PART I. FINANCIAL INFORMATION
Item 1. Financial Statements and Supplementary Data

ESSA Pharma Inc.
CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS
(Unaudited)
(Expressed in United States dollars)
FOR THE THREE MONTHS ENDED DECEMBER 31, 2023
8
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
For the quarterly period ended
or
For the transition period fromto
Commission file number:
(Exact name of registrant as specified in its charter)
(State or other jurisdiction of | (I.R.S. Employer |
incorporation or organization) | Identification Number) |
(Address of principal executive offices, including zip code) | |
Registrant’s telephone number, including area code: ( | |
Securities registered pursuant to Section 12(b) of the Act: | |
Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | ☐ | Accelerated filer | ☐ | |
☒ | Smaller reporting company | |||
Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Securities Exchange Act of 1934). Yes
The number of outstanding Common Shares of the registrant, no par value per share, as of February 12, 2024 was
ESSA PHARMA INC.
QUARTERLY REPORT ON FORM 10-Q
For the Quarter Ended December 31, 2023
Table of Contents
2
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Quarterly Report on Form 10-Q includes “forward-looking statements” within the meaning of the U.S. Private Securities Litigation Reform Act of 1995 and “forward-looking information” within the meaning of Canadian securities laws, or collectively, forward-looking statements. Forward-looking statements include statements that may relate to our plans, objectives, goals, strategies, future events, future revenue or performance, capital expenditures, financing needs and other information that is not historical information. Many of these statements appear, in particular, under the headings “Business,” “Risk Factors,” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations”. Forward-looking statements can often be identified by the use of terminology such as “subject to”, “believe,” “anticipate,” “plan,” “expect,” “intend,” “estimate,” “project,” “may,” “might,” “will,” “should,” “would,” “could,” “hope,” “can,” the negatives thereof, variations thereon and similar expressions, or by discussions of strategy. Such forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause the Company’s actual results, performance or achievements to be materially different from any future results, performance or achievements that may be expressed or implied by such forward-looking statements. Examples of such forward looking statements include, but are not limited to statements related to:
| ● | our ability to advance our product candidate and potential future product candidates through, and successfully complete, clinical trials; |
| ● | our ability to recruit sufficient numbers of patients for future clinical trials, and the benefits expected therefrom; |
| ● | our ability to establish and maintain relationships with collaborators with acceptable development, regulatory and commercialization expertise and the benefits to be derived from such collaborative efforts; |
| ● | our ability to obtain funding for operations, including research funding, and the timing and potential sources of such funding; |
| ● | the initiation, timing, cost, location, progress and success of, strategy and plans with respect to our research and development programs (including research programs and related milestones with regards to next-generation drug candidates and compounds), preclinical studies and clinical trials; |
| ● | the therapeutic benefits, properties, effectiveness, pharmacokinetic profile and safety of our product candidate and potential future product candidates, if any, including the expected benefits, properties, effectiveness, pharmacokinetic profile and safety of our next-generation Aniten compounds; |
| ● | our ability to protect our intellectual property and operate our business without infringing upon the intellectual property rights of others; |
| ● | developments relating to our competitors and our industry, including the success of competing therapies that are or may become available; |
| ● | our ability to achieve profitability; |
| ● | the grant (“CPRIT Grant”) under the Cancer Prevention and Research Institute of Texas (“CPRIT”) and payments thereunder, including any residual obligations; |
| ● | sales of our Common Shares, no par value (the “Common Shares”) under the Open Market Sale AgreementSM between the Company and Jefferies LLC, effective November 3, 2023 (the “ATM Sales Agreement”); |
| ● | our intended use of proceeds from past and future offerings of our securities; |
| ● | the implementation of our business model and strategic plans, including strategic plans with respect to patent applications and strategic collaborations and partnerships; |
| ● | our ability to identify, develop and commercialize product candidates; |
| ● | our commercialization, marketing and manufacturing capabilities and strategy; |
| ● | our expectations regarding federal, state, provincial and foreign regulatory requirements, including our plans with respect to anticipated regulatory filings; |
| ● | whether we will receive, and the timing and costs of obtaining, regulatory approvals in the United States, Canada and other jurisdictions; |
| ● | the accuracy of our estimates of the size and characteristics of the markets that may be addressed by our product candidate and potential future product candidates, if any; |
3
| ● | our ability to maintain operations, development programs, preclinical studies, clinical trials and raise capital as a result of global macroeconomic factors including inflation, supply chain issues and any current or future impact related to widespread health concerns, pandemics, or epidemics, and other outbreaks of illness; |
| ● | the rate and degree of market acceptance and clinical utility of our potential future product candidates, if any; |
| ● | the timing of, and our ability and our collaborators’ ability, if any, to obtain and maintain regulatory approvals for our product candidate and potential future product candidates, if any; |
| ● | our expectations regarding market risk, including inflation, interest rate changes and foreign currency fluctuations; |
| ● | our ability to engage and retain the employees required to grow our business; |
| ● | the compensation that is expected to be paid to our employees; |
| ● | our future financial performance and projected expenditures; and |
| ● | estimates of our financial condition, expenses, future revenue, capital requirements and our need for additional financing and potential sources of capital and funding. |
Such statements reflect our current views with respect to future events, are subject to risks and uncertainties and are necessarily based upon a number of estimates and assumptions that are inherently subject to significant medical, scientific, business, economic, competitive, political and social uncertainties and contingencies. Many factors could cause our actual results, performance or achievements to be materially different from any future results, performance, or achievements that may be expressed or implied by such forward-looking statements, including those described under “Risk Factors” in our Annual Report on Form 10-K. All forward-looking statements included in this Quarterly Report on Form 10-Q, are based upon our current expectations and various assumptions. Certain assumptions made in preparing the forward-looking statements include, but are not limited to:
| ● | our ability to conduct clinical studies involving our product candidate and to identify any future product candidates; |
| ● | our ability to obtain regulatory and other approvals to commence clinical trials involving any future product candidates; |
| ● | our ability to obtain positive results from research and development activities, including clinical trials; |
| ● | the availability of sufficient financing on reasonable terms; |
| ● | our ability to obtain required regulatory approvals; |
| ● | our ability to protect patents and proprietary rights; |
| ● | our ability to successfully out-license or sell future products, if any, and in-license and develop new products; |
| ● | the absence of material adverse changes in our industry or the global economy; |
| ● | our ability to attract and retain key personnel; |
| ● | our continued compliance with third-party license terms and non-infringement of third-party intellectual property rights; |
| ● | our ability to maintain good business relationships with our strategic partners; and |
| ● | our ability to understand and predict market competition. |
We believe there is a reasonable basis for our current expectations, views and assumptions, but they are inherently uncertain. We may not realize our expectations and our views and assumptions may not prove correct. Actual results could differ materially from those described or implied by such forward-looking statements. In evaluating forward-looking statements, investors should specifically consider the following uncertainties and factors, among others (including those set forth under the heading “Risk Factors” in our Annual Report on Form 10-K), that could affect future performance and cause actual results to differ materially from those matters expressed in or implied by forward-looking statements
RISK FACTOR SUMMARY
Below is a summary of material factors that make an investment in our Common Shares speculative or risky. Importantly, this summary does not address all of the risks and uncertainties that we face. Additional discussion of the risks and uncertainties summarized in this risk factor summary, as well as other risks and uncertainties that we face, can be found under “Cautionary Note Regarding Forward-Looking Statements” and Part I, Item 1A. “Risk Factors” in our Annual Report on Form 10-K. The below summary is qualified in its entirety by those more complete discussions of such risks and uncertainties. You should consider carefully the risks and uncertainties described under Part I, Item 1A. “Risk Factors”
4
in our Annual Report on Form 10-K as part of your evaluation of an investment in our Common Shares. Important factors that could cause such differences include, among other things, the following:
| ● | risks related to clinical trial development and our ability to conduct the clinical trial of our product candidate and the predictive value of our current or planned clinical trials; |
| ● | risks related to our future success being dependent primarily on identification through preclinical studies, clinical studies, regulatory approval for commercialization of a single product candidate; |
| ● | risks related to our license agreement with third parties; |
| ● | uncertainty related to our ability to obtain required regulatory approvals for our proposed products; |
| ● | risks related to the Company’s ability to conduct a clinical trial or submit a future NDA/NDS or IND/CTA (each, as defined herein); |
| ● | risks related to our ability to successfully commercialize future product candidates; |
| ● | risks related to the possibility that our product candidate and potential future product candidates, if any, may have undesirable side effects when used alone or in combination with other drugs; |
| ● | risks related to our ability to enroll subjects in clinical trials; |
| ● | risks that the FDA may not accept data from trials conducted in locations outside the United States; |
| ● | risks related to our ongoing obligations and continued regulatory review; |
| ● | risks related to potential administrative or judicial sanctions; |
| ● | the risk of increased costs associated with prolonged, delayed or terminated clinical trials; |
| ● | risks related to clinical trials being conducted by third parties under collaboration and clinical supply agreements, including combination studies, using the Company’s product candidate, studies which the Company may not control, and ensuing reputational risk related to clinical trial results; |
| ● | the risk that third parties may not carry out their contractual duties or terminate the relationship; |
| ● | risks related to our lack of experience manufacturing product candidates on a large clinical or commercial scale and our lack of manufacturing facility; |
| ● | risks inherent in foreign operations, including related to foreign sourced raw materials, manufacturing or clinical trials; |
| ● | risks related to disruptions in domestic and foreign supply chains that the Company relies on for the production and shipment of raw materials and clinical trial materials; |
| ● | risks related to our failure to obtain regulatory approval in international jurisdictions; |
| ● | risks related to recently enacted and future legislation in the United States and internationally that may increase the difficulty and cost for us to obtain marketing approval of, and commercialize, our product candidate and potential future products, if any, and affect the prices we may obtain; |
| ● | risks related to new legislation, new regulatory requirements, and the continuing efforts of governmental and third-party payors to contain or reduce the costs of healthcare; |
| ● | uncertainty as to our ability to raise additional funding; |
| ● | risks related to our ability to raise additional capital on favorable terms and the impact of dilution from incremental financing; |
| ● | risks of a deemed default on any residual obligations of the agreement providing for the CPRIT Grant and having to reimburse all of the CPRIT Grant, if such deemed default is not waived by CPRIT; |
| ● | risks related to our incurrence of significant losses in every quarter since inception and our anticipation that it will continue to incur significant losses in the future; |
| ● | risks related to our limited operating history; |
| ● | risks related to our reliance on proprietary technology; |
| ● | risks related to our ability to protect our intellectual property rights throughout the world; |
| ● | risks related to claims by third parties asserting that we, or our employees, contractors or consultants have misappropriated their intellectual property, or claiming ownership of what we regard as our intellectual property; |
| ● | risks related to our ability to comply with governmental patent agency requirements in order to maintain patent protection; |
| ● | risks related to computer system failures or security breaches and increasing cyber threats; |
| ● | risks related to business disruptions that could seriously harm our future revenues and financial condition and increase our costs and expenses; |
5
| ● | risks related to our ability to attract and maintain highly qualified personnel; |
| ● | risks relating to the possibility that third-party coverage and reimbursement and health care cost containment initiatives and treatment guidelines may constrain our future revenues; |
| ● | risks related to potential conflicts of interest between us and our directors and officers; |
| ● | risks related to competition from other biotechnology and pharmaceutical companies; |
| ● | risks related to movements in foreign currency exchange rates, interest rates and rate of inflation; |
| ● | risks related to our ability to convince public payors and hospitals to include our product candidate and potential future products, if any, on their approved formulary lists; |
| ● | risks related to our ability to establish an effective sales force and marketing infrastructure, or enter into acceptable third-party sales and marketing or licensing arrangements; |
| ● | risks related to our ability to manage growth; |
| ● | risks related to our ability to achieve or maintain expected levels of market acceptance for our products; |
| ● | risks related to our ability to realize benefits from acquired businesses or products or form strategic alliances in the future; |
| ● | risks related to collaborations with third parties; |
| ● | risks that employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could cause significant liability for us and harm our reputation; |
| ● | risks related to product liability lawsuits; |
| ● | risks related to compulsory licensing and/or generic competition; |
| ● | risks related to the costs of and management time devoted to operating as a public company; |
| ● | risks related to being a smaller reporting company; |
| ● | risks related to the possibility that laws and regulations governing international operations may preclude us from developing, manufacturing and selling certain product candidates outside of the United States and Canada and require us to develop and implement costly compliance programs; |
| ● | risks related to laws that govern fraud and abuse and patients’ rights; |
| ● | risks related to our ability to comply with environmental, health and safety laws and regulations; |
| ● | risks related to us being a “passive foreign investment company”; |
| ● | risks related to United States investors’ ability to effect service of process or enforcement of actions against us; |
| ● | risks related to market price and trading volume volatility; |
| ● | risks related to our dividend policy; |
| ● | risks associated with future sales of our securities; |
| ● | risks related to our ability to maintain an active trading market for the Company's Common Shares; |
| ● | risks related to widespread health concerns, pandemics, or epidemics and other outbreaks of illness; |
| ● | risks related to our ability to implement and maintain effective internal controls; |
| ● | risks related to provisions in our charter documents and Canadian law affecting corporate governance; and |
| ● | risks related to analyst coverage. |
If one or more of these risks or uncertainties or a risk that is not currently known to us, materialize, or if our underlying assumptions prove to be incorrect, actual results may vary materially from those expressed or implied by forward-looking statements. The forward-looking statements represent our expectations, plans, estimates and views as of the date of this document. We do not undertake and specifically decline any obligation to update, republish or revise forward-looking statements to reflect future events or circumstances or to reflect the occurrences of unanticipated events, except as required by law. Investors are cautioned that we cannot guarantee future results, events, levels of activity, performance or achievements and that forward-looking statements are inherently uncertain. Accordingly, investors are cautioned not to put undue reliance on forward-looking statements. We advise you that these cautionary remarks expressly qualify in their entirely all forward-looking statements attributable to us or persons acting on our behalf.
6
We express all amounts in this Quarterly Report on Form 10-Q in U.S. dollars, except where otherwise indicated. References to “$” and “US$” are to U.S. dollars and references to “C$” are to Canadian dollars. Except as otherwise indicated, references in this Quarterly Report on Form 10-Q to “ESSA,” “the Company,” “we,” “us” and “our” refer to ESSA Pharma Inc. and its subsidiary.
7
PART I. FINANCIAL INFORMATION
Item 1. Financial Statements and Supplementary Data
ESSA Pharma Inc.
CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS
(Unaudited)
(Expressed in United States dollars)
FOR THE THREE MONTHS ENDED DECEMBER 31, 2023
8
ESSA PHARMA INC.
CONDENSED CONSOLIDATED INTERIM BALANCE SHEETS
(Unaudited)
(Expressed in United States dollars)
AS OF
December 31, | September 30, | |||||
| 2023 |
| 2023 | |||
ASSETS |
|
|
|
| ||
Current |
|
|
|
| ||
Cash and cash equivalents | $ | |
| $ | | |
Short-term investments (Note 4) | | | ||||
Receivables |
| |
| | ||
Prepaids (Note 5) |
| |
| | ||
| |
| | |||
Deposits | |
| | |||
Operating lease right-of-use assets (Note 7) |
| |
| | ||
Total assets | $ | |
| $ | | |
LIABILITIES AND SHAREHOLDERS’ EQUITY |
|
|
|
| ||
Current |
|
|
|
| ||
Accounts payable and accrued liabilities (Note 6) | $ | |
| $ | | |
Current portion of operating lease liabilities (Note 7) |
| |
| | ||
| |
| | |||
Operating lease liabilities (Note 7) |
| |
| | ||
Total liabilities |
| |
| | ||
Shareholders’ equity |
|
|
|
| ||
Authorized |
|
|
|
| ||
Unlimited common shares, without par value |
|
|
|
| ||
Unlimited preferred shares, without par value | ||||||
Common shares |
| |
| | ||
Additional paid-in capital (Note 8) |
| |
| | ||
Accumulated other comprehensive loss |
| ( |
| ( | ||
Accumulated deficit |
| ( |
| ( | ||
| |
| | |||
Total liabilities and shareholders’ equity | $ | |
| $ | | |
Nature of operations (Note 1)
The accompanying notes are an integral part of these condensed consolidated interim financial statements.
9
ESSA PHARMA INC.
CONDENSED CONSOLIDATED INTERIM STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(Unaudited)
(Expressed in United States dollars)
FOR THE THREE MONTHS ENDED DECEMBER 31,
For the three months ended | ||||||
December 31, | ||||||
| 2023 |
| 2022 | |||
OPERATING EXPENSES |
|
|
|
| ||
Research and development | $ | |
| $ | | |
Financing costs |
| — |
| | ||
General and administration |
| |
| | ||
Total operating expenses |
| ( |
| ( | ||
Foreign exchange |
| ( |
| | ||
Investment and other income |
| |
| | ||
Net loss for the period | ( | ( | ||||
OTHER COMPREHENSIVE INCOME | ||||||
Unrealized gain on short-term investments (Note 4) |
| |
| | ||
Loss and comprehensive loss for the period | $ | ( |
| $ | ( | |
Basic and diluted loss per common share | $ | ( |
| $ | ( | |
Weighted average number of common shares outstanding |
| |
| | ||
The accompanying notes are an integral part of these condensed consolidated interim financial statements.
10
ESSA PHARMA INC.
CONDENSED CONSOLIDATED INTERIM STATEMENTS OF CASH FLOWS
(Unaudited)
(Expressed in United States dollars)
FOR THE THREE MONTHS ENDED DECEMBER 31,
| 2023 |
| 2022 | |||
CASH FLOWS FROM OPERATING ACTIVITIES |
|
|
|
| ||
Net loss for the period | $ | ( |
| $ | ( | |
Items not affecting cash and cash equivalents: |
|
|
|
| ||
Amortization of right-of-use asset |
| |
| | ||
Accretion of premiums/discounts on short-term investments, net | ( |
| ( | |||
Accretion of lease liability |
| |
| | ||
Accrued Investment income | ( | ( | ||||
Unrealized foreign exchange |
| ( |
| ( | ||
Unrealized loss on short-term investments | ||||||
Share-based payments |
| |
| | ||
Changes in non-cash working capital items: |
|
|
|
| ||
Receivables |
| ( |
| ( | ||
Prepaids |
| ( |
| | ||
Accounts payable and accrued liabilities |
| |
| | ||
Operating lease liabilities | ( | | ||||
Net cash used in operating activities |
| ( |
| ( | ||
CASH FLOWS FROM INVESTING ACTIVITIES |
|
|
|
| ||
Purchase of short-term investments | ( | ( | ||||
Proceeds from short-term investments sold | | | ||||
Net cash provided by (used in) investing activities |
| |
| ( | ||
CASH FLOWS FROM FINANCING ACTIVITIES |
|
|
|
| ||
Options exercised | | | ||||
Shares purchased through employee share purchase plan | | | ||||
Lease payments |
| |
| ( | ||
Net cash provided by financing activities |
| |
| | ||
Effect of foreign exchange on cash and cash equivalents |
| |
| ( | ||
Change in cash and cash equivalents for the period |
| |
| ( | ||
Cash and cash equivalents, beginning of period |
| |
| | ||
Cash and cash equivalents, end of period | $ | |
| $ | | |
Supplemental disclosure of non-cash investing and finance items: | ||||||
Leased assets obtained in exchange for operating lease liabilities | $ | | $ | | ||
The accompanying notes are an integral part of these condensed consolidated interim financial statements.
11
ESSA PHARMA INC.
CONDENSED CONSOLIDATED INTERIM STATEMENT OF CHANGES IN SHAREHOLDERS’ EQUITY
(Unaudited)
(Expressed in United States dollars)
FOR THE THREE MONTHS ENDED DECEMBER 31, 2023 AND 2022
Accumulated | |||||||||||||||||
Additional | other | ||||||||||||||||
Number | Common | paid-in | comprehensive |
| |||||||||||||
| of shares |
| shares |
| capital |
| loss |
| Deficit |
| Total | ||||||
Balance, September 30, 2022 |
| | $ | | $ | | $ | ( | $ | ( |
| $ | | ||||
Shares issued through employee share purchase plan | | | ( | — | — |
| | ||||||||||
Share-based payments |
| — | — | | — | — |
| | |||||||||
Loss and comprehensive loss for the period |
| — | — | — | | ( |
| ( | |||||||||
Balance, December 31, 2022 |
| | $ | | $ | | $ | ( | $ | ( |
| $ | | ||||
Accumulated | |||||||||||||||||
Additional | other | ||||||||||||||||
Number | Common | paid-in | comprehensive |
| |||||||||||||
of shares |
| shares |
| capital |
| loss |
| Deficit |
| Total | |||||||
Balance, September 30, 2023 |
| | $ | | $ | | $ | ( | $ | ( |
| $ | | ||||
Options exercised | | | ( | — | — |
| | ||||||||||
Shares issued through employee share purchase plan | | | ( | — | — |
| | ||||||||||
Share-based payments |
| — | — | | — | — |
| | |||||||||
Loss and comprehensive loss for the period |
| — | — | — | | ( |
| ( | |||||||||
Balance, December 31, 2023 |
| | $ | | $ | | $ | ( | $ | ( |
| $ | | ||||
The accompanying notes are an integral part of these condensed consolidated interim financial statements.
12
ESSA PHARMA INC.
NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS
(Unaudited)
(Expressed in United States dollars)
FOR THE THREE MONTHS ENDED DECEMBER 31, 2023
1. NATURE OF OPERATIONS
Nature of Operations
The Company was incorporated under the laws of the Province of British Columbia on January 6, 2009. The Company’s head office address is Suite 720 – 999 West Broadway, Vancouver, BC, V5Z 1K5. The registered and records office address is the Suite 3500, The Stack, 1133 Melville Street, Vancouver, BC, V6E 4E5. The Company is listed on the Nasdaq Capital Market (“Nasdaq”) under the symbol “EPIX”.
The Company is focused on the development of small molecule drugs for the treatment of prostate cancer. The Company has acquired a license to certain patents (“NTD”) which were the joint property of the British Columbia Cancer Agency and the University of British Columbia. As of December 31, 2023, no products are in commercial production or use.
2. BASIS OF PRESENTATION
Basis of Presentation
These accompanying unaudited condensed consolidated interim financial statements, including comparatives, have been prepared in accordance with United States’ Generally Accepted Accounting Principles (“U.S. GAAP”) and pursuant to the rules and regulations of the United States Securities and Exchange Commission (“SEC”) for interim financial information. Accordingly, these condensed consolidated interim financial statements do not include all of the information and footnotes required for complete consolidated financial statements and should be read in conjunction with the audited consolidated financial statements and notes for the year ended September 30, 2023 and included in the Company’s 2023 Annual Report on Form 10-K filed with the SEC and with the securities commissions in British Columbia, Alberta and Ontario on December 12, 2023.
These unaudited condensed consolidated interim financial statements reflect all adjustments, consisting of normal recurring adjustments, which, in the opinion of management, are necessary for a fair presentation of results for the interim periods presented. The results of operations for the three months ended December 31, 2023 and 2022 are not necessarily indicative of results that can be expected for a full year. These unaudited condensed consolidated interim financial statements follow the same significant accounting policies as those described in the notes to the audited consolidated financial statements of the Company included in the Company’s 2023 Annual Report on Form 10-K for the year ended September 30, 2023, with the exception of any policies described in Note 3. Certain prior period amounts in the unaudited condensed consolidated interim statements of cash flows have been reclassified to conform to the current period presentation.
These accompanying unaudited condensed consolidated interim financial statements include the accounts of the Company and its wholly owned subsidiaries. Inter-company transactions, balances and unrealized gains or losses on transactions are eliminated upon consolidation.
The accompanying condensed consolidated interim financial statements have been prepared on a historical cost basis except for certain financial assets measured at fair value.
All amounts expressed in these accompanying condensed consolidated interim financial statements and the accompanying notes are expressed in United States dollars, except per share data and where otherwise indicated. References to “$” are to United States dollars and references to “C$” are to Canadian dollars.
13
Use of Estimates
The preparation of the accompanying condensed consolidated interim financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions about future events that affect the reported amounts of assets, liabilities, expenses, contingent assets and contingent liabilities as of the end of, or during, the reporting period. Actual results could significantly differ from those estimates. Significant areas requiring management to make estimates include the valuation of equity instruments issued for services. Further details of the nature of these assumptions and conditions may be found in the relevant notes to these condensed consolidated interim financial statements.
The effect of a change in an accounting estimate is recognized prospectively by including it in comprehensive income in the period of the change, if the change affects that period only, or in the period of the change and future periods, if the change affects both. Estimates and assumptions are reviewed quarterly.
3. RECENT ACCOUNTING PRONOUNCEMENTS
Recent accounting pronouncements not yet adopted
Management does not believe that any recently issued, but not yet effective, accounting standards, if currently adopted, would have a material effect on the Company’s condensed consolidated interim financial statements.
Recent accounting pronouncements issued by the FASB, including its Emerging Issues Task Force, the American Institute of Certified Public Accountants, and the Securities and Exchange Commission did not or are not believed by management to have a material impact on the Company’s present or future consolidated financial statement presentation or disclosures.
14
4. SHORT-TERM INVESTMENTS
Short-term investments consist of guaranteed investment certificates (“GICs”) held at financial institutions purchased in accordance with the Company’s treasury policy. These GICs and term deposits bear interest at
Short-term investments also consist of U.S. treasury securities, corporate debt securities and commercial paper. The Company has classified these investments as available-for-sale, as the sale of such investments may be required prior to maturity to implement management strategies, and therefore has classified all investment securities as current assets. Those investments with maturity dates of three months or less at the date of purchase are presented as cash equivalents in the accompanying balance sheets. Short-term investments are carried at fair value with the unrealized gains and losses included in accumulated other comprehensive loss as a component of shareholders’ equity (deficit) until realized. Any premium or discount arising at purchase is amortized or accreted to investment income as an adjustment to yield using the straight-line method over the life of the instrument. The Company records an allowance for credit losses when unrealized losses are due to credit-related factors. Realized gains and losses are calculated using the specific identification method and recorded as investment income.
As of December 31, 2023 | |||||||||||||||
Amortized | Unrealized | Estimated | |||||||||||||
| Cost |
| Gains |
| Losses |
| Investment Income |
| Fair Value | ||||||
U.S. Treasury securities | $ | |
| $ | — |
| $ | ( |
| $ | |
| $ | | |
GICs and Term deposits | | — | — | | | ||||||||||
Corporate debt securities | — | — | — | — | — | ||||||||||
Balance, end of period | $ | |
| $ | — |
| $ | ( |
| $ | |
| $ | | |
As of December 31, 2023, short-term investments have an aggregate fair market value of $
5. PREPAIDS
December 31, | September 30, | |||||
| 2023 |
| 2023 | |||
Prepaid insurance | $ | |
| $ | | |
Prepaid CMC and clinical expenses and deposits |
| |
| | ||
Other deposits and prepaid expenses |
| |
| | ||
Balance, end of period | $ | |
| $ | | |
15
6. ACCOUNTS PAYABLE AND ACCRUED LIABILITIES
December 31, | September 30, | |||||
| 2023 |
| 2023 | |||
Accounts payable | $ | |
| $ | | |
Accrued expenses |
| |
| | ||
Accrued vacation |
| |
| | ||
Balance, end of period | $ | |
| $ | | |
7. OPERATING LEASE
Operating lease right-of-use assets |
|
| |
Balance, September 30, 2023 | $ | | |
Addition |
| | |
Amortization | ( | ||
Balance, December 31, 2023 | $ | | |
Operating lease liabilities |
|
| |
Balance, September 30, 2023 | $ | | |
Addition | | ||
Cost of operating lease | ( | ||
Balance, December 31, 2023 | $ | | |
Operating lease liabilities with expected life of less than one year |
| $ | |
Operating lease liabilities with expected life greater than one year |
| $ | |
The Company recognizes a right-of-use asset for the right to use the underlying asset for the lease term, and a lease liability, which represents the present value of the Company’s obligation to make payments over the lease term. The present value of the lease payments is calculated using an incremental borrowing rate as the Company’s leases do not provide an implicit interest rate. At December 31, 2023, the Company’s incremental borrowing rate was
As at December 31, 2023, the maturity of the Company’s operating lease liability was as follows:
| Operating lease | ||
Within 1 year | $ | | |
1 to 2 years |
| | |
2 to 3 years | | ||
3 to 4 years | | ||
| |||
Less: | |||
Imputed interest | ( | ||
Operating lease liability | $ | | |
16
8. SHAREHOLDERS’ EQUITY
Authorized
Unlimited common shares, without par value.
Unlimited preferred shares, without par value.
Omnibus Incentive Plan
On February 25, 2021, the Company adopted an omnibus incentive plan (“Omnibus Plan”) consistent with the policies and rules of Nasdaq. Pursuant to the Omnibus Plan, the Company may issue stock options, share appreciation rights, restricted shares, restricted share units and other share-based awards. As of September 30, 2023, the Company has not issued any instruments other than stock options under the Omnibus Plan.
Prior to the adoption of the Omnibus Plan, the Company issued equity compensation pursuant to the Company’s amended and restated stock option plan (the “Legacy Option Plan”), Amended and Restated Restricted Share Unit Plan (the “RSU Plan”) and Employee Stock Purchase Plan. Since the adoption of the Omnibus Plan, no further grants have been made under the Legacy Option Plan or RSU Plan, though existing grants under the Legacy Option Plan will continue in effect in accordance with their terms.
As of December 31, 2023, the Omnibus Plan has a maximum of
Employee Share Purchase Plan
The Company has adopted an Employee Share Purchase Plan (“ESPP”) under which qualifying employees may be granted purchase rights (“Purchase Rights”) to the Company’s common shares at not less of
Eligible employees are able to contribute up to
During the three months ended December 31, 2023, the Company issued
For the three months ended | ||||||
December 31, | ||||||
| 2023 |
| 2022 | |||
Research and development expense | $ | |
| $ | | |
General and administrative |
| |
| | ||
$ | | $ | | |||
17
The Company measures the purchase rights based on their estimated grant date fair value using the Black-Scholes option pricing model and the estimated number of shares that can be purchased. The following weighted average assumptions were used for the valuation of purchase rights:
For the three months ended | |||||
December 31, | |||||
2023 |
| 2022 |
| ||
Risk-free interest rate |
| | % | | % |
Expected life of share purchase rights |
|
|
| ||
Expected annualized volatility |
| | % | | % |
Dividend |
| |
| |
|
Stock options
Pursuant to the Legacy Option Plan and Omnibus Plan, options were previously or may be granted, respectively, with expiry terms of up to
Stock option transactions are summarized as follows:
|
| Weighted | |||
Number | Average | ||||
of Options | Exercise Price* | ||||
| |||||
Balance, September 30, 2023 |
| | $ | | |
Options granted |
| |
| | |
Options exercised |
| ( |
| ( | |
Options expired/forfeited |
| ( |
| ( | |
Balance outstanding, December 31, 2023 |
| | $ | | |
Balance exercisable, December 31, 2023 |
| | $ | | |
| * | Options exercisable in Canadian dollars as of December 31, 2023 are translated at current rates to reflect the current weighted average exercise price in U.S. dollars for all outstanding options. |
18
At December 31, 2023, options were outstanding enabling holders to acquire common shares as follows:
| Weighted average remaining | |||||
Exercise price | Number of options | contractual life (years) | ||||
$ | | |||||
$ | | |||||
$ | | |||||
$ | |
| ||||
$ | |
| ||||
$ | |
| * | |||
$ | |
| ||||
$ | |
| ||||
$ | |
| ||||
$ | |
| ||||
$ | |
| ||||
$ | |
| ||||
$ | |
| ||||
$ | |
| ||||
$ | |
| ||||
$ | |
| ||||
$ | |
| ||||
$ | |
| ||||
$ | |
| ||||
$ | |
| ||||
C$ | |
| ||||
C$ | |
| ||||
|
|
*
Share-based compensation
During the three months ended December 31, 2023, the Company granted a total of
The Company recognized share-based payments expense for options granted and vesting, net of recoveries on cancellations of unvested options, during the period ended December 31, 2023 and 2022 with allocations to its functional expense as follows:
For the three months ended | ||||||
December 31, | ||||||
2023 |
| 2022 | ||||
Research and development expense | $ | |
| $ | | |
General and administrative |
| |
| | ||
$ | |
| $ | | ||
19
The following weighted average assumptions were used for the Black-Scholes option-pricing model valuation of stock options granted:
2023 |
| 2022 | |||
Risk-free interest rate |
| | % | | % |
Expected life of options |
| years | years | ||
Expected annualized volatility |
| | % | | % |
Dividend |
| |
| |
Warrants
Warrant transactions are summarized as follows:
|
| Weighted | |||
Number | Average | ||||
of Warrants | Exercise Price | ||||
| |||||
Balance, September 30, 2023 |
| | $ | | |
Warrants expired |
| ( |
| ( | |
Balance outstanding and exercisable, December 31, 2023 |
| | $ | | |
At December 31, 2023, warrants were outstanding enabling holders to acquire common shares as follows:
Number |
|
| ||||
of Warrants | Exercise Price | Expiry Date | ||||
$ | |
| ||||
|
|
|
| |||
9. RELATED PARTY TRANSACTIONS
Included in accounts payable and accrued liabilities at December 31, 2023 is $
10. SEGMENTED INFORMATION
The Company works in
11. FINANCIAL INSTRUMENTS AND RISK
The Company’s financial instruments consist of cash and cash equivalents, short-term investments, receivables and accounts payable and accrued liabilities. The fair value of cash and cash equivalents, GICs and term deposists included in short-term investments, receivables and accounts payable and accrued liabilities approximates their carrying values due to their short term to maturity. The fair value of U.S. treasury securities, corporate debt securities and commercial paper included in short-term investments and the fair value of the money market funds included in cash equivalents are measured using Level 2 inputs based on standard observable inputs, including reported trades, broker/dealer quotes, and bids and/or offers (Note 4).
20
Fair value estimates of financial instruments are made at a specific point in time, based on relevant information about financial markets and specific financial instruments. As these estimates are subjective in nature, involving uncertainties and matters of judgment, they cannot be determined with precision. Changes in assumptions can significantly affect estimated fair values.
Financial Risk Factors
The Company’s risk exposures and the impact on the Company’s financial instruments are summarized below:
Credit risk
Financial instruments that potentially subject the Company to a significant concentration of credit risk consist primarily of cash and cash equivalents, short-term investments and receivables. The Company limits its exposure to credit loss by placing its cash, to the extent possible in segregated funds with major financial institutions. The Company considers highly liquid investments with a maturity of up to twelve months when purchased to be short-term investments. Short-term investments includes investments that may have maturity dates exceeding one year at the date of purchase; however, the Company may liquidate investment positions prior to maturity to implement management strategies. The Company maintains an investment policy which requires certain minimum investment grades over its investment instruments.
As of December 31, 2023, cash and cash equivalents consisted of cash in Canada and the United States, money market funds in the United States and investments in certain instruments which have a maturity of less than three months at the date of purchase. Balances in cash accounts exceed amounts insured by the Canada Deposit Insurance Corporation for up to C$
Liquidity risk
The Company’s approach to managing liquidity risk is to ensure that it will have sufficient liquidity to meet liabilities when due. As of December 31, 2023, the Company had working capital of $
Market risk
Market risk is the risk of loss that may arise from changes in market factors such as interest rates, and foreign exchange rates.
As of December 31, 2023, the Company has cash and cash equivalents balances and short-term investments which are interest bearing. Interest income is not central to the Company’s capital management strategy and not significant to the Company’s projected operational budget. Interest rate fluctuations are not significant to the Company’s risk assessment.
The Company’s foreign currency risk exposure relates to net monetary assets denominated in Canadian dollars and Euro. The Company maintains its cash and cash equivalents in U.S. dollars and converts on an as needed basis to discharge Canadian denominated expenditures. A 10% change in the foreign exchange rate between the Canadian dollar and U.S. dollar in relation to Canadian dollar held at December 31, 2023 would result in a fluctuation of $
21
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion should be read in conjunction with the attached financial statements and notes thereto included in Part I, Item 1 of this Quarterly Report on Form 10-Q, as well as our audited financial statements and related notes thereto and management’s discussion and analysis of financial condition and results of operations for the fiscal year ended September 30, 2023 included in our Annual Report on Form 10-K filed with the U.S. Securities and Exchange Commission (the “SEC”) on December 12, 2023. This Quarterly Report on Form 10-Q, including the following sections, contains forward-looking statements within the meaning of the U.S. Private Securities Litigation Reform Act of 1995 and “forward-looking information” within the meaning of Canadian securities laws, or collectively, forward-looking statements. These statements are subject to risks and uncertainties that could cause actual results and events to differ materially from those expressed or implied by such forward-looking statements. For a detailed discussion of these risks and uncertainties, see “Risk Factors” in our Annual Report on Form 10-K. We caution the reader not to place undue reliance on these forward-looking statements, which reflect management’s analysis only as of the date of this Quarterly Report on Form 10-Q. We undertake no obligation to update forward-looking statements to reflect events or circumstances occurring after the date of this Quarterly Report on Form 10-Q.
Overview
ESSA is a clinical stage pharmaceutical company, focused on developing novel therapies for the treatment of prostate cancer with a primary focus on patients whose disease is still predominantly driven by the androgen axis. ESSA’s development of proprietary small molecule inhibitors of the N-terminal domain (“NTD”) of the androgen receptor (“AR”) is focused on the treatment of these patients in combination with second-generation antiandrogen drugs such as abiraterone, enzalutamide, apalutamide, and darolutamide. The Company believes its latest series of investigational compounds, including its product candidate masofaniten (formerly known as EPI-7386), have the potential to significantly expand the interval of time in which patients with earlier stage prostate cancer can benefit from anti-hormone-based therapies. Specifically, the compounds are designed to disrupt the androgen receptor AR signaling pathway, the primary pathway that drives prostate cancer growth and prevent AR activation through selective binding to the NTD of the AR. In this respect, the Company’s compounds differ mechanistically from classical non-steroid antiandrogens. These classic antiandrogens interfere either with androgen synthesis (i.e., abiraterone), or with the binding of androgens to the ligand-binding domain (“LBD”), located at the opposite end of the receptor from the NTD (i.e., “lutamides”). A functional NTD is essential for the functionality of the AR; blocking the NTD inhibits AR-driven transcription and therefore androgen-driven biology.
The Company believes that the transcription inhibition mechanism of its preclinical compounds is unique and has the potential advantage of bypassing several of the identified mechanisms of resistance to the antiandrogens currently used in the treatment of castration-resistant prostate cancer (“CRPC”). The Company has been granted by the United States Adopted Names (“USAN”) Council a unique USAN stem “-Aniten” to recognize this new first-in-class mechanistic class. The Company refers to this series of proprietary investigational compounds as the “Aniten” series. In preclinical studies, blocking the NTD has demonstrated the capability to prevent AR-driven gene expression. A previously completed Phase 1 clinical trial of ESSA’s first-generation agent, ralaniten acetate (“EPI-506”) administered to patients with metastatic CRPC (“mCRPC”) refractory to current standard of care therapies demonstrated prostate-specific antigen (“PSA”) declines, a sign of inhibition of AR-driven biology. This inhibition, however, was neither deep nor sustained enough to confer clinical benefit and the Company made the decision to develop a more potent next generation drug which would also have a longer half-life. The Company has done so and is now in clinical trial with this next generation Aniten, masofaniten (EPI-7386), focusing on the treatment of earlier stage, more homogeneously androgen-driven tumors, in combination with one or another of the current latest generation classic antiandrogens.
According to the American Cancer Society, in the United States, prostate cancer is the second most frequently diagnosed cancer among men, behind skin cancer. Approximately one-third of all prostate cancer patients who have been treated for local disease with curative intent will subsequently have rising serum levels of PSA, which is an indication of recurrent disease with or without development of distant metastasis. Patients with recurrent disease as indicated by rising PSA or nodal or bone metastasis usually undergo initial androgen ablation therapy using analogues of luteinizing hormone releasing hormone or surgical castration; this approach is termed “androgen deprivation therapy” (“ADT”). Most of these patients initially respond to ADT; however, many experience a recurrence in tumor growth despite the reduction of
22
testosterone to castrate levels, and at that point are considered to have CRPC. Following diagnosis of CRPC, patients have been generally treated with antiandrogens that block the binding of androgens (enzalutamide) to the AR, or inhibit synthesis of androgens (abiraterone). More recently, significant improvements in progression free survival and overall survival have been achieved by utilizing this latest generation of antiandrogens in combination with ADT earlier in the disease natural history (i.e., metastatic hormone-sensitive prostate cancer (“mHSPC”) and non-metastatic castration-resistant prostate cancer (“nmCRPC”)).
Since the mid-20th century, it has been recognized that the growth of prostate tumors is in large part mediated by an activated AR. Generally, there are three means of activating the AR. First, androgens, such as dihydrotestosterone can activate AR by binding to its LBD. Second, CRPC can be driven by variants of AR that lack an LBD, are constitutively activated, and consequently do not require androgen for activation. A third mechanism, of less certain clinical significance, may involve certain signaling pathways that activate AR independent of androgen activity. Generally, current drugs for the treatment of prostate cancer are directed against the first mechanism by either (i) interfering with the production of androgen, or (ii) preventing androgen from binding to the LBD. Over time, these approaches eventually fail due to mechanisms of resistance which involve the LBD end of the receptor, whether at the DNA (AR amplification or LBD mutations) or RNA level (emergence of AR splice variants). With respect to the development of alternative pathway mechanisms of AR activation, tumors may also become insensitive to antiandrogen activity. Finally, in patients who have been treated for years with various antiandrogen therapies, genomic changes may lead to additional, non-AR-related oncogenic drivers, also insensitive to inhibition of AR biology.
The Company believes that through their potential to block androgen-driven gene transcription by using a unique mechanism involving the NTD and thereby bypassing these known mechanisms of resistance to current antiandrogens, the Aniten series of compounds hold the potential to be effective in cases where LBD-based mechanisms of resistance to second generation antiandrogens in otherwise AR-driven disease are operating. The results from both extensive preclinical studies and the initial clinical experience support the Company’s belief. In preclinical studies, the Aniten series of compounds has been observed to shrink AR-dependent prostate cancer xenografts, including tumors both sensitive and resistant to the second-generation antiandrogens, such as enzalutamide. Plasma PSA level declines and increases in PSA doubling time as well as declines in circulating tumor DNA and decreases in radiographic tumor measurements were observed in a subset of patients enrolled in the Phase 1 study of masofaniten (EPI-7386) as described below. Importantly with respect to the potential clinical application of NTD inhibition during the natural history of the disease, recent studies by the Company and its collaborators have also suggested the potential advantage for combinations of the Company’s Aniten compounds with currently approved antiandrogens to inhibit AR-driven biology more completely than AR inhibition from either end of the receptor alone. This hypothesis is supported by the clinical trial results obtained in recent years of the superior overall survival obtained in the hormone-sensitive prostate cancer (“HSPC”) setting by combining ADT and the latest generation antiandrogens earlier in the course of the disease versus the administration of these two therapies sequentially.
While the potential importance of the NTD as a drug target has been appreciated for more than two decades, for technical reasons this has been a difficult target for therapeutic agent development. The NTD of the AR is flexible with a high degree of intrinsic disorder making it difficult for use in classic crystal structure-based drug design. The Company is not currently aware of any clinical-stage NTD AR inhibitors that are in development by other drug development companies. The nature of the highly specific binding of the Aniten compounds to the NTD, and the biological consequences of that binding, have been defined in scientific studies. The selectivity of the binding, based on in vivo imaging as well as in vitro studies, has been consistent with the favorable toxicological results observed in preclinical studies of the first-generation EPI-506 and the subsequent safety results observed in the Phase 1 trial of EPI-506. Subsequent to this trial and following the decision to pursue masofaniten (EPI-7386) as the Company’s lead product candidate, the Company completed a series of biophysical and biological studies revealing the interaction and binding of masofaniten (EPI-7386) to the NTD of the AR and presented these findings at several medical conferences in 2021. See “Completed Phase 1 Clinical Study of EPI-506” and “Next generation Aniten molecules” below.
23
The incidence of both metastatic and non-metastatic CRPC continues to rise. Using a dynamic progression model, Scher et al.1 projected a 2020 incidence of 546,955 and prevalence of 3,072,480.The Company believes that the Aniten series of compounds could ultimately hold potential benefit for many of those patients. In its early clinical development, the Company focused on patients who have failed second-generation antiandrogen therapies (i.e., abiraterone and/or lutamides) for the following reasons:
| ● | CRPC treatment remains a prostate cancer market segment with an apparent and significant unmet therapeutic need and is a potentially large market; |
| ● | the Company believes that the unique mechanism of action of its Aniten compounds is well suited to treat those patients who have failed AR LBD focused therapies and whose biological characterization reveals that their tumors are still largely driven by AR biology; and |
| ● | the Company expects that the relatively large number of patients with an apparent unmet therapeutic need in this area will facilitate timely enrollment in its clinical trials. |
The Company believes that the demonstration of favorable safety and tolerability in the initial Aniten Phase 1 clinical trial, together with the compelling preclinical rationale, enabled and emphasized the importance of the study of the combination of masofaniten (EPI-7386) with second-generation antiandrogens.Furthermore, the Company believes that this application of two independent, complementary mechanisms of AR transcription inhibition may result in greater suppression of androgen activity and the delay or prevention of drug resistance. Recent progress in the clinical treatment of prostate cancer has resulted from the earlier utilization of antiandrogens in combination with classic ADT, consistent with the premise that more effective androgen suppression may yield clinical benefit. The Company believes that the introduction of NTD inhibitors, such as masofaniten (EPI-7386), therefore has the potential to improve androgen suppression, delay the emergence of resistance, and result in improved clinical benefit.
Completed Phase 1 Clinical Study of EPI-506
The Company conducted an initial proof-of-concept Phase 1 clinical study utilizing the first-generation Aniten compound, EPI-506 from 2015 to 2017. The objective of the EPI-506 Phase 1 clinical trial was to explore the safety, tolerability, maximum tolerated dose and pharmacokinetics of EPI-506, in addition to anti-tumor activity in asymptomatic or minimally symptomatic patients with mCRPC who were no longer responding to either abiraterone or enzalutamide treatments, or both. Efficacy endpoints, such as PSA reduction, and other disease progression criteria were evaluated. Details relating to the design of the Phase 1/2 clinical trial of EPI-506 are available on the U.S. National Institutes of Health clinical trials website (see https://clinicaltrials.gov under identifier NCT02606123).
The Investigational New Drug (“IND”) application to the FDA for EPI-506 to begin a Phase 1 clinical trial, was allowed in September 2015, with the first clinical patient enrolled in November 2015. The Company’s Clinical Trial Application (“CTA”) submission to Health Canada was subsequently also cleared. Based on allometric scaling, an initial dose level of EPI-506 of 80 mg was determined. However, following the enrollment of the initial cohorts, it became apparent that EPI-506 exposure was much lower in humans than projected. EPI-506 dosing was escalated aggressively to allow patients in the clinical study greater exposure to the drug. The highest dose patients ultimately received was 3600 mg of EPI-506, administered in a single dose or split into two doses daily. The initial data from the Phase 1 clinical trial was presented at the European Society of Medical Oncology meeting in September 2017.
Conducted at five sites in the United States and Canada, the open-label, single-arm, dose-escalation study evaluated the safety, pharmacokinetics, maximum-tolerated dose and anti-tumor activity of EPI-506 in men with end-stage mCRPC who had progressed after prior enzalutamide and/or abiraterone treatment and who may have received one prior line of chemotherapy. Twenty-eight patients were available for analysis, with each patient having received four or more prior therapies for prostate cancer at the time of study entry. Patients self-administered oral doses of EPI-506 ranging from 80 mg to 3600 mg, with a mean drug exposure of 85 days (range of eight to 535 days). Four patients underwent prolonged treatment (with a median of 318 days; and a range of 219 to 535 days at data cut-off), following intra-patient dose
1 Scher HI, Solo K, Valant J, Todd MB, Mehra M (2015) Prevalence of Prostate Cancer Clinical States and Mortality
in the United States: Estimates Using a Dynamic Progression Model. PLoS ONE 10(10): e0139440.
doi:10.1371/journal.pone.013944
24
escalation. PSA declines, a measure of potential efficacy, ranging from 4% to 37% were observed in five patients, which occurred predominantly in the higher dose cohorts (≥1280 mg).
EPI-506 was generally well-tolerated with favorable safety results observed across all doses up to 2400 mg. At a dose of 3600 mg, gastrointestinal adverse events (nausea, vomiting and abdominal pain) were observed in two patients: one patient in the once-daily (“QD”) dosing cohort and one patient in the 1800 mg twice-daily dosing cohort, leading to study discontinuation and a dose-limiting toxicity (“DLT”) due to more than 25% of doses being missed in the 28-day safety reporting period. A separate patient in the 3600 mg QD cohort experienced a transient Grade 3 increase in liver enzymes (AST/ALT), which also constituted a DLT, and enrollment was consequently concluded in this cohort.
Although the Company believes that the safety results and possible signs of anti-tumor activity observed at higher dose levels support the concept that inhibiting the AR-NTD may provide a clinical benefit to mCRPC patients, the pharmacokinetic and metabolic studies revealed the limitations of the first generation agent EPI-506. Through its discovery research the Company had concluded that it should be feasible to develop a next generation of NTD inhibitor which would demonstrate greater potency, reduced metabolism and other improved pharmaceutical properties. As a result, the Company announced on September 11, 2017 its decision to discontinue the further clinical development of EPI-506 and to implement a corporate restructuring plan to focus research and development resources on its next-generation Anitens targeting the AR-NTD. This next generation Aniten compound includes significantly more potent drugs designed to exhibit increased resistance to metabolism and therefore a longer predicted circulating half-life. The Company’s lead product candidate masofaniten (EPI-7386) has demonstrated these and other favorable characteristics in extensive preclinical characterization and clinical studies which the Company has presented in a series of poster presentations at scientific meetings.
Next generation Aniten molecules
The Company’s family of next-generation investigational Aniten compounds incorporate multiple chemical scaffold changes to the first-generation drugs which in preclinical studies retain NTD inhibition of the AR. In addition, they have shown improvement in a range of attributes when compared to the first-generation compound, EPI-506, in preclinical studies. In in vitro assays measuring inhibition of AR transcriptional activity, these product candidates demonstrated 20 times higher potency than EPI-506 or its active metabolite, EPI-002. In addition, the compounds have demonstrated increased metabolic stability in preclinical studies, suggesting the potential for longer half-lives in humans. Lastly, the compounds have demonstrated more favorable pharmaceutical properties relative to EPI-506. The Company believes that these product candidates, if successfully developed and approved, may offer advancements in ease and cost of large-scale manufacture, drug product stability, and suitability for commercialization globally. Of these next-generation Anitens, masofaniten (EPI-7386) was selected for IND filing and a Phase 1 clinical trial.
Our Strategy
In developing possible therapeutics that involve binding to the NTD, the Company’s strategic approach involves:
| ● | combining Aniten compounds with second generation antiandrogens in earlier lines of therapy. The Company, with industry partners, has been conducting clinical trials of combinations of masofaniten (EPI-7386) and second-generation antiandrogens in patients with nmCRPC, mCRPC, mHSPC and neo-adjuvant prostate cancer surgical therapy in earlier lines of treatment; |
| ● | completing the initial Phase 1 clinical development of masofaniten (EPI-7386) as a monotherapy treatment for patients with mCRPC, who are resistant to the current standard of care, to demonstrate the drug’s characteristics as a single agent as completely as possible, with regards to safety, tolerability, and efficacy together with detailed pharmacological and biological studies. The Company’s assessment of this clinical data will determine its clinical development of masofaniten (EPI-7386) as a single agent therapy and also as a combination therapy, whilst considering the impact of such treatment against the size of the patient population whose tumors have progressed and are prevalently driven by the AR pathway despite heavy pre-treatment of the latest generation antiandrogens; and |
25
| ● | continuing preclinical studies including work on other Aniten molecules and other potential applications for AR NTD inhibitors. |
The identification and characteristics of masofaniten (EPI-7386)
The purpose of the next-generation program has been to identify drug candidates with increased potency, reduced metabolic susceptibility and superior pharmaceutical properties compared to ESSA’s first-generation compounds. Structure-activity relation studies conducted on the chemical scaffold of ESSA’s first-generation compounds have resulted in the generation of a new series of compounds that have demonstrated higher potency and predicted longer half-lives. Multiple changes in the chemical scaffold have also been incorporated with the goal of improving ADME (absorption, distribution, metabolism, and excretion) and pharmaceutical properties of the chemical class.
Several next-generation Aniten molecules met prespecified preclinical target product profile goals regarding potency, stability, selectivity and pharmaceutical properties. On March 26, 2019, the Company announced the nomination of masofaniten (EPI-7386) as its lead clinical candidate for the treatment of mCRPC through inhibition of the NTD of the androgen receptor. In preclinical studies, masofaniten (EPI-7386) has displayed activity in vitro in numerous AR-dependent prostate cancer models including models where second-generation antiandrogens are inactive. In addition, masofaniten (EPI-7386) is significantly more potent, metabolically stable and more effective in preclinical studies compared to ESSA’s first-generation compound, EPI-506. Lastly, masofaniten (EPI-7386) has demonstrated a favorable tolerability profile in all animal studies of the compound conducted to date.
From this series of next-generation compounds, masofaniten (EPI-7386) was selected as the lead candidate for the initial clinical development in mCRPC. An IND was submitted to the FDA on March 30, 2020 and was allowed by the FDA on April 30, 2020. A CTA was filed with Health Canada in April 2020 and clearance was subsequently received. Clinical testing of masofaniten (EPI-7386) commenced in July 2020, allowing for accommodations to the planned timeline as a result of the impact of COVID-19 at clinical trial sites (see “Risk Factors - Widespread health concerns, pandemics or epidemics, and other outbreaks of illness may negatively affect the Company’s ability to maintain operations and execute its business plan” in our Annual report on Form 10-K).
26
Advancing masofaniten (EPI-7386) through clinical development
The Company is advancing masofaniten (EPI-7386) through two clinical trials: EPI-7386-CS-001 and EPI-7386-CS-010. The clinical trial of EPI-7386-CS-001 has two arms that represent a monotherapy and combination component of the study schema, as outlined below:

Notes: “mCRPC” means metastatic castration-resistant prostate cancer; “AAP” means abiraterone acetate/prednisone; “mHSPC” means metastatic hormone-sensitive prostate; and “nmCRPC” means non-metastatic castration-resistant prostate cancer
The clinical trial of EPI-7386-CS-010 is a combination trial with enzalutamide with a Phase 1 dose equilibration component and Phase 2 head-to-head comparison component, as outlined in the study schema below:
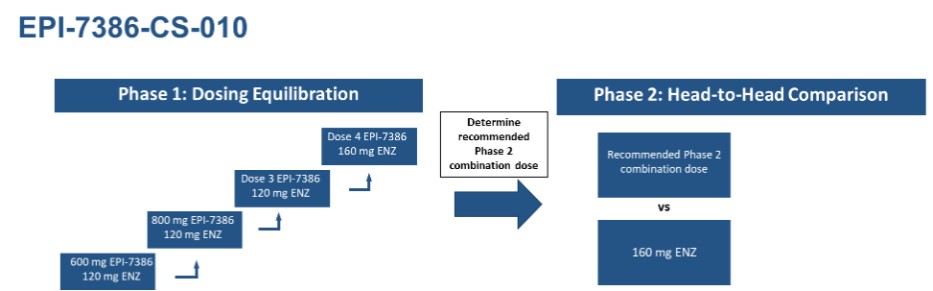
Notes: “ENZ” means enzalutamide
Phase 1 Clinical Trial - EPI-7386-CS-001- Monotherapy Treatment
The Phase 1 clinical trial of masofaniten (EPI-7386) “Oral EPI-7386 in Patients With Castration-Resistant Prostate Cancer (EPI-7386)” is currently actively enrolling nmCRPC patients naïve to second generation antiandrogens and mCRPC patients who are refractory to standard of care treatments at clinical sites in the U.S. and Canada (www.clinicaltrials.gov under identifier NCT04421222). In September 2021, the Company amended the protocol to allow testing in the Phase 1a for a 800 mg/day and 1200 mg/day dosages, administered as either 400 mg or 600 mg dosed twice daily (“BID”), respectively. In addition, the protocol amendment focused further monotherapy development in less heavily pretreated patients with mCRPC, i.e., patients who have received a maximum of three prior approved systemic therapies for mCRPC, including at least one second-generation antiandrogen and excluded patients with visceral metastasis.
27
Part A Monotherapy - Phase 1a – Dose Escalation
The open-label, dose-escalation Phase 1a clinical trial was designed to determine the safety, tolerability, pharmacokinetics, maximum tolerated dose and/or a recommended Phase 2 range of doses in line with the FDA’s Project Optimus, and to access preliminary anti-tumor activity of the drug.
The design of the Phase 1 clinical trial included the standard 3+3 design per dose cohort for the Part 1a dose escalation phase, with subjects receiving a daily oral dose of masofaniten (EPI-7386) once a day QD until there is objective evidence of clinical disease progression, and or occurrence of an unacceptable toxicity.
The dose escalation Part 1a of the study has completed enrollment. Patients enrolled in the Part 1a of the study were selected clinically, on the basis of having progressive mCRPC as exemplified by rising PSA values and/or radiological disease progression despite latest generation antiandrogen treatment. However, all patients were also retrospectively biologically characterized for underlying tumor genomic characteristics, for evidence of AR pathway activation as well as non-AR oncogenic pathways and during the conduct of the trial, for dose-related biological, pharmacological and pharmacodynamic effects.
The protocol amendments filed with the FDA in September 2021 and July 2022 allow for monotherapy development in less heavily pretreated patients (as described above) in whom the androgen receptor pathway is more likely to be the primary driver of tumor growth. The Company’s goal has been to establish, one or more doses/schedules to be tested in the expansion Phase 1b study in alignment with the FDA Project Optimus guidance, based on multiple inputs, including pharmacokinetic and biological observations, in addition to clinical experience. Two dose levels have been advanced to Phase 1b dose expansion testing: 600 mg QD and 600 mg BID.
Part A Monotherapy - Phase 1b – Dose Expansion
The primary objective of Phase 1b is to further evaluate the safety, tolerability, pharmacokinetics, and preliminary anti-tumor activity (as measured by changes in tumor burden measured by imaging and changes in PSA levels over time) of masofaniten (EPI-7386) at 600 mg BID and 600 mg QD in a patient population enrolled under eligibility criteria similar to the one adopted for the Phase 1a with a focus on chemo-naïve mCRPC patients whose diseases have progressed after two lines of treatment including at least one line of second-generation antiandrogens. The 600 mg BID cohort of 12 patients has been fully enrolled and the 600 mg QD cohort of 12 patients is completing enrollment.
Demonstration of the favorable safety and tolerability profile of masofaniten (EPI-7386) in the Phase 1a, together with clinical evidence for its mechanism of action and efficacy, were necessary to enable the study of patient populations with less advanced and less heavily pre-treated prostate cancer. The experience in the initial Phase 1a trial provided evidence for both an antiandrogen biological effect as well as some clinically relevant anti-tumor activity. The biological characterization of these patients also demonstrated favorable safety profiles.
The Company’s preclinical data and other evidence suggest earlier patient populations are more homogeneously AR-driven, and the favorable safety profile demonstrated in the Phase 1a dose escalation trial justified the study of the combination of masofaniten (EPI-7386) with classic antiandrogens. As a result the Company, together with its collaborators, are conducting a series of clinical trials in this regard. The first Phase 1/2 study involves masofaniten (EPI-7386) evaluated in combination with enzalutamide in patients with mCRPC naïve to second generation antiandrogens. Additional clinical trials involving the combination of masofaniten (EPI-7386) with apalutamide, abiraterone acetate, or darolutamide are in various stages of implementation.
Combination studies – developing a new standard of care for the treatment of prostate cancer
An activated AR is required for the growth and survival of most prostate cancer. Unlike current antiandrogen therapies which can only inhibit full-length AR, NTD inhibition of AR-directed biology occurs both in full length AR and splice variant ARs. Therefore, the Company believes that the AR-NTD is an ideal target for next-generation antiandrogen hormone therapy. If ESSA’s masofaniten (EPI-7386) is successful in treating CRPC patients, it is reasonable to expect
28
that such clinical candidate may be effective in treating earlier stage patients. Preclinical studies suggest particular value to the use of Anitens in combination with the currently widely used second-generation antiandrogens. The Company is conducting clinical trials in line with this strategy.
Clinical Trial - EPI-7386-CS-010 – Combination Treatment with Enzalutamide
The Company has been running a Phase 1/2 study of masofaniten (EPI-7386) in combination with enzalutamide compared with enzalutamide alone in patients with mCRPC. Phase 1 of the study is a single-arm dose escalation study of masofaniten (EPI-7386) in combination with a fixed dose of enzalutamide.
A collaboration and supply agreement with Astellas Pharma Inc. (“Astellas”) to evaluate masofaniten (EPI-7386) in combination with Astellas and Pfizer Inc.’s (“Pfizer”) AR inhibitor, enzalutamide, in patients with mCRPC was announced on February 24, 2021. ESSA is paying for and is operationally conducting this trial, with an initial Phase 1 dose equilibration followed by a randomized Phase 2 trial involving a planned 120 patients. The enzalutamide for this trial is supplied by Astellas. The first patient in this Phase 1/2 study was dosed in January 2022 and the safety, tolerability, pharmacokinetics, and initial PSA responses were originally reported in the June 2022 clinical update in poster presentations at the Prostate Cancer Foundation Retreat in October 2022 and the American Society of Clinical Oncology Genitourinary Cancers Symposium in February 2023. The clinical trial is a two part study: a Phase 1 dose equilibration stage followed by a Phase 2 open label randomized study. Masofaniten (EPI-7386) is being evaluated at escalating dose levels including 600 mg QD, 800 mg QD and 600 mg BID in combination with 120 mg and 160 mg enzalutamide in patients with mCRPC naïve to second generation antiandrogens. The Phase 1 part of the study has completed enrollment. The recommended Phase 2 combination doses for the Phase 2 randomized phase is 600 mg BID masofaniten (EPI-7386) with 160 mg enzalutamide (the highest dose levels tested). The Phase 2 study is currently enrolling patients.
Clinical Trial - EPI-7386-CS-001 – Combination Treatments with Abiraterone and with Apalutamide
The first collaboration, with Janssen Research & Development, LLC (“Janssen”), to study in clinical trials the safety and potential benefit of the combination of masofaniten (EPI-7386) with abiraterone acetate with prednisone as well as the combination of masofaniten (EPI-7386) with apalutamide in patients with mCRPC, was announced on January 13, 2021. Under the collaboration agreement with Janssen, Janssen would pay for and conduct a clinical trial with masofaniten (EPI-7386) and in separate cohorts each of their antiandrogens, apalutamide and abiraterone acetate. This combination trial was initiated in March 2022. Enrollment was suspended by Janssen in October 2022 due to operational recruitment challenges. On April 12, 2023, ESSA announced that it had entered into a clinical trial support agreement with Janssen, with ESSA paying for and conducting a study of the combinations, in an earlier patient population, and with Janssen supplying apalutamide and abiraterone acetate.
The Company’s most recent protocol amendment in June 2023, modifies the protocol design of this study by adding combination treatment with second-generation antiandrogens. Specifically, the amended protocol consists of two parts: a Part A Monotherapy study and a Part B Combination study. Part A has two phases: a Phase 1a Dose Escalation and a Phase 1b Dose Expansion, as discussed above, with Part B conducted in two cohorts, Cohort 1 evaluating masofaniten (EPI-7386) in combination with abiraterone acetate/prednisone for patients with mHSPC or mCRPC who receive abiraterone acetate/prednisone as part of their standard of care treatment and Cohort 2, previously the “window of opportunity cohort”, evaluating single agent masofaniten (EPI-7386) for 12 weeks in patients with nmCRPC before apalutamide is added.
Part B Combination - Cohort 1 – Combination with Abiraterone acetate/prednisone
The Company will evaluate the combination of masofaniten (EPI-7386) with abiraterone acetate/prednisone (AAP) in patients with mHSPC or mCRPC. AAP will be provided by Janssen under a clinical trial support agreement as described above. This study is currently enrolling patients.
29
Part B Combination - Cohort 2 – Window of opportunity with clinical endpoints followed by combination with Apalutamide
The primary objective of Cohort 2 is to assess the anti-tumor activity (as measured by changes of PSA over time) of masofaniten (EPI-7386) administered at 600 mg BID for a limited window of time (up to 12 weeks before patients start standard of care therapy) in nmCRPC patients whose disease is unperturbed by previous second-generation antiandrogen therapies or chemotherapy. Following the dosage of masofaniten (EPI-7386) as a single agent after the 12-week window, the Company will evaluate the combination of masofaniten (EPI-7386) with apalutamide. Apalutamide will be provided by Janssen under the same clinical trial support agreement.
In addition to the agreements announced with Pfizer, and Janssen, a third collaboration has been announced with Bayer. Bayer will pay for and conduct a Phase 1/2 clinical trial with masofaniten (EPI-7386) to evaluate masofaniten (EPI-7386) in combination with darolutamide in earlier line mCRPC patients. ESSA will provide masofaniten (EPI-7386) for the combination trial. This clinical trial has not yet been initiated. The Company continues to evaluate potential collaborations that could enhance the value of its prostate cancer program and allow it to leverage the expertise of such strategic collaborators such as those with Janssen, Astellas, and Bayer.
Preclinical Development of Anitens and other indications
The Company continues preclinical work on other emerging potential clinical applications for NTD inhibitors. As part of the preclinical work on Aniten compounds, the Company has also studied NTD degraders and presented data for its first generation of AR ANITAC NTD degraders at the AACR annual meeting on April 10, 2022 in a poster titled “Androgen receptor (AR) N-Terminal Domain degraders can degrade AR full length and AR splice variants in CRPC preclinical models”.
Recent Developments
The Company has presented preclinical and clinical scientific data relative to masofaniten (EPI-7386) in a number of presentations at scientific meetings and other forums.
2024
On January 25-27, 2024, the Company presented updated dose escalation data from its Phase 1/2 study evaluating masofaniten (EPI-7386) in combination with enzalutamide at the 2024 ASCO Genitourinary Cancers Symposium.
In patients evaluable for safety (n=18), masofaniten combined with enzalutamide, continues to be well-tolerated at the dose levels tested through 25 cycles of dosing in some patients. Most frequent adverse events were Grades 1 and 2, related to either AR inhibition or gastrointestinal tract irritation. In Cohort 4, one patient experienced a Grade 3 rash, which was observed immediately following administration of masofaniten combined with enzalutamide and deemed probably related, resulting in the expansion of the cohort from four to seven patients. No additional dose-limiting toxicities were observed, therefore the maximum tolerated dose was not reached. The recommended Phase 2 combination doses were identified as masofaniten 600 mg BID in combination with enzalutamide 160 QD.
In the patients evaluable for efficacy (n=16), rapid, deep and durable reductions in PSA were observed, regardless of previous chemotherapy status, including in patients who received lower than the full dose of enzalutamide (120 mg). Across all dose cohorts, 88% of patients (14 of 16) achieved PSA50, 81% of patients (13 of 16) achieved PSA90, 69% of patients (11 of 16) achieved PSA90 in less than 90 days, and 63% of patients (10 of 16) achieved PSA <0.2ng/mL. While the data for disease PSA progression are still maturing with a current median follow up of 11.1 months, the median time to PSA progression is at 16.6 months.
The randomized, open-label, two arm, Phase 2 dose expansion portion of the study is underway and is designed to evaluate the combination of masofaniten and enzalutamide versus single agent enzalutamide in patients with mCRPC naïve to second generation anti-androgens. The study is currently enrolling at approximately 25 sites in the USA, Canada and Australia. Expansion to European sites is in progress.
30
2023
On October 26-28, 2023, the Company presented an update to the poster previously presented at the European Society of Medical Oncology (ESMO) 2023 Congress for its Phase 1/2 study evaluating masofaniten (EPI-7386) in combination with enzalutamide at the 30th Annual Prostate Cancer Foundation Scientific Retreat.
The data presented were from the four cohorts of patients in the Phase 1 dose escalation portion of the study. The data indicated that masofaniten (EPI-7386) had no effect on enzalutamide exposure, thus allowing the use of full dose per label (160mg) of enzalutamide in combination. It also indicated that enzalutamide reduces masofaniten (EPI-7386) exposure but twice daily dosing of masofaniten (EPI-7386) appears to mitigate the reduction and maintains clinically relevant drug exposures.
In patients evaluable for safety (n=18), masofaniten (EPI-7386) combined with enzalutamide, was well-tolerated at the doses tested through 21 cycles of dosing in some patients. The most frequent adverse events were Grade 1 and 2, related to either AR inhibition or gastrointestinal tract irritation. In Cohort 4, one patient experienced a Grade 3 rash, which was observed immediately following administration of masofaniten (EPI-7386) combined with enzalutamide and deemed probably related.
In the patients evaluable for efficacy (n=16), rapid, deep and durable reductions in PSA were observed, regardless of previous chemotherapy status, including in patients who received lower than the full dose of enzalutamide (120 mg). In the first three cohorts, 90% of patients (9 of 10) achieved PSA50 and PSA90, 80% of patients (8 of 10) achieved PSA90 in less than 90 days, and 70% of patients (7 of 10) achieved PSA <0.2ng/mL. Across all dose cohorts including patients in the recently enrolled Cohort 4, 88% of patients (14 of 16) achieved PSA50, 81% of patients (13 of 16) achieved PSA90, 69% of patients (11 of 16) achieved PSA90 in less than 90 days, and 56% of patients (9 of 16) achieved PSA <0.2ng/mL. The randomized Phase 2 dose expansion portion of the study was reported to be enrolling.
On October 20-24, 2023, the Company presented updated dose escalation data from its Phase 1/2 study evaluating masofaniten (EPI-7386) in combination with enzalutamide at the European Society of Medical Oncology (ESMO) 2023 Congress.
In patients evaluable for safety (n=18), masofaniten combined with enzalutamide, continued to be well-tolerated at the doses tested through 21 cycles of dosing in some patients. Most frequent adverse events were Grade 1 and 2, related to either AR inhibition or gastrointestinal tract irritation. In Cohort 4, one patient experienced a Grade 3 rash, which was observed immediately following administration of masofaniten combined with enzalutamide and deemed probably related.
In the patients evaluable for efficacy (n=16), rapid, deep and durable reductions in PSA were observed, regardless of previous chemotherapy status, including in patients who received lower than the full dose of enzalutamide (120 mg). In the first three cohorts, 90% of patients (9 of 10) achieved PSA50 and PSA90, 80% of patients (8 of 10) achieved PSA90 in less than 90 days, and 70% of patients (7 of 10) achieved PSA <0.2ng/mL. Across all dose cohorts including patients in the recently enrolled cohort four, 88% of patients (14 of 16) achieved PSA50, 69% of patients (11 of 16) achieved PSA90, 63% of patients (10 of 16) achieved PSA90 in less than 90 days, and 56% of patients (9 of 16) achieved PSA <0.2ng/mL. The randomized Phase 2 dose expansion portion of the study was reported to be enrolling.
On October 3, 2023, the Company filed a prospectus supplement to its registration statement on Form S-3, including a base prospectus, with the SEC. Further to this, on November 6, 2023, the Company announced that it had entered into the ATM Sales Agreement with Jefferies LLC, effective as of November 3, 2023. Under the ATM Sales Agreement, ESSA may, within the period that the ATM Sales Agreement is in effect, sell its Common Shares from time to time for up to US$50.0 million in aggregate sales proceeds. No offers or sales of Common Shares will be made in Canada, to anyone known by Jefferies LLC to be a resident of Canada or on or through the facilities of any stock exchange or trading markets in Canada.
On September 18, 2023, the Company announced the initiation of the Phase 2 portion of its Phase 1/2 study evaluating its lead candidate, masofaniten (EPI-7386), in combination with Astellas and Pfizer's enzalutamide in patients with mCRPC naïve to second-generation antiandrogens.
31
On August 31, 2023, the Company announced the establishment of Automatic Securities Disposition Plans for its President and Chief Executive Officer, David R. Parkinson and its Executive Vice President and Chief Operating Officer, Peter Virsik.
On June 6, 2023, the Company appointed Lauren Merendino to its Board of Directors (the “Board”).
On April 12, 2023, the Company announced it had entered into a clinical trial support agreement with Janssen. ESSA will sponsor and conduct a Phase 1 clinical trial evaluating the safety, pharmacokinetics, drug-drug interactions, and preliminary anti-tumor activity of masofaniten (EPI-7386) when administered in combination with either apalutamide or abiraterone acetate plus prednisone. Janssen will supply apalutamide and abiraterone acetate.
On February 16-19, 2023, the Company presented analyses of initial clinical data from two Phase 1 studies of masofaniten (EPI-7386) in patients with mCRPC at the American Society of Clinical Oncology Genitourinary Cancers Symposium. The Company presented an update to the Phase 1 monotherapy study demonstrating that masofaniten (EPI-7386) single agent showed a favorable safety profile and was well-tolerated up to a daily dose of 1200 mg (600 mg BID), achieved target clinical exposures and showed preliminary signals of anti-tumor activity in heavily pretreated mCRPC patients. The second poster presented preliminary results to the Phase 1/2 trial of masofaniten (EPI-7386) in combination with Astellas and Pfizer’s AR inhibitor, enzalutamide. Ten patients had been enrolled in the first three cohorts: three in cohort 1 (600 mg QD masofaniten (EPI-7386) and 120 mg QD enzalutamide), four in cohort 2 (800 mg QD masofaniten (EPI-7386) and 120 mg QD enzalutamide) and three in cohort 3 (600 mg BID masofaniten (EPI-7386) and 120 mg QD enzalutamide). At that time, the DLT period had not cleared for cohort 3. For the first 2 cohorts that cleared the DLT period, no DLTs were observed, and the safety profile was consistent with second-generation antiandrogens (e.g., Grade 1 or 2 AEs of fatigue and hot flushes). Pharmacokinetic results from cohorts 1 and 2 had demonstrated that enzalutamide exposure was minimally impacted by masofaniten (EPI-7386), while, as expected, masofaniten (EPI-7386) exposure was reduced by approximately 60% by enzalutamide (a well established CYP3A4 inducer). The observed masofaniten (EPI-7386) exposures remained in the clinically relevant range suggested by pre-clinical xenograph studies. Five out of six evaluable patients enrolled in the first two cohorts showed a PSA decrease >90% regardless of the patients previous chemotherapy status, and four out of six evaluable patients PSA levels reached < 0.2 ng/mL. All five patients that experienced biochemical responses showed stable disease by imaging.
2022
On October 31, 2022, the Company announced that Janssen Research and Development is suspending enrollment into the Phase 1 clinical study of masofaniten (EPI-7386) with apalutamide and masofaniten (EPI-7386) with abiraterone acetate plus prednisone in mCRPC patients as a result of operational recruitment challenges. Initial clinical activity was observed in some patients, with two of the three patients achieving a PSA reduction of 90% (“PSA90”) within 12 weeks. The Company is in discussions with Janssen to supply abiraterone acetate and apalutamide for an ESSA-sponsored combination study.
On October 26, 2022, the Company announced the presentation of preclinical data for its lead first generation AR ANITen bAsed Chimera (“ANITAC”™) NTD degrader in a poster session at the 34th EORTC-NCI-AACR Annual Symposium on Molecular Targets and Cancer Therapeutics.
On October 26, 2022, the Company announced the presentation of updated clinical data from the first two cohorts of the Phase 1/2 study of ESSA's lead candidate masofaniten (EPI-7386) in combination with enzalutamide at the 2022 Prostate Cancer Foundation Scientific Retreat. In the multicenter, open-label Phase 1/2 dose escalation study, seven mCRPC patients naïve to second generation antiandrogens were enrolled in the first two cohorts, with escalating doses of masofaniten (EPI-7386) and a fixed 120 mg once a day QD dose of enzalutamide. The study permitted one prior line of chemotherapy. Pharmacokinetic results from these first two cohorts demonstrated that enzalutamide exposure was minimally impacted by masofaniten (EPI-7386) while exposures of masofaniten (EPI-7386) were reduced by coadministration with enzalutamide, but remained in the clinically relevant range as suggested by preclinical xenograft studies. The safety of the combination was favorable with a safety profile consistent with second-generation antiandrogens and no dose limiting toxicities were observed. One of the patients in the first cohort discontinued after one cycle of dosing due to a strong CYP3A inducer concomitant medication which lowered exposures to both masofaniten (EPI-7386) and
32
enzalutamide and was therefore not evaluable for efficacy. Anti-tumor activity in the remaining six patients enrolled demonstrated that four of six of these patients achieved a PSA90 by 12 weeks of dosing and five of six patients to date have achieved a PSA90.
On September 13, 2022, the Company appointed Philip Kantoff to its Board.
On June 30, 2022, the Company announced the establishment of Automatic Securities Disposition Plans for its President and Chief Executive Officer, David R. Parkinson and its Executive Vice President and Chief Operating Officer, Peter Virsik.
On June 27, 2022, the Company presented, by conference call and webcast, a clinical update on masofaniten (EPI-7386) monotherapy and combination therapy clinical development. The update on the Phase 1a dose escalation study showed initial data from 36 patients that demonstrated that masofaniten (EPI-7386) was well-tolerated, exhibited a favorable pharmacokinetic profile, and demonstrated initial anti-tumor activity in a heavily pretreated group of patients. The Company believes the favorable safety and tolerability profile, good pharmaceutical characteristics together with both antiandrogen biological and anti-tumor activity support the Company’s decision to move into earlier lines of therapy and study masofaniten (EPI-7386) in combination with second-generation antiandrogens. The update also noted that ctDNA molecular analysis in the heavily pretreated population has provided a detailed profile of genetic alterations, which reveals the biological complexity of late-stage mCRPC patients and also allows for the continued refinement of the population of prostate cancer patients whose tumors are still primarily driven by the androgen receptor, and therefore most likely to respond to an androgen receptor inhibitor.
The update detailed that in the multi-center, open-label Phase 1a dose escalation study, 31 patients received masofaniten (EPI-7386) as oral tablets once a day QD in cohorts with 200 milligram increments from 200 milligrams up to 1000 milligrams. Patients in this QD group were heavily pretreated, with a median of seven lines of prior therapy for prostate cancer and four lines of therapy for mCRPC. Almost 60% of patients had been treated with prior chemotherapy. Patients entered the trial with rapidly progressive disease, as evidenced by a median PSA doubling time of only 2.1 months and a median ctDNA percent of 29%. Almost a third of the patients had lung, liver, or brain metastases, and an overlapping third of patients had overt neuroendocrine differentiation. The ctDNA analysis revealed that tumors in these patients had extensive non-AR associated genomic changes denoting the presence of multiple non-AR oncogenic drivers associated with late-stage prostate cancer. Subsequent to a protocol amendment, the experience was also presented for the five initial patients enrolled in a twice daily dose regimen in 400 mg and 600 mg BID cohorts. The amendment excluded patients who had been treated with more than three prior lines of therapy, excluded patients with visceral metastases, and permitted only one prior line of chemotherapy.
The key safety results from both QD and BID patients, as of June 1, 2022, showed that masofaniten (EPI-7386) was safe and well-tolerated at all dose levels and schedules tested, with no dose-limiting toxicities, treatment related adverse events were limited to Grade 1 or Grade 2, with one Grade 3 occurrence of anemia ultimately deemed unlikely to be treatment related, and that there was no apparent dose dependency in any of the side effects.
Antiandrogen response was assessed by changes in circulating PSA levels, changes in ctDNA levels, and radiographic changes in disease burden measured by both traditional RECIST criteria as well as by total lesion volumetric quantification using the AIQ Solutions platform.
The key response findings in both QD and BID patients, as of June 1, 2022, demonstrated that tumor volume decreased in five patients out of 10 patients who had measurable disease and were on therapy for more than 12 weeks. PSA decrease or PSA stabilization was observed in a clinical subset of patients with no visceral disease, fewer DNA genomic aberrations in non-AR oncogenic pathways, and fewer than 3 lines of therapy. This provides further information to support refining the monotherapy development program patient population. In 17 patients with measurable ctDNA levels at baseline, ctDNA declines were observed in patients harboring AR point mutations, AR gain/amplification and AR truncations, suggesting masofaniten’s (EPI-7386) potential activity against these tumors.
The update also described the planned Phase 1b study, the planned window of opportunity cohort and the status of the combination study of masofaniten (EPI-7386) with enzalutamide. The Phase 1b study will evaluate a patient population
33
of mCRPC similar to the one treated under the Phase 1a BID cohort but with the additional exclusion of prior chemotherapy. Up to 12 patients per each dose/schedule (600 mg QD and either 400 mg or 600 mg BID) will be evaluated to gain additional information about safety, tolerability, exposure and anti-tumor activity of masofaniten (EPI-7386) in a less heavily pretreated patient population.
The update also described the planned window of opportunity cohort as part of the Phase 1b expansion in which a separate group of non-metastatic CRPC will be enrolled into a 12-week study with a clinical endpoint (i.e. PSA changes) to assess the anti-tumor activity of masofaniten (EPI-7386) in a patient population in which the disease is mainly AR-driven and the tumor biology has not been affected by second-generation antiandrogen therapy.
The clinical update also provided the status of the combination studies evaluating masofaniten (EPI-7386) in earlier lines of therapy in Phase 1/2 trials which combine masofaniten (EPI-7386) with approved second-generation antiandrogens. In the Phase 1/2 study being conducted by the Company of masofaniten (EPI-7386) in combination with Astellas Pharma Inc.’s and Pfizer Inc.’s AR inhibitor, enzalutamide, in patients with mCRPC who have not been treated with second-generation antiandrogens, the first cohort had cleared the 28 day DLT period with no safety issues and when reported the trial was currently enrolling the second cohort of patients. The preliminary data from the first cohort in the Phase 1/2 combination trial with enzalutamide suggests that the drugs can be combined safely and based upon clinical and preclinical data predicted to be active. The early data, in addition to preclinical studies, support masofaniten’s (EPI-7386) potential in combination with second-generation antiandrogens to suppress androgen receptor biology and induce a potent anti-tumor response.
The Company also described the anticipated initiation later in 2022 of a Phase 2 investigator-sponsored neoadjuvant study which will evaluate darolutamide compared to masofaniten (EPI-7386) + darolutamide in patients undergoing prostatectomy for high-risk localized prostate cancer.
At the AACR annual meeting on April 10, 2022, in a poster titled “Androgen receptor (AR) N-Terminal Domain degraders can degrade AR full length and AR splice variants in CRPC preclinical models,” the Company presented preclinical data for its first generation of androgen receptor (AR) ANITen bAsed Chimera (ANITAC™) N-terminal domain (NTD) degraders. The preclinical data demonstrated the potential of ESSA’s ANITAC degraders as a new approach to AR pathway inhibition. The intrinsically disordered nature of the NTD region of the AR has meant it has generally been considered undruggable. The preclinical studies have shown that through their unique ability to bind to the NTD of AR, ANITACs have the ability to inhibit NTD-mediated AR transcription while also degrading AR protein including resistant forms of AR which are commonly associated with CRPC. The preclinical results demonstrate that ANITAC degraders utilize the ubiquitin proteasome system and can degrade many forms of AR including full length, mutant and splice variants which are often expressed in CRPC patients. Specifically, the ANITAC degraders show robust potency in inhibiting AR transcriptional activity driven by AR-FL, AR-V7, or AR-V567es. In addition, the orally bioavailable ANITAC degraders exhibit high potency in inhibiting AR-dependent transcription and reducing viability of AR-dependent prostate cancer cells. The Company continues to design and test ANITAC degraders with a focus on improving selectivity.
On January 19, 2022, the Company announced the first patient dosed in the Company-sponsored Phase 1/2 study to evaluate the safety, tolerability and preliminary efficacy of ESSA’s lead product candidate, masofaniten (EPI-7386), a first-in-class N-terminal domain androgen receptor inhibitor, in combination with Astellas and Pfizer Inc.’s ligand-binding domain androgen receptor inhibitor, enzalutamide, in patients with mCRPC. This combination trial investigates the potential clinical benefit of inhibiting the androgen receptor through two independent pathways in the treatment of patients with mCRPC who have not yet received treatment with a second-generation antiandrogen drug. In preclinical models, the combination of masofaniten(EPI-7386) with lutamides by simultaneously targeting both ends of the AR resulted in deeper and broader inhibition of androgen biology.
The Phase 1/2 clinical trial (NCT05075577) is a two part study. Phase 1 evaluates the safety and tolerability of the drug combination to establish the recommended Phase 2 range of doses for masofaniten (EPI-7386) and enzalutamide when dosed in combination. This Phase of the study is expected to enroll up to 30 mCRPC patients who have not yet been treated with second-generation antiandrogen therapies. As described below on June 27, 2022, the results of the initial experience with the first cohort were presented, demonstrating the safety and tolerability of the combination in this first cohort, along with the accompanying pharmacokinetic and PSA reduction information. In Phase 2, single agent enzalutamide is
34
compared to the combination of enzalutamide and masofaniten (EPI-7386) in the same patient population. The goal of Phase 2 is to evaluate the safety, tolerability and anti-tumor activity of masofaniten (EPI-7386) in combination with a fixed dose of enzalutamide compared with enzalutamide as a single agent. This part of the study is expected to enroll 120 mCRPC patients who have not yet been treated with second-generation antiandrogen therapies.
Future Clinical Development Program
Phase 2/3 Clinical Trial Design for treating CRPC patients
Combination Therapy Development
The wealth of preclinical data developed by the Company, as well as the continued emergence of evidence that while earlier stages of prostate cancer are more homogeneously AR pathway driven, the later stages of the disease are much more biologically complex. The Company is conducting the combination of masofaniten (EPI-7386) with current antiandrogens in earlier patients as reflected in several clinical trial collaborations established with leading pharmaceutical companies. Following demonstration of the practical ability to combine masofaniten (EPI-7386) with each of these antiandrogens, further clinical development will require conduct of randomized clinical trials in earlier patient populations (potentially ranging from newly diagnosed through non-metastatic and metastatic hormone-sensitive or pre-latest generation antiandrogen CRPC populations).
Single Agent Development
Depending on the results of the Phase 1 study, a Phase 2 single arm clinical trial evaluating the activity of masofaniten (EPI-7386) as a single agent in a larger group of biologically characterized mCRPC patients might be conducted. While the decision as to whether or not to conduct such a trial has not been formally reached, embarking on the conduct of such a trial will depend on several considerations. The considerations related to the decision to conduct such a trial relate to the company’s ability to identify those patients who, having progressed on one or another late generation antiandrogen, have tumors which are still predominantly AR-driven, as well as the size of that patient population. Our experience during the conduct of the Phase 1 dose escalation trial has revealed that many such patients will have tumors driven predominantly by other oncogenic drivers. Those patients whose tumors remain predominantly AR-driven are the population most likely to benefit clinically from Aniten therapy, and the size of this population will weigh in the decision as to whether or not to pursue a single agent registration strategy.
In order to ultimately obtain full single agent regulatory approval, the Company expects that at least one Phase 3 clinical trial would be required, most likely in patients similar to the population of mCRPC patients who will have been enrolled in the planned Phase 1/2 clinical trial. However, the results of the Phase 1/2 clinical trial may also suggest modification of the initial patient population based on anti-tumor response and biomarker assessment. In a Phase 3 clinical trial, the key end-point is expected to be progression-free survival or overall survival relative to patients receiving the standard of care. It is expected that such a Phase 3 clinical trial would be conducted at numerous sites around the world.
Competition
The competition in the prostate cancer market is very high, many of the companies against which we compete or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Several pharmaceutical therapies already have approved and many new molecules are being tested for their effect in this patient population. In addition, generic forms of Zytiga (abiraterone acetate) are now approved and commercially available in the U.S.
35
Currently approved therapies include:
GENERIC/PROGRAM | BRAND NAME | COMPANY NAME(S) | STAGE | |||
Enzalutamide | Xtandi | Astellas and Pfizer | Marketed | |||
Abiraterone acetate | Zytiga | Johnson & Johnson | Marketed | |||
Abiraterone acetate | Yonsa | Sun Pharma | Marketed | |||
Sipuleucel-T | Provenge | Valeant | Marketed | |||
Docetaxel | n/a | Sanofi and various | Marketed | |||
Cabazitaxel | Jevtana | Sanofi | Marketed | |||
Radium-233 | Xofigo | Bayer | Marketed | |||
Apalutamide (ARN-509) | Erleada | Johnson & Johnson | Marketed | |||
Darolutamide | Nubeqa | Bayer | Marketed | |||
Pembrolizumab | Keytruda | Merck | Marketed | |||
Olaparib | Lynparza | AstraZeneca | Marketed | |||
Rucaparib | Rubraca | Clovis Oncology | Marketed | |||
Vipivotide tetraxetan | Pluvicto | Novartis | Marketed | |||
Niraparib/abiraterone acetate | Akeega | Johnson & Johnson | Marketed | |||
Talazaparib (w/enzalutamide) | Talzenna | Pfizer | Marketed |
In this market, ESSA believes that its competitive position is strong because its product candidate, if successful, involves a mechanistically unique, differentiated approach to prostate cancer treatment involving the therapeutic modality that has been shown to make the biggest difference to the survival of recurrent prostate cancer patients: blocking AR activation. Since Anitens have been shown to directly bind to AR-NTD and prevent AR-mediated transcription, they have the potential to bypass the AR-dependent resistance pathways (discussed above) that may develop as a result of treatment with current hormone-related therapies that target the AR LBD. If successful, ESSA believes this could represent a significant step forward in the treatment of prostate cancer. To ESSA’s knowledge, no other antagonist to the AR-NTD is currently undergoing clinical trials for prostate cancer or any other indication. Other approaches to interfering with AR signaling include potentially complementary strategies to degrade the AR such as that being pursued by Arvinas, Inc.
Patents and Proprietary Rights
License Agreement with UBC and the BCCA
ESSA has in-licensed intellectual property embodied in issued patents, pending patents applications and know-how relating to compounds that modulate AR activity. ESSA refers to these intellectual property rights as the “Licensed IP”.
The Company is party to a license agreement with the British Columbia Cancer Agency and the University of British Columbia (the “Licensors”) dated December 22, 2010, as amended on February 10, 2011, May 27, 2014, and May 25, 2021 (the “License Agreement”), which provides the Company with exclusive world-wide rights to develop and commercialize products based on the Licensed IP.
ESSA paid a minimum annual royalty of C$85,000 in 2017, 2018 and 2019 and must continue to pay a minimum of C$85,000 for each year thereafter. For a First Compound entering clinical development, C$50,000 was paid upon enrollment of a patient in a Phase 2 clinical trial. Additionally, C$900,000 must be paid upon enrollment of a patient in a Phase 3 clinical trial.
The Licensors may terminate the License Agreement upon ESSA’s insolvency, or the License Agreement may be terminated by either party for certain material breaches by the other party. ESSA is required to allocate reasonable time to the development and commercialization of the Licensed IP and to use reasonable efforts to promote, market and sell products covered by the Licensed IP. The terms of the License Agreement required ESSA to issue to the Licensors,
36
1,000,034 pre-Consolidation Common Shares, in lieu of payment of an initial license fee. If ESSA develops products covered by the Licensed IP in the future, it will be required to pay certain development and regulatory milestone payments up to an aggregate of C$2.4 million for the first drug product developed under the license and up to an aggregate of C$510,000 for each subsequent product. ESSA must also pay the Licensors low single-digit royalties based on aggregate worldwide net sales of products covered by the Licensed IP and a percentage of sublicensing revenue in the low teens. The License Agreement will expire on the later of 20 years after the date of the License Agreement or the expiry of the last issued patent included in the Licensed IP.
ESSA’s Intellectual Property Strategy
The Company currently retains all commercial rights for its Aniten series drug portfolio and believes it has developed a strong and defensive intellectual property position for the Aniten structural classes. ESSA has licensed certain patent rights, with respect to some of its compounds that modulate AR activity, from the Licensors. ESSA has the right to acquire ownership of the licensed patents and patent applications upon specified payment to the Licensors, and providing that payments required under the License Agreement continue to be made.
As of December 2023, ESSA owns rights to a patent portfolio that includes 71 issued patents, including 19 issued U.S. patents, that are in force and cover multiple EPI- and Aniten structural classes of compounds with different structural motifs/analogues. Patent applications are also pending in the United States and in contracting states to the Patent Cooperation Treaty for the Aniten next-generation NTD inhibitors, with expected expiration dates between 2036 to 2043.
The patent portfolio includes issued patents that are in force and pending patent applications that cover the masofaniten (EPI-7386) compound, pharmaceutical compositions comprising the masofaniten (EPI-7386) compound, or its methods of use, and are expected to provide protection until the expiration dates, ranging from 2036 to 2043.
Both ESSA and the broader pharmaceutical industry attach significant importance to patents for the protection of new technologies, products and processes. Accordingly, ESSA’s success depends, in part, on its ability to obtain patents or rights thereto, to protect commercial secrets and carry on activities without infringing the rights of third parties. Disputes may arise as to the inventorship of and/or ownership interest in the Company’s or the Licensors’ patents, including for example, former or current employees of the Company or the Licensors pursuing ownership rights of patents owned by or licensed to the Company. The Company may have limited ability to impact any internal disputes between the Licensors and their employees or former employees. See “Risk Factors” in our Annual Report on Form 10-K. Where appropriate, and consistent with management’s objectives, ESSA will continue to seek patents in relation to components or concepts of its technology that it perceives to be important.
Regulatory Environment
The production and manufacture of ESSA’s product candidate and potential future product candidates and its R&D activities are subject to regulation for safety, efficacy, quality and ethics by various governmental authorities around the world. In the United States, drugs and biological products are subject to regulation by the FDA. In Canada, these activities are regulated by the Food and Drugs Act and the rules and regulations thereunder, which are enforced by the TPD. Drug approval laws require registration of manufacturing facilities, carefully controlled research and testing of product candidates, government review and approval of experimental results prior to giving approval to sell drug products. Regulators also require that rigorous and specific standards such as cGMP, good laboratory practices (“GLP”) and current good clinical practices (“GCP”) are followed in the manufacture, testing and clinical development respectively of any drug product. See “Risk Factors” in our Annual Report on Form 10-K.
The process of obtaining regulatory approvals and the corresponding compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval may subject an applicant or sponsor to a variety of administrative or judicial sanctions, including refusal by the FDA to approve pending applications, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters and other types of enforcement letters, product recalls, product seizures, total or partial suspension of production or
37
distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement of profits, or civil or criminal investigations and penalties brought by the FDA and the Department of Justice or other governmental entities.
Drug Products Development Process
An applicant seeking approval to market and distribute a new drug product in the United States must typically undertake the following:
| ● | completion of extensive nonclinical, sometimes referred to as preclinical laboratory tests, and preclinical animal trials in compliance with applicable requirements for the humane use of laboratory animals and formulation studies, including GLPs; |
| ● | submission to the FDA of an IND, which must take effect before human clinical trials may begin; |
| ● | approval by an institutional review board (“IRB”), representing each clinical site before each clinical trial may be initiated; |
| ● | performance of adequate and well-controlled human clinical trials according to the FDA’s regulations commonly referred to as GCP regulations and any additional requirements for the protection of human research subjects and their health information, to establish the safety and efficacy of the proposed drug product for its intended use; |
| ● | preparation and submission to the FDA of a New Drug Application (“NDA”); |
| ● | review of the product by an FDA advisory committee, where appropriate or if applicable; |
| ● | satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities at which the product, or components thereof, are produced to assess compliance with cGMP requirements and to assure that the facilities, methods and controls are adequate to preserve the product’s identity, strength, quality and purity; |
| ● | payment of user fees and securing FDA approval of the NDA; and |
| ● | compliance with any post-approval requirements, including Risk Evaluation and Mitigation Strategies (“REMS”) and post-approval studies required by the FDA. |
Preclinical Studies
Preclinical studies are conducted in vitro and in animals to evaluate pharmacokinetics, metabolism and possible toxic effects to provide evidence of the safety of the product candidate prior to its administration to humans in clinical studies and throughout development. The conduct of preclinical studies is subject to federal regulations and requirements, including GLP regulations. The results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and plans for clinical studies, among other things, are submitted to the FDA as part of an IND. Some long-term preclinical testing, such as animal tests of reproductive adverse events and carcinogenicity, may continue after the IND is submitted.
Initiation of Human Testing
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include, among other things, the requirement that all research subjects provide their informed consent in writing before their participation in any clinical trial. Clinical trials are conducted under written trial protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to a proposed clinical trial and places the trial on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. In Canada, this application is called a CTA. An IND/CTA application must be filed and accepted by the FDA or TPD, as applicable, before human clinical trials may begin. In addition, an IRB representing each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution, and the IRB must conduct continuing review and reapprove the trial at least annually. The IRB must review
38
and approve, among other things, the trial protocol and informed consent information to be provided to trial subjects. An IRB must operate in compliance with FDA regulations.
Two key factors influencing the rate of progression of clinical trials are the rate at which patients can be enrolled to participate in the research program and whether effective treatments are currently available for the disease that the drug is intended to treat. Patient enrollment is largely dependent upon the incidence and severity of the disease, the treatments available and the potential side effects of the drug to be tested and any restrictions for enrollment that may be imposed by regulatory agencies.
Phase 1 Clinical Trials
Phase 1 clinical trials for cancer therapeutics are typically conducted on a small number of patients to evaluate safety, dose limiting toxicities, tolerability, pharmacokinetics and to determine the dose for Phase 2 clinical trials in humans.
Phase 2 Clinical Trials
Phase 2 clinical trials typically involve a larger patient population than Phase 1 clinical trials and are conducted to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of a product candidate for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule.
Phase 3 Clinical Trials
Phase 3 clinical trials typically involve testing an experimental drug on a much larger population of patients suffering from the targeted condition or disease – in ESSA’s case, CRPC. These studies involve testing the experimental drug in an expanded patient population at geographically dispersed test sites (multi-center trials) to establish clinical safety and effectiveness. These trials also generate information from which the overall risk-benefit relationship relating to the drug can be determined.
In most cases FDA requires two adequate and well controlled Phase 3 clinical trials to demonstrate the efficacy of the drug. A single Phase 3 trial with other confirmatory evidence may be sufficient in rare instances where the trial is a large multicenter trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible.
New Drug Application
Assuming successful completion of required clinical testing and other requirements, the results of the preclinical and clinical studies, together with detailed information relating to the product’s chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA, or the TPD as part of a New Drug Submission (“NDS”), requesting approval to market the drug product for one or more indications. The NDS or NDA is then reviewed by the applicable regulatory body for approval to market the drug.
The FDA conducts a preliminary review of an NDA within 60 days of its receipt and informs the sponsor by the 74th day after the FDA’s receipt of the submission to determine whether the application is sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA has agreed to specified performance goals in the review process of NDAs. Most such applications are meant to be reviewed within ten months from the date of filing, and most applications for “priority review” products are meant to be reviewed within nine months of filing. The review process may be extended by the FDA for three additional months to consider new information or clarification provided by the applicant to address an outstanding deficiency identified by the FDA following the original submission.
39
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is or will be manufactured. These pre-approval inspections cover all facilities associated with an NDA submission, including drug component manufacturing (such as Active Pharmaceutical Ingredients), finished drug product manufacturing, and control testing laboratories. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP.
The cost of preparing and submitting an NDA is substantial. The submission of most NDAs is additionally subject to a substantial application user fee, currently exceeding $2,500,000 and the manufacturer or sponsor under an approved new drug application are also subject to significant annual program and establishment user fees. These fees are typically increased annually.
On the basis of the FDA’s evaluation of the NDA and accompanying information, including the results of the inspection of the manufacturing facilities, the FDA may issue an approval letter or a complete response letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or nine months depending on the type of information included. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
If the FDA approves a product, it may limit the approved indications for use for the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess the drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-market studies or surveillance programs. After approval, many types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Post-Approval Requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, significant changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new
40
safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
| ● | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| ● | fines, warning letters or holds on post-approval clinical trials; |
| ● | refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product license approvals; |
| ● | product seizure or detention, or refusal to permit the import or export of products; or |
| ● | injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act, or PDMA, which regulates the distribution of drugs and drug samples at the federal level, and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution.
Orphan Designation and Exclusivity
ESSA may, in the future, seek orphan drug designation for its product candidates. Under the Orphan Drug Act, the FDA may designate a drug product as an “orphan drug” if it is intended to treat a rare disease or condition (generally meaning that it affects fewer than 200,000 individuals in the United States, or more in cases in which there is no reasonable expectation that the cost of developing and making a drug product available in the United States for treatment of the disease or condition will be recovered from sales of the product). A company must request orphan product designation before submitting an NDA. If the request is granted, the FDA will disclose the identity of the therapeutic agent and its potential use. Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product with orphan status receives the first FDA approval for the disease or condition for which it has such designation, the product generally will receive orphan product exclusivity. Orphan product exclusivity means that the FDA may not approve any other applications for the same product for the same indication for seven years, except in certain limited circumstances. Competitors may receive approval of different products for the indication for which the orphan product has exclusivity and may obtain approval for the same product but for a different indication. If a drug or drug product designated as an orphan product ultimately receives marketing approval for an indication broader than what was designated in its orphan product application, it may not be entitled to exclusivity.
41
Selected Quarterly Financial Information
The following table sets forth ESSA’s unaudited consolidated financial data for each of the last eight quarters, prepared in accordance with U.S. GAAP. The Company has not earned any revenues or declared dividends as of December 31, 2023:
For the Quarters Ended | ||||||||||||
December 31, | September 30, | June 30, | March 31, | |||||||||
| 2023 |
| 2023 |
| 2023 |
| 2023 | |||||
Research and development expense |
| $ | 5,376,764 |
| $ | 5,226,231 |
| $ | 6,271,186 |
| $ | 4,480,863 |
General and administration |
| 2,217,868 |
| 1,922,382 |
| 2,639,381 |
| 3,730,692 | ||||
Comprehensive loss |
| (5,944,198) |
| (5,479,603) |
| (7,256,184) |
| (7,119,354) | ||||
Basic and diluted loss per share |
| (0.14) |
| (0.12) |
| (0.17) |
| (0.16) | ||||
Cash and cash equivalents | 35,344,517 | 33,701,912 | 38,466,991 | 44,300,677 | ||||||||
Short-term investments | 106,775,273 | 114,374,489 | 114,001,923 | 112,743,037 | ||||||||
Total assets |
| 144,489,279 |
| 149,122,131 |
| 153,650,198 |
| 158,504,040 | ||||
Long-term liabilities |
| 275,445 |
| - |
| - |
| 19,777 | ||||
Working capital |
| 140,337,994 |
| 145,301,807 |
| 149,882,729 |
| 155,967,246 | ||||
For the Quarters Ended | ||||||||||||
December 31, | September 30, | June 30, | March 31, | |||||||||
| 2022 |
| 2022 |
| 2022 |
| 2022 | |||||
Research and development expense |
| $ | 5,344,250 |
| $ | 4,351,494 |
| $ | 6,394,534 |
| $ | 7,649,459 |
General and administration |
| 2,519,119 |
| 2,769,678 |
| 2,895,542 |
| 3,817,370 | ||||
Comprehensive loss |
| (6,712,455) |
| (6,330,969) |
| (8,829,694) |
| (10,903,335) | ||||
Basic and diluted loss per share |
| (0.15) |
| (0.14) |
| (0.20) |
| (0.25) | ||||
Cash and cash equivalents | 51,220,602 | 57,076,475 | 67,868,096 | 86,235,830 | ||||||||
Short-term investments | 111,850,895 | 110,161,029 | 106,727,807 | 94,782,609 | ||||||||
Total assets |
| 165,003,340 |
| 169,505,295 |
| 175,660,846 |
| 182,609,005 | ||||
Long-term liabilities |
| 48,274 |
| 76,418 |
| 111,273 |
| 145,268 | ||||
Working capital |
| 161,652,689 |
| 166,748,942 |
| 171,150,678 |
| 178,353,354 | ||||
ESSA has never been profitable and has incurred net losses since inception. ESSA’s comprehensive losses were $5,944,198 and $6,712,455 for the three months ended December 31, 2023, and 2022 respectively. ESSA expects to incur losses for the foreseeable future, and it expects these losses to increase as it continues the development of, and seek regulatory approvals for, its product candidate. Because of the numerous risks and uncertainties associated with product development, ESSA is unable to predict the timing or amount of increased expenses or when, or if, it will be able to achieve or maintain profitability.
42
Results of Operations for the Three Months Ended December 31, 2023 and 2022
There was no revenue in any of the periods ended as reported. The Company incurred a comprehensive loss of $5,944,198 for the three months ended December 31, 2023 compared to a comprehensive loss of $6,712,455 for the three months ended December 31, 2022. Variations in ESSA’s expenses and net loss for the periods resulted primarily from the following factors:
Research and Development Expenditures
Research and development expense included the following major expenses by nature:
Three months ended | ||||||
| December 31, 2023 |
| December 31, 2022 | |||
Clinical |
| $ | 1,827,247 |
| $ | 1,379,393 |
Preclinical and data analysis | 1,248,687 | 1,492,268 | ||||
Salaries and benefits |
| 649,702 |
| 576,912 | ||
Share-based payments |
| 526,241 |
| 791,192 | ||
Manufacturing |
| 423,116 |
| 843,151 | ||
Legal patents and license fees |
| 365,118 |
| 105,061 | ||
Other |
| 124,715 |
| 28,628 | ||
Consulting |
| 103,755 |
| 89,573 | ||
Travel and other |
| 71,717 |
| 38,072 | ||
Royalties |
| 36,466 |
| - | ||
Total |
| $ | 5,376,764 |
| $ | 5,344,250 |
The overall research and development expense for the three months ended December 31, 2023 was $5,376,764 compared to $5,344,250 for the three months ended December 31, 2022. R&D expense in the three month periods ended December 31, 2023 and 2022 reflects the ongoing clinical trial of masofaniten (EPI-7386) which commenced in July 2020.
Clinical costs of $1,827,247 (2022 - $1,379,393) were generated in relation to expenditures associated with the Company’s clinical research organizations conducting the Phase 1 clinical trial of masofaniten (EPI-7386).
Preclinical and data analysis costs of $1,248,687 (2022 – $1,492,268) were generated in relation to expenditures for pharmacokinetic data analysis on data from the clinical trial related to the Phase 1 study and work on preclinical pipeline and Anitac compounds.
Salaries and benefits have increased to $649,702 (2022 - $576,912) as a result of an increased number of preclinical and clinical staff.
The share-based payments expense of $526,241 (2022 - $791,192), which is a non-cash expense, relates to the value assigned to stock options and employee share purchase rights granted to key management and consultants of the Company. The expense is recognized in relation to the grant and vesting of these equity instruments, net of expiries and forfeitures, and allocated to research and development, general and administration and financing expenditures relative to the activity of the underlying optionee.
Manufacturing costs of $423,116 (2022 - $843,151) includes amount for cGMP manufacturing of masofaniten (EPI-7386) drug supply to support the ongoing clinical trial as well as costs incurred in formulation and chemistry work around the Company’s pharmaceutical characteristics of masofaniten (EPI-7386).
Legal patents and license fees for the period totaled $365,118 (2022 - $105,061). The Company has adopted a tiered patent strategy to protect its intellectual property as the pharmaceutical industry places significant importance on patents for the protection of new technologies, products and processes. The costs reflect that ongoing investment and the timing of associated maintenance costs. The Company anticipates that there will be continued investment into patent applications.
43
Consulting costs were $103,755 for the three months ended December 31, 2023 (2022 - $89,573) relating to contract project management services.
General and Administration Expenditures
General and administrative expenses include the following major expenses by nature:
Three months ended | ||||||
| December 31, 2023 |
| December 31, 2022 | |||
Salaries and benefits | $ | 765,455 | $ | 699,782 | ||
Professional fees |
| 396,236 |
| 175,612 | ||
Insurance |
| 348,323 |
| 456,486 | ||
Share-based payments |
| 277,177 |
| 772,419 | ||
Investor relations |
| 203,428 |
| 118,032 | ||
Office, insurance, IT and communications |
| 113,943 |
| 134,549 | ||
Director fees |
| 104,000 |
| 95,250 | ||
Travel and other |
| 101,065 |
| 63,740 | ||
Regulatory fees and transfer agent |
| 50,007 |
| 63,664 | ||
Consulting and subcontractor fees |
| 35,042 |
| - | ||
Operating lease liabilities and rent |
| 30,296 |
| 3,366 | ||
Accretion of short-term investments |
| (207,104) | (63,781) | |||
Total |
| $ | 2,217,868 | $ | 2,519,119 | |
The overall general and administration expense was $2,217,868 for the three months ended December 31, 2023 compared to $2,519,119 for the three months ended December 31, 2022 and includes non-cash expense related to share-based payments of $277,177 (2022 - $772,419). This non-cash expense relates to the value assigned to stock options and employees share purchase rights granted to key management and consultants of the Company. The expense is recognized in relation to the grant and vesting of these equity instruments, net of expiries and forfeitures, and allocated to research and development, general and administration and financing expenditures relative to the activity of the underlying optionee.
Salaries and benefits have increased to $765,455 (2022 - $699,782) as a result of an increased number of corporate staff and relative to wage adjustments.
Professional fees of $396,236 (2022 - $175,612) were incurred for legal and accounting services in conjunction with ongoing corporate activities.
Insurance expense of $348,323 (2022 - $456,486) relates to the cost of insurance coverage for directors and officers of the Company.
Director fees of $104,000 (2022 - $95,250) were incurred for remuneration paid to directors for their membership on the Board based on an annual fee structure.
44
Liquidity and Capital Resources
ESSA is a clinical stage company and does not currently generate revenue.
As of December 31, 2023, the Company has working capital of $140,337,994 (September 30, 2023 - $145,301,807). Operational activities during the three months ended December 31, 2023 were financed mainly by proceeds from the financings completed in July 2020 and February 2021. At December 31, 2023, the Company had available cash reserves and short-term investments of $142,119,790 (September 30, 2023 - $148,076,401) to settle current liabilities of $3,530,895 (September 30, 2023 - $3,495,071). At December 31, 2023, the Company believed that it had sufficient capital to satisfy its obligations as they became due and execute its planned expenditures for more than twelve months.
ESSA’s future cash requirements may vary materially from those now expected due to a number of factors, including the costs associated with future preclinical work and to take advantage of strategic opportunities, such as partnering collaborations or mergers and acquisitions activities. In the future, it may be necessary to raise additional funds. These funds may come from sources such as entering into strategic collaboration arrangements, the issuance of shares from treasury, or alternative sources of financing. However, there can be no assurance that ESSA will successfully raise funds to continue its operational activities. See “Risk Factors” in our Annual Report on Form 10-K.
Critical Accounting Policies and Estimates
The Company makes estimates and assumptions about the future that affect the reported amounts of assets and liabilities. Estimates and judgments are continually evaluated based on historical experience and other factors, including expectations of future events that are believed to be reasonable under the circumstances. In the future, actual experience may differ from these estimates and assumptions.
The effect of a change in an accounting estimate is recognized prospectively by including it in comprehensive income in the period of the change, if the change affects that period only, or in the period of the change and future periods, if the change affects both.
The critical accounting policies are those polices that require the most significant judgments and estimates in the preparation of our condensed consolidated interim financial statements. A summary of the critical accounting policies is presented in Note 3 of the annual consolidated financial statements for the year ended September 30, 2023 filed with the SEC and with the securities commissions in British Columbia, Alberta and Ontario on December 12, 2023.
Trend Information
ESSA is a clinical development stage company and does not currently generate revenue. The Company is focused on the development of small molecule drugs for the treatment of prostate cancer. The Company has acquired a license to certain Licensed IP. As of the date of this Quarterly Report on Form 10-Q, no products are in commercial production or use. The Company’s financial success will be dependent upon its ability to continue development of its compounds through preclinical and clinical stages to commercialization.
Off-Balance Sheet Arrangement
ESSA has no material undisclosed off-balance sheet arrangements that have, or are reasonably likely to have, a current or future effect on its results of operations, financial condition, revenues or expenses, liquidity, capital expenditures or capital resources that is material to investors.
Outstanding Share Data
As of February 12, 2024, our authorized share capital consisted of an unlimited number of common shares, each without par value, of which 44,238,147 were issued and outstanding, and an unlimited number of preferred shares, each without par value, none of which were issued and outstanding. As of February 12, 2024, we had 2,920,000 common shares issuable pursuant to 2,920,000 common share purchase warrants pursuant to full cash exercise, 6,705,629 common shares issuable
45
pursuant to 6,705,629 exercisable outstanding stock options, 1,488,312 common shares issuable pursuant to 1,488,312 outstanding options that were not exercisable at that date, and no outstanding restricted stock units.
Safe Harbor
See “Cautionary Note Regarding Forward-Looking Statements” in the introduction to this Quarterly Report.
46
Item 3. Quantitative and Qualitative Disclosures About Market Risk
We are a smaller reporting company as defined by Rule 12b-2 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”) and are not required to provide the information required under this item.
Item 4. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
As of end of the period covered by this Quarterly Report on Form 10Q, our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the design and operating effectiveness of our disclosure controls and procedures as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act. Our disclosure controls and procedures are designed to ensure that information required to be disclosed in the reports that the Company files or submits under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. Any such information is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate, to allow timely decisions regarding required disclosure.
Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on our evaluation of our disclosure controls and procedures as of the end of the period covered by this report, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were, in design and operation, effective at the reasonable assurance level.
Management’s Annual Report on Internal Control over Financial Reporting
Our management, with the participation of our Chief Executive Officer and our Chief Financial Officer, is responsible for establishing and maintaining adequate internal control over our financial reporting, defined in Rule 13a-15(f) and Rule 15d-15(f) of the Exchange Act.
The effectiveness of any system of internal control over financial reporting, including ours, is subject to inherent limitations, including the exercise of judgment in designing, implementing, operating, and evaluating the controls and procedures, and the inability to eliminate misconduct completely. Accordingly, any system of internal control over financial reporting, including ours, no matter how well designed and operated, can only provide reasonable, not absolute, assurances. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate. Management has assessed the effectiveness of our internal control over financial reporting as of December 31, 2023. In making its assessment, management used the criteria set forth in the internal control – integrated framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 COSO framework) to evaluate the effectiveness of our internal control over financial reporting. Based on this evaluation, management has concluded that our internal control over financial reporting was effective as of December 31, 2023.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting during the quarter ended December 31, 2023 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
47
PART II. OTHER INFORMATION
Item 1. Legal Proceedings
From time to time, we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business. As of December 31, 2023, we are not a party to any legal proceedings that, in the opinion of our management, would reasonably be expected to have a material adverse effect on our business, financial condition, operating results or cash flows if determined adversely to us. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Item 1A. Risk Factors
There have been no material changes in our risk factors from those disclosed in our Annual Report on Form 10-K for the year ended September 30, 2023.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds
None.
Item 3. Defaults Upon Senior Securities
None.
Item 4. Mine Safety Disclosures
Not applicable.
Item 5. Other Information
During the three months ended December 31, 2023, no director or officer of the Company adopted or terminated a “Rule 10b5-1 trading arrangement” or “non-Rule 10b5-1 trading arrangement,” as each term is defined in Item 408(a) of Regulation S-K.
48
Item 6. Exhibits
Exhibit No.
3.1 | |
4.1 | |
31.1 | |
31.2 | |
32.1 | |
101.INS | Inline XBRL Instance Document – The instance document does not appear in the interactive data file because its XBRL tags are embedded within the Inline XBRL document. * |
101.SCH | Inline XBRL Taxonomy Extension Schema Document * |
101.CAL | Inline XBRL Taxonomy Extension Calculation Linkbase Document * |
101.LAB | Inline XBRL Taxonomy Extension Label Linkbase Document * |
101.PRE | Inline XBRL Taxonomy Extension Presentation Linkbase Document * |
101.DEF | Inline XBRL Taxonomy Extension Definition Linkbase Document * |
104 | Cover page from the Company’s Annual Report on Form 10-K for the year ended September 30, 2023 formatted in Inline XBRL (included in Exhibit 101). |
* Filed herewith.
49
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Dated: February 13, 2024
ESSA PHARMA INC. | ||
(Registrant) | ||
By: | /S/ DAVID PARKINSON | |
Name: | David Parkinson | |
Title: | Chief Executive Officer | |
By: | /S/ DAVID WOOD | |
Name: | David Wood | |
Title: | Chief Financial Officer | |
50