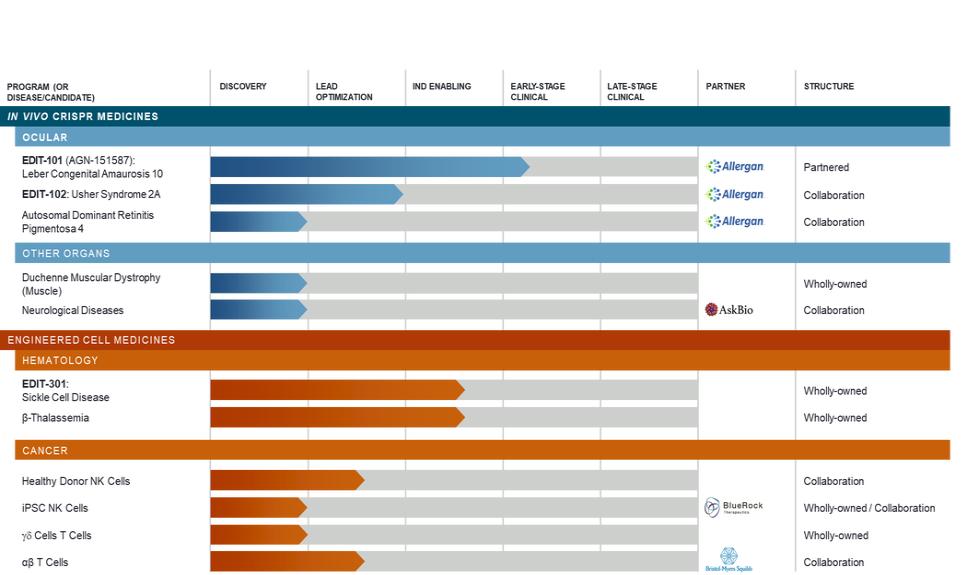
question, for example, we cannot be certain that there is no invalidating prior art, of which we or our licensing partners and the patent examiner were unaware during prosecution. If a third party were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our technology or platform, or any product candidates that we develop. Such a loss of patent protection would have a material adverse impact on our business, financial condition, results of operations, and prospects.
The intellectual property landscape around genome editing technology, including CRISPR, is highly dynamic, and third parties may initiate legal proceedings alleging that we are infringing, misappropriating, or otherwise violating their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
The field of genome editing, especially in the area of CRISPR technology, is still in its infancy, and no such products have reached the market. Due to the intense research and development that is taking place by several companies, including us and our competitors, in this field, the intellectual property landscape is in flux, and it may remain uncertain for the coming years. There may be significant intellectual property related litigation and proceedings relating to our owned and in-licensed, and other third party, intellectual property and proprietary rights in the future.
Our commercial success depends upon our ability and the ability of our collaborators to develop, manufacture, market, and sell any product candidates that we develop and use our proprietary technologies without infringing, misappropriating, or otherwise violating the intellectual property and proprietary rights of third parties. The biotechnology and pharmaceutical industries are characterized by extensive litigation regarding patents and other intellectual property rights. We are subject to and may in the future become party to, or threatened with, adversarial proceedings or litigation regarding intellectual property rights with respect to our technology and any product candidates we develop, including interference, re-examination, post-grant review, inter partes review, and derivation proceedings before the USPTO and similar proceedings in foreign jurisdictions such as oppositions before the EPO. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future, regardless of their merit. We are aware of certain third party patents and patent applications in this landscape that may be asserted to encompass our CRISPR/Cas9 technology. In particular, we are aware of several separate families of U.S. patents and/or U.S. patent applications and foreign counterparts which relate to CRISPR/Cas9 technology, where the earliest priority dates of each family pre-date the priority dates of our in-licensed patents and patent applications, including PCT Publication No. WO 2013/141680 (and its related U.S. Patent No. 9,637,739 and other related U.S. patent applications and foreign counterparts including European Patent No. EP 2,828,386 B1, which is being opposed by at least one party) filed by Vilnius University (which is reported to have exclusively licensed its rights to DuPont Pioneer, which is reported to have licensed certain rights to Caribou Biosciences, which is reported to have non-exclusively licensed certain rights to Intellia Therapeutics and CRISPR Therapeutics), WO 2013/176772 (and its related U.S. Patents including U.S. Patent Nos. 10,000,772, 10,113,167, 10,227,611, and 10,266,850, 10,301,651, 10,308,961, 10,337,029, 10,351,878, 10,358,658, and 10,358,659 among others, and other related U.S. patent applications and foreign counterparts including European Patent Nos. EP 2,800,811 B1, EP 3,241,902 B1, and EP 3,401,400 B1 which are being opposed by several parties) filed by the University of California, the University of Vienna (both of which are reported to have exclusively licensed their rights to Caribou Biosciences, which is reported to have exclusively licensed certain rights to Intellia Therapeutics), and Emmanuelle Charpentier (who is reported to have exclusively licensed her rights to CRISPR Therapeutics, ERS Genomics and TRACR Hematology), WO 2014/065596 (and its related U.S. patent applications and foreign counterparts including European Patent No. EP 2,912,175 B1 which is being opposed by several parties) filed by ToolGen, and WO 2014/089290 (and its related U.S. patent applications and foreign counterparts including European Patent Nos. EP 2,928,496 B1, EP 3,138,910 B1, EP 3,138,911 B1, EP 3,138,912 B1, EP 3,360,964 B1 and EP 3,363,902 B1, which are being opposed by several parties) filed by Sigma-Aldrich Co. LLC. Each of these patent families are owned by a different third party and contain claims that may be construed to cover components and uses of CRISPR/Cas9 technology. If we are not able to obtain or maintain a license on commercially reasonable terms to any third-party patents that cover our product candidates or activities, such third parties could potentially assert infringement claims against us, which could have a material adverse effect on the conduct of our business.
Even if we believe third-party intellectual property claims are without merit, there is no assurance that a court would find in our favor on questions of infringement, validity, enforceability, or priority. A court of competent