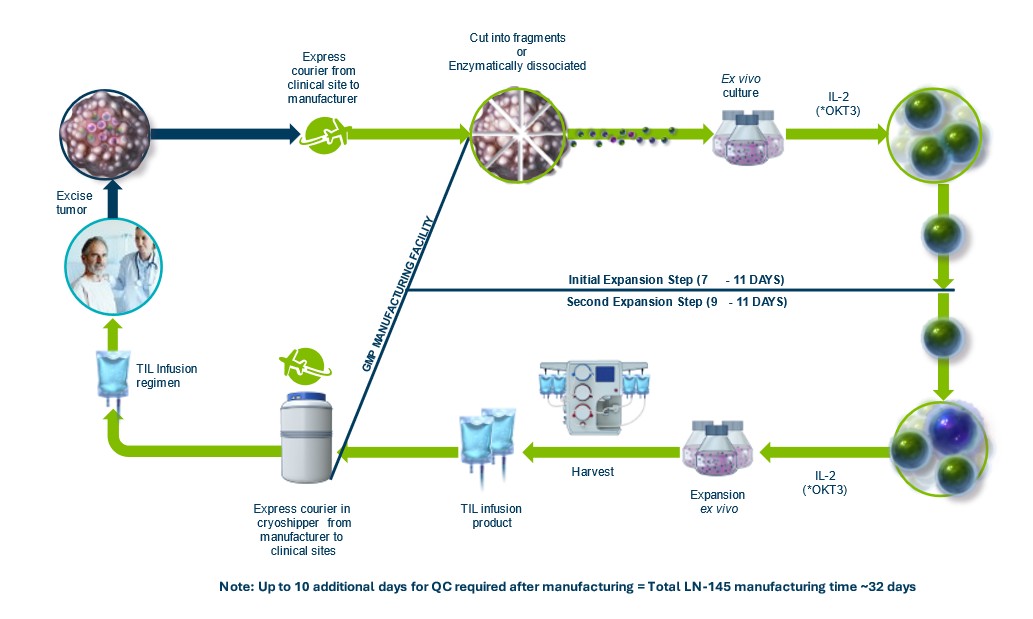
Our Iovance Cell Therapy Center, or iCTC, for Large-Scale Centralized Manufacturing
Our iCTC is the first centralized and scalable cGMP manufacturing facility dedicated to producing TIL as a potential therapy for patients with solid tumors. At 136,000 square feet, the iCTC is among the largest cell therapy manufacturing facilities in existence. It currently supplies investigational TIL therapy for Iovance clinical trials, with commercial manufacturing expected to begin after initial product approval. The proximity of the iCTC to multiple airports facilitates delivery of TIL therapies to treatment centers, with the iCTC expected to cover logistics and delivery of TIL therapy in both North America and Europe. This location may also offer future tax savings under Pennsylvania’s Keystone Opportunity Improvement Zone, or KOIZ.
Clinical Manufacturing
In 2021, we completed the commissioning activities at the iCTC and successfully initiated manufacturing of clinical batches of lifileucel and LN-145. In addition, we have entered into MSAs with WuXi Advanced Therapies, Inc., or WuXi, and Moffitt pursuant to which they have agreed to manufacture, package, ship and handle quality assurance and quality control of certain clinical trials for our TIL products working closely with our employees. In the future, we may rely on them or other third parties, or our own manufacturing capabilities, for the manufacturing and processing of TIL-based product candidates for our clinical trials.
Commercial Manufacturing Preparations
To meet projected needs for commercial quantities, we continue our launch readiness and scale up activities to supply commercial TIL upon potential BLA approval. We expect to rely primarily on our own iCTC facility for commercial supply, with flexibility to use CMOs to meet anticipated clinical trial and, if approved, commercial demands. For example, we expect to be able to use two suites for clinical or commercial manufacturing at WuXi in Philadelphia if needed based on demand. If we are unable to meet manufacturing capabilities and demand at our own commercial manufacturing facility, we may need to rely on CMOs, including both current and alternate suppliers, to ensure sufficient capacity is available for commercial purposes. Our CMO relationship is described in more detail in the Research, Development, Manufacturing and License agreements section of this Annual Report on Form 10-K.
Cell processing activities are conducted at all facilities under cGMP, using qualified equipment and materials. We believe that all materials and components utilized in the production of the final TIL product are readily available from qualified suppliers.
Intellectual Property
We aim to lead in the field of T-cell-based immunotherapy by building and augmenting the patent rights for our proprietary TIL technology platform, which we have developed internally and licensed from third parties. Intellectual property is of importance in our field and in biotechnology generally. We seek to protect and enhance proprietary technology, inventions, and improvements that are commercially important to the development of our business by seeking, maintaining, and defending patent rights. We also plan to rely on regulatory protection afforded through Orphan Drug Designation, or ODD, available regulatory exclusivities and patent term extensions where available. To achieve this objective, our strategic focus has been to develop our own intellectual property, while also identifying and licensing patents from third parties that provide protection and serve as an optimal platform to enhance our intellectual property and technology base. We expect to further develop our patent portfolio as a strategic focus in 2023.
We currently own more than 60 U.S. patents related to TIL therapy, including patents directed to compositions and methods of treatment in a broad range of cancers, such as U.S. Patent Nos. 10,130,659; 10,166,257; 10,272,113; 10,363,273; 10,398,734; 10,420,799; 10,463,697; 10,517,894; 10,537,595; 10,639,330; 10,646,517; 10,653,723; 10,695,372; 10,894,063; 10,905,718; 10,918,666; 10,925,900; 10,933,094; 10,946,044; 10,946,045; 10,953,046; 10,953,047; 11,007,225; 11,007,226; 11,013,770; 11,026,974; 11,040,070; 11,052,115; 11,052,116; 11,058,728; 11,083,752; 11,123,371; 11,141,438 11,168,303; 11,168,304; 11,179,419; 11,202,803; 11,202,804; 11,241,456; 11,254,913; 11,266,694; 11,273,180; 11,273,181; 11,291,687; 11,304,979; 11,304,980; 11,311,578; 11,337,998; 11,344,579; 11,344,580; 11,344,581; 11,351,197; 11,351,198; 11,351,199; 11,364,266; 11,369,637; 11,384,637; 1,433,097; 11,529,372; and 11,541,077. More than 35 of these patents are related to our Gen 2 TIL manufacturing processes and have terms that we anticipate will extend to January 2038, not including any patent term extensions or adjustments that may be available. Our owned and licensed intellectual property portfolio also includes patents and patent applications relating to TIL, MIL, and PBL therapies; frozen tumor-based TIL technologies; remnant TIL and digest TIL compositions, methods and processes; methods of manufacturing TIL, MIL, and PBL therapies; the use of costimulatory and T-cell modulating molecules in TIL therapy and manufacturing; stable and transient genetically-modified TIL therapies, including genetic knockouts of immune