0001548187--12-312019FYfalseP3YP5YP5YP4YP1YP1YP4Y0M0DP5Y0M0DP5YP5YP5YP4Y00015481872019-01-012019-12-31iso4217:USD00015481872019-06-30xbrli:shares00015481872020-02-2000015481872019-12-3100015481872018-12-31iso4217:USDxbrli:shares0001548187us-gaap:SeriesAPreferredStockMember2018-12-310001548187us-gaap:SeriesAPreferredStockMember2019-12-310001548187us-gaap:SeriesBPreferredStockMember2018-12-310001548187us-gaap:SeriesBPreferredStockMember2019-12-3100015481872018-01-012018-12-310001548187us-gaap:PreferredStockMemberus-gaap:SeriesAPreferredStockMember2017-12-310001548187us-gaap:PreferredStockMemberus-gaap:SeriesBPreferredStockMember2017-12-310001548187us-gaap:CommonStockMember2017-12-310001548187us-gaap:AdditionalPaidInCapitalMember2017-12-310001548187us-gaap:RetainedEarningsMember2017-12-3100015481872017-12-310001548187us-gaap:AdditionalPaidInCapitalMember2018-01-012018-12-310001548187soly:StockIssuanceExcludingPipeDealMemberus-gaap:CommonStockMember2018-01-012018-12-310001548187us-gaap:AdditionalPaidInCapitalMembersoly:StockIssuanceExcludingPipeDealMember2018-01-012018-12-310001548187soly:StockIssuanceExcludingPipeDealMember2018-01-012018-12-310001548187us-gaap:RetainedEarningsMember2018-01-012018-12-310001548187us-gaap:PreferredStockMemberus-gaap:SeriesAPreferredStockMember2018-12-310001548187us-gaap:PreferredStockMemberus-gaap:SeriesBPreferredStockMember2018-12-310001548187us-gaap:CommonStockMember2018-12-310001548187us-gaap:AdditionalPaidInCapitalMember2018-12-310001548187us-gaap:RetainedEarningsMember2018-12-310001548187us-gaap:AdditionalPaidInCapitalMember2019-01-012019-12-310001548187us-gaap:PreferredStockMemberus-gaap:SeriesAPreferredStockMember2019-01-012019-12-310001548187us-gaap:PreferredStockMemberus-gaap:SeriesBPreferredStockMember2019-01-012019-12-310001548187us-gaap:CommonStockMember2019-01-012019-12-310001548187soly:JunePrivateInvestmentInPublicEquityOfferingMemberus-gaap:CommonStockMember2019-01-012019-12-310001548187soly:JunePrivateInvestmentInPublicEquityOfferingMemberus-gaap:AdditionalPaidInCapitalMember2019-01-012019-12-310001548187soly:JunePrivateInvestmentInPublicEquityOfferingMember2019-01-012019-12-310001548187soly:OctoberPrivateInvestmentInPublicEquityOfferingMemberus-gaap:CommonStockMember2019-01-012019-12-310001548187soly:OctoberPrivateInvestmentInPublicEquityOfferingMemberus-gaap:AdditionalPaidInCapitalMember2019-01-012019-12-310001548187soly:OctoberPrivateInvestmentInPublicEquityOfferingMember2019-01-012019-12-310001548187soly:StockIssuanceExcludingPipeDealMemberus-gaap:CommonStockMember2019-01-012019-12-310001548187us-gaap:AdditionalPaidInCapitalMembersoly:StockIssuanceExcludingPipeDealMember2019-01-012019-12-310001548187soly:StockIssuanceExcludingPipeDealMember2019-01-012019-12-310001548187us-gaap:RetainedEarningsMember2019-01-012019-12-310001548187us-gaap:PreferredStockMemberus-gaap:SeriesAPreferredStockMember2019-12-310001548187us-gaap:PreferredStockMemberus-gaap:SeriesBPreferredStockMember2019-12-310001548187us-gaap:CommonStockMember2019-12-310001548187us-gaap:AdditionalPaidInCapitalMember2019-12-310001548187us-gaap:RetainedEarningsMember2019-12-3100015481872019-02-192019-02-190001548187soly:PrivateInvestmentInPublicEquityOfferingMember2019-06-162019-06-160001548187soly:June19Member2018-01-012018-12-310001548187soly:PrivateInvestmentInPublicEquityOfferingMember2019-10-102019-10-100001548187soly:October19Member2018-01-012018-12-31soly:segment00015481872019-02-190001548187soly:ConversionOfPreferredStockIntoCommonStockMember2019-02-192019-02-19xbrli:pure0001548187soly:WarrantsIssuedInPrivateInvestmentInPublicEquityOfferingMember2019-06-1600015481872019-06-162019-06-160001548187soly:PrivateInvestmentInPublicEquityOfferingMember2019-06-160001548187soly:WarrantsIssuedInPrivateInvestmentInPublicEquityOfferingMember2019-06-190001548187soly:WarrantsIssuedInPrivateInvestmentInPublicEquityOfferingMember2019-10-100001548187soly:WarrantsIssuedInPrivateInvestmentInPublicEquityOfferingMember2019-10-102019-10-100001548187us-gaap:SubsequentEventMember2020-02-110001548187srt:MinimumMember2019-01-012019-12-310001548187srt:MaximumMember2019-01-012019-12-310001548187us-gaap:TrademarksMember2019-12-310001548187us-gaap:TrademarksMember2018-12-310001548187us-gaap:PatentsMember2018-01-012018-12-3100015481872019-02-280001548187us-gaap:EmployeeStockOptionMember2019-01-012019-12-310001548187us-gaap:RestrictedStockMember2019-01-012019-12-310001548187us-gaap:ConvertibleDebtSecuritiesMember2019-01-012019-12-310001548187us-gaap:EmployeeStockOptionMember2018-01-012018-12-310001548187soly:ConversionOfPreferredStockIntoCommonStockMember2018-01-012018-12-310001548187us-gaap:RestrictedStockMember2018-01-012018-12-310001548187us-gaap:ConvertibleDebtSecuritiesMember2018-01-012018-12-310001548187us-gaap:ComputerEquipmentMember2019-12-310001548187us-gaap:ComputerEquipmentMember2018-12-310001548187soly:ResearchAndDevelopmentEquipmentMember2019-12-310001548187soly:ResearchAndDevelopmentEquipmentMember2018-12-310001548187us-gaap:EquipmentMember2019-12-310001548187us-gaap:EquipmentMember2018-12-310001548187us-gaap:LeaseholdImprovementsMember2019-12-310001548187us-gaap:LeaseholdImprovementsMember2018-12-310001548187us-gaap:FurnitureAndFixturesMember2019-12-310001548187us-gaap:FurnitureAndFixturesMember2018-12-310001548187us-gaap:ConvertibleDebtMembersoly:FirstNoteMembersoly:ASingleRelatedPartyMembersoly:ConversionOfConvertibleDebtToCommonStockMember2017-01-182017-01-180001548187us-gaap:ConvertibleDebtMembersoly:FirstNoteMember2017-01-182017-01-180001548187us-gaap:ConvertibleDebtMembersoly:FirstNoteMembersoly:ConversionOfConvertibleDebtToCommonStockMember2017-01-182017-01-180001548187us-gaap:ConvertibleDebtMembersoly:FirstNoteMember2017-01-180001548187us-gaap:ConvertibleDebtMembersoly:ASingleRelatedPartyMembersoly:ConversionOfConvertibleDebtToCommonStockMembersoly:FirstAmendmentMember2017-06-190001548187us-gaap:ConvertibleDebtMembersoly:ASingleRelatedPartyMembersoly:ConversionOfConvertibleDebtToCommonStockMembersoly:FirstAmendmentMember2017-06-192017-06-190001548187us-gaap:ConvertibleDebtMembersoly:FirstNoteMembersoly:ASingleRelatedPartyMembersoly:ConversionOfConvertibleDebtToCommonStockMember2017-01-012017-12-310001548187us-gaap:ConvertibleDebtMembersoly:FirstNoteMembersoly:ASingleRelatedPartyMembersoly:ConversionOfConvertibleDebtToCommonStockMember2019-01-012019-12-310001548187us-gaap:ConvertibleDebtMembersoly:ASingleRelatedPartyMembersoly:SecondNoteMembersoly:ConversionOfConvertibleDebtToCommonStockMember2017-11-012017-11-010001548187us-gaap:ConvertibleDebtMembersoly:SecondNoteMember2017-11-012017-11-010001548187us-gaap:ConvertibleDebtMembersoly:FirstNoteMembersoly:ASingleRelatedPartyMembersoly:ConversionOfConvertibleDebtToCommonStockMember2017-11-012017-11-010001548187us-gaap:ConvertibleDebtMembersoly:SecondNoteMember2017-11-010001548187us-gaap:ConvertibleDebtMembersoly:SecondNoteMember2017-11-092017-11-090001548187us-gaap:ConvertibleDebtMembersoly:SecondNoteMember2017-12-012017-12-010001548187us-gaap:ConvertibleDebtMembersoly:SecondNoteMember2017-12-262017-12-260001548187us-gaap:ConvertibleDebtMembersoly:SecondNoteMember2018-01-082018-01-080001548187us-gaap:ConvertibleDebtMembersoly:SecondNoteMember2018-01-252018-01-250001548187us-gaap:ConvertibleDebtMembersoly:SecondNoteMember2018-02-132018-02-130001548187us-gaap:ConvertibleDebtMembersoly:SecondNoteMember2017-11-092018-02-130001548187us-gaap:ConvertibleDebtMembersoly:ASingleRelatedPartyMembersoly:SecondNoteMember2019-12-310001548187us-gaap:ConvertibleDebtMembersoly:ASingleRelatedPartyMembersoly:SecondNoteMembersoly:ConversionOfConvertibleDebtToCommonStockMember2019-01-012019-12-310001548187us-gaap:ConvertibleDebtMembersoly:ThirdNoteMember2018-08-100001548187us-gaap:ConvertibleDebtMembersoly:ThirdNoteMember2018-08-102018-08-100001548187us-gaap:ConvertibleDebtMembersoly:ASingleRelatedPartyMembersoly:ConversionOfConvertibleDebtToCommonStockMembersoly:ThirdNoteMember2019-01-012019-12-310001548187us-gaap:ConvertibleDebtMembersoly:NonRelatedPartyInvestorsMembersoly:ThirdNoteMember2019-12-310001548187us-gaap:ConvertibleDebtMembersoly:ASingleRelatedPartyMembersoly:ThirdNoteMember2019-12-310001548187us-gaap:ConvertibleDebtMembersoly:NonRelatedPartyInvestorsMembersoly:ConversionOfConvertibleDebtToCommonStockMembersoly:ThirdNoteMember2019-01-012019-12-310001548187us-gaap:ConvertibleDebtMembersoly:NonRelatedPartyInvestorsMembersoly:ConversionOfConvertibleDebtToCommonStockMembersoly:ThirdNoteMember2019-08-012019-09-300001548187us-gaap:ConvertibleDebtMembersoly:NonRelatedPartyInvestorsMembersoly:ConversionOfConvertibleDebtToCommonStockMembersoly:ThirdNoteMember2019-12-312019-12-310001548187us-gaap:ConvertibleDebtMembersoly:FourthNoteMember2018-04-170001548187us-gaap:ConvertibleDebtMembersoly:FourthNoteMember2019-12-310001548187us-gaap:ConvertibleDebtMemberus-gaap:InvestorMembersoly:FourthNoteMember2019-12-310001548187us-gaap:ConvertibleDebtMembersoly:NonRelatedPartyInvestorsMembersoly:FourthNoteMember2019-12-310001548187us-gaap:ConvertibleDebtMembersoly:NonRelatedPartyInvestorsMembersoly:FourthNoteMembersoly:ConversionOfConvertibleDebtToCommonStockMember2019-01-012019-12-310001548187soly:WarrantIssuedUnderFourthNoteMember2019-12-310001548187soly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2018-08-070001548187soly:WarrantIssuedWithFifthNoteMember2018-08-070001548187soly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2018-08-072018-08-070001548187soly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2018-08-312018-08-310001548187soly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2018-12-212018-12-210001548187soly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2018-12-210001548187soly:RelatedPartyMembersoly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2018-10-012018-10-310001548187soly:RelatedPartyMembersoly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2019-02-012019-02-280001548187soly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2019-02-152019-02-150001548187soly:WarrantsIssuedInConnectionWithFifthNoteMember2018-01-012018-12-310001548187soly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2018-12-310001548187soly:RelatedPartyMembersoly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2018-12-310001548187soly:NonRelatedPartyInvestorsMembersoly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2018-12-310001548187soly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2019-01-012019-12-310001548187soly:WarrantsIssuedInConnectionWithFifthNoteMember2019-01-012019-01-310001548187soly:WarrantsIssuedInConnectionWithFifthNoteMember2019-02-012019-02-280001548187soly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2019-12-310001548187soly:NonRelatedPartyInvestorsMembersoly:FifthNoteMembersoly:NonconvertiblePromissoryNotesMember2019-12-3100015481872019-06-012019-06-300001548187soly:MdAndersonMember2019-01-012019-12-310001548187soly:MdAndersonMember2018-01-012018-12-310001548187soly:OfficeSpaceLeaseArrangementMember2016-02-010001548187soly:OfficeSpaceLeaseArrangementMember2016-02-012016-02-010001548187soly:ConversionOfPreferredStockToCommonStockMember2019-02-192019-02-190001548187us-gaap:SeriesAPreferredStockMember2019-01-012019-12-310001548187us-gaap:SeriesBPreferredStockMember2019-01-012019-12-310001548187us-gaap:SeriesBPreferredStockMember2019-02-190001548187soly:ConversionOfAccruedDividendsIntoCommonStockMember2019-02-192019-02-190001548187soly:LongTermIncentivePlan2012Member2012-11-300001548187soly:LongTermIncentivePlan2012Member2019-12-310001548187soly:StockPlan2018Member2019-12-310001548187us-gaap:RestrictedStockMember2017-12-310001548187us-gaap:RestrictedStockMember2018-01-012018-12-310001548187us-gaap:RestrictedStockMember2018-12-310001548187us-gaap:RestrictedStockMember2019-01-012019-12-310001548187us-gaap:RestrictedStockMember2019-12-310001548187soly:ThreeConsultantsMemberus-gaap:RestrictedStockMember2019-05-082019-05-080001548187us-gaap:ShareBasedCompensationAwardTrancheOneMemberus-gaap:RestrictedStockMember2019-05-082019-05-080001548187us-gaap:ShareBasedCompensationAwardTrancheTwoMemberus-gaap:RestrictedStockMember2019-05-082019-05-080001548187us-gaap:ShareBasedCompensationAwardTrancheTwoMemberus-gaap:RestrictedStockMember2019-01-012019-12-310001548187us-gaap:EmployeeStockOptionMemberus-gaap:ShareBasedCompensationAwardTrancheOneMember2019-09-192019-09-190001548187us-gaap:RestrictedStockMember2019-05-092019-12-3100015481872017-01-012017-12-310001548187soly:EmployeesMember2018-01-012018-12-310001548187us-gaap:EmployeeStockOptionMember2018-01-012018-12-310001548187us-gaap:ShareBasedCompensationAwardTrancheOneMemberus-gaap:RestrictedStockMember2018-01-012018-12-310001548187us-gaap:EmployeeStockOptionMembersrt:MinimumMember2018-01-012018-12-310001548187us-gaap:EmployeeStockOptionMembersrt:MaximumMember2018-01-012018-12-310001548187us-gaap:EmployeeStockOptionMembersrt:MinimumMember2018-12-3100015481872019-01-012019-01-310001548187soly:CertainIndividualsMember2019-01-012019-01-310001548187us-gaap:EmployeeStockOptionMember2019-01-012019-01-310001548187us-gaap:EmployeeStockOptionMemberus-gaap:ShareBasedCompensationAwardTrancheTwoMember2019-01-012019-01-310001548187us-gaap:EmployeeStockOptionMemberus-gaap:ShareBasedCompensationAwardTrancheOneMember2019-01-012019-01-310001548187us-gaap:EmployeeStockOptionMembersrt:MinimumMember2019-01-310001548187soly:CertainIndividualsMember2019-01-012019-12-310001548187us-gaap:EmployeeStockOptionMember2019-01-012019-12-310001548187us-gaap:EmployeeStockOptionMemberus-gaap:ShareBasedCompensationAwardTrancheOneMember2019-01-012019-12-310001548187us-gaap:EmployeeStockOptionMemberus-gaap:ShareBasedCompensationAwardTrancheTwoMember2019-01-012019-12-310001548187us-gaap:EmployeeStockOptionMembersrt:MinimumMember2019-01-012019-12-310001548187us-gaap:EmployeeStockOptionMembersrt:MaximumMember2019-01-012019-12-310001548187us-gaap:EmployeeStockOptionMembersrt:MinimumMember2019-12-310001548187us-gaap:EmployeeStockOptionMembersrt:MaximumMember2019-12-310001548187us-gaap:EmployeeStockOptionMember2019-12-310001548187soly:WarrantIssuedUnderFourthAndFifthNoteMember2018-12-310001548187soly:FifthNoteMember2019-01-310001548187soly:FifthNoteMember2019-02-280001548187soly:WarrantsIssuedToUnderwritersMember2019-02-190001548187us-gaap:MeasurementInputDiscountRateMembersrt:MinimumMember2019-02-190001548187us-gaap:MeasurementInputDiscountRateMembersrt:MaximumMember2019-02-190001548187us-gaap:MeasurementInputPriceVolatilityMembersrt:MinimumMember2019-02-190001548187us-gaap:MeasurementInputPriceVolatilityMembersrt:MaximumMember2019-02-190001548187us-gaap:MeasurementInputExpectedDividendPaymentMember2019-02-190001548187us-gaap:MeasurementInputSharePriceMember2019-02-190001548187us-gaap:ConvertibleDebtMembersoly:FifthNoteMember2019-02-190001548187us-gaap:MeasurementInputDiscountRateMembersoly:WarrantsIssuedInPrivateInvestmentInPublicEquityOfferingMember2019-06-160001548187us-gaap:MeasurementInputExpectedTermMembersoly:WarrantsIssuedInPrivateInvestmentInPublicEquityOfferingMember2019-06-160001548187us-gaap:MeasurementInputPriceVolatilityMembersoly:WarrantsIssuedInPrivateInvestmentInPublicEquityOfferingMember2019-06-160001548187us-gaap:MeasurementInputExpectedDividendPaymentMembersoly:WarrantsIssuedInPrivateInvestmentInPublicEquityOfferingMember2019-06-160001548187us-gaap:MeasurementInputSharePriceMembersoly:WarrantsIssuedInPrivateInvestmentInPublicEquityOfferingMember2019-06-1600015481872019-10-100001548187us-gaap:MeasurementInputDiscountRateMember2019-10-100001548187us-gaap:MeasurementInputPriceVolatilityMembersrt:MinimumMember2019-10-100001548187us-gaap:MeasurementInputSharePriceMember2019-10-100001548187us-gaap:SubsequentEventMembersoly:CertainIndividualsMember2020-02-042020-02-040001548187us-gaap:EmployeeStockOptionMemberus-gaap:SubsequentEventMember2020-02-042020-02-040001548187us-gaap:EmployeeStockOptionMemberus-gaap:SubsequentEventMember2020-02-04
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C., 20549
FORM 10-K
| | | | | |
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2019
OR
| | | | | |
| ☐ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _________________ to ___________________
Commission File Number: 001-38815
SOLITON, INC.
(Exact Name of Registrant as Specified in its Charter)
| | | | | | | | | | | | | | |
| Delaware | | 3841 | | 36-4729076 |
| (State or Other Jurisdiction of | | (Primary Standard Industrial | | (I.R.S. Employer Identification No.) |
| Incorporation or Organization) | | Classification Code Number) | | |
5304 Ashbrook Drive
Houston, Texas 77081
(Address of Principal Executive Offices) (Zip Code)
Registrant’s Telephone Number, including Area Code:
(844) 705-4866
Securities Registered Pursuant to Section 12(b) of the Act:
| | | | | | | | |
| Title of Each Class | Trading Symbol(s) | Name of Each Exchange on which Registered |
Common Stock, par value $0.001 per share | SOLY | The Nasdaq Stock Market |
Securities Registered Pursuant to Section 12(g) of the Act:
None
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter periods as the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (check one)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Large accelerated filer | ☐ | | Accelerated filer | ☐ | | Non-accelerated filer | ☒ | | Smaller reporting company | ☒ |
| | | | | | | | | Emerging growth company | ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
As of June 30, 2019, the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of the voting stock held by non-affiliates of the registrant was approximately $78,221,596 based on the closing sale price of the common stock as reported on the The Nasdaq Stock Market on June 30, 2019.
The number of shares of the registrant’s common stock outstanding as of February 20, 2020 was 16,932,184.
TABLE OF CONTENTS
References in this Form 10-K to “we”, “us”, ”its”, “our” or the “Company” are to Soliton, Inc. (“Soliton”), as appropriate to the context.
Cautionary Statement About Forward-Looking Statements
We make forward-looking statements under the “Risk Factors,” “Business,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and in other sections of this Form 10-K. In some cases, you can identify these statements by forward-looking words such as “may,” “might,” “should,” “would,” “could,” “expect,” “plan,” “anticipate,” “intend,” “believe,” “estimate,” “predict,” “potential” or “continue,” and the negative of these terms and other comparable terminology. These forward-looking statements, which are subject to known and unknown risks, uncertainties and assumptions about us, may include projections of our future financial performance based on our growth strategies and anticipated trends in our business. These statements are only predictions based on our current expectations and projections about future events. There are important factors that could cause our actual results, level of activity, performance or achievements to differ materially from the results, level of activity, performance or achievements expressed or implied by the forward-looking statements. In particular, you should consider the numerous risks and uncertainties described under “Risk Factors.”
While we believe we have identified material risks, these risks and uncertainties are not exhaustive. Other sections of this Form 10-K describe additional factors that could adversely impact our business and financial performance. Moreover, we operate in a very competitive and rapidly changing environment. New risks and uncertainties emerge from time to time, and it is not possible to predict all risks and uncertainties, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements.
Although we believe the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, level of activity, performance or achievements. Moreover, neither we nor any other person assumes responsibility for the accuracy or completeness of any of these forward-looking statements. You should not rely upon forward-looking statements as predictions of future events. We are under no duty to update any of these forward-looking statements after the date of this Form 10-K to conform our prior statements to actual results or revised expectations, and we do not intend to do so.
Forward-looking statements include, but are not limited to, statements about:
•our ability to obtain additional funding to commercialize our Rapid Acoustic Pulse (“RAP”) for tattoo removal, develop the RAP device for other indications and develop our dermatological technologies;
•the need to obtain regulatory approval, and the timing of such approval, for our Generation 1 RAP device, and the potential to obtain an additional approval when we modify the Generation 1 RAP device to become our Generation 2 device before our commercial launch and to become our Generation 3 device;
•the success of our future clinical trials;
•compliance with obligations under our intellectual property license with The University of Texas M.D. Anderson Cancer Center (“MD Anderson”);
•market acceptance of the RAP device;
•competition from existing products or new products that may emerge;
•potential product liability claims;
•our dependency on third-party manufacturers to supply or manufacture our products;
•our ability to establish or maintain collaborations, licensing or other arrangements;
•our ability and third parties’ abilities to protect intellectual property rights;
•our ability to adequately support future growth;
•our ability to attract and retain key personnel to manage our business effectively;
•risks associated with our identification of material weaknesses in our control over financial reporting;
•natural disasters affecting us, our primary manufacturer or our suppliers;
•our ability to establish relationships with health care professionals and organizations;
•general economic uncertainty that adversely effects spending on cosmetic procedures;
•volatility in the market price of our stock;
•potential dilution to current stockholders from the issuance of equity awards.
We caution you not to place undue reliance on the forward-looking statements, which speak only as of the date of this Form 10-K in the case of forward-looking statements contained in this Form 10-K. Except for ongoing obligations to disclose material information under the federal securities laws, we expressly disclaim any obligation or undertaking to release publicly any updates or revisions to any such statement to reflect any change in our expectations or any change in events, conditions or circumstances on which any such statement is based.
You should not rely upon forward-looking statements as predictions of future events. Our actual results and financial condition may differ materially from those indicated in the forward-looking statements. We qualify all of our forward-looking statements by these cautionary statements. Although we believe that the expectations reflected in the forward looking-statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. Therefore, you should not rely on any of the forward-looking statements. In addition, with respect to all of our forward-looking statements, we claim the protection of the safe harbor for forward-looking statements contained in the Private Securities Litigation Reform Act of 1995.
PART I
Item 1. Business
Overview
We are a medical device company with a novel and proprietary platform technology licensed from The University of Texas MD Anderson Cancer Center ("MD Anderson"). Our Rapid Acoustic Pulse (“RAP”) device uses rapid pulses of designed acoustic shockwaves to disrupt cellular and subcellular structures in the dermis and subcutaneous tissue. The uniqueness of our designed shockwave allows us to target the changes in stiffness between cellular structures and generate a shearing effect that we believe represents a platform technology potentially useful in tattoo removal, cellulite reduction, fibrotic scar treatment and other indications. We believe the high repetition rate, rapid rise and fall of the wave, and significant peak pressure delivered in a non-focused manner make our shockwave significantly different from other available shockwave technologies. Importantly, our technology allows the disruption of targeted structures within the skin without significant pain and without treatment-related downtime.
We received clearance for our RAP device for tattoo removal from the U.S. Food and Drug Administration ("FDA") in May 2019 allowing our device to be used as an accessory to a 1064 nm Q-switched laser for tattoo removal on patients with skin tones on the Fitzpatrick scale between I and III. When used in conjunction with existing lasers for tattoo removal, our technology allows a doctor to treat a patient multiple times in a single office visit and significantly reduces the number of office visits required to remove a tattoo, allowing a dramatic acceleration of the tattoo removal process.
We plan to launch our RAP device for tattoo removal in mid-2020 into select dermatologist offices. We expect to generate revenue from both the initial sale of the device and from the recurring sales of disposable cartridges that are required by the device. We refer to this as our "razor and blade" recurring revenue model. Cartridges are designed to be specific to the intended indication (for example, tattoo cartridges will be different from potential future cellulite cartridges, if approved) and each treatment session would require one or more cartridges. We expect that one tattoo cartridge will facilitate up to five standard laser treatments in a single office visit for the average-sized tattoo (about five square centimeters). Therefore, a patient with an average-sized tattoo that requires three office visits will require the use of three cartridges.
We also have ongoing clinical programs in several indications, which, if successful, will allow us to expand commercialization of our products into additional markets. Importantly, we are undertaking ongoing clinical trials of our RAP device to support an application with the FDA for the treatment of cellulite. As a stand-alone device, we believe our RAP device has the potential to reduce the effects of fibrosis and stimulate beneficial fibroblast behavior. This capability enables the targeting of cellulite and fibrotic (keloid and hypertrophic) scars, as well as smoothing and tightening skin. We also intend to pursue regulatory approval in international markets and we are currently developing a regulatory strategy for these additional markets.
Our Technology
Our RAP device is composed of three parts: a console, a hand piece and a disposable cartridge. The console houses a pulse power system used to provide high voltage power to a pair of electrodes housed within the cartridge. The cartridge is snapped in and out of the hand piece for easy replacement and forms the basis for our planned “razor and blade” recurring revenue model. The proprietary nature of our technology is supported by eight patent families and over 100 patents issued or pending.
Our RAP device uses electrohydraulics to generate designed acoustic shockwaves at a rate of up to 100 per second to effectively target differences in stiffness at the cellular level. The first two indications we are targeting with our RAP technology are tattoo removal and cellulite reduction.
•In tattoo removal, our RAP device is used in conjunction with a laser and disperses both tattoo ink particles and the superficial and dermal vacuoles that are formed when a laser interacts with the ink particles during the use of the laser. Removing these vacuoles allows for subsequent laser treatments in the same treatment session, thereby rapidly accelerating the tattoo removal process.
•In cellulite reduction, our RAP device is used as a stand-alone device that disrupts the stiff, sclerotic septa structures that run through the subcutaneous fat layer causing the dimples and ridges associated with cellulite. Importantly, it
does this without breaking the skin. We call this “acoustic subcision” and the disruption of these structures allows cellulite dimples and ridges to be released and the appearance of the cellulite to be improved. Until now, such disruption of sclerotic septa could only be achieved using surgical procedures that require penetrating the skin and involve significant pain and treatment-related downtime.
Our RAP device is also in clinical development for treating certain fibrotic conditions. We have completed a proof-of-concept study for the treatment of keloid and hypertophic scars and early data suggests that our technology can impact the overactive fibroblasts that generate these scars. This supports our belief that our technology can potentially have an impact on a much broader set of fibrotic conditions. Scientific publications suggest that fibroblasts become over-active when they are located in a stiffened environment and that disrupting the stiff environment may lead to fibroblast apoptosis, ultimately resulting in a resolution of the fibrosis. On this basis, we believe that our technology could have efficacy in a number of fibrotic diseases, including within the extracellular matrix, such as radiation induced fibrosis and capsular contracture, and in other systems of the body such as peripheral artery disease and even Liver Fibrosis. To date, other than the proof-of-concept study for the treatment of keloid and hypertophic scars, we have not begun any substantive pre-clinical work on these fibrotic diseases.
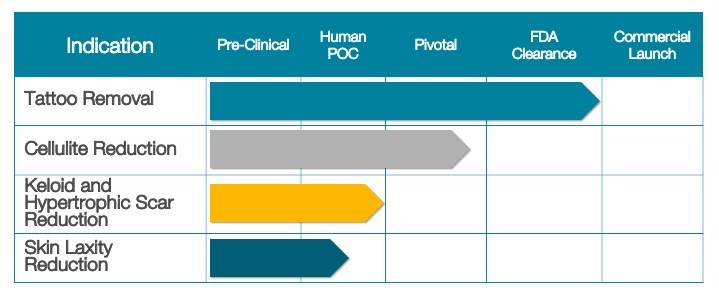
Our Clinical Pipeline
Set forth below is a table presenting the current status of our clinical pipeline:
Our Market Opportunity
Tattoo Removal
Approximately one-third of all adults in the United States have a tattoo. In 2015, we commissioned our own survey of individuals with one or more tattoos in an effort to better understand their interest in, motivations for and concerns about tattoo removal. This survey was designed to be representative of the US population with 95% confidence (+/- 3%) and indicated that 63% of individuals with tattoos were interested in some form of removal. At these rates, an estimated 44 million Americans are interested in tattoo removal. In fact, based upon third party market research, the global tattoo removal market is estimated to be approximately $4 billion by 2023.
The current standard of care for tattoo removal is to use a Q-switched (pulsed) laser to ablate the tattoo ink particles into pieces small enough for the body’s natural processes to remove them. Unfortunately, this current method is highly inefficient, requiring, on average, 10 or more office visits to achieve acceptable results. An independent clinical trial has demonstrated that using our RAP device in conjunction with a Q-switched laser has the potential to achieve removal of an average-sized tattoo in just 2 to 3 office visits. We believe this “Soliton” method can not only dramatically accelerate tattoo removal, but also has the potential to lower removal cost for patients, while increasing profitability to practitioners, and to reduce the potential for unwanted scarring and ghosting (a lingering silhouette image of the tattoo).
Cellulite Reduction
Between 80-90% of women suffer from cellulite. Based on third party market research, the global market for cellulite treatment was estimated to be approximately $2.4 billion in 2018 and is expected to grow to approximately $4 billion by 2025.
This is a significant addressable market, but we believe currently available treatment options are limited. A 2015 review of a variety of studies into the effectiveness of different non-surgical techniques for treating cellulite indicated that either the procedures did not work or the research methodology was flawed. Furthermore, most of these non-surgical techniques offer only a temporary reduction in the appearance of cellulite. The American Academy of Dermatology (AAD) reviewed a number of surgical techniques that may be successful in reducing the appearance of cellulite by cutting the bands of connective tissue under the skin's surface. However, these techniques are often painful and expensive. As a non-invasive technique, if a version of our device is capable of reducing the appearance of cellulite with results that approach those of the surgical techniques, we believe this could become an important new indication for our technology.
Keloid and Hypertrophic Scars
Keloids are a type of raised scar. They typically occur where the skin has healed after an injury. They can grow to be much larger than the original injury that caused the scar. A hypertrophic scar is a cutaneous condition characterized by deposits of excessive amounts of collagen which gives rise to a raised scar, but not to the degree observed with keloids. Like keloids, they form most often at the sites of pimples, body piercings, cuts and burns.
The American Osteopathic College of Dermatology estimates that keloids affect around 10% of people, whereas hypertrophic scars are more common. Keloid scars are more prevalent among populations with darker skin pigmentation. Hypertrophic scars affect men and women from any racial group equally, although people between 10 and 30 years old are more likely to be affected. Based on third party market research, the global market for hypertrophic and keloid scar treatment was estimated to be approximately $4.8 billion in 2017 and is expected to grow at a CAGR of 9.9% through 2025.
Planned Commercialization
We intend to begin selling our RAP device in the United States in mid-2020 for the removal of tattoos by marketing to a narrow group of key dermatologists. We are diligently working with our sole manufacturer, Sanmina Corporation, to complete the design and build of the device that will be used in our initial commercial market launch. We will be conservative in the building of our sales team during the initial stages of the launch and intend to grow this team as we achieve traction in the marketplace.
Should we have favorable results with our pivotal cellulite FDA trial and receive FDA clearance for the cellulite reduction indication, we intend to introduce this indication to the marketplace through the sale of new cartridges designed specifically for treating cellulite.
Why Soliton?
•Large addressable markets with significant growth potential.
•Platform technology that is non-invasive and that dermatologists will be able to use across multiple indications.
•“Razor and blade” revenue model with a true consumable.
•Targeted indications are cash pay.
•In clinical trials, treatment across indications has shown no significant or adverse events and has been well-tolerated by patients.
Our Growth Strategy
Our goal is to provide safe, effective, and science-based solutions to the marketplace that truly improve our patients’ lives. We believe the following strategies will enable us to achieve this goal and help us to succeed and grow.
•Focus on launching aesthetic indications that are treated in the dermatologist office and are cash-pay.
•Execute on our clinical plan to expand our indications and support the clinical proposition in further medical indications.
•Recognize the importance of building awareness of our product offerings in both the physician and patient communities and plan our marketing strategies and spending accordingly.
•Build our specialized sales force and practice development managers gradually as we grow to support the success of each device placed in service.
The Rapid Acoustic Pulse (RAP) Device
Description of Technology
The RAP device uses electrohydraulics to generate the designed acoustic shockwaves at a rate of up to 100 per second to effectively disperse ink particles and superficial and dermal vacuoles. The RAP device for commercial launch is composed of three parts: a console, a hand piece and a disposable cartridge. The console houses a pulse power system used to provide high voltage power to a pair of electrodes housed within the cartridge. Additionally, the console contains a fluid management system that circulates saline through the cartridge. The cartridge is snapped in and out of the hand piece for easy replacement and forms the basis for our planned “razor and blade” recurring revenue model.
Figure 1

Our RAP device generates high-energy designed acoustic shockwaves when electricity is applied to the electrodes immersed in the circulating saline contained in the cartridge enclosure. An electrical arc with a very short duration of 100 to 200 nanoseconds is formed within the saline between the electrodes. When this arc is formed, a small amount of water is vaporized between the electrodes creating a nearly instantaneous expansion and collapse of a plasma bubble. This creates a shockwave that propagates outward through the saline, most of which is reflected off a curved surface surrounding the electrodes designed to form a shockwave front that passes through the cartridge’s acoustically transparent window. This window is placed against the patient’s skin above the tattoo to be removed allowing the acoustic energy to penetrate to a depth of 1 to 2 mm, which corresponds with the typical depth of tattoo pigment. These shockwaves are generated at a rate of up to 100 times per second.
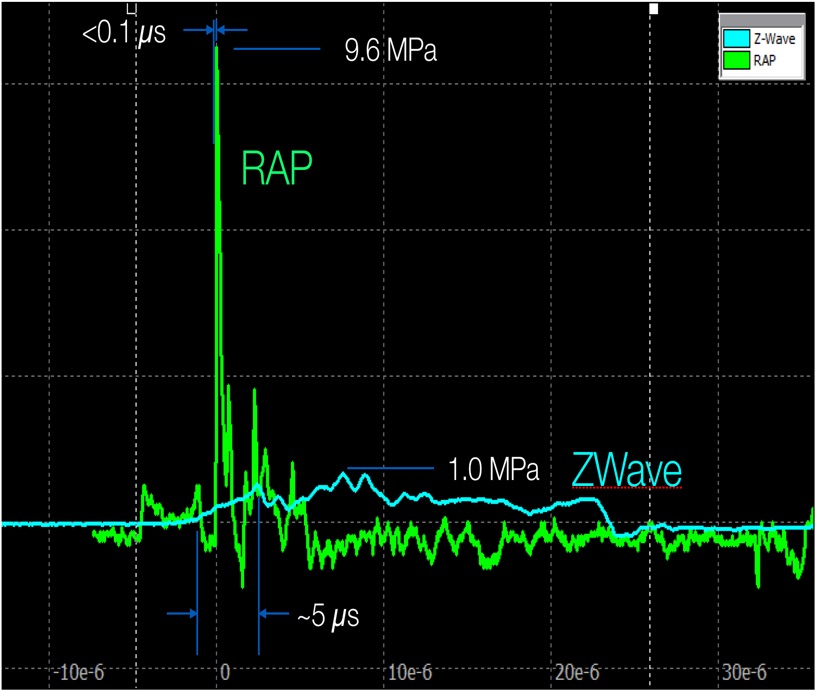
The high repetition rate of Soliton shockwaves is a key component of our patent-pending technology. Specifically, a single shockwave from our RAP device is delivering .25 to 12 MPa (Megapascals) of acoustic pressure. Although this is a significant level of pressure, a single shockwave will pass through a typical skin cell with relatively little disruption. This is because the general elasticity of the cell is capable of deforming slightly to absorb that single impact and then returning to its original shape. The rate at which the cell returns to its normal shape is referred to as its “relaxation rate,” and this rate is well understood in the field of biomechanics. By increasing the repetition rate of Soliton shockwaves above approximately 25 times per second, we begin to exceed the relaxation rate of skin cells, which triggers their natural “viscoelastic” property and causes them to stiffen. In that stiffened state, the cells are quite vulnerable and shear waves created by the interaction of subsequent shockwaves with the tattoo ink particles in macrophages is now enough to rupture the cell membranes and disperse the particles.
The graphic below (Figure 2) is a graphical representation of hydrophone measurements and compares the Soliton wave form with a competing shockwave technology that is cleared by the FDA as a massage device. Many dermatologists have this particular device in their practice and use it in conjunction with Coolsculpting to massage the treatment area post treatment. The difference in height of the two ways represents the difference in acoustic pressure or the strength of the pulse delivered.
Figure 2
The shockwaves generated by our RAP device are designed and proprietary, comprised of high acoustic energy delivered with a very short rise time (less than 5 nano seconds). Very high electrical energy (approximately 3000 volts at 3000 amps) is discharged in the treatment head with nanosecond precision to minimize unwanted acoustic frequencies (which helps minimize pain and collateral tissue damage and extend electrode life). A proprietary custom-shaped reflector designed through finite element computer simulation technology directs the bulk of the acoustic energy to the patient’s skin in uniform waves that are nearly planar (perpendicular) to the surface of the skin but slightly diverging in order to deliver maximum acoustic pressure to the depth of a typical tattoo, but then rapidly dissipate beyond that distance. The pressure mapping diagram in Figure 3 provides an example of how our reflector design controls energy density at varying treatment depths. The brighter yellow colors indicate maximum pressure at tattoo ink depth (top layer) and the darker red colors indicate lower pressures deeper in the skin (lower levels).
Figure 3
While our RAP device designed acoustic shockwaves are measured in the ultrasound spectrum, they should not be confused with typical therapeutic ultrasound that is focused and creates significant heat through cavitation (bubble formation) within the skin. In contrast Soliton designed acoustic shockwaves are deliberately unfocused and produce little to no heat within the skin. The specific frequency and rise time of Soliton shockwaves allow them to pass harmlessly through normal skin cells but when encountering a significant mass differential like that of tattoo ink particles, they create shear waves that break apart macrophage structures containing the particles and dissipate dermal vacuoles resulting from laser treatment.
Figure 4
Given the high level of energy involved with each electrical discharge and the high repetition rate (up to 100 times per second), the tungsten electrodes in the treatment head have a limited life, hence the need for a replaceable cartridge. The cartridge designed for tattoo removal (Figure 4) is capable of delivering as many as 120,000 shockwaves before replacement, which we believe is enough to treat an average sized tattoo throughout one office visit. This length of service life is only possible through the use of a proprietary drive mechanism for feeding electrode material into the electrical arc without changing the focal point established by the cartridge’s reflector.
In total, we have eight patent families pending relating to the technologies that makes our RAP device and certain variations possible, as well as various applications of our RAP device, with still more potential patent applications under way. As of December 31, 2019, our patent portfolio is comprised of 11 pending U.S. patent applications, 28 granted and 59 pending foreign counterpart patent applications, and three pending PCT patent applications, each of which we either own directly or we are the exclusive licensee.
Approved Indications
Tattoo Removal
The RAP device is initially being commercialized to be used in conjunction with the 1064 nm Q-switched laser to enable effective multiple pass laser treatments in a single office session to accelerate removal of tattoos on the arms, legs and torso in Fitzpatrick Skin Type I-III individuals. Our animal testing suggests that the RAP device is as effective on other tattoo ink colors using alternate wavelength lasers and analytical modeling supports the expectation that RAP should also work well with Pico-switched lasers. Use of the device on other colors and with a Pico-switched laser would be considered an off-label
use until further FDA clearance is achieved. The RAP device uses repeated, rapidly rising acoustic waves to both disrupt pigment laden cells and provide dermal clearing of both superficial and dermal vacuoles generated during the laser process. The clearing of these vacuoles allows for multiple laser treatments within one office visit and animal testing data suggests that remaining agglomerations of ink particles will be dispersed providing greater access for subsequent laser passes.
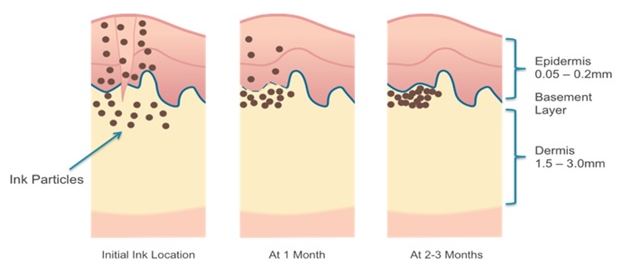
Understanding Tattoos
Tattooing involves the placement of pigment into the skin's dermis, the layer of dermal tissue underlying the epidermis. As illustrated in Figure 5, ink particles are typically injected by being placed on the tips of needles that puncture the skin with the ink particles being left behind as the needles are withdrawn. While the keratinaceous cycle will eventually remove pigment particles from the epidermis, with the dermis pigment particles are consumed by and remain trapped within macrophages, ultimately concentrating in a layer just below the dermis/epidermis boundary as the macrophage becomes pigment laden and immobile. Its presence there is stable, but in the long term (decades) the pigment tends to migrate deeper into the dermis, accounting for the degraded detail of old tattoos.
Figure 5
As macrophages collect individual ink particles, many are carried away by the circulatory and lymphatic systems and it has been estimated that more than half of the injected ink particles are carried away within the first several months after a tattoo is applied. However, many macrophages over consume ink particles to the point where they can no longer be absorbed into the circulatory and lymphatic systems. These “pigment laden macrophages” thereby form the relatively permanent tattoo that remains.
Current Standard of Care for Tattoo Removal
Tattoo removal has been performed with various tools during the history of tattooing. While tattoos were once considered permanent, it is now possible to remove them, fully or partially, with treatments. Non-laser tattoo removal methods include dermabrasion, TCA (Trichloroacetic acid, an acid that removes the top layers of skin, reaching as deep as the layer in which the tattoo ink resides), salabrasion (scrubbing the skin with salt), cryosurgery and excision that is sometimes still used along with skin grafts for larger tattoos. Tattoo removal by laser was performed with continuous-wave lasers initially, later with Q-switched (short-pulse) lasers, which became commercially available in the early 1990s, and more recently with Pico-switched lasers that deliver shorter pulse bursts of energy than Q-switched lasers. Today, "laser tattoo removal" usually refers to the non-invasive removal of tattoo pigments using (primarily or most commonly) Q-switched lasers with some increasing use of the Pico-switched lasers.
This “laser tattoo removal” is further described as using lasers to fragment pigment particles, as well as break-apart pigment laden macrophages resulting in the dispersion of the ink particles they contain. The fragmented ink particles are then absorbed by the body, repeating the same natural immune response by macrophages that accounted for the loss of 50% or more of the ink originally injected when the tattoo was applied.
All tattoo pigments have specific light absorption spectra. A tattoo removal laser must be capable of emitting adequate energy within the given absorption spectrum of the pigment to provide an effective treatment. To specifically target tattoos, laser wavelength and pulse duration must be chosen appropriately. Certain tattoo pigments, such as yellows, greens and fluorescent inks, are more challenging to treat with a Q-switched laser than darker blacks and blues because they have absorption spectra that fall outside or on the edge of the emission spectra available in the device.
There are several types of short-pulse lasers appropriate for tattoo removal, with one differentiating factor being the color spectrum for which it is optimized. Q-switched lasers can provide multiple wavelengths and are used to treat a much broader range of tattoo pigments than previous lasers. The more recently developed Pico-switched lasers claim to be more effective on those colors that present the greatest challenge for Q-switched lasers and are used either in conjunction with or replacement of Q-switched lasers. The amount of energy to be delivered is determined prior to each treatment, as well as the spot size and treatment speed. Light is optically scattered in the skin, like automobile headlights in fog. Larger spot sizes slightly increase the effective penetration depth of the laser light, thus enabling more effective targeting of deeper tattoo pigments, and can also help make treatments faster by covering a larger area with each pulse.
Laser tattoo removal can be described as ranging from uncomfortable to quite painful. The pain is often described to be similar to that of hot oil on the skin, or a "slap" from an elastic band. To mitigate pain one common method is to cool the area during treatment with a medical-grade chiller/cooler and to use a topical anesthetic. Pre-treatment options include the application of an anesthetic cream under occlusion for 45 to 90 minutes prior to the laser treatment session. In other cases, anesthesia is administered locally by injections of 1% to 2% lidocaine, sometimes including epinephrine. The addition of epinephrine to the injection must be done with careful consideration as the drug restricts blood flow, and reduced blood flow makes it more difficult for the body to remove the residual heat from the laser.
A common risk for patients treated with lasers for tattoo removal is the appearance of darkening of the normal skin pigmentation (hyperpigmentation). These changes may resolve in 6 to 12 months but may also be permanent. Hyperpigmentation is more commonly related to patients with darker skin tone. Another common risk is scarring as a result of collateral tissue damage caused by the residual heat caused by lasers. The potential for more extreme keloid scarring also increases with darker skin tone. The standard measure for skin tone is called the Fitzpatrick Scale, a scale from I to VI, with I being extremely fair and VI being extremely dark. Generally speaking, great care must be used when treating patients who are Fitzpatrick IV and above to avoid hyperpigmentation and keloid scarring, and as a result, clinicians generally use lower energy settings, which in turn means each treatment is likely to be less effective and more treatments are likely to be needed for satisfactory tattoo removal.
As illustrated in Figure 6, “complete” laser tattoo removal usually involves numerous treatment sessions typically spaced at least six to eight weeks apart. Treating more quickly than six weeks increases the risk of adverse effects and does not necessarily increase the rate of tattoo fading. At each session, some, but not all, of the tattoo pigment particles are fragmented, and the body removes the smallest fragments over the course of several weeks. The result is that the tattoo is lightened over time. Remaining large agglomerations of tattoo pigment are then targeted at subsequent treatment sessions, causing further lightening. The number of sessions and spacing between treatments depends on various parameters, including the area of the body treated and skin color. Tattoos located on the extremities, such as the ankle, require even more treatments. As tattoos fade, clinicians may recommend that patients wait many months between treatments to facilitate fragmented ink particle absorption and minimize unwanted side effects.
Figure 6
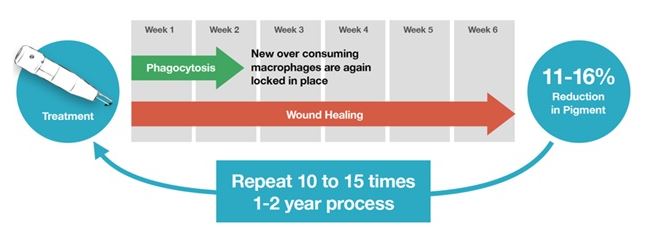
We believe the amount of time or the number of treatments required to “completely” remove a tattoo is a critical hurdle to tattoo owner adoption of the current laser tattoo removal procedure. The Wall Street Journal reported a research study conducted at a laser surgery center in Milan, Italy, from 1995 through 2010. There were 352 people in the study, of which 201 were men, with a median age of 30 years old. Overall, the study found about 47% of people had their tattoos successfully removed after 10 laser treatments and it took 15 treatments to remove tattoos from 75% of patients. Black and red pigments in tattoos were most easily removed. The researchers also found that the amount of time between Q-switched laser treatment sessions was important to the technique's success. Treatment intervals of eight weeks or less were found to be less effective for
tattoo removal. Patient frustration and dissatisfaction with removal success and with the time to achieve success results in a significant number of patients discontinuing treatments, or “dropping out.”
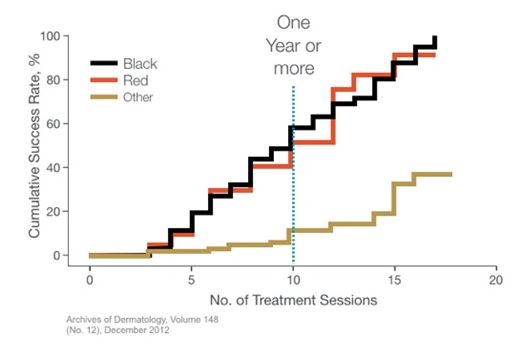
A more recent study of 237 patients treated with Q-switched lasers showed very similar results, which are plotted below. As can be seen on the graph in Figure 7, only about half of the patients with black or red tattoos achieved complete removal after 10 treatments, which if spaced only six weeks apart will still require over a year’s worth of time-consuming and uncomfortable office visits.
Many studies accepted by the FDA deem 75% or greater removal to be a "successful removal," while others simply do not define what a successful removal is, using the word “complete” without clarification. Many successful removals do not remove all traces of the original tattoo, but instead reduce the visible tattoo to the point where it is difficult to see with the naked eye. Generally speaking, we consider a removal procedure to be complete when 75% or more of the visible ink is gone and the patient and the physician are satisfied that whatever residual ink particles remain are likely to be absorbed by the body through natural immune, healing, and skin remodeling processes.
Figure 7
How the RAP Device Makes Laser Tattoo Removal More Effective
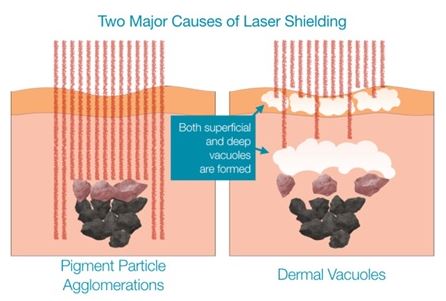
Our marketing research has shown that, for most patients interested in tattoo removal, the poor efficacy of the standard of care presents too much of a barrier for them to move forward with tattoo removal. Our laboratory research into the problem of tattoo removal has led us to the conclusion that laser shielding is a major cause of this poor efficacy. This laser shielding can be broken down into two subtypes: Particle Shielding and Vacuole Shielding, as depicted below in Figure 8.
Figure 8
Particle Shielding
Lasers are essentially “line of sight” dependent, meaning the laser light pulses can only ablate particles that are directly in their path. Because tattoo ink particles tend to aggregate into clusters within the skin, the particles at the top of the clusters (closest to the surface of the skin) effectively shield the rest of the particles from the laser energy (particle shielding). This leads to two conclusions: each laser pass only affects a small percentage of ink particles, explaining why multiple passes are important, and, if we can spread these particles out, each subsequent laser pass has an opportunity to hit more targets.
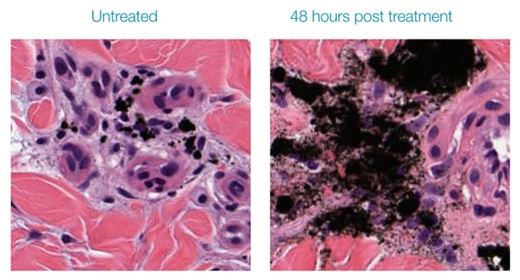
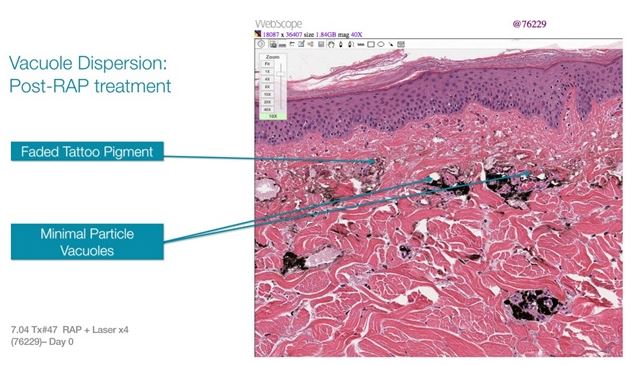
Much of our research utilized tattooed pig skin, because pig skin is considered the most like human skin when it comes to dermatology treatments. Biopsies from pigs with mature tattoos allow us to see the effect the RAP device has on pigment particle agglomerations. A microscopic histological comparison in Figure 9 shows an untreated tattoo on the left with intact tightly formed (macrophage) agglomerations of tattoo ink and a similar tattoo on the right treated with the RAP device. The result of the RAP device treatment is a noticeable destruction of the macrophages and dispersal of the pigment particles. We have effectively created more targets for the laser to hit.
Figure 9
Vacuole Shielding
A second, and more limiting problem arises the moment that the laser light contacts ink particles within its path. Almost instantly, a plasma event occurs that quickly results in the formation of steam vacuoles. These vacuoles appear white in color and result in “optical scattering” that immediately blocks any additional laser energy from reaching ink particles below the vacuoles. Until those vacuoles are gone, subsequent laser passes will have very little effect. In the picture shown below in Figure 10, you can see the emergence of a white frost or crust that forms immediately with each pulse of the laser.
Figure 10
Several efforts have been made to address these vacuole formations in an attempt to facilitate multiple laser passes in a single office visit, but they have failed to gain traction for lack of sufficient improvement in results or due to their relative impracticality in practice.
A relatively new treatment protocol has been studied, referred to as the “R20 method.” The “R” stands for Repeating, while the “20” represents 20 minutes. The R20 Method suggests administering a single pass of the laser every 20 minutes, with up to 4 passes, providing effectively 4 removal treatments during one office visit. The 20-minute pause between passes of the laser allows the epidermal or surface vacuoles to dissipate, presumably increasing the ability of the laser to reach more pigment with each subsequent pass.
The R20 method has not been heavily adopted by the medical community as the “wait” time between treatments presents two hurdles: the recommended 20-minute wait between treatments in practice grows to an hour or more between treatments as the physician moves to treat other patients during the “wait,” and keeping the patient properly anesthetized for the entire treatment session becomes a challenge. While the level of improved results has not justified this cumbersome routine, data varies as to the number of R20 treatment sessions required to successfully remove a tattoo; most seem to center on 6-8 laser passes, or 2 treatment sessions (likely separated by at least eight weeks).
A company called OnLight (recently acquired by Merz Pharma) introduced a transparent patch infused with a clear chemical called Perfluorodecalin (PFD), which they claimed was capable of reducing the formation of surface vacuoles, thereby enabling multiple laser passes in succession. And, while a study has shown that the PFD patch appears to enable 3 to 4 laser passes in a single office visit (without long interruptions between treatments), any improvement in tattoo fading only occurred in about 2 out of 3 of patients and, in most of those patients, the degree of improvement was only marginal.
Data from our research presented at the American Society for Laser Medicine & Surgery in April 2017 offers an explanation. A histology image (Figure 7) of a biopsy taken 2 hours after laser treatment reveal that, while the surface vacuoles have dissipated, deeper “dermal vacuoles” persist and continue to shield the remaining particles from subsequent laser passes. And, our studies have shown that these deep dermal vacuoles persist for up to 48 hours. The histology image in Figure 11 shows the presence of these vacuoles 2 hours after laser treatment, well beyond what the R20 method could hope to avoid, and importantly, below the reach of Perfluorodecalin in the PFD patch, which cannot penetrate below the epidermis and into the dermis where these vacuoles occur.
Figure 11
However, if you apply the RAP device immediately following a laser treatment (Figure 12), histology reveals that these deep dermal vacuoles are dispersed, allowing lasers to again have line of sight access to pigment particles.
Figure 12
With traditional laser treatment tattoo removal, efficacy is limited by particle shielding resulting from the natural clustering or agglomeration of pigment particles and the formation of laser-induced dermal vacuoles, both of which block access of laser energy to the particles being targeted (see Particle Shielding and Vacuole Shielding above). Importantly, the dermal vacuoles inhibit any additional passes of the laser from effectively reaching the remaining tattoo pigment agglomerations due to optical scattering. The shape, frequency and repetition rate of the RAP device’s acoustic shockwave pulses are designed to increase dispersion of ink particles and to diffuse and disperse both superficial and dermal vacuoles, while minimizing damage to adjacent non-pigmented tissue as well as pain perceived by the patient. With RAP dermal clearing, loss of laser efficacy due to optical scattering is thereby minimized. In addition, we believe more ink is exposed to each successive laser pass due to increased particle dispersion. As a result, effective, fast, multi-pass laser treatment of tattoo sites in a single office session may be realized.
Market for RAP Tattoo Removal
Over the past two decades or so, the tattoo has become an attractive, artistic expression among many people. The popularity of tattoos continues to rise as they become more accepted in popular culture. Approximately one-third of all adults in the United States have a tattoo. People 18-29 years old have the most tattoos, according to a 2010 study by the Pew Research Center with 38% of that age group having at least one. Nearly half of this group with tattoos have between two and five tattoos, while 18% have six or more. Among other generations, the following indicates the percentages by age with at least one tattoo:
•30-45 year-olds: 32%;
•46-64 year-olds: 15%; and
•> 65 years old: 6%
Currently Americans spend $3.4 billion per year on tattoos, and as social acceptance of body art steadily increases spending on tattoos will likely continue to grow. With the tremendous growth in the number of people getting tattoos, there is a corresponding increase in demand for tattoo removal. Estimates of the size of the tattoo removal market vary widely. One independent source estimates that, globally, the market for tattoo removal is expected to grow at the rate of about 15.6% from 2017 to 2023 and that the global market for tattoo removal is expected to reach several billion in revenue by 2023. Our own research and analysis suggests that regardless of its potential, the current tattoo removal market is significantly underdeveloped.
Tattoo removal is a process of removing a permanent tattoo from the skin. The removal process is undertaken by using laser, surgery, creams, and various other processes. The use of laser techniques for tattoo removal is the predominant tattoo removal process with 66% of the market. Different type of lasers such as Q-switched ruby laser, Q-Switched Nd:YAG laser, and Q-Switched Alexandrite laser are used to remove black as well as colored tattoos. The other options available for tattoo removal include surgical excision, tattoo removal creams, dermabrasion, plastic surgery, and others. Creams are less painful than laser and surgical procedures to remove tattoos, but the use is time consuming and inefficient.
Laser tattoo removal is an elective, private pay procedure performed on an outpatient basis. The procedure is primarily performed at laser centers and dermatology clinics with laser centers performing 60.9% of the procedures in 2016. Because the cost of tattoo removal is many times the cost of tattoo application, the procedure only attracts those who can pay. Laser tattoo removal practitioners charge a premium for their time. Each treatment is generally priced from $100 to $500, and most patients require 10 or more treatments, depending on the size and complexity of the tattoo, to achieve comprehensive removal. Because tattoo removal is a painful, time-consuming and expensive process, patients need to be very motivated for removal. Here are some of the most common reasons people seek tattoo removal:
•Tattoo includes the name of a former spouse or significant other;
•Limited clothing options to hide tattoo;
•Do not want their children to see it;
•Curtails job prospects;
•Poor quality tattoo;
•Tattoo has faded; and
•The importance of getting the tattoo has lessened.
We commissioned our own survey of individuals with one or more tattoos in an effort to better understand their interest in, motivations for and concerns about tattoo removal. This survey was designed to be representative of the US population with 95% confidence (+/- 3%) and indicated that 63% of individuals with tattoos were interested in some form of removal. Importantly, a majority of these individuals didn’t regret having tattoos, they simply wanted to make a change. From this observation we conclude that the total available market in the US alone could be calculated as 63% of the estimated 70 million US adults with one or more tattoos (29% of the 2016 US population), or 44 million potential customers.
In this survey we also asked what barriers prevented these individuals from taking action to have a tattoo removed. The primary reasons were cost, pain and efficacy (time required for removal). With this in mind, we believe the dramatic reduction in the number of office visits required for tattoo removal using the Soliton method may be sufficient to motivate many individuals who have been considering tattoo removal to finally take action, which we, in turn believe may result in a material acceleration of the current rate of growth for tattoo removal.
Clinical Trial Results
Our RAP device has received institutional review board (IRB) approval as a non-significant risk device. Subsequent to receiving this status, we have conducted several human clinical trials to study the use of the RAP device to accelerate tattoo fading.
Human Correlation Trial - 1 (HCT-1)
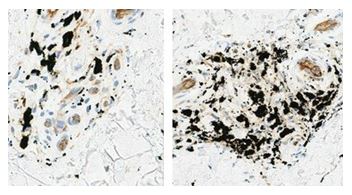
An initial human clinical trial was conducted to demonstrate the dispersion of tattoo pigment. In the first part of the HCT-1 study, three patients with black tattoos in various locations (lower back, lower leg and shoulder) were selected. Two tattoo sites on each patient were treated with a single pass of the RAP device. One site was treated and then immediately biopsied and the other was treated with a biopsy taken 24 hours post treatment. All biopsies in all patients demonstrated pigment dispersion from macrophages. As seen in Figure 13, the images present the tattoo site untreated (left image) and 24 hours post-treatment with the RAP device (right image). Note the significant dispersion of the tattoo ink pigment at 24 hours post treatment in the right image.
Figure 13
In the second part of the HCT-1 study, six patients were selected for a single treatment session to demonstrate tattoo fading. For each patient, a single black tattoo was selected and divided into three adjacent areas. Two of the areas were treated (i.e. test areas) and the third area remained untreated as a control for comparison to the test areas. One test area was treated with a single laser treatment (Laser Only). The other test area was treated with multiple laser passes, with each laser pass followed by a treatment with the RAP device (Laser+RAP). After each laser pass, the laser was adjusted to increase the laser fluence.
Dermal vacuolization was immediately identified in all tattoos treated with a laser. Minimal dermal clearing was detected 5 minutes post treatment in the Laser Only treatment areas. Significant dermal clearing was immediately identified in the Laser+RAP treatment areas. The Laser+RAP treated test area, demonstrated accelerated tattoo fading at 24 hours post treatment when compared to the non-treated tattoo test site and to tattoos treated with Laser Only.
The trial also offered important conclusions to the treatment therapy. The importance of preventing thermal damage to the tattoo site resulting from multiple laser passes is critical and includes avoiding the use of epinephrine, maintaining the hydrogel dressing throughout the procedure, and titrating the increase of laser fluence and spot size with each laser pass (titrating these increases can be done by listening for a treatment ‘snap’ during the laser treatment process or by watching for new vacuole formation).
Human Correlation Trial- 2 (HCT-2)
To further demonstrate accelerated tattoo fading in a single office session when the RAP device is used as an accessory to the 1064 nm Q-switched laser, the multi-pass method was again tested in humans in a pivotal clinical trial (HCT-2). The RAP device was evaluated in a single-center (Skin Care Physicians, Chestnut Hill, MA), prospective study.
A total of 32 black tattoos, from 22 participants, were divided into three zones. Two zones in each tattoo, separated by a control zone, were treated with either multiple laser passes, each separated by RAP device applications (“Laser + RAP”) or a single-pass laser treatment (“Laser Only”). The treatment sites were assessed for the number of laser passes and adverse events immediately following the treatment as well as at six weeks and 12 weeks following the treatment session. The treatment sites were also assessed for the degree of fading at 12 weeks post treatment using blinded review.
The HCT-2 study confirmed the feasibility of using the RAP device to enable safe, multi-pass laser treatments in a single session. The observed mean number of laser passes in the Laser + RAP treated participants was 4.16. Studies of the PFD Patch demonstrated an ability to achieve a mean of 3.7 passes with use of the patch. The average number of deliverable passes in a single treatment session of the RAP device, as an alternative accessory device instead of the PFD Patch, was determined to be at least comparable to the average number of deliverable passes in a single treatment session of the PFD Patch. Based on these results, the primary objective of this study was considered met.
The secondary objective was to assess the degree of tattoo fading from a single treatment session for both the Laser + RAP treatment and the Laser Only treatment. Assessment by blinded reviewers at 12 weeks indicated that there was accelerated fading for Laser + RAP in comparison to Laser Only. Specifically, 72% of the tattoos treated with the Laser + RAP had a good, excellent or complete response (>25% fading) compared to 40% of the tattoos treated with Laser Only. Furthermore, 41% of the tattoos treated with the Laser + RAP had an excellent or complete response (≥50% fading) compared to 12% of the tattoos treated with Laser Only. Finally, 19% of the tattoos treated with the Laser + RAP had a complete response (>75% fading) compared to 3% of the tattoos treated with Laser Only.
As an additional comparison, assessment of tattoo fading at 12 weeks was performed by the treating physicians (non-blinded reviewers). The non-blinded reviewers scored 81% of the tattoos treated with the Laser + RAP as having a good, excellent, or complete response (>25% fading) compared to 16% of the tattoos treated with Laser Only. On average, the tattoos treated with the Laser + RAP had 49% fading in a single treatment session, as compared with only 16% for the tattoos treated with Laser Only. The difference between the blinded and non-blinded reviewers in terms of fading scores is believed to be a result of the non-blinded reviewers’ direct examination the tattoos at 12 weeks compared to the blinded reviewers’ use of photographs only. However, the differences were not statistically significant using chi-square analysis.
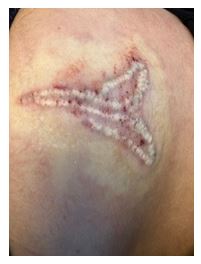
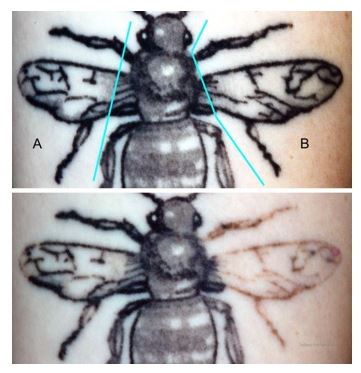
A representative cross-polarized images of one participant’s tattoo, before treatment and 12 weeks after treatment, are shown in Figure 14. In these images, the tattoo zone marked with ‘A’ was treated with Laser Only and the tattoo zone marked with ‘B’ was treated with Laser+RAP. As can be seen with these images, after 12 weeks, the tattoo zone treated with Laser+RAP demonstrated a significant degree of fading in comparison with the tattoo zone treated with Laser Only.
The conclusion of the HCT-2 study was that the RAP device, as an accessory to the 1064 nm Q-switched laser, safely enables multiple laser treatments in a single office visit. More importantly, the RAP device enables accelerated tattoo fading in a single treatment session.
Figure 14
Human Correlation Trial - 3 (HCT-3)
HCT-3 built upon HCT-2 by bringing back 10 HCT-2 subjects (12 tattoos) for up to an additional two separate treatment sessions. The first session performed as part of the HCT-2 multi-pass laser treatment study was followed by a second session 20 weeks after the first session. The third and final session (where needed) was performed 28 weeks after the first session (eight weeks after the second session). As described for the HCT-2 study above, each test site was treated with either Laser + RAP or Laser Only. The test sites were assessed for degree of fading at 40 weeks following the first session (12 weeks following the third session).
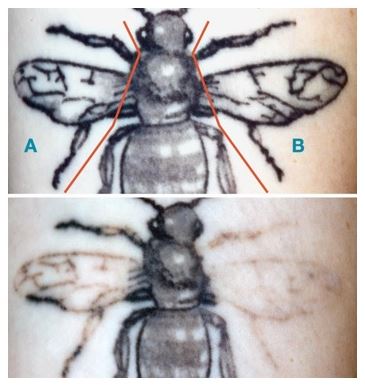
The Laser + RAP in HCT-3 again outperformed Laser Only, with subjects showing an average of 80% fading after only two visits vs. 44% for Laser Only. After 3 “Soliton” treatments, 100% of the treated tattoos had a ‘Complete’ (76-100% faded) response; in comparison, only 16% of the tattoos treated with the Laser Only had a ‘Complete’ response.
The same representative image from Figure 14 is shown in Figure 15 before treatment and a new image taken after three treatment sessions is shown below it. Hence, the top photo in Figure 15 is taken before any treatments began and the bottom photo is taken at week 40--12 weeks post the third treatment. In the top photo, the section marked with an "A" was treated with Laser Only and the section marked with "B" was treated with Laser + RAP. As can be seen with these images, after 40 weeks, the tattoo zone treated with Laser + RAP demonstrated a significant degree of fading in comparison with the tattoo zone treated with Laser Only.
The conclusion from HCT-3 was that RAP, used as an accessory to the 1064 nm Q-Switched laser, enabled accelerated tattoo fading in just three office visits.
Figure 15
Research and Development
While we are initially targeting the tattoo removal market, higher-energy versions of our technology also show promise in a number of other indications. We have conducted animal studies and some limited human trials in some of these other indications as discussed below. The results observed in our proof-of-concept cellulite and keloid studies underlie our belief that our technology may impact a much broader set of fibrotic conditions. Scientific publications suggest that fibroblasts become over-active when they are located in a stiffened environment and that disrupting the stiff environment may lead to fibroblast apoptosis, ultimately resulting in a resolution of the fibrosis. On this basis, we believe that our technology could have efficacy in a number of fibrotic diseases both in the extracellular matrix, such as radiation induced fibrosis, and in other systems of the body such as peripheral artery disease and even non-alcoholic SteatoHepatitis ("NASH").
Clinical Stage Indications
Reduction of Cellulite
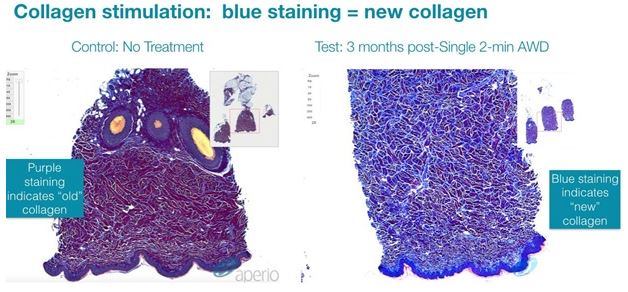
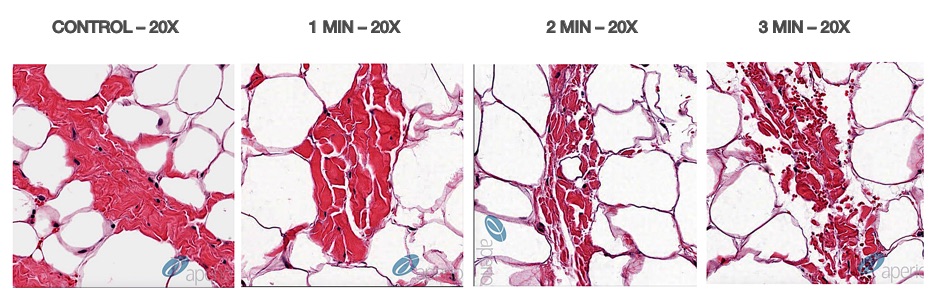
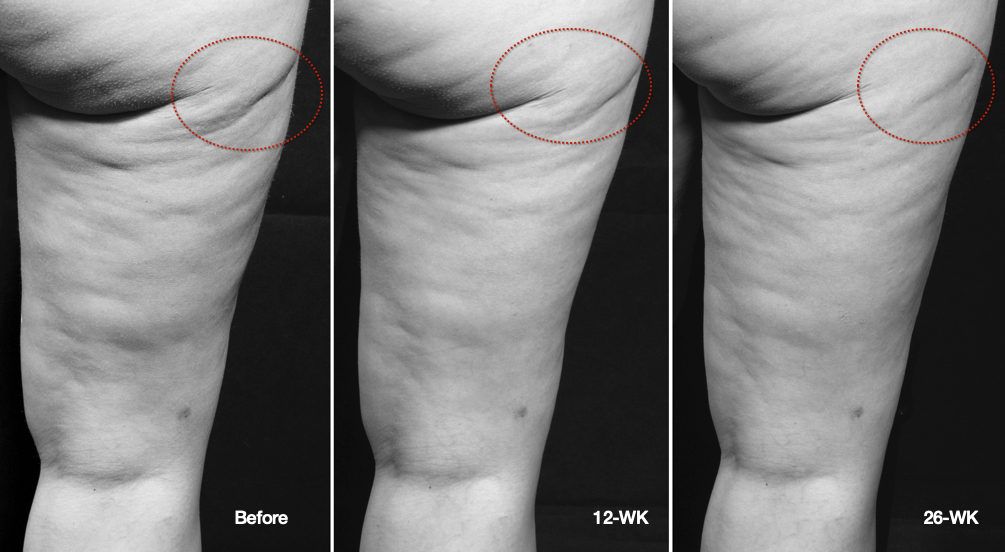
Cellulite is a condition that primarily affects women, usually occurring in the buttock and thigh area, where the skin has a dimpled or lumpy appearance. Between 80 and 90 percent of women will probably experience cellulite sometime in their lives. There is a very large global market for cellulite treatment. In the U.S. alone, women spend roughly one billion dollars a year on cellulite therapy, with approximately 85% of U.S. women reporting concerns about cellulite. Based on third party market research, the global market for cellulite treatment was estimated to be approximately $2.4 billion in 2018 and is expected to grow to approximately $4 billion by 2025. There are numerous treatments available, but the effect is mostly temporary. A 2015 review of a variety of studies into the effectiveness of different techniques indicated that either the procedures did not work, or the research methodology was flawed. The American Academy of Dermatology (AAD) reviewed a number of surgical techniques that may be successful in reducing the appearance of cellulite by breaking up the bands of connective tissue under the skin's surface. As a non-invasive technique, if a version of our device is capable of reducing the appearance of cellulite with results that approach those of the surgical techniques, we believe this could become an important new indication for our technology.
Cellulite is characterized by relief alterations (lumpiness) of the skin surface, which give the skin an orange peel, cottage cheese, or mattress-like appearance. Some factors leading to the appearance of cellulite are believed to include sclerotic septa connecting the dermis to the fascia below the subcutaneous fat layer and inadequate collagen in the dermis leading to a weak dermal extracellular matrix (ECM). Excess subcutaneous fat can then protrude into pockets formed between the sclerotic septa within the weakened ECM resulting in a mottled or lumpy appearance to the skin. This same weakening of the ECM can also be associated with skin laxity whereby the skin appears loose, wrinkled and creped.
We believe it may be possible to reduce the appearance of cellulite and skin laxity by both severing sclerotic septa and strengthening the ECM. Existing methods for treating cellulite include physically cutting septa through invasive methods, however, we believe it may be possible to do this non-invasively with high-energy acoustic pulses. In addition, existing independent research suggests that weakened ECM can be strengthened by inducing the fibroblasts in the skin to produce more collagen. One approach to inducing collagen production is to apply an external force to pre-stress fibroblasts by applying external pulsed acoustic shockwaves at high repetition rates. Given the viscoelastic nature of fibroblasts, we believe external acoustic waves applied at repetition rates faster than the relaxation rate of the fibroblasts will cause the cells to stiffen and become “pre-stressed.” In this pre-stressed state, fibroblasts become more susceptible to external forces and if the external forces are great enough, we believe the fibroblast will then produce collagen.
Soliton’s RAP device produces designed acoustic shockwaves at pulse rates between 50 and 100 Hz. We believe our device at this high pulse rate may be capable of “pre-stressing” fibroblasts so that they are sensitized to the external forces from the acoustic shockwaves. As an initial proof-of-concept we have demonstrated in a pig model that a version of our device is capable of consistently forming new collagen within the ECM of the dermis. As seen in Figure 16, the histological image on the right demonstrates the stimulation of new collagen growth in pig skin after a single 2-minute application of version of our device (i.e., increase in blue staining) in comparison to the histological image on the left from non-treated skin.
Figure 16
Acoustic Subcision
We consider cellulite, at its core, to be a fibrotic disorder. In normal skin structure, septa run through a layer of subcutaneous fat connecting the dermis to the muscle layer of the body. In certain situations, these fibrous septa become stiff (“sclerotic”) and inflexible. As a result, when subcutaneous fat pushes up, the sclerotic fibrous septa pull the skin down causing the appearance of cellulite with deep dimples. Surgically severing the fibrotic septa is currently the only viable permanent means to remove these dimples.
As discussed previously with regard to tattoo removal, the specific frequency and rise time of Soliton shockwaves allow them to pass harmlessly through normal skin cells but when encountering a significant differential in structural stiffness, they create shear waves. In the case of cellulite treatment, this enables our technology to differentiate between fibrotic septa and the surrounding fat cells, which appears to result in the disruption and breaking apart of these septa, a process we call "acoustic subcision."
Preclinical studies have provided us with biopsy results that demonstrate the ability of the RAP device to deliver increased disruption of the fibrotic septa with increased treatment time, implying a dose response to the therapy. Shown in Figure 17 below are multiple biopsy slides, with the image on the left being an untreated septa and the three images to the right demonstrating one, two and three minute treatments with the RAP device.
Figure 17
Human Cellulite Proof of Concept Trial - 1 (HCPOCT-1)
We began our first human clinical trial for the cellulite indication during 2018. The Soliton proof of concept trial involved a study of five patients with moderate to severe cellulite, each treated on both of their thighs, with a higher-powered version of Soliton’s recently cleared RAP device intended to assist in tattoo removal. Three blinded reviewers, who are trained in the use of the Cellulite Severity Score ("CSS") and the Global Aesthetic Improvement Scale ("GAIS") scoring systems, scored the before and after photos,
Patient follow-up visits were conducted at 12 and 26 weeks. At 12 weeks, the range of improvement in the CSS was 20 to 47% from the single non-invasive treatment, with the average improvement being 29%. At 26 weeks, patients were reassessed. The GAIS 5-point scale was used to evaluate changes between the 12-week and 26-week time points and patients improved, on average, a full point on this scale. At the 26-week time point, CSS continued to improve with an average improvement compared to baseline of 31%. The average improvement for all patients was 1.24 and 1.31 on the 0 to 5-point CSS scale, for the 3-month and 6-month time points, respectively.
Importantly, the treatments required no anesthesia, caused no bruising, swelling or infection, and were
evaluated by the trial participants as a “0” on a pain scale of 0-10 in 97% of the treatments. None of the study participants experienced any post-treatment downtime.
Figure 18 below shows one of the patients treated in the study with the image on the left taken prior to treatment, the image on the middle taken at 12 weeks post the single RAP treatment, and the image on the right taken at 26 weeks post the single RAP treatment.
Figure 18
Human Cellulite Pivotal Trial - 1 (HCPT-1)
We began our pivotal clinical trial for the reduction of cellulite in the second half of 2019. The study had four clinical sites located in Phoenix, Boston, Washington, D.C. and Chicago. There were a total of 67 patients treated across the four clinical sites. The protocol called for patients between 18 and 50 years old with a BMI (Body Mass Index) of less than 30. The patients were to be treated in a single 20-30 minute session with therapy focused on dimples and ridges in the treatment area. The apparent dimples and ridges were located on the treatment area, marked for the physician and then treated for approximately one minute per marked location in the treatment area.
A single follow-up visit at 12 weeks post treatment was conducted for each patient with photographs taken using a camera designed to capture measurements of changes in the skin. Before and after photos for each patient will be provided to three independent reviewers, who are trained in the use of the CSS and GAIS scoring system, to score the photos before and after treatment.
12 week follow up visits have been conducted and photographic data will be scored by the independent physicians during the first quarter of 2020.
Treatment of Fibrotic Scars
Fibrotic scars, such as keloid and hypertrophic scars, represent wound healing gone awry. A typical example would be a post-surgical scar that grows beyond its boundaries. Existing published research suggests that factors relating to the wound-healing environment (including tension at the boundary of the scar) can cause fibroblasts to become stuck in a hyper-productive loop, unable to stop the production of collagen that leads to the thickened, raised and dense structures often associated with these fibrotic scars.
The American Osteopathic College of Dermatology estimates that keloids affect around 10% of people, whereas hypertrophic scars are more common. Keloid scars more prevalent among populations with darker skin pigmentation.
Hypertrophic scars affect men and women from any racial group equally, although people between 10 and 30 years old are more likely to be affected.
There are few treatment options available for fibrotic scars, which in addition to being disfiguring, can also cause significant discomfort. Current treatment methods include surgical excision of the scar, direct corticosteroid injections, laser treatment and cryotherapy. Some patients even receive radiation to help prevent the return of the scar. The most common treatment is the direct injection of steroids into the scar, however this is painful, can require multiple injections, and may not be a permanent solution.
Our preclinical and early clinical studies combined with published literature on the behavior of fibrotic tissue have suggested that our acoustic shockwaves may be capable of disrupting stiff, sclerotic structures created by unwanted fibrosis, of which fibrotic scars are just one example. Beyond this, we may also be able to help reset the targeted tissue to more normal fibroblast activity for lasting effects.
Human Fibrotic Scar Proof of Concept Trial - 1 (HFPOCT-1)
In mid-2019 we began a proof-of-concept study for the treatment of fibrotic scars. We treated 10 people, each of whom received just a single 6-minute treatment with our RAP device. Before treatment, we photographed each scar with a 3D imaging device that allows for precise measurement of both the volume and height of the raised scar. Six weeks after that single treatment we took another set of 3D images, which then allowed for an objective comparison of before and after data. We conducted the same assessment at 12 weeks post treatment.
The six-week results (for nine scars as one patient was not able to come in for this six-week time point) have been analyzed using quantitative data captured with the 3D imaging system. There was an average reduction in volume of over 27%, and a reduction in height of almost 17% for the nine patients. Figure 19 below provide a graph depicting the average change in scar volume at six weeks.
Figure 19
Importantly, there were no unexpected treatment-related adverse events and patients reported little pain from the treatment, so with the primary endpoint of this study being to establish safety and tolerability of RAP for this indication, we believe we have met that endpoint.
Reduction of Subcutaneous Fat
The aesthetic device market for subcutaneous fat reduction is dominated by a technology branded as CoolSculpting®, which is owned by Allergan. The CoolSculpting technology centers around a process Allergan calls Cryolipolysis® and utilizes cooling plates against which a patient’s skin is held by vacuum. The objective of this method is to cause the death of subcutaneous fat cells, which are then absorbed by the body over a period of 90 days, resulting in an overall reduction in fat volume. While this method has enjoyed market success, its efficacy has been limited by the relative percentage of fat reduction it can achieve (about 20% to 25% as reported by Allergan) and the uniformity or smoothness of the resulting skin area after treatment.
Following the success of the CoolSculpting procedure, competing methods of reducing subcutaneous fat have also been introduced. One of the more successful competing technologies has been a procedure called SculpSure® from the Cynosure division of Hologic, a leading laser manufacturer. SculpSure relies on the use of heat generated from laser energy rather than Cryolipolysis.
In vitro and in vivo testing with higher-energy versions of our acoustic shockwave device suggests that Soliton shockwaves may have an effect on subcutaneous fat cells that may be beneficial to the current method of subcutaneous fat reduction. In light of this, we have entered into a series of small clinical trials with a large global aesthetics company to test whether or not this is the case in human subjects. These trials are early stage and intended as a proof-of-concept to determine if expanded human trials are warranted.
Reduction of Skin Laxity
We also believe our mechanism of action may play a role in reducing skin laxity, adding yet another important potential new indication for our technology.
Another of our animal studies shows the apparent potential of our higher-energy device treatments to strengthen the ECM (extracellular matrix) in pig skin, which independent research has suggested may lead to increased skin stiffness and uniformity in humans. As shown in Figure 20, the septa in the adipose layer demonstrate increased thickening over time with repeated acoustic shockwave treatments. The histology image on the left was before any treatment. The image in the middle was after a single acoustic shockwave treatment. The histology image on the far right was after multiple acoustic shockwave treatments. We believe the increase in septa thickening should lead to increased skin stiffness and uniformity.
Figure 20
In addition to the physician assessment that will be done for cellulite improvement during our pivotal cellulite study described above, we are also measuring and assessing improvement in skin laxity for these patients.
Potential Indications
Other Fibrotic Disorders
The results observed in our cellulite and keloid proof-of-concept study support our belief that our technology may potentially have an impact on a much broader set of fibrotic conditions. Fibrosis plays an important role in many different pathologies. It results from tissue injury, chronic inflammation,autoimmune reactions and genetic alterations, and it is characterized by the excessive growth of extracellular matrix ("ECM") components. Scientific publications suggest that fibroblasts become over-active when they are located in a stiffened environment and that disrupting the stiff environment may lead to fibroblast apoptosis, ultimately resulting in a resolution of the fibrosis.
In normal wound healing, myofibroblasts are required for tissue repair. To repair, regenerate and restore equilibrium after injury, tissue-resident fibroblasts are activated and transform into myofibroblasts. However, in certain conditions, activated myofibroblasts become the critical effectors of fibrotic disorders. In fibrotic disease progression, mechanical stresses in the surrounding microenvironment are a key mediator in the differentiation of myofibroblasts.
For fibroblasts and myofibroblasts, mechanical stress can regulate the production of ECM proteins indirectly, by stimulating the release of a paracrine growth factor, or directly, by triggering an intracellular signaling pathway that activates the genes that produce ECM proteins and growth factors. Focal adhesions at the cellular surface allow mechanical tension generated in the system to be transduced to the cytoskeletal network. These changes create a sensitivity to mechanical tension that transmits to the cell via signaling that ultimately triggers fibroblast differentiation to myofibroblast, and wound contraction with excess collagen.
The alteration in the ECM biomechanical properties, stiffness in particular, may be an important therapeutic target that is able to modulate myofibroblast formation and fibrosis. Studies suggest that fibroblasts cultured on low modulus substrates can maintain a normal phenotype. However, when cultured on stiffer substrates they are activated to myofibroblasts. Importantly, when cultured on a flexible or less stiff substrate, the myofibroblast activation was reversible.
The unfocused, non-cavitating, rapid pulse acoustic shockwaves of our RAP technology, when applied to tissue, cause a disruption in tissue structures. This disruption of tissue structures results in a loss of mechanical stiffness in the treated structure. As a result, based on published studies, the activated myofibroblasts found in fibrotic tissue can be pushed into a apoptotic state leading to a reduction of fibrosis.
On this basis, we believe that our technology could have efficacy in a number of fibrotic diseases both in the extracellular matrix, such as Radiation Induced Fibrosis and Capsular Contracture, and in other systems of the body such as Peripheral Artery Disease and even Non-Alcoholic SteatoHepatitis ("NASH"). To date, we have not begun any substantive pre-clinical work on these indications.
Capsular Contracture
Breast augmentation is one of the most commonly performed cosmetic procedures. As with any surgery, implant based breast augmentation has been associated with a number of risks and complications. The most common complication is capsular contracture as identified in a 25 years longitudinal study by Handel et al. Introduction of non-biologic materials into the body always induces formation of a capsule, but in the breast this may be particularly severe. Capsular contracture is a local complication thought to occur due to an excessive fibrotic foreign body reaction to the implant. It is thought to be an inflammatory reaction which causes fibrosis through the production of collagen, leading to excessively firm and painful breasts If severe enough, this can require reoperation
Individual studies have published incidence rates of capsular contracture ranging from 2.8% to 20.4%. A systematic review published a combined overall rate of 3.6% following augmentation surgery.
We believe that the RAP technology could be used to break up the fibrotic capsule and aid in the reversal of the contracture. We have initiated in vitro testing on stand-alone implants to determine whether our device causes any disruption in the integrity of the implant itself.
Radiation Induced Fibrosis
Radiation-induced fibrosis ("RIF") is a long-term side effect of external beam radiation therapy for the treatment of cancer. It results in a multitude of symptoms that significantly impact quality of life. RIF is the result of a misguided wound healing response. In addition to causing direct DNA damage, ionizing radiation generates reactive oxygen and nitrogen species
that lead to localized inflammation. This inflammatory process ultimately evolves into a fibrotic one characterized by increased collagen deposition, poor vascularity, and scarring.
We believe the mechanism of action that has driven a response to our technology in fibrotic scars may have a similar affect on fibrosis induced by radiation therapy. To date, we have not begun any substantive pre-clinical work on this indication.
Peyronie's Disease
Peyronie’s disease ("PD"), described by and named after Francois Gigot de la Peyronie, is a localized connective tissue disorder that arises from plaque formation, caused by the deposition of collagen and fibrin in the tunica albuginea of the penis. This fibrous plaque replaces the normally elastic fibers and can result in penile deformity. The disease is characterized by an initial or acute inflammatory phase which usually lasts about 12–18 months where the clinical hallmarks are unstable penile deformity and pain on erection. The stable or chronic phase begins when the acute phase subsides and it is characterized by stable penile deformity. The etiology of PD has not been fully elucidated, but one hypothesis is that PD is a disorder of wound healing.
More recently, a web-based survey of a large (n = 11,420) probability-based panel of research subjects representative of the full US population estimated the prevalence of PD to range from 0.5% (the percentage of surveyed subjects with PD diagnosis) to 13% (percentage with diagnosis, treatment, or penile symptoms of PD).
We believe the mechanism of action that has driven a response to our technology in fibrotic scars may have a similar affect on fibrosis seen in Peyronie's Disease. To date, we have not begun any substantive pre-clinical work on this indication.
Peripheral artery disease ("PAD")
Peripheral artery disease is a circulatory disease in which plaque builds up in the arteries carrying blood from heart to legs, arms, and other limbs. Increase in incidence of population suffering from diabetes and high blood pressure which poses a high risk factor for PAD drives the increase in suffering population.
Independent research indicates that the global PAD Market was valued at $3.1 billion in 2016, and is estimated to reach $4.9 billion by 2023, growing at a CAGR of 6.8% from 2017 to 2023.
We believe the mechanism of action that has driven a response to our technology in fibrotic scars may have a similar affect on fibrosis seen in calcified PAD. To date, we have not begun any substantive pre-clinical work on this indication.
Liver Fibrosis
Liver fibrosis occurs when repetitive or long-lasting injury or inflammation causes excessive amounts of scar tissue to build up in the organ. Most types of chronic liver disease can eventually cause fibrosis. Scar tissue from fibrosis can also block or limit the flow of blood within the liver. This can starve and eventually kill healthy liver cells, creating more scar tissue in the process. Treatment tends to involve clearing infections, making lifestyle changes, and taking certain medications. This can often reverse the damage of mild to moderate liver fibrosis. If inflammation continues, possibly because a person has not received treatment, liver fibrosis can develop into more serious liver conditions.
The most common causes of liver fibrosis in the U.S. are:
•chronic alcohol abuse
•viral hepatitis C or B
•nonalcoholic fatty liver disease (NAFLD)
•NASH, a subtype of NAFLD
For example, NASH is a form of liver disease that develops in patients who are not alcoholic or consume little alcohol. NASH is one of the common liver diseases, often called silent liver disease. About 20% of people with NASH will go on to develop scarring (fibrosis) of the liver, which is known as cirrhosis when it becomes severe enough to affect the liver’s function. Independent research indicates that the global NASH market generated $1.1 billion in 2017, and is projected to reach $21.5 billion by 2025, growing at a CAGR of 58.4% from 2021 to 2025.
We believe the mechanism of action that has driven a response to our technology in fibrotic scars may have a similar affect on fibrosis seen in Liver Fibrosis. To date, we have not begun any substantive pre-clinical work on this indication.
Patents and Proprietary Technology
To establish and protect our proprietary technologies and products, we rely on a combination of patent, copyright, trademark, and trade-secret laws, as well as confidentiality provisions in our contracts. We have implemented a patent strategy designed to protect our technology and facilitate commercialization of our current and future products. In total, we have eight patent families pending relating to the technologies that make our RAP device and certain variations possible, as well as various applications of our technology, with still more potential patent applications under way. As of December 31, 2019, our patent portfolio is comprised of 11 pending U.S. patent applications, 28 granted and 59 pending foreign counterpart patent applications, and three pending PCT patent applications, each of which we either own directly or we are the exclusive licensee. Our intellectual property portfolio for our core RAP technology was built through the combination of licensing patents from third parties and the issuance or filing of new patent applications by us as the result of our ongoing development activities. Our pending patents were exclusively licensed from MD Anderson and generally relate to early variations of our core technology relating to our acoustic shockwave platform. In general, patents have a term of 20 years from the application filing date or earliest claimed priority date.
We also rely on trade secrets, technical know-how, contractual arrangements, and continuing innovation to protect our intellectual property and maintain our competitive position. We have a policy to enter into confidentiality agreements with third parties, employees, and consultants. We also have a policy that our employees and consultants sign agreements requiring that they assign to us their interests in intellectual property such as patents and copyrights arising from their work for us. It is our policy that all employees sign an agreement not to compete unfairly with us during their employment and upon termination of their employment through the misuse of confidential information, soliciting employees, and soliciting customers.
We have registered “Soliton” as a trademark in the United States, “soliton.com” is a URL registered in the name of Soliton, Inc. and our logo and product designs are protected by copyright. Additionally, we have also applied to register the “Soliton” trademark in 11 other foreign countries. These trademark applications have been allowed in the United States and registered in thirteen other countries. We have also registered "Acoustic Subcision" as a trademark in the United States.
MD Anderson License Agreement
On April 5, 2012, we entered into a Patent and Technology License Agreement with MD Anderson. Pursuant to the agreement, we obtained a royalty-bearing, worldwide, exclusive license to intellectual property including patent rights related to the patents and technology we use. Under the agreement, we agreed to pay a nonrefundable license documentation fee in the high-five digits 30 days after the effective date of the agreement. Additionally, we agreed to pay a nonrefundable annual maintenance fee starting on the third anniversary of the effective date of the agreement, which escalates each anniversary and is currently in the high-five digits. Additionally, we agreed to a running royalty percentage of net sales in the mid-single digits. We also agreed to make certain milestone payments in the low to mid-six digits and sublicensing payments, including a $250,000 milestone payment made in June 2019 after we received FDA clearance for our RAP device for tattoo removal. The specific patents initially subject to the agreement expire between 2031 and 2032.
MD Anderson has the right to terminate the agreement upon advanced notice in the event of a default by Soliton. The agreement will expire upon the expiration of the licensed intellectual property. The rights obtained by us pursuant to the agreement are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between MD Anderson and the U.S. government. To the extent that is the case, our license agreement with, and the intellectual property rights we have licensed from, MD Anderson are subject to such a funding agreement and any superior rights that the U.S. government may have with respect to the licensed intellectual property. Therefore, there is a risk that the intellectual property rights we have licensed from MD Anderson may be non-exclusive or void if a funding agreement related to the licensed technology between MD Anderson and the U.S. government does exist and depending on the terms of such an agreement. Notwithstanding the foregoing, we do not believe our RAP technology received any federal funding. All out-of-pocket expenses incurred by MD Anderson in filing, prosecuting and maintaining the licensed patents have been and shall continue to be assumed by the Company.
Manufacturing
We currently partner with outsourced engineering and manufacturing companies for the development and commercialization of the RAP device. Our manufacturing partner, Sanmina, is one of the world’s largest medical device manufacturers. We have worked with Sanmina on the development of the device and will partner with their engineering team and other outside contractors as we make changes to the device to insure ease of manufacturing before our commercial launch. Once we have launched the device, our intent is that Sanmina will continue to function as our contract manufacturer.
Employees
As of December 31, 2019, we had ten employees, including nine full-time employees and one part-time employee, and accordingly, a high percentage of the work performed for our development projects is outsourced to qualified independent contractors. None of our employees are unionized.
Competitors
The medical device industry is subject to intense competition. Our products will compete against stand-alone laser treatments offered by offered by Hologic (Cynosure), Cutera, Lumenis, Candela and Laserscope, as well as several smaller highly-specialized companies. We intend to compete primarily on the basis of improved time to remove, reduced pain, reduced chance of scarring and reduced trips to the doctor. In addition, competition among providers of devices for the aesthetic market is characterized by extensive research efforts and rapid technological progress. To compete effectively, we must demonstrate that our products are attractive alternatives to other laser-only methods for tattoo removal. Additionally, there are many companies, both public and private, that are developing devices that use both laser-based and alternative technologies for the conditions treated by our products that may prove to be more effective, safer or less costly than our products. Many of these competitors have significantly greater financial and human resources than we do and have established reputations as well as worldwide distribution channels that are more effective than ours. Additional competitors may enter the market, and we are likely to compete with new companies in the future. We expect to encounter potential customers that, due to existing relationships with our competitors, are committed to or prefer the products offered by these competitors. We expect that competitive pressures may result in price reductions, reduced margins and loss of market share. There can be no assurance that competitors, many of which have made substantial investments in competing technologies, will not prevent, limit or interfere with our ability to make, use or sell our products either in the United States or in international markets.
A company called OnLight (recently acquired by Merz Pharma) introduced a transparent patch infused with a clear chemical called Perfluorodecalin (“PFD”). The DESCRIBE® PFD Patch is a single-use, optical clearing device accessory for use in laser-assisted tattoo removal procedures and is now marketed by Merz Aesthetics. Side effects, including pain, erythema and edema were reported during laser tattoo removal. The DESCRIBE® PFD Patch is available only through licensed physicians. They claim to speed the time to clearance of a tattoo by absorbing laser-induced whitening and allowing for immediate re-treatment.
Some patients may choose to have their tattoo surgically excised by a plastic surgeon or dermatologist. As of December 31, 2019, the FDA has not approved or cleared any do-it-yourself tattoo removal ointments or creams.
The cellulite removal market is highly competitive and has numerous device companies in the space. The technologies currently being used vary significantly in approach, efficacy and invasiveness to the patient.
A technology called Cellfina, owned by Merz Pharma, is currently being marketed as a long-term solution for the dimples caused by cellulite. The treatment requires injected anesthesia be given to the patient prior to the portion of the skin to be treated being pulled upward utilizing a suctioning plate. A lance is inserted into the side of the treatment area which is used to slice through the septa that are causing the dimple(s). The patient is required to rest for the 24 hours following the procedure and is often bruised and discolored for significantly longer.
Cellulaze, a technology owned by Cynosure, delivers a therapy similar to Cellfina using laser energy. A very small cannula (or tube about the size of the tip of a pen) is inserted under the skin. The laser fiber delivers energy directly under the skin. This is intended to increase the thickness and quality of the patient's skin, while simultaneously releasing the septa. As with Cellfina, patients will experience bruising and discomfort, as well as fluid drainage from the incision sites.
BTL Aesthetics owns a technology called Emtone™, which is intended to provide a temporary reduction in the appearance of cellulite. Emtone simultaneously emits both radiofrequency and targeted pressure energy. There are a number of other radiofrequency devices in the market intended to provide a temporary improvement in the appearance of cellulite.
Regulation of Our Business
Our product candidate and operations are subject to extensive and rigorous regulation by the U.S. Food and Drug Administration (“FDA”), under the Federal Food, Drug, and Cosmetic Act (“FDCA”), and it’s implementing regulations, guidance documentation, and standards. Our RAP device is regulated by the FDA as a medical device. The FDA regulates the design, development, research, testing, manufacturing, safety, labeling, storage, record keeping, promotion, distribution, sale and advertising of medical devices in the United States to ensure that medical products distributed domestically are safe and effective for their intended uses. The FDA also regulates the export of medical devices manufactured in the United States to international markets. Any violations of these laws and regulations could result in a material adverse effect on our business, financial condition and results of operations. In addition, if there is a change in law, regulation or judicial interpretation, we
may be required to change our business practices, which could have a material adverse effect on our business, financial condition and results of operations.
Unless an exemption applies, before we can commercially distribute medical devices in the United States, we must obtain, depending on the type of device, either prior premarket clearance or premarket approval, ('PMA"), from the FDA. The FDA classifies medical devices into one of three classes:
•Class I devices, which are subject to only general controls (e.g., labeling, medical devices reporting, and prohibitions against adulteration and misbranding) and, in some cases, to the premarket clearance requirements;
•Class II devices, generally requiring premarket clearance before they may be commercially marketed in the United States; and
•Class III devices, consisting of devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a predicate device, generally requiring submission of a PMA supported by clinical trial data.
Our current product candidates, including the RAP device, are all class II devices and will require submission of a premarket notification.
510(k) Clearance Pathway
When a 510(k) clearance is required, we must submit a premarket notification demonstrating that our proposed device is substantially equivalent to a previously cleared 510(k) device or a device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for the submission of PMAs. By regulation, the FDA is required to clear or deny a 510(k) premarket notification within 90 days of submission of the application. As a practical matter, clearance may take longer. The FDA may require further information, including clinical data, to make a determination regarding substantial equivalence.
Any modification to a 510(k)-cleared device that would constitute a major change in its intended use, or any change that could significantly affect the safety or effectiveness of the device, requires a new 510(k) clearance and may even, in some circumstances, require a PMA, if the change raises complex or novel scientific issues or the product has a new intended use. The FDA requires every manufacturer to make the determination regarding the need for a new 510(k) submission in the first instance, but the FDA may review any manufacturer's decision.
Premarket Approval (“PMA”) Pathway
A PMA must be submitted to the FDA if the device cannot be cleared through the 510(k) process. A PMA must be supported by extensive data, including but not limited to, technical, preclinical, clinical trials, manufacturing and labeling to demonstrate to the FDA's satisfaction the safety and effectiveness of the device for its intended use. During the review period, the FDA will typically request additional information or clarification of the information already provided. Also, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. The FDA may or may not accept the panel's recommendation. In addition, the FDA will generally conduct a pre-approval inspection of the manufacturing facility or facilities to ensure compliance with the QSRs.
New PMAs or PMA supplements are required for modifications that affect the safety or effectiveness of the device, including, for example, certain types of modifications to the device's indication for use, manufacturing process, labeling and design. PMA supplements often require submission of the same type of information as a PMA, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA and may not require as extensive clinical data or the convening of an advisory panel.
de novo Classification
Medical device types that the FDA has not previously classified as Class I, II or III are automatically classified into Class III regardless of the level of risk they pose. The Food and Drug Administration Modernization Act of 1997 established a new route to market for low to moderate risk medical devices that are automatically placed into Class III due to the absence of a predicate device, called the “Request for Evaluation of Automatic Class III Designation,” or the de novo classification procedure. This procedure allows a manufacturer whose novel device is automatically classified into Class III to request down-classification of its medical device into Class I or Class II on the basis that the device presents low or moderate risk, rather than requiring the submission and approval of a PMA application. Prior to the enactment of the Food and Drug Administration
Safety and Innovation Act of 2012, (the "FDASIA"), a medical device could only be eligible for de novo classification if the manufacturer first submitted a 510(k) premarket notification and received a determination from the FDA that the device was not substantially equivalent. FDASIA streamlined the de novo classification pathway by permitting manufacturers to request de novo classification directly without first submitting a 510(k) premarket notification to the FDA and receiving a not substantially equivalent determination. Under FDASIA, the FDA is required to classify the device within 120 days following receipt of the de novo application. If the manufacturer seeks reclassification into Class II, the manufacturer must include a draft proposal for special controls that are necessary to provide a reasonable assurance of the safety and effectiveness of the medical device. In addition, the FDA may reject the reclassification petition if it identifies a legally marketed predicate device that would be appropriate for a 510(k) or determines that the device is not low to moderate risk or that general controls would be inadequate to control the risks and special controls cannot be developed.
Clinical Trials
Clinical trials are generally required to support a PMA application and are sometimes required for 510(k) or de novo clearance. Such trials generally require an investigational device exemption application, ("IDE"), approved in advance by the FDA for a specified number of patients and study sites, unless the product is deemed a nonsignificant risk device eligible for more abbreviated IDE requirements. Clinical trials are subject to extensive monitoring, record keeping and reporting requirements. Clinical trials must be conducted under the oversight of an institutional review board, ("IRB"), for the relevant clinical trial sites and must comply with FDA regulations, including but not limited to those relating to good clinical practices. To conduct a clinical trial, we also are required to obtain the patients' informed consent in form and substance that complies with both FDA requirements and state and federal privacy and human subject protection regulations. We, the FDA or the IRB could suspend a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits. Even if a trial is completed, the results of clinical testing may not adequately demonstrate the safety and efficacy of the device or may otherwise not be sufficient to obtain FDA approval to market the product in the U.S. Similarly, in Europe the clinical study must be approved by a local ethics committee and in some cases, including studies with high-risk devices, by the ministry of health in the applicable country.
Pervasive and Continuing Regulation
After a device is placed on the market, numerous regulatory requirements apply. These include:
•Product listing and establishment registration, which helps facilitate FDA inspections and other regulatory action;
•Quality System Regulation, ("QSR"), which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the manufacturing process;
•labeling regulations and FDA prohibitions against the promotion of products for uncleared, unapproved or off-label use or indication;
•clearance of product modifications that could significantly affect safety or efficacy or that would constitute a major change in intended use of one of our cleared devices;
•approval of product modifications that affect the safety or effectiveness of one of our approved devices;
•medical device reporting regulations, which require that manufacturers comply with FDA requirements to report if their device may have caused or contributed to a death or serious injury, or has malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction of the device or a similar device were to recur;
•post-approval restrictions or conditions, including post-approval study commitments;
•post-market surveillance regulations, which apply when necessary to protect the public health or to provide additional safety and effectiveness data for the device;
•the FDA's recall authority, whereby it can ask, or under certain conditions order, device manufacturers to recall from the market a product that is in violation of governing laws and regulations;
•regulations pertaining to voluntary recalls; and
•notices of corrections or removals.
Advertising and promotion of medical devices, in addition to being regulated by the FDA, are also regulated by the Federal Trade Commission and by state regulatory and enforcement authorities. Recently, promotional activities for FDA-
regulated products of other companies have been the subject of enforcement action brought under healthcare reimbursement laws and consumer protection statutes. In addition, under the federal Lanham Act and similar state laws, competitors and others can initiate litigation relating to advertising claims. In addition, we are required to meet regulatory requirements in countries outside the U.S., which can change rapidly with relatively short notice. If the FDA determines that our promotional materials or training constitutes promotion of an unapproved use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions.
Furthermore, our products could be subject to voluntary recall if we or the FDA determine, for any reason, that our products pose a risk of injury or are otherwise defective. Moreover, the FDA can order a mandatory recall if there is a reasonable probability that our device would cause serious adverse health consequences or death.
The FDA has broad post-market and regulatory enforcement powers. Once we have a marketed product, we will be subject to unannounced inspections by the FDA to determine our compliance with the QSR and other regulations, and these inspections may include the manufacturing facilities of some of our subcontractors. Failure by us or by our suppliers to comply with applicable regulatory requirements can result in enforcement action by the FDA or other regulatory authorities, which may result in sanctions including, but not limited to:
•untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties;
•unanticipated expenditures to address or defend such actions
•customer notifications for repair, replacement, refunds;
•recall, detention or seizure of our products;
•operating restrictions or partial suspension or total shutdown of production;
•refusing or delaying our requests for premarket clearance or premarket approval of new products or modified products;
•operating restrictions;
•withdrawing premarket clearances or PMA approvals that have already been granted;
•refusal to grant export approval for our products; or
•criminal prosecution.
Available Information
Our Internet address is www.soliton.com. On this Web site, we post the following filings as soon as reasonably practicable after they are electronically filed with or furnished to the U.S. Securities and Exchange Commission (“SEC”): our Annual Reports on Form 10-K; our Quarterly Reports on Form 10-Q; our Current Reports on Form 8-K; our proxy statements related to our annual stockholders’ meetings; and any amendments to those reports or statements. All such filings are available on our Web site free of charge. The charters of our audit, nominating and governance and compensation committees and our Code of Business Conduct and Ethics Policy are also available on our Web site and in print to any stockholder who requests them. The content on our Web site is not incorporated by reference into this Form 10-K.
Item 1A. Risk Factors.
An investment in our securities involves a high degree of risk. You should consider carefully all of the material risks described below, together with the other information contained in this Form 10-K. If any of the following events occur, our business, financial condition, results of operations and cash flows may be materially adversely affected.
RISK FACTORS RELATING TO OUR BUSINESS AND THE INDUSTRY IN WHICH WE OPERATE
We will require substantial additional funding, which may not be available to us on acceptable terms, or at all, and, if not so available, may require us to delay, limit, reduce or cease our operations.
We have used the proceeds from our IPO and recent financings to advance our initially approved RAP device through the FDA clearance process and commercial development in preparation for commercial launch with a Generation 2 device. The Generation 2 device that we intend to offer in our initial commercial launch, will have significant changes from the Generation 1 device we have submitted for FDA review and clearance. The changes made to our device from Generation 1 to Generation 2 necessitated the filing of an additional 510(k) before being launched. We cannot be certain that the changes we deem appropriate to make to future RAP devices prior to the launch of any of these device will not require another 510(k) filing. Commercializing and launching medical device products can be expensive. Our current available cash is not sufficient to complete the commercialization and launch of our product. We will require substantial additional future capital in order to complete commercialization, launch and market the device nationwide, build out a sales force and manufacture the device. We will continue to require substantial additional capital to continue commercialization activities.
We have determined there is substantial doubt about our ability to continue as a going concern; as a result, we could have difficulty finding additional financing.
Our financial statements have been prepared assuming that we will continue as a going concern. We have not generated any revenue from our main operations since inception and have accumulated losses. Our ability to continue our operations depends on our ability to complete equity or debt financings or generate profitable operations. Such financings may not be available or may not be available on reasonable terms. Our financial statements do not include any adjustments that could result from the outcome of this uncertainty. If we are unable to raise sufficient capital when needed, our business, financial condition and results of operations will be materially and adversely affected, and we will need to significantly modify our operational plans to continue as a going concern. If we are unable to continue as a going concern, we might have to liquidate our assets and the values we receive for our assets in liquidation or dissolution could be significantly lower than the values reflected in our financial statements. The inclusion of a going concern explanatory paragraph by our auditors, our lack of cash resources and our potential inability to continue as a going concern may materially adversely affect our share price and our ability to raise new capital or to enter into critical contractual relations with third parties.
We anticipate needing additional financing over the longer term to execute our business plan and fund operations, which additional financing may not be available on reasonable terms or at all.
As of December 31, 2019, we had total assets of $13,140,973, including cash, cash equivalents and restricted cash of $12,076,425. We have an accumulated deficit as of December 31, 2019, of $56,043,371. Our cash on hand of $10,081,593 at February 11, 2020 is expected to fund our operations into the third quarter of 2020, but not beyond. We believe that we will require additional capital to mount a major sales and marketing effort and execute our business plan. We cannot give any assurance that we will be able to obtain all the necessary funding that we may need. We may pursue additional funding through various financing sources, including additional public offerings, the issuance of debt securities, fees associated with licensing some or all of our technology, joint ventures with capital partners and project type financing. There can be no assurance that funds will be available on commercially reasonable terms, if at all. If financing is not available on satisfactory terms, we may be unable to further pursue our business plan and we may be unable to continue operations, in which case you may lose some or all of your investment. Alternatively, we may consider changes in our business plan that might enable us to achieve aspects of our business objectives and lead to some commercial success with a smaller amount of capital, but we cannot assure that changes in our business plan will result in revenues or maintain any value in your investment.
We have a limited operating history and we expect a number of factors to cause our operating results to fluctuate on an annual basis, which may make it difficult to predict our future performance.
We formed our corporation in 2012 without a working RAP prototype. During the first 5 years of operations, we focused on research and development of a fully-integrated working prototype of the RAP device to remove tattoos. During the past 2 years, we have focused our efforts on developing a commercial device that would receive FDA clearance to sell. We applied for FDA clearance in March 2019; after such application our efforts will be focused on refining our commercial device to improve ease of use features necessary for adoption in dermatological settings. Developing this commercial device for our market launch is anticipated to cost at least $2.6 million and is expected to be completed in the first half of 2020. Further refinement to the device to develop the GEN 3.0 device is anticipated to cost at least an additional $2.5 million and take another year of additional work. Additionally, a high percentage of our expenses will be associated with pre-launch marketing activities as well as fixed costs. We have not yet sold any products, and we may never achieve commercial success with RAP technology. We have limited historical financial data upon which we may base our projected revenue and operating expenses. Our limited operating history makes it difficult for potential investors to evaluate our technology or prospective operations and business prospects. As a pre-commercialization stage company, we are subject to all the risks inherent in business development, financing, unexpected expenditures, and complications and delays that often occur in a new business. Investors should evaluate an investment in us in light of the uncertainties encountered by developing companies in a competitive environment. There can be no assurance that our efforts will be successful or that we will ultimately be able to attain profitability.
RAP utilizes potentially dangerous energy levels and we could face liability for claims related to the RAP device that would be costly and would damage our reputation.
The acoustic shockwaves generated by our RAP device are the result of producing and directing electrical energy within the device's hand piece approaching 3,000 volts at 3,000 amps of current. Although the RAP device has been designed in accordance, and has been independently tested and found to comply, with the electrical and other safety requirements for comparable medical devices, we cannot be certain that such design and testing measures have identified every possible mode of failure. An unanticipated failure mode or misuse of the RAP device could potentially expose the operator or patient to hazardous and potentially lethal electrical shock and we could face liability for claims of injury or death and our ability to commercialize the RAP device could be materially harmed. In addition, such claims would damage our reputation and hinder our ability to commercialize the RAP device.
We cannot assure you that we will generate revenue or become profitable in the future.
Our products may never be cleared by the FDA or become commercially viable or accepted for use. We have incurred significant losses since our inception and expect to experience operating losses and negative cash flow for the foreseeable future. We expect to expend significant resources on hiring of personnel, continued scientific and product research and development, product testing and preclinical and clinical investigation, intellectual property development and prosecution, marketing and promotion, capital expenditures, working capital, general and administrative expenses, and fees and expenses associated with our capital raising efforts. We expect to incur costs and expenses related to consulting costs, hiring of scientists, engineers, science and other operational personnel, and the continued development of relationships with strategic partners.
The use of lasers to remove tattoos has inherent dangers and our device will be used in conjunction with lasers for tattoo removal.
Our RAP device uses rapid pulses of designed acoustic shockwaves to dramatically accelerate the removal of tattoos when used in conjunction with existing lasers. Specifically, our technology allows a doctor to treat a patient multiple times in a single office visit and significantly reduce the overall time it takes to remove a tattoo. Recognized and published (see "Complications of Tattoos and Tattoo Removal: Stop and Think Before you ink;" Khunger, Molpariya, & Khunger, 2015) adverse events of Q-switched laser tattoo removal include: pain; blistering; crusting; pinpoint hemorrhage; urticarial reaction; hypopigmentation; hyperpigmentation; leukotrichia; local-papule; plaques; darkening of tattoos; photoallergic reactions; systemic reactions; residual pigmentation; ghost images; scarring; and textural changes. These adverse events may be increased when multiple laser passes are used to remove a tattoo in a single session.
Because we have not yet launched the RAP device, we have been using our available capital resources for development of the commercial units and have not yet generated any revenues; therefore, we may not be able to continue as a going concern.
We are a pre-revenue stage medical device company, and do not expect to generate any revenues until our commercial RAP device, cleared by the FDA, has been updated for use in the field, had those updates cleared by the FDA, and are sold. Our ability to continue as a going concern is dependent upon our generating cash flow from sales that are sufficient to fund operations or finding adequate financing to support our operations. To date, we have had no revenues and have relied on equity-based financing from the sale of securities in private placements, the issuance of convertible and non-convertible notes, and proceeds from our IPO and private placements. Our sales plan may not be successful in achieving a sustainable business and revenues. We have no arrangements in place for all the anticipated required financing to be able to fully implement our business plan. If we are unable to continue as planned currently, we may have to curtail some or all of our business plan and operations. In such case, investors may lose some or all of their investment.
Our clinical experience with the RAP device is limited to black tattoos with one type of laser, and future trials may not result in similar results.
To date, our clinical trial data is limited to the use of the RAP device in conjunction with Q-Switched lasers treating primarily black tattoos. We do not have clinical data indicating the efficacy of the RAP device in conjunction with shorter pulse “Pico-Switched” lasers or in treating tattoo ink colors other than black. Although, based on animal and theoretical models, we believe RAP has the potential to be similarly effective in such instances, we cannot be certain. If it is not as effective in such instances, our ability to successfully commercialize the RAP device could be materially harmed.
Clinical trials may be necessary to support future product submissions to FDA. These clinical trials will be expensive and will require the enrollment of large numbers of patients, and suitable patients may be difficult to identify and recruit. Delays or failures in our clinical trials will prevent us from commercializing any modified or new products and will adversely affect our business, operating results and prospects.
Initiating and completing clinical trials necessary to support any future PMA applications, and additional safety and efficacy data beyond that typically required for a 510(k) clearance, for our possible future product candidates, will be time consuming and expensive and the outcome uncertain. Moreover, the results of early clinical trials are not necessarily predictive of future results, and any product we advance into clinical trials may not have favorable results in later clinical trials.
Conducting successful clinical studies will require the enrollment of large numbers of patients, and suitable patients may be difficult to identify and recruit. Patient enrollment in clinical trials and completion of patient participation and follow-up depends on many factors, including the size of the patient population, the nature of the trial protocol, the attractiveness of, or the discomforts and risks associated with, the treatments received by enrolled subjects, the availability of appropriate clinical trial investigators, support staff, and proximity of patients to clinical sites and able to comply with the eligibility and exclusion criteria for participation in the clinical trial and patient compliance. For example, patients may be discouraged from enrolling in our clinical trials if the trial protocol requires them to undergo extensive post-treatment procedures or follow-up to assess the safety and effectiveness of our products or if they determine that the treatments received under the trial protocols are not attractive or involve unacceptable risks or discomforts.
Development of sufficient and appropriate clinical protocols to demonstrate safety and efficacy are required and we may not adequately develop such protocols to support clearance and approval. Further, the FDA may require us to submit data on a greater number of patients than we originally anticipated and/or for a longer follow-up period or change the data collection requirements or data analysis applicable to our clinical trials. Delays in patient enrollment or failure of patients to continue to participate in a clinical trial may cause an increase in costs and delays in the approval and attempted commercialization of our products or result in the failure of the clinical trial. In addition, despite considerable time and expense invested in our clinical trials, FDA may not consider our data adequate to demonstrate safety and efficacy. Such increased costs and delays or failures could adversely affect our business, operating results and prospects.
If the third parties on which we rely to conduct our clinical trials and to assist us with pre-clinical development do not perform as contractually required or expected, we may not be able to obtain regulatory clearance or approval for or commercialize our products.
We do not have the ability to independently conduct our pre-clinical and clinical trials for our product candidates and future products and we must rely on third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories to conduct such trials. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if these third parties need to be replaced, or if the quality or
accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our pre-clinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize, our products on a timely basis, if at all, and our business, operating results and prospects may be adversely affected. Furthermore, our third-party clinical trial investigators may be delayed in conducting our clinical trials for reasons outside of their control.
The results of our clinical trials may not support our product candidate claims or may result in the discovery of adverse side effects.
Even though our first clinical trials are completed, we cannot be certain that their results will support our product candidate claims or that the FDA will agree with our conclusions regarding them. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and pre-clinical studies. The clinical trial process may fail to demonstrate that our product candidates are safe and effective for the proposed indicated uses, which could cause us to abandon a product candidate and may delay development of others. Any delay or termination of our clinical trials will delay the filing of our product submissions and, ultimately, our ability to commercialize our product candidates and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of the product candidate’s profile.
We may be required to suspend or discontinue clinical trials due to side effects or other safety risks that could preclude approval of our products.
Our clinical trials may be suspended at any time for a number of reasons. We may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to participants. In addition, regulatory agencies may order the temporary or permanent discontinuation of our clinical trials at any time if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements or that they present an unacceptable safety risk to participants.
We utilize a single manufacturer, Sanmina Corporation, for the manufacture of the RAP device and expect to continue to do so for commercial devices. Risks associated with the manufacturing of our products could reduce our gross margins and negatively affect our operating results.
We do not have any manufacturing facilities or direct manufacturing personnel. We currently rely, and expect to continue to rely, on Sanmina Corporation for the manufacture of the RAP device for commercial manufacture. Although Sanmina is a large contract manufacturer of medical devices, we are subject to numerous risks relating to our reliance on their manufacturing capabilities. If they encounter problems in manufacturing the RAP device then our business could be significantly impacted. These problems include:
•inability to secure product components in a timely manner, in sufficient quantities or on commercially reasonable terms;
•failure to increase production of the RAP device to meet demand;
•inability to modify production lines to enable us to efficiently produce future products or implement changes in current products in response to regulatory requirements;
•difficulty identifying and qualifying alternative manufacturers in a timely manner;
•inability to establish agreements with future third-party manufacturers or to do so on acceptable terms; or
•potential damage to or destruction of our manufacturers' equipment or facilities.
As demand for our products increases, our manufacturer will need to invest additional resources to purchase components, hire and train employees, and enhance their manufacturing processes. If they fail to increase production capacity efficiently, our sales may not increase in line with our expectations and our operating margins could fluctuate or decline. The RAP device has many parts that are specialized high-voltage components and many of these components are only produced by one supplier and the loss of any of these suppliers, or their inability to provide Sanmina with an adequate supply of materials, could harm our business. For our business strategy to be successful, Sanmina must be able to provide us with components in sufficient quantities, in compliance with regulatory requirements and quality control standards, in accordance with agreed upon specifications, at acceptable costs and on a timely basis. Future increases in sales of the RAP device could strain the ability of Sanmina to deliver an increasingly large supply of components and RAP systems in a manner that meets these various requirements. We do not have a long-term agreement with Sanmina and contract with Sanmina on a project-to-project basis
utilizing a separate purchase order for each project. As such, there is no assurance that Sanmina will continue to provide us with manufacturing services in the future.
We have limited experience in assembling and testing our products and may encounter problems or delays in the assembly of our products or fail to meet certain regulatory requirements which could result in an adverse effect on our business and financial results.
We have limited experience in assembling and testing our RAP device, and no experience in doing so on a commercial scale. To become profitable, we must assemble and test the RAP device in commercial quantities in compliance with regulatory requirements and at an acceptable cost. Increasing our capacity to assemble and test our products on a commercial scale will require us to improve internal efficiencies. We may encounter a number of difficulties in increasing our assembly and testing capacity, including:
•managing production yields;
•maintaining quality control and assurance;
•providing component and service availability;
•maintaining adequate control policies and procedures;
•hiring and retaining qualified personnel; and
•complying with state, federal and foreign regulations.
If we are unable to satisfy commercial demand for our RAP device due to our inability to assemble and test our RAP device, our ability to generate revenue would be impaired, market acceptance of our products could be adversely affected and customers may instead purchase or use, our competitors’ products.
Certain parts used in the manufacturing of our equipment may experience shortages in global supply which could impact our ability to manufacture our device for customers or maintain research and development timelines.
There are a number of component parts used in the manufacture of our device that are used by many manufacturers in a variety of products. We will compete with other manufacturers for the supply of these components. Additionally, certain parts that are currently in our design may be discontinued by our supplier requiring us to find alternative parts. This issue may require us to change the design of our device or purchase significant inventories of these parts in order to protect against manufacturing delays. We may not be able to procure alternative components or adequate raw material inventories which would result in an inability to produce our device.
We have limited sales, marketing, and distribution capabilities or arrangements, and will need to substantially build out these capabilities as we move towards commercialization of our products.
We do not yet have sales, marketing, and distribution capabilities or arrangements. To be able to commercialize our potential products, we will need to develop all of the foregoing. We have limited experience in establishing these capabilities, and therefore, we may be unsuccessful in achieving commercialization and earning revenues. We believe that setting up the commercialization parts of the Company will take substantial capital and commitment of time and effort. We may seek development and marketing partners for RAP technology and license technology that is complementary, but not directly associated with RAP technology to others in order to avoid our having to provide the marketing, manufacturing and distribution capabilities within our organization. There can be no assurance that we will find any development and marketing partners or companies that are interested in licensing our technology. If we are unable to establish and maintain adequate sales, marketing, manufacturing and distribution capabilities, independently or with others, we will not be able to generate product revenue, and may not become profitable.
Achieving and maintaining market acceptance of the RAP device for tattoo removal could be negatively impacted by many factors, which may prevent us from successfully commercializing the RAP device.
Even though the RAP device is cleared by the FDA, we may not be successful achieving market acceptance of the RAP device for tattoo removal. Many factors could negatively impact our ability to achieve or maintain market acceptance, including:
•the failure of the RAP device to achieve wide acceptance among people who regret having one or more tattoos or have a tattoo they would like to modify (prospective clients), dermatologists, and key opinion leaders in the tattoo removal community;
•possible reluctance by dermatologists to change their current practices because of perceived liability risks arising from the use of new products;
•perceived risks associated with the use of the RAP device or similar products or technologies generally;
•the introduction of competitive products and the rate of acceptance of those products as compared to the RAP device;
•adverse results of future clinical trials relating to the RAP device or similar competitive products; and
•adverse publicity or other adverse events including any product liability lawsuits.
If we are not successful in convincing prospective clients and dermatologists of the benefits of the RAP device then our sales potential, strategic objectives and profitability could be negatively impacted, which would adversely affect our business, financial condition and operating results.
If important assumptions we have made about what prospective clients want and are willing to purchase are inaccurate, our business and operating results may be adversely affected.
Our business strategy was developed based on a number of important assumptions about prospective clients, including their desire to have one or more tattoos removed, their reasons for not taking action to remove those tattoos to date and their willingness to pay for an improved method of removing their tattoos. These assumptions were based on published secondary research, as well as primary research commissioned by us. This research may be flawed and/or any of the resulting assumptions may prove to be inaccurate. If so, our efforts to commercialize the RAP device, even though cleared by the FDA, may fall short of expectations and you could lose some or all of your investment.
The sizes of the markets for our RAP device have not been established with precision, and may be smaller than we estimate.
Our estimates of the annual total addressable markets for our products, including those under development, are based on a number of internal and third-party estimates. While we believe our assumptions and the data underlying our estimates are reasonable, these assumptions and estimates may not be correct and the conditions supporting our assumptions or estimates may change at any time, thereby reducing the predictive accuracy of these underlying factors. If the actual number of people who would benefit from our products, the price at which we can sell our products, or the annual total addressable market for our products is smaller than we have estimated, it may impair our sales growth and have an adverse impact on our business.
All of our near-term indications are elective procedures that will be not be reimbursable and to the extent there is a general reduction in discretionary spending that could result in a reduction in the demand for these services.
The decision to undergo a procedure from our systems will be driven by consumer demand. Procedures performed using our systems will be elective procedures, the cost of which must be borne by the patient and are not reimbursable through government or private health insurance. In times of economic uncertainty or recession, individuals often reduce the amount of money that they spend on discretionary items, including aesthetic procedures. The general economic difficulties being experienced and the lack of availability of consumer credit for some of our customers' patients could adversely affect the markets in which we will operate.
We expect to operate in a highly competitive market, we may face competition from large, well-established medical device and product manufacturers with significant resources, and we may not be able to compete effectively.
A method for facilitating multiple laser passes in a single office visit by applying a chemically infused patch (PFD Patch) to the skin was introduced to the market within the last several years. Although we believe, based on currently available published clinical data for the PFD Patch, that the Soliton method is more effective than the PFD Patch, the company that owns the PFD Patch, Merz Pharma, has substantially more resources than Soliton. Furthermore, we have made this assessment based on separate clinical trials with differing protocols, not on a direct head-to-head comparison between the PFD Patch and the Soliton method, so we cannot be certain that the Soliton method is more effective. Also, there are currently a number of laser companies such as Lumenis, Cynosure (Hologic) and Cutera that market their lasers for tattoo removal and all of these companies have substantially more resources than Soliton. Furthermore, our clinical trials have demonstrated clinically significant improvement in tattoo fading over laser alone. Since we are pursuing FDA clearance for the RAP device to treat tattoos in conjunction with lasers, some of these companies may view our product as a competitive threat.
Also, there may be numerous companies of which we are not aware that may be working on separate technology for tattoo fading or removal. As well, the broader market for energy-based devices in the aesthetic market is becoming more competitive. Over time, we believe this field will become subject to more rapid change and new devices and products will emerge. We may find ourselves in competition with companies that have competitive advantages over us, such as:
•greater name recognition;
•established relations with dermatologists;
•established distribution networks;
•additional lines of products, and the ability to offer rebates, higher discounts or incentives to gain a competitive advantage; and
•greater financial and human resources for product development, sales and marketing, and patent litigation.
As a result, we may not be able to compete effectively against these companies or their devices and products.
Rapidly changing technology in life sciences could make the products we are developing obsolete.
The medical device and life-science industry in general is characterized by rapid and significant technological changes, frequent new product introductions and enhancements and evolving industry standards. Our future success will depend on our ability to continually develop and then improve the products that we design and to develop and introduce new products that address the evolving needs of our customers on a timely and cost-effective basis.
If we do not enhance our product offerings through our research and development efforts on a timely basis, we may fail to effectively compete or become profitable.
In order to capture and grow market share in the tattoo removal market, we will need to enhance and broaden our product offerings to meet the evolving demands of patients and dermatologists, as well as compete against new technologies. The success of the RAP device or future versions of the RAP device will depend on numerous factors, including our ability to:
•identify product enhancements that improve performance of tattoo removal and clinicians’ ability to use the device and successfully incorporate those features into our products;
•develop and introduce future generations of the RAP device in a timely manner;
•offer products at a price that is competitive with other products then available; and
•adequately protect our intellectual property and avoid infringing upon the intellectual property rights of third-parties.
We have in the past experienced, and we may in the future experience, delays in various phases of product development and commercial launch, including engineering, manufacturing, and marketing. Any delays in our anticipated product launches may significantly impede our ability to successfully compete in our markets. In particular, such delays could cause customers to delay or forego purchases of our products. Even if we are able to successfully develop the RAP device or future versions of the RAP device when anticipated, these products may not produce sales in excess of the costs of development, and they may be quickly rendered obsolete by the changing preferences of dermatologists and patients, or the introduction by our competitors of products embodying new technologies or features.
Potential complications from the RAP device or future versions of the RAP device may not be revealed by our clinical experience or other testing. Undetected errors or defects in the RAP device or future versions of the RAP device could harm our reputation, decrease the market acceptance of the RAP device or expose us to product liability claims.
Our RAP device is a highly complex device with many potential areas for undetected errors, defects or other complications. We cannot be certain that our clinical and other safety and efficacy testing has revealed all such complications. If such complications emerge in the future, we may not have sufficient resources to address them and our commercialization plans could be materially adversely affected.
If we lose key management personnel, or if we fail to recruit additional highly skilled personnel, our ability to expand our operations and increase the size of our company will be impaired, and we may experience loss of markets or market share and we may become less competitive.
As of December 31, 2019, we had ten employees, including nine full-time employees and one part-time employee. Because of our small size, growth in accordance with our business plan will place a significant strain on our financial, technical, operational and management resources. As we advance our product candidates through commercial development, launch and post-launch activities, we will need to increase our product development, scientific and administrative headcount to manage these programs.
We are highly dependent upon the principal members of our management team, scientific advisory board and consultants. These persons have significant experience not only in development, regulatory, commercialization and business development activities, but also with the RAP system, acoustic energy and the biology of tattoos. If we lose one or more of our executive officers or key employees or consultants, our ability to implement our business strategy successfully could be seriously harmed. Any of our executive officers or key employees or consultants may terminate their employment at any time. Replacing executive officers, key employees and consultants may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to develop, gain regulatory approval of and commercialize products successfully. Competition to hire and retain employees and consultants from this limited pool is intense, and we may be unable to hire, train, retain or motivate these additional key personnel and consultants. Our failure to retain key personnel or consultants could materially harm our business.
In addition, we have scientific and clinical advisors and consultants who assist us in formulating our regulatory and clinical strategies. These advisors are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us and typically they will not enter into non-compete agreements with us. If a conflict of interest arises between their work for us and their work for another entity, we may lose their services. In addition, our advisors may have arrangements with other companies to assist those companies in developing products or technologies that may compete with ours.
In addition, to meet our obligations as a public company, we may need to increase our general and administrative capabilities. Our management, personnel and systems currently in place may not be adequate to support this future growth. If we are unable to successfully manage this growth and increased complexity of operations, our business may be adversely affected.
If we are unable to establish good relationships with physicians, our business could be negatively affected.
Our business model will depend on the distribution of our RAP device into the offices of practicing dermatologists and other physicians. This will require us to build and maintain good relationships with physicians who will have a significant source of patients that will generate treatment revenues for both the physician and the Company. If we are unable to establish good relationships with physicians and maintain them, it will jeopardize both device and replaceable component revenues.
We may become subject to legal proceedings that could have a material adverse impact on our business, results of operations and financial condition.
From time to time and in the ordinary course of our business, we may become involved in various legal proceedings. All such legal proceedings are inherently unpredictable and, regardless of the merits of the claims, litigation may be expensive, time-consuming and disruptive to our operations and distracting to management. If resolved against us, such legal proceedings could result in excessive verdicts, injunctive relief or other equitable relief that may affect how we operate our business. Similarly, if we settle such legal proceedings, it may affect how we operate our business. Future court decisions, alternative dispute resolution awards, business expansion or legislative activity may increase our exposure to litigation and regulatory investigations. In some cases, substantial non-economic remedies or punitive damages may be sought. Although we maintain liability insurance coverage, there can be no assurance that such coverage will cover any particular verdict, judgment or settlement that may be entered against us, that such coverage will prove to be adequate or that such coverage will continue to remain available on acceptable terms, if at all. If we incur liability that exceeds our insurance coverage or that is not within the scope of the coverage in legal proceedings brought against us, it could have a material adverse effect on our business, results of operations and financial condition.
Economic uncertainty or economic deterioration could adversely affect us.
While the global economy is improving, there are still uncertainties surrounding the strength and duration of the recovery that may continue to drive stock market and interest rate volatility and adversely impact consumer confidence, product demand, and our ability to refinance our debt. Economic conditions, along with our operating performance, may also materially and adversely impact our ability to access the financial markets. Accordingly, our future business and financial results are subject to uncertainty. If economic conditions deteriorate in the future, our future revenues and financial results could be materially and adversely affected.
We have limited brand awareness and there is no assurance that we will be able to achieve brand awareness.
We have achieved limited brand awareness with respect to our RAP technology. There is no assurance that we will be able to achieve brand awareness. In addition, we must develop a successful market for our products in order to complete sales. If we are not able to develop successful markets for our products, then such failure will have a material adverse effect on our business, financial condition and operating results.
We may expend our limited resources to pursue a particular product or indication and fail to capitalize on products or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we focus on specific products, indications and discovery programs. As a result, we may forgo or delay pursuit of other opportunities with others that could have had greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular potential product, we may relinquish valuable rights to that potential product through future collaborations, licenses and other similar arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such potential product.
Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.
Our operations could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or man-made disasters or business interruptions, for which we are predominantly self-insured. We rely on third-party manufacturers to produce our products. Our ability to obtain clinical supplies of our products could be disrupted if the operations of these suppliers were affected by a man-made or natural disaster or other business interruption. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses.
RISK RELATED TO REGULATORY ISSUES
There is no guarantee that the FDA will grant 510(k) or de novo clearance or PMA approval of our future products and failure to obtain necessary clearances or approvals for our future products would adversely affect our ability to grow our business.
Our lead product candidate, as well as some of our future products, will require FDA clearance of a 510(k) or de novo application or may require FDA approval of a PMA. The FDA may not approve or clear these products for the indications that are necessary or desirable for successful commercialization. Indeed, the FDA may refuse our requests for premarket clearance or premarket approval of new products, new intended uses or modifications to existing products. Failure to receive clearance or approval for our products would have an adverse effect on our ability to continue or expand our business.
If we fail to obtain and maintain regulatory approvals and clearances, or are unable to obtain, or experience significant delays in obtaining, FDA clearances or approvals for our RAP device, our future products or product enhancements, our ability to commercially distribute and market these products could suffer.
Our products are subject to rigorous regulation by the FDA and numerous other federal, state and foreign governmental authorities. The FDA classifies medical devices into one of three classes on the basis of the intended use of the device, the risk associated with the use of the device for that indication, as determined by the FDA, and on the controls deemed by the FDA to be necessary to reasonably ensure their safety and effectiveness. Class I devices, which have the lowest level of risk associated with them, are subject to general controls. Class II devices are subject to general controls and special controls, including performance standards. Class III devices, which have the highest level of risk associated with them, are subject to general controls and premarket approval. Most Class I devices and some Class II devices are exempt from a requirement that
the manufacturer submit a premarket notification, or 510(k), and receive clearance from the FDA which is otherwise a premarketing requirement for a Class II device. Class III devices may not be commercialized until a premarket approval application, (" PMA"), is submitted to and approved by the FDA.
The process of obtaining regulatory clearances or approvals to market a medical device can be costly and time consuming, and we may not be able to obtain these clearances or approvals on a timely basis, if at all. The FDA will clear marketing of a lower risk medical device through the 510(k) process if the manufacturer demonstrates that the new product is substantially equivalent to other 510(k)-cleared products or through a de novo process if substantial equivalence is not available. High risk devices deemed to pose the greatest risk, such as life-sustaining, life-supporting, or implantable devices, or devices not deemed substantially equivalent to a previously cleared device, require the approval of a PMA. The PMA process is more costly, lengthy and uncertain than the 510(k) or de-novo clearance processes. A PMA application must be supported by extensive data, including, but not limited to, technical, preclinical, clinical trial, manufacturing and labeling data, to demonstrate to the FDA’s satisfaction the safety and efficacy of the device for its intended use. FDA also allows the submission of a direct de-novo petition. This procedure allows a manufacturer whose novel device is automatically classified into Class III to request down-classification of its medical device into Class I or Class II on the basis that the device presents low or moderate risk, rather than requiring the submission and approval of a PMA application. Prior to the enactment of the Food and Drug Administration Safety and Innovation Act of 2012 (“FDASIA”), a medical device could only be eligible for de-novo classification if the manufacturer first submitted a 510(k) premarket notification and received a determination from the FDA that the device was not substantially equivalent. FDASIA streamlined the de-novo classification pathway by permitting manufacturers to request de-novo classification directly without first submitting a 510(k) premarket notification to the FDA and receiving a not substantially equivalent determination. We believe our Generation 2 device will require clearance through the 510(k) or de-novo process, If the FDA determines that the Generation 2 of the RAP device should be considered a Class III device for treatment of future indications more medical in nature, we may be required to pursue a PMA, which could consume several years of additional approval time and considerable unanticipated expense.
U.S. legislative or FDA regulatory reforms or changes in internal FDA policies and procedures may make it more difficult and costly for us to obtain regulatory approval of our product candidates and to manufacture, market and distribute our products after approval is obtained.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulatory approval, manufacture and marketing of regulated products or the reimbursement thereof. In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of future products. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted, or FDA regulations, guidance or interpretations changed, and what the impact of such changes, if any, may be. Any change in the laws or regulations that govern the clearance and approval processes relating to our current and future products could make it more difficult and costly to obtain clearance or approval for new products, or to produce, market and distribute existing products. Significant delays in receiving clearance or approval, or the failure to receive clearance or approval for our new products would have an adverse effect on our ability to expand our business.
Our financial performance may be adversely affected by medical device tax provisions in the healthcare reform legislation.
The imposition of the 2.3% medical device excise tax enacted as part of the Affordable Care Act could adversely affect our financial results. Although the suspension of the excise tax was extended to the end of 2019, we do not know whether the suspension will continue beyond 2019. We may not be able to pass along the cost of the tax to our customers or offset the cost of the tax through higher sales volumes resulting from the expansion of health insurance coverage. Ongoing implementation of this legislation could have a material adverse effect on our business, financial condition and results of operations.
Modifications to our products may require new regulatory clearances or approvals or may require us to recall or cease marketing our products until clearances or approvals are obtained.
Modifications to our products may require new regulatory approvals or clearances, including 510(k) clearances or premarket approvals, or require us to recall or cease marketing the modified devices until these clearances or approvals are obtained. The FDA requires device manufacturers to initially make and document a determination of whether or not a modification requires a new approval, supplement or clearance. A manufacturer may determine that a modification could not significantly affect safety or efficacy and does not represent a major change in its intended use, so that no new 510(k) clearance is necessary. However, the FDA can review a manufacturer's decision and may disagree. The FDA may also, on its own initiative, determine that a new 510(k) clearance, de-novo submission or PMA approval is required. Once we have a
commercialized product, we may make modifications in the future that we believe do not or will not require additional clearances or approvals. If the FDA disagrees and requires new clearances or approvals for these modifications, we may be required to recall and to stop marketing our products as modified, which could require us to redesign our products and harm our operating results. In these circumstances, we may be subject to significant enforcement actions.
Where we determine that modifications to our products require a new 510(k) or de-novo clearance or PMA application, we may not be able to obtain those additional clearances or approvals for the modifications or additional indications in a timely manner, or at all. Obtaining clearances and approvals can be a time-consuming process, and delays in obtaining required future clearances or approvals would adversely affect our ability to introduce new or enhanced products in a timely manner, which in turn would harm our future growth.
The Generation 1 device, which has been cleared by the FDA for tattoo removal, is not ready for commercial launch and we will need to modify this device prior to commercial launch, which modifications may be unsuccessful or costly.
We conducted our clinical trials for the acceleration of tattoo removal with and have applied for premarket clearance based on a device that is not optimized for commercial launch. We have made certain modifications and we expect to make additional modifications to this Generation 1 device that include improvements in user interface, improvements to extend the life and ease of replacement of the consumable treatment head cartridges and general aesthetics, which will be made via a next genration devices intended for commercial market launch, respectively. The changes made to our device from Generation 1 to Generation 2 will necessitate the filing of an additional 510(k) before being launched. We cannot be certain that the changes we deem appropriate to make to the future RAP device prior to that product's launch will not require another 510(k) filing. While we believe these changes will not affect the therapy delivered by our RAP device, we may be unsuccessful or experience delays in making these changes and/or the FDA may require additional 510(k) submissions to properly document these changes.
Even if our products are cleared or approved by the FDA, if we or our suppliers fail to comply with ongoing FDA requirements, or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any product for which we obtain clearance or approval, and the manufacturing processes, reporting requirements, post-approval clinical data and promotional activities for such product, will be subject to continued regulatory review, oversight and periodic inspections by the FDA and other domestic and foreign regulatory bodies. In particular, we and our suppliers are required to comply with FDA’s Quality System Regulations, ("QSR"), which covers the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage and shipping of any product for which we obtain clearance or approval. FDA enforces the QSR and other regulations through periodic inspections. The failure by us or one of our suppliers to comply with applicable statutes and regulations administered by the FDA, or the failure to timely and adequately respond to any adverse inspection observations or product safety issues, could result in, among other things, any of the following enforcement actions:
•untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties;
•unanticipated expenditures to address or defend such actions;
•customer notifications for repair, replacement, refunds;
•recall, detention or seizure of our products;
•operating restrictions or partial suspension or total shutdown of production;
•refusing or delaying our requests for premarket clearance or premarket approval of new products or modified products;
•operating restrictions;
•withdrawing premarket clearances on PMA approvals that have already been granted;
•refusal to grant export approval for our products; or
•criminal prosecution.
If any of these actions were to occur, it would harm our reputation and cause our product sales and profitability to suffer and may prevent us from generating revenue. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with all applicable regulatory requirements which could result in our failure to produce our products on a timely basis and in the required quantities, if at all.
Even if regulatory clearance or approval of a product is granted, such clearance or approval may be subject to limitations on the intended uses for which the product may be marketed and reduce our potential to successfully commercialize the product and generate revenue from the product. If the FDA determines that our promotional materials, labeling, training or other marketing or educational activities constitute promotion of an unapproved use, it could request that we cease or modify our training or promotional materials or subject us to regulatory enforcement actions. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our training or other promotional materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement.
In addition, we may be required to conduct costly post-market testing and surveillance to monitor the safety or effectiveness of our products, and we must comply with medical device reporting requirements, including the reporting of adverse events and malfunctions related to our products. Later discovery of previously unknown problems with our products, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements such as QSR, may result in changes to labeling, restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recalls, a requirement to repair, replace or refund the cost of any medical device we manufacture or distribute, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties which would adversely affect our business, operating results and prospects.
Our products may in the future be subject to product recalls that could harm our reputation, business and financial results.
The FDA and similar foreign governmental authorities have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture. In the case of the FDA, the authority to require a recall must be based on an FDA finding that there is a reasonable probability that the device would cause serious injury or death. Manufacturers may, under their own initiative, recall a product if any material deficiency in a device is found. A government-mandated or voluntary recall by us or one of our distributors could occur as a result of component failures, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our products would divert managerial and financial resources and have an adverse effect on our financial condition and results of operations. The FDA requires that certain classifications of recalls be reported to FDA within 10 working days after the recall is initiated. Companies are required to maintain certain records of recalls, even if they are not reportable to the FDA. We may initiate voluntary recalls involving our products in the future that we determine do not require notification of the FDA. If the FDA disagrees with our determinations, they could require us to report those actions as recalls. A future recall announcement could harm our reputation with customers and negatively affect our sales. In addition, the FDA could take enforcement action for failing to report the recalls when they were conducted.
If our products cause or contribute to a death or a serious injury, or malfunction in certain ways, we will be subject to medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions.
Under the FDA medical device reporting regulations, medical device manufacturers are required to report to the FDA information that a device has or may have caused or contributed to a death or serious injury or has malfunctioned in a way that would likely cause or contribute to death or serious injury if the malfunction of the device or one of our similar devices were to recur. If we fail to report these events to the FDA within the required time frames, or at all, FDA could take enforcement action against us. Any such adverse event involving our products also could result in future voluntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection or enforcement action. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require the dedication of our time and capital, distract management from operating our business, and may harm our reputation and financial results.
RISKS RELATED TO OUR LICENSE AGREEMENT AND INTELLECTUAL PROPERTY
We have licensed certain intellectual property rights for our technology from MD Anderson, and if our license agreement with MD Anderson is terminated, our business will be materially harmed.
We are a party to a royalty-bearing, worldwide, exclusive license agreement for certain intellectual property rights, including patent rights related to RAP technology, with MD Anderson. If we become insolvent, cannot meet commercial diligence requirements contained in the license agreement, fail to make annual maintenance fee payments, or otherwise materially default without curing the default, the license agreement could be terminated. Furthermore, if we are successful in commercializing and selling the RAP device, we will owe milestone and royalty payments pursuant to this license agreement. If we fail to make those payments in accordance with the license agreement, our license could be terminated. If our license agreement with MD Anderson is terminated, our business will be materially harmed.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
We may from time to time seek to enforce our intellectual property rights against infringers when we determine that a successful outcome is probable and may lead to an increase in the value of the intellectual property. The validity of our patents and the patents we have licensed may be challenged in court and in administrative proceedings before the U.S. Patent and Trademark Office (“USPTO”) and other patent offices outside the United States. These lawsuits and proceedings are expensive and would consume time and resources and divert the attention of managerial and scientific personnel even if we were successful in stopping the infringement of such patents. In addition, there is a risk that the court will decide that such patents are not valid and that we do not have the right to stop the other party from using the inventions. There is also the risk that, even if the validity of such patents is upheld, the court will refuse to stop the other party on the ground that such other party's activities do not infringe our intellectual property rights.
If we are unable to protect the intellectual property used in our products, others may be able to copy our innovations which may impair our ability to compete effectively in our markets.
The strength of our patents involves complex legal and scientific questions and can be uncertain. We have eight families of patents. As of December 31, 2019, our patent portfolio is comprised of 11 pending U.S. patent applications, 28 granted and 59 pending foreign counterpart patent applications, and three pending PCT patent applications, each of which we either own directly or we are the exclusive licensee. These patent applications may be challenged or fail to result in issued patents, or if issued, these patents and our existing patents may be too narrow to prevent third-parties from developing or designing around our intellectual property and in that event, we may lose competitive advantage, which could result in harm to our business.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the medical device industry, we employ individuals who were previously employed at other medical device companies, including our competitors or potential competitors. We may be subject to claims that these employees, or we, have used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected.
In addition to patented technology, we rely upon, among other things, unpatented proprietary technology, processes, trade secrets and know-how. Any involuntary disclosure to or misappropriation by third-parties of our confidential or proprietary information could enable competitors to duplicate or surpass our technological achievements, potentially eroding our competitive position in our market. We seek to protect confidential or proprietary information in part by confidentiality agreements with our employees, consultants and third-parties. While we require all of our employees, consultants, advisors and any third-parties who have access to our proprietary know-how, information and technology to enter into confidentiality agreements, we cannot be certain that this know-how, information and technology will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. These agreements may be terminated or breached, and we may not have adequate remedies for any such termination or breach. Furthermore, these agreements may not provide meaningful protection for our trade secrets and know-how in the event of unauthorized use or disclosure.
If third parties claim that our products infringe their intellectual property rights, we may be forced to expend significant financial resources and management time defending against such actions and our financial condition and our results of operations could suffer.
Third parties may claim that our products infringe their patents and other intellectual property rights. Identifying third-party patent rights can be particularly difficult because, in general, patent applications can be maintained in secrecy for at least 18 months after their earliest priority date. Historically, there has been substantial litigation regarding patients and other intellectual property rights in the medical device and related industries. If a competitor were to challenge our patents, licenses or other intellectual property rights, or assert that our products infringe its patent or other intellectual property rights, we could incur substantial litigation costs, be forced to make expensive changes to our product design, pay royalties or other fees to license rights in order to continue manufacturing and selling our products, or pay substantial damages. Third-party infringement claims, regardless of their outcome, would not only consume our financial resources but also divert our management’s time and effort.
Cyber security risks and cyber incidents could adversely affect our business and disrupt operations.
Cyber incidents can result from deliberate attacks or unintentional events. These incidents can include, but are not limited to, gaining unauthorized access to digital systems for purposes of misappropriating assets or sensitive information, corrupting data, or causing operational disruption. The result of these incidents could include, but are not limited to, disrupted operations, misstated financial data, liability for stolen assets or information, increased cyber security protection costs, litigation and reputational damage adversely affecting customer or investor confidence. We have implemented systems and processes to focus on identification, prevention, mitigation and resolution. However, these measures cannot provide absolute security, and our systems may be vulnerable to cyber-security breaches such as viruses, hacking, and similar disruptions from unauthorized intrusions. In addition, we may rely on third party service providers to perform certain services, such as payroll and tax services. Any failure of our systems or third-party systems may compromise our sensitive information and/or personally identifiable information of our employees. While we have secured cyber insurance to potentially cover certain risks associated with cyber incidents, there can be no assurance the insurance will be sufficient to cover any such liability.
RISKS RELATING TO OUR COMMON STOCK
Our stock price has been and may continue to be sporadically traded and volatile, which could result in substantial losses for investors.
Since our IPO in February 2019, our stock price has ranged from a high of $29.00 to a low of $4.12, the trading volume in our stock has been limited and sporadic, and the market price of our common stock is likely to continue to be highly volatile and could fluctuate widely in response to various factors, many of which are beyond our control. In addition, the securities markets have from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. These market fluctuations may also significantly affect the market price of our common stock.
Your ownership may be diluted if additional capital stock is issued to raise capital, to finance acquisitions or in connection with strategic transactions.
We have in the past and intend in the future to raise funds, finance acquisitions or develop strategic relationships by issuing equity or convertible debt securities, which will reduce the percentage ownership of our existing stockholders. Our board of directors has the authority, without action or vote of the stockholders, to issue all or any part of our authorized but unissued shares of common stock. We are authorized to issue up to 100,000,000 shares of common stock. Future issuances of common stock would reduce your influence over matters on which stockholders vote and would be dilutive to earnings per share.
Shares issuable upon the exercise of outstanding options or warrants may substantially increase the number of shares available for sale in the public market and depress the price of our common stock.
As of December 31, 2019, we had a material number of outstanding options and warrants to purchase shares of common stock. To the extent any of these options or warrants are exercised and any additional options or warrants are granted and exercised, there will be further dilution to stockholders and investors. Until the options and warrants expire, these holders will have an opportunity to profit from any increase in the market price of our common stock without assuming the risks of ownership. Holders of options and warrants may convert or exercise these securities at a time when we could obtain additional capital on terms more favorable than those provided by the options or warrants. The exercise of the options and warrants will dilute the voting interest of the owners of presently outstanding shares by adding a substantial number of additional shares of our common stock.
The concentration of our common stock ownership by a single shareholder will limit your ability to influence corporate matters.
Our largest shareholder, Remeditex Ventures, LLC (Remeditex), beneficially owns and will be able to vote in the aggregate a majority of our outstanding common stock. As such, Remeditex, will continue to have the ability to exert significant influence over all corporate activities, including the election or removal of directors and the outcome of tender offers, mergers, proxy contests or other purchases of common stock that could give our stockholders the opportunity to realize a premium over the then-prevailing market price for their shares of common stock. This concentrated control will limit your ability to influence corporate matters and, as a result, we may take actions that shareholders do not view as beneficial. In addition, such concentrated control could discourage others from initiating changes of control. In such cases, the perception of our prospects in the market may be adversely affected and the market price of our common stock may decline.
Certain provisions in our organizational documents could enable our board of directors to prevent or delay a change of control.
Our organizational documents contain provisions that may have the effect of discouraging, delaying or preventing a change of control of, or unsolicited acquisition proposals, that a stockholder might consider favorable. These include provisions:
•prohibiting the stockholders from acting by written consent;
•requiring advance notice of director nominations and of business to be brought before a meeting of stockholders;
•requiring a majority vote of the outstanding shares of common stock to amend the bylaws; and
•limiting the persons who may call special stockholders’ meetings.
In addition, Delaware law makes it difficult for stockholders that recently have acquired a large interest in a corporation to cause the merger or acquisition of the corporation against the directors’ wishes. Under Section 203 of the Delaware General Corporation Law, a Delaware corporation may not engage in any merger or other business combination with an interested stockholder for a period of three years following the date that the stockholder became an interested stockholder except in limited circumstances, including by approval of the corporation’s board of directors.
We have no intention of declaring dividends in the foreseeable future.
The decision to pay cash dividends on our common stock rests with our board of directors and will depend on our earnings, unencumbered cash, capital requirements and financial condition. We do not anticipate declaring any dividends in the foreseeable future, as we intend to use any excess cash to fund our operations. Investors in our common stock should not expect to receive dividend income on their investment, and investors will be dependent on the appreciation of our common stock to earn a return on their investment.
Failure to maintain effective internal control over our financial reporting in accordance with Section 404 of the Sarbanes-Oxley Act could cause our financial reports to be inaccurate.
We are required pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, to maintain internal control over financial reporting and to assess and report on the effectiveness of those controls. This assessment includes disclosure of any material weaknesses identified by our management in our internal control over financial reporting. Our management concluded that our internal controls over financial reporting were, and continue to be, ineffective as of December 31, 2019. We identified material weaknesses in our internal controls due to the lack of segregation of duties, the
limitations of our financial accounting system to properly segregate duties and the absence of internal staff with extensive knowledge of SEC financial and GAAP reporting. While management is working to remediate these material weaknesses, there is no assurance that such changes, when economically feasible and sustainable, will remediate the identified material weaknesses or that the controls will prevent or detect future material weaknesses. If we are not able to maintain effective internal control over financial reporting, our financial statements, including related disclosures, may be inaccurate, which could have a material adverse effect on our business.
Failure to continue improving our accounting systems and controls could impair our ability to comply with the financial reporting and internal controls requirements for publicly traded companies.
As a public company, we operate in an increasingly demanding regulatory environment, which requires us to comply with the Sarbanes-Oxley Act of 2002, and the related rules and regulations of the SEC. Company responsibilities required by the Sarbanes-Oxley Act include establishing corporate oversight and adequate internal control over financial reporting and disclosure controls and procedures. Effective internal controls are necessary for us to produce reliable financial reports and are important to help prevent financial fraud.
Management did elect to perform an annual assessment as of December 31, 2019 of the effectiveness of our internal control over financial reporting for our first annual report. Our management concluded that our internal control over financial reporting was, and continues to be, ineffective as of December 31, 2019, due to material weaknesses in our internal controls resulting from a lack of segregation of duties, the limitations of our financial accounting system, and the absence of internal staff with extensive knowledge of SEC financial and GAAP reporting. This annual assessment was performed earlier than required. Section 404(a) of the Sarbanes-Oxley Act requires annual management assessments of the effectiveness of our internal control over financial reporting, starting with the second annual report that we would expect to file with the SEC. However, for as long as we remain an “emerging growth company” as defined in the JOBS Act, we have and intend to consider to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act. We may continue to take advantage of these reporting exemptions until we are no longer an “emerging growth company.” If we cannot provide reliable financial reports or prevent fraud, our business and results of operations could be harmed and investors could lose confidence in our reported financial information.
As an “emerging growth company” under the Jumpstart Our Business Startups Act, or JOBS Act, we are permitted to, and intend to, rely on exemptions from certain disclosure requirements.
As an “emerging growth company” under the JOBS Act, we are permitted to, and intend to, rely on exemptions from certain disclosure requirements. We are an emerging growth company until the earliest of:
•the last day of the fiscal year during which we have total annual gross revenues of $1.07 billion or more;
•the last day of the fiscal year following the fifth anniversary of our IPO;
•the date on which we have, during the previous 3-year period, issued more than $1 billion in non-convertible debt; or
•the date on which we are deemed a “large accelerated issuer” as defined under the federal securities laws.
For so long as we remain an emerging growth company, we will not be required to:
•have an auditor report on our internal control over financial reporting pursuant to the Sarbanes-Oxley Act of 2002;
•comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements (auditor discussion and analysis);
•submit certain executive compensation matters to shareholders advisory votes pursuant to the “say on frequency” and “say on pay” provisions (requiring a non-binding shareholder vote to approve compensation of certain executive officers) and the “say on golden parachute” provisions (requiring a non-binding shareholder vote to approve golden parachute arrangements for certain executive officers in connection with mergers and certain other business combinations) of the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010;
•include detailed compensation discussion and analysis in our filings under the Securities Exchange Act of 1934, as amended, and instead may provide a reduced level of disclosure concerning executive compensation;
•present more than two years of audited financial statements or two years of related Management’s Discussion and Analysis of Financial Condition and Results of Operations ("MD&A"); and
•immediately adopt new or revised financial accounting standards under §107 of the JOBS Act; instead we are eligible to claim longer phase-in periods.
We intend to take advantage of all of these reduced reporting requirements and exemptions, including the longer phase-in periods for the adoption of new or revised financial accounting standards under §107 of the JOBS Act. We have elected to avail ourselves of this exemption from new or revised accounting standards, and, therefore, will not be subject to the same new or revised accounting standards as public companies that are not emerging growth companies.
Certain of these reduced reporting requirements and exemptions were already available to us due to the fact that we also qualify as a “smaller reporting company” under SEC rules. For instance, smaller reporting companies are not required to obtain an auditor attestation and report regarding management’s assessment of internal control over financial reporting; are not required to provide a compensation discussion and analysis; are not required to provide a pay-for-performance graph or CEO pay ratio disclosure; and may present only two years of audited financial statements and related MD&A disclosure.
Under the JOBS Act, we may take advantage of the above-described reduced reporting requirements and exemptions for up to five years after our initial sale of common equity pursuant to a registration statement declared effective under the Securities Act of 1933, or such earlier time that we no longer meet the definition of an emerging growth company. Further, under current SEC rules, we will continue to qualify as a “smaller reporting company” for so long as we have a public float (i.e., the market value of common equity held by non-affiliates) of less than $250 million as of the last business day of our most recently completed second fiscal quarter.
We cannot predict if investors will find our securities less attractive due to our reliance on these exemptions.
Sales of a substantial amount of our common stock in the public market, particularly sales by our directors, executive officers and significant stockholders, or the perception that these sales could occur, could cause the market price of our common stock to decline.
Sales of a substantial number of shares of our common stock in the public market, particularly sales by our directors, executive officers and principal stockholders, or the perception that these sales might occur, could cause the market price of our common stock to decline. The 2,172,591 shares of our common stock sold in our IPO are freely tradable in the public market without restrictions or further registration except for any shares held by our affiliates, as defined in Rule 144 under the Securities Act). With respect to the remaining shares, our directors, executive officers and the holders of substantially all of our common stock outstanding prior to our IPO (or issued upon conversion of convertible securities in connection with our IPO) entered into lock-up agreements with us that, for a period of at least 90 days from the date of our IPO and ending one year from our IPO, or February 19, 2020, subject to certain exceptions, prohibit them from offering for sale, selling, contracting to sell, granting any option for the sale of, transferring or otherwise disposing of any shares of our common stock and of any securities convertible into or exercisable for our common stock. Shares held by directors, executive officers, and other affiliates will also be subject to volume limitations under Rule 144 under the Securities Act. As the applicable lock-up periods described above have expired, our security holders that were subject to a lock-up agreement are able to sell shares of our common stock in the public market. Sales of a substantial number of such shares or the perception that such sales may occur could cause our market price to fall or make it more difficult for you to sell your common stock at a time and price that you deem appropriate.
Techniques employed by short sellers have in the past and may in the future drive down the market price of our common stock.
Short selling is the practice of selling securities that the seller does not own but rather has borrowed from a third-party with the intention of buying identical securities back at a later date to return to the lender. The short seller hopes to profit from a decline in the value of the securities between the sale of the borrowed securities and the purchase of the replacement shares, as the short seller expects to pay less in that purchase than it received in the sale. As it is in the short seller’s best interests for the price of the stock to decline, many short sellers publish, or arrange for the publication of, negative opinions regarding the relevant issuer and its business prospects in order to create negative market momentum and generate profits for themselves after selling a stock short. These short attacks have led to selling of shares in the market. Issuers such as Soliton, that have common stock with limited trading volumes and/or have been susceptible to relatively high volatility levels, can be particularly vulnerable to such short seller attacks. In May 2019, our common stock was the subject of a report by a short seller
that contained incorrect and misleading information, which report led to a severe decline in our stock price. Although we timely responded to these false and misleading allegations, we cannot assure you that such similar false and misleading articles will not be published again in the future. The publication of any such articles regarding us in the future may bring about a temporary, or possibly long term, decline in the market price of our common stock. If we continue to be the subject of unfavorable allegations, we may have to expend a significant amount of resources to investigate such allegations and/or defend ourselves. While we would strongly defend against any such short seller attacks, we may be constrained in the manner in which we can proceed against the relevant short seller by applicable state law or issues of commercial confidentiality. Such a situation could be costly and time-consuming, and could be distracting for our management team.
Item 1B. Unresolved Staff Comments.
None.
Item 2. Properties.
We do not own any real property. Our corporate and executive offices are in located in a leased facility in Houston, Texas. The current lease terminates in 2021. We believe that our facilities are sufficient to meet the current needs and that suitable space will be available as and when needed.
Item 3. Legal Proceedings.
From time to time in the ordinary course of our business, we may be involved in legal proceedings, the outcomes of which may not be determinable. The results of litigation are inherently unpredictable. Any claims against us, whether meritorious or not, could be time consuming, result in costly litigation, require significant amounts of management time and result in diversion of significant resources. However, we are currently not a party to any pending legal actions. We have insurance policies covering any potential losses where such coverage is cost effective.
Item 4. Mine Safety Disclosures.
Not applicable.
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Our common stock has been listed on the The Nasdaq Capital Market ("Nasdaq") under the symbol “SOLY” since our initial public offering on February 19, 2019.
Holders of Common Equity
As of February 20, 2020, we had approximately 91 stockholders of record of our common stock. This does not include beneficial owners of our common stock.
Dividends
We have not paid any dividends on our common stock in the two most recent fiscal years. Dividends on our preferred stock, which accrued until the closing of our IPO, were converted into common shares at $5.00 per share. Accrued dividends of $4,773,480 were converted into 954,696 shares of our common stock. The payment of dividends in the future will be contingent upon our revenues and earnings, if any, capital requirements and general financial condition. It is the present intention of our Board of Directors to retain all earnings, if any, for use in our business operations and, accordingly, our Board of Directors does not anticipate declaring any dividends in the foreseeable future.
Recent Sales of Unregistered Securities
All information related to equity securities sold by us during the period covered by this report that were not registered under the Securities Act have been included in our Form 10-Q filings or in a Form 8-K filing.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
We did not repurchase any of our equity securities during the year ended December 31, 2019.
Equity Compensation Plan Information
See Part III, Item 12 to this Form 10-K for information relating to securities authorized for issuance under our equity compensation plans.
Stock Performance Graph
Soliton is a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and is not required to provide the information required under this item.
Item 6. Selected Financial Data.
Soliton is a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and is not required to provide the information required under this item.
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
You should read the following discussion and analysis of our financial condition and results of operations in conjunction with the financial statements and the related notes appearing elsewhere in this Form 10-K. This discussion contains forward-looking statements reflecting our current expectations that involve risks and uncertainties, including those set forth under “Cautionary Statement About Forward-Looking Statements.” Actual results and experience could differ materially from the anticipated results and other expectations expressed in our forward-looking statements as a result of a number of factors, including but not limited to those discussed in this Item and in Item 1A - “Risk Factors.” Actual results and the timing of events could differ materially from those discussed in our forward-looking statements as a result of many factors, including those set forth under “Risk Factors” and elsewhere in this Form 10-K.
Overview
Soliton, Inc. was incorporated in the state of Delaware on March 27, 2012. We are a medical technology company focused on developing and commercializing products utilizing our proprietary designed acoustic shockwave technology platform referred to as Rapid Acoustic Pulse ("RAP"). We are a pre-revenue stage company with our first product preparing for launch for the removal of tattoos. We received clearance for our initial device from the FDA on May 24, 2019 allowing our device to be used as an accessory to a 1064 Q-switch laser for tattoo removal in patients with Fitzpatrick Scale I-III skintones. Our product will need to receive clearance from the Food and Drug Administration ("FDA"), in order to be marketed for other indications in the United States. We also intend to secure regulatory approval in international markets and are currently developing a regulatory strategy for these markets.
Our business model anticipates generating revenue from the sale of our RAP console to dermatologists, plastic surgeons, and other physician offices, as well as medi-spas under the supervision of a doctor. Our model contemplates recurring revenues generated by the sale of disposable cartridges that are utilized with each patient visit and treatment. We believe additional revenues will result from maintenance services to our customers. Our system comprises a console with a hand piece and our consumable treatment cartridges, which are designed to allow a physician to perform a single office visit involving multiple laser passes on an average-sized tattoo or, if our cellulite reduction indication is approved in the future, a single stand-alone treatment for cellulite reduction. In simple terms, we expect this to translate into approximately one treatment cartridge per patient, per visit for tattoo and two cartridges per patient, per visit for cellulite, if approved.
Our ongoing research and development activities are primarily focused on finalizing the commercial device and cartridge design for tattoo, obtaining FDA clearance for our system for the treatment of cellulite, and then developing our system and treatment head for additional indications. In addition to these development activities, we are exploring additional uses of RAP technology for the dermatology, plastic surgery, and aesthetic markets, as well as new methods for improving the safety and efficacy of laser-based devices. We have completed proof-of-concept clinical trials for the reduction of cellulite and the treatment of fibrotic scars. We have also initiated a pivotal study for the reduction of cellulite and expect to conclude this study in the first quarter of 2020.
The medical technology and aesthetic product markets are highly competitive and dynamic and are characterized by rapid and substantial technological development and product innovations. We will compete with many other technologies for consumer demand. Further, the aesthetic industry in which we will operate is particularly vulnerable to economic trends. The decision to undergo a procedure from our systems will be driven by consumer demand. Procedures performed using our systems will be elective procedures, the cost of which must be borne by the patient and are not reimbursable through government or private health insurance. In times of economic uncertainty or recession, individuals often reduce the amount of money that they spend on discretionary items, including aesthetic procedures. The general economic difficulties being experienced and the lack of availability of consumer credit for some of our customers' patients could adversely affect the markets in which we will operate.
Recent Developments
We completed the 12-week follow up visit for our proof-of-concept trial targeting keloid and other hypertrophic scars and presented the results of this visit at the Maui Derm conference in January 2020. 3D scar assessment of the pre- and post-treatment photographs of 11 treated scars demonstrated an average reduction in volume of 29.6% (p<0.01) and an average reduction in height of 14.6% (p<0.005).
We completed all of the patient 12-week follow up visits in our cellulite pivotal trial intended to support our 510(k) application for the treatment of cellulite. Before and after photos are being reviewed by independent physicians for scoring of this data.
On February 4, 2020, we granted options to employees and executives to purchase 381,800 shares of the Company’s common stock for a term of 10 years, an exercise price of $11.71, and a vesting period of 25% annually over a four year period. The options had an aggregated grant date fair value of $3,193,468 that was calculated using the Black-Scholes option-pricing model. Variables used in the Black-Scholes option-pricing model include: (1) a discount rate of 1.42% based on the daily yield curve rates for U.S. Treasury obligations, (2) expected life of 6.25 years based on the simplified method (vesting plus contractual term divided by two), (3) expected volatility of 83.12% based on the historical volatility of comparable companies' stock, (4) no expected dividends and (5) fair market value of the Company's stock of $11.71 per share.
Results of Operations for the Year Ended December 31, 2019 Compared to the Year Ended December 31, 2018
Below is a summary of the results of operations:
| | | | | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, | | | | | | |
| 2019 | | 2018 | | Change $ | | Change % |
| Operating expenses: | | | | | | | |
| | | | | | | |
| Research and development | $ | 5,108,407 | | | $ | 4,669,747 | | | 438,660 | | | 9.39 | % |
| Sales and marketing | 189,394 | | | 304,601 | | | (115,207) | | | (37.82) | % |
| Depreciation and amortization | 216,737 | | | 120,488 | | | 96,249 | | | 79.88 | % |
| General and administrative | 7,428,850 | | | 3,107,813 | | | 4,321,037 | | | 139.04 | % |
| Total operating expenses | 12,943,388 | | | 8,202,649 | | | 4,740,739 | | | 57.80 | % |
| | | | | | | |
| Other (income) expense: | | | | | | | |
| | | | | | | |
| Interest expense | 822,858 | | | 1,115,501 | | | (292,643) | | | (26.23) | % |
| Other income | (14,369) | | | (3,214) | | | (11,155) | | | 347.08 | % |
| Total other expenses | 808,489 | | | 1,112,287 | | | (303,798) | | | (27.31) | % |
| Net Loss | $ | 13,751,877 | | | $ | 9,314,936 | | | $ | 4,436,941 | | | 47.63 | % |
Research and development. R&D expenses increased by $438,660 compared to the same period in 2018, primarily due to increases in expenses for clinical cellulite trials of $512,313, license costs and other spending on intellectual property of $453,735, which included a $250,000 milestone payment after we received FDA clearance for our RAP device for tattoo removal, salaries and related expenses of $101,961, travel and supplies of $92,163, and animal research of $66,714. These increases were offset by decreases in contract engineering expenses of $788,226.
Sales and marketing. S&M expenses decreased by $115,207 compared to the same period in 2018, primarily due to decreases in expenses related to social media marketing of $113,226 and our Scientific Advisory Board (“SAB”) and other conference related expenses and meetings of $1,981. We include our SAB fees in S&M because they primarily advise on our product launch and marketing decisions related to dermatologists and prospective patients.
General and administrative. G&A expenses increased by $4,321,037 compared to same period in 2018 primarily due to increases in expenses for salaries and related expenses of $1,203,867, which included the payout of bonuses and the accrual of management incentives (versus a prior year reversal) of $1,226,041 offset by a decrease in salaries and wages of $22,174. The increase was also driven by increases in expenses for investor relations of $482,839, insurance of $317,933, board related fees of $223,542, legal of $196,748, travel and related expenses of $141,053, membership fees of $85,927, office related expenses of $75,764, accounting and other professional fees of $25,859 and information technology of $17,636. Further contributing to the increase in G&A were the increases in non-cash expenses related to stock options, restricted stock and acceleration of restricted stock vesting due to our IPO and grants during 2019 of $1,549,869. The investor relations, board fees, insurance, membership fee, accounting and other professional fee and information technology increases relate primarily to our becoming a public company.
Other (income) expenses. Other (income) expenses decreased by $303,798 compared to the same period in 2018 mainly due to a decrease in interest expense of $292,643, which was a result of the conversion of debt upon our IPO in February 2019.
Liquidity and Capital Resources
Since our inception, we have financed our operations through private placements of common stock, convertible preferred stock, convertible and non-convertible bridge notes, and our IPO. Our cash, cash equivalents and restricted cash as of December 31, 2019 was $12,076,425, comprised of $11,876,425 in cash and $200,000 in restricted cash collateralizing a letter of credit benefiting our contract manufacturer.
On February 19, 2019, we consummated our IPO. In the IPO, we sold a total of 2,172,591 shares of common stock at a purchase price of $5.00 per share for gross proceeds of $10,862,955 and net proceeds of $9,714,198. In connection with the closing of the IPO, our convertible notes (and related accrued interest) of $11,784,987 were converted into 6,825,391 shares of our common stock, and accrued dividends of $4,773,480 were converted into 954,696 shares of our common stock. We repaid non-convertible notes and accrued interest from our IPO proceeds in the amount of $1,005,038. In August and September 2019, the remaining $47,781 of convertible notes were converted to 273,034 shares of our common stock. Additionally, we utilized approximately $2,000,000 to pay outstanding liabilities with vendors.
On June 16, 2019, we entered into a private placement with certain institutional and accredited investors for the sale by us of 675,000 units (each a "June Unit") at $14.00 per June Unit for total gross proceeds of $9,450,000. Each June Unit consisted of (i) one share of our common stock and (ii) a warrant to purchase 0.7 shares (a total of 472,500 shares) of common stock (each a "June Warrant") at $16.00 per share. The June Warrants included in the June Units will expire on August 23, 2024. On July 1, 2019, we filed a Registration Statement on Form S-1 to register for resale the common stock underlying the June Units sold in the June 2019 private offering. The net proceeds from the closing of the sale of the June Units on June 19, 2019 was $8,643,302 after deducting the placement agent fees and estimated offering expenses payable us.
On October 10, 2019, we entered into a private placement with certain institutional and accredited investors for the sale by us of 485,250 units (each an “October Unit”) at $12.88 per October Unit for total gross proceeds of $6,250,020. Each October Unit consisted of (i) one share of our common stock and (ii) a warrant to purchase 1.1 shares (a total of 533,775 shares) of common stock (each an “October Warrant”). The October Warrants included in the October Units are exercisable at a price of $12.88 per share commencing on the date of issuance and will expire on October 10, 2024. On November 8, 2019, we filed a Registration Statement on Form S-1 to register for resale the common stock underlying the October Units sold in the October 2019 private offering. The net proceeds from the closing of the sale of the October Units on October 11, 2019 was $5,738,111 after deducting the placement agent fees and estimated offering expenses payable by us.
We expect to continue to invest in our research and development efforts to support our current initiatives. We will not generate revenue until our commercial RAP units receive clearance from the FDA for the changes made to the device from the initial device cleared by the FDA on May 28, 2019, and we have initiated sales of the units.
We estimate our current cash, cash equivalents and restricted cash resources of $12,076,425 at December 31, 2019, is sufficient to fund our operations into but not beyond the third quarter of 2020. We also recognize we will need to raise additional capital in order to continue to execute our business plan, including obtaining regulatory clearance for our products currently under development and commercializing and generating revenues from products under development. There are no assurances that additional financing will be available when needed or that management will be able to obtain financing on terms acceptable to us. A failure to raise sufficient capital, generate sufficient product revenues, control expenditures and regulatory matters, among other factors, will adversely impact our ability to meet our financial obligations as they become due and payable and to achieve our intended business objectives. If we are unable to raise sufficient additional funds, we will have to scale back our operations.
Summary of Cash Flows
The following table summarizes our cash flows for the year ended December 31, 2019 and 2018, respectively:
| | | | | | | | | | | |
| For the Year ended December 31, | | |
| 2019 | | 2018 |
| Net cash used in operating activities | $ | (10,605,073) | | | $ | (4,599,677) | |
| Net cash used in investing activities | (842,510) | | | (29,980) | |
| Net cash provided by financing activities | 23,390,573 | | | 4,744,680 | |
| Net increase in cash and cash equivalents | $ | 11,942,990 | | | $ | 115,023 | |
Cash Flows for the Years ended December 31, 2019 and 2018
Operating activities. Net cash used in operating activities was $10,605,073 during the year ended December 31, 2019, and consisted of a net loss of $13,751,877 and a net change in operating assets and liabilities of $215,604 offset by non-cash items of $3,362,408. The change in operating assets included an increase in prepaid expenses of $85,777 offset by a net decrease in operating liabilities of $129,827, comprised of a decrease in accounts payable of $369,574 offset by increases in accrued liabilities of $83,463 and accrued interest - related party and non-related party of $156,284. The increase in prepaid expenses was largely driven by new insurance policies and reporting software for public companies. The decrease in accounts payable was largely due to payments to several vendors, previously on extended payment terms, as a result of our IPO closing and returning to consistent terms with vendors. The increase in accrued liabilities was driven primarily by an increase in accruals for clinical trial costs incurred but not yet billed. The increase in accrued interest-related party is due to the issuance of related party convertible notes and the calculation of interest thereon. Non-cash items consisted of stock-based compensation of $2,488,053, amortization of debt discount of $664,953, depreciation and amortization expense of $216,737 and deferred rent of $7,335.
Net cash used in operating activities was $4,599,677 during the year ended December 31, 2018, and consisted of a net loss of $9,314,936, which was offset by a net change in operating assets and liabilities of $3,528,428 and by non-cash items of $1,186,831. The change in operating assets included an increase in prepaid expenses of $2,787 offset by a net increase in liabilities of $3,531,215, comprised of increases in accounts payable of $1,387,383, accrued liabilities of $1,143,139 and accrued interest - related party and non-related party of $1,000,693. The increase in accounts payable was largely due to extended payment terms established with several vendors during our IPO process. The increase in accrued liabilities was driven primarily by salary deferrals for management of $406,875 that was enacted to conserve operating cash and the remaining balance is attributed to accruals for various vendors. The increase in accrued interest-related party is due to the issuance of the related party convertible notes and the calculation of interest thereon. Non-cash items consisted of stock-based compensation of $938,184, depreciation and amortization expense of $120,488, amortization of debt discount of $111,537, impairment of intangible assets of $19,138, and deferred rent of $2,516.
Investing activities. Net cash used in investing activities for the year ended December 31, 2019 was $842,510 compared to $29,980 for the same comparable period in 2018. For the years ended December 31, 2019 and 2018, $829,896 and $17,626, respectively, was utilized towards the purchase of property and equipment primarily as a result of the investment in our research equipment, including $780,000 in lab equipment in the field at clinical trial sites and held by a vendor for final testing. We invested $12,614 and $12,354 towards the acquisition of intangibles (trademarks) in 2019 and 2018, respectively.
Financing activities. Net cash provided by financing activities during the year ended December 31, 2019 was $23,390,573. We received cash proceeds from our IPO of $9,714,198, net of deal costs and other expenses, $8,643,302 from the proceeds related to our PIPE offering from June 2019, $5,738,111 from the proceeds related to our PIPE offering from October 2019, and cash proceeds of $300,000 from the issuance of non-convertible notes payable to non-related parties. These amounts were offset by a use of cash for the payment of non-convertible notes and accrued interest to both related and non-related parties for $1,005,038.
Net cash provided by financing activities for the year ended December 31, 2018, was $4,744,680, which was related to proceeds of $2,397,000 from convertible note issuances to related parties, proceeds of $1,814,240 from convertible note issuances to non-related parties, proceeds of $560,000 from non-convertible note issuances-non related party and proceeds of $125,000 from non-convertible note issuances-related party, offset by $151,560 for a use of cash for financing cost related to our proposed offering.
Contractual Obligations and Commitments
On April 5, 2012, we entered into a Patent and Technology License Agreement with MD Anderson. Pursuant to the agreement, we obtained a royalty-bearing, worldwide, exclusive license to intellectual property including patent rights related to the patents and technology we use. Under the agreement, we agreed to pay a nonrefundable license documentation fee in the high-five digits 30 days after the effective date of the agreement. Additionally, we agreed to pay a nonrefundable annual maintenance fee starting on the third anniversary of the effective date of the agreement, which escalates each anniversary and is currently in the mid-five digits. Additionally, we agreed to a running royalty percentage of net sales in the mid-single digits. We also agreed to make certain milestone payments in the low to mid-six digits and sublicensing payments, including a $250,000 milestone payment made in June 2019 after we received FDA clearance for our RAP device for tattoo removal. The specific patents initially subject to the agreement expire between 2031 and 2032.
MD Anderson has the right to terminate the agreement upon advanced notice in the event of a default by us. The agreement will expire upon the expiration of the licensed intellectual property. The rights obtained by us pursuant to the agreement are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between MD Anderson and the U.S. government. To the extent that is the case, our license agreement with, and the intellectual property rights we have licensed from, MD Anderson are subject to such a funding agreement and any superior rights that the U.S. government may have with respect to the licensed intellectual property. Therefore, there is a risk that the intellectual property rights we have licensed from MD Anderson may be non-exclusive or void if a funding agreement related to the licensed technology between MD Anderson and the U.S. government does exist and depending on the terms of such an agreement. Notwithstanding the foregoing, we do not believe our RAP technology received any federal funding. All out-of-pocket expenses incurred by MD Anderson in filing, prosecuting and maintaining the licensed patents have been and shall continue to be assumed by us.
As the inventor of the intellectual property licensed from MD Anderson, Dr. Capelli, our Chief Executive Officer, is entitled to 50% of the license income (which is determined after MD Anderson recoups any costs associated therewith) that we are required to pay to MD Anderson pursuant to our license agreement with MD Anderson. For the years ended December 31, 2019 and 2018, Dr. Capelli received $187,500 and $27,500 respectively from MD Anderson. In addition, Dr. Capelli is entitled to 50% of the proceeds (after the recoupment of any costs associated therewith) from the sale by MD Anderson of 175,000 shares issued to MD Anderson in connection with the license agreement.
On November 20, 2019, we entered into a cooperative development addendum ("Addendum") to our engineering and development services master agreement with Emphysys, Inc. ("Emphysys”). The Addendum states that Emphysys will provide us with engineering and design services related to shockwave technology for use in dermatology and aesthetics fields for a three-year period.
During the term of the Addendum, we agreed to certain minimum annual expenditures. If we fail to spend such minimum annual amounts or if we terminate the Addendum without cause, we will be required to pay Emphysys a termination fee ranging in the low to mid-six digits. In the event that all or substantially all of the stock or assets of either party are sold then, at the request of other party, the Addendum may be terminated (without the requirement to pay a termination fee) and the obligation of Emphysys to provide future services to us shall terminate. Pursuant to the Addendum, with certain exceptions, Emphysys covenanted that it will not perform or agree to perform services with any company other than Soliton in the area of arc-discharge driven acoustical shockwave generation for medical dermatological or aesthetic dermatological indications during the term of the Addendum or any extension thereof, and for a period of six months after the termination of the Addendum.
Lease Commitments
We lease space for our corporate office, which provides for a 63 month term beginning on February 1, 2016, for rent payments of $7,867 per month.
Future minimum lease payments under the operating leases as of December 31, 2019 were as follows:
| | | | | | | | |
| Year Ending December, 31 | | Amount |
| 2020 | | $ | 106,153 | |
| 2021 | | 35,751 | |
| Total future minimum lease payments | | $ | 141,904 | |
Purchase Commitments
As of December 31, 2019, we had purchase obligations of $3,937,500 to a single engineering service provider. This commitment is for services used in the ordinary course of business and do not represent excess commitments or loss contracts. This commitment can be terminated with a penalty payment of no more than $500,000.
| | | | | | | | |
| Year Ending December, 31 | | Amount |
| 2020 | | $ | 1,575,000 | |
| 2021 | | 1,575,000 | |
| 2022 | | 787,500 | |
| Total future minimum purchase commitments | | $ | 3,937,500 | |
Employment Agreements
We have agreements with key employees to provide certain benefits in the event of termination where the base salary and certain other benefits would aggregate $1,775,035 using the rate of compensation in effect at December 31, 2019.
Unrecognized Tax Benefits
As of December 31, 2019, we have not recorded a provision for income taxes in our financial statements as we have been in a loss position since inception and we cannot be more certain than not that we will be able to recognize the income tax benefit from our net operating loss carry forward in the future.
Off-balance Sheet Arrangements
As of December 31, 2019, we did not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes.
JOBS Act Accounting Election
The Jumpstart Our Business Startups Act of 2012 (" the JOBS Act") exempts an “emerging growth company” such as us from being required to comply with new or revised financial accounting standards until private companies are required to comply with the new or revised financial accounting standards. The JOBS Act provides that a company can elect to opt out of the extended transition period and comply with the requirements that apply to non-emerging growth companies but any such election to opt out is irrevocable. We elected not to opt out of such extended transition period which means that when a standard is issued or revised and it has different application dates for public or private companies, we, as an emerging growth company, can adopt the new or revised standard at the time private companies adopt the new or revised standard. This may make comparison of our financial statements with another public company which is neither an emerging growth company nor an emerging growth company which has opted out of using the extended transition period difficult or impossible because of the potential differences in accounting standards used.
Critical Accounting Policies and Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates, assumptions and judgments that affect the amounts reported in the financial statements, including the notes thereto. We consider critical accounting policies to be those that require more significant judgments and estimates in the preparation of our financial statements, including the following: research and development expenses, long lived assets; intangible assets valuations, accrued liabilities, income tax valuations, warrants, and stock-based compensation. Management relies on historical experience and other assumptions believed to be reasonable in making its judgment and estimates. Actual results could differ materially from those estimates.
Management believes its application of accounting policies, and the estimates inherently required therein, are reasonable. These accounting policies and estimates are periodically reevaluated, and adjustments are made when facts and circumstances dictate a change.
Our accounting policies are more fully described under the heading “Summary of Significant Accounting Policies” in Note 2 to our Financial Statements included in this Form 10-K.
We believe that the following accounting policies are the most critical to aid in fully understanding and evaluating our reported financial results, and they require our most difficult, subjective or complex judgments, resulting from the need to make estimates about the effect of matters that are inherently uncertain.
Research and Development Costs
We record accrued expenses for estimated costs of our research and development activities conducted by third-party service providers, which include the conducting of pre-clinical studies, preparation for and conducting of clinical trials and contract engineering activities. We record the estimated costs of research and development activities based upon the estimated amount of services provided but not yet invoiced, and we include these costs in accrued liabilities in the balance sheets and within research and development expense in the statements of operations. These costs are a significant component of our research and development expenses. We record accrued expenses for these costs based on the estimated amount of work completed and in accordance with agreements established with these third parties.
We estimate the amount of work completed through discussions with internal personnel and external service providers as to the progress or stage of completion of the services and the agreed-upon fee to be paid for such services. We make significant judgments and estimates in determining the accrued balance in each reporting period. As actual costs become known, we adjust our accrued estimates. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed may vary from our estimates and could result in us reporting amounts that are too high or too low in any particular period. Our accrued expenses are dependent, in part, upon the receipt of timely and accurate reporting from clinical research organizations, engineering firms and other third-party service providers. To date, there have been no material differences from our accrued expenses to actual expenses.
Impairment of Long-Lived Assets
Management reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount may not be realizable, or in the case of trademarks, at a minimum annually during the fourth quarter of the year. If an evaluation is required, the estimated future undiscounted cash flows associated with the asset are compared to the asset’s carrying value to determine if an impairment of such asset is necessary. The effect of any impairment would be to expense the difference between the fair value of such asset and its' carrying value.
Components of our Results of Operations and Financial Condition
Operating expenses
We classify our operating expenses into four categories: (i) research and development; (ii) sales and marketing; (iii) general and administrative; and (iv) depreciation.
Research and development. Research and development expenses consist primarily of:
•costs incurred to conduct research, such as animal research;
•costs related to the design and development of our technology, including fees paid to contract engineering firms and contract manufacturers;
•salaries and expenses related to our employees primarily engaged in research and development activities;
•fees paid to clinical consultants, clinical trial sites and vendors, including clinical research organizations, in preparation for clinical trials and our applications with the FDA; and
•costs related to compliance with regulatory requirements.
We recognize all research and development costs as they are incurred. Pre-clinical costs, contract engineering and design costs, patent costs and other development costs incurred by third parties are expensed as the contracted work is performed.
We expect our research and development expenses to increase in the future as we advance our product into and through clinical trials, pursue additional regulatory approvals of our product in the United States, and continue commercial development of our RAP device and replaceable cartridge. The process of conducting the necessary clinical research to obtain regulatory approval is costly and time-consuming. The actual probability of success for our technology may be affected by a variety of factors including: the quality of our product, early clinical data, investment in our clinical program, competition, manufacturing capability and commercial viability. We may not succeed in achieving all necessary regulatory approvals for any of our product candidates. As a result of the uncertainties discussed above, we are unable to determine the duration and completion costs of our research and development process or when and to what extent, if any, we will generate revenue from the commercialization and sale of our device.
Sales and Marketing
Sales and marketing expenses consist of marketing, conferences, web development, advisory boards and other miscellaneous expenses. We expect our sales and marketing expense to increase due to the anticipated growth of our business and related infrastructure as well as expanding our sales personnel, web development and other costs associated with becoming a public company.
General and administrative
General and administrative expenses consist of personnel related costs, which include salaries, as well as the costs of professional services, such as accounting and legal, facilities, insurance, travel costs, information technology and other administrative expenses. We expect our general and administrative expense to increase due to the anticipated growth of our business and related infrastructure as well as accounting, insurance, investor relations and other costs associated with becoming a public company.
Depreciation
Depreciation expense consists of depreciation on our property and equipment. We depreciate our assets over their estimated useful lives. We estimate research and development equipment and lab equipment to have a five year life; computer equipment and software to have a three year life; furniture to have a three year life; and leasehold improvements to be depreciated over the shorter of the remaining lease term or useful lives of the asset.
Accounting for warrants
We issued warrants to purchase shares of common stock related to (i) bridge notes issued prior to its IPO, (ii) private investment public equity ("PIPE") deals, and (iii) as part of underwriter compensation in 2019 and 2018. We accounted for such warrants in accordance with Accounting Standards Codification (ASC) Topic 480-10, Distinguishing Liabilities from Equity, which identifies three categories of freestanding financial instruments that are required to be accounted for as a liability. Based on this guidance, we determined, for each issuance, that warrants did not need to be accounted for as a liability. Accordingly, the warrants were classified as equity and are not subject to remeasurement at each balance sheet date. In addition, we account for issuance costs of warrants issued with debt instruments in accordance with ASC 470-20, Debt with Conversion and Other Options, which states proceeds from the sale of a debt instrument with stock purchase warrants (detachable call options) are allocated to elements based on the relative fair values of the debt instrument without the warrants and of the warrants themselves at time of issuance. The portion of the proceeds so allocated to the warrants are accounted for as paid-in capital. The remainder of the proceeds are allocated to the debt instrument, which may result in a discount or premium.
Registration rights agreements are accounted for in accordance with Financial Accounting Standards Board ("FASB") Accounting Standards Codification ("ASC") Topic 450-20, Loss Contingencies, which requires measurement of the contingent liability when an entity would be required to deliver shares under a registration payment arrangement, the transfer of consideration is probable and the number of shares to be delivered can be reasonably estimated. Accordingly, there is no liability under the payment arrangement requiring disclosure or recognition.
The fair value of warrants is estimated using the Black-Scholes option pricing model, based on the market value of the underlying common stock at the measurement dates, the contractual terms of the warrants, risk-free interest rates and expected volatility of the price of the underlying common stock. There are no expected dividends.
Stock-based compensation
Stock-based compensation transactions are recognized as compensation expense in the statements of operations based on their fair values on the date of the grant, with the compensation expense recognized over the period in which a grantee is required to provide service in exchange for the award. The expense for equity awards vested during the reporting period is recognized over the applicable vesting period of the stock award using either the straight-line method or the accelerated method, depending on the vesting structure, and is included in general and administrative expenses. We estimate the fair value of options granted using the Black-Scholes option valuation model. This estimate uses assumptions regarding a number of inputs that require us to make significant estimates and judgments. Because we are a new publicly traded common stock the expected volatility assumption was based on industry peer information.
Item 7A. Quantitative and Qualitative Disclosure About Market Risk.
We are a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and are not required to provide the information required under this item.
Item 8. Financial Statements and Supplementary Data.
Index to Financial Statements
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors of Soliton, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheet of Soliton, Inc. (the “Company”) as of December 31, 2019, the related statements of operations, changes in stockholders’ equity (deficit) and cash flows for the year then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2019, and the results of its operations and its cash flows for the year then ended, in conformity with U.S. generally accepted accounting principles.
Substantial Doubt about the Company’s Ability to Continue as a Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has incurred an accumulated deficit of $56 million since inception, has not generated revenue from operations and does not expect to experience positive cash flows from operating activities in the near term. These conditions, along with other matters as set forth in Note 1, raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
| | |
| /s/ Dixon Hughes Goodman LLP |
|
| We have served as the Company’s auditor since 2019. |
|
| Atlanta, Georgia |
| March 2, 2020 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Soliton, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheet of Soliton, Inc. (the “Company”) as of December 31, 2018, the related statements of operations, changes in stockholders’ deficit and cash flows for the year then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2018, and the results of its operations and its cash flows for the year then ended, in conformity with accounting principles generally accepted in the United States of America.
Explanatory Paragraph – Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As more fully described in Note 1, the Company has suffered recurring losses from operations and does not expect to generate positive cash flows from operating activities in the near future. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
| | |
| /s/ Marcum LLP |
|
| We have served as the Company's auditor from 2017 to 2019. |
|
| Marcum LLP |
| Houston, Texas |
| March 29, 2019 |
SOLITON, INC.
BALANCE SHEETS
| | | | | | | | | | | |
| As of | | |
| December 31,
2019 | | December 31,
2018 |
| ASSETS | | | |
| Current assets: | | | |
| Cash and cash equivalents | $ | 11,876,425 | | | $ | 133,435 | |
| Restricted cash | 200,000 | | | — | |
| Total cash, cash equivalents and restricted cash | 12,076,425 | | | 133,435 | |
| Prepaid expenses and other current assets | 96,310 | | | 10,533 | |
| Total current assets | 12,172,735 | | | 143,968 | |
| Deferred direct issuance costs - offering | — | | | 276,560 | |
| Property and equipment, net of accumulated depreciation | 847,399 | | | 1,014,240 | |
| Intangible assets, net of accumulated amortization | 97,556 | | | 84,942 | |
| Other assets | 23,283 | | | 23,283 | |
| Total assets | $ | 13,140,973 | | | $ | 1,542,993 | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) | | | |
| Current liabilities: | | | |
| Accounts payable | $ | 1,338,262 | | | $ | 2,737,836 | |
| Accrued liabilities | 1,513,274 | | | 1,863,874 | |
| Dividends payable | — | | | 4,613,260 | |
| Accrued interest | — | | | 133,804 | |
| Accrued interest - related party | — | | | 1,162,719 | |
| Convertible notes payable, net | — | | | 1,784,976 | |
| Convertible notes payable - related party | — | | | 8,422,000 | |
| Notes payable, net | — | | | 293,568 | |
| Notes payable – related party, net | — | | | 65,479 | |
| Deferred rent - current portion | 11,745 | | | 7,106 | |
| Total current liabilities | 2,863,281 | | | 21,084,622 | |
| Deferred rent | 4,282 | | | 16,256 | |
| Total liabilities | 2,867,563 | | | 21,100,878 | |
| Commitments and contingencies (see Note 6) | | | |
| Stockholders’ equity (deficit): | | | |
Series A preferred stock, $0.001 par value, liquidation value of $1,999,997, 416,666 shares designated, issued and outstanding at December 31, 2018 | — | | | 417 | |
Series B preferred stock, $0.001 par value, liquidation value of $14,000,641, 2,118,100 shares designated, issued and outstanding at December 31, 2018 | — | | | 2,118 | |
Common stock, $0.001 par value, 100,000,000 authorized, 16,932,184 shares issued and outstanding at December 31, 2019 and 1,998,056 shares issued and outstanding at December 31, 2018 | 16,932 | | | 1,998 | |
| Additional paid-in capital | 66,299,849 | | | 22,568,857 | |
| Accumulated deficit | (56,043,371) | | | (42,131,275) | |
| Total stockholders’ equity (deficit) | 10,273,410 | | | (19,557,885) | |
| Total liabilities and stockholders’ equity (deficit) | $ | 13,140,973 | | | $ | 1,542,993 | |
See accompanying notes to the financial statements.
SOLITON, INC.
STATEMENTS OF OPERATIONS
| | | | | | | | | | | |
| For the Year Ended December 31, | | |
| 2019 | | 2018 |
| Revenue | $ | — | | | $ | — | |
| | | |
| Operating expenses: | | | |
| Research and development | 5,108,407 | | | 4,669,747 | |
| Sales and marketing | 189,394 | | | 304,601 | |
| Depreciation and amortization expense | 216,737 | | | 120,488 | |
| General and administrative expenses | 7,428,850 | | | 3,107,813 | |
| Total operating expenses | 12,943,388 | | | 8,202,649 | |
| | | |
| Loss from operations | (12,943,388) | | | (8,202,649) | |
| | | |
| Other (expense) income: | | | |
| Interest expense | (822,858) | | | (1,115,501) | |
| Other income | 14,369 | | | 3,214 | |
| Total other expense | (808,489) | | | (1,112,287) | |
| | | |
| Loss before income taxes | (13,751,877) | | | (9,314,936) | |
| Income tax expense | — | | | — | |
| | | |
| Net loss | $ | (13,751,877) | | | $ | (9,314,936) | |
| | | |
| Accrued dividends to Series A and Series B preferred stockholders | (160,219) | | | (1,280,000) | |
| Net loss attributable to common stockholders | $ | (13,912,096) | | | $ | (10,594,936) | |
| | | |
| Net loss per common share, basic and diluted | $ | (1.00) | | | $ | (5.64) | |
| | | |
| Weighted average number of common shares outstanding, basic and diluted | 13,841,884 | | | 1,877,775 | |
See accompanying notes to the financial statements.
SOLITON, INC.
STATEMENTS OF CHANGES IN
STOCKHOLDERS’ EQUITY (DEFICIT)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Series A Preferred Stock | | | | Series B Preferred Stock | | | | Common Stock | | | | Additional Paid-In Capital | | Accumulated Deficit | | Total |
| Shares | | Par | | Shares | | Par | | Shares | | Par | | | | | | |
| | | | | | | | | | | | | | | | | |
| Balance December 31, 2017 | 416,666 | | | $ | 417 | | | 2,118,100 | | | $ | 2,118 | | | 1,820,556 | | | $ | 1,821 | | | $ | 21,031,388 | | | $ | (31,536,339) | | | $ | (10,500,595) | |
| | | | | | | | | | | | | | | | | |
| Share-based compensation | — | | | — | | | — | | | — | | | — | | | — | | | 938,184 | | | — | | | 938,184 | |
| Debt discount on convertible notes and notes payable – issuance of warrants | — | | | — | | | — | | | — | | | — | | | — | | | 466,754 | | | — | | | 466,754 | |
| Issuance of common shares | — | | | — | | | — | | | — | | | 177,500 | | | 177 | | | (177) | | | — | | | — | |
| Accrued preferred dividends | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (1,280,000) | | | (1,280,000) | |
| Net loss | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (9,314,936) | | | (9,314,936) | |
| Debt forgiveness | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 132,708 | | | | — | | | | 132,708 | |
| | | | | | | | | | | | | | | | | |
| Balance December 31, 2018 | 416,666 | | | $ | 417 | | | 2,118,100 | | | $ | 2,118 | | | 1,998,056 | | | $ | 1,998 | | | $ | 22,568,857 | | | $ | (42,131,275) | | | $ | (19,557,885) | |
| | | | | | | | | | | | | | | | | |
| Share-based compensation | — | | | — | | | — | | | — | | | — | | | — | | | 2,488,053 | | | — | | | 2,488,053 | |
| Debt discount on convertible notes and notes payable – issuance of warrants | — | | | — | | | — | | | — | | | — | | | — | | | 145,974 | | | — | | | 145,974 | |
| Payment of deferred direct issuance costs | — | | | — | | | — | | | — | | | — | | | — | | | (186,029) | | | — | | | (186,029) | |
| Issuance of common shares for extinguishment of preferred shares | (416,666) | | | (417) | | | (2,118,100) | | | (2,118) | | | 2,534,766 | | | 2,535 | | | — | | | — | | | — | |
| Issuance of common shares for extinguishment of convertible debt | — | | | — | | | — | | | — | | | 7,098,425 | | | 7,098 | | | 11,825,670 | | | — | | | 11,832,768 | |
| Issuance of common shares for extinguishment of dividends payable | — | | | — | | | — | | | — | | | 954,696 | | | 955 | | | 4,772,525 | | | — | | | 4,773,480 | |
| Issuance of common shares for IPO, net of costs | — | | | — | | | — | | | — | | | 2,172,591 | | | 2,173 | | | 9,871,494 | | | — | | | 9,873,667 | |
| Issuance of common shares for accelerated vesting | — | | | — | | | — | | | — | | | 127,500 | | | 127 | | | (127) | | | — | | | — | |
| Issuance of common shares for June PIPE offering, net of costs | — | | | — | | | — | | | — | | | 675,000 | | | 675 | | | 8,642,627 | | | — | | | 8,643,302 | |
| Issuance of common shares for October PIPE offering, net of costs | — | | | — | | | — | | | — | | | 485,250 | | | 485 | | | 5,737,626 | | | — | | | 5,738,111 | |
| Issuance of common shares | — | | | — | | | — | | | — | | | 885,900 | | | 886 | | | (886) | | | — | | | — | |
| Accrued preferred dividends | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (160,219) | | | (160,219) | |
| Net loss | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (13,751,877) | | | (13,751,877) | |
| Debt forgiveness | — | | | — | | | — | | | — | | | — | | | — | | | 434,065 | | | — | | | 434,065 | |
| | | | | | | | | | | | | | | | | |
| Balance December 31, 2019 | — | | | $ | — | | | — | | | $ | — | | | 16,932,184 | | | $ | 16,932 | | | $ | 66,299,849 | | | $ | (56,043,371) | | | $ | 10,273,410 | |
See accompanying notes to the financial statements.
SOLITON, INC.
STATEMENTS OF CASH FLOWS
| | | | | | | | | | | |
| For the Years Ended December 31, | | |
| 2019 | | 2018 |
| CASH FLOWS FROM OPERATING ACTIVITIES: | | | |
| Net loss | $ | (13,751,877) | | | $ | (9,314,936) | |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | |
| Depreciation and amortization | 216,737 | | | 120,488 | |
| Share-based compensation | 2,488,053 | | | 938,184 | |
| Write-down of intangible assets | — | | | 19,138 | |
| Amortization of debt discount | 664,953 | | | 111,537 | |
| Deferred rent | (7,335) | | | (2,516) | |
| Changes in operating assets – (Increase)/Decrease: | | | |
| Prepaid expenses and other current assets | (85,777) | | | (2,787) | |
| Changes in operating liabilities – Increase/(Decrease): | | | |
| Accounts payable | (369,574) | | | 1,387,383 | |
| Accrued liabilities | 83,463 | | | 1,143,139 | |
| Accrued interest – non-related party | 10,617 | | | 133,804 | |
| Accrued interest – related party | 145,667 | | | 866,889 | |
| | | | | |
| NET CASH USED IN OPERATING ACTIVITIES: | (10,605,073) | | | (4,599,677) | |
| | | |
| CASH FLOWS FROM INVESTING ACTIVITIES: | | | |
| Payments for the purchase of property and equipment | (829,896) | | | (17,626) | |
| Payments for acquisition of intangibles | (12,614) | | | (12,354) | |
| NET CASH USED IN INVESTING ACTIVITIES: | (842,510) | | | (29,980) | |
| | | |
| CASH FLOWS FROM FINANCING ACTIVITIES: | | | |
| Payment of non-convertible notes payable - related party and non-related party | (985,000) | | | — | |
| Payment of non-convertible notes payable accrued interest - related party and non-related party | (20,038) | | | — | |
| Proceeds from the issuance of non-convertible notes payable - non-related party | 300,000 | | | — | |
| Proceeds from initial public offering, net of costs | 9,714,198 | | | — | |
| Proceeds from private investment in public equity offering - June PIPE, net of costs | 8,643,302 | | | — | |
| Proceeds from private investment in public equity offering - October PIPE, net of costs | 5,738,111 | | | — | |
| Proceeds from issuance of convertible notes – related party | — | | | 2,397,000 | |
| Proceeds from issuance of convertible notes, net | — | | | 1,814,240 | |
| Proceeds from issuance of non-convertible notes – related party | — | | | 125,000 | |
| Proceeds from issuance of non-convertible notes | — | | | 560,000 | |
| Payment of deferred direct issuance costs – proposed offering | — | | | (151,560) | |
| NET CASH PROVIDED BY FINANCING ACTIVITIES: | 23,390,573 | | | 4,744,680 | |
| | | |
| Net increase (decrease) in cash, cash equivalents and restricted cash | 11,942,990 | | | 115,023 | |
| Cash, cash equivalents and restricted cash, beginning of period | 133,435 | | | 18,412 | |
| Cash, cash equivalents and restricted cash, end of period | $ | 12,076,425 | | | $ | 133,435 | |
| | | |
| Supplemental cash flow disclosures: | | | |
| Cash paid for interest | $ | 20,038 | | | $ | — | |
| Cash paid for income taxes | $ | — | | | $ | — | |
| | | |
| Non-cash investing and financing activities: | | | |
| Accrued direct issuance costs – offering | $ | (276,560) | | | $ | 125,000 | |
| Property and equipment acquired through accounts payable | $ | — | | | $ | 780,000 | |
| Capital contributions – debt forgiveness | $ | 434,065 | | | $ | 132,708 | |
| Accrued preferred dividends | $ | 160,219 | | | $ | 1,280,000 | |
| Debt discount on convertible notes and notes payable – issuance of warrants | $ | 145,974 | | | $ | 466,754 | |
| | | | | | | | | | | |
| Issuance of common stock for extinguishment of convertible note payable - related party and non-related party | $ | 10,400,000 | | | $ | — | |
| Issuance of common stock for extinguishment of convertible note payable accrued interest - related party and non-related party | $ | 1,432,768 | | | $ | — | |
| Issuance of common stock for extinguishment of dividends payable | $ | 4,773,480 | | | $ | — | |
| Issuance of common stock for extinguishment of preferred stock A and preferred stock B | $ | 2,535 | | | $ | — | |
See accompanying notes to the financial statements.
SOLITON, INC.
NOTES TO THE FINANCIAL STATEMENTS
1. Background, Organization and Going Concern
Soliton, Inc. (the “Company”) was organized under the laws of the State of Delaware on March 27, 2012. The Company operates in one segment as a medical device company organized to develop and commercialize products utilizing a proprietary Rapid Acoustic Pulse ("RAP") technology platform. The Company is a pre-revenue stage medical device company with a novel and proprietary platform technology licensed from The University of Texas M.D. Anderson Cancer Center ("MD Anderson"). The Company's first product being developed will be for the removal of tattoos. In addition, the Company completed proof-of-concept clinical trials for the reduction of cellulite and the treatment of hypertrophic scars and has initiated a four-site pivotal trial for the reduction of cellulite. The Company is based in Houston, Texas. Upon completion of the development of its products and regulatory clearances to market such products, the Company anticipates revenue will be driven by the sale of its RAP console and disposable cartridges to dermatologists, plastic surgeons and other physician offices, as well as medi-spas under the supervision of a doctor.
Initial Public Offering
On February 19, 2019, the Company consummated its initial public offering (“IPO”). In the IPO, the Company sold a total of 2,172,591 shares of common stock at a purchase price of $5.00 per share for gross proceeds of $10,862,955 and net proceeds of $9,714,198. In connection with the closing of the IPO, the Company's convertible notes (and related accrued interest) of $11,784,987 were converted into 6,825,391 shares of the Company's common stock, accrued dividends of $4,773,480 were converted into 954,696 shares of the Company's common stock, and preferred stock, both Series A and Series B, were converted into 2,534,766 shares of the Company's common stock. In addition, 127,500 shares of unvested restricted stock grants were immediately vested upon the completion of the IPO. Total shares of common stock outstanding at the closing of the IPO amounted to 14,613,000. Upon the closing of the IPO, certain notes were to be automatically converted according to their terms into the Company’s common stock to the extent and provided that certain holders of these notes are not permitted to convert such notes to the extent that the holders or any of its affiliates would beneficially own in excess of 4.99% of the Company’s common stock after such conversion. Due to this 4.99% limitation, principal representing $47,781 of these notes remained outstanding and were converted into 273,034 shares of its common stock in August and September 2019 when the conversion did not result in the holders and any of its affiliates to own more than 4.99% of the Company's outstanding common shares.
Private Investment in Public Entity Offerings ("PIPE")
On June 16, 2019, the Company entered into a private offering with certain institutional and accredited investors for the sale by the Company of 675,000 units (each a “June Unit”) of common stock issued at $14.00 per June Unit for total gross proceeds of $9,450,000. Each June Unit consisted of (i) one share of the Company’s common stock, and (ii) a warrant to purchase 0.7 shares (a total of 472,500) of common stock (each a “June Warrant”) (collectively, "June PIPE"). The offering price of the June Units was $14.00 per Unit. The June Warrants included in the June Units are exercisable at a price of $16.00 per share commencing on the date of issuance and will expire on August 23, 2024, pursuant to which the resale of the shares of common stock underlying the June Warrants are registered. On July 1, 2019, the Company filed a Registration Statement on Form S-1 to register for resale the common stock underlying the June Units sold with the Company's June 2019 private offering. The Company estimates the net proceeds from the closing of the sale of the June Units on June 19, 2019 was $8,643,302 after deducting the placement agent fees and estimated offering expenses payable by the Company.
On October 10, 2019, the Company entered into a private placement with certain institutional and accredited investors for the sale by the Company of 485,250 units (each an “October Unit”) of common stock issued at $12.88 per October Unit for total gross proceeds of $6,250,020. Each October Unit consisted of (i) one share of the Company’s common stock and (ii) a warrant to purchase 1.1 shares (a total of 533,775 shares) of common stock (each an “October Warrant”) (collectively, "October PIPE"). The October Warrants included in the October Units are exercisable at a price of $12.88 per share commencing on the date of issuance and will expire on October 10, 2024. On November 8, 2019, the Company filed a Registration Statement on Form S-1 to register for resale the common stock underlying the October Units sold with the Company's October 2019 private offering. The Company estimates the net proceeds from the closing of the sale of the October Units on October 11, 2019 was $5,738,111 after deducting the placement agent fees and estimated offering expenses payable by the Company.
Going Concern
The Company is an early stage and emerging growth company and has not generated any revenues to date. As such, the Company is subject to all of the risks associated with early stage and emerging growth companies. Since inception, the Company has incurred losses and negative cash flows from operating activities. The Company does not expect to generate positive cash flows from operating activities in the near future.
For the years ended December 31, 2019 and 2018, the Company incurred net losses of $13,751,877 and $9,314,936, respectively, and had net cash flows used in operating activities of $10,605,073 and $4,599,677, respectively. At December 31, 2019, the Company had an accumulated deficit of $56,043,371, positive working capital of $9,309,454 and cash of $12,076,425. The Company does not expect to experience positive cash flows from operating activities in the near future, if at all. The Company anticipates incurring operating losses for the next several years as it completes the development of its products and seeks requested regulatory clearances to market such products. These factors raise substantial doubt about the Company's ability to continue as a going concern within one year after the date the financial statements are issued. The accompanying financial statements have been prepared on a going concern basis and do not include any adjustments that might be necessary if the Company is unable to continue as a going concern.
The Company’s cash, cash equivalents and restricted cash on hand of $10,081,593 as of February 11, 2020 is sufficient to fund its operations through the third quarter of 2020 but not beyond. The Company also believes it will need to raise additional capital in order to continue to execute its business plan, including obtaining additional regulatory clearance for its products currently under development and commercializing and generating revenues from products already cleared. There is no assurance that additional financing will be available when needed or that management will be able to obtain financing on terms acceptable to the Company. A failure to raise sufficient capital will adversely impact the Company’s ability to meet its financial obligations as they become due and payable and to achieve its intended business objectives. If the Company is unable to raise sufficient additional funds, it will have to scale back its operations.
2. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying annual financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”) and the rules and regulations of the U.S. Securities and Exchange Commission (“SEC”).
Segments
The Company operates in one reportable segment based on management’s view of its business for purposes of evaluating performance and making operating decisions.
Use of Estimates in Financial Statement Presentation
The preparation of these financial statements and accompanying notes in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. The Company's significant estimates and assumptions include estimated work performed but not yet billed by contract manufacturers, engineers and research organizations, the valuation of equity related instruments, depreciable lives of long-lived assets (including property and equipment and intangible assets), and the valuation allowance related to deferred taxes. Some of these judgments can be subjective and complex, and, consequently, actual results could differ from those estimates. Although the Company believes that its estimates and assumptions are reasonable, they are based upon information available at the time the estimates and assumptions were made. Actual results could differ from those estimates.
Cash, Cash Equivalents and Restricted Cash
The Company considers all highly liquid accounts with original maturities of three months or less to be cash equivalents. The Company participates in an insured cash sweep program through its bank that sweeps cash balances exceeding the FDIC insured limit of $250,000 into multiple accounts. Periodically in the ordinary course of business, the Company may carry cash balances at financial institutions in excess of the insured limits of $250,000.
Restricted cash consists of amounts held in deposit with the Company’s bank to collateralize a letter of credit which supports the Company's obligations to pay or perform according to the requirements of an underlying agreement with a certain vendor. Such letter of credit has an initial term of one year, renews automatically and can only be modified or canceled with the approval of the beneficiary. As of December 31, 2019, the letter of credit was not used.
Property and Equipment
Property and equipment are stated at historical cost and depreciated on a straight-line basis over the estimated useful lives, generally three to five years. Leasehold improvements are depreciated over the shorter of the remaining lease term or useful lives of the assets. Upon disposition of the assets, the costs and related accumulated depreciation are removed from the accounts and any resulting gain or loss is included in the results of operations. Repairs and maintenance costs are included as expense in the accompanying statement of operations.
Intangible Assets
Intangible assets include trademarks. At December 31, 2019 and 2018, the Company had trademarks of $97,556 and $84,942, respectively. Trademarks are determined to have an indefinite useful life are not amortized, but instead are tested for impairment at least annually or sooner if events or changes in circumstances indicate that the asset may be impaired. During the year ended December 31, 2018, the Company wrote off its patents, included in the research and development line item in the accompanying statement of operations, with net book value of $19,138. During the year ended December 31, 2019, the costs for filing and prosecuting patent applications and patents filed by the Company were expensed as incurred and were classified as research and development expenses. Amortization expense for the years ended December 31, 2019 and 2018 was $0 and $376, respectively.
Long-Lived Assets
The Company evaluates its long-lived assets, including property and equipment, for impairment whenever events or changes in circumstances indicate that the carrying value of these assets may not be recoverable. Recoverability of these assets is measured by comparison of the carrying amount of each asset to the future undiscounted cash flows expected to result from the use of the asset and its eventual disposition. If the asset is considered impaired, the amount of any impairment is measured as the difference between the carrying value and the fair value of the impaired assets.
Deferred Rent
Deferred rent is recorded and amortized to the extent the total minimum rental payments allocated to the current period on a straight-line basis differ from the cash payments required.
Convertible Debt
When conversion terms related to convertible debt would be triggered by future events not controlled by the Company, the Company accounts for the conversion feature as contingent conversion options. Recognition of the intrinsic value of the conversion option is recognized only upon the occurrence of a triggering event.
Fair Value Measurements
Fair value is defined as the price which would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. A three-tier fair value hierarchy which prioritizes the inputs used in the valuation methodologies, as follows:
Level 1 Inputs - Unadjusted quoted prices in active markets for identical assets or liabilities that the reporting entity has the ability to access at the measurement date.
Level 2 Inputs - Inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly or indirectly. These might include quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active, inputs other than quoted prices that are observable for the asset or liability (such as interest rates, volatilities, prepayment speeds, credit risks, etc.) or inputs that are derived principally from or corroborated by market data by correlation or other means.
Level 3 Inputs - Unobservable inputs for determining the fair values of assets or liabilities that reflect an entity’s own assumptions about the assumptions that market participants would use in pricing the assets or liabilities.
At December 31, 2019 and 2018, the carrying amounts of the Company's financial instruments, including cash, cash equivalents and restricted cash, convertible notes payable, notes payable and accounts payable, approximate their respective fair value due to the short-term nature of these instruments.
At December 31, 2019 and 2018, the Company does not have any assets or liabilities required to be measured at fair value on a recurring basis.
Deferred Direct IPO Issuance Costs – Offering
The Company had capitalized offering costs of $276,560, consisting of legal, accounting and other fees and costs related to the IPO, which were reclassified to additional paid-in capital ("APIC") as a reduction of the proceeds upon the closing of the IPO in February 2019.
Warrants to Purchase Common Stock
The Company issued warrants to purchase shares of common stock related to (i) bridge notes issued prior to its IPO, (ii) private investment in public equity ("PIPE") deals, and (iii) as part of underwriter compensation in 2019 and 2018. The Company accounted for such warrants in accordance with ASC Topic 480-10, Distinguishing Liabilities from Equity, which identifies three categories of freestanding financial instruments that are required to be accounted for as a liability. Based on this guidance, the Company determined, for each issuance, that its warrants did not need to be accounted for as a liability. Accordingly, the warrants were classified as equity and are not subject to remeasurement at each balance sheet date. In addition, the Company accounts for issuance costs of warrants issued with debt instruments in accordance with ASC 470-20, Debt with Conversion and Other Options, which states proceeds from the sale of a debt instrument with stock purchase warrants (detachable call options) are allocated to elements based on the relative fair values of the debt instrument without the warrants and of the warrants themselves at time of issuance. The portion of the proceeds so allocated to the warrants are accounted for as additional paid-in capital. The remainder of the proceeds are allocated to the debt instrument, which may result in a discount or premium.
On July 1, 2019, the Company filed a Registration Statement on Form S-1 to register for resale the common stock underlying the June Units sold with the Company's June 2019 private offering. On November 8, 2019, the Company filed a Registration Statement on Form S-1 to register for resale the common stock underlying the October Units sold with the Company's October 2019 private offering. Related registration rights agreements are accounted for in accordance with ASC Topic 450-20, Loss Contingencies, which requires measurement of the contingent liability when an entity would be required to deliver shares under a registration payment arrangement, the transfer of consideration is probable and the number of shares to be delivered can be reasonably estimated. Accordingly, there is no liability under the payment arrangement requiring disclosure or recognition.
The fair value of warrants is estimated using the Black-Scholes option pricing model, based on the market value of the underlying common stock at the measurement dates, the contractual terms of the warrants, risk-free interest rates and expected volatility of the price of the underlying common stock. There are no expected dividends.
Research and Development Expenses
Research and development expenses are recognized as incurred and include the costs related to the Company's various contract research service providers, suppliers, engineering services, supplies, outsourced testing and consulting, clinical costs, and salaries and related costs of employees working directly on research activities.
Stock-Based Compensation
Stock-based compensation expense includes the estimated fair value of equity awards vested during the reporting period. The expense for equity awards vested during the reporting period is determined based upon the grant date fair value of the award and is recognized over the applicable vesting period of the stock award using either the straight-line method or an accelerated method, depending on the vesting structure, and is included in general and administrative expenses. Forfeitures are recognized as they are incurred.
Income Taxes
The Company uses the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial reporting and the tax bases of reported assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company must then assess the likelihood that the resulting deferred tax assets will be realized. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized. Tax rate changes are reflected in income during the period such changes are enacted. All of the Company's tax years remain subject to examination by the tax authorities.
In assessing the realization of deferred tax assets, management considers whether it is more likely than not that some portion or all of deferred assets will not be realized. The ultimate realization of the deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. The Company has recorded a full valuation allowance against its net total deferred tax assets as of December 31, 2019 and 2018 because management determined that it is not more-likely-than not that those assets will be realized. Accordingly, there was no income tax benefit for all periods presented.
Management has evaluated and concluded that there were no material uncertain tax positions requiring recognition in the Company's financial statements as of December 31, 2019. The Company does not expect any significant changes in the unrecognized tax benefits within twelve months of the reporting date.
The Company's policy is to classify interest expense and any related penalties related to income tax uncertainties as a component of income tax expense. No interest or penalties have been recognized in 2019 and 2018.
Net Loss per Common Share
Basic net loss per common share is computed by dividing net loss available to common stockholders by the weighted-average number of common shares outstanding during the period. Diluted net loss per common share is determined using the weighted-average number of common shares outstanding during the period, adjusted for the dilutive effect of common stock equivalents. The Company's unvested stock awards that contain non-forfeitable rights to dividends or dividend equivalents, whether paid or unpaid, are considered participating securities and are contemplated in the computations of basic and diluted earnings or loss per share. These securities do not participate in losses and accordingly no such allocation has been made in the periods presented. In periods when losses are reported, the weighted-average number of common shares outstanding excludes common stock equivalents, because their inclusion would be anti-dilutive.
As of December 31, 2019, potentially dilutive securities included options to purchase 2,883,550 common shares, unvested restricted stock of 158,336 shares and warrants to purchase 1,374,608 common shares.
As of December 31, 2018, potentially dilutive securities included options to purchase 2,235,000 common shares, preferred stock convertible to 2,534,766 common shares, accrued preferred stock dividend convertible at a price determined by the Company's Board of Directors (the "Board"), unvested restricted stock of 127,500 shares, warrants to purchase 776,350 common shares and notes and accrued interest convertible to common shares upon a future financing.
JOBS Act Accounting Election
The Company is an emerging growth company ("EGC"), as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”). The JOBS Act exempts emerging growth companies from being required to comply with new or revised financial accounting standards until private companies are required to comply with the new or revised financial accounting standards. The JOBS Act provides that a company can elect to opt out of the extended transition period and comply with the requirements that apply to non-emerging growth companies but any such election to opt out is irrevocable. The Company has elected not to opt out of such extended transition period which means that when a standard is issued or revised and it has different application dates for public or private companies, the Company, as an EGC, can adopt the new or revised standard at the time private companies adopt the new or revised standard. This may make comparison of the Company’s financial statements with another public company which is neither an EGC nor an EGC which has opted out of using the extended transition period difficult or impossible because of the potential differences in accounting standards used.
Subsequent Events
The Company’s management reviewed all material events through the date that the financial statements were issued for subsequent event disclosure consideration as discussed in Note 9.
Recent Accounting Standards
In February 2016, the FASB issued Accounting Standards Update ("ASU") No. 2016-02, “Leases (Topic 842)”, which establishes a right-of-use (“ROU”) model requiring a lessee to recognize a ROU asset and a lease liability for all leases with terms greater-than 12 months. Leases will be classified as either finance or operating, with classification affecting the pattern of expense recognition in the income statement. This guidance is currently effective, for public EGC companies like the Company, for fiscal years beginning after December 15, 2020 and may include interim periods within those fiscal years. The modified retrospective transition approach applies to leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements, with certain practical expedients available. The Company has the option to instead apply the provisions at the effective date without adjusting the comparative periods presented. The Company is currently evaluating the impact of this guidance on its financial position, results of operations, and cash flows.
In June 2018, the FASB issued ASU No. 2018-07, “Compensation Stock Compensation (Topic 718), Improvements to Non-Employee Share-Based Payment Accounting.” Under legacy guidance, the accounting for non-employee share-based payments differs from that applied to employee awards, particularly with regard to the measurement date and the impact of performance conditions. ASU No. 2018-07 provides that existing employee guidance will apply to non-employee share-based transactions (as long as the transaction is not effectively a form of financing), with the exception of specific guidance related to the attributions of compensation cost. The cost of non-employee awards will continue to be recorded as if the grantor had paid cash for the goods or services. In addition, the contractual term will be able to be used in lieu of an expected term in the option-pricing model for non-employee awards. The Company adopted the standard as of January 1, 2019 and it did not have an impact on the Company's financial statements, as non-employee stock compensation is nominal relative to the Company's total expenses for the year ended December 31, 2019.
The Company does not believe that any other recently issued effective standards, or standards issued but not yet effective, if adopted, would have a material effect on the accompanying financial statements.
Reclassifications
In certain instances, amounts reported in the prior year financial statements have been reclassified to conform to the current financial statement presentation. Such reclassifications had no effect on previously reported shareholders' equity (deficit) or net loss.
3. Prepaid Expenses and Other Current Assets
Prepaid expenses and other current assets consisted of the following:
| | | | | | | | | | | |
| December 31, 2019 | | December 31, 2018 |
| Prepaid insurance | $ | 93,950 | | | $ | 9,453 | |
| Other prepaids and receivables | 2,360 | | 1,080 |
| Total prepaid expenses and other current assets | $ | 96,310 | | | | $ | 10,533 | |
4. Property and Equipment
Property and equipment consisted of the following:
| | | | | | | | | | | |
| December 31,
2019 | | December 31,
2018 |
| Computer equipment and software | $ | 126,643 | | | $ | 105,704 | |
| Research and development equipment | 244,480 | | | 244,480 | |
| Lab equipment | 780,000 | | | 780,000 | |
| Leasehold improvements | 271,124 | | | 242,167 | |
| Furniture | 19,893 | | | 19,893 | |
| Subtotal | 1,442,140 | | | 1,392,244 | |
| Less: accumulated depreciation | (594,741) | | | (378,004) | |
| Total property and equipment | $ | 847,399 | | | $ | 1,014,240 | |
As of December 31, 2019, the Company had $689,000 of lab equipment in the field at clinical trial sites and held by a vendor for final testing. Depreciation of this equipment started when it was placed in service in June 2019.
Depreciation expense for the years ended December 31, 2019 and 2018 was $216,737 and $120,488, respectively.
5. Notes Payable
On February 19, 2019, the Company consummated its IPO. In connection with the closing of the IPO, the Company's convertible notes (and related accrued interest) of $11,784,987 were converted into 6,825,391 shares of the Company's common stock. Upon the closing of the IPO, certain notes were to be automatically converted according to their terms into the Company’s common stock to the extent and provided that certain holders of these notes are not permitted to convert such notes to the extent that the holders or any of its affiliates would beneficially own in excess of 4.99% of the Company’s common stock after such conversion. Due to this 4.99% limitation, principal representing $47,781 of these notes remained outstanding and were converted into 273,034 shares of its common stock in August and September 2019 when the conversion did not result in the holders and any of its affiliates owning more than 4.99% of the Company's outstanding common shares.
On January 18, 2017, the Board approved a note purchase agreement (the "First Note") allowing the Company to sell an aggregate of $3,000,000 of convertible bridge notes. The notes were convertible into either the Company’s preferred or common stock (depends on the equity securities offered in the equity financing) at 75% of the price paid per share in a subsequent equity financing where the Company receives gross proceeds of not less than $5,000,000 or at 85% of the per share price determined by dividing the equity value of the Company that is expected to be available for distribution to the Company’s stockholders by the aggregate number of the Company’s fully-diluted common shares upon the closing of a sale, liquidation, merger, or change of control of the Company. The notes bore interest at 8.25% per annum and initially matured on January 31, 2018, which date was extended as discussed below. At maturity, the interest rate increased to 12.0% per annum. On June 19, 2017, the Company entered into the first amendment ("First Amendment") to the First Note to allow for the sale and issuance of an additional $3,250,000 of Notes up to an aggregated amount of $6,250,000. The total amount of issuances under the Company's First Note and First Amendment as of December 31, 2018 amounted to $5,000,000 and were issued to a single related party, who is a major stockholder of the Company. As a result of the Company’s IPO on February 19, 2019, the principal amount of $5,000,000 and accrued interest of $944,063 were converted into 1,585,086 shares of the Company’s common stock.
On November 1, 2017 the Board approved a second note purchase agreement (the "Second Note") allowing the Company to sell an aggregate of $1,900,000 of notes. The notes were convertible into either the Company’s preferred or common stock (depends on the equity securities offered in the equity financing) at 75% of the price paid per share in a subsequent equity financing where the Company receives gross proceeds of not less than $5,000,000 or at 85% of the per share price determined by dividing the equity value of the Company that is expected to be available for distribution to the Company’s stockholders by the aggregate number of the Company’s fully-diluted common shares upon the closing of a sale, liquidation, merger, or change of control of the Company. The notes bore interest at 8.25% per annum and initially matured on June 29, 2018, which date was extended as discussed below. At maturity, the interest rate increased to 12.0% per annum.
The Company closed the initial tranche of the Second Note on November 9, 2017 for $400,000, followed by a tranche on December 1, 2017, for $375,000, a third tranche on December 26, 2017 for $250,000, a fourth tranche on January 8, 2018 for $250,000, a fifth tranche on January 25, 2018 for $250,000 and a final tranche on February 13, 2018 for $375,000 for a total of $1,900,000.
On June 29, 2018, the Company and the related party modified the maturity date of the Notes entered into under the First Note and Second Note to April 30, 2019.
The total amount of issuance under the Second Note amounted to $1,900,000 and was issued to a single related party, who is a major stockholder of the Company. As a result of the Company’s IPO, the principal amount of $1,900,000 and accrued interest of $223,368 were converted into 566,235 shares of the Company’s common stock.
On April 2, 2018, the Board approved a note purchase agreement (the "Third Note"), which was amended on August 10, 2018, allowing the Company to sell an aggregate of $500,000 of notes. The Third Note provided that, on the closing date of the IPO, the outstanding principal and accrued, but unpaid, interest would be converted into common stock at the conversion price of $0.175. However, certain notes holders were not permitted to convert their notes when the holders or any of its affiliates would beneficially owned in excess of 4.99% of the Company’s common stock after such conversion. The holders of the Company’s outstanding preferred shares agreed to waive the adjustment to the preferred stock conversion price triggered by the Third Note. The notes bore interest at 10.0% per annum and were to mature on April 2, 2020 but were settled as a result of the Company's IPO on February 19, 2019.
The total amount of issuance under the Third Note amounted to $500,000. The Company issued $250,000 to a single related party, who is a major stockholder of the Company, and $250,000 to four non-related party investors. As a result of the Company’s IPO, principal amount of $452,219 and accrued interest of $43,562 were converted into 2,833,034 shares of the Company’s common stock. In August and September 2019, the remaining principal amount of $47,781 was converted into 273,034 shares of the Company's common stock. As of December 31, 2019, the amount outstanding under the Third Note was fully converted for a total of 3,106,068 shares of the Company's common stock.
On April 17, 2018, the Board approved a note purchase agreement (the "Fourth Note") allowing the Company to sell an aggregate of $3,000,000 of notes. The Fourth Note provided that on the closing date of the IPO, the outstanding principal and accrued, but unpaid, interest would be converted into common stock at the conversion price of $1.75. The holders of the Company’s outstanding preferred shares agreed to waive the adjustment to the preferred stock conversion price triggered by the Fourth Note. The notes bore interest at 10% per annum and matured 2 years from the note issuance date but were settled as a result of the Company's IPO on February 19, 2019.
The total amount of issuance under the Fourth Note amounted to $3,000,000. The Company issued $1,272,000 in principal amount of such notes to related party investors and $1,728,000 to non-related party investors. As a result of the Company’s IPO, the principal amount of $3,000,000 and accrued interest of $221,775 were converted into 1,841,036 shares of the Company’s common stock.
The Company incurred issuance costs relating to the Fourth Note in the amount of $163,760, which were being amortized over 24 months but were accelerated as a result of the Company’s IPO closing, resulting in the remaining $118,492 being expensed during the year ended December 31, 2019.
The Company also issued warrants to purchase 91,350 shares of common stock at a price of $1.75 per share to placement agents in connection with the notes issued under the Fourth Note. For additional information, see Note 7. The value of these warrants were $103,006 which was being amortized over 24 months months but was accelerated as a result of the Company’s IPO closing, resulting in the remaining $74,532 being expensed during the year ended December 31, 2019.
On August 7, 2018, the Company's Board authorized it to commence a new offering for up to $485,000 10% non-convertible promissory notes, which were accompanied by a five year warrant to purchase one share of common stock with an exercise price of $1.75 per share for each dollar in principal amount of notes purchased (collectively, the "Fifth Note") that can be exercised (i) at any time on or after the issuance of the notes and (ii) on or prior to the close of business on the five year anniversary of the issuance of the notes. Mr. Klemp, Dr. Capelli, Ms. Bisson and other members of management collectively purchased $125,000 of such notes and warrants. The principal and interest on the Fifth Note were due on the earlier of one-year from the date of issuance or upon successful completion of the IPO.
On August 31, 2018, the Company's Board approved a $200,000 increase to the Fifth Note authorized on August 7, 2018. On December 21, 2018, the Company's Board approved an additional $300,000 increase to the Fifth Note authorized on August 7, 2018 up to a maximum of $985,000. From October 2018 to February 2019, the Company issued $125,000 and $860,000 of the Fifth Note to related parties and non-related parties, respectively. On February 15, 2019, the Company paid $985,000 in principal and 20,038 in accrued interest to the note holders to repay the Fifth Note in full.
The Company issued 685,000 warrants in connection with the issuances of the Fifth Note in 2018. These warrants were valued at $775,616. Proceeds of $363,748 (of which $66,423 was for related party and $297,325 was for non-related party) were allocated to issuance cost based on the relative fair value of these warrants. These issuance costs were being amortized over 24 months but were accelerated as a result of the Company’s IPO closing, resulting in the remaining balance of $325,955 being expensed during the year ended December 31, 2019.
The Company issued 300,000 warrants in connection with the issuances of the Fifth Note in January and February 2019. These warrants were valued at $285,234. Proceeds of $145,974 (of which all was for non-related party) were allocated to issuance cost based on the relative fair value of these warrants. These issuance costs were being amortized over 24 months but were accelerated as a result of the Company’s IPO closing, resulting in the entire balance of $145,974 being expensed during the year ended December 31, 2019.
6. Commitments and Contingencies
On April 5, 2012, the Company entered into a Patent and Technology License Agreement with MD Anderson. Pursuant to the agreement, the Company obtained a royalty-bearing, worldwide, exclusive license to intellectual property including patent rights related to the patents and technology the Company uses. Under the agreement, the Company agreed to pay a nonrefundable license documentation fee in the high-five digits 30 days after the effective date of the agreement. Additionally, the Company agreed to pay a nonrefundable annual maintenance fee starting on the third anniversary of the effective date of the agreement, which escalates each anniversary and is currently in the mid-five digits. Additionally, the Company agreed to a running royalty percentage of net sales in the mid-single digits. The Company also agreed to make certain milestone payments in the low to mid-six digits and sublicensing payments, including a $250,000 milestone payment made in June 2019 after the Company received U.S. Food & Drug Administration ("FDA") clearance for our RAP device for tattoo removal. The specific patents initially subject to the agreement expire between 2031 and 2032.
MD Anderson has the right to terminate the agreement upon advanced notice in the event of a default by Soliton. The agreement will expire upon the expiration of the licensed intellectual property. The rights obtained by the Company pursuant to the agreement are made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between MD Anderson and the U.S. government. To the extent that is the case, the Company's license agreement with, and the intellectual property rights it has licensed from MD Anderson, are subject to such a funding agreement and any superior rights that the U.S. government may have with respect to the licensed intellectual property. Therefore, there is a risk that the intellectual property rights the Company has licensed from MD Anderson may be non-exclusive or void if a funding agreement related to the licensed technology between MD Anderson and the U.S. government does exist and depending on the terms of such an agreement. Notwithstanding the foregoing, the Company does not believe our RAP technology received any federal funding. All out-of-pocket expenses incurred by MD Anderson in filing, prosecuting and maintaining the licensed patents have been and shall continue to be assumed by the Company. For the years ended December 31, 2019 and 2018, the Company paid $65,000 and $55,000, respectively, for expenses related to this agreement.
As the inventor of the intellectual property licensed from MD Anderson, Dr. Capelli, the Company's Chief Executive Officer, is entitled to 50% of the license income (which is determined after MD Anderson recoups any costs associated therewith) that the Company is required to pay to MD Anderson pursuant to the Company's license agreement with MD Anderson. For the years ended December 31, 2019 and 2018, Dr. Capelli received $187,500 and $27,500 respectively from MD Anderson. In addition, Dr. Capelli is entitled to 50% of the proceeds (after the recoupment of any costs associated therewith) from the sale by MD Anderson of 175,000 shares issued to MD Anderson in connection with the license agreement.
Lease Commitments
The Company leases space for its corporate office, which provides for a 63 month term beginning on February 1, 2016, for rent payments of $7,867 per month. Total rent expense under this office space lease arrangement for the years ended December 31, 2019 and 2018 was $95,519 and $89,643, respectively.
Future minimum lease payments as of December 31, 2019 were as follows:
| | | | | | | | |
| Year Ending December 31, | | Amount |
| 2020 | | $ | 106,153 | |
| 2021 | | 35,751 | |
| Total future minimum lease payments | | $ | 141,904 | |
Purchase Commitments
On November 20, 2019, the Company entered into a cooperative development addendum ("Addendum") to its engineering and development services master agreement with Emphysys, Inc. ("Emphysys”). The Addendum states that Emphysys will provide the Company with engineering and design services related to shockwave technology for use in dermatology and aesthetics fields for a three year period.
During the term of the Addendum, the Company agreed to certain minimum annual expenditures. If the Company fails to spend such minimum annual amounts or if the Company terminates the Addendum without cause, the Company will be required to pay Emphysys a termination fee ranging in the low to mid-six digits. In the event that all or substantially all of the stock or assets of either party are sold then, at the request of other party, the Addendum may be terminated (without the requirement to pay a termination fee) and the obligation of Emphysys to provide future services to the Company shall terminate. Pursuant to the Addendum, with certain exceptions, Emphysys covenanted that it will not perform or agree to
perform services with any company other than Soliton in the area of arc-discharge driven acoustical shockwave generation for medical dermatological or aesthetic dermatological indications during the term of the Addendum or any extension thereof, and for a period of six months after the termination of the Addendum.
As of December 31, 2019 the Company had purchase obligations of $3,937,500 to a single engineering service provider. This commitment is for services used in the ordinary course of business and does not represent excess commitments or loss contracts. This commitment can be terminated with a penalty payment of no more than $500,000.
| | | | | |
| Year Ending December, 31 | Amount |
| 2020 | $ | 1,575,000 | |
| 2021 | 1,575,000 | |
| 2022 | 787,500 | |
| Total future minimum purchase commitments | $ | 3,937,500 | |
Letters of Credit
The Company has an irrevocable letter of credit which supports its obligations to pay or perform according to the requirements of an underlying agreement with a certain vendor. Such letter of credit has an initial term of one year, renews automatically for an additional year and can only be modified or canceled with the approval of the beneficiary. As of December 31, 2019, the letter of credit was not used.
Legal Proceedings
In the normal course of business, from time-to-time, the Company may be subject to claims in legal proceedings. However, the Company does not believe it is currently a party to any pending legal actions. Notwithstanding, legal proceedings are subject-to inherent uncertainties, and an unfavorable outcome could include monetary damages, and in such event, could result in a material adverse impact on the Company's business, financial position, results of operations, or cash flows.
Employment Agreements
The Company has agreements with certain employees to provide certain benefits in the event of termination where the base salary and certain other benefits would aggregate $1,775,035 using the rate of compensation in effect at December 31, 2019.
7. Stockholders’ (Deficit) Equity
Preferred Stock
Until amending the Company's certificate of incorporation in February 2019, the Company was authorized to issue 2,534,766 shares of preferred stock with a par value of $0.001 per share with such designation, rights, and preferences as may be determined from time-to-time by the Company's Board. As of December 31, 2019 and 2018, there were 0 and 416,666 Series A preferred stock and 0 and 2,118,100 Series B preferred stock issued and outstanding, respectively. Dividends accrued at a rate of 8.00% per annum based on $4.80 per Series A preferred share, the dividends were cumulative but non-compounding.
The Series B preferred stock had similar rights as Series A preferred stock except that the dividends were based on $6.61 per Series B preferred share and Series B preferred stock was convertible into common stock at a rate of $6.61 divided by a conversion price initially set at $6.61. As of the Company’s IPO date of February 19, 2019 and December 31, 2018, accrued dividends for preferred stock were $4,773,480 and $4,613,261, respectively. The holder of the Series A and Series B preferred stock agreed to convert the preferred stock into common stock upon the completion of the Company's IPO. The holders of the Company’s outstanding shares of preferred stock agreed to waive the adjustment to the conversion price of the preferred stock upon the issuances of the Third and Fourth Note.
On February 19, 2019, all outstanding shares of Series A and Series B preferred stock and accrued dividends on these shares were converted into 2,534,766 and 954,696 shares of common stock, respectively, upon the closing of the Company’s IPO. The Company amended its articles of incorporation on February 19, 2019 eliminating the preferred shares authorized under the amended certificate of incorporation.
Adoption of 2012 Long Term Incentive Plan
In November 2012, the Company’s Board and stockholders adopted the 2012 Long Term Incentive Plan (the “2012 Stock Plan”). The 2012 Stock Plan is designed to enable the Company to offer employees, officers, directors and consultants, as defined, an opportunity to acquire a proprietary interest in the Company. The types of awards that may be granted under the 2012 Stock Plan include stock options, stock appreciation rights, restricted stock, and other stock-based awards subject to limitations under applicable law. All awards are subject to approval by the Company’s Board. The 2012 Stock Plan reserves shares of common stock for issuance in accordance with the 2012 Stock Plan’s terms. Total number of shares reserved and available for issuance under the plan were 789,745 shares. As of December 31, 2019, 14,745 shares remained under the 2012 Stock Plan. The Company does not intend to utilize the 2012 Stock Plan and instead intends to utilize the 2018 Stock Plan.
Adoption of 2018 Stock Plan
In June 2018, the Company’s Board and stockholders adopted the 2018 Stock Plan. The 2018 Stock Plan is designed to enable the Company to offer employees, officers, directors and consultants, as defined, an opportunity to acquire a proprietary interest in the Company. The types of awards that may be granted under the 2018 Stock Plan include stock options, stock appreciation rights, restricted stock, and other stock-based awards subject to limitations under applicable law. All awards are subject to approval by the Company’s Board. The 2018 Stock Plan reserves shares of common stock for issuance in accordance with the 2018 Stock Plan’s terms. Total number of shares reserved and available for issuance under the plan is 3,400,000 shares. As of December 31, 2019, 531,450 shares remained available for grant under the 2018 Stock Plan.
Restricted Stock
Restricted stock activity for the year ended December 31, 2019 and 2018 is summarized as follows:
| | | | | | | | | | | |
| Number of Shares | | Weighted-Average Grant Date Fair Value |
| Outstanding at December 31, 2017 | 305,000 | | | $ | 3.21 | |
| Vested | (177,500) | | | 3.21 | |
| Outstanding at December 31, 2018 | 127,500 | | | $ | 3.21 | |
| Granted | 200,000 | | | 11.54 | |
| Vested | (169,164) | | | 5.26 | |
| Outstanding at December 31, 2019 | 158,336 | | | $ | 11.54 | |
| | | |
On May 8, 2019, the Company granted and issued 200,000 shares of restricted common stock to three consultants in connection with the provision of services pursuant to agreements entered into in April 2019. The consultants were each accredited investors. 25,000 shares vested within 4 months of the approval date of the agreement. The remaining 175,000 shares vest over 42 months, beginning on September 19, 2019. As of December 31, 2019, 41,664 shares have vested and 158,336 remain unvested.
During the years ended December 31, 2019 and 2018, the Company recorded $889,539 and $553,552, respectively, in stock-based compensation for the restricted shares previously issued. During the year ended December 31, 2018, 177,500 shares vested and 127,500 shares remained unvested, which immediately vested upon completion of the Company’s IPO.
As of December 31, 2019, there was $1,682,917 of unrecognized compensation expense related to restricted shares.
Stock Options
The following table summarizes stock option activities for the years ended December 31, 2019 and 2018:
| | | | | | | | | | | | | | | | | | | | | | | |
| Number of Shares | | Weighted Average Exercise Price | | Weighted Average Remaining Life (in Years) | | Aggregate Intrinsic Value |
| Outstanding, December 31, 2017 | 15,000 | | | $ | 0.13 | | | 9.75 | | $ | — | |
| Granted | 2,220,000 | | | 1.75 | | | — | | | — | |
| Outstanding, December 31, 2018 | 2,235,000 | | | | 1.74 | | | 9.44 | | $ | 23,100 | |
| Granted | 648,550 | | | | 6.03 | | | — | | | — | |
| Outstanding, December 31, 2019 | 2,883,550 | | | $ | 2.70 | | | 8.62 | | $ | 23,861,981 | |
| | | | | | | |
| Exercisable, December 31, 2019 | 867,563 | | $ | 1.73 | | | 8.66 | | $ | 8,025,827 | |
During the year ended December 31, 2018, the Company granted its employees 2,220,000 options to purchase the Company’s common stock with an exercise price of $1.75 per share, for a term of 10 years, and a vesting period of four years. The options have an aggregated grant date fair value of $2,694,567 that was calculated using the Black-Scholes option-pricing model. Variables used in the Black-Scholes option-pricing model include: (1) discount rate of 2.77% based on the daily yield curve rates for U.S. Treasury obligations, (2) expected life of 6.25 years based on the simplified method provided in Staff Accounting Bulletin, (3) expected volatility range from 84.5% to 84.7% based on the historical volatility of comparable companies' stock, (4) no expected dividends and (5) fair market value of the Company's stock at $1.67 per share which value was determined by the Company's Board after reviewing and considering, among other factors, a valuation report issued by an independent appraisal firm.
Officer Debt Forgiveness - Stock Options
In January 2019, certain individuals agreed to the extinguishment of $484,065 in deferred compensation, including $434,065 for individuals still with the Company, that had been earned through September 30, 2018 and was to be repaid out of the proceeds from the Company's IPO. In recognition of this extinguishment of deferred compensation, during the three months ended March 31, 2019, the Company granted these individuals options to purchase 401,750 shares of the Company’s common stock with an exercise price of $1.75 per share, for a term of 10 years, and a vesting period of 25% per quarter over one year. The options have an aggregated grant date fair value of $456,961 that was calculated using the Black-Scholes option-pricing model. Variables used in the Black-Scholes option-pricing model include: (1) discount rate of 2.53% based on the daily yield curve rates for U.S. Treasury obligations, (2) expected life of 5.27 years based on the simplified method (vesting plus contractual term divided by two), (3) expected volatility of 84.3% based on the historical volatility of comparable companies' stock, (4) no expected dividends and (5) fair market value of the Company's stock at $1.67 per share which value was determined by the Company's Board after reviewing and considering, among other factors, a valuation report issued by an independent appraisal firm.
In addition, during the year ended December 31, 2019, the Company granted certain individuals options to purchase 246,800 shares of the Company’s common stock with an average exercise price of $13.00 per share, for a term of 10 years, and a vesting period ranging from one year to 25% per year over four years. The options have an aggregated grant date fair value of $2,762,693 that was calculated using the Black-Scholes option-pricing model. Variables used in the Black-Scholes option-pricing model include: (1) discount rates ranging from 1.65% to 2.12% based on the daily yield curve rates for U.S. Treasury obligations, (2) expected lives ranging from 5.50 to 6.25 years based on the simplified method (vesting plus contractual term divided by two), (3) expected volatility ranging from 82.99% to 85.06% based on the historical volatility of comparable companies' stock, (4) no expected dividends and (5) fair market value of the Company's stock ranging from $5.73 to $17.50 per share.
All options issued and outstanding are being amortized over their respective vesting periods. The unrecognized compensation expense at December 31, 2019 was $3,485,140. During the years ended December 31, 2019 and 2018, the Company recorded $1,598,514 and $404,632, respectively, of stock-based compensation expense for options.
Warrants
During the year ended December 31, 2018, the Company issued warrants to purchase 776,350 shares of common stock at an exercise price of $1.75. The warrants expire five years from the date of issuance. The warrants were issued to placement agents and investors in connection with notes issued under the Fourth and Fifth Notes.
In January and February 2019, the Company issued warrants to purchase 300,000 shares of common stock at an exercise price of $1.75 on various dates. The warrants were issued to investors in connection with notes issued under the Fifth Note.
On February 19, 2019, the Company issued warrants to the underwriters of the Company's IPO to purchase 152,081 shares of common stock at an exercise price of $6.00. The warrants expire five years from the date of issuance.
The total grant date fair value of all these 1,228,431 warrants was $1,636,232, which was determined utilizing the Black-Scholes option pricing model. Variables used in the Black-Scholes option-pricing model include (1) discount rates in the range of 2.5% to 2.8% based on the daily yield curve rates for U.S. Treasury obligations, (2) expected term of five years based on the term of the warrants, (3) expected volatilities of 84.09% to 85.80% based on the historical volatility of comparable companies' stock, (4) no expected dividends, and (5) fair value of the Company's stock at $1.67 per share for warrants issued prior to the IPO, a value determined by the Company's Board after reviewing and considering, among other factors, a valuation report issued by an independent appraisal firm, or the fair market value of the Company's stock at the closing of its' IPO on February 19, 2019 of $4.87 for warrants on that day.
The fair value amount was included in discounts on convertible notes payable and was amortized over the life of the convertible notes payable. As a result of the Company’s IPO closing on February 19, 2019, all $664,953 of unamortized discount on convertible notes payable was accelerated and recorded as expense.
PIPE Offerings
On June 16, 2019, the Company entered into a private offering with certain institutional and accredited investors for the sale by the Company in a private placement of 675,000 units (each a “June Unit”) of common stock issued at $14.00 per June Unit for total gross proceeds of $9,450,000. Each June Unit consisted of (i) one share of its common stock, and (ii) a warrant to purchase 0.7 shares (a total of 472,500) of common stock (each a “June Warrant”) (collectively, "June PIPE"). The June Warrants included in the June Units are exercisable at a price of $16.00 per share commencing on the date of issuance and will expire on August 23, 2024, pursuant to which the resale of the shares of common stock underlying the June Warrants are registered. On July 1, 2019, the Company filed a Registration Statement on Form S-1 to register for resale the common stock underlying the June Units sold with the Company's June 2019 private offering. The Company estimates the net proceeds from the closing of the sale of the June Units on June 19, 2019 was $8,643,302 after deducting the placement agent fees and estimated offering expenses payable by the Company.
The grant date fair value of these 472,500 June Warrants was $4,420,503, which was determined utilizing the Black-Scholes option pricing model. Variables used in the Black-Scholes option-pricing model include (1) discount rate of 1.85% based on the daily yield curve rates for U.S. Treasury obligations, (2) expected term of five years based on the term of the warrants, (3) expected volatility of 85% based on the historical volatility of comparable companies' stock, (4) no expected dividends, and (5) fair value of the Company's stock at $14.30 per share.
On October 10, 2019, the Company entered into a second private placement of 485,250 units (each an “October Unit”) of common stock issued at $12.88 per October Unit for total gross proceeds of $6,250,020. Each October Unit consisted of (i) one share of the Company’s common stock, and (ii) a warrant to purchase 1.1 shares (a total of 533,775) of common stock (each an “October Warrant”) (collectively, "October PIPE"). The October Warrants included in the October Units are exercisable at a price of $12.88 per share commencing on the date of issuance and will expire on October 10, 2024. On November 8, 2019, the Company filed a Registration Statement on Form S-1 to register for resale the common stock underlying the October Units sold with the Company's October 2019 private offering. The Company estimates the net proceeds from the closing of the sale of the October Units on October 11, 2019 was $5,738,111 after deducting the placement agent fees and estimated offering expenses payable by the Company.
The grant date fair value of these 533,775 October Warrants was $4,537,648, which was determined utilizing the Black-Scholes option pricing model. Variables used in the Black-Scholes option-pricing model include (1) discount rate of 1.59% based on the daily yield curve rates for U.S. Treasury obligations, (2) expected term of five years based on the term of the warrants, (3) expected volatility of 82.92% based on the historical volatility of comparable companies' stock, (4) no expected dividends, and (5) fair value of the Company's stock at $12.88 per share.
The fair value amount of these PIPE transaction warrants were included in additional paid-in-capital as deal costs.
The following table summarizes warrant activities for the years ended December 31, 2019 and 2018:
| | | | | | | | | | | | | | | | | | | | | | | |
| Number of Shares | | Weighted Average Exercise Price | | Weighted Average Remaining Contractual Term (in Years) | | Aggregate Intrinsic Value |
| Outstanding, December 31, 2017 | — | | | $ | — | | | — | | | $ | — | |
| Granted | 776,350 | | | 1.75 | | | 4.80 | | — | |
| Outstanding, December 31, 2018 | 776,350 | | | $ | 1.75 | | | 4.80 | | $ | — | |
| Granted | 1,458,356 | | | 10.88 | | | — | | | — | |
| Exercised | (685,900) | | | 2.12 | | | — | | | 6,073,855 | |
| Forfeited (cashless exercise) | (174,198) | | | 3.99 | | | — | | | — | |
| Outstanding December 31, 2019 | 1,374,608 | | | $ | 10.97 | | | 4.43 | | $ | 13,591 | |
| | | | | | | | |
| Exercisable, December 31, 2019 | 1,374,608 | | | $ | 10.97 | | | 4.43 | | $ | 13,591 | |
Officer Debt Forgiveness - Warrants
During the year ended December 31, 2018, certain executives agreed to forgive bonuses totaling $132,708 that were previously approved by the Company’s Board. The bonuses related to services for 2015 and were included in accrued liabilities. The Company recorded the forgiveness as capital contributions in 2018 as the executives are considered related parties.
8. Income Taxes
The Company files U.S. federal and various U.S. state income tax returns. Due to the Company’s losses, there was no income tax expense for the years ended December 31, 2019 and 2018.
The income tax provision differs from the amount using the statutory federal income tax rate of 21% for 2019 and 2018 for the following reasons:
| | | | | | | | | | | | | | | | | | | | | | | |
| December 31,
2019 | | | | December 31,
2018 | | |
| Amount | | % | | Amount | | % |
| Tax benefit at the U.S. federal statutory rate | $ | (2,887,894) | | | (21.00) | % | | $ | (1,956,137) | | | (21.00) | % |
| Tax rate change | — | | | — | | | — | | | — | % |
| Permanent differences | 24,939 | | | 0.18 | % | | 119,266 | | | 1.28 | % |
| Return to provision | (24,608) | | | (0.18) | % | | (94,219) | | | (1.01) | % |
| Valuation allowance | 2,887,563 | | | 21.00 | % | | 1,931,090 | | | 20.73 | % |
| Effective income tax rate | $ | — | | | — | % | | $ | — | | | — | % |
The effective income tax rate varied from the statutory rate in 2019 and 2018 primarily due to the increase in the valuation allowance.
Deferred tax assets and liabilities consist of the following:
| | | | | | | | | | | |
| December 31,
2019 | | December 31,
2018 |
| Assets related to: | | | |
| Accounts payable and accrued liabilities | $ | 588,037 | | | $ | 1,224,599 | |
| Net operating losses and start-up costs | 9,891,122 | | | 6,240,595 | |
| Total deferred tax assets | 10,479,159 | | | 7,465,194 | |
| Valuation allowance for deferred tax assets | (10,297,430) | | | (7,409,867) | |
| Net deferred tax | 181,729 | | | 55,327 | |
| Liabilities related to: | | | |
| Accounts receivable and prepaid expenses | (20,226) | | | (27,976) | |
| Depreciation and amortization | (161,503) | | | (27,351) | |
| Net deferred tax liabilities | (181,729) | | | (55,327) | |
| | | |
| Net deferred tax assets | $ | — | | | $ | — | |
As of December 31, 2018, the Company’s filed tax returns include federal net operating loss (“NOL”) carryforwards of $29,602,639, of which $24,727,679 begin expiring in 2036 through 2037. Additionally, the Company estimates an NOL carryforward of $17,460,252 for the year ended December 31, 2019. Under the new Tax Cuts and Jobs Act from 2018, carryforwards do not expire, but can only offset 80% of taxable income in the year the loss carryforward is used.
The Company has recorded a full valuation allowance against its net total deferred tax assets as of December 31, 2019 and 2018 because management determined that it is not more-likely-than not that those assets will be realized. In assessing the realization of deferred tax assets, management considers whether it is more likely than not that some portion or all of deferred assets will not be realized. The ultimate realization of the deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible.
During the year ended December 31, 2019 and 2018, the valuation allowance increased by $2,879,647 and $1,931,090, respectfully, due to additional net operating losses.
Note 9 - Subsequent Events
On February 4, 2020, the Company granted options to employees and executives to purchase 381,800 shares of the Company’s common stock for a term of 10 years, an exercise price of $11.71, and a vesting period of 25% annually over a four year period. The options had an aggregated grant date fair value of $3,193,468 that was calculated using the Black-Scholes option-pricing model. Variables used in the Black-Scholes option-pricing model include: (1) a discount rate of 1.42% based on the daily yield curve rates for U.S. Treasury obligations, (2) expected life of 6.25 years based on the simplified method (vesting plus contractual term divided by two), (3) expected volatility of 83.12% based on the historical volatility of comparable companies' stock, (4) no expected dividends and (5) fair market value of the Company's stock of $11.71 per share.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosures.
On April 9, 2019, our Audit Committee dismissed Marcum LLP (“Marcum”) as our independent registered public accounting firm, effective as of such date.
The report of Marcum on our consolidated financial statements as of December 31, 2018 and for the year then ended did not contain an adverse opinion or disclaimer of opinion, and was not qualified or modified as to uncertainty, audit scope, or accounting principles, other than an explanatory paragraph relating to our ability to continue as a going concern. During the year ended December 31, 2018 and through April 1, 2019 there were no: (1) disagreements (as defined in Item 304(a)(1)(iv) of Regulation S-K and the related instructions to Item 304 of Regulation S-K) with Marcum on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedures, which disagreements, if not resolved to the satisfaction of Marcum, would have caused Marcum to make reference to the matter in its report on the consolidated financial statements for such year.
On April 9, 2019, the Audit Committee approved the appointment of Dixon Hughes Goodman LLP (“DHG”) as our independent registered public accounting firm for the fiscal year ended December 31, 2019.
During our last two fiscal years and through April 1, 2019, neither we nor anyone on our behalf consulted with DHG with respect to either (i) the application of accounting principles to a specific transaction, either completed or proposed, or the type of audit opinion that might be rendered on our financial statements, and neither written nor oral advice was provided to us that DHG concluded was an important factor considered by us in reaching a decision as to any accounting, auditing or financial reporting issue; or (ii) any matter that was either the subject of disagreement (as defined in Item 304(a)(1)(iv) of Regulation S-K and the related instructions to Item 304 of Regulation S-K) or a reportable event (as described in Item 304(a)(1)(v) of Regulation S-K).
We had no disagreements on accounting and financial disclosure matters with our independent registered public accountants to report under this Item 9.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Our management, including our Chief Executive Officer (“CEO”), who serves as our principal executive officer, and our Chief Financial Officer (“CFO”), who serves as our principal financial officer, evaluated the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act), as of the end of the period covered by this Form 10-K. Based on this evaluation, our CEO and our CFO, concluded that as a result of the material weaknesses in our internal control over financial reporting discussed below, our disclosure controls and procedures were not effective at ensuring that information required to be disclosed in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC's rules and forms and that such information is accumulated and communicated to our management, including our CEO and CFO, or persons performing similar functions, as appropriate to allow timely decisions regarding disclosure.
Attestation Report of the Registered Public Accounting Firm
Our independent registered public accounting firm will not be required to formally attest to the effectiveness of our internal controls over financial reporting for as long as we are an “emerging growth company” pursuant to the provisions of the Jumpstart Our Business Startups Act.
Management’s Report on Internal Control Over Financial Reporting
Our CEO and our CFO are responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Management conducted an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2019. In making this assessment, management used the criteria described in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). Our management concluded that our internal control over financial reporting was, and continues to be ineffective, as of December 31, 2019, due to material weaknesses in our internal controls from the lack of segregation of duties, the limitations of our financial accounting system to properly segregate duties, and the minimal internal staff with extensive knowledge of SEC financial and GAAP reporting.
A material weakness is a control deficiency (within the meaning of the Public Company Accounting Oversight Board (“PCAOB”) Auditing Standard 1305) or combination of control deficiencies that result in more than a remote likelihood that a material misstatement of the annual or interim financial statements will not be prevented or detected.
It should be noted that any system of controls, however well designed and operated, can provide only reasonable and not absolute assurance that the objectives of the system are met. In addition, the design of any control system is based in part upon certain assumptions about the likelihood of certain events. Because of these and other inherent limitations of control systems, there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions, regardless of how remote.
Due to our size and nature, segregation of all conflicting duties may not always be possible and may not be economically feasible. However, to the extent possible, the initiation of transactions, the custody of assets and the recording of transactions should be performed by separate individuals. Management evaluated the impact of our failure to maintain effective segregation of duties on our assessment of our internal control over financial reporting and has concluded that the control deficiency represents a material weakness. In April 2019, an additional experienced staff was hired in the accounting and finance department. Experienced personnel will be hired in the accounting and finance department, appropriate consultants will be retained, and our accounting system will be upgraded as soon as it becomes economically feasible and sustainable. In addition, management added additional mitigating controls with regards to cash disbursements; changes were made in our authorization processes to improve segregation of duties; and we performed additional analysis and other post-closing procedures to ensure our financial statements were prepared in accordance with generally accepted accounting principles. Accordingly, we believe that the financial statements included in this report fairly present, in all material respects, our financial condition, results of operations and cash flows for the periods presented.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the fourth quarter of the fiscal year covered by this Annual Report on Form 10-K that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information.
Our certificate of incorporation provides that the Court of Chancery of the State of Delaware shall be the sole and exclusive forum for (i) any derivative action or proceeding brought our behalf, (ii) any action asserting a claim of breach of a fiduciary duty owed by any of our directors or officers to us or our stockholders, (iii) any action asserting a claim against us arising pursuant to any provision of the Delaware General Corporation Law, or our certificate of incorporation or the bylaws, and (iv) any action asserting a claim against us governed by the internal affairs doctrine. This provision would not apply to suits brought to enforce a duty or liability created by the Exchange Act or Securities Act.
This choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and employees. Alternatively, a court could find these provisions of our certificate of incorporation to be inapplicable or unenforceable in respect of one or more of the specified types of actions or proceedings, which may require us to incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect our business and financial.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The following table sets forth the names and ages of all of our directors and executive officers as of February 13, 2020. Our officers are appointed by, and serve at the pleasure of, the Board of Directors.
| | | | | | | | | | | | | | |
| Name | | Age | | Position |
| Walter V. Klemp | | 60 | | | Executive Chairman |
| Christopher Capelli | | 60 | | | Chief Executive Officer, President and Chief Science Officer |
| Lori Bisson | | 49 | | | Chief Financial Officer |
| Joe Tanner | | 73 | | | Chief Operating Officer |
| Jonathan P. Foster | | 56 | | | Director |
| Bradley Hauser | | 42 | | | Director |
| Danika Harrison | | 44 | | | Director |
Walter V. Klemp - Founder and Executive Chairman. Mr. Klemp is a co-founder of our company and has served as our executive chairman since July 2018. He previously served as our Chief Executive Officer. From 2006 until 2016, Mr. Klemp has served as the chairman, co-founder and part-time chief executive officer of Moleculin, LLC, and since 2016 Mr. Klemp has served as chairman and chief executive officer of Moleculin Biotech, Inc., a clinical stage pharmaceutical company focused on the development of oncology drug candidates. Mr. Klemp served as president and chief executive officer of Zeno Corporation from 2004 to April 2011, where he developed and marketed dermatology devices from concept through FDA approval and market launch. From 1987 to 2000, Mr. Klemp served as chief executive officer and chairman of Drypers Corporation, a publicly traded multinational consumer products company that was listed as #1 on the INC 500 List of America’s Fastest Growing Companies. We believe that Mr. Klemp’s history with our company and background, coupled with his extensive experience in the medical field, provide him with the qualifications to serve as a director. Mr. Klemp earned a B.A. degree from Lewis & Clark. Mr. Klemp currently provides services as needed by us, which we estimate does not exceed 10 hours per week.
Christopher Capelli, M.D. - Founder, Chief Executive Officer, President and Chief Science Officer. Dr. Capelli is a co-founder of our company and has served as our chief executive officer since August 2018, president since March 2018 and chief science officer since September 2015. From September 2014 through August 2015. Dr. Capelli was a consultant to the company. Dr. Capelli is the lead inventor of Soliton’s RAP technology. From March 2005 through August 2014, Dr. Capelli served as the vice president in the office of Technology Based Ventures at The University of Texas M. D. Anderson Cancer Center. From March 2001 through February 2005, Dr. Capelli served the director of the Office of Technology Management at the University of Pittsburgh. From 1987 through 1998, Dr. Capelli served the president and was the founder of BioInterface Technologies, Inc. which developed new a silver-based antimicrobial technology for use in wound care. Dr. Capelli is a graduate of Massachusetts Institute of Technology with a Bachelor of Science degree in Mechanical Engineering. Dr. Capelli earned his MD from the University of Wisconsin Medical School and maintains a medical license in the State of Wisconsin. We believe that Dr. Capelli's history with our company as a founder and as the creator of our technology provide him with the qualifications to serve as a director.
Lori Bisson - Executive Vice President and Chief Financial Officer. Ms. Bisson has served as our chief financial officer since January 2015. Prior to joining Soliton, Ms. Bisson worked as a financial and business development consultant as a Shareholder in Condon & Company, PC, from 2009 through December 2014, where she advised a number of life science companies. From 2005 to 2009, Ms. Bisson served as the CFO and Vice-President of Operations for Zeno Corporation, a medical device company focused on new technology in the aesthetics area. Ms. Bisson previously served as the CFO of Gulfstream Trading, Ltd., an international oil trading organization from 2001 to 2005. From 1995 to 2001, Ms. Bisson held various positions with Drypers Corporation, a publicly traded multinational consumer products company, where she ultimately held the title of Vice President of Integrated Solutions and oversaw accounting, information technology, and logistics for the U.S. operation. Ms. Bisson began her career at Arthur Andersen, LLP as an auditor focused on consumer products companies. Ms. Bisson also serves as an advisor to Moleculin Biotech, Inc., a clinical stage pharmaceutical company focused on the development of oncology drug candidates. Ms. Bisson is a Certified Public Accountant and holds a BBA in Accounting from Baylor University.
Joe Tanner - Chief Operating Officer. Mr. Tanner has served as Soliton’s Chief Operating Officer since October 2014. Since 2000, Mr. Tanner has served as co-owner and part time co-manager of a chain of convenience stores in Washington State. Mr. Tanner served as Chief Operating Officer of Zeno Corporation from 2005 to 2011, a company that developed and marketed dermatology devices from concept through FDA approval and market launch. From 1993 to 2000, Mr. Tanner served as Chief Operating Officer of Drypers Corporation’s International Division, comprised of manufacturing facilities in 6 countries and sales teams in many other counties. Mr. Tanner has an undergraduate degree from Harvard University and a law degree from the University of Texas.
Jonathan P. Foster - Director. Mr. Jonathan P. Foster joined our Board of Directors effective as of June 15, 2018. Mr. Foster currently serves as the Executive Vice President and Chief Financial Officer for Moleculin Biotech, Inc., a clinical stage pharmaceutical company focused on the development of oncology drug candidates. Prior to his tenure at Moleculin, Mr. Foster served as the Executive Vice President and Chief Financial Officer of InfuSystem Holdings, Inc., a medical technology company providing pumps for hospital use, from 2012 to 2016. Prior to InfuSystem, Mr. Foster served as a consultant to the Chief Financial Officer of LSG Sky Chefs, USA, Inc., a subsidiary of Deutsche Lufthansa AG and the world's largest provider of airline catering and in-flight services. Prior to that, from 2000-2012, he was President, CFO and majority owner of United Credit, Inc. & Advance Today, Inc., a privately-owned consumer finance company with multiple locations. From 1996-2000, Mr. Foster served as Executive Vice President and Chief Financial Officer of Drypers Corporation, a publicly traded global consumer products company with more than 2,000 employees internationally and $460 million in revenue. He previously served as Chief Financial Officer of Dickson Weatherproof Nail Company, Controller & Treasurer of divisions of Schlumberger Industries, and as a Manager in the Middle Market Group of Deloitte & Touche. He has also served on the State of South Carolina Board of Financial Institutions and the Board of Directors for the Easley Baptist Hospital Foundation. Mr. Foster has a BS in Accounting from Clemson University, is a Certified Public Accountant and AICPA Chartered Global Management Accountant. We believe that Mr. Foster's experience as a chief financial officer in the biotechnology industry and his extensive accounting experience provide him with the qualifications to serve as a director.
Brad Hauser - Director. Mr. Bradley Hauser, also known as Brad, joined our Board of Directors effective as of June 15, 2018. Mr. Hauser has served as the Vice President, R&D and General Manager for CoolSculpting at Allergan Pharmaceuticals since ZELTIQ Aesthetics, Inc. was acquired by Allergan in April 2017. Previously, he served as the Senior Vice President of Research and Development at ZELTIQ Aesthetics, Inc. from January 2017 to April 2017 and as its Vice President of Research and Development from July 2015 to January 2017. Mr. Hauser joined ZELTIQ in December 2013 as Vice President of Product and Clinical Strategy. Prior to joining ZELTIQ, he held multiple roles in the aesthetic industry, including Executive Vice President of Commercial Operations for Cutera, Director of Research and Development at Medicis and Managing Director of Product and Clinical Marketing at Solta Medical. Mr. Hauser received his Bachelor of Arts in Human Biology from Stanford University. We believe that Mr. Hauser's experience in the aesthetic industry provide him with the qualifications to serve as a director.
Danika Harrison - Director. Ms. Danika R. Harrison joined our Board of Directors effective as of June 15, 2018. Ms. Harrison has been the Chief Marketing Officer of HintMD since May 2019. Prior to that, she served as President and CEO of Elira Therapeutics, Inc. from September 2017. Ms. Harrison served as Senior Vice President of Global Marketing at ZELTIQ Aesthetics, Inc. from January 2017, serving as its Vice President of Global Marketing from February 2016 and as VP of Consumer and Brand Marketing from November 2014, until the acquisition of Zeltiq by Allergan in April 2017. Ms. Harrison served as Senior Vice President of Direct Marketing & Innovation at TRIA Beauty, Inc. from December 2013 to June 2014, serving previously as Senior Vice President of Global Marketing from December 2011, and as VP/GM of North America from March 2011. From April 2006 to March 2011, Ms. Harrison worked at Rosetta, a consulting-centered interactive agency, where she was most recently a Partner leading the relationship marketing group consulting for leading brands like Dannon, Johnson’s Baby and Rogers to develop direct and digital marketing programs throughout the United States and Canada. Ms. Harrison holds a B.S. from Georgetown University and an M.B.A. from the Kellogg School of Management at Northwestern University. We believe that Ms. Harrison's experience in the aesthetic industry provide her with the qualifications to serve as a director.
Delinquent Section 16(a) Reports
Section 16(a) of the Securities Exchange Act of 1934, as amended, requires our executive officers and directors, and persons who own more than ten percent of our common stock, to file reports of ownership and changes in ownership of our common stock with the SEC. Officers, directors, and greater-than-ten-percent stockholders are required by the SEC’s regulations to furnish us with copies of all Section 16(a) forms that they file. Based solely on the Company's review of the copies of such forms prepared by it or received by it with respect to the fiscal year ended December 31, 2019, all reports were filed on a timely basis, other than the Form 3 for Mr. Tanner and for Remeditex Ventures LLC.
Audit Committee
The members of the Audit Committee are Jonathan P. Foster (Chairperson), Brad Hauser and Danika Harrison. Each member of the Audit Committee is independent as defined by The Nasdaq Stock Market ("Nasdaq") Rules. In addition, each member of the Audit Committee satisfies the additional requirements of the SEC and Nasdaq Rules for audit committee membership, including the additional independence requirements and the financial literacy requirements. The Board has determined that at least one member of the Audit Committee, Mr. Foster, is an “audit committee financial expert” as defined in the SEC’s rules and regulations. The primary purpose of the Audit Committee is to oversee the quality and integrity of our accounting and financial reporting processes and the audit of our financial statements. The Audit Committee is responsible for selecting, compensating, overseeing and terminating the selection of our independent registered public accounting firm.
Nomination of Director Candidates
The Nominating and Governance Committee will consider stockholder recommendations for candidates for the board of directors.
Qualifications for consideration as a Board nominee may vary according to the particular areas of expertise being sought as a complement to the existing board composition. However, minimum qualifications include high level leadership experience in business activities, breadth of knowledge about issues affecting the Company, experience on other boards of directors, preferably public company boards, and time available for meetings and consultation on Company matters. Our Nominating and Governance Committee does not have a formal policy with regard to the consideration of diversity in identifying director candidates but seeks a diverse group of candidates who possess the background, skills and expertise to make a significant contribution to the Board, to the Company and our stockholders. Candidates whose evaluations are favorable are recommended by our Nominating and Corporate Governance Committee to the full Board for consideration. The full Board selects and recommends candidates for nomination as directors for stockholders to consider and vote upon at the annual meeting.
A stockholder wishing to nominate a candidate for election to our Board of Directors at any annual meeting at which the Board of Directors has determined that one or more directors will be elected must submit a written notice of his or her nomination of a candidate to the Chairperson of the Nominating and Governance Committee (c/o the Corporate Secretary), providing the candidates name, biographical data and other relevant information together with a consent from the nominee. Pursuant to our Bylaws, the submission must be received at our principal executive offices 120 days prior to the anniversary date of the mailing date of our previous year’s proxy statement so as to permit the Board of Directors time to evaluate the qualifications of the nominee.
We have not employed an executive search firm, or paid a fee to any other third party, to locate qualified candidates for director positions.
Code of Business Conduct and Ethics
We have adopted a written code of business conduct and ethics that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. A copy of the code is made available on the Corporate Governance section of our website, which is located at www.soliton.com. If we make any substantive amendments to, or grant any waivers from, the code of business conduct and ethics for any officer or director, we will disclose the nature of such amendment or waiver on our website or in a current report on Form 8-K filed with the SEC.
Item 11. Executive Compensation
The following table and the related notes set forth information relating to the compensation earned by each of the named executive officers during the last two fiscal years.
Summary Compensation Table - 2019 and 2018
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Name and Principal Position | | Year | | Salary (1)
($) | | Bonus (1)($) | | Non-Equity Incentive Plans
($) | | Option
Awards (1),(2)
($) | | Total
($) |
| Walter V. Klemp, Executive Chairman | | 2019 | | $ | 212,500 | | | $ | 135,000 | | | $ | 100,000 | | | $ | 412,958 | | | $ | 860,458 | |
| | 2018 | | $ | 255,208 | | | $ | 18,750 | | | $ | — | | | $ | 880,002 | | | $ | 1,153,960 | |
| Christopher Capelli, Chief Executive Officer, President & Chief Science Officer | | 2019 | | $ | 442,187 | | | $ | 135,000 | | | $ | 150,000 | | | $ | 384,007 | | | $ | 1,111,194 | |
| | 2018 | | $ | 300,000 | | | $ | 37,500 | | | $ | — | | | $ | 880,002 | | | $ | 1,217,502 | |
| Lori Bisson, Chief Financial Officer | | 2019 | | $ | 320,000 | | | $ | 120,000 | | | $ | 100,000 | | | $ | 318,939 | | | $ | 858,939 | |
| | 2018 | | $ | 250,000 | | | $ | 45,000 | | | $ | — | | | $ | 218,483 | | | $ | 513,483 | |
| Joe Tanner, Chief Operating Officer | | 2019 | | $ | 283,008 | | | $ | 75,000 | | | $ | 90,000 | | | $ | 181,548 | | | $ | 629,556 | |
| | 2018 | | $ | 238,907 | | | $ | 28,125 | | | $ | — | | | $ | 194,207 | | | $ | 461,239 | |
(1)In 2017, Mr. Klemp, Dr. Capelli, and Mr. Tanner agreed to defer $31,250, $31,250, and $23,440, respectively, of their compensation, and in 2018, Mr. Klemp, Dr. Capelli, Ms. Bisson and Mr. Tanner agreed to defer $37,500, $75,000, $90,000 and $56,250, respectively, of their compensation until we had additional funding. Additionally, we agreed to pay a 50% premium on the amount deferred in both years. Of this deferred compensation and 50% premium, $103,125, $131,250, $101,250 and $98,440 were extinguished in January 2019 by Mr. Klemp, Dr. Capelli, Ms. Bisson and Mr. Tanner, respectively, in exchange for the issuance of options during 2019. The amounts represented by the 2018 deferred compensation are included in the Salary column for 2018 for each named executive officer, and the amounts represented by the 50% premium on the 2018 deferred compensation are included in the Bonus column for 2018 for each named executive officer. In addition, the fair market value of the options issued to Mr. Klemp, Dr. Capelli and Mr. Tanner to extinguish the above amounts was in excess of the actual deferred salary and 50% premium payable to Mr. Klemp, Dr. Capelli, and Mr. Tanner. The excess amounts of the options issued to Mr. Klemp, Dr. Capelli and Mr. Tanner were $39,863, $10,911 and $7,472, respectively, and are included in the Option Awards column for 2019. The fair market value of the options issued to Ms. Bisson was less than actual deferred salary and 50% premium for 2018 and, as such, the total amount of the options issued was excluded from the Option Award column for 2019.
(2)Represents the full grant date fair value of the stock option grant calculated in accordance with FASB ASC Topic 718. The measurement objective of FASB ASC Topic 718 is to estimate the fair value at the grant date of the equity instruments that the entity is obligated to issue when employees have rendered the requisite service and satisfied any other conditions necessary to earn the right to benefit from the instruments (for example, to exercise share options). That estimate is based on the share price and other pertinent factors, such as in the case of stock options the expected volatility at the grant date. The grant date fair value of an award reflects the accounting expense and may not represent the actual value that will be realized. For a summary of the assumptions made in the valuation of these awards, please see Note 7 to our financial statements included elsewhere in this Annual Report on Form 10-K.
Narrative to Summary Compensation Table
We review compensation annually for all employees, including our executives. In setting executive base salaries and bonuses and granting equity incentive awards, we consider compensation for comparable positions in the market, the individual executive’s performance as compared to our expectations and objectives, our desire to motivate our employees to achieve short and long-term results that are in the best interests of our stockholders and a long-term commitment to our company. We do not target a specific competitive position or a specific mix of compensation among base salary, bonus or long-term incentives. Our Compensation Committee typically reviews and discusses management’s proposed compensation with the Chief Executive
Officer for all executives other than the Chief Executive Officer. Based on those discussions and its discretion, the Compensation Committee then determines the compensation for each executive officer. Our Compensation Committee, without members of management present, discusses and ultimately approves the compensation of our executive officers. In 2019, the Compensation Committee retained Pay Governance, a compensation consulting firm, to evaluate our executive compensation program. Pay Governance’s engagement included assisting the Compensation Committee with the selection of a peer group of companies for benchmarking purposes, and an analysis of our existing executive compensation. The consultant serves at the pleasure of the Compensation Committee rather than us, and the consultant’s fees are approved by the Compensation Committee.
Annual Base Salary
For 2019, the base salaries for Mr. Klemp, Dr. Capelli, Ms. Bisson and Mr. Tanner were $200,000, $425,000, $300,000 and $250,000, respectively. Effective January 1, 2020, the base salaries for Mr. Klemp, Dr. Capelli, Ms. Bisson and Mr. Tanner were adjusted to $225,000, $450,000, $325,000 and $265,000, respectively.
Annual Bonus and Non-Equity Incentive Plan Compensation
We seek to motivate and reward our executives for achievements relative to our corporate goals and objectives, and with respect to their respective individual goals, for each fiscal year. For 2019, the target bonus for Mr. Klemp, Dr. Capelli, Ms. Bisson and Mr. Tanner was approximately 50%, 35%, 38% and 36%, respectively, of their base salary. For 2020, the target bonus for Mr. Klemp, Dr. Capelli, Ms. Bisson and Mr. Tanner will be approximately 56%, 39%, 38% and 38%, respectively, of their base salary.
The actual performance-based annual bonus paid is calculated by multiplying the executive’s annual base salary, target bonus percentage, the percentage attainment of the corporate goals established by the Board for such year, which represents 75% of the potential bonus payable, and the percentage attainment of the individual goals approved by our Compensation Committee, which represents 25% of the potential bonus payable. However, the Compensation Committee is not required to calculate bonuses in this manner and retains discretion in the amounts it awards and the factors it takes into consideration in determining bonus amounts. At the end of the year, the Compensation Committee reviews our performance against our goals and objectives and approves the extent to which we achieved each of our corporate and individual goals and objectives, and, for each named executive officer, the amount of the bonus awarded.
For 2019, bonuses were awarded based on our achievement of specified corporate goals, including our clinical trial development and progress, our FDA clearance progress, the improvement of our internal controls and our ability to maintain sufficient funding to maintain our planned clinical activities, and individual goals, as applicable. Based on the level of achievement, our Compensation Committee awarded each of our named executive officers the full amount of their potential bonuses for 2019. These actual bonus amounts are reflected in the “Non-Equity Incentive Plan Compensation” column of the Summary Compensation Table above.
For 2020, bonuses will be awarded based on our achievement of specified corporate goals, including our clinical trial development and progress, our FDA clearance progress related to new indications, the success of our commercial launch, the improvement of our internal controls and our ability to maintain sufficient funding to maintain our planned clinical and commercial activities, and individual goals, as applicable.
We completed our IPO in February 2019. Upon the completion of our IPO, our Compensation Committee awarded discretionary bonuses to our named executive officers. For 2018, bonuses were accrued for the 50% premium, which was earned on the amount of salaries deferred by the named officers. These actual bonus amounts are reflected in the “Bonus” column of the Summary Compensation Table above.
Long-Term Incentives
Our 2018 Stock Plan (the “2018 Plan”) provides for the grant of stock options, stock awards, stock unit awards and stock appreciation rights to key employees, non-employee directors and consultants.
Each year our Compensation Committee establishes a value for the option grant payable to each of our named executive officers. For 2019, the fair value of the option grants for Mr. Klemp, Dr. Capelli, Ms. Bisson and Mr. Tanner were $750,000, $750,000, $450,000 and $350,000, respectively. These options were issued in February 2020 and are not reflected in the Summary Compensation Table. For 2020, the fair value of the option grants for Mr. Klemp, Dr. Capelli, Ms. Bisson and Mr. Tanner will be targeted at $800,000, $800,000, $475,000 and $385,000, respectively. We set the option exercise price, and
grant date fair value based on the closing price of our common stock on Nasdaq on the date of grant. The shares underlying options typically vest in four equal annual installments.
Employment Agreements
As discussed in the Form 1-A we filed in connection with our IPO, we stated that we intended to enter into employment agreements with our named executive officers upon the closing of our IPO on the following terms: (i) Mr. Klemp - base salary: $200,000; cash bonus target for 2019: 50%; option grant value target for 2019: $750,000; (ii) Dr. Capelli - base salary: $425,000; cash bonus target for 2019: 35%; option grant value target for 2019: $750,000; (iii) Ms. Bisson - base salary: $265,000; cash bonus target for 2019: 38%; option grant value target for 2019: $450,000; and (iv) Mr. Tanner - base salary: $250,000; cash bonus target for 2019: 36%; option grant value target for 2019: $350,000.
On February 25, 2019, we entered into employment agreements with each of the foregoing named executive officers. Each employment agreement provides for an initial term of one year, which will be automatically renewed for additional one-year terms unless either party chooses not to renew the employment agreement. The employment agreements provide for an initial base salary, cash bonus target, and option grant value target in accordance with the above disclosure set forth in our Form 1-A. Notwithstanding the targeted bonus and option amounts, the final determination on the amount of the bonus or option grant, if any, will be made by the Compensation Committee of the Board of Directors, based on criteria established by the Compensation Committee.
Pursuant to the employment agreements, if the named executive officer is terminated at our election without “cause” (as defined in the employment agreement), or by the named executive officer for “good reason” (as defined in the employment agreement), the named executive officer shall be entitled to receive severance payments equal to twelve months base salary, with respect to Mr. Klemp and Dr. Capelli, and nine months base salary, with respect to Ms. Bisson and Mr. Tanner, and, in each case, a pro rata portion of the target bonus, if any, for the year in which such termination occurs. Pursuant to the employment agreements, the named executive officers agreed not to compete with us until twelve months after the termination of their employment, with respect to Mr. Klemp and Dr. Capelli, and nine months after termination of their employment, with respect to Ms. Bisson and Mr. Tanner.
Equity Awards
The following table sets forth certain information concerning our outstanding equity awards for our named executive officers at December 31, 2019.
Outstanding Equity Awards at Fiscal Year-End-2019
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Name | | Option Awards | | | | | | |
| | Number of
Securities
Underlying
Unexercised
Options (#) exercisable | | Number of Securities Underlying Unexercised Options (#) Unexercisable | | Option Exercise Price ($) | | Option
Expiration
Date |
| Walter V. Klemp (1) | | 181,250 | | | 543,750 | | | $1.75 | | | 6/7/2028 |
| | 63,375 | | | 21,125 | | | $1.75 | | | 2/4/2029 |
| | — | | | 35,150 | | | $14.62 | | | 6/27/2029 |
| Christopher Capelli (2) | | 181,250 | | | 543,750 | | | $1.75 | | | 6/7/2028 |
| | 81,375 | | | 27,125 | | | $1.75 | | | 2/4/2029 |
| | | | 35,150 | | | $14.62 | | | 6/27/2029 |
| Lori Bisson (3) | | 45,000 | | | 135,000 | | | $1.75 | | | 6/7/2028 |
| | 62,625 | | | 20,875 | | | $1.75 | | | 2/4/2029 |
| | | | 21,100 | | | $14.62 | | | 6/27/2029 |
| Joe Tanner (4) | | 40,000 | | | 120,000 | | | $1.75 | | | 6/7/2028 |
| | 60,563 | | | 20,188 | | | $1.75 | | | 2/4/2029 |
| | | | 16,400 | | | $14.62 | | | 6/27/2029 |
(1)At December 31, 2019, Mr. Klemp owned the following options: (i) unvested stock options to purchase 543,750 shares of Common Stock, which vest in three remaining annual installments of 181,250 on each annual anniversary date of June 7, 2020, June 7, 2021 and June 7, 2022; (ii) unvested stock options to purchase 21,125 shares of Common Stock, which vest in one remaining quarterly installment on February 3, 2020; and (iii) unvested stock options to purchase 35,150 shares of Common Stock, which vest in four annual installments over a four year period, with 1/4 vesting on June 27, 2020, and thereafter 1/4 vesting on each annual anniversary date of the issuance.
(2)At December 31, 2019, Mr. Capelli owned the following options: (i) unvested stock options to purchase 543,750 shares of Common Stock, which vest in three remaining annual installments of 181,250 on each annual anniversary date of June 7, 2020, June 7, 2021 and June 7, 2022; (ii) unvested stock options to purchase 27,125 shares of Common Stock, which vest in one remaining quarterly installment on February 3, 2020; and (iii) unvested stock options to purchase 35,150 shares of Common Stock, which vest in four annual installments over a four year period, with 1/4 vesting on June 27, 2020, and thereafter 1/4 vesting on each annual anniversary date of the issuance.
(3)At December 31, 2019, Ms. Bisson owned the following options: (i) unvested stock options to purchase 135,000 shares of Common Stock, which vest in three remaining annual installments of 45,000 on each annual anniversary date of June 7, 2020, June 7, 2021 and June 7, 2022; (ii) unvested stock options to purchase 20,875 shares of Common Stock, which vest in one remaining quarterly installment on February 3, 2020; and (iii) unvested stock options to purchase 21,100 shares of Common Stock, which vest in four annual installments over a four year period, with 1/4 vesting on June 27, 2020, and thereafter 1/4 vesting on each annual anniversary date of the issuance.
(4)At December 31, 2019, Mr. Tanner owned the following options: (i) unvested stock options to purchase 120,000 shares of Common Stock, which vest in three remaining annual installments of 40,000 on each annual anniversary date of June 7, 2020, June 7, 2021 and June 7, 2022; (ii) unvested stock options to purchase 20,188 shares of Common Stock, which vest in one remaining quarterly installment on February 3, 2020; and (iii) unvested stock options to purchase 16,400 shares of Common Stock, which vest in four annual installments over a four year period, with 1/4 vesting on June 27, 2020, and thereafter 1/4 vesting on each annual anniversary date of the issuance.
Director Compensation
The following table sets forth the total compensation earned by our non-employee directors in 2019 (Mr. Klemp and Dr. Capelli, our non-independent board members, do not receive any additional compensation for serving as directors):
Director Compensation – 2019
| | | | | | | | | | | | | | | | | | | | |
| Name | | Fees earned or paid in cash ($) | | Option Awards ($) (1) | | Total ($) |
| Jonathan P. Foster | | $ | 78,819 | | | $ | 152,359 | | | $ | 231,178 | |
| Danika Harrison | | $ | 70,208 | | | $ | 152,359 | | | $ | 222,567 | |
| Brad Hauser | | $ | 74,514 | | | $ | 152,359 | | | $ | 226,873 | |
(1) Represents the full grant date fair value of the stock option grant calculated in accordance with FASB ASC Topic 718. The measurement objective of FASB ASC Topic 718 is to estimate the fair value at the grant date of the equity instruments that the entity is obligated to issue when employees have rendered the requisite service and satisfied any other conditions necessary to earn the right to benefit from the instruments (for example, to exercise share options). That estimate is based on the share price and other pertinent factors, such as in the case of stock options the expected volatility at the grant date. The grant date fair value of an award reflects the accounting expense and may not represent the actual value that will be realized. For a summary of the assumptions made in the valuation of these awards, please see Note 7 to our financial statements included elsewhere in this Annual Report on Form 10-K.
Upon the completion of our IPO, our Board established the following compensation policy for non-employee directors, effective February 26, 2019:
l Each independent director shall receive annual cash compensation of $35,000. In addition, the chair-person of the Audit Committee, Compensation Committee and Nominating and Governance Committee shall receive an annual compensation of $15,000, $10,000 and $7,500, respectively; the other members of such committees shall receive an annual compensation of $7,500, $5,000 and $5,000, respectively. In addition, since the independent directors had not received any compensation during 2018, the Board approved a one-time cash payment to each independent director of $25,000.
l Upon the initial appointment (or election) of independent directors to the Board, the director will be issued a 10-year option to purchase 30,000 shares of our common stock, under our incentive stock plan, with four-year annual vesting and an exercise price equal the closing price of our common stock on the date of the appointment (or election). Since our current independent directors received such a grant upon joining, no additional option grants were made to such directors.
l Annually, on the date of our annual meeting, each independent director that is re-elected at the annual meeting will be issued, upon a motion and approval of the Board of Directors, a 10-year option to purchase 15,000 shares of our common stock, under our incentive stock plan, with a one-year vesting period and an exercise price equal the closing price of our common stock on the date of the annual meeting.
Effective January 1, 2020, the compensation policy for non-employee directors was modified to the following:
l Each independent director shall receive annual cash compensation of $38,000. In addition, the chair-person of the Audit Committee, Compensation Committee and Nominating and Governance Committee shall receive an annual compensation of $20,000, $13,800 and $9,500, respectively; the other members of such committees shall receive an annual compensation of $9,000, $6,000 and $5,000, respectively.
l Upon the initial appointment (or election) of independent directors to the Board, the director will be issued a 10-year option to purchase 30,000 shares of our common stock, under our incentive stock plan, with four-year annual vesting and an exercise price equal the closing price of our common stock on the date of the appointment (or election). Since our current independent directors received such a grant upon joining, no additional option grants were made to such directors.
l Commencing in 2021, annually, on the date of our annual meeting, each independent director that is re-elected at the annual meeting will be issued, upon a motion and approval of the Board of Directors, a 10-year option to purchase 20,000 shares of our common stock, under our incentive stock plan, with a one-year vesting period and an exercise price equal the closing price of our common stock on the date of the annual meeting.
Scientific Advisory Board
Our executive team is supported by our scientific advisory board, the members of which include dermatologists experienced in the fields in which we pursue. The members of our Scientific Advisory Board are compensated based on our utilization of their time. The chairman of our Scientific Advisory Board is on retainer. The retainer provides that the chairman of our Scientific Advisory Board will receive compensation of $12,500 per fiscal quarter and a one-time issuance of an option to purchase 15,000 shares. Dr. Kaminer is affiliated with Skin Care Physicians, which was the site for our HCT-2 clinical trial and served as the investigator for such trial. Skin Care Physicians was also a site for our pivotal cellulite study, for which Dr. Kaminer also served as the investigator.
Michael S. Kaminer, M.D., chair of Soliton’s Scientific Advisory Board, is known as a leader, innovator and talented skin cancer and cosmetic surgeon in the Boston area. Dr. Kaminer is one of the pre-eminent educators in cosmetic surgery in the nation, having lectured at many national meetings, including national meetings of the American Academy of Dermatology and the American Society for Dermatologic Surgery, the Hawaii Dermatology Conference, and the American Society for Laser Medicine and Surgery. He has also lectured at numerous international meetings and symposia, recently serving as Co-Chairman of the Anti-Aging World Congress in Paris, France.
E. Victor Ross, M.D., is a dermatologist specializing in laser surgery of the skin. Presently, he is the director of the Scripps Clinic Laser and Cosmetic Dermatology Center and a frequent lecturer at national and international meetings on cutaneous laser medicine. He also serves on the editorial board of two major dermatologic journals.
Roy G. Geronemus, M.D., Director of the Laser & Skin Surgery Center of New York®, graduated from Harvard University and pursued his medical education at the University of Miami School of Medicine. He is a Clinical Professor of Dermatology at New York University Medical Center where he founded its laser program and served nine years as chief of dermatologic and laser surgery. He is past president of the American Society for Dermatologic Surgery and the American Society for Laser Medicine & Surgery.
Mathew M. Avram, M.D., J.D. is the director of the MGH Dermatology Laser & Cosmetic Center. He is the Faculty Director for Procedural Training in the Department of Dermatology, Harvard Medical School. Dr. Avram attended college at Princeton and completed his residency training at Harvard, where he served as chief resident.
Dr. Elizabeth Tanzi is a board-certified dermatologist proudly serving men and women in the Washington D.C. area. After 15 years of practicing cosmetic dermatology in Washington D.C., she founded Capital Laser & Skin Care. Capital Laser & Skin Care was one of four sites in our pivotal cellulite study for which Dr. Tanzi served as Principal Investigator.
Dr. Jeffrey Dover graduated as the silver medalist, Magna cum Laude with an M.D. degree from the University of Ottawa. He now co-directs SkinCare Physicians of Chestnut Hill, a comprehensive facility specializing in dermatology, laser and cosmetic surgery, and he is Associate Professor of Clinical Dermatology at Yale University School of Medicine, and Associate Professor of Dermatology at Brown Medical School. Dr. Dover is Past President of both the American Society of Dermatologic Surgery and the American Society for Lasers in Medicine and Surgery.
Dr. Eric F. Bernstein is Director of Laser Surgery and Cosmetic Dermatology Centers and one of the world’s leading experts on laser medicine and surgery. As a result of Dr. Bernstein’s research and development work, he often is among the very first in the world to utilize new laser applications for patient treatment.
Dr. Christopher Zachary, Professor and Chair of the Department of Dermatology at the University of California, Irvine, heads up one of the world’s premier laser and skin surgery facilities. He has been the program director for the Mohs College and the American Society for Laser Surgery and Medicine annual meetings. He is a Past President of the Association of Academic Dermatologic Surgeons.
2012 Stock Plan
In March 2012, the Company’s board of directors and stockholders adopted the 2012 Long Term Incentive Plan (the “2012 Stock Plan”). The 2012 Stock Plan was designed to enable the Company to offer employees, officers, directors and consultants, as defined, an opportunity to acquire a proprietary interest in the Company. The types of awards that may be granted under the 2012 Stock Plan include stock options, stock appreciation rights, restricted stock, and other stock-based awards subject to limitations under applicable law. All awards are subject to approval by the Company’s board of directors. The 2012 Stock Plan reserves shares of common stock for issuance in accordance with the 2012 Stock Plan’s terms. We are no longer using the 2012 Stock Plan and are utilizing our 2018 Stock Plan discussed below. The following is a summary of the materials terms of the 2012 Stock Plan.
Administration. The 2012 Stock Plan is administered by our board of directors, and, once constituted, will be administered by the Compensation Committee of the board of directors (we refer to body administering the 2012 Stock Plan as the “Committee”). The Committee will have full authority to select the individuals who will receive awards under the 2012 Stock Plan, determine the form and amount of each of the awards to be granted and establish the terms and conditions of awards.
Number of shares of common stock. The number of shares of the common stock that may be issued under the 2012 Stock Plan is 789,745. As of December 31, 2019, we had issued an option to purchase 15,000 shares of common stock under the 2012 Stock Plan and had granted 760,000 shares of restricted stock under the 2012 Stock Plan. Shares issuable under the 2012 Stock Plan may be authorized but unissued shares or treasury shares. If there is a lapse, forfeiture, expiration, termination or cancellation of any award made under the 2012 Stock Plan for any reason, the shares subject to the award will again be available for issuance. Any shares subject to an award that are delivered to us by a participant, or withheld by us on behalf of a participant, as payment for an award or payment of withholding taxes due in connection with an award will not again be available for issuance, and all such shares will count toward the number of shares issued under the 2012 Stock Plan. The number of shares of common stock issuable under the 2012 Stock Plan is subject to adjustment, in the event of any reorganization, recapitalization, stock split, stock distribution, merger, consolidation, split-up, spin-off, combination, subdivision, consolidation or exchange of shares, any change in the capital structure of the company or any similar corporate transaction. In each case, the Committee has the discretion to make adjustments it deems necessary to preserve the intended benefits under the 2012 Stock Plan. No award granted under the 2012 Stock Plan may be transferred, except by will, the laws of descent and distribution.
Eligibility. All officers and employees, and other persons who provide services to us, including directors are eligible to receive awards under the 2012 Stock Plan. On December 31, 2018, eight employees and all non-employee directors were eligible to participate in the 2012 Stock Plan.
Awards to participants. The 2012 Stock Plan provides for discretionary awards of stock options, stock awards and stock unit awards to participants. Each award made under the 2012 Stock Plan will be evidenced by a written award agreement specifying the terms and conditions of the award as determined by the Committee in its sole discretion, consistent with the terms of the 2012 Stock Plan.
Stock options. The Committee has the discretion to grant non-qualified stock options or incentive stock options to participants and to set the terms and conditions applicable to the options, including the type of option, the number of shares subject to the option and the vesting schedule; provided that the exercise price of each stock option will be the fair market value of the common stock on the date on which the option is granted, each option will expire not later than 10 years from the date of grant.
Stock awards. The Committee has the discretion to grant stock awards to participants. Stock awards will consist of shares of common stock granted without any consideration from the participant or shares sold to the participant for appropriate consideration as determined by the Board. The number of shares awarded to each participant, and the restrictions, terms and conditions of the award, will be at the discretion of the Committee. Subject to the restrictions, a participant will be a shareholder with respect to the shares awarded to him or her and will have the rights of a shareholder with respect to the shares, including the right to vote the shares and receive dividends on the shares.
Payment for stock options and withholding taxes. The Committee may make one or more of the following methods available for payment of any award, including the exercise price of a stock option, and for payment of the minimum required tax obligation associated with an award: (i) cash; (ii) cash received from a broker-dealer to whom the holder has submitted an exercise notice together with irrevocable instructions to deliver promptly to us the amount of sales proceeds from the sale of the shares subject to the award to pay the exercise price or withholding tax; (iii) by directing us to withhold shares of common stock otherwise issuable in connection with the award having a fair market value equal to the amount required to be withheld; and (iv) by delivery of previously acquired shares of common stock that are acceptable to the Committee and that have an aggregate fair market value on the date of exercise equal to the exercise price or withholding tax, or certification of ownership by attestation of such previously acquired shares.
Provisions relating to a “change in control” of the Company. Notwithstanding any other provision of the 2012 Stock Plan or any award agreement, in the event of a “Change in Control” of the Company, the Committee has the discretion to provide that all outstanding awards will become fully exercisable, all restrictions applicable to all awards will terminate or lapse, and performance goals applicable to any stock awards will be deemed satisfied at the highest target level. In addition, upon such Change in Control, the Committee has sole discretion to provide for the purchase of any outstanding stock option for cash equal to the difference between the exercise price and the then fair market value of the common stock subject to the option had the option been currently exercisable, make such adjustment to any award then outstanding as the Committee deems appropriate to reflect such Change in Control and cause any such award then outstanding to be assumed by the acquiring or surviving corporation after such Change in Control.
Amendment of award agreements; Amendment and termination of the plan; Term of the plan. The Committee may amend any award agreement at any time, provided that no amendment may adversely affect the right of any participant under any agreement in any material way without the written consent of the participant, unless such amendment is required by applicable law, regulation or stock exchange rule. The Board may terminate, suspend or amend the 2012 Stock Plan, in whole or in part, from time to time, without the approval of the shareholders, unless such approval is required by applicable law, regulation or stock exchange rule, and provided that no amendment may adversely affect the right of any participant under any outstanding award in any material way without the written consent of the participant, unless such amendment is required by applicable law, regulation or rule of any stock exchange on which the shares are listed.
No awards may be granted under the 2012 Stock Plan on or after the tenth anniversary of the effective date of the Plan.
2018 Stock Plan
In June 2018, the Company’s board of directors adopted the Soliton, Inc. 2018 Stock Plan (the “2018 Plan”) for issuances to the Company’s employees, officers, directors and consultants, subject to shareholder approval of the plan. The 2018 Plan is a stock-based compensation plan that provides for discretionary grants of stock options, stock awards, stock unit awards and stock appreciation rights to key employees, non-employee directors and consultants. The material features of the 2018 Plan, as amended, are outlined below. The following description of the 2018 Plan is a summary only and is qualified in its entirety by reference to the complete text of the 2018 Plan.
Administration. The 2018 Plan will be administered by our board of directors or, once established, the compensation committee of the board of directors (we refer to the body administering the 2018 Plan as the “Committee”). The Committee has full authority to select the individuals who will receive awards under the 2018 Plan, determine the form and amount of each of the awards to be granted and establish the terms and conditions of awards.
Limit on Non-Employee Director Compensation. Under the 2018 Plan, the following limits will apply to non-employee directors. The aggregate value of all compensation granted or paid, as applicable, to any individual for service as a non-employee director with respect to any calendar year, including awards granted under the 2018 Plan and cash fees paid to such non-employee director, will not exceed $300,000 in total value. For purposes of these limitations, the value of awards is calculated based on the grant date fair value of such awards for financial reporting purposes.
Number of Shares of Common Stock. The number of shares of the common stock that may be issued under the 2018 Plan is 3,400,000. As of December 31, 2019, we had issued options to purchase 2,868,550 shares of common stock under the 2018 Plan. Shares issuable under the 2018 Plan may be authorized but unissued shares or treasury shares. If there is a lapse, forfeiture, expiration, termination or cancellation of any award made under the 2018 Plan for any reason, the shares subject to the award will again be available for issuance. Any shares subject to an award that are delivered to us by a participant, or withheld by us on behalf of a participant, as payment for an award or payment of withholding taxes due in connection with an award will not again be available for issuance, and all such shares will count toward the number of shares issued under the 2018 Plan. The number of shares of common stock issuable under the 2018 Plan is subject to adjustment, in the event of any reorganization, recapitalization, stock split, stock distribution, merger, consolidation, split-up, spin-off, combination, subdivision, consolidation or exchange of shares, any change in the capital structure of the company or any similar corporate transaction. In each case, the Committee has the discretion to make adjustments it deems necessary to preserve the intended benefits under the 2018 Plan. No award granted under the 2018 Plan may be transferred, except by will, the laws of descent and distribution.
Eligibility. All employees designated as key employees for purposes of the 2018 Plan, all non-employee directors and consultants are eligible to receive awards under the 2018 Plan. As of December 31, 2019, ten employees and all non-employee directors were eligible to participate in the 2018 Plan.
Awards to Participants. The 2018 Plan provides for discretionary awards of stock options, stock awards, stock unit awards and stock appreciation rights to participants. Each award made under the 2018 Plan will be evidenced by a written award agreement specifying the terms and conditions of the award as determined by the Committee in its sole discretion, consistent with the terms of the 2018 Plan.
Stock Options. The Committee has the discretion to grant non-qualified stock options or incentive stock options to participants and to set the terms and conditions applicable to the options, including the type of option, the number of shares subject to the option and the vesting schedule; provided that the exercise price of each stock option will be the closing price of the common stock on the date on which the option is granted (“fair market value”), each option will expire 10 years from the date of grant and no dividend equivalents may be paid with respect to stock options.
In addition, an incentive stock option granted to a key employee is subject to the following rules: (i) the aggregate fair market value (determined at the time the option is granted) of the shares of common stock with respect to which incentive stock options are exercisable for the first time by a key employee during any calendar year (under all incentive stock option plans of the company and its subsidiaries) cannot exceed $100,000, and if this limitation is exceeded, that portion of the incentive stock option that does not exceed the applicable dollar limit will be an incentive stock option and the remainder will be a non-qualified stock option; (ii) if an incentive stock option is granted to a key employee who owns stock possessing more than 10% of the total combined voting power of all class of stock of the Company, the exercise price of the incentive stock option will be 110% of the closing price of the common stock on the date of grant and the incentive stock option will expire no later than five years from the date of grant; and (iii) no incentive stock option can be granted after 10 years from the date the 2018 Plan was adopted.
Stock Appreciation Rights. The Committee has the discretion to grant stock appreciation rights to participants. The Committee determines the exercise price for a stock appreciation right, which cannot be less than 100% of the fair market value of our common stock on the date of grant. Upon the exercise of a stock appreciation right, we will pay the participant in common stock or in cash, at our discretion, an amount equal to the product of (1) the excess of the per share fair market value of our common stock on the date of exercise over the exercise price, multiplied by (2) the number of shares of common stock with respect to which the stock appreciation right is exercised. The Committee has the discretion to set the terms and conditions applicable to the award, including the number of shares subject to the stock appreciation right and the vesting schedule, provided that each stock appreciation right will expire not more than 10 years from the date of grant and no dividends or dividend equivalents shall be paid with respect to any stock appreciation right prior to the exercise of the stock appreciation right.
Stock Awards. The Committee has the discretion to grant stock awards to participants. Stock awards will consist of shares of common stock granted without any consideration from the participant or shares sold to the participant for appropriate consideration as determined by the Board. The number of shares awarded to each participant, and the restrictions, terms and
conditions of the award, will be at the discretion of the Committee. Subject to the restrictions, a participant will be a shareholder with respect to the shares awarded to him or her and will have the rights of a shareholder with respect to the shares, including the right to vote the shares and receive dividends on the shares; provided that dividends otherwise payable on any stock award subject to restrictions will be held by us and will be paid to the holder of the stock award only to the extent the restrictions on such stock award lapse.
Stock Units. The Committee has the discretion to grant stock unit awards to participants. Each stock unit entitles the participant to receive, on a specified date or event set forth in the award agreement, one share of common stock or cash equal to the fair market value of one share on such date or event, as provided in the award agreement. The number of stock units awarded to each participant, and the terms and conditions of the award, will be at the discretion of the Committee. Unless otherwise specified in the award agreement, a participant will not be a shareholder with respect to the stock units awarded to him prior to the date they are settled in shares of common stock. The award agreement may provide that until the restrictions on the stock units lapse, the participant will be paid an amount equal to the dividends that would have been paid had the stock units been actual shares; provided that such dividend equivalents will be held by us and paid only to the extent the restrictions lapse.
Payment for Stock Options and Withholding Taxes. The Committee may make one or more of the following methods available for payment of any award, including the exercise price of a stock option, and for payment of the tax obligation associated with an award: (i) cash; (ii) cash received from a broker dealer to whom the holder has submitted an exercise notice together with irrevocable instructions to deliver promptly to us the amount of sales proceeds from the sale of the shares subject to the award to pay the exercise price or withholding tax; (iii) by directing us to withhold shares of common stock otherwise issuable in connection with the award having a fair market value equal to the amount required to be withheld; and (iv) by delivery of previously acquired shares of common stock that are acceptable to the Committee and that have an aggregate fair market value on the date of exercise equal to the exercise price or withholding tax, or certification of ownership by attestation of such previously acquired shares.
Provisions Relating to a “Change in Control” of the Company. Notwithstanding any other provision of the 2018 Plan or any award agreement, in the event of a “Change in Control” of the Company, the Committee has the discretion to provide that all outstanding awards will become fully exercisable, all restrictions applicable to all awards will terminate or lapse, and performance goals applicable to any stock awards will be deemed satisfied at the target level. In addition, upon such Change in Control, the Committee has sole discretion to provide for the purchase of any outstanding stock option for cash equal to the difference between the exercise price and the then fair market value of the common stock subject to the option had the option been currently exercisable, make such adjustment to any award then outstanding as the Committee deems appropriate to reflect such Change in Control and cause any such award then outstanding to be assumed by the acquiring or surviving corporation after such Change in Control.
Amendment of Award Agreements; Amendment and Termination of the 2018 Plan; Term of the 2018 Plan. The Committee may amend any award agreement at any time, provided that no amendment may adversely affect the right of any participant under any agreement in any material way without the written consent of the participant, unless such amendment is required by applicable law, regulation or stock exchange rule.
The Board may terminate, suspend or amend the 2018 Plan, in whole or in part, from time to time, without the approval of the stockholders, unless such approval is required by applicable law, regulation or stock exchange rule, and provided that no amendment may adversely affect the right of any participant under any outstanding award in any material way without the written consent of the participant, unless such amendment is required by applicable law, regulation or rule of any stock exchange on which the shares are listed.
Notwithstanding the foregoing, neither the 2018 Plan nor any outstanding award agreement can be amended in a way that results in the repricing of a stock option. Repricing is broadly defined to include reducing the exercise price of a stock option or stock appreciation right or cancelling a stock option or stock appreciation right in exchange for cash, other stock options or stock appreciation rights with a lower exercise price or other stock awards. (This prohibition on repricing without stockholder approval does not apply in case of an equitable adjustment to the awards to reflect changes in the capital structure of the company or similar events.)
No awards may be granted under the 2018 Plan on or after the tenth anniversary of the initial effective date of the 2018 Plan.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The following table sets forth information, as of February 13, 2020, regarding beneficial ownership of our common stock by:
•each of our directors and director nominees;
•each of our executive officers;
•all directors and executive officers as a group; and
•each person, or group of affiliated persons, known by us to beneficially own more than five percent of our shares of common stock.
Beneficial ownership is determined according to the rules of the SEC, and generally means that person has beneficial ownership of a security if he or she possesses sole or shared voting or investment power of that security and includes options that are currently exercisable or exercisable within 60 days. Each director or officer, as the case may be, has furnished us with information with respect to beneficial ownership. Except as otherwise indicated, we believe that the beneficial owners of common stock listed below, based on the information each of them has given to us, have sole investment and voting power with respect to their shares, except where community property laws may apply. Except as otherwise noted below, the address for each person or entity listed in the table is c/o Soliton, Inc., 5304 Ashbrook Drive, Houston, Texas, 77081.
| | | | | | | | |
| Shares beneficially owned | Percentage owned (1) |
| Name and Address of Beneficial Owner | | |
| Directors and Executive Officers | | |
| Walter V. Klemp | 718,667 | | 4.2 | % |
| Christopher Capelli, M.D. (2) | 756,107 | | 4.5 | % |
| Lori Bisson | 203,500 | | 1.2 | % |
| Joe Tanner | 200,750 | | 1.2 | % |
| Jonathan P. Foster | 20,000 | | * |
| Danika Harrison | 7,500 | | * |
| Brad Hauser | 7,500 | | * |
| Directors and Executive Officers as a Group (7 persons) | 1,914,024 | | 11.3 | % |
| 5% or greater shareholders | | |
| Remeditex Ventures (3) | 9,514,604 | | 56.2 | % |
* Less than 1%.
(1) Based on 16,932,184 shares of common stock outstanding as of February 13, 2020. Shares of Common Stock subject to options and warrants held by any person that are currently exercisable or are exercisable within 60 days of February 13, 2020 are deemed outstanding for purposes of computing the percentage ownership of such person but are not deemed outstanding for purposes of computing the percentage ownership of any other person.
(2) Includes 175,000 currently held by MD Anderson that were issued pursuant to our license agreement with MD Anderson. As the inventor of the intellectual property we license from MD Anderson, Dr. Capelli is entitled to 50% of the proceeds (after the recoupment of any costs associated therewith) from the sale by MD Anderson of the shares issued to the MD Anderson in connection with the license agreement as licensing consideration. Notwithstanding Dr. Capelli pecuniary interest in the shares held by MD Anderson, Dr. Capelli has no right to vote or sell the shares held by MD Anderson.
(3) Remeditex Ventures LLC is the record and beneficial owner of the securities set forth in the table, and shares voting and dispositive power over such securities with Malachite Trust, the majority owner of Remeditex Ventures LLC and Lyda Hill. Ms. Hill is the Trustee of the Malachite Trust. By reason of such relationships, Ms. Hill, the Malachite Trust and Remeditex Ventures LLC may be deemed to share voting and dispositive power over the securities owned directly by Remeditex Ventures LLC. Remeditex Ventures LLC, the Malachite Trust and Lyda Hill each disclaims beneficial ownership of the reported securities except to the extent of its or her pecuniary interest therein.
Securities Authorized for Issuance under Equity Compensation Plans
The following table sets forth information regarding our equity compensation plans at December 31, 2019:
| | | | | | | | | | | | | | | | | | | | |
| Plan category | | Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) | | Weighted-average exercise price of outstanding options, warrants and rights (b) | | Number of securities (by class) remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (c) |
| Equity compensation plans approved by security holders (1) | | 2,883,550 | | | $2.70 | | | 531,450 | |
| Equity compensation plans not approved by security holders | | — | | | $— | | | — | |
(1) Represents shares of common stock issuable upon exercise of outstanding stock options and rights under our 2012 Stock Plan and 2018 Plan.
Item 13. Certain Relationships and Related Transactions, and Director Independence
Director Independence
The rules of the Nasdaq, or the Nasdaq Rules, require a majority of a listed company’s board of directors to be composed of independent directors. In addition, the Nasdaq Rules require that, subject to specified exceptions, each member of a listed company’s audit, compensation and nominating and governance committees be independent. Under the Nasdaq Rules, a director will only qualify as an independent director if, in the opinion of our board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. The NasdaqRules also require that audit committee members satisfy independence criteria set forth in Rule 10A-3 under the Securities Exchange Act of 1934, as amended ("the Exchange Act"). In order to be considered independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors, or any other board committee, accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries or otherwise be an affiliated person of the listed company or any of its subsidiaries. In considering the independence of compensation committee members, the Nasdaq Rules require that our board of directors must consider additional factors relevant to the duties of a compensation committee member, including the source of any compensation we pay to the director and any affiliations with the company.
Our current board of directors undertook a review of the composition of our current board of directors and the independence of each director. Our board of directors has determined that Messrs. Foster and Hauser, and Ms. Harrison are independent as defined under the Nasdaq rules. Messrs. Klemp and Capelli are not independent.
Related Party Transactions
We have a license agreement with MD Anderson for certain of the patents, patent applications and related intellectual property on which we base our research and product development. MD Anderson is a stockholder of our Company as a result of the shares issued to acquire the license agreement.
As the inventor of the intellectual property we license from MD Anderson, Dr. Capelli is entitled to 50% of the license income (which is determined after MD Anderson recoups any costs associated therewith) that we are required to pay to MD Anderson pursuant to our license agreement with MD Anderson. For the years ended December 31, 2019 and 2018, Dr. Capelli received $187,500 and $27,500 respectively from MD Anderson. In addition, Dr. Capelli is entitled to 50% of the proceeds (after the recoupment of any costs associated therewith) from the sale by MD Anderson of 175,000 shares issued to MD Anderson in connection with the license agreement.
As of December 31, 2018, we had convertible bridge notes outstanding with Remeditex Ventures LLC, our largest stockholder, in the amount of $8,400,000, consisting of $6,900,000 in principal amount of 8.25% convertible notes and
$1,500,000 in principal amount of 10.00% convertible notes. The 8.25% convertible notes were originally due on January 31, 2018 with respect to $5,000,000 in principal amount of notes and June 29, 2018 with respect to $1,900,000 in principal amount of notes; the maturity date of the notes had been extended to April 30, 2019 and the interest rate on such notes increased from 8.25% to 12.00% commencing on the original due date. Upon the closing of our IPO, these notes included accrued interest (calculated through an assumed date of February 14, 2019 of $1,279,932) converted into 4,470,482 shares of common stock.
On June 16, 2019, we entered into a securities purchase agreement with investors, including Remeditex Ventures LLC, for the issuance and sale of an aggregate of 675,000 units (of which Remeditex Ventures LLC purchased 357,143 units), each unit consisting of (i) one share of our common stock, and (ii) a warrant to purchase 0.7 shares (a total of 472,500) of our common stock. The offering price of the units was $14.00 per unit. The warrants included in the units are exercisable at a price of $16.00 per share and will expire on August 23, 2024, pursuant to which the resale of the shares of common stock underlying the June Warrants are registered. The closing of the issuance and sale of these securities was consummated on June 19, 2019. On July 1, 2019, we filed a Registration Statement on Form S-1 to register for resale the common stock underlying the June Units sold with our June 2019 private offering.
On October 10, 2019, we entered into a securities purchase agreement with investors, including Remeditex Ventures LLC, for the issuance and sale of an aggregate of 485,250 units (of which Remeditex Ventures LLC purchased 155,280 units), each unit consisting of (i) one share of our common stock, and (ii) a warrant to purchase 1.1 shares (a total of 533,775 shares) of our common stock. The offering price of the units was $12.88 per unit. The warrants included in the units are exercisable at a price of $12.88 per share, and expire on October 10, 2024. The closing of the issuance and sale of these securities was consummated on October 11, 2019. On November 8, 2019, we filed a Registration Statement on Form S-1 to register for resale the common stock underlying the October Units sold with our October 2019 private offering.
As of February 14, 2019, we had a convertible bridge note outstanding with Christopher Capelli, our chief executive officer, president and chief science officer, in the amount of $22,000. Upon the closing of our IPO, these notes included the accrued interest (calculated through an assumed date of February 14, 2019 of $1,519) converted into 13,440 shares of common stock.
From October 2018 through February 2019, we issued $985,000 in principal amount of 10% nonconvertible promissory notes. Upon the closing of our IPO, the principal and accrued interest was due (calculated through an assumed date of February 14, 2019 of $20,038). For each dollar in principal amount of notes purchase by investors, we issued the investors a five year warrant to purchase one share of common stock at an exercise price of $1.75 per share. Mr. Klemp, Dr. Capelli, Ms. Bisson and other members of management collectively purchased $125,000 of these notes and warrants on the same terms as described above. On February 15, 2019, the Company paid $985,000 in principal and $20,038 in accrued interest to the note holders to repay the above notes in full.
Policies and Procedures for Related Party Transactions
Our Audit Committee charter provides that our Audit Committee will be responsible for reviewing and approving in advance any related party transaction. This will cover, with certain exceptions set forth in Item 404 of Regulation S-K under the Securities Act, any transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships in which we were or are to be a participant, where the amount involved exceeds $120,000 and a related person had or will have a direct or indirect material interest, including, without limitation, purchases of goods or services by or from the related person or entities in which the related person has a material interest, indebtedness, guarantees of indebtedness and employment by us of a related person. All of the transactions described in this section occurred prior to the creation of our audit committee and the adoption of this policy.
Item 14. Principal Accounting Fees and Services
Aggregate fees for professional services by Dixon Hughes Goodman, LLP and Marcum LLP for their respective services for the fiscal years ended December 31, 2019 and 2018, respectively, were as follows:
| | | | | | | | |
| 2019 | 2018 |
| Audit fees * | $ | 117,668 | | $ | 79,000 | |
| Audit-related fees * | 101,331 | | 63,900 | |
| Tax fees | — | | — | |
| All other fees | — | | — | |
| | |
| Total | $ | 218,999 | | $ | 142,900 | |
*The amounts disclosed for 2018 include final billings in 2019 for the work completed with respect to fiscal year 2018. The amounts disclosed for 2019 include expected final billings in 2020 for the work completed with respect to fiscal year 2019.
Audit Fees. Consist of fees billed for professional services rendered for the audits of our financial statements, reviews of our interim financial statements included in quarterly reports, and services performed in connection with our regular filings with the SEC for the fiscal years ended December 30, 2019 and 2018 and in connection with our other statutory and regulatory filings for such periods.
Audit-Related Fees. This category consists of fees related to services rendered in connection with registration statements, including comfort letters and consents.
Audit Committee Pre-Approval Policies and Procedures
The Audit Committee on an annual basis reviews audit and non-audit services performed by the independent auditors. All audit and non-audit services are pre-approved by the Audit Committee, which considers, among other things, the possible effect of the performance of such services on the auditors’ independence.
PART IV
Item 15. Exhibits, Financial Statement Schedules
(a) The following documents are filed or furnished as part of this Form 10-K:
1. Financial Statements
Reference is made to the Index to Financial Statements under Item 8, Part II hereof.
2. Financial Statement Schedules
The Financial Statement Schedules have been omitted either because they are not required or because the information has been included in the financial statements or the notes thereto included in this Annual Report on Form 10-K.
3. Exhibits
Exhibit Index
| | | | | | | | |
Exhibit Number | | Description of Document |
| 3.1 | | | Amended and Restated Certificate of Incorporation dated February 19, 2019 (incorporated by reference to exhibit 3.1 of the Form 8-K filed February 22, 2019) |
| | |
| 3.2 | | | Amended and Restated Bylaws of Soliton, Inc. (incorporated by reference to exhibit 2.3 of the Form 1-A, file number 024-10854) |
| | |
| 4.1 | | Form of Common Stock certificate (incorporated by reference to exhibit 4.1 of the Form 10-K filed March 29, 2019) |
| | |
| 4.2 | | | Form of Warrant issuable in October 2018 Offering (incorporated by reference to exhibit 6.10 of the Form 1-A, file number 024-10854) |
| | |
| 4.3 | | Form of Warrant Agreement issued in June 2019 PIPE offering (incorporated by reference to exhibit 4.1 of the Company's Form 8-K filed June 18, 2019) |
| | |
| 4.4 | | Form of Warrant Agreement issued in October 2019 offering (incorporated by reference to exhibit 4.1 of the Company's Form 8-K filed October 15, 2019) |
| | |
| 10.1 | | | Patent and Technology License Agreement between Soliton, Inc. and The Board of Regents of The University of Texas System dated April 5, 2012 (incorporated by reference to exhibit 6.1 of the Form 1-A, file number 024-10854) |
| | |
| 10.2 ** | | Soliton, Inc. 2012 Long Term Incentive Plan (incorporated by reference to exhibit 6.2 of the Form 1-A, file number 024-10854) |
| | |
| 10.3 ** | | 2018 Stock Plan of Soliton, Inc., as amended, and forms of award agreements thereunder (incorporated by reference to exhibit 99.1 of the Company's Form S-8, file number 333-232636) |
| | |
| 10.4 | | | Lease Agreement between Soliton, Inc. and Ashbrook Land, Ltd. dated July 16, 2015 (incorporated by reference to exhibit 6.6 of the Form 1-A, file number 024-10854) |
| | |
| 10.5 ** | | Employment Agreement by and between Soliton, Inc. and Walter Kemp, effective February 25, 2019 (incorporated by reference to exhibit 10.1 of the Form 10-Q filed March 1, 2019) |
| | |
| 10.6 ** | | Employment Agreement by and between Soliton, Inc. and Christopher Capelli, effective February 25, 2019 (incorporated by reference to exhibit 10.2 of the Form 10-Q filed March 1, 2019) |
| | |
| 10.7 ** | | Employment Agreement by and between Soliton, Inc. and Joe Tanner, effective February 25, 2019 (incorporated by reference to exhibit 10.3 of the Form 10-Q filed March 1, 2019) |
| | |
| 10.8 ** | | Employment Agreement by and between Soliton, Inc. and Lori Bisson, effective February 25, 2019 (incorporated by reference to exhibit 10.4 of the Form 10-Q filed March 1, 2019) |
| | |
| 10.9 | | Form of Registration Rights Agreement dated June 16, 2019 by and among Soliton, Inc. and the investors in the June 2019 PIPE offering (incorporated by reference to exhibit 10.2 of the Company's Form 8-K filed June 18, 2019) |
| | |
| 10.10 | | Form of Registration Rights Agreement dated October 10, 2019 by and among Soliton, Inc. and the investors in the October 2019 offering (incorporated by reference to exhibit 10.2 of the Company's Form 8-K filed June 18, 2019) |
| | |
| 10.11 + | | Cooperative Development Addendum to Engineering and Development Services Master Agreement between Soliton, Inc. and Emphysys, Inc. dated November 20, 2019 (incorporated by reference to exhibit 10.1 of the Company's Form 8-K filed November 22, 2019) |
| | |
| 23.1* | | |
| | |
| 23.2* | | |
| | |
| | |
| | |
| | |
| | | | | | | | |
| 31.1* | | |
| | |
| 31.2* | | |
| | |
| 32.1* | | |
| | |
| 32.2* | | |
| | |
| 101.INS* | | XBRL Instance Document |
| | |
| 101.SCH* | | XBRL Taxonomy Extension Schema Document |
| | |
| 101.CAL* | | XBRL Taxonomy Extension Calculation Linkbase Document |
| | |
| 101.DEF* | | XBRL Taxonomy Extension Definition Linkbase Document |
| | |
| 101.LAB* | | XBRL Taxonomy Extension Label Linkbase Document |
| | |
| 101.PRE* | | XBRL Taxonomy Extension Presentation Linkbase Document |
_______________
* Filed herewith
** Management contract or compensatory plan, contract or arrangement.
+ Pursuant to Item 601(b)(10)(iv) of Regulation S-K promulgated by the SEC, certain portions of this exhibit have been redacted. The Company hereby agrees to furnish supplementally to the SEC, upon its request, an unredacted copy of this exhibit.
Item 16. 10-K Summary
None.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | | | | |
| SOLITON, INC. | |
| | |
| Date: March 2, 2020 | By: | /s/ CHRISTOPHER CAPELLI |
| | Christopher Capelli Chief Executive Officer, President and Director (Principal Executive Officer) |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacity and on the dates indicated.
| | | | | | | | |
| Date: March 2, 2020 | By: | /s/ CHRISTOPHER CAPELLI |
| | Christopher Capelli Chief Executive Officer, President and Director (Principal Executive Officer) |
| | |
| Date: March 2, 2020 | | /s/ LORI BISSON |
| | Lori Bisson Chief Financial Officer (Principal Financial and Accounting Officer) |
| | |
| Date: March 2, 2020 | | /s/ WALTER KLEMP |
| | Walter Klemp Executive Chairman of the Board Director |
| | |
| Date: March 2, 2020 | | /s/ JONATHAN FOSTER |
| | Jonathan Foster Director |
| | |
| Date: March 2, 2020 | | /s/ DANIKA HARRISON |
| | Danika Harrison Director |
| | |
| Date: March 2, 2020 | | /s/ BRAD HAUSER |
| | Brad Hauser Director |