Intrinsic |
|
||||
|
|
Shares |
|
|
Price |
|
|
Term |
|
|
Value |
|
||||
|
|
|
|
|
|
|
|
(in years) |
|
|
|
|
||||
Outstanding as of December 31, 2021 |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
Granted |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Exercised |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
Canceled / forfeited |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
Outstanding as of December 31, 2022 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Options exercisable as of December 31, 2022 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Options vested or expected to vest as of December 31, 2022 |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
The aggregate intrinsic value of stock options is calculated as the difference between the exercise price of the stock options and the fair value of the Company’s Class A common stock for those stock options that have exercise prices lower than the fair value of the Company’s Class A common stock.
The total fair value of options vested during the years ended December 31, 2022 and 2021 was $
As of December 31, 2022, the total unrecognized stock compensation expense was $
Between 2010 and 2013, a former executive took several partial recourse notes totaling $
F-23
15. Income Taxes
The components of the income tax expense (benefit) consisted of the following for the years ended December 31, 2022, 2021, and 2020:
|
|
Year Ended December 31, |
|
||||||||||||
|
|
2022 |
|
|
|
|
2021 |
|
|
|
2020 |
|
|||
Income tax expense (benefit): |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Current tax expense (benefit) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Federal |
|
$ |
|
|
|
|
$ |
- |
|
|
|
$ |
( |
) |
|
State |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Foreign |
|
|
|
|
|
|
|
( |
) |
|
|
|
|
||
Total current tax expense |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Deferred tax expense (benefit) |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Federal |
|
|
|
|
|
|
|
( |
) |
|
|
|
|
||
State |
|
|
( |
) |
|
|
|
|
( |
) |
|
|
|
- |
|
Foreign |
|
|
- |
|
|
|
|
|
- |
|
|
|
|
|
|
Total deferred tax expense (benefit) |
|
|
|
|
|
|
|
( |
) |
|
|
|
|
||
Total income tax expense (benefit) |
|
$ |
|
|
|
|
$ |
( |
) |
|
|
$ |
|
||
On a periodic basis, the Company reassesses the valuation allowance on its deferred income tax assets, weighing positive and negative evidence to assess the recoverability of the deferred tax assets. In the fourth quarter of fiscal year 2021, the Company assessed the valuation allowance and considered positive evidence, including significant cumulative consolidated income over the three years ended December 31, 2021, revenue growth and expectations of future profitability, and negative evidence, including the impact of a negative change in the economic climate, significant risks and uncertainties in the business and restrictions on tax loss utilization in certain state jurisdictions. After assessing both the positive evidence and the negative evidence, the Company determined it was more likely than not that its deferred tax assets would be realized in the future and released the valuation allowance on its net deferred tax assets as of December 31, 2021, resulting in a benefit from income taxes of $
As of December 31, 2022, the Company had available for the reduction of future years’ federal taxable income, net operating loss carry-forwards of approximately $
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes.
|
|
December 31, |
|
|||||||
|
|
2022 |
|
|
|
|
2021 |
|
||
Net operating loss carryforwards |
|
|
|
|
|
|
|
|
||
Federal |
|
$ |
|
|
|
|
$ |
|
||
State |
|
|
|
|
|
|
|
|
||
Foreign |
|
|
|
|
|
|
|
|
||
Other |
|
|
|
|
|
|
|
|
||
Capitalized R&D |
|
|
|
|
|
|
|
- |
|
|
Stock-based compensation |
|
|
|
|
|
|
|
|
||
Finance leases |
|
|
|
|
|
|
|
|
||
Operating leases |
|
|
|
|
|
|
|
|
||
Fixed assets |
|
|
|
|
|
|
|
|
||
Net deferred tax assets before valuation allowance |
|
|
|
|
|
|
|
|
||
Valuation allowance |
|
|
- |
|
|
|
|
|
- |
|
ROU assets |
|
|
( |
) |
|
|
|
|
( |
) |
Intangibles |
|
|
( |
) |
|
|
|
|
( |
) |
Net deferred tax assets |
|
$ |
|
|
|
|
$ |
|
||
F-24
The Company’s subsidiary in Switzerland is carrying a deferred tax asset of approximately $
Ownership changes, as defined in the Internal Revenue Code, may limit the amount of net operating losses and research and development tax credit carryforwards that can be utilized annually to offset future taxable income. Subsequent ownership changes could further affect the limitation in future years. The Company completed an analysis in 2021 and determined that it had not experienced an ownership change during the periods 2001 through 2021.
The differences between income taxes expected at the U.S. federal statutory income tax rate of
|
|
December 31, |
||||||||||||||
|
|
2022 |
|
|
2021 |
|
2020 |
|
|
|||||||
U.S. federal statutory income tax rate |
|
|
|
% |
|
|
|
|
% |
|
|
|
% |
|||
Federal valuation allowance |
|
|
- |
|
% |
|
|
|
( |
) |
% |
|
|
( |
) |
% |
State valuation allowance |
|
|
- |
|
% |
|
|
|
( |
) |
% |
|
|
( |
) |
% |
Return to provision and other adjustments |
|
|
( |
) |
% |
|
|
|
- |
|
|
|
|
- |
|
|
Prior period correction |
|
|
( |
) |
% |
|
|
|
- |
|
|
|
|
- |
|
|
State and local income taxes |
|
|
|
% |
|
|
|
|
% |
|
|
|
% |
|||
Nondeductible expenses |
|
|
|
% |
|
|
|
|
% |
|
|
|
% |
|||
Executive compensation limited by 162(m) |
|
|
|
% |
|
|
|
- |
|
|
|
|
- |
|
|
|
Foreign rate differential |
|
|
|
% |
|
|
|
- |
|
|
|
|
- |
|
|
|
Uncertain tax position reserves |
|
|
|
% |
|
|
|
|
% |
|
|
|
% |
|||
Research and development credits |
|
|
|
% |
|
|
|
|
% |
|
|
|
% |
|||
Effective income tax rate |
|
|
|
% |
|
|
|
( |
) |
% |
|
|
|
% |
||
The Company recognizes the tax benefit from an uncertain tax position only if it is more-likely-than-not that the tax position will be sustained on examination by taxing authorities, based on the technical merits of the position.
A tabular roll forward of the Company’s uncertainties in its income tax provision liability is presented below:
|
|
Year Ended December 31, |
|
||||||||||||
|
|
2022 |
|
|
|
|
2021 |
|
|
|
2020 |
|
|||
Gross balance at beginning of year |
|
$ |
|
|
|
|
$ |
|
|
|
$ |
|
|||
Additions based on tax positions related to the current period |
|
|
|
|
|
|
|
|
|
|
|
|
|||
Reductions for tax positions of prior years |
|
|
( |
) |
|
|
|
|
( |
) |
|
|
|
( |
) |
Gross balance at end of year |
|
$ |
|
|
|
|
$ |
|
|
|
$ |
|
|||
The Company files income tax returns in the U.S. federal and state jurisdictions and Switzerland. With limited exceptions, the Company is no longer subject to federal, state, local or foreign examinations for years prior to December 31, 2018. However, carryforward attributes that were generated prior to December 31, 2018 may still be adjusted upon examination by state or local tax authorities if they either have been or will be used in a future period.
The Company recognizes interest and penalty-related expenses in tax expenses. $
16. Earnings (Loss) per Share (EPS)
Basic EPS is calculated by dividing net income (loss) by the weighted-average number of shares outstanding during the period. Diluted EPS is calculated by dividing net income (loss) by the weighted-average number of shares outstanding plus the dilutive effect,
F-25
if any, of outstanding equity awards using the treasury stock method which includes consideration of unrecognized compensation expenses as additional proceeds.
A reconciliation of the numerator and denominator used in the calculation of the basic and diluted net income (loss) attributable to the Class A common stockholders is as follows:
|
|
Year Ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
Numerator: |
|
|
|
|
|
|
|
|
|
|||
Net Income |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Denominator: |
|
|
|
|
|
|
|
|
|
|||
Weighted average common shares outstanding —basic |
|
|
|
|
|
|
|
|
|
|||
Dilutive effect of restricted stock units |
|
|
|
|
|
|
|
|
|
|||
Dilutive effect of options |
|
|
|
|
|
|
|
|
|
|||
Weighted-average common shares outstanding—diluted |
|
|
|
|
|
|
|
|
|
|||
Earnings per share—basic |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Earnings per share—diluted |
|
$ |
|
|
$ |
|
|
$ |
|
|||
For the year ended December 31, 2022, 2021, and 2020, outstanding stock-based awards of
17. Leases
The Company’s leases consist primarily of real estate, equipment and vehicle leases.
The Company leases real estate for office, lab, warehouse and production space under noncancelable leases that expire at various dates through 2035, subject to the Company’s options to terminate or renew certain leases for an additional to
The Company leases vehicles under operating leases for certain employees and has fleet services agreements for service on these vehicles. The minimum lease term for each newly leased vehicle is
The Company determines if an arrangement is a lease at lease inception. The options to extend or terminate a lease are included in the lease terms when it is reasonably certain that the Company will exercise the options. Operating leases are included in operating lease right-of-use assets and operating lease obligations on the consolidated balance sheets. Finance lease right-of-use assets are included in property and equipment, net, and the related liabilities are included in finance lease obligations on the consolidated balance sheets.
Right-of-use assets represent the Company’s right to use an underlying asset for the lease term and lease liabilities represent the Company’s obligation to make lease payments arising from the leases. Right-of-use assets and lease liabilities are recognized based on the present value of the fixed lease payments over the lease term at the commencement date. The right-of-use assets also include any initial direct costs incurred and lease payments made at or before the commencement date and are reduced by lease incentives. The Company uses its incremental borrowing rate as the discount rate to determine the present value of the lease payments for leases that do not have a readily determinable implicit discount rate. The Company’s incremental borrowing rate is the rate of interest that it would have to borrow on a collateralized basis over a similar term and amount in a similar economic environment. The Company determines the incremental borrowing rates for its leases by adjusting the risk-free interest rate with a credit risk premium corresponding to the Company’s credit rating.
The Company records rent expense for its operating leases on a straight-line basis from the lease commencement date until the end of the lease term. The Company records finance lease cost as a combination of the depreciation expense for the right-of-use assets and interest expense for the outstanding lease liabilities using the discount rate discussed above. Variable lease payments are primarily related to the office and fleet leases which include but are not limited to taxes, insurance, common area maintenance and maintenance programs for leased vehicles. Variable lease payments are based on the occurrence or usage; therefore, they are not included as part of the initial right-of-use assets and liabilities calculation.
On January 1, 2013, the Company entered into finance lease arrangements with 65 Dan Road SPE, LLC, 85 Dan Road Associates, LLC, Dan Road Equity I, LLC and 275 Dan Road SPE, LLC for office and laboratory space in Canton, Massachusetts. 65 Dan Road SPE, LLC, 85 Dan Road Associates, LLC, Dan Road Equity I, LLC and 275 Dan Road SPE, LLC are related parties as the owners of these entities are also directors, former directors and / or stockholders of the Company. In August 2021, the Company purchased the building (the “275 Dan Road Building”) under the lease with 275 Dan Road SPE, LLC for $
F-26
which was terminated in August 2021, the remaining three leases were set to terminate on December 31, 2022 and each contained a renewal option for a five-year period with a rental rate at the greater of (i) rent for the last year of the prior term, or (ii) the then fair market value. The Company exercised the option to extend the leases for an additional five years in November 2021. It remeasured the lease assets and liabilities based on its best estimate of the market rental rate in the renewal period and reassessed the classification for these leases according to ASC 842-10-25-1 Lease Classification. As a result, these leases were reclassified from finance leases to operating leases. The related finance lease assets and liabilities were reclassified to operating lease right-of-use assets and operating lease obligations on the consolidated balance sheet as of December 31, 2021. In December 2022, the Company and the landlord finalized the market rental rate in the renewal period for these properties, resulting in an additional $
The Company owes some accrued but unpaid lease obligations under the aforementioned leases as detailed in the section below. Effective April 1, 2019, the Company agreed to accrue interest on the accrued but unpaid lease obligations at an interest rate equal to the rate charged under the 2019 Credit Agreement. In connection with the purchase of the 275 Dan Road Building in August 2021, the Company paid
The accrued but unpaid lease obligations as well as the related interest accruals are shown below.
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Principal portion of rent in arrears |
|
$ |
|
|
$ |
|
||
Unpaid operating and common area maintenance costs |
|
|
- |
|
|
|
|
|
Total accrued but unpaid lease obligations |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
Accrued interest on accrued but unpaid lease obligations |
|
$ |
|
|
$ |
|
||
The principal portion of rent in arrears was included in the short-term portion of operating lease obligations other than the balance related to the 275 Dan Road Building that was included in accrued expenses and other current liabilities on the consolidated balance sheets as of December 31, 2022 and 2021. The unpaid operating and common area maintenance costs, and the accrued interest on the accrued but unpaid lease obligations were included in accrued expenses and other current liabilities on the consolidated balance sheets as of December 31, 2022 and 2021.
The components of lease cost were as follows:
|
|
Classification |
|
|
Year Ended |
|
|
Year Ended |
|
||
Finance lease |
|
|
|
|
|
|
|
|
|
||
Amortization of right-of-use assets |
|
COGS and SG&A |
|
|
$ |
|
|
$ |
|
||
Interest on lease liabilities |
|
Interest Expense |
|
|
|
|
|
|
|
||
Total Finance lease cost |
|
|
|
|
|
|
|
|
|
||
Operating lease cost |
|
COGS, R&D, SG&A |
|
|
|
|
|
|
|
||
Short-term lease cost |
|
COGS, R&D, SG&A |
|
|
|
|
|
|
|
||
Variable lease cost |
|
COGS, R&D, SG&A |
|
|
|
|
|
|
|
||
Total lease cost |
|
|
|
|
$ |
|
|
$ |
|
||
Supplemental balance sheet information related to finance leases was as follows:
|
|
December 31, |
|
|
December 31, |
|
||
|
|
2022 |
|
|
2021 |
|
||
Property and equipment, gross |
|
$ |
|
|
$ |
|
||
Accumulated depreciation |
|
|
( |
) |
|
|
( |
) |
|
$ |
- |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
|
$ |
- |
|
|
$ |
|
||
Finance lease long-term obligations |
|
|
- |
|
|
|
- |
|
Total finance lease liabilities |
|
$ |
- |
|
|
$ |
|
|
F-27
Supplemental cash flow information related to leases was as follows:
|
|
Year Ended |
|
|
Year Ended |
|
||
Cash paid for amounts included in the measurement of lease liabilities: |
|
|
|
|
|
|
||
Operating cash flows for operating leases |
|
$ |
|
|
$ |
|
||
Operating cash flows for finance leases |
|
$ |
|
|
$ |
|
||
Financing cash flows for finance leases |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
Right-of-use assets obtained in exchange for lease obligations |
|
|
|
|
|
|
||
Operating leases |
|
$ |
|
|
$ |
|
||
Finance leases |
|
$ |
- |
|
|
$ |
- |
|
|
|
December 31, |
|
|
December 31, |
|
||
|
|
2022 |
|
|
2021 |
|
||
Weighted-average remaining lease term |
|
|
|
|
|
|
||
Finance leases |
|
|
- |
|
|
|
|
|
Operating leases |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
|
|
December 31, |
|
|
December 31, |
|
||
|
|
2022 |
|
|
2021 |
|
||
Weighted-average discount rate |
|
|
|
|
|
|
||
Finance leases |
|
|
- |
|
|
|
% |
|
Operating leases |
|
|
% |
|
|
% |
||
As of December 31, 2022, the maturities of lease liabilities were as follows:
|
|
Operating leases |
|
|
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
Thereafter |
|
|
|
|
Total lease payments |
|
|
|
|
Less: interest |
|
|
( |
) |
Total lease liabilities |
|
$ |
|
|
18. Commitments and Contingencies
Royalties
The Company entered into a license agreement with a university for certain patent rights related to the development, use and production of one of its advanced wound care products. Under this agreement, the Company incurred a royalty based on a percentage of net product sales, for the use of these patents until the patents expired, which was in November 2006. Accrued royalties totaled $
In October 2017, the Company entered into a license agreement with a third party. Under the license agreement, the Company is required to pay royalties based on a percentage of net sales of the licensed product that occur, after December 31, 2017, through the expiration of the underlying patent in October 2026, subject to minimum royalty payment provisions. The Company recorded royalty expense of $
F-28
As part of the NuTech Medical acquisition, the Company inherited certain product development and consulting agreements for ongoing consulting services and royalty payments based on a percentage of net sales on certain products over a period of
Legal Matters
In conducting its activities, the Company, from time to time, is subject to various claims and also has claims against others. In management’s opinion, the ultimate resolution of such claims would not have a material effect on the financial position, operating results or cash flows of the Company. The Company accrues for these claims when amounts due are probable and estimable. The Company accrued $
The purchase price for NuTech Medical acquired in 2017 included $
19. Related Party Transactions
Lease obligations to affiliates, including accrued but unpaid lease obligations, purchase of an asset under a finance lease with an affiliate, and renewal of leases with affiliates are further described in Note “17. Leases”.
In 2010, the Company’s Board of Directors approved a loan program that permitted the Company to make loans to three executives of the Company (the “Employer Loans”) to (i) provide them with liquidity (“Liquidity Loans”) and (ii) fund the exercise of vested stock options (“Option Loans”). Two of the executives left the Company in 2014. The Employer Loans matured with all principal and accrued interest due on the tenth anniversary of the issuance date of each subject loan. Interest on the Employer Loans was at various rates ranging from
20. Employee Benefit Plan
The Company maintains a 401(k) Savings Plan (the “Plan”) for the U.S. employees. Under the Plan, eligible employees may contribute, subject to statutory limitations, a percentage of their salary to the Plan. Contributions made by the Company are made at the discretion of the Board of Directors and vest immediately. During the years ended December 31, 2022, 2021, and 2020, the Company made employer contributions of $
21. Subsequent Events
The Company has performed an evaluation of subsequent events through the time of filing this Annual Report on Form 10-K with the SEC.
On February 3, 2023, the Company announced a reduction in force in response to macroeconomic conditions. The reduction in force reduced the Company’s headcount by
F-29
In the first quarter of 2023, options to purchase
At the time of the submission of this Annual Report on Form 10-K, the Company's products are subject to the Infrastructure Investment and Jobs Act and rebate obligation that took effect on January 1, 2023. Consequently, the Company may owe rebates to the federal government prospectively, on Apligraf, Dermagraft (when marketed), and PuraPly products and possibly other products if more than a certain percentage of a single-use product is not administered to a patient and is discarded by providers.
F-30