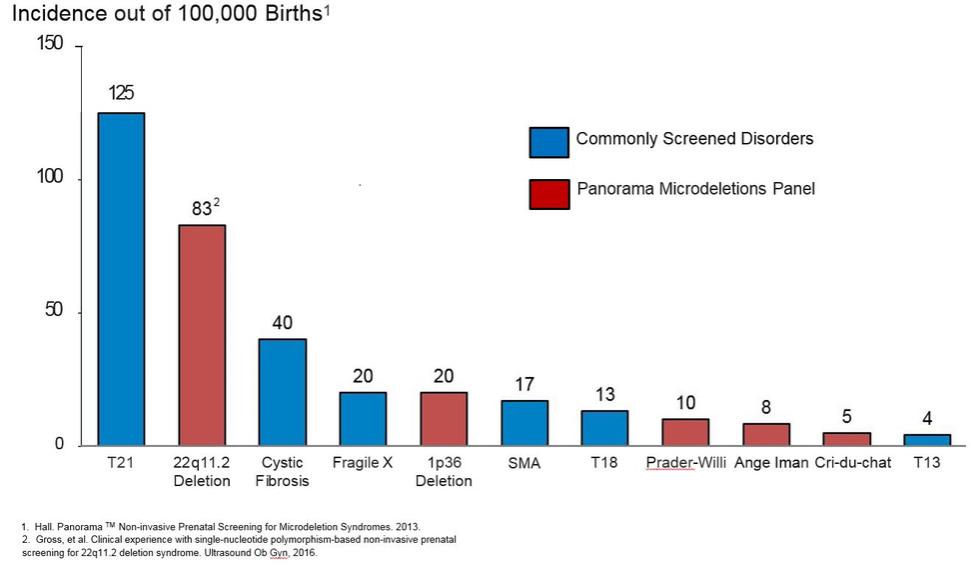
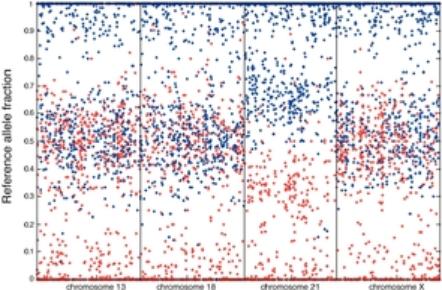
An illustration of the resolution that can be achieved with our mmPCR capability is provided below. The figures display data from our approximately 20,000 primer mmPCR assay, where each primer targets one SNP. On the left, the assay is applied to a large genomic DNA sample from a child. On the right, the assay is applied to a single cell from the same child. Each dot represents data from a particular SNP location on a chromosome. The assay measures the amount of each of the two possible sequences of nucleotides, or alleles, at each SNP. The plots below show the relative proportion of the two alleles, plotted along the vertical axis, for each of the approximately 20,000 SNPs, arranged sequentially along the vertical axis. The two alleles are arbitrarily labeled A and B, and each dot is colored according to the allelic contribution of the mother—red (A) or blue (B). Those SNPs where both copies of DNA in the child contain only the A allele are red and are found at the very top of the plot, and those SNPs where both copies of DNA in the child contain only the B allele are blue and are found at the very bottom of the plot. The SNPs where the fetus contains at least one copy of the A allele and one copy of the B allele are found near the center of the plot. The four vertical bars separated by dotted lines display data from chromosomes 13, 18, 21 and X. For chromosomes 13, 18 and X, the middle band is centered on 0.5; which indicates that for those SNPs, the child has one copy of the A allele and one copy of a B allele (and therefore a relative proportion of 0.5), and, therefore, has the right number of chromosomes—two. In this sample, an additional chromosome is present at chromosome 21, which indicates the presence of trisomy 21. For chromosome 21, the bands centered at 0.33 and 0.66 signal the additional nucleotides contributed by the mother. The band centered at 0.33 represents SNPs where the child has two copies of the B allele and one copy of the A allele, and the band centered at 0.66 represents SNPs where the child has two copies of the A allele and one copy of the B allele. The assay clearly quantifies the difference between single molecules of a particular allele at each SNP. The images demonstrate our ability to derive actionable information from tiny quantities of DNA, as the data from a single cell in the image on the right is nearly as informative as the data from a large genomic sample in the image on the left.
|
|
|

Our bioinformatics technology complements our molecular technology to deliver a risk assessment with high sensitivity and specificity. We use proprietary statistical techniques to combine the measurements of our molecular assays with our internal databases and the vast and growing sources of publicly available genomic information to build highly detailed models of the genome of interest. This process includes the use of a statistical technique known as maximum likelihood estimation, or MLE, which is widely used in other industries, such as in the conversion of a noisy transmitted analog communications signal to a digital format. However, it is computationally complex to leverage this technique to combine genomic information from the patient's sample and information from the databases of the broader scientific community. We have issued U.S. patents claiming methods to do so and pending applications in the United States and abroad. We also maintain trade secrets on our processes and practices. Our proprietary solution using MLE enables us to continuously improve the performance of our existing tests and efficiently develop new ones. As our patient volumes grow, our internal database of samples with genetic mutations and corresponding clinical outcomes further enhances our ability to interpret the clinical significance of complex genetic mutations. As the genomic data from the scientific community, such as from the Cosmic Database and the Cancer Genome Atlas, becomes richer, we can seamlessly integrate new clinical knowledge into our bioinformatics algorithm, driving further improvement in our tests.
7