As filed with the Securities and Exchange Commission on July 21, 2015
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S-1
REGISTRATION STATEMENT
UNDER THE SECURITIES ACT OF 1933
VistaGen Therapeutics, Inc.
(Exact Name of Registrant as Specified in its Charter)
|
Nevada
|
2086
|
20-5093315
|
||
|
(State or Other Jurisdiction of
Incorporation or Organization)
|
(Primary Standard Industrial
Classification Code Number)
|
(I.R.S. Employer
Identification Number)
|
343 Allerton Avenue
South San Francisco, California 94080
(650) 577-3600
(Address, including zip code and telephone number, including area code, of registrant’s principal executive offices)
Copy of correspondence to:
Daniel W. Rumsey, Esq.
Disclosure Law Group
One American Plaza
600 West Broadway, Suite 700
San Diego, CA 92101
(619) 795-1134
From time to time after the effective date of this Registration Statement.
(Approximate date of commencement of proposed sale to the public)
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box. [X]
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b2 of the Exchange Act.
|
Large accelerated filer
|
[ ]
|
Accelerated filer
|
[ ]
|
|
Non-accelerated filer
|
[ ]
|
Smaller reporting company
|
[X]
|
(do not check if a smaller reporting company )
CALCULATION OF REGISTRATION FEE
|
Title of Each Class of Securities to be Registered
|
Amount to be Registered(2)
|
Proposed Maximum
Aggregate
Offering Price (3)
|
Amount of
Registration Fee (3) (4)
|
|||||||||
|
Common Stock, $0.001 par value per share
|
3,623,341
|
(1)
|
$
|
36,233,410
|
$
|
4,210.32
|
||||||
|
(1)
|
Consists of up to 2,668,305 shares of common stock issuable upon conversion of shares of Series B 10% Convertible Preferred Stock (“Series B Preferred”), and up to 955,036 shares of common stock, $0.001 par value per share, issuable upon exercise of warrants to purchase common stock (“Warrants”), issued in private placement transactions, first consummated on May 12, 2015 (the “Private Placements”).
|
|
|
(2)
|
In the event of a stock split, stock dividend or similar transaction involving the common stock of the Registrant, in order to prevent dilution, the number of shares registered shall be automatically increased to cover additional shares in accordance with Rule 416(a) under the Securities Act of 1933, as amended (“Securities Act”).
|
|
|
(3)
|
Estimated solely for the purpose of calculating the registration fee in accordance with Rule 457(c) under the Securities Act.
|
|
| (4) |
A registration fee in the aggregate amount of $4,404.96 was previously paid by the registrant in connection with the filing of a Registration Statement on Form S-1 (Registration No. 333-195901), first filed on May 12, 2014 and subsequently withdrawn prior to the sale of any securities thereunder. Pursuant to Rule 457(p) under the Securities Act, the Registrant hereby applies $4,210.32 of the previously paid filing fee against amounts due herewith.
|
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The information in this prospectus is not complete and may be changed. The Selling Stockholders may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting offers to buy these securities in any state where the offer or sale is not permitted.
PRELIMINARY PROSPECTUS
(Subject to Completion)
Dated _________ __, 2015
3,623,341 Shares of Common Stock
VISTAGEN THERAPEUTICS, INC.
We are registering for resale up to 3,623,341 shares of common stock, $0.001 per share, of VistaGen Therapeutics, Inc. (“we,” “us,” or the “Company”), held by the selling stockholders listed beginning on page 106 of this prospectus (“Selling Stockholders”). All of the shares being offered for resale by the Selling Stockholders. The shares of common stock registered for resale under this prospectus include:
|
·
|
up to 2,668,305 shares of common stock issuable upon conversion of shares of Series B 10% Convertible Preferred Stock (“Series B Preferred”) issued in a series of private placement transactions, the first of which was consummated on May 12, 2015 (the “Private Placements”); and
|
|
·
|
up to 955,036 shares of common stock issuable upon exercise of warrants to purchase common stock (“Warrants”) issued in connection with the Private Placements.
|
We will not receive any proceeds from the resale of any shares of common stock by the Selling Stockholders under this prospectus. However, if Warrants are exercised by the Selling Stockholders who hold them, we will receive the exercise price of any such Warrant exercises. We will pay the expenses of registering the shares of common stock for resale by the Selling Stockholders under this prospectus. See “Selling Stockholders” beginning on page 106 of this prospectus for a list of the Selling Stockholders.
The shares of common stock are being registered to permit the Selling Stockholders to resell the shares from time to time, in amounts and at prices and on terms determined at the time of the offering. The Selling Stockholders may resell the shares of our common stock covered by this prospectus in a number of different ways and at prevailing market prices or privately negotiated transactions. We provide more information about how the Selling Stockholders may resell the shares in the section entitled “Plan of Distribution” beginning on page 109 of this prospectus.
Our common stock is quoted on the OTCQB under the symbol “VSTA.” The last reported sale price of our common stock on July 17, 2015 was $10.00 per share.
No underwriter or other person has been engaged to facilitate the resale of shares of common stock or exercise of Warrants by the Selling Stockholders under this prospectus.
You should rely only on the information contained in this prospectus. We have not, and the Selling Stockholders have not, authorized anyone to provide you with different information. No dealer, salesperson or other person is authorized to give any information or to represent anything not contained in this prospectus. You must not rely on any unauthorized information or representations. If anyone provides you with different information, you should not rely on it. We are not, and the Selling Stockholders are not, making an offer to sell these securities in any jurisdiction where the offer or sale is not permitted. You should assume that the information contained in this prospectus is accurate only as of the date on the front cover of this prospectus. Our business, financial condition, results of operations and prospects may have changed since that date.
Investing in our common stock involves a high degree of risk. See “Risk Factors” beginning on page 3 of this prospectus.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
The date of this prospectus is ______ __, 2015.
VistaGen Therapeutics, Inc.
TABLE OF CONTENTS
|
Page
|
||
| 2 | ||
| 2 | ||
| 3 | ||
| 33 | ||
| 33 | ||
| 34 | ||
| 73 | ||
| 74 | ||
| 75 | ||
| 75 | ||
| 86 | ||
| 86 | ||
| 91 | ||
| 98 | ||
| 99 | ||
| 105 | ||
| 106 | ||
| 109 | ||
| 109 | ||
| 110 | ||
| 111 | ||
| 111 | ||
| 111 | ||
| F-1 |
You should rely only on the information contained in this prospectus. We have not authorized anyone to provide you with additional information or information different from that contained in this prospectus. The information contained in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or any sale of our common stock.
We own or have rights to use a number of common law trademarks and trade names that we use in connection with our business, including VistaGen Therapeutics, Inc., VistaGen, our logo, Better Cells Lead to Better Medicine, Human Clinical Trials in a Test Tube, CardioSafe 3D and LiverSafe 3D. Solely for convenience, the trademarks and trade names referred to in certain portions of this prospectus may have been included without the TM symbol, but any such references are not intended to indicate in any way that we will not assert to the fullest extent under applicable law our rights to use those trademarks and trade names. All other trademarks, service marks and trade names referred to in this prospectus, if any, are, to our knowledge, the property of their respective owners.
Unless the context otherwise requires, the words “VistaGen Therapeutics, Inc.” “VistaGen,” “we,” “the Company,” “us” and “our” refer to VistaGen Therapeutics, Inc., a Nevada corporation. “VistaGen California” refers to VistaGen Therapeutics, Inc., a California corporation and our wholly owned subsidiary.
FORWARD-LOOKING STATEMENTS
This prospectus, including the information incorporated by reference, contains forward-looking statements as defined in the Private Securities Litigation Reform Act of 1995. The use of any statements containing the words “intend,” “believe,” “estimate,” “project,” “expect,” “anticipate,” “plan,” “should” or similar expressions are intended to identify such statement. Forward-looking statements inherently involve risks and uncertainties that could cause actual results to differ materially from the forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, changes in demand for our products and services, changes in the level of operating expenses, our ability to execute our business and operating plan, changes in general economic conditions that impact government spending, regulatory issues, dependence on third party suppliers, and other risks detailed in this prospectus under the heading “Risk Factors” and in our periodic report filings with the Securities and Exchange Commission (the “SEC”).
Forward-looking statements are subject to numerous assumptions, risks and uncertainties, which change over time. Forward-looking statements speak only as of the date they are made, and we assume no duty to and do not undertake to update forward-looking statements. These forward-looking statements may not meet the safe harbor for forward-looking statements pursuant to Sections 21E or 27A of the Securities Act. Actual results could differ materially from those anticipated in forward-looking statements and future results could differ materially from historical performance.
PROSPECTUS SUMMARY
This summary highlights information contained elsewhere in this prospectus. This summary does not contain all the information you should consider before buying our common stock. You should read the following summary together with the more detailed information appearing in this prospectus, including our consolidated financial statements and related notes, and our risk factors beginning on page 3, before deciding whether to purchase shares of our common stock.
Overview
We are a clinical-stage biopharmaceutical company committed to developing and commercializing innovative product candidates for patients with depression, other diseases and various disorders related to the central nervous system (“CNS”), as well as cancer.
More than one billion people worldwide suffer from CNS disorders. Recently, the economic burden of these disorders was estimated at $2.0 trillion in the U.S. and European Union alone, a figure that is expected to triple by 2030. The World Health Organization estimates that 350 million people worldwide are affected by depression. According to the U.S. National Institutes of Health (“NIH”), major depression is one of the most common mental disorders in the U.S. In 2012, the NIH estimated 16 million adults aged 18 or older in the U.S. had at least one major depressive episode. This represented approximately 7 percent of all U.S. adults.
Our lead product candidate, AV-101, is an orally available small molecule prodrug in Phase 2 clinical development for Major Depressive Disorder (“MDD”). AV-101’s mechanism of action (“MOA”), as an N-methyl-D-aspartate receptor (“NMDAR”) antagonist binding selectively at the glycine-binding (“GlyB”) co-agonist site of the NMDAR, is fundamentally different from all antidepressants currently approved by the U.S. Food and Drug Administration (“FDA”). In four preclinical studies utilizing well-validated animal models of depression, AV-101 was shown to induce fast-acting, dose-dependent, persistent and statistically significant antidepressant-like responses, following a single treatment, which was equivalent to responses seen with a control single sub-anesthetic dose of ketamine (sometimes used by clinicians off-label to treat suicidal behavior). In the same studies, fluoxetine (Prozac) did not induce rapid onset antidepressant-like responses. Preclinical studies also support the hypothesis that AV-101 has potential to treat several additional CNS disorders, including chronic neuropathic pain, epilepsy and neurodegenerative diseases, such as Parkinson’s disease and Huntington’s disease where modulation of the NMDAR may have therapeutic benefit.
Following two successful randomized, double-blind, placebo-controlled Phase 1 safety studies funded by the NIH, in February 2015, we entered into a Cooperative Research and Development Agreement (“CRADA”) with the U.S. National Institute of Mental Health (“NIMH”), part of the NIH. Under the CRADA, we will collaborate with the NIMH on the initial Phase 2 clinical study of AV-101 in subjects with treatment-resistant MDD. Pursuant to the CRADA, the study will be conducted at the NIMH and be fully funded by the NIMH. It is contemplated that this clinical study will begin in Fall 2015 under the direction of Dr. Carlos Zarate, Jr., the NIMH’s Chief of Experimental Therapeutics & Pathophysiology Branch and of the Section on Neurobiology and Treatment of Mood and Anxiety Disorders.
In addition to developing AV-101 for MDD and other CNS indications, we are applying our stem cell technology for drug rescue programs intended to identify and develop proprietary new chemical entities (“NCEs”) for our internal drug candidate pipeline. Drug rescue involves (1) using our customized in vitro bioassay systems to predict potential heart and liver toxicity of NCEs, (2) leveraging prior investments by pharmaceutical companies and others related to screening large-scale compound libraries, optimizing and testing for efficacy NCEs that were terminated before FDA approval due to heart or liver toxicity and are now available in the public domain, and (3) applying modern medicinal chemistry to produce safer NCEs for our internal development pipeline. Our CardioSafe 3D™ bioassay system uses our human pluripotent stem cell (“hPSC”)-derived cardiomyocytes, or human heart cells. We believe CardioSafe 3D is more comprehensive and clinically predictive than the hERG assay, which is currently the only in vitro cardiac safety assay required by FDA guidelines. We use our hPSC-derived hepatocytes, or human liver cells, in our LiverSafe 3D™ bioassay system to predict potential liver toxicity of NCEs, including potential drug metabolism issues and adverse drug-drug interactions. CardioSafe 3D and LiverSafe 3D offer a new paradigm for evaluating and predicting potential heart and liver toxicity of NCEs, including drug rescue NCEs, early in the development process, long before costly, high risk animal studies and human clinical trials. We intend to develop internally for our pipeline each lead drug rescue NCE we produce.
THE OFFERING
|
Securities Offered by the Selling Stockholders
|
3,623,341 shares of common stock.
|
|
Common Stock Outstanding as of July 17, 2015
|
1,594,461 shares.
|
|
Use of Proceeds
|
We will not receive any of the proceeds of the shares of common stock offered for resale by the Selling Stockholders. If Warrants held by certain of the Selling Stockholders are exercised, we will receive the exercise price from such exercises. The shares of common stock that may be resold by the Selling Stockholders under this prospectus are issuable upon the conversion of securities sold by us to the Selling Stockholders in private placement transactions, or upon exercise of Warrants.
|
|
Risk Factors
|
Prior to making an investment decision, you should carefully consider all of the information in this prospectus and, in particular, you should evaluate the risk factors set forth under the caption “Risk Factors” beginning on page 3.
|
|
Trading Symbol
|
VSTA
|
RISK FACTORS
Investing in our securities involves a high degree of risk. You should consider carefully the risks and uncertainties described below, together with all of the other information in this Prospectus before investing in our securities. The risks described below are not the only risks facing our Company. Additional risks and uncertainties not currently known to us or that we currently deem to be immaterial may also materially adversely affect our business, financial condition and/or operating results. If any of the following risks are realized, our business, financial condition and results of operations could be materially and adversely affected.
Risks Related to Product Development, Regulatory Approval and Commercialization
We depend heavily on the success of AV-101. We cannot be certain that we will be able to obtain regulatory approval for, or successfully commercialize AV-101, or any product candidate.
We currently have no drug products for sale and may never be able to develop and commercialize marketable drug products. Our business depends heavily on the successful non-clinical and clinical development, regulatory approval and commercialization of AV-101 for depression, including Major Depressive Disorder (“MDD”), and various other diseases and disorders involving the central nervous system (“CNS”) , as well as our ability to produce, develop and commercialize new chemical entities (“NCEs”) from our drug rescue programs. AV-101 will require substantial additional Phase 2 and Phase 3 clinical development, testing and regulatory approval before we are permitted to commence its commercialization. Each drug rescue NCE will require substantial non-clinical development, all phases of clinical development, and regulatory approval before we are permitted to commence its commercialization. The non-clinical studies and clinical trials of our product candidates are, and the manufacturing and marketing of our product candidates will be, subject to extensive and rigorous review and regulation by numerous government authorities in the United States and in other countries where we intend to test and, if approved, market any product candidate. Before obtaining regulatory approvals for the commercial sale of any product candidate, we must demonstrate through non-clinical studies and clinical trials that the product candidate is safe and effective for use in each target indication. Drug development is a long, expensive and uncertain process, and delay or failure can occur at any stage of any of our non-clinical studies or clinical trials. This process can take many years and may also include post-marketing studies and surveillance, which will require the expenditure of substantial resources beyond the proceeds we have raised to date. Of the large number of drugs in development in the United States, only a small percentage will successfully complete the U.S. Food and Drug Administration, or FDA, regulatory approval process and will be commercialized. Accordingly, even if we are able to obtain the requisite financing to continue to fund our non-clinical studies and clinical trials, we cannot assure you that AV-101 or any other product candidate will be successfully developed or commercialized.
We are not permitted to market our product candidates in the United States until we receive approval of a New Drug Application, or an NDA, from the FDA, or in any foreign countries until we receive the requisite approval from such countries. We recently received FDA clearance to initiate the initial Phase 2 clinical trial involving AV-101, to study its safety, tolerability and efficacy in patients with MDD. If our Phase 2 clinical trial of AV-101 is successful, we expect the FDA to require us to complete at least one additional Phase 2 clinical trial and at least one pivotal Phase 3 clinical trial in order to submit an NDA for AV-101 as a treatment for MDD. However, the FDA may require that we conduct more than one additional Phase 2 clinical study and more than one Phase 3 pivotal trial of AV-101 before we can submit an NDA. The FDA may also require that we conduct additional toxicity studies and additional non-clinical studies before submitting an NDA for AV-101.
Obtaining FDA approval of an NDA is a complex, lengthy, expensive and uncertain process, and the FDA may delay, limit or deny approval of AV-101 or any of our product candidates for many reasons, including, among others:
|
·
|
we may not be able to demonstrate that the product candidate is safe and effective in treating a human disease or disorder, to the satisfaction of the FDA;
|
|
·
|
the results of our non-clinical studies and clinical trials may not meet the level of statistical or clinical significance required by the FDA for marketing approval;
|
|
·
|
the FDA may disagree with the number, design, size, conduct or implementation of our non-clinical studies and clinical trials;
|
|
·
|
the FDA may require that we conduct additional non-clinical studies and clinical trials;
|
|
·
|
the FDA or the applicable foreign regulatory agency may not approve the formulation, labeling or specifications of any of our product candidates;
|
|
·
|
the contract research organizations, or CROs, that we retain to conduct our non-clinical studies and clinical trials may take actions outside of our control that materially adversely impact our non-clinical studies and clinical trials;
|
|
·
|
the FDA may find the data from non-clinical studies and clinical trials insufficient to demonstrate that our product candidates’ clinical and other benefits outweigh their safety risks;
|
|
·
|
the FDA may disagree with our interpretation of data from our non-clinical studies and clinical trials;
|
|
·
|
the FDA may not accept data generated at our non-clinical studies and clinical trial sites;
|
|
·
|
if our NDA, if and when submitted, is reviewed by an advisory committee, the FDA may have difficulties scheduling an advisory committee meeting in a timely manner or the advisory committee may recommend against approval of our application or may recommend that the FDA require, as a condition of approval, additional non-clinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions;
|
|
·
|
the FDA may require development of a Risk Evaluation and Mitigation Strategy, or REMS, as a condition of approval or post-approval;
|
|
·
|
the FDA or the applicable foreign regulatory agency may determine that the manufacturing processes or facilities of third-party contract manufacturers with which we contract do not conform to applicable requirements, including current Good Manufacturing Practices, or cGMPs; or
|
|
·
|
the FDA or applicable foreign regulatory agency may change its approval policies or adopt new regulations.
|
Any of these factors, many of which are beyond our control, could jeopardize our ability to obtain regulatory approval for and successfully market AV-101 or any other product candidate we may develop, including drug rescue NCEs. Any such setback in our pursuit of regulatory approval would have a material adverse effect on our business and prospects.
A Fast Track designation by the FDA may not actually lead to a faster development or regulatory review or approval process.
We intend to seek FDA Fast Track designation for AV-101, and we may do so for other product candidates as well. If a product candidate is intended for the treatment of a serious or life-threatening condition and the product candidate demonstrates the potential to address unmet medical needs for this condition, the sponsor may apply for the FDA Fast Track designation. The FDA has broad discretion whether or not to grant this designation, and even if we believe AV-101 and other product candidates are eligible for this designation, we cannot be sure that the review or approval will compare to conventional FDA procedures. Even if granted, the FDA may withdraw Fast Track designation if it believes that the designation is no longer supported by data from our clinical development programs.
The number of patients suffering from MDD has not been established with precision. If the actual number of patients with MDD is smaller than we anticipate, we may encounter difficulties in enrolling patients in our AV-101 clinical trials, including our impending NIH-funded Phase 2 clinical study of AV-101 in MDD, thereby delaying or preventing clinical development. Further, if AV-101 is approved for treatment of MDD, and the market for this indication is smaller than we anticipate, our ability to achieve profitability could be limited.
Results of earlier clinical trials may not be predictive of the results of later-stage clinical trials.
The results of preclinical studies and early clinical trials of AV-101 and other product candidates may not be predictive of the results of later-stage clinical trials. AV-101 or other product candidates in later stages of clinical trials may fail to show the desired safety and efficacy results despite having progressed through preclinical studies and initial clinical trials. Many companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to adverse safety profiles or lack of efficacy, notwithstanding promising results in earlier studies. Similarly, our future clinical trial results may not be successful for these or other reasons.
This drug candidate development risk is heightened by any changes in planned clinical trials compared to completed clinical trials. As product candidates are developed through preclinical to early and late stage clinical trials towards approval and commercialization, it is customary that various aspects of the development program, such as manufacturing and methods of administration, are altered along the way in an effort to optimize processes and results. While these types of changes are common and are intended to optimize the product candidates for later stage clinical trials, approval and commercialization, such changes do carry the risk that they will not achieve these intended objectives.
For example, the results of planned clinical trials may be adversely affected if we or our collaborator seek to optimize and scale-up production of a product candidate. In such case, we will need to demonstrate comparability between the newly manufactured drug substance and/or drug product relative to the previously manufactured drug substance and/or drug product. Demonstrating comparability may cause us to incur additional costs or delay initiation or completion of our clinical trials, including the need to initiate a dose escalation study and, if unsuccessful, could require us to complete additional preclinical or clinical studies of our product candidates.
If serious adverse events or other undesirable side effects are identified during the use of AV-101 in investigator-sponsored trials, it may adversely effect our development of AV-101 for MDD and other CNS indications.
AV-101 will be tested in an NIH investigator sponsored Phase 2 clinical trial for the treatment of MDD and may be subjected to testing in the future for other CNS indications in additional investigator sponsored clinical trials. If serious adverse events or other undesirable side effects, or unexpected characteristics of AV-101 are observed in investigator sponsored clinical trials of AV-101 or our clinical trials, it may adversely affect or delay our clinical development of AV-101, and the occurrence of these events would have a material adverse effect on our business.
Positive results from early non-clinical studies and clinical trials of AV-101 or other product candidates are not necessarily predictive of the results of later non-clinical studies and clinical trials of such product candidates. If we cannot replicate the positive results from our earlier non-clinical studies and clinical trials of AV-101 or other product candidates in our later non-clinical studies and clinical trials, we may be unable to successfully develop, obtain regulatory approval for and commercialize our product candidates.
Positive results from non-clinical studies of our product candidates, and any positive results we may obtain from early clinical trials of our product candidates, may not necessarily be predictive of the results from required later non-clinical studies and clinical trials. Similarly, even if we are able to complete our planned non-clinical studies or clinical trials of our product candidates according to our current development timeline, the positive results from our non-clinical studies and clinical trials of our product candidates may not be replicated in subsequent non-clinical studies or clinical trial results. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials after achieving positive results in early-stage development, and we cannot be certain that we will not face similar setbacks. These setbacks have been caused by, among other things, non-clinical findings made while clinical trials were underway or safety or efficacy observations made in non-clinical studies and clinical trials, including previously unreported adverse events. Moreover, non-clinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in non-clinical studies and clinical trials nonetheless failed to obtain FDA approval. We have not yet completed any Phase 2 clinical trial for AV-101, and if we fail to produce positive results in our NIH-sponsored Phase 2 clinical trial of AV-101 in MDD, the development timeline and regulatory approval and commercialization prospects for AV-101 and, correspondingly, our business and financial prospects, would be materially adversely affected.
Failures or delays in the commencement or completion of our planned clinical trials of our product candidates could result in increased costs to us and could delay, prevent or limit our ability to generate revenue and continue our business.
We and the NIH are preparing to commence an NIH-funded Phase 2 clinical trial of AV-101 as a treatment for MDD. We will need to complete at least two additional large clinical trials prior to the submission of an NDA for AV-101 as a treatment for MDD. Successful completion of our clinical trials is a prerequisite to submitting an NDA to the FDA and, consequently, the ultimate approval and commercial marketing of AV-101 for MDD and any other product candidates we may develop. We do not know whether the NIH-funded Phase 2 study of AV-101 or any of our future-planned clinical trials will begin or be completed on schedule, if at all, as the commencement and completion of clinical trials can be delayed or prevented for a number of reasons, including, among others:
|
·
|
the FDA may deny permission to proceed with our planned clinical trials or any other clinical trials we may initiate, or may place a clinical trial on hold;
|
|
·
|
delays in filing or receiving approvals of additional INDs that may be required;
|
|
·
|
negative results from our ongoing non-clinical studies;
|
|
·
|
delays in reaching or failing to reach agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
|
|
·
|
inadequate quantity or quality of a product candidate or other materials necessary to conduct clinical trials, for example delays in the manufacturing of sufficient supply of finished drug product;
|
|
·
|
difficulties obtaining Institutional Review Board, or IRB, approval to conduct a clinical trial at a prospective site or sites;
|
|
·
|
challenges in recruiting and enrolling patients to participate in clinical trials, including the proximity of patients to trial sites;
|
|
·
|
eligibility criteria for the clinical trial, the nature of the clinical trial protocol, the availability of approved effective treatments for the relevant disease and competition from other clinical trial programs for similar indications;
|
|
·
|
severe or unexpected drug-related side effects experienced by patients in a clinical trial;
|
|
·
|
delays in validating any endpoints utilized in a clinical trial;
|
|
·
|
the FDA may disagree with our clinical trial design and our interpretation of data from clinical trials, or may change the requirements for approval even after it has reviewed and commented on the design for our clinical trials;
|
|
·
|
reports from non-clinical or clinical testing of other CNS therapies that raise safety or efficacy concerns; and
|
|
·
|
difficulties retaining patients who have enrolled in a clinical trial but may be prone to withdraw due to rigors of the clinical trials, lack of efficacy, side effects, personal issues or loss of interest.
|
Clinical trials may also be delayed or terminated as a result of ambiguous or negative interim results. In addition, a clinical trial may be suspended or terminated by us, the FDA, the IRBs at the sites where the IRBs are overseeing a clinical trial, a data and safety monitoring board, or DSMB, overseeing the clinical trial at issue or other regulatory authorities due to a number of factors, including, among others:
|
·
|
failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols;
|
|
·
|
inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities that reveals deficiencies or violations that require us to undertake corrective action, including the imposition of a clinical hold;
|
|
·
|
unforeseen safety issues, including any that could be identified in our ongoing non-clinical carcinogenicity studies, adverse side effects or lack of effectiveness;
|
|
·
|
changes in government regulations or administrative actions;
|
|
·
|
problems with clinical supply materials; and
|
|
·
|
lack of adequate funding to continue clinical trials.
|
Changes in regulatory requirements, FDA guidance or unanticipated events during our non-clinical studies and clinical trials of our product candidates may occur, which may result in changes to non-clinical studies and clinical trial protocols or additional non-clinical studies and clinical trial requirements, which could result in increased costs to us and could delay our development timeline.
Changes in regulatory requirements, FDA guidance or unanticipated events during our non-clinical studies and clinical trials may force us to amend non-clinical studies and clinical trial protocols or the FDA may impose additional non-clinical studies and clinical trial requirements. Amendments or changes to our clinical trial protocols would require resubmission to the FDA and IRBs for review and approval, which may adversely impact the cost, timing or successful completion of clinical trials. Similarly, amendments to our non-clinical studies may adversely impact the cost, timing, or successful completion of those non-clinical studies. If we experience delays completing, or if we terminate, any of our non-clinical studies or clinical trials, or if we are required to conduct additional non-clinical studies or clinical trials, the commercial prospects for our product candidates may be harmed and our ability to generate product revenue will be delayed.
We rely, and expect that we will continue to rely, on third parties to conduct any clinical trials for our product candidates. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates and our business could be substantially harmed.
We do not have the ability to independently conduct clinical trials. We rely on medical institutions, clinical investigators, contract laboratories and other third parties, such as contract research organizations, or CROs, to conduct clinical trials on our product candidates. We enter into agreements with third-party CROs to provide monitors for and to manage data for our clinical trials. We rely heavily on these parties for execution of clinical trials for our product candidates and control only certain aspects of their activities. As a result, we have less direct control over the conduct, timing and completion of these clinical trials and the management of data developed through clinical trials than would be the case if we were relying entirely upon our own staff. Communicating with outside parties can also be challenging, potentially leading to mistakes as well as difficulties in coordinating activities. Outside parties may:
|
·
|
have staffing difficulties;
|
|
·
|
fail to comply with contractual obligations;
|
|
·
|
experience regulatory compliance issues;
|
|
·
|
undergo changes in priorities or become financially distressed; or
|
|
·
|
form relationships with other entities, some of which may be our competitors.
|
These factors may materially adversely affect the willingness or ability of third parties to conduct our clinical trials and may subject us to unexpected cost increases that are beyond our control. Nevertheless, we are responsible for ensuring that each of our clinical trials is conducted in accordance with the applicable protocol, legal, regulatory and scientific requirements and standards, and our reliance on CROs or the NIH does not relieve us of our regulatory responsibilities. We and our CROs and the NIH are required to comply with regulations and guidelines, including current Good Clinical Practices, or cGCPs, for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate, and that the trial patients are adequately informed of the potential risks of participating in clinical trials. These regulations are enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area and comparable foreign regulatory authorities for any products in clinical development. The FDA enforces cGCP regulations through periodic inspections of clinical trial sponsors, principal investigators and trial sites. If we or our CROs fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, the FDA will determine that any of our clinical trials comply with cGCPs. In addition, our clinical trials must be conducted with product candidates produced under cGMPs regulations and will require a large number of test patients. Our failure or the failure of our CROs to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process and could also subject us to enforcement action up to and including civil and criminal penalties.
Although we intend to design our clinical trials for our product candidates, we plan to have CROs, and in the case of our initial AV-101 Phase 2 study in MDD, the NIH, conduct all of the clinical trials. As a result, many important aspects of our drug development programs are outside of our direct control. In addition, the CROs or the NIH, as the case may be, may not perform all of their obligations under arrangements with us or in compliance with regulatory requirements, but we remain responsible and are subject to enforcement action that may include civil penalties up to and including criminal prosecution for any violations of FDA laws and regulations during the conduct of our clinical trials. If the NIH or CROs do not perform clinical trials in a satisfactory manner, breach their obligations to us or fail to comply with regulatory requirements, the development and commercialization of AV-101 and other product candidates may be delayed or our development program materially and irreversibly harmed. We cannot control the amount and timing of resources these CROs or the NIH devote to our program or our clinical products. If we are unable to rely on clinical data collected by our CROs or the NIH, we could be required to repeat, extend the duration of, or increase the size of our clinical trials and this could significantly delay commercialization and require significantly greater expenditures.
If any of our relationships with these third-party CROs or the NIH terminate, we may not be able to enter into arrangements with alternative CROs or collaborators. If CROs or the NIH do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, any clinical trials such CROs or the NIH are associated with may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. As a result, we believe that our financial results and the commercial prospects for our product candidates in the subject indication would be harmed, our costs could increase and our ability to generate revenue could be delayed.
We rely completely on third-party suppliers to manufacture our clinical drug supplies for our product candidates, and we intend to rely on third parties to produce non-clinical, clinical and commercial supplies of any future product candidate.
We do not currently have, nor do we plan to acquire, the infrastructure or capability to internally manufacture our clinical drug supply of AV-101 or any other product candidates, for use in the conduct of our non-clinical studies and clinical trials, and we lack the internal resources and the capability to manufacture any product candidates on a clinical or commercial scale. The facilities used by our contract manufacturers to manufacture the active pharmaceutical ingredient and final drug product must complete a pre-approval inspection by the FDA and other comparable foreign regulatory agencies to assess compliance with applicable requirements, including cGMPs, after we submit our NDA or relevant foreign regulatory submission to the applicable regulatory agency.
We do not control the manufacturing process of, and are completely dependent on, our contract manufacturers to comply with cGMPs for manufacture of both active drug substances and finished drug products. If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or applicable foreign regulatory agencies, they will not be able to secure and/or maintain regulatory approval for their manufacturing facilities. In addition, we have no direct control over our contract manufacturers’ ability to maintain adequate quality control, quality assurance and qualified personnel. Furthermore, all of our contract manufacturers are engaged with other companies to supply and/or manufacture materials or products for such companies, which exposes our third-party contract manufacturers to regulatory risks for the production of such materials and products. As a result, failure to satisfy the regulatory requirements for the production of those materials and products may affect the regulatory clearance of our contract manufacturers’ facilities generally. If the FDA or an applicable foreign regulatory agency determines now or in the future that these facilities for the manufacture of our product candidates are noncompliant, we may need to find alternative manufacturing facilities, which would adversely impact our ability to develop, obtain regulatory approval for or market our product candidates. Our reliance on contract manufacturers also exposes us to the possibility that they, or third parties with access to their facilities, will have access to and may appropriate our trade secrets or other proprietary information.
We do not have long-term supply agreements in place with our contract manufacturers and each batch of our product candidates is individually contracted under a quality and supply agreement. If we engage new contract manufacturers, such contractors must complete an inspection by the FDA and other applicable foreign regulatory agencies. We plan to continue to rely upon contract manufacturers and, potentially, collaboration partners, to manufacture commercial quantities AV-101 and other product candidates, if approved. Our current scale of manufacturing for AV-101 is adequate to support our currently planned needs for non-clinical studies and clinical trial supplies.
Even if we receive marketing approval for our product candidates in the United States, we may never receive regulatory approval to market our product candidates outside of the United States.
We have not yet selected any markets outside of the United States where we intend to seek regulatory approval to market our product candidates. In order to market any product outside of the United States, however, we must establish and comply with the numerous and varying safety, efficacy and other regulatory requirements of other countries. Approval procedures vary among countries and can involve additional product candidate testing and additional administrative review periods. The time required to obtain approvals in other countries might differ from that required to obtain FDA approval. The marketing approval processes in other countries may implicate all of the risks detailed above regarding FDA approval in the United States as well as other risks. In particular, in many countries outside of the United States, products must receive pricing and reimbursement approval before the product can be commercialized. Obtaining this approval can result in substantial delays in bringing products to market in such countries. Marketing approval in one country does not ensure marketing approval in another, but a failure or delay in obtaining marketing approval in one country may have a negative effect on the regulatory process in others. Failure to obtain marketing approval in other countries or any delay or other setback in obtaining such approval would impair our ability to market our product candidates in such foreign markets. Any such impairment would reduce the size of our potential market, which could have a material adverse impact on our business, results of operations and prospects.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell our product candidates, we may not be able to generate any revenue.
We do not currently have an infrastructure for the sales, marketing and distribution of pharmaceutical products. In order to market our product candidates, if approved by the FDA or any other regulatory body, we must build our sales, marketing, managerial and other non-technical capabilities or make arrangements with third parties to perform these services. If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, or if we are unable to do so on commercially reasonable terms, our business, results of operations, financial condition and prospects will be materially adversely affected.
Even if we receive marketing approval for our product candidates, our product candidates may not achieve broad market acceptance, which would limit the revenue that we generate from their sales.
The commercial success of our product candidates, if approved by the FDA or other applicable regulatory authorities, will depend upon the awareness and acceptance of our product candidates among the medical community, including physicians, patients and healthcare payors. Market acceptance of our product candidates, if approved, will depend on a number of factors, including, among others:
|
·
|
the efficacy of our product candidates as demonstrated in clinical trials, and, if required by any applicable regulatory authority in connection with the approval for the applicable indications, to provide patients with incremental health benefits, as compared with other available CNS therapies;
|
|
·
|
limitations or warnings contained in the labeling approved for our product candidates by the FDA or other applicable regulatory authorities;
|
|
·
|
the clinical indications for which our product candidates are approved;
|
|
·
|
availability of alternative treatments already approved or expected to be commercially launched in the near future;
|
|
·
|
the potential and perceived advantages of our product candidates over current treatment options or alternative treatments, including future alternative treatments;
|
|
·
|
the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies;
|
|
·
|
the strength of marketing and distribution support and timing of market introduction of competitive products;
|
|
·
|
publicity concerning our products or competing products and treatments;
|
|
·
|
pricing and cost effectiveness;
|
|
·
|
the effectiveness of our sales and marketing strategies;
|
|
·
|
our ability to increase awareness of our product candidates through marketing efforts;
|
|
·
|
our ability to obtain sufficient third-party coverage or reimbursement; or
|
|
·
|
the willingness of patients to pay out-of-pocket in the absence of third-party coverage.
|
If our product candidates are approved but do not achieve an adequate level of acceptance by patients, physicians and payors, we may not generate sufficient revenue from our product candidates to become or remain profitable. Before granting reimbursement approval, healthcare payors may require us to demonstrate that our product candidates, in addition to treating these target indications, also provide incremental health benefits to patients. Our efforts to educate the medical community and third-party payors about the benefits of our product candidates may require significant resources and may never be successful.
Our product candidates may cause undesirable side effects that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt non-clinical studies and clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other regulatory authorities.
Further, clinical trials by their nature utilize a sample of the potential patient population. With a limited number of patients and limited duration of exposure, rare and severe side effects of our product candidates may only be uncovered with a significantly larger number of patients exposed to the product candidate. If our product candidates receive marketing approval and we or others identify undesirable side effects caused by such product candidates (or any other similar products) after such approval, a number of potentially significant negative consequences could result, including:
|
·
|
regulatory authorities may withdraw or limit their approval of such product candidates;
|
|
·
|
regulatory authorities may require the addition of labeling statements, such as a “boxed” warning or a contraindication;
|
|
·
|
we may be required to change the way such product candidates are distributed or administered, conduct additional clinical trials or change the labeling of the product candidates;
|
|
·
|
we may be subject to regulatory investigations and government enforcement actions;
|
|
·
|
we may decide to remove such product candidates from the marketplace;
|
|
·
|
we could be sued and held liable for injury caused to individuals exposed to or taking our product candidates; and
|
|
·
|
our reputation may suffer.
|
We believe that any of these events could prevent us from achieving or maintaining market acceptance of the affected product candidates and could substantially increase the costs of commercializing our product candidates and significantly impact our ability to successfully commercialize our product candidates and generate revenues.
Even if we receive marketing approval for our product candidates, we may still face future development and regulatory difficulties.
Even if we receive marketing approval for our product candidates, regulatory authorities may still impose significant restrictions on our product candidates, indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies. Our product candidates will also be subject to ongoing FDA requirements governing the labeling, packaging, storage and promotion of the product and record keeping and submission of safety and other post-market information. The FDA has significant post-marketing authority, including, for example, the authority to require labeling changes based on new safety information and to require post-marketing studies or clinical trials to evaluate serious safety risks related to the use of a drug. The FDA also has the authority to require, as part of an NDA or post-approval, the submission of a REMS. Any REMS required by the FDA may lead to increased costs to assure compliance with new post-approval regulatory requirements and potential requirements or restrictions on the sale of approved products, all of which could lead to lower sales volume and revenue.
Manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMPs and other regulations. If we or a regulatory agency discover problems with our product candidates, such as adverse events of unanticipated severity or frequency, or problems with the facility where our product candidates are manufactured, a regulatory agency may impose restrictions on our product candidates, the manufacturer or us, including requiring withdrawal of our product candidates from the market or suspension of manufacturing. If we, our product candidates or the manufacturing facilities for our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may, among other things:
|
·
|
issue warning letters or untitled letters;
|
|
·
|
seek an injunction or impose civil or criminal penalties or monetary fines;
|
|
·
|
suspend or withdraw marketing approval;
|
|
·
|
suspend any ongoing clinical trials;
|
|
·
|
refuse to approve pending applications or supplements to applications submitted by us;
|
|
·
|
suspend or impose restrictions on operations, including costly new manufacturing requirements; or
|
|
·
|
seize or detain products, refuse to permit the import or export of products, or require that we initiate a product recall.
|
Competing therapies could emerge adversely affecting our opportunity to generate revenue from the sale of our product candidates.
The biopharmaceuticals industry is highly competitive. There are many public and private biopharmaceutical companies, universities, governmental agencies and other research organizations actively engaged in the research and development of products that may be similar to our product candidates or address similar markets. It is probable that the number of companies seeking to develop products and therapies similar to our products will increase.
Currently, management is unaware of any FDA-approved therapy for MDD with the mechanism of action of AV-101. However, products approved for other indications, for example, the anesthetic ketamine, are being or may be used off-label for treatment of MDD, as well as other CNS indications for which AV-101 may have therapeutic potential. Additionally, other treatment options, such psychotherapy and electroconvulsive therapy, or ECT, are sometimes used before or instead of antidepressant medications to treat patients with MDD.
In the field of new generation antidepressants focused on modulation of the NMDA receptor, our principal competitor is Naurex, Inc., which is developing GLYX-13 and NRX-1074 for treatment-resistant MDD. Although each of these drug candidates is a peptide and may not be orally active in MDD patients (GLYX-13 is only administered intravenously and, we believe, NRX-1074 has not yet been administered orally to MDD patients), both are new generation NMDA modulators focused, as is AV-101, on the glycine binding site of the NMDAR.
Many of our potential competitors, alone or with their strategic partners, have substantially greater financial, technical and human resources than we do and significantly greater experience in the discovery and development of product candidates, obtaining FDA and other regulatory approvals of treatments and the commercialization of those treatments. We believe that a range of pharmaceutical and biotechnology companies have programs to develop small molecule drug candidates for the treatment of depression, including MDD, epilepsy, neuropathic pain, Parkinson’s disease and other neurological conditions and diseases, including, but not limited to, Abbott Laboratories, Actavis, Astra Zeneca, Eli Lilly, GlaxoSmithKline, Johnson & Johnson, Lundbeck, Merck, Novartis, Otsuka, Pfizer, Roche, Sumitomo Dainippon, and Takeda. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market
We may seek to establish collaborations, and, if we are not able to establish them on commercially reasonable terms, we may have to alter our development and commercialization plans.
Our drug development programs and the potential commercialization of our product candidates will require substantial additional cash to fund expenses. For some of our product candidates, we may decide to collaborate with pharmaceutical and biotechnology companies for the development and potential commercialization of those product candidates.
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA or similar regulatory authorities outside the United States, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such collaboration could be more attractive than the one with us for our product candidate. The terms of any collaboration or other arrangements that we may establish may not be favorable to us.
We may also be restricted under existing collaboration agreements from entering into future agreements on certain terms with potential collaborators. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
We may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of the product candidate for which we are seeking to collaborate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidates or bring them to market and generate product revenue.
In addition, any future collaborations that we enter into may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborators generally have significant discretion in determining the efforts and resources that they will apply to these collaborations. Disagreements between parties to a collaboration arrangement regarding clinical development and commercialization matters can lead to delays in the development process or commercializing the applicable product candidate and, in some cases, termination of the collaboration arrangement. These disagreements can be difficult to resolve if neither of the parties has final decision-making authority. Collaborations with pharmaceutical or biotechnology companies and other third parties often are terminated or allowed to expire by the other party. Any such termination or expiration would adversely affect us financially and could harm our business reputation.
We may not be successful in our efforts to identify or discover additional product candidates or we may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
The success of our business depends primarily upon our ability to identify, develop and commercialize biopharmaceutical product candidates. Although AV-101 is in Phase 2 clinical development, our research programs, we may fail to identify other potential product candidates for clinical development for a number of reasons. Our research methodology may be unsuccessful in identifying potential product candidates or our potential product candidates may be shown to have harmful side effects or may have other characteristics that may make the products unmarketable or unlikely to receive marketing approval.
Because we have limited financial and management resources, we focus on a limited number of research programs and product candidates and are currently focused on AV-101 and stem cell technology-based drug rescue . As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial drugs or profitable market opportunities. Our spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable drugs. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through future collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate.
If any of these events occur, we may be forced to abandon our development efforts for a program or programs, which would have a material adverse effect on our business and could potentially cause us to cease operations. Research programs to identify new product candidates require substantial technical, financial and human resources. We may focus our efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful.
We are subject to healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
Although we do not currently have any products on the market, once we begin commercializing our products, we may be subject to additional healthcare statutory and regulatory requirements and enforcement by the federal government and the states and foreign governments in which we conduct our business. Healthcare providers, physicians and others will play a primary role in the recommendation and prescription of our product candidates, if approved. Our future arrangements with third-party payors will expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our product candidates, if we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations include the following:
|
·
|
The federal anti-kickback statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal healthcare programs such as Medicare and Medicaid.
|
|
·
|
The federal False Claims Act imposes criminal and civil penalties, including those from civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease, or conceal an obligation to pay money to the federal government.
|
|
·
|
The federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program and also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information.
|
|
·
|
The federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services.
|
|
·
|
The federal transparency requirements, sometimes referred to as the “Sunshine Act,” under the Patient Protection and Affordable Care Act, require manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid, or the Children’s Health Insurance Program to report to the Department of Health and Human Services information related to physician payments and other transfers of value and physician ownership and investment interests.
|
|
·
|
Analogous state laws and regulations, such as state anti-kickback and false claims laws and transparency laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers, and some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance.
|
|
·
|
guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures and drug pricing.
|
Ensuring that our future business arrangements with third parties comply with applicable healthcare laws and regulations could be costly. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations, including anticipated activities to be conducted by our sales team, were found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines and exclusion from government funded healthcare programs, such as Medicare and Medicaid, any of which could substantially disrupt our operations. If any of the physicians or other providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
The FDA and other regulatory agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. If we are found to have improperly promoted off-label uses, we may become subject to significant liability.
The FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products, such as AV-101, if approved. In particular, a product may not be promoted for uses that are not approved by the FDA or such other regulatory agencies as reflected in the product’s approved labeling. For example, if we receive marketing approval for AV0-101 as a treatment for MDD, physicians may nevertheless prescribe AV-101 to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed. If we cannot successfully manage the promotion of our product candidates, if approved, we could become subject to significant liability, which would materially adversely affect our business and financial condition.
Even if approved, reimbursement policies could limit our ability to sell our product candidates.
Market acceptance and sales of our product candidates will depend on reimbursement policies and may be affected by healthcare reform measures. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels for those medications. Cost containment is a primary concern in the U.S. healthcare industry and elsewhere. Government authorities and these third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. We cannot be sure that reimbursement will be available for our product candidates and, if reimbursement is available, the level of such reimbursement. Reimbursement may impact the demand for, or the price of, our product candidates. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize our product candidates.
In some foreign countries, particularly in Canada and European countries, the pricing of prescription pharmaceuticals is subject to strict governmental control. In these countries, pricing negotiations with governmental authorities can take six months or longer after the receipt of regulatory approval and product launch. To obtain favorable reimbursement for the indications sought or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidates with other available therapies. If reimbursement for our product candidates is unavailable in any country in which we seek reimbursement, if it is limited in scope or amount, if it is conditioned upon our completion of additional clinical trials, or if pricing is set at unsatisfactory levels, our operating results could be materially adversely affected.
Even if we have obtained orphan drug designation for one or more of our product candidates, there may be limits to the regulatory exclusivity afforded by such designation.
Even if we obtain orphan drug designation from the FDA for one or more of our product candidates, there are limitations to exclusivity afforded by such designation. In the United States, the company that first obtains FDA approval for a designated orphan drug for the specified rare disease or condition receives orphan drug marketing exclusivity for that drug for a period of seven years. This orphan drug exclusivity prevents the FDA from approving another application, including a full NDA to market the same drug for the same orphan indication, except in very limited circumstances, including when the FDA concludes that the later drug is safer, more effective or makes a major contribution to patient care. For purposes of small molecule drugs, the FDA defines “same drug” as a drug that contains the same active moiety and is intended for the same use as the drug in question. To obtain orphan drug exclusivity for a drug that shares the same active moiety as an already approved drug, it must be demonstrated to the FDA that the drug is safer or more effective than the approved orphan designated drug, or that it makes a major contribution to patient care. In addition, a designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. In addition, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the drug to meet the needs of patients with the rare disease or condition or if another drug with the same active moiety is determined to be safer, more effective, or represents a major contribution to patient care.
Our future growth may depend, in part, on our ability to penetrate foreign markets, where we would be subject to additional regulatory burdens and other risks and uncertainties.
Our future profitability may depend, in part, on our ability to commercialize our product candidates in foreign markets for which we may rely on collaboration with third parties. If we commercialize our product candidates in foreign markets, we would be subject to additional risks and uncertainties, including:
|
·
|
our customers’ ability to obtain reimbursement for our product candidates in foreign markets;
|
|
·
|
our inability to directly control commercial activities because we are relying on third parties;
|
|
·
|
the burden of complying with complex and changing foreign regulatory, tax, accounting and legal requirements;
|
|
·
|
different medical practices and customs in foreign countries affecting acceptance in the marketplace;
|
|
·
|
import or export licensing requirements;
|
|
·
|
longer accounts receivable collection times;
|
|
·
|
longer lead times for shipping;
|
|
·
|
language barriers for technical training;
|
|
·
|
reduced protection of intellectual property rights in some foreign countries;
|
|
·
|
the existence of additional potentially relevant third party intellectual property rights;
|
|
·
|
foreign currency exchange rate fluctuations; and
|
|
·
|
the interpretation of contractual provisions governed by foreign laws in the event of a contract dispute.
|
Foreign sales of our product candidates could also be adversely affected by the imposition of governmental controls, political and economic instability, trade restrictions and changes in tariffs.
We are a development stage biopharmaceutical company with no current revenues or approved products, and limited experience developing new drug, biological and/or regenerative medicine candidates, including conducting clinical trials and other areas required for the successful development and commercialization of therapeutic products, which makes it difficult to assess our future viability.
We are a development stage biopharmaceutical company. Although our lead drug candidate is in Phase 2 development, we currently have no approved products and generate no revenues, and we have not yet fully demonstrated an ability to overcome many of the fundamental risks and uncertainties frequently encountered by development stage companies in new and rapidly evolving fields of technology, particularly biotechnology. To execute our business plan successfully, we will need to accomplish the following fundamental objectives, either on our own or with strategic collaborators:
|
·
|
produce product candidates;
|
|
·
|
develop and obtain required regulatory approvals for commercialization of products we produce;
|
|
·
|
maintain, leverage and expand our intellectual property portfolio;
|
|
·
|
establish and maintain sales, distribution and marketing capabilities, and/or enter into strategic partnering arrangements to access such capabilities;
|
|
·
|
gain market acceptance for our products; and
|
|
·
|
obtain adequate capital resources and manage our spending as costs and expenses increase due to research, production, development, regulatory approval and commercialization of product candidates.
|
Our future success is highly dependent upon our ability to successfully develop and commercialize AV-101 and produce, develop and commercialize proprietary NCEs using our stem cell technology, human cells derived from stem cells, our customized human cell-based bioassay systems and medicinal chemistry, and we cannot provide any assurance that we will successfully develop and commercialize AV-101 or drug rescue NCEs , or that, if produced, AV-101 or any drug rescue NCE will be successfully commercialized.
Research programs designed to identify and produce drug rescue NCEs require substantial technical, financial and human resources, whether or not any NCEs are ultimately identified and produced. In particular, our drug rescue programs may initially show promise in identifying potential NCEs, yet fail to yield a lead NCE suitable for preclinical, clinical development or commercialization for many reasons, including the following:
|
·
|
our drug rescue research methodology may not be successful in identifying potential drug rescue NCEs;
|
|
·
|
competitors may develop alternatives that render our drug rescue NCEs obsolete;
|
|
·
|
a drug rescue NCE may, on further study, be shown to have harmful side effects or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory criteria;
|
|
·
|
a drug rescue NCE may not be capable of being produced in commercial quantities at an acceptable cost, or at all; or
|
|
·
|
a drug rescue NCE may not be accepted as safe and effective by regulatory authorities, patients, the medical community or third-party payors.
|
In addition, we do not have a sales or marketing infrastructure, and we, including our executive officers, do not have any significant pharmaceutical sales, marketing or distribution experience. We may seek to collaborate with others to develop and commercialize AV-101, drug rescue NCEs and/or other product candidates if and when they are developed. If we enter into arrangements with third parties to perform sales, marketing and distribution services for our products, the resulting revenues or the profitability from these revenues to us are likely to be lower than if we had sold, marketed and distributed our products ourselves. In addition, we may not be successful in entering into arrangements with third parties to sell, market and distribute AV-101, any drug rescue NCEs or other product candidates or may be unable to do so on terms that are favorable to us. We likely will have little control over such third parties, and any of these third parties may fail to devote the necessary resources and attention to sell, market and distribute our products effectively. If we do not establish sales, marketing and distribution capabilities successfully, in collaboration with third parties, we will not be successful in commercializing our product candidates.
We have limited operating history with respect to drug development, including our anticipated focus on the identification and assessment of potential drug rescue NCEs and no operating history with respect to the production of drug rescue NCEs, and we may never be able to produce a drug rescue NCE.
If we are unable to develop and commercialize AV-101 or produce suitable drug rescue NCEs for internal development or out-license to pharmaceutical companies and others, we may not be able to generate sufficient revenues to execute our business plan, which likely would result in significant harm to our financial position and results of operations, which could adversely impact our stock price.
There are a number of factors, in addition to the utility of CardioSafe 3D, that may impact our ability to identify and produce, develop or out-license and commercialize drug rescue NCEs, independently or with strategic partners, including:
|
·
|
our ability to identify potential drug rescue candidates in the public domain, obtain sufficient quantities of them, and assess them using our bioassay systems;
|
|
·
|
if we seek to rescue drug rescue candidates that are not available to us in the public domain, the extent to which third parties may be willing to out-license or sell certain drug rescue candidates to us on commercially reasonable terms;
|
|
·
|
our medicinal chemistry collaborator’s ability to design and produce proprietary drug rescue NCEs based on the novel biology and structure-function insight we provide using CardioSafe 3D or LiverSafe 3D; and
|
|
·
|
financial resources available to us to develop and commercialize lead drug rescue NCEs internally, or, if we out-license them to strategic partners, the resources such partners choose to dedicate to development and commercialization of any drug rescue NCEs they license from us.
|
Even if we do produce proprietary drug rescue NCEs, we can give no assurance that we will be able to develop and commercialize them as a marketable drug, on our own or in a strategic collaboration. Before we generate any revenues from AV-101 and/or additional drug rescue NCEs we or our potential strategic collaborator must complete preclinical and clinical developments, submit clinical and manufacturing data to the FDA, qualify a third party contract manufacturer, receive regulatory approval in one or more jurisdictions, satisfy the FDA that our contract manufacturer is capable of manufacturing the product in compliance with cGMP, build a commercial organization, make substantial investments and undertake significant marketing efforts ourselves or in partnership with others. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the FDA or comparable foreign regulatory authorities, and we may never receive such regulatory approval for any of our product candidates.
If CardioSafe 3D fails to predict accurately and efficiently the cardiac effects, both toxic and nontoxic, of drug rescue candidates and drug rescue NCEs, then our drug rescue programs will be adversely affected.
Our success is highly dependent on our ability to use CardioSafe 3D to identify and predict, accurately and efficiently, the potential toxic and nontoxic cardiac effects of drug rescue candidates and drug rescue NCEs. If CardioSafe 3D is not capable of providing physiologically relevant and clinically predictive information regarding human cardiac biology, our drug rescue business will be adversely affected.
We have not yet fully validated LiverSafe 3D for potential drug rescue applications, and we may never do so.
We have developed proprietary protocols for controlling the differentiation of human pluripotent stem cells and producing functional, mature, adult liver cells we believe are superior to primary (cadaver) hepatocytes used in in vitro testing. However, we have not yet fully validated our ability to use the human liver cells we produce for LiverSafe 3D to predict important biological effects, both toxic and nontoxic, of reference drugs, drug rescue candidates or drug rescue NCEs on the human liver, including drug-induced liver injury and adverse drug-drug interactions. Furthermore, we may never be able to do so, which could adversely affect our business and the potential applications of LiverSafe 3D for drug discovery, drug rescue and regenerative medicine.
CardioSafe 3D, and, if validated, LiverSafe 3D may not be meaningfully more predictive of the behavior of human cells than existing methods.
The success of our drug rescue business is highly dependent, in the first instance, upon CardioSafe 3D, and, in the second instance, if validated, LiverSafe 3D, being more accurate, efficient and clinically predictive than long-established surrogate safety models, including animal cells and live animals, and immortalized, primary and transformed cells, currently used by pharmaceutical companies and others. We cannot give assurance that CardioSafe 3D, and, when validated, LiverSafe 3D, will be more efficient or accurate at predicting the heart or liver safety of new drug candidates than the testing models currently used. If CardioSafe 3D and LiverSafe 3D fail to provide a meaningful difference compared to existing or new models in predicting the behavior of human heart and liver cells, respectively, their utility for drug rescue will be limited and our drug rescue business will be adversely affected.
We may invest in producing drug rescue NCEs for which there proves to be no demand.
To generate revenue from our drug rescue activities, we must produce proprietary drug rescue NCEs for which there proves to be demand within the healthcare marketplace, and, if we intend to out-license a particular drug rescue NCE for development and commercialization prior to market approval, then also among pharmaceutical companies and other potential strategic collaborators. However, we may produce drug rescue NCEs for which there proves to be no or limited demand in the healthcare market and/or among pharmaceutical companies and others. If we misinterpret market conditions, underestimate development costs and/or seek to rescue the wrong drug rescue candidates, we may fail to generate sufficient revenue or other value, on our own or in collaboration with others, to justify our investments, and our drug rescue business may be adversely affected.
We may experience difficulty in producing human cells and our future stem cell technology research and development efforts may not be successful within the timeline anticipated, if at all.
Our human pluripotent stem cell technology is technically complex, and the time and resources necessary to develop various human cell types and customized bioassay systems are difficult to predict in advance. We might decide to devote significant personnel and financial resources to research and development activities designed to expand, in the case of drug rescue, and explore, in the case of drug discovery and regenerative medicine, potential applications of our stem cell technology platform. In particular, we may conduct research and development programs related to producing and using functional, mature adult liver cells to validate LiverSafe 3D as a novel bioassay system for drug rescue, as well as exploratory nonclinical regenerative medicine programs involving blood, bone, cartilage, heart, and liver. Although we and our collaborators have developed proprietary protocols for the production of multiple differentiated cell types, we could encounter difficulties in differentiating particular cell types, even when following these proprietary protocols. These difficulties could result in delays in production of certain cells, assessment of certain drug rescue candidates and drug rescue NCEs, design and development of certain human cellular assays and performance of certain exploratory nonclinical regenerative medicine studies. In the past, our stem cell research and development projects have been significantly delayed when we encountered unanticipated difficulties in differentiating human pluripotent stem cells into heart and liver cells. Although we have overcome such difficulties in the past, we may have similar delays in the future, and we may not be able to overcome them or obtain any benefits from our future stem cell technology research and development activities. Any delay or failure by us, for example, to produce functional, mature blood, bone, cartilage, and liver cells could have a substantial and material adverse effect on our potential drug discovery, drug rescue and regenerative medicine business opportunities and results of operations.
Restrictions on research and development involving human embryonic stem cells and religious and political pressure regarding such stem cell research and development could impair our ability to conduct or sponsor certain potential collaborative research and development programs and adversely affect our prospects, the market price of our common stock and our business model.
Some of our ongoing and planned research and development programs involve the use of human cells derived from our controlled differentiation of human embryonic stem cells (“hESCs”). Some believe the use of hESCs gives rise to ethical and social issues regarding the appropriate use of these cells. Our research related to differentiation of hESCs may become the subject of adverse commentary or publicity, which could significantly harm the market price of our common stock. Although now substantially less than in years past, certain political and religious groups in the United States and elsewhere voice opposition to hESC technology and practices. We use hESCs derived from excess fertilized eggs that have been created for clinical use in in vitro fertilization (“IVF”) procedures and have been donated for research purposes with the informed consent of the donors after a successful IVF procedure because they are no longer desired or suitable for IVF. Certain academic research institutions have adopted policies regarding the ethical use of human embryonic tissue. These policies may have the effect of limiting the scope of future collaborative research opportunities with such institutions, thereby potentially impairing our ability to conduct certain research and development in this field that we believe is necessary to expand the drug rescue capabilities of our technology, which would have a material adverse effect on our business.
The use of embryonic or fetal tissue in research (including the derivation of hESCs) in other countries is regulated by the government, and varies widely from country to country. Government-imposed restrictions with respect to use of hESCs in research and development could have a material adverse effect on us by harming our ability to establish critical collaborations, delaying or preventing progress in our research and development, and causing a decrease in the market interest in our stock. These potential ethical concerns do not apply to induced pluripotent stem cells (iPSCs), or our plans to pursue studies involving human cells derived from iPSCs, because their derivation does not involve the use of embryonic tissues.
We have assumed that the biological capabilities of induced pluripotent stem cells (“iPSCs”) and hESCs are likely to be comparable. If it is discovered that this assumption is incorrect, our exploratory research and development activities focused on potential regenerative medicine applications of our stem cell technology platform could be harmed.
We use both hESCs and iPSCs to produce human cells for our customized in vitro assays for drug discovery and drug rescue purposes. However, we anticipate that our future exploratory research and development focused on potential regenerative medicine applications of our stem cell technology platform primarily will involve iPSCs. With respect to iPSCs, we believe scientists are still somewhat uncertain about the clinical utility, life span, and safety of such cells, and whether such cells differ in any clinically significant ways from hESCs. If we discover that iPSCs will not be useful for whatever reason for potential regenerative medicine programs, this would negatively affect our ability to explore expansion of our platform in that manner, including, in particular, where it would be preferable to use iPSCs to reproduce rather than approximate the effects of certain specific genetic variations.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain workers' compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions, which could have a material adverse effect on our operations.
To the extent our research and development activities involve using induced pluripotent stem cells, we will be subject to complex and evolving laws and regulations regarding privacy and informed consent. Many of these laws and regulations are subject to change and uncertain interpretation, and could result in claims, changes to our research and development programs and objectives, increased cost of operations or otherwise harm the Company.
To the extent that we pursue research and development activities involving iPSCs, we will be subject to a variety of laws and regulations in the United States and abroad that involve matters central to such research and development activities, including obligations to seek informed consent from donors for the use of their blood and other tissue to produce, or have produced for us, iPSCs, as well as state and federal laws that protect the privacy of such donors. United States federal and state and foreign laws and regulations are constantly evolving and can be subject to significant change. If we engage in iPSC-related research and development activities in countries other than the United States, we may become subject to foreign laws and regulations relating to human subjects research and other laws and regulations that are often more restrictive than those in the United States. In addition, both the application and interpretation of these laws and regulations are often uncertain, particularly in the rapidly evolving stem cell technology sector in which we operate. These laws and regulations can be costly to comply with and can delay or impede our research and development activities, result in negative publicity, increase our operating costs, require significant management time and attention and subject us to claims or other remedies, including fines or demands that we modify or cease existing business practices.
Legal, social and ethical concerns surrounding the use of iPSCs, biological materials and genetic information could impair our operations.
To the extent that our future stem cell research and development activities involve the use of iPSCs and the manipulation of human tissue and genetic information, the information we derive from such iPSC-related research and development activities could be used in a variety of applications, which may have underlying legal, social and ethical concerns, including the genetic engineering or modification of human cells, testing for genetic predisposition for certain medical conditions and stem cell banking. Governmental authorities could, for safety, social or other purposes, call for limits on or impose regulations on the use of iPSCs and genetic testing or the manufacture or use of certain biological materials involved in our iPSC-related research and development programs. Such concerns or governmental restrictions could limit our future research and development activities, which could have a material adverse effect on our business, financial condition and results of operations.
Our human cellular bioassay systems and human cells we derive from human pluripotent stem cells, although not currently subject to regulation by the FDA or other regulatory agencies as biological products or drugs, could become subject to regulation in the future.
The human cells we produce from HPSCs and our customized bioassay systems using such cells, including CardioSafe 3D and LiverSafe 3D, are not currently sold, for research purposes or any other purpose, to biotechnology or pharmaceutical companies, government research institutions, academic and nonprofit research institutions, medical research organizations or stem cell banks, and they are not therapeutic procedures. As a result, they are not subject to regulation as biological products or drugs by the FDA or comparable agencies in other countries. However, if, in the future, we seek to include human cells we derive from hPSCs in therapeutic applications or product candidates, such applications and/or product candidates would be subject to the FDA’s pre- and post-market regulations. For example, if we seek to develop and market human cells we produce for use in performing cell therapy or for other regenerative medicine applications, such as tissue engineering or organ replacement, we would first need to obtain FDA pre-market clearance or approval. Obtaining such clearance or approval from the FDA is expensive, time-consuming and uncertain, generally requiring many years to obtain, and requiring detailed and comprehensive scientific and clinical data. Notwithstanding the time and expense, these efforts may not result in FDA approval or clearance. Even if we were to obtain regulatory approval or clearance, it may not be for the uses that we believe are important or commercially attractive.
General Company-Related Risks
If we fail to attract and retain senior management and key scientific personnel, we may be unable to successfully produce, develop and commercialize AV-101, drug rescue NCEs, other potential product candidates and other commercial applications of our stem cell technology.
Our success depends in part on our continued ability to attract, retain and motivate highly qualified management and scientific and technical personnel. We are highly dependent upon our Chief Executive Officer, President and Chief Scientific Officer and Chief Financial Officer, as well as other employees, consultants and scientific collaborators. As of the date of this prospectus, we have nine full-time employees, which may make us more reliant on our individual employees than companies with a greater number of employees. The loss of services of any of these individuals could delay or prevent the successful development of AV-101, drug rescue NCEs, other product candidates, and other potential applications of our stem cell technology, including our production and assessment of potential drug recuse NCEs or disrupt our administrative functions.
Although we have not historically experienced unique difficulties attracting and retaining qualified employees, we could experience such problems in the future. For example, competition for qualified personnel in the biotechnology and pharmaceuticals field is intense. We will need to hire additional personnel as we expand our research and development and administrative activities. We may not be able to attract and retain quality personnel on acceptable terms.
In addition, we rely on a diverse range of consultants and advisors, including scientific and clinical development advisors, to assist us in designing our research and development strategies, including our AV-101 development and drug rescue strategies and plans. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us.
We may encounter difficulties in managing our growth and expanding our operations successfully.
As we seek to advance development of AV-101 for MDD and other CNS-related conditions, as well as cell production, bioassay development, drug discovery, drug rescue and development unrelated to AV-101, and stem cell technology-related regenerative medicine programs, we will need to expand our research and development capabilities or contract with third parties to provide these capabilities for us. As our operations expand, we expect that we will need to manage additional relationships with various strategic partners and other third parties. Future growth will impose significant added responsibilities on members of management. Our future financial performance and our ability to develop and commercialize our product candidates and to compete effectively will depend, in part, on our ability to manage any future growth effectively. To that end, we must be able to manage our research and development efforts effectively and hire, train and integrate additional management, administrative and technical personnel. The hiring, training and integration of new employees may be more difficult, costly and/or time-consuming for us because we have fewer resources than a larger organization. We may not be able to accomplish these tasks, and our failure to accomplish any of them could prevent us from successfully growing the company.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
If we develop AV-101, drug rescue NCEs, other product candidates, or regenerative medicine product candidates, either on our own or in collaboration with others, we will face an inherent risks of product liability as a result of the required clinical testing of such product candidates, and will face an even greater risk if we or our collaborators commercialize any such product candidates. For example, we may be sued if AV-101, any drug rescue NCE, other product candidate, or regenerative medicine product candidate we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability, and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
|
·
|
decreased demand for products that we may develop;
|
|
·
|
injury to our reputation;
|
|
·
|
withdrawal of clinical trial participants;
|
|
·
|
costs to defend the related litigation;
|
|
·
|
a diversion of management's time and our resources;
|
|
·
|
substantial monetary awards to trial participants or patients;
|
|
·
|
product recalls, withdrawals or labeling, marketing or promotional restrictions;
|
|
·
|
loss of revenue;
|
|
·
|
the inability to commercialize our product candidates; and
|
|
·
|
a decline in our stock price.
|
Our inability to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products we develop. Although we maintain liability insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
As we continue to grow, we will need to hire additional qualified accounting and financial personnel with appropriate public company experience.
As we continue to grow our organization and seek to obtain listing of our common stock on a national securities market, we will need to establish and maintain more elaborate disclosure and financial controls and make changes in our corporate governance practices. We will need to hire additional accounting and financial personnel with appropriate public company experience and technical accounting knowledge, and it may be difficult to recruit and retain such personnel. Even if we are able to hire appropriate personnel, our existing operating expenses and operations will increase by the direct costs of their employment and the indirect consequences related to the diversion of management resources from product development efforts.
Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets. The recent global financial crisis caused extreme volatility and disruptions in the capital and credit markets. A severe or prolonged economic downturn, such as the recent global financial crisis, could result in a variety of risks to our business, including, weakened demand for our product candidates and our ability to raise additional capital when needed on acceptable terms, if at all. A weak or declining economy could also strain our suppliers, possibly resulting in supply disruption, or cause our customers to delay making payments for our services. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate and financial market conditions could adversely impact our business.
We or the third parties upon whom we depend may be adversely affected by natural disasters and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
Natural disasters could severely disrupt our operations, and have a material adverse effect on our business, results of operations, financial condition and prospects. If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as the manufacturing facilities of our third-party CMOs, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans we have in place may prove inadequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which, could have a material adverse effect on our business.
Our internal computer systems, or those of our third-party CROs or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product candidates’ development programs.
Despite the implementation of security measures, our internal computer systems and those of our third-party CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any such system failure, accident, or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our programs. For example, the loss of clinical trial data for our product candidates could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications or other data or applications relating to our technology or product candidates, or inappropriate disclosure of confidential or proprietary information, we could incur liabilities and the further development of our product candidates could be delayed.
We may acquire businesses or products, or form strategic alliances, in the future, and we may not realize the benefits of such acquisitions.
We may acquire additional businesses or products, form strategic alliances or create joint ventures with third parties that we believe will complement or augment our existing business. If we acquire businesses with promising markets or technologies, we may not be able to realize the benefit of acquiring such businesses if we are unable to successfully integrate them with our existing operations and company culture. We may encounter numerous difficulties in developing, manufacturing and marketing any new products resulting from a strategic alliance or acquisition that delay or prevent us from realizing their expected benefits or enhancing our business. We cannot assure you that, following any such acquisition, we will achieve the expected synergies to justify the transaction.
Risks Related to Our Financial Position and Need for Capital
We have incurred significant net losses since inception and we will continue to incur substantial operating losses for the foreseeable future. We may never achieve or sustain profitability, which would depress the market price of our common stock, and could cause you to lose all or a part of your investment.
We have incurred significant net losses in each fiscal year since our inception in 1998, including net losses of $13.9 million and $3.0 million during the fiscal years ending March 31, 2015 and 2014, respectively. As of March 31, 2015, we had an accumulated deficit of $84.5 million. We do not know whether or when we will become profitable. Substantially all of our operating losses have resulted from costs incurred in connection with our research and development programs and from general and administrative costs associated with our operations. We expect to incur increasing levels of operating losses over the next several years and for the foreseeable future. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders’ deficit and working capital. We expect our research and development expenses to significantly increase in connection with non-clinical studies and clinical trials of our product candidates. In addition, if we obtain marketing approval for our product candidates, we will incur significant sales, marketing and outsourced-manufacturing expenses. As a public company, we incur additional costs associated with operating as a public company. As a result, we expect to continue to incur significant and increasing operating losses for the foreseeable future. Because of the numerous risks and uncertainties associated with developing pharmaceutical products, we are unable to predict the extent of any future losses or when we will become profitable, if at all. Even if we do become profitable, we may not be able to sustain or increase our profitability on a quarterly or annual basis.
Our ability to become profitable depends upon our ability to generate revenues. To date, although we have generated approximately $16.4 million in revenues, we have not commercialized any products or generated any revenues from product sales, and we do not know when, or if, we will generate any revenue from product sales. We do not expect to generate significant revenue unless and until we obtain marketing approval of, and begin to sell, AV-101, or we enter into one or more strategic development and commercialization agreements with respect to AV-101 or another product candidate. Our ability to generate revenue depends on a number of factors, including, but not limited to, our ability to:
|
·
|
initiate and successfully complete clinical trials that meet their clinical endpoints;
|
|
·
|
initiate and successfully complete all safety studies required to obtain U.S. and foreign marketing approval for our product candidates;
|
|
·
|
commercialize our product candidates, if approved, by developing a sales force or entering into collaborations with third parties; and
|
|
·
|
achieve market acceptance of our product candidates in the medical community and with third-party payors.
|
|
·
|
Absent our entering into a strategic development and commercialization collaboration or partnership agreement, we expect to incur significant sales and marketing costs as we prepare to commercialize our product candidates. Even if we initiate and successfully complete pivotal clinical trials of our product candidates, and our product candidates are approved for commercial sale, and despite expending these costs, our product candidates may not be a commercially successful drug. We may not achieve profitability soon after generating product sales, if ever. If we are unable to generate product revenue, we will not become profitable and may be unable to continue operations without continued funding.
|
Our independent auditors have expressed substantial doubt about our ability to continue as a going concern.
Our consolidated financial statements for the year ended March 31, 2015 included in this prospectus have been prepared assuming we will continue to operate as a going concern. However, due to our ongoing operating losses and our accumulated deficit, in their opinion on our audited financial statements for our fiscal year ended March 31, 2015, our auditors indicated that there is substantial doubt about our ability to continue as a going concern. Because we continue to experience net operating losses, our ability to continue as a going concern is subject to our ability to obtain necessary funding from outside sources, including obtaining additional funding from the sale of our securities or obtaining loans and grants from financial institutions and/or government agencies where possible. Our continued net operating losses increase the difficulty in completing such sales or securing alternative sources of funding, and there can be no assurances that we will be able to obtain such funding on favorable terms or at all. If we are unable to obtain sufficient financing from the sale of our securities or from alternative sources, we may be required to reduce, defer, or discontinue certain of our research and development activities or we may not be able to continue as a going concern.
We will require substantial additional financing to achieve our goals, and a failure to obtain this necessary capital when needed could force us to delay, limit, reduce or terminate our product development or commercialization efforts or other operations.
Since our inception, most of our resources have been dedicated to research and development of AV-101 and the drug rescue capabilities of our human pluripotent stem cell technology platform. In particular, we have expended substantial resources advancing AV-101 through preclinical development and Phase 1 clinical safety studies, and developing CardioSafe 3D and LiverSafe 3D for drug rescue applications, and we will continue to expend substantial resources for the foreseeable future developing and commercializing AV-101, validating LiverSafe 3D, and developing drug rescue NCEs. These expenditures will include costs associated with general and administrative costs, facilities costs, research and development, acquiring new technologies, manufacturing product candidates, conducting preclinical experiments and clinical trials and obtaining regulatory approvals, as well as commercializing any products approved for sale.
At March 31, 2015, our existing cash and cash equivalents were not sufficient to fund our current operations for the next 12 months. Although, in February 2015, we entered into a Cooperative Research and Development Agreement (“CRADA”) with the U.S. National Institutes of Health (“NIH”), under which CRADA the NIH will fully fund and conduct the initial Phase 2 clinical efficacy and safety of AV-101 in Major Depressive Disorder at no cost to us, we have no current source of revenue to sustain our present activities, and we do not expect to generate revenue until, and unless, we (i) out-license or sell AV-101, a drug rescue NCE, another drug candidate unrelated to AV-101 to third-parties, (ii) enter into license arrangements involving our stem cell technology, including customized drug discovery and predictive toxicology assays and services and/or regenerative medicine opportunities with a third party, or (iii) obtain approval from the FDA or other regulatory authorities and successfully commercialize, on our own or through a future collaboration, one or more of our compounds. As the outcome of our proposed drug rescue and AV-101 development activities and future anticipated clinical trials is highly uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of our product candidates, on our own or in collaboration with others. In addition, other unanticipated costs may arise. As a result of these and other factors, we will need to seek additional capital in the near term to meet our future operating requirements, including capital necessary to obtain regulatory approval for, and to commercialize, our product candidates, and may seek additional capital in the event there exists favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. We are considering a range of potential sources of funding, including public or private equity or debt financings, government or other third-party funding, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements or a combination of these approaches, and we intend to complete additional financing arrangements prior to the end of 2015. Raising funds in the current economic environment may present additional challenges. Even if we believe we have sufficient funds for our current or future operating plans, we may seek additional capital if market conditions are favorable or if we have specific strategic considerations.
Our future capital requirements depend on many factors, including:
|
·
|
the number and characteristics of the product candidates we pursue, including AV-101 or drug rescue NCEs;
|
|
·
|
the scope, progress, results and costs of researching and developing our product candidates, and conducting preclinical and clinical studies;
|
|
·
|
the timing of, and the costs involved in, obtaining regulatory approvals for our product candidates;
|
|
·
|
the cost of commercialization activities if any of our product candidates are approved for sale, including marketing, sales and distribution costs;
|
|
·
|
the cost of manufacturing our product candidates and any products we successfully commercialize;
|
|
·
|
our ability to establish and maintain strategic partnerships, licensing or other arrangements and the financial terms of such agreements;
|
|
·
|
market acceptance of our products;
|
|
·
|
the effect of competing technological and market developments;
|
|
·
|
our ability to obtain government funding for our programs;
|
|
·
|
the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims necessary to preserve our freedom to operate in the stem cell industry, including litigation costs associated with any claims that we infringe third-party patents or violate other intellectual property rights and the outcome of such litigation;
|
|
·
|
the timing, receipt and amount of potential future licensee fees, milestone payments, and sales of, or royalties on, our future products, if any; and
|
|
·
|
the extent to which we acquire or invest in businesses, products and technologies, although we currently have no commitments or agreements relating to any of these types of transactions.
|
Any additional fundraising efforts will divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates. In addition, we cannot guarantee that future financing will be available in sufficient amounts, in a timely manner, or on terms acceptable to us, if at all, and the terms of any financing may adversely affect the holdings or the rights of our stockholders and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our shares to decline. The sale of additional equity or convertible securities would also dilute all of our stockholders. The incurrence of indebtedness could result in increased fixed payment obligations and we could be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. We could also be required to seek funds through arrangements with collaborative partners or otherwise at an earlier stage than otherwise would be desirable and we may be required to relinquish rights to some of our technologies or product candidate or otherwise agree to terms unfavorable to us, any of which may have a material adverse effect on our business, operating results and prospects.
If we are unable to obtain funding on a timely basis and on acceptable terms, we may be required to significantly curtail, delay or discontinue one or more of our research or product development programs or the commercialization of any product candidate or be unable to continue or expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition and results of operations.
Raising additional capital will cause dilution to our existing stockholders, and may restrict our operations or require us to relinquish rights.
We intend to seek additional capital through a combination of private and public equity offerings, debt financings, collaborations and strategic and licensing arrangements. To the extent that we raise additional capital through the sale of common stock or securities convertible or exchangeable into common stock, your ownership interest in our company will be diluted. In addition, the terms of any such securities may include liquidation or other preferences that materially adversely affect your rights as a stockholder. Debt financing, if available, would increase our fixed payment obligations and may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through collaboration, strategic partnerships and licensing arrangements with third parties, we may have to relinquish valuable rights to our product candidates, our intellectual property, future revenue streams or grant licenses on terms that are not favorable to us.
Some of our programs have been partially supported by government grants, which may not be available to us in the future.
Since inception, we have received substantial funds under grant award programs funded by state and federal governmental agencies, such as the NIH, the NIH’s National Institute of Neurological Disease and Stroke and the California Institute for Regenerative Medicine. To fund a portion of our future research and development programs, we may apply for additional grant funding from such or similar governmental organizations. However, funding by these governmental organizations may be significantly reduced or eliminated in the future for a number of reasons. For example, some programs are subject to a yearly appropriations process in Congress. In addition, we may not receive funds under future grants because of budgeting constraints of the agency administering the program. Therefore, we cannot assure you that we will receive any future grant funding from any government organization or otherwise. A restriction on the government funding available to us could reduce the resources that we would be able to devote to future research and development efforts. Such a reduction could delay the introduction of new products and hurt our competitive position.
Our ability to use net operating losses to offset future taxable income is subject to certain limitations.
As of March 31, 2015, we had federal and state net operating loss carryforwards of $58.7 million and $53.1 million, respectively, which begin to expire in fiscal 2016. Under Section 382 of the Internal Revenue Code of 1986, as amended, or the Code, changes in our ownership may limit the amount of our net operating loss carryforwards that could be utilized annually to offset our future taxable income, if any. This limitation would generally apply in the event of a cumulative change in ownership of our company of more than 50% within a three-year period. Any such limitation may significantly reduce our ability to utilize our net operating loss carryforwards and tax credit carryforwards before they expire. Any such limitation, whether as the result of future offerings, prior private placements, sales of our common stock by our existing stockholders or additional sales of our common stock by us in the future, could have a material adverse effect on our results of operations in future years. We have not completed a study to assess whether an ownership change for purposes of Section 382 has occurred, or whether there have been multiple ownership changes since our inception, due to the significant costs and complexities associated with such study.
Risks Related to Our Intellectual Property Rights
If we are unable to adequately protect our proprietary technology, or obtain and maintain issued patents that are sufficient to protect our product candidates, others could compete against us more directly, which would have a material adverse impact on our business, results of operations, financial condition and prospects.
We strive to protect and enhance the proprietary technologies that we believe are important to our business, including seeking patents intended to cover our products and compositions, their methods of use and any other inventions we consider are important to the development of our business. We also rely on trade secrets to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection.
Our success will depend significantly on our ability to obtain and maintain patent and other proprietary protection for commercially important technology, inventions and know-how related to our business, defend and enforce our patents, should they issue, preserve the confidentiality of our trade secrets and operate without infringing the valid and enforceable patents and proprietary rights of third parties. We also rely on know-how, continuing technological innovation and in-licensing opportunities to develop, strengthen and maintain the proprietary position of our product candidates. Our owned and licensed patents and patent applications relate to AV-101 and, in general, human pluripotent stem cell technology.
We currently have no issued patents covering AV-101. We cannot provide any assurances that any of our multiple pending U.S. and foreign patent applications relating to AV-101 will mature into issued patents and, if they do, that such patents will include, claims with a scope sufficient to protect AV-101 or otherwise provide any competitive advantage. Moreover, other parties may have developed technologies that may be related or competitive to our approach, and may have filed or may file patent applications and may have received or may receive patents that may overlap or conflict with our patent applications, either by claiming the same methods or formulations or by claiming subject matter that could dominate our patent position. Such third-party patent positions may limit or even eliminate our ability to obtain patent protection for certain inventories.
The patent positions of biotechnology and pharmaceutical companies, including our patent position, involve complex legal and factual questions, and, therefore, the issuance, scope, validity and enforceability of any patent claims that we may obtain cannot be predicted with certainty. Patents, if issued, may be challenged, deemed unenforceable, invalidated, or circumvented. U.S. patents and patent applications may also be subject to interference proceedings, ex parte reexamination, or inter partes review proceedings, supplemental examination and challenges in district court. Patents may be subjected to opposition, post-grant review, or comparable proceedings lodged in various foreign, both national and regional, patent offices. These proceedings could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such proceedings may be costly. Thus, any patents, should they issue, that we may own or exclusively license may not provide any protection against competitors. Furthermore, an adverse decision in an interference proceeding can result in a third party receiving the patent right sought by us, which in turn could affect our ability to develop, market or otherwise commercialize our product candidates.
Furthermore, though a patent, if it were to issue, is presumed valid and enforceable, its issuance is not conclusive as to its validity or its enforceability and it may not provide us with adequate proprietary protection or competitive advantages against competitors with similar products. Even if a patent issues and is held to be valid and enforceable, competitors may be able to design around our patents, such as using pre-existing or newly developed technology. Other parties may develop and obtain patent protection for more effective technologies, designs or methods. We may not be able to prevent the unauthorized disclosure or use of our technical knowledge or trade secrets by consultants, vendors, former employees and current employees. The laws of some foreign countries do not protect our proprietary rights to the same extent as the laws of the United States, and we may encounter significant problems in protecting our proprietary rights in these countries. If these developments were to occur, they could have a material adverse effect on our sales.
Our ability to enforce our patent rights depends on our ability to detect infringement. It is difficult to detect infringers who do not advertise the components that are used in their products. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product. Any litigation to enforce or defend our patent rights, even if we were to prevail, could be costly and time-consuming and would divert the attention of our management and key personnel from our business operations. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded if we were to prevail may not be commercially meaningful.
In addition, proceedings to enforce or defend our patents, if and when issued, could put our patents at risk of being invalidated, held unenforceable, or interpreted narrowly. Such proceedings could also provoke third parties to assert claims against us, including that some or all of the claims in one or more of our patents are invalid or otherwise unenforceable. If any of our patents, if and when issued, covering our product candidates are invalidated or found unenforceable, our financial position and results of operations would be materially and adversely impacted. In addition, if a court found that valid, enforceable patents held by third parties covered our product candidates, our financial position and results of operations would also be materially and adversely impacted.
The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
|
·
|
any of our AV-101 or other pending patent applications, if issued, will include claims having a scope sufficient to protect AV-101 or any other products or product candidates;
|
|
·
|
any of our pending patent applications will issue as patents at all;
|
|
·
|
we will be able to successfully commercialize our product candidates, if approved, before our relevant patents expire;
|
|
·
|
we were the first to make the inventions covered by each of our patents and pending patent applications;
|
|
·
|
we were the first to file patent applications for these inventions;
|
|
·
|
others will not develop similar or alternative technologies that do not infringe our patents;
|
|
·
|
others will not use pre-existing technology to effectively compete against us;
|
|
·
|
any of our patents, if issued, will be found to ultimately be valid and enforceable;
|
|
·
|
any patents issued to us will provide a basis for an exclusive market for our commercially viable products, will provide us with any competitive advantages or will not be challenged by third parties;
|
|
·
|
we will develop additional proprietary technologies or product candidates that are separately patentable; or
|
|
·
|
that our commercial activities or products will not infringe upon the patents or proprietary rights of others.
|
We rely upon unpatented trade secrets, unpatented know-how and continuing technological innovation to develop and maintain our competitive position, which we seek to protect, in part, by confidentiality agreements with our employees and our collaborators and consultants. It is possible that technology relevant to our business will be independently developed by a person that is not a party to such an agreement. Furthermore, if the employees and consultants who are parties to these agreements breach or violate the terms of these agreements, we may not have adequate remedies for any such breach or violation, and we could lose our trade secrets through such breaches or violations. Further, our trade secrets could otherwise become known or be independently discovered by our competitors.
We may infringe the intellectual property rights of others, which may prevent or delay our product development efforts and stop us from commercializing or increase the costs of commercializing our product candidates, if approved.
Our success will depend in part on our ability to operate without infringing the intellectual property and proprietary rights of third parties. We cannot assure you that our business, products and methods do not or will not infringe the patents or other intellectual property rights of third parties.
The pharmaceutical industry is characterized by extensive litigation regarding patents and other intellectual property rights. Other parties may allege that our product candidates or the use of our technologies infringes patent claims or other intellectual property rights held by them or that we are employing their proprietary technology without authorization. As we continue to develop and, if approved, commercialize our current product candidates and future product candidates, competitors may claim that our technology infringes their intellectual property rights as part of business strategies designed to impede our successful commercialization. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our product candidates. Because patent applications can take many years to issue, third parties may have currently pending patent applications which may later result in issued patents that our product candidates may infringe, or which such third parties claim are infringed by our technologies. The outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, we would need to demonstrate that our product candidates, products or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid, and we may not be able to do this. Even if we are successful in these proceedings, we may incur substantial costs and the time and attention of our management and scientific personnel could be diverted in pursuing these proceedings, which could have a material adverse effect on us. In addition, we may not have sufficient resources to bring these actions to a successful conclusion.
Patent and other types of intellectual property litigation can involve complex factual and legal questions, and their outcome is uncertain. Any claim relating to intellectual property infringement that is successfully asserted against us may require us to pay substantial damages, including treble damages and attorney’s fees if we are found to be willfully infringing another party’s patents, for past use of the asserted intellectual property and royalties and other consideration going forward if we are forced to take a license. In addition, if any such claim were successfully asserted against us and we could not obtain such a license, we may be forced to stop or delay developing, manufacturing, selling or otherwise commercializing our product candidates.
Even if we are successful in these proceedings, we may incur substantial costs and divert management time and attention in pursuing these proceedings, which could have a material adverse effect on us. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, defend an infringement action or challenge the validity of the patents in court, or redesign our products. Patent litigation is costly and time-consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, intellectual property litigation or claims could force us to do one or more of the following:
|
·
|
cease developing, selling or otherwise commercializing our product candidates;
|
|
·
|
pay substantial damages for past use of the asserted intellectual property;
|
|
·
|
obtain a license from the holder of the asserted intellectual property, which license may not be available on reasonable terms, if at all; and
|
|
·
|
in the case of trademark claims, redesign, or rename, some or all of our product candidates to avoid infringing the intellectual property rights of third parties, which may not be possible and, even if possible, could be costly and time-consuming.
|
Any of these risks coming to fruition could have a material adverse effect on our business, results of operations, financial condition and prospects.
We may be subject to claims challenging the inventorship or ownership of our patents and other intellectual property.
We enter into confidentiality and intellectual property assignment agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors. These agreements generally provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us. For example, even if we have a consulting agreement in place with an academic advisor pursuant to which such academic advisor is required to assign any inventions developed in connection with providing services to us, such academic advisor may not have the right to assign such inventions to us, as it may conflict with his or her obligations to assign all such intellectual property to his or her employing institution.
Litigation may be necessary to defend against these and other claims challenging inventorship or ownership. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The U.S. Patent and Trademark Office, or U.S. PTO, and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case.
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time-consuming and unsuccessful.
Even if the patent applications we own or license are issued, competitors may infringe these patents. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours or our licensors is not valid, is unenforceable and/or is not infringed, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Interference proceedings provoked by third parties or brought by us may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our licensors. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our defense of litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common stock.
Issued patents covering our product candidates could be found invalid or unenforceable if challenged in court.
If we or one of our licensing partners initiated legal proceedings against a third party to enforce a patent, if and when issued, covering one of our product candidates, the defendant could counterclaim that the patent covering our product candidate is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge include alleged failures to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for unenforceability assertions include allegations that someone connected with prosecution of the patent withheld relevant information from the U.S. PTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before administrative bodies in the U.S. or abroad, even outside the context of litigation. Such mechanisms include re-examination, post grant review and equivalent proceedings in foreign jurisdictions, e.g., opposition proceedings. Such proceedings could result in revocation or amendment of our patents in such a way that they no longer cover our product candidates or competitive products. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to validity, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our product candidates. Such a loss of patent protection would have a material adverse impact on our business.
We will not seek to protect our intellectual property rights in all jurisdictions throughout the world and we may not be able to adequately enforce our intellectual property rights even in the jurisdictions where we seek protection.
Filing, prosecuting and defending patents on product candidates in all countries and jurisdictions throughout the world is prohibitively expensive, and our intellectual property rights in some countries outside the United States could be less extensive than those in the United States, assuming that rights are obtained in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. The statutory deadlines for pursuing patent protection in individual foreign jurisdictions are based on the priority date of each of our patent applications. For the patent applications relating to AV-101, as well as for many of the patent families that we own or license, the relevant statutory deadlines have not yet expired. Thus, for each of the patent families that we believe provide coverage for our lead product candidates or technologies, we will need to decide whether and where to pursue protection outside the United States.
Competitors may use our technologies in jurisdictions where we do not pursue and obtain patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing. Even if we pursue and obtain issued patents in particular jurisdictions, our patent claims or other intellectual property rights may not be effective or sufficient to prevent third parties from so competing.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to biotechnology. This could make it difficult for us to stop the infringement of our patents, if obtained, or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries.
Furthermore, proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
We are dependent, in part, on licensed intellectual property. If we were to lose our rights to licensed intellectual property, we may not be able to continue developing or commercializing our product candidates, if approved. If we breach any of the agreements under which we license the use, development and commercialization rights to our product candidates or technology from third parties or, in certain cases, we fail to meet certain development or payment deadlines, we could lose license rights that are important to our business.
We are a party to a number of license agreements under which we are granted rights to intellectual property that are or could become important to our business, and we expect that we may need to enter into additional license agreements in the future. Our existing license agreements impose, and we expect that future license agreements will impose on us, various development, regulatory and/or commercial diligence obligations, payment of fees, milestones and/or royalties and other obligations. If we fail to comply with our obligations under these agreements, or we are subject to a bankruptcy, the licensor may have the right to terminate the license, in which event we would not be able to develop or market products which could be covered by the license. Our business could suffer, for example, if any current or future licenses terminate, if the licensors fail to abide by the terms of the license, if the licensed patents or other rights are found to be invalid or unenforceable, or if we are unable to enter into necessary licenses on acceptable terms. See “Business—Licenses” for a description of our license agreements, which includes a description of the termination provisions of these agreements.
As we have done previously, we may need to obtain licenses from third parties to advance our research or allow commercialization of our product candidates, and we cannot provide any assurances that third-party patents do not exist that might be enforced against our current product candidates or future products in the absence of such a license. We may fail to obtain any of these licenses on commercially reasonable terms, if at all. Even if we are able to obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. In that event, we may be required to expend significant time and resources to develop or license replacement technology. If we are unable to do so, we may be unable to develop or commercialize the affected product candidates, which could materially harm our business and the third parties owning such intellectual property rights could seek either an injunction prohibiting our sales, or, with respect to our sales, an obligation on our part to pay royalties and/or other forms of compensation.
Licensing of intellectual property is of critical importance to our business and involves complex legal, business and scientific issues. Disputes may arise between us and our licensors regarding intellectual property subject to a license agreement, including:
|
·
|
the scope of rights granted under the license agreement and other interpretation-related issues;
|
|
·
|
whether and the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement;
|
|
·
|
our right to sublicense patent and other rights to third parties under collaborative development relationships;
|
|
·
|
our diligence obligations with respect to the use of the licensed technology in relation to our development and commercialization of our product candidates, and what activities satisfy those diligence obligations; and
|
|
·
|
the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners.
|
If disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates.
We have entered into several licenses to support our various programs. We may enter into additional license(s) to third-party intellectual property that are necessary or useful to our business. Our current licenses and any future licenses that we may enter into impose various royalty payment, milestone, and other obligations on us. For example, the licensor may retain control over patent prosecution and maintenance under a license agreement, in which case, we may not be able to adequately influence patent prosecution or prevent inadvertent lapses of coverage due to failure to pay maintenance fees. If we fail to comply with any of our obligations under a current or future license agreement, our licensor(s) may allege that we have breached our license agreement and may accordingly seek to terminate our license with them. In addition, future licensor(s) may decide to terminate our license at will. Termination of any of our current or future licenses could result in our loss of the right to use the licensed intellectual property, which could materially adversely affect our ability to develop and commercialize a product candidate or product, if approved, as well as harm our competitive business position and our business prospects.
In addition, if our licensors fail to abide by the terms of the license, if the licensors fail to prevent infringement by third parties, if the licensed patents or other rights are found to be invalid or unenforceable, or if we are unable to enter into necessary licenses on acceptable terms our business could suffer.
Some intellectual property which we have licensed may have been discovered through government funded programs and thus may be subject to federal regulations such as “march-in” rights, certain reporting requirements, and a preference for U.S. industry. Compliance with such regulations may limit our exclusive rights, subject us to expenditure of resources with respect to reporting requirements, and limit our ability to contract with non-U.S. manufacturers.
Some of the intellectual property rights we have licensed or license in the future may have been generated through the use of U.S. government funding and may therefore be subject to certain federal regulations. As a result, the U.S. government may have certain rights to intellectual property embodied in our current or future product candidates pursuant to the Bayh-Dole Act of 1980, or Bayh-Dole Act. These U.S. government rights in certain inventions developed under a government-funded program include a non-exclusive, non-transferable, irrevocable worldwide license to use inventions for any governmental purpose. In addition, the U.S. government has the right to require us to grant exclusive, partially exclusive, or non-exclusive licenses to any of these inventions to a third party if it determines that: (i) adequate steps have not been taken to commercialize the invention; (ii) government action is necessary to meet public health or safety needs; or (iii) government action is necessary to meet requirements for public use under federal regulations (also referred to as “march-in rights”). The U.S. government also has the right to take title to these inventions if we fail, or the applicable licensor fails, to disclose the invention to the government and fail to file an application to register the intellectual property within specified time limits. In addition, the U.S. government may acquire title to these inventions in any country in which a patent application is not filed within specified time limits. Intellectual property generated under a government funded program is also subject to certain reporting requirements, compliance with which may require us, or the applicable licensor, to expend substantial resources. In addition, the U.S. government requires that any products embodying the subject invention or produced through the use of the subject invention be manufactured substantially in the U.S. The manufacturing preference requirement can be waived if the owner of the intellectual property can show that reasonable but unsuccessful efforts have been made to grant licenses on similar terms to potential licensees that would be likely to manufacture substantially in the U.S. or that under the circumstances domestic manufacture is not commercially feasible. This preference for U.S. manufacturers may limit our ability to contract with non-U.S. product manufacturers for products covered by such intellectual property.
In the event we apply for additional U.S. government funding, and we discover compounds or drug candidates as a result of such funding, intellectual property rights to such discoveries may be subject to the applicable provisions of the Bayh-Dole Act.
If we do not obtain additional protection under the Hatch-Waxman Amendments and similar foreign legislation by extending the patent terms and obtaining data exclusivity for our product candidates, our business may be materially harmed.
Depending upon the timing, duration and specifics of FDA marketing approval of our product candidates, one or more of the U.S. patents we own or license may be eligible for limited patent term restoration under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, we may not be granted an extension because of, for example, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. For example, we may not be granted an extension if the active ingredient of AV-101 is used in another drug company’s product candidate and that product candidate is the first to obtain FDA approval. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extension or restoration or the term of any such extension is less than we request, our competitors may obtain approval of competing products following our patent expiration, and our ability to generate revenues could be materially adversely affected.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other biotechnology companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biotechnology industry involve both technological and legal complexity, and is therefore costly, time-consuming and inherently uncertain. In addition, the United States has recently enacted and is currently implementing wide-ranging patent reform legislation: the Leahy-Smith America Invents Act, referred to as the America Invents Act. The America Invents Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. It is not yet clear what, if any, impact the America Invents Act will have on the operation of our business. However, the America Invents Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of any patents that may issue from our patent applications, all of which could have a material adverse effect on our business and financial condition.
In addition, recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. The full impact of these decisions is not yet known. For example, on March 20, 2012 in Mayo Collaborative Services, DBA Mayo Medical Laboratories, et al. v. Prometheus Laboratories, Inc., the Court held that several claims drawn to measuring drug metabolite levels from patient samples and correlating them to drug doses were not patentable subject matter. The decision appears to impact diagnostics patents that merely apply a law of nature via a series of routine steps and it has created uncertainty around the ability to obtain patent protection for certain inventions. Additionally, on June 13, 2013 in Association for Molecular Pathology v. Myriad Genetics, Inc., the Court held that claims to isolated genomic DNA are not patentable, but claims to complementary DNA molecules are patent eligible because they are not a natural product. The effect of the decision on patents for other isolated natural products is uncertain. However, on March 4, 2014, the U.S. PTO issued a memorandum to patent examiners providing guidance for examining claims that recite laws of nature, natural phenomena or natural products under the Myriad and Prometheus decisions. This guidance did not limit the application of Myriad to DNA but, rather, applied the decision to other natural products.
In addition to increasing uncertainty with regard to our ability to obtain future patents, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on these and other decisions by the U.S. Congress, the federal courts and the U.S. PTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce any patents that may issue in the future.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Certain of our current employees have been, and certain of our future employees may have been, previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We also engage advisors and consultants who are concurrently employed at universities or who perform services for other entities.
Although we are not aware of any claims currently pending or threatened against us, we may be subject to claims that we or our employees, advisors or consultants have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third party. We have and may in the future also be subject to claims that an employee, advisor or consultant performed work for us that conflicts with that person’s obligations to a third party, such as an employer, and thus, that the third party has an ownership interest in the intellectual property arising out of work performed for us. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. If we fail in defending such claims, in addition to paying monetary claims, we may lose valuable intellectual property rights or personnel. A loss of key personnel or their work product could hamper or prevent our ability to commercialize our product candidates, which would materially adversely affect our commercial development efforts.
Numerous factors may limit any potential competitive advantage provided by our intellectual property rights.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, provide a barrier to entry against our competitors or potential competitors, or permit us to maintain our competitive advantage. Moreover, if a third party has intellectual property rights that cover the practice of our technology, we may not be able to fully exercise or extract value from our intellectual property rights. The following examples are illustrative:
|
·
|
others may be able to develop and/or practice technology that is similar to our technology or aspects of our technology but that is not covered by the claims of patents, should such patents issue from our patent applications;
|
|
·
|
we might not have been the first to make the inventions covered by a pending patent application that we own;
|
|
·
|
we might not have been the first to file patent applications covering an invention;
|
|
·
|
others may independently develop similar or alternative technologies without infringing our intellectual property rights;
|
|
·
|
pending patent applications that we own or license may not lead to issued patents;
|
|
·
|
patents, if issued, that we own or license may not provide us with any competitive advantages, or may be held invalid or unenforceable, as a result of legal challenges by our competitors;
|
|
·
|
third parties may compete with us in jurisdictions where we do not pursue and obtain patent protection;
|
|
·
|
we may not be able to obtain and/or maintain necessary or useful licenses on reasonable terms or at all;
|
|
·
|
the patents of others may have an adverse effect on our business.
|
Should any of these events occur, they could significantly harm our business and results of operations.
If we seek to leverage prior discovery and development of drug rescue candidates under in-license arrangements with academic laboratories, biotechnology companies, the NIH, pharmaceutical companies or other third parties, it is uncertain what ownership rights, if any, we will obtain over intellectual property we derive from such licenses to drug rescue NCEs we may produce or develop in connection with any such third-party licenses.
If, instead of identifying drug rescue candidates based on information available to us in the public domain, we seek to in-license drug rescue candidates from biotechnology, medicinal chemistry and pharmaceutical companies, academic, governmental and nonprofit research institutions, including the NIH, or other third-parties, there can be no assurances that we will obtain material ownership or economic participation rights over intellectual property we may derive from such licenses or similar rights to the drug rescue NCEs we may produce and develop. If we are unable to obtain ownership or substantial economic participation rights over intellectual property related to drug rescue NCEs we produce and develop, our business may be adversely affected.
Risks Related to our Common Stock
There is no assurance that an active, liquid and orderly trading market will develop for our common stock or what the market price of our common stock will be and, as a result, it may be difficult for you to sell your shares of our common stock.
Since we became a publicly-traded company in May 2011, there has been a limited public market for shares of our common stock on the OTCQB Marketplace (“OTCQB”). We do not yet meet the initial listing standards of the New York Stock Exchange, the NASDAQ Capital Market, or other similar national securities exchanges. Until our common stock is listed on a broader exchange, we anticipate that it will remain quoted on the OTC Markets. In that venue, investors may find it difficult to obtain accurate quotations as to the market value of our common stock. In addition, if we fail to meet the criteria set forth in SEC regulations, various requirements would be imposed by law on broker-dealers who sell our securities to persons other than established customers and accredited investors. Consequently, such regulations may deter broker-dealers from recommending or selling our common stock, which may further affect liquidity. This could also make it more difficult to raise additional capital.
We cannot predict the extent to which investor interest in our company will lead to the development of a more active trading market on the OTC Markets, whether we will meet the initial listing standards of the New York Stock Exchange, the NASDAQ Capital Market, or other similar national securities exchanges, or how liquid that market might become. If an active trading market does not develop, you may have difficulty selling any of the shares of our common stock that you buy.
Market volatility may affect our stock price and the value of your investment.
The market price for our common stock, similar to other biopharmaceutical companies, is likely to be volatile. The market price of our common stock may fluctuate significantly in response to a number of factors, most of which we cannot control, including, among others:
|
·
|
plans for, progress of or results from non-clinical studies and clinical trials of our product candidates;
|
|
·
|
the failure of the FDA to approve our product candidates;
|
|
·
|
announcements of new products, technologies, commercial relationships, acquisitions or other events by us or our competitors;
|
|
·
|
the success or failure of other CNS therapies;
|
|
·
|
regulatory or legal developments in the United States and other countries;
|
|
·
|
failure of our product candidates, if approved, to achieve commercial success;
|
|
·
|
fluctuations in stock market prices and trading volumes of similar companies;
|
|
·
|
general market conditions and overall fluctuations in U.S. equity markets;
|
|
·
|
variations in our quarterly operating results;
|
|
·
|
changes in our financial guidance or securities analysts’ estimates of our financial performance;
|
|
·
|
changes in accounting principles;
|
|
·
|
our ability to raise additional capital and the terms on which we can raise it;
|
|
·
|
sales of large blocks of our common stock, including sales by our executive officers, directors and significant stockholders;
|
|
·
|
additions or departures of key personnel;
|
|
·
|
discussion of us or our stock price by the press and by online investor communities; and
|
|
·
|
other risks and uncertainties described in these risk factors.
|
Future sales of our common stock may cause our stock price to decline.
Sales of a substantial number of shares of our common stock in the public market or the perception that these sales might occur could significantly reduce the market price of our common stock and impair our ability to raise adequate capital through the sale of additional equity securities.
The stock market in general, and biotechnology-based companies like ours in particular, has from time to time experienced volatility in the market prices for securities that often has been unrelated to the operating performance of the underlying companies. These broad market and industry fluctuations may adversely affect the market price of our common stock, regardless of our operating performance. In certain recent situations in which the market price of a stock has been volatile, holders of that stock have instituted securities class action litigation against such company that issued the stock. If any of our stockholders were to bring a lawsuit against us, the defense and disposition of the lawsuit could be costly and divert the time and attention of our management and harm our operating results. Additionally, if the trading volume of our common stock remains low and limited there will be an increased level of volatility and you may not be able to generate a return on your investment.
A significant portion of our total outstanding shares are restricted from immediate resale but may be sold into the market in the near future. Future sales of shares by existing stockholders could cause our stock price to decline, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market could occur at any time. These sales, or the perception in the market that the holders of a large number of shares intend to sell shares, could reduce the market price of our common stock. Prior to this date of this prospectus, there has been a limited public market for shares of our common stock on the OTC Markets. Future sales of a substantial number of shares of our common stock in the public market, including shares issued upon the exchange of our Series A Preferred Stock, and Series B Preferred Stock under this prospectus, and exercise of outstanding options and warrants for common stock, including shares of common stock under this prospectus which are issuable upon exercise of warrants, in the public market, or the perception that these sales might, occur, could significantly reduce the market price for our common stock and impair our ability to raise adequate capital through the sale of equity securities.
Our principal institutional stockholders may continue to have substantial control over us and could limit your ability to influence the outcome of key transactions, including changes in control.
Certain of our current institutional stockholders and their respective affiliates own a substantial portion of our outstanding capital stock, including our common stock, Series A Preferred Stock and Series B 10% Convertible Preferred Stock, which preferred stock is convertible into a substantial number of shares of common stock. Accordingly, these stockholders may exert significant influence over us and over the outcome of any corporate actions requiring approval of holders of our common stock, including the election of directors and amendments to our organizational documents, such as increases in our authorized shares of common stock, any merger, consolidation or sale of all or substantially all of our assets or any other significant corporate transactions. These stockholders may also delay or prevent a change of control of us, even if such a change of control would benefit our other stockholders. The significant concentration of stock ownership may adversely affect the trading price of our common stock due to investors’ perception that conflicts of interest may exist or arise. Furthermore, the interests of our principal institutional stockholders may not always coincide with your interests or the interests of other stockholders may act in a manner that advances its best interests and not necessarily those of other stockholders, including seeking a premium value for its common stock, which might affect the prevailing market price for our common stock.
If equity research analysts do not publish research or reports about our business or if they issue unfavorable commentary or downgrade our common stock, the price of our common stock could decline.
The trading market for our common stock relies in part on the research and reports that equity research analysts publish about us and our business. We do not control these analysts. The price of our common stock could decline if one or more equity research analysts downgrade our common stock or if analysts issue other unfavorable commentary or cease publishing reports about us or our business.
There may be additional issuances of shares of preferred stock in the future.
Following approval by our stockholders in October 2011, our Articles of Incorporation permit us to issue up to 10.0 million shares of preferred stock. Our Board of Directors has authorized the issuance of both 500,000 shares of Series A Preferred, all of which shares are currently issued and outstanding, and 4.0 million shares of Series B 10% Convertible Preferred stock, of which approximately 3.1 million shares are issued and outstanding. Our Board of Directors could authorize the issuance of additional series of preferred stock in the future and such preferred stock could grant holders preferred rights to our assets upon liquidation, the right to receive dividends before dividends would be declared to holders of our common stock, and the right to the redemption of such shares, possibly together with a premium, prior to the redemption of the common stock. In the event and to the extent that we do issue additional preferred stock in the future, the rights of holders of our common stock could be impaired thereby, including without limitation, with respect to liquidation.
We do not intend to pay dividends on our common stock and, consequently, your ability to achieve a return on your investment will depend on appreciation in the price of our common stock.
We have never declared or paid any cash dividend on our common stock and do not currently intend to do so in the foreseeable future. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends in the foreseeable future. Therefore, the success of an investment in shares of our common stock will depend upon any future appreciation in their value. There is no guarantee that shares of our common stock will appreciate in value or even maintain the price at which you purchased them.
USE OF PROCEEDS
We will not receive any of the proceeds of the shares of common stock offered and resold by the Selling Stockholders under this prospectus. However, we may receive proceeds upon exercise of Warrants (discussed below). The shares of common stock that will be resold under this prospectus are issuable upon the conversion of certain securities issued by us during the Private Placements, or upon exercise of Warrants. The funds that may be received by us upon exercise of Warrants, estimated to be up to approximately $5.8 million, if all Warrants are exercised, will be used for general working capital purposes.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus includes forward-looking statements that relate to future events or our future financial performance and involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to differ materially from any future results, levels of activity, performance or achievements expressed or implied by these forward-looking statements. Words such as, but not limited to, “believe,” “expect,” “anticipate,” “estimate,” “intend,” “plan,” “targets,” “likely,” “will,” “would,” “could,” and similar expressions or phrases identify forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
|
·
|
our ability to implement our business strategy;
|
|
·
|
anticipated trends and challenges in our business and the markets in which we operate;
|
|
·
|
our expected future financial performance;
|
|
·
|
our expectations regarding our operating expenses;
|
|
·
|
our ability to anticipate market needs or develop new or enhanced products to meet those needs;
|
|
·
|
our expectations regarding market acceptance of our products;
|
|
·
|
our ability to compete in our industry and innovation by our competitors;
|
|
·
|
our ability to protect our confidential information and intellectual property rights;
|
|
·
|
our ability to obtain additional financing; and
|
|
·
|
our ability to manage growth.
|
All forward-looking statements involve risks, assumptions and uncertainties. The occurrence of the events described, and the achievement of the expected results, depend on many events, some or all of which are not predictable or within our control. Actual results may differ materially from expected results. See the section titled “Risk Factors” and elsewhere in this prospectus for a more complete discussion of these risks, assumptions and uncertainties and for other risks and uncertainties. These risks, assumptions and uncertainties are not necessarily all of the important factors that could cause actual results to differ materially from those expressed in any of our forward-looking statements. Other unknown or unpredictable factors also could harm our results. In light of these risks, uncertainties and assumptions, the forward-looking events discussed in this prospectus might not occur.
Readers are cautioned not to place undue reliance on forward-looking statements, as there can be no assurance that the plans, intentions or expectations upon which they are based will occur. By their nature, forward-looking statements involve numerous assumptions, known and unknown risks and uncertainties, both general and specific, that contribute to the possibility that the predictions, forecasts, projections and other things contemplated by the forward-looking statements will not occur. Forward-looking statements in this prospectus are based on management’s beliefs and opinions at the time the statements are made. The forward-looking statements contained in this prospectus are expressly qualified in their entirety by this cautionary statement. The forward-looking statements included in this prospectus are made as of the date of this prospectus and we undertake no obligation to publicly update or revise any forward-looking statements to reflect new information, future events or otherwise, except as required by applicable securities laws.
BUSINESS
Company Overview
We are a clinical-stage biopharmaceutical company committed to developing and commercializing innovative product candidates for patients with depression, other diseases and various disorders related to the central nervous system (“CNS”), as well as cancer.
More than one billion people worldwide suffer from CNS disorders. Recently, the economic burden of these disorders was estimated at $2.0 trillion in the U.S. and European Union alone, a figure that is expected to triple by 2030. The World Health Organization estimates that 350 million people worldwide are affected by depression. According to the U.S. National Institutes of Health (“NIH”), major depression is one of the most common mental disorders in the U.S.. In 2012, the NIH estimated 16 million adults aged 18 or older in the U.S. had at least one major depressive episode. This represented 6.9 percent of all U.S. adults.
Our lead product candidate, AV-101, is an orally available small molecule prodrug in Phase 2 clinical development for Major Depressive Disorder (“MDD”). AV-101’s mechanism of action (“MOA”), as an N-methyl-D-aspartate receptor (“NMDAR”) antagonist binding selectively at the glycine-binding (“GlyB”) co-agonist site of the NMDAR, is fundamentally different from all antidepressants currently approved by the U.S. Food and Drug Administration (“FDA”). In four preclinical studies utilizing well-validated animal models of depression, AV-101 was shown to induce fast-acting, dose-dependent, persistent and statistically significant antidepressant-like responses, following a single treatment, which was equivalent to responses seen with a control single sub-anesthetic dose of ketamine (sometimes used by clinicians off-label to treat suicidal behavior). In the same studies, fluoxetine (Prozac) did not induce rapid onset antidepressant-like responses. Preclinical studies also support the hypothesis that AV-101 has potential to treat several additional CNS disorders, including chronic neuropathic pain, epilepsy and neurodegenerative diseases, such as Parkinson’s disease and Huntington’s disease where modulation of the NMDAR may have therapeutic benefit.
Following two successful randomized, double-blind, placebo-controlled Phase 1 safety studies funded by the NIH, in February 2015, we entered into a Cooperative Research and Development Agreement (“CRADA”) with the U.S. National Institute of Mental Health (“NIMH”), part of the NIH. Under the CRADA, we will collaborate with the NIMH on the initial Phase 2 clinical study of AV-101 in subjects with treatment-resistant MDD. Pursuant to the CRADA, the study will be conducted at the NIMH and be fully funded by the NIMH. It is contemplated that this clinical study will begin in Fall 2015 under the direction of Dr. Carlos Zarate, Jr., the NIMH’s Chief of Experimental Therapeutics & Pathophysiology Branch and of the Section on Neurobiology and Treatment of Mood and Anxiety Disorders.
In addition to developing AV-101 for MDD and other CNS indications, we apply our stem cell technology for drug rescue programs intended to identify and develop proprietary new chemical entities (“NCEs”) for our internal drug candidate pipeline. Drug rescue involves (1) using our customized in vitro bioassay systems to predict potential heart and liver toxicity of NCEs, (2) leveraging prior investments by pharmaceutical companies and others related to screening large-scale compound libraries, optimizing and testing for efficacy NCEs that were terminated before FDA approval due to heart or liver toxicity and are now available in the public domain, and (3) applying modern medicinal chemistry to produce safer NCEs for our internal development pipeline. Our CardioSafe 3D™ bioassay system uses our human pluripotent stem cell (“hPSC”)-derived cardiomyocytes, or human heart cells. We believe CardioSafe 3D is more comprehensive and clinically predictive than the hERG assay, which is currently the only in vitro cardiac safety assay required by FDA guidelines. We use our hPSC-derived hepatocytes, or human liver cells, in our LiverSafe 3D™ bioassay system to predict potential liver toxicity of NCEs, including potential drug metabolism issues and adverse drug-drug interactions. CardioSafe 3D and LiverSafe 3D offer a new paradigm for evaluating and predicting potential heart and liver toxicity of NCEs, including drug rescue NCEs, early in the development process, long before costly, high risk animal studies and human clinical trials. We intend to develop internally for our pipeline each lead drug rescue NCEs we produce.
Our Strategy
Our strategy is to develop, and commercialize innovative small molecule drugs that address unmet medical needs related to CNS disorders and cancer. We have assembled a management team and a team of scientific, clinical, and regulatory advisors, including recognized experts in the fields of depression and other CNS disorders, with significant industry experience to lead the development and commercialization of our product opportunities. Key elements of our strategy are to:
|
·
|
Develop and commercialize our lead product candidate, AV-101, for depression, including MDD. We are pursuing MDD as our lead indication for AV-101. We are preparing to launch our initial MDD Phase 2 clinical study in collaboration with the NIH in the second half of 2015. We intend to develop AV-101 internally, through Phase 3 clinical studies and submission of our NDA. If approved by the FDA, we plan to commercialize AV-101 for this indication in the U.S. either by (A) establishing or contracting for a specialty U.S. sales force focused primarily on psychiatrists and long-term care physicians who are high prescribers of currently-approved antidepressants or (B) collaborating with a pharmaceutical company with an extensive presence in U.S. CNS markets. Outside the U.S., we may choose to commercialize AV-101 in selected markets by establishing one or more strategic alliances.
|
|
·
|
Leverage the commercial potential AV-101 by expanding to additional CNS-related disorders. We intend to pursue the development and commercialization of AV-101 in MDD and additional CNS-related indications that are underserved by currently available medicines and represent large unmet medical needs. Based on AV-101 preclinical studies, and by leveraging our AV-101 IND and successful Phase 1 clinical studies, we now have the opportunity to explore Phase 2 development of AV-101 as a potential treatment for chronic neuropathic pain, epilepsy and neurodegenerative diseases such as Parkinson’s disease and Huntington’s disease.
|
|
·
|
Grow our internal development pipeline by pursuing drug rescue opportunities using our stem cell technology. We are using our stem cell technology to screen and develop proprietary new chemical entities (“NCEs”) through drug rescue programs intended to produce proprietary NCEs for our internal drug development pipeline. We will focus on NCEs with established therapeutic and commercial potential. Our ability to build on that valuable head start with our biological and electrophysiological insights regarding cardiac and liver safety effects of NCEs obtained using CardioSafe 3D and, eventually, LiverSafe 3D, we believe will help us produce and then optimize patentable drug rescue NCEs for our internal pipeline without incurring many of the substantial costs and risks typically inherent in new drug discovery and nonclinical drug development.
|
|
·
|
Pursue other product candidates, including product candidates for treatment of CNS-related disorders. While our resources are currently focused on developing AV-101 and producing drug rescue NCEs, we may pursue additional product candidates in the future. These may be directed at CNS-related disorders and may be developed independently or in partnerships. We believe that a diversified portfolio will mitigate risks inherent in drug development and increase the likelihood of our success.
|
Our Product Opportunities
AV-101 (L-4-cholorkyurenine or 4-Cl-KYN)
Overview and Mechanism of Action
AV-101 is an orally available, clinical-stage prodrug candidate that readily gains access to the CNS after systemic administration and is rapidly converted in vivo to its active metabolite 7-chlorokynurenic acid (“7-Cl-KYNA”), a well-characterized, potent and highly selective antagonist of then N-methyl-D-aspartate receptor (“NMDAR”) at the glycine-binding co-agonist (“GlyB”) site.
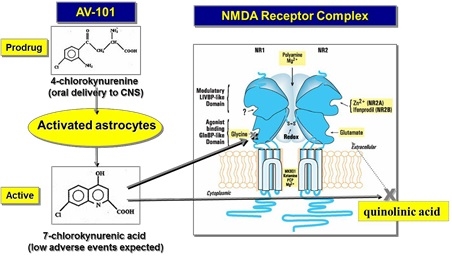
Current evidence suggests that AV-101’s antagonism of NMDAR signaling may provide fast-acting antidepressant effects in the treatment of Major Depressive Disorder (“MDD”). In addition, as confirmed in our AV-101 Phase 1 clinical studies, targeting the GlyB site of the NMDAR does not have the adverse effects typically associated with classic NMDAR antagonists, such as ketamine, and other NMDA channel blockers.
We believe Phase 2 clinical development of AV-101 for MDD and multiple CNS-related indications is supported by strong scientific rationale, significant IND-enabling nonclinical data, and two successful Phase 1 clinical safety studies. To date, the U.S. National Institutes of Health (“NIH”) has awarded us $8.8 million of grant funding for our pre-Phase 2 development of AV-101. We are currently preparing to launch our initial Phase 2 clinical trial of AV-101 in MDD. This Phase 2 study will be fully funded by the NIH.
Major Depressive Disorder
Depression is a serious medical illness and a global public health concern. The World Health Organization estimates that depression is the leading cause of disability worldwide, and is a major contributor to the global burden of disease, affecting 350 million people globally. According to the U.S. Centers for Disease Control and Prevention, approximately one in every 10 Americans aged 12 and over takes antidepressant medication.
While most people will experience depressed mood at some point during their lifetime, MDD is different. MDD is the chronic, pervasive feeling of utter unhappiness and suffering, which impairs daily functioning. Symptoms of MDD include diminished pleasure in activities, changes in appetite that result in weight changes, insomnia or oversleeping, psychomotor agitation, loss of energy or increased fatigue, feelings of worthlessness or inappropriate guilt, difficulty thinking, concentrating or making decisions, and thoughts of death or suicide and attempts at suicide. Suicide is estimated to be the cause of death in up to 15% of individuals with MDD.
Current Antidepressants
For many people, depression cannot be controlled for any length of time without treatment. Current medications available in the multi-billion dollar global antidepressant market, including commonly-prescribed selective serotonin reuptake inhibitors (“SSRIs”) and serotonin-norepinephrine reuptake inhibitors (“SNRIs”), have limited effectiveness, and, because of their mechanism of action, must be taken for several weeks or months before patients experience any significant benefit. In addition, most current antidepressant medications have an FDA-required “Black Box” safety warning due to a risk of worsening depression and an increased risk of suicidal thoughts and behaviors during treatment, a property not expected to occur with AV-101. Only about 33% of depression sufferers benefit from their initial treatment with current antidepressants, and the likelihood of achieving remission of depressive symptoms declines with each successive treatment attempt. Even after multiple treatment attempts, about 33% of depression sufferers still fail to find an effective therapy. In addition, this trial and error process and the systemic effects of the various antidepressant medications involved, increases the risks of patient tolerability issues and serious side effects, including suicidal thoughts and behaviors.
Ketamine and NIH Clinical Studies in Major Depressive Disorder
Ketamine hydrochloride (“ketamine”) is an FDA-approved, rapid-acting general anesthetic. The use of ketamine (an NMDA receptor antagonist which acts as an NMDA channel blocker) to treat MDD has been studied in several clinical trials conducted by depression experts at the U.S. National Institute of Mental Health (“NIMH”), part of the NIH, including Dr. Carlos Zarate, Jr., the NIMH’s Chief of Experimental Therapeutics & Pathophysiology Branch and of the Section on Neurobiology and Treatment of Mood and Anxiety Disorders. In randomized, placebo-controlled, double-blind clinical trials reported by Dr. Zarate and others at the NIMH, a single intravenous dose of ketamine (0.5 mg/kg over 40 minutes) produced robust and rapid antidepressant effects in MDD patients who had not responded to currently-approved medications. These results were in contrast to the slow onset of currently FDA-approved antidepressant medications, which usually require many weeks or months of chronic usage to achieve similar antidepressant effects. The potential for widespread therapeutic use of ketamine, and FDA Schedule III drug, is limited by its potential for abuse, dissociative and psychosis-like side effects, and by practical challenges associated with its intravenous administration in a medical center. Notwithstanding these limitations, the discovery of ketamine’s fast-acting antidepressant effects revolutionized thinking about the MDD treatment paradigm. The discovery also increased interest in the development of a new generation of antidepressants with a fast-acting mechanism of action similar to ketamine’s. Our orally available AV-101 is among the new generation of antidepressants with potential to deliver ketamine-like antidepressant effects, without ketamine’s side effects or required intravenous administration.
AV-101 and Major Depressive Disorder
AV-101 is an orally available prodrug candidate that produces, in the brain, 7 Cl KYNA, one of the most potent and selective antagonists of the GlyB site of the NMDAR, resulting in the down-regulation of NMDAR signaling. Growing evidence suggests that the glutamatergic system is central to the neurobiology and treatment of MDD and other mood disorders.
AV-101’s mechanism of action is fundamentally different from currently available antidepressants, placing it among a new generation of glutamatergic antidepressants with potential to treat millions of MDD sufferers worldwide who are poorly served by SSRIs, SNRIs and other current depression therapies. AV-101 is functionally similar to ketamine in that both are NMDAR antagonists. However, AV-101 modulates (down-regulated) NMDAR channel activity and ketamine blocks it. AV-101 accomplishes this NMDAR modulation by selectively binding to the functionally required GlyB site of the NMDAR, thus down-regulating the NMDAR in a dose-dependent manner. We believe targeting the GlyB site of the NMDAR and modulating NMDAR activity rather than blocking it can bypass adverse effects that result when ketamine blocks the NMDA ion channel, without affecting the robust efficacy observed in previous clinical studies conducted by the NIH and others using ketamine to treat MDD. This NMDAR modulation by AV-101 may then result in the “glutamate surge,” and the increase in neuronal connections, that has been associated with the fast-acting antidepressant effects of ketamine.
In preclinical studies, AV-101 has demonstrated the antidepressant-like activity of ketamine, including rapid onset and long duration of effect, without ketamine’s serious side effects. In two NIH-funded randomized, double-blind, placebo-controlled Phase 1 safety studies, AV-101 was safe, well-tolerated and not associated with any severe adverse events. There were no signs of sedation, hallucinations or schizophrenia-like side effects often associated with ketamine and traditional NMDAR channel blockers.
Building on over $8.8 million of prior non-dilutive funding from the NIH for preclinical and Phase 1 clinical development of AV-101, in February 2015, we entered a Cooperative Research and Development Agreement (“CRADA”) with the NIMH. Under the CRADA, we will collaborate with Dr. Carlos Zarate and the NIMH on a Phase 2 clinical study of AV-101 in subjects with treatment-resistant MDD. Pursuant to the CRADA, this study will be conducted at the NIMH by Dr. Zarate and fully-funded by the NIH. The primary objective of the NIH-sponsored Phase 2 study will be to evaluate the ability of AV-101 to improve overall depressive symptomatology in subjects with MDD, specifically whether subjects with MDD have a greater and more rapid decrease in depressive symptoms when treated with AV-101 than with placebo. We currently anticipate commencement of the study in the second half of 2015.
AV-101 Nonclinical Studies in Chronic Neuropathic Pain, Epilepsy, and Parkinson’s and Huntington’s diseases
In addition to well-established nonclinical models of depression, AV-101 nonclinical data in other CNS-related disorders support our hypothesis that it may have therapeutic and commercial potential beyond treatment of depression.
Chronic Neuropathic Pain and Acute Tissue Injury Hyperalgesia
The effect of AV-101 on chronic neuropathic pain due to inflammation and nerve damage was assessed in rats by using the Chung nerve ligation model. AV-101 effects were compared to either saline, MK-801 or gabapentin controls. Similarly to what was observed in the formalin and thermal hyperalgesia test systems, AV-101 had a positive effect on chronic neuropathic pain in the Chung model, with no observed adverse behavioral effects. The efficacy observed for AV-101 in both the acute and chronic neuropathic pain model systems was dose dependent, and the drug response was not associated with any side effects within the range of doses administered.
The antihyperalgesic effect of AV-101 has been evaluated in two standard tissue injury model systems: inflammatory thermal hyperalgesia and the formalin paw test. AV-101 was compared to two positive controls, the classic NMDAR antagonist MK-801 (discontinued in preclinical development by Merck due to neurotoxicity) and the anticonvulsant gabapentin. A significant drug response was defined as a response that was greater than or equal to 2 standard deviations (“SD”) from the response produced by vehicle. Animal behavior and motor function were observed and evaluated throughout the study.
In the formalin hyperalgesia model, MK-801 caused significant spontaneous locomotor activity that prevented assessment of its analgesic activity. However, AV-101 displayed dose-dependent antihyperpathic effects in the absence of behavioral deficits for both Phase 1 (acute nociceptive pain) and Phase 2 (chronic and neuropathic pain) of hyperalgesia. In contrast, gabapentin did not have a significant anti-hyperalgesia response at any dose during Phase 1, but showed a significant positive response during Phase 2.
For the carrageenan inflammatory thermal hyperalgesia model, neither MK-801, gabapentin, nor AV-101 had an effect on acute thermal nociception, but produced a dose dependent block of the carrageenan-induced hyperalgesia that were greater than 2 SD of the vehicle: There were no behavioral changes observed at any AV-101 dose, but signs of behavioral and motor dysfunction were observed for gabapentin and MK-801 treated animals. The profile of analgesic activity observed for AV-101 in the formalin and inflammatory thermal hyperalgesia model systems supports the conclusion that AV-101 demonstrates anti-hyperalgesia activity in validated models of facilitated pain processing produced by peripheral tissue inflammation.
Epilepsy
AV-101 has been shown to protect against seizures and neuronal damage in animal models of epilepsy, providing preclinical support for its potential as a novel treatment of epilepsy. Epilepsy is one of the most prevalent neurological disorders, affecting almost 1% of the worldwide population. Approximately 2.5 million Americans have epilepsy. Nearly half of the people suffering from epilepsy are not effectively treated with currently available medications. In addition, the anticonvulsants used today can cause significant side effects, which frequently interfere with compliance.
Glutamate is a neurotransmitter that is critically involved in the pathophysiology of epilepsy. Through its stimulation of the NMDAR subtype, glutamate has been implicated in the neuropathology and clinical symptoms of the disease. In support of this, NMDAR antagonists are potent anticonvulsants. However, classic NMDAR antagonists are limited by adverse effects, such as neurotoxicity, declining mental status, and the onset of psychotic symptoms following administration of the drug. The endogenous amino acid glycine modulates glutamatergic neurotransmission by stimulating the GlyB co-agonist site of the NMDA receptor. GlyB site antagonists inhibit NMDAR function and are therefore anticonvulsant and neuroprotective. Importantly, GlyB site antagonists have fewer and less severe side effects than classic NMDAR antagonists and other antiepileptic agents, making them a safer potential alternative to, and one expected to be associated with greater patient compliance than, available anticonvulsant medications.
AV-101 has two additional therapeutically important properties as a drug candidate for treatment of epilepsy:
|
1.
|
AV-101 is preferentially converted to 7-Cl-KYNA in brain areas related to neuronal injury. This is because astrocytes, which are responsible for the enzymatic transamination of 4-Cl-KYN prodrug to active 7-Cl-KYNA, are focally activated at sites of neuronal injury. Due to AV-101’s highly focused site of conversion, local concentrations of newly formed 7-Cl-KYNA are greatest at the site of therapeutic need. In addition to delivering the drug where it is needed, this reduces the chance of systemic and dangerous side effects with long-term use of the drug; and
|
|
2.
|
An active metabolite of AV-101, 4-Cl-3-hydroxyanthranilic acid, inhibits the synthesis of quinolinic acid, an endogenous NMDAR agonist that causes convulsions and excitotoxic neuronal damage.
|
AV-101’s ability to activate astrocytes for focal delivery of an anti-epileptic principle, and its dual action as a NMDAR GlyB antagonist and quinolinic acid synthesis inhibitor, make AV-101 a potential Phase 2 development candidate for treatment of epilepsy.
Parkinson’s Disease
AV-101 has been shown to activate ventral tegmental area (“VTA”) dopaminergic (“DA”) neurons. Kynurenic acid (“KYNA”) is an endogenous NMDA receptor antagonist, as well as a blocker of the 7-nicotinic acid receptor. Mounting evidence suggests that this compound participates in the pathophysiology of schizophrenia. Preclinical studies have shown that elevated levels of endogenous KYNA are associated with increased firing of midbrain DA neurons. AV-101 is converted to the selective NMDAR GlyB antagonist 7-Cl-KYNA, which is 20 times more potent and selective than KYNA in binding the GlyB site. Utilizing extra cellular single unit cell recording techniques, we have shown that AV-101, which is converted to the selective NMDAR GlyB antagonist 7-Cl-KYNA, significantly increases the firing rate and percent burst firing activity of VTA DA neurons. These results have potential therapeutic implications for Parkinson’s disease.
Huntington’s Disease
Working together with metabotropic glutamate receptors, the NMDAR ensures the establishment of long-term potentiation (“LTP”), a process believed to be responsible for the acquisition of information. These functions are mediated by calcium entry through the NMDAR-associated channel, which in turn influences a wide variety of cellular components, like cytoskeletal proteins or second- messenger synthases. However, over activation at the NMDAR triggers an excessive entry of calcium ions, initiating a series of cytoplasmic and nuclear processes that promote neuronal cell death through necrosis as well as apoptosis, and these mechanisms have been implicated in several neurodegenerative diseases.
Huntington’s disease (“HD”), a chronic neurodegenerative disorder, is caused by an expansion in the number of glutamine repeats beyond 35 at the amino terminal end of a protein termed “huntingtin.” Such a mutation in huntingtin leads to a sequence of progressive cellular changes in the brain that result in neuronal loss and other characteristic neuropathological features of HD. These are most prominent in the neostriatum and in the cerebral cortex, but also observed in other brain areas.
The tissue levels of two neurotoxic metabolites of the kynurenine pathway of tryptophan degradation, quinolinic acid (“QUIN”) and 3-hydroxykynurenine (3-HK) are increased in the striatum and neocortex, but not in the cerebellum, in early stage HD. QUIN and 3-HK and especially the joint action of these two metabolites, have long been associated with the neurodegenerative and other features of the pathophysiology of HD. The neuronal death caused by QUIN and 3-HK is due to both free radical formation and NMDA receptor overstimulation (excitotoxicity).
Based on the hypothesis that 3-HK and QUIN are involved in the progression of HD, early intervention aimed at affecting the kynurenine pathway in the brain may present a promising treatment strategy. We believe the ability of AV-101 to reduce the brain levels of neurotoxic OUIN and to potentially produce significant local concentrations of 7-Cl-KYNA on chronic administration, presents an exciting opportunity for Phase 2 clinical investigation of AV-101 as a potential chronic treatment of HD.
Summary of Additional AV-101 Nonclinical Information
A comprehensive nonclinical pharmacology, pharmacokinetic (“PK”)/toxicokinetic (“TK”), and toxicology program has been conducted to support the clinical use of AV-101 in multiple CNS-related indications. The primary pharmacological activity of AV-101 has been investigated in a series of in vitro and in vivo studies. Pharmacology (absorption, distribution, metabolism, and excretion), PK/TK, and toxicology studies have been conducted with AV-101 in rats, dogs, and monkeys. The excellent safety profile of AV-101 was confirmed by pilot tolerability, single-dose range-finding, and repeated-dose toxicology studies in rats, dogs and monkeys. The genotoxic potential of AV-101 and its active metabolite, 7-Cl-KYNA, was assessed in multiple in vitro genotoxicity studies (bacterial mutation, chromosomal aberration, mouse lymphoma TK+/-, and micronucleus tests).
The behavioral effects of AV-101 assessed in a Good Laboratory Practice (“GLP”) Irwin test in rats show it to have no adverse effect on the CNS following single oral administration at doses up to 2,000 mg/kg. Although AV-101 inhibited the human ether à-go-go–related gene (“hERG”) current in a dose-dependent manner (median concentration that causes 50% inhibition for the inhibitory effect [IC50] of 70.5 μM), its active metabolite, 7-Cl-KYNA, showed no inhibitory effect on the hERG channel current. Electrocardiograms (“ECGs”) recorded during in vivo dog toxicology studies showed no AV-101–related adverse cardiovascular effects. Furthermore, in a pivotal GLP dog 14-day toxicology study, no treatment-related effects on ECGs, including QT interval and QTc, at dose levels up to 120 mg/kg/d. No evidence of any treatment-related adverse effects on the respiratory system has been noted with AV-101.
Oral administration of AV-101 to Sprague-Dawley rats and mice was shown to result in rapid absorption of AV-101 (rats: time to maximum plasma concentration [Tmax], approximately 0.25 to 0.5 hours), adequate bioavailability (rats: approximately 39% to 94%), and plasma elimination half-life (rats: t1/2 approximately 1 to 3 hours). Furthermore, in rats 7-Cl-KYNA was detected in the plasma and reached the maximum plasma concentration (“Cmax”) approximately 0.25 to 0.5 hours after oral administration, suggesting a rapid conversion of AV-101 to 7-Cl-KYNA. Pharmacokinetic analyses were conducted in many of the toxicology studies in rats, dogs, and monkeys. These analyses showed that the AV-101-related clinical signs observed in dogs (versus monkeys) were associated with a similar, and at some does a significantly higher, exposure. Furthermore, although AUC and Cmax values increased non-proportionately with dose level in dogs, AUC values only marginally increased with dose in monkeys, with little change in Cmax values.
Low levels of potential metabolites of AV-101 were detected following in vitro incubations with hepatocytes from the mouse, rat, dog, monkey, and humans, indicating little concern with liver metabolism issues. No appreciable conversion of AV-101 to D-4-Cl-KYN during these hepatocyte incubations was noted. Results from cytochrome P-450 (“CYP”) inhibition and induction studies showed that AV-101 was not a potent inhibitor or inducer of the human CYP isoforms evaluated.
Single-dose studies in rats and monkeys did not show clear evidence of toxicity at maximal doses of 2,000 mg/kg. In dogs, consistent with the expected drug mechanism of action, oral administration of AV-101 resulted in CNS-related clinical signs, including decreased activity, abnormal gait/stance, ataxia, and prostration at the maximum tolerated dose.
A repeated-dose (14-day) ocular toxicity study in Sprague-Dawley rats (unpigmented) and brown Norway rats (pigmented) at dose levels up to 2,000 mg/kg/d did not reveal any signs of retinal degeneration at any dose level or rat strain. A subsequent pivotal GLP 14-day repeated-dose toxicity study in Sprague-Dawley rats showed no treatment-related ocular findings after daily dosing of AV-101 for 14 consecutive days at dose levels up to 2,000 mg/kg/d.
A GLP 14-day repeated-dose CNS toxicity study conducted in dogs, at dose levels up to 100 mg/kg/d showed no treatment-related lesions in the brain of any animal. The pivotal GLP 14-day repeated-dose toxicity study in Beagle dogs, also showed no treatment-related CNS findings after daily dosing of AV-101 for 14 consecutive days at dose levels up to 120 mg/kg/d.
The genotoxic potential of AV-101 and 7-Cl-KYNA was assessed in multiple in vitro genotoxicity studies (bacterial reverse mutation, chromosomal aberration, mouse lymphoma TK+/-, and micronucleus tests), and the overall results confirmed that both AV-101 and 7-Cl-KYNA are not mutagenic.
A rat Olney lesion study was conducted to assess the potential CNS toxicity. No lesions were observed in the brain after a single oral dose of AV-101 at doses up to 2,000 mg/kg.
Nonclinical Pharmacology Studies
Primary Pharmacodynamics
Much of the nonclinical pharmacology information of AV-101 is derived from many published research results on L-4-Cl-KYN or 7-Cl-KYNA. Primary pharmacodynamic studies conducted in rodent models for neuropathic pain demonstrated AV-101’s antihyperalgesic activity in models of facilitated pain processing, its analgesic properties, its ability to provide neuroprotection from excitotoxic death, its ability to reduce seizures, and its activity in multiple preclinical models of depression.
Nonclinical Absorption, Distribution, Metabolism and Excretion Studies
In rats, area under the concentration-time curve from time of dosing extrapolated to infinity (AUC0-∞) values were proportional to dose for AV-101, but Cmax was less than proportional to dose, suggesting a saturation of absorption rate. 7-Cl-KYNA Cmax was less than proportional to dose, and generally females tended to have a higher exposure to AV-101 than males, but no sex difference was noted for 7-Cl-KYNA exposure. In the repeated-dose studies, D-4-Cl-KYN, L-4-Cl-KYN, and 7-Cl-KYNA mean area under the concentration-time curves from time of dosing to the last sampling time (AUC0-t) and AUC0-∞ values were higher on Day 14 than on Day 1 in both sexes of most treatment groups, indicating that exposure increased following daily repeated dosing of AV-101. Sex differences were noted for D-4-Cl-KYN and L-4-Cl-KYN, with mean AUC0-t and AUC0-∞ estimates higher in females relative to males for most treatment groups. Conversely, mean AUC0-t and AUC0-∞ values of 7-Cl-KYNA were generally higher in males relative to females.
In dogs, AUC0-∞ values were slightly less than proportional to dose up to 100 mg/kg AV-101 and Cmax values were less than proportional to dose, suggesting a saturation of absorption. No consistent sex differences were noted for Cmax or AUC values. AUC0-∞ and Cmax values for 7-Cl-KYNA were less than proportional to dose. In the repeated-dose study, D-4-Cl-KYN, L-4-Cl-KYN, and 7-Cl-KYNA showed a proportional increase in Cmax with the administered dose level of AV-101 in both sexes. There was no evidence of plasma accumulation for any of the analytes. Sex differences were noted for D-4-Cl-KYN, with slightly higher mean AUC0-t and AUC0-∞ estimates in females relative to males on Day 1 and Day 14, in all treatment groups. For 7-Cl-KYNA, mean Cmax was elevated in females relative to males at all dose levels on Days 1 and Day 14, and mean AUC0-t and AUC0-∞ estimates were also generally higher in females relative to males at all dose levels. No clear sex differences were noted for L-4-Cl-KYN.
In monkeys, AUC0-∞ values were relatively proportional to dose, but Cmax values were not proportional to dose (comparable or lower Cmax with increasing doses). The AUC0-∞ and Cmax values for 7-Cl-KYNA were less than proportional to dose, and no major sex differences were noted.
Nonclinical Toxicology Studies
The safety profile of AV-101 was determined in single-dose, range-finding, and repeated-dose toxicology studies in rats and dogs, and in a single-dose study in monkeys. A GLP CNS safety pharmacology study in rats that included a microscopic evaluation for Olney lesions was also conducted. Additionally, pivotal GLP 14-day repeated-dose toxicology studies in rats and dogs have been conducted. The genotoxic potentials of AV-101 and 7-Cl-KYNA were assessed in multiple in vitro and in vivo genotoxicity studies, including bacterial reverse mutation, chromosomal aberration, mouse lymphoma TK+/-, and micronucleus tests. Neither were determined to be mutagenic.
Local tolerance studies have not been conducted with AV-101. However, no lesions in the gastrointestinal tract were observed after oral administration of AV-101 in the repeated-dose toxicity studies in the rat and dog.
The results of the pivotal 14-day studies show the dog to be the most sensitive species. The dog NOAEL was determined to be the highest dose level (120 mg/kg/d), and therefore the maximum recommended starting dose (MRSD) would be 6.5 mg/kg (12 mg/kg/d × 0.54 [conversion factor]) or 390 mg per subject for a 60-kg person. As a further added margin of safety for the clinical use of AV-101, VistaGen applied an additional safety factor to the calculated MRSD, and set the starting dose in the proposed Phase 1a clinical trial at 0.5 mg/kg (i.e., 30 mg for 60 kg subjects).
Summary of AV-101 Phase 1 Clinical Safety Studies
The safety data from two NIH-funded AV-101 Phase 1 clinical safety studies indicate that AV-101 was safe and well-tolerated at all three doses tested. Overall, 57 mild to moderate AEs were reported by 34 subjects, with 17 AEs (29.8%) occurring in the placebo group. There was a higher rate of AEs reported from subjects that received placebo than from subjects that received AV-101. A total of 40 AEs were reported by 24 of 37 (64.9%) subjects receiving AV-101, and 17 AEs were reported by 10 of 13 (76.9%) subject receiving placebo. Additionally, 49 of the 57 total AEs (85.9%) were considered mild, and the remaining 8 AEs (14.0%) were considered moderate. Of these AEs, headache was the most commonly reported preferred term. All of the AEs were completely resolved.
Overall, the safety results indicate AV-101 is safe and well-tolerated in healthy subjects. Subjects receiving AV-101 reported a lower percentage of AEs relative to subjects receiving placebo. Moreover, there were no AEs reported by subjects that received AV-101 that were graded as probably related to study drug. The type and distribution of AEs reported by subjects in this study was considered to be typical for a study in healthy volunteers.
A total of 40 AEs were reported by 24 of 37 (64.9%) subjects receiving AV-101, and 17 AEs were reported by 10 of 13 (76.9%) subject receiving placebo. The frequency of AEs was similar among the treatment groups. Thirty-four subjects experienced a total of 57 AEs, with 16 (28.1% of the total AEs) in the 360-mg group, 14 (24.6% of the total AEs) in the 1,040-mg group, 10 (17.5% of the total AEs) in the 1,440-mg group, and 17 (29.8% of the total AEs) in the placebo group. All of the AEs were completely resolved.
Although the Phase 1 safety and pharmacokinetic studies were not designed to measure or evaluate the potential antidepressant effects of AV-101, approximately 9% (5/57) of the subjects receiving AV-101 and 0% of the 31 subjects receiving placebo reported “feelings of well-being” (coded as euphoric mood), similar to the fast-acting antidepressant effects reported in the literature with ketamine.
Phase 1 Clinical Safety Trials
Phase 1a Study (VSG-CL-101)
A phase 1a, randomized, double-blind, placebo-controlled study to evaluate the safety and PK of single doses of AV-101 in healthy volunteers was conducted (VSG-CL-001). Seven cohorts (30, 120, 360, 720, 1,080, 1,440, and 1,800 mg) with six subjects per cohort (1:1, AV-101: placebo) were to be enrolled in the study. For the first five cohorts (30, 120, 360, 710 and 1,080 mg) only two subjects were dosed at a time as a pair (1:1, AV-101: placebo) on Day 1. The safety and tolerability of AV-101 in each pair of subjects was assessed by the investigator before proceeding to the next pair within the dose cohort of the study. If no safety concerns were found after analysis of the laboratory samples, physical assessments, and results of the neurological and ophthalmological examinations, the next two subjects in the cohort were dosed, but no sooner than 48 hours after the previous pair of subjects. The next cohort was dosed when the investigator and medical monitor agreed that it was safe to proceed based on review of the previous dose group’s preliminary safety information. In addition, PK assessments were to be reviewed for each cohort starting with the 720 mg through the 1,800 mg dose cohort. A minimum of four evaluable subjects (two AV-101 and two placebo) were required for determination of tolerability and safety of a dose level. The PK stopping criteria would be reached when the L-4-C1-KYN mean AUC0-t reaches 900,486 ng·h/mL, or a mean Cmax of 81,633 ng/mL, or a PK extrapolation predicts exceeding one of these values in the next cohort.
All the subjects from the 1,440 mg cohort were dosed during a single day (3 subjects receiving active drug and 3 subjects receiving placebo). The safety and tolerability of AV-101 in the 1,440 mg dose cohort was to be assessed by the investigator and medical monitor before proceeding to the 1,800 mg dose cohort. If no safety concerns were found after analysis of the laboratory samples including the PK results, physical assessments, and results of the neurological and ophthalmological examinations for the 1,440 mg cohort, the 1,800-mg cohort was to be dosed. However, the PK stopping criteria were reached at the 1,440-mg cohort, and the study was stopped and did not proceed to the planned 1,800 mg cohort.
Phase 1a Study Pharmacokinetics Summary
Validated bioanalytical methods were used to measure plasma analyte concentrations. These assays had lower limits of quantification of 2 ng/mL for 7-Cl-KYNA and 5 ng/mL for L-4-Cl-KYN and D-4-Cl- KYN. Pharmacokinetic parameters were calculated by using WinNonlin Pro v. 5.2. Parameters calculated included observed maximal concentration (Cmax), observed time to Cmax (Tmax), area under the concentration-time curve to the last sample collected (AUC0-t) or extrapolated to infinity (AUC0-∞), and half-life (t1/2). Concentrations of all three analytes were measurable in both plasma and urine after administration of each of the six dose levels: 30, 120, 360, 720, 1,080 and 1,440 mg.
Concentration-time data were obtained after dosing of the six cohorts. Three subjects received AV-101 and three received placebo in each cohort. Plasma concentrations of 4-Cl-KYN (AV-101) and 7-Cl-KYNA were obtained in addition to urine concentrations of these two analytes. Plasma and urine concentrations of D-4-Cl-KYN also were determined, but will be reported only for the first two cohorts.
This study was conducted under dose escalation stopping criteria as determined by the FDA of 4-Cl-KYN mean Cmax and AUC limits of 81,633 ng/mL and 900,486 ng∙h/mL, respectively. Although these criteria were not met for the mean data of the 1,440-mg dose, one subject had a Cmax that was slightly greater than the limit of 81,633 ng/mL. Therefore, dose escalation to the planned seventh cohort of 1,800 mg of AV-101 did not occur in this study. However, from a safety perspective, a maximum tolerable dose was not achieved. Also, maximum AUC values at the highest dose level remained substantially lower than the limit.
Concentrations of all three analytes were measurable in both plasma and urine after administration of all dose levels, although many of the samples from the 30-mg dose group had concentrations below the limit of quantification for 7-Cl-KYNA. Plasma concentration-time profiles were consistent with rapid absorption of the oral dose and first-order elimination. The plasma concentration-time profiles were well defined for 4-Cl-KYN at all dose levels. Maximum concentrations occurred fairly rapidly, with individual values of Tmax ranging from 0.5 to 2 hours, with greater values tending to be in the higher dose groups. Individual t1/2 values were fairly consistent within cohorts, and mean values ranged from 1.80 to 3.33 hours. Mean t1/2 values also tended to increase with increasing dose. Mean Cmax and AUC0-∞ values appeared to be approximately dose proportional except for those of the highest dose group.
The 7-Cl-KYNA plasma concentration-time profiles were not well defined for the 30-mg dose. Most samples for the 30-mg dose cohort had concentrations below the lower limits of quantification, and t1/2 values could not be calculated; however, profiles were sufficient after the 120-mg and greater doses to calculate all parameters.
In general, 7-Cl-KYNA maximum concentrations occurred at the same time or later than those for 4-Cl-KYN, as may be expected since 7-Cl-KYNA is a metabolite of 4-Cl-KYN. Individual values of Tmax ranged from 0.5 to 2 hours for both analytes. Individual 7-Cl-KYNA t1/2 values were fairly consistent within cohorts, and mean values ranged from 2.17 to 3.19 hours. Mean t1/2 values did not appear to be dose-related. Mean 7-Cl-KYNA Cmax values were somewhat dose proportional for the two initial dose groups, but tended to increase in a more than dose-proportional manner. Similarly, mean 7-Cl-KYNA AUC0-t values for all dose groups and AUC0-∞ values for dose groups of 120 mg or greater tended to increase in a more than dose-proportional manner. Mean plasma concentrations of 4-Cl-KYN (Figure 1) and 7-Cl-KYNA (Figure 2) are depicted for all six cohorts.
As with the 120-mg dose cohort, the plasma concentration-time profiles were well defined for both 4-Cl-KYN and 7-Cl-KYNA at the four higher dose levels. Interestingly, the mean concentration-time profiles suggest that maximum concentrations were lower than expected, particularly for 7-Cl-KYNA.

Figure-1. Mean plasma concentrations of 4-Cl-KYN after oral administration of a single dose of AV-101.

Figure 2. Mean plasma concentrations of 7-Cl-KYNA after oral administration of a single dose of AV-101.
Assessment of Dose Proportionality
For 4-Cl-KYN, mean Cmax and AUC0-∞ values appeared to be approximately dose proportional except for those of the highest dose group. These values are presented by dose in Figure 3 (Cmax) and in Figure 4 (AUC0-∞) below. Figure 3 indicates that for 4-Cl-KYN the mean Cmax values are approximately dose linear and proportional up to a dose of 1,080 mg of AV-101. After a dose of 1,440 mg, the mean Cmax values increased only 8.8% while the dose increased by 33.3%. This is evident in the deviation of the graph from linearity at the highest dose.
Although the 4-Cl-KYN mean Cmax values were not linear after the 1,080-mg dose, AUC0-∞ values are approximately linear and dose proportional throughout the dose range. The nonlinearity of Cmax values at the highest dose could be a result of an outlier or simply variability in a small number of subjects (Cmax values of 44,600, 54,900, and 89,500 ng/mL were observed after the dose of 1,040-mg AV-101), it suggests that the rate or extent of absorption could be limited. The fact that AUC0-∞ values were linear throughout the dose range suggests that the extent of absorption was not a limitation, but the rate of absorption may be limited at doses above 1,080 mg.
The lack of linearity of the 4-Cl-KYN mean Cmax values would be expected to have a similar effect on the 7-Cl-KYNA mean Cmax values. Similarly, because the extent of absorption of 4-Cl-KYN was linear throughout the dose range, exposure to 7-Cl-KYNA would be expected to also be linear. Mean values of 7-Cl-KYNA are presented by dose in Figure 5 (Cmax) and in Figure 6 (AUC0-∞).


Figure 4. Mean AUC0-∞ values of 4-Cl-KYN by dose of AV-101.
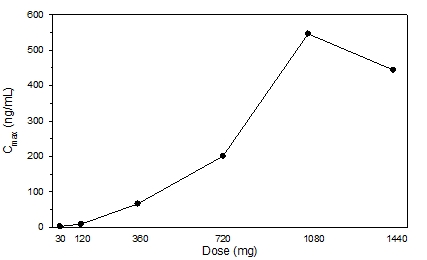

Phase 1a Safety Summary
Nine subjects experienced 10 AEs, with four of the AEs occurring in subjects in the placebo group and two of the AEs occurring for one subject receiving 30 mg AV-101. For the AEs occurring in the AV-101–treated subjects, there were no meaningful differences in the number of AEs observed at the 30-mg dose (2 AEs) when compared with that at the 120-mg dose (1 AE), 360-mg dose (1 AE), 720-mg dose (0 AEs), 1,080-mg dose (0 AEs), or 1,440-mg dose (2 AEs). Eight of 10 AEs (80%) were considered mild, and two (20%, headache and gastroenteritis) were considered moderate. Four subjects on AV-101, one each in Cohorts 1 through 4 and two subjects on placebo in Cohort 5 reported AEs of headaches. Five headaches were mild with no concomitant treatment, and one was moderate with concomitant drug therapy administered. Most completely resolved the same day as onset and were considered not serious. One headache started the day after dosing and resolved approximately one week later on the same day as the concomitant drug therapy was administered. One case of contact dermatitis bilateral lower extremities was reported in Cohort 2 on placebo that was ongoing. One of the subjects with the headache also reported an AE of gastroenteritis that was unrelated to AV-101. This AE was considered moderate but did not require any drug therapy and was completely resolved within 2 days of onset. This AE was also considered not serious.
Even though these safety studies were not designed to quantitatively assess effects on mood, during the interviews 2 out of 3 subjects who received the highest dose (1440 mg) of AV-101, voluntarily acknowledged positive effects on mood. Similar comments were not made by any of the 18 placebo group subjects. One incident lasted approximately 15 minutes after study drug dosing, and the other event of euphoria lasted approximately 3 hours after study drug dosing. There were no other reported AEs for this cohort. The events resolved and were considered not serious.
Table 1. Summary of Adverse Events, Phase 1a
|
Placebo
(n = 18)
|
Cohorts (mg)
|
|||||||
|
MedDRA SOC and Preferred Term
|
30
(n = 3)
|
120
(n = 3)
|
360
(n = 3)
|
720
(n = 3)
|
1,080
(n = 3)
|
1,440
(n = 3)
|
Overall
(n = 36)
|
|
|
Infections and Infestations
|
0
(0%)
|
1
(33.3%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
1 (2.8%)
|
|
Gastroenteritis
|
0
(0%)
|
1
(33.3%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
1 (2.8%)
|
|
Nervous System Disorders
|
1
(5.6%)
|
1
(33.3%)
|
1
(33.3%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
3 (8.3%)
|
|
Headache
|
1
(5.6%)
|
1
(33.3%)
|
1
(33.3%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
3 (8.3%)
|
|
Psychiatric Disorder
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
2
(66.7%)
|
2 (5.6%)
|
|
Euphoric mood
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
2
(66.7%)
|
2 (5.6%)
|
|
Skin and Subcutaneous Tissue Disorder
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0 (0%)
|
|
Dermatitis contact
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0
(0%)
|
0 (0%)
|
MedDRA = Medical Dictionary for Regulatory Activities; SOC = system organ class.
Phase 1b Study (VSG-CL-102)
A Phase 1b clinical study was conducted as a single-site, dose-escalating study to evaluate the safety, tolerability, and PK of multiple doses of AV-101 administered daily in healthy volunteers. The antihyperalgesic effect of AV-101 on capsaicin-induced hyperalgesia was also assessed. Subjects were sequentially enrolled into one of three cohorts (360 mg, 1,080 mg, and 1,440 mg) and were randomized to AV-101 or placebo at a 12:4 (AV-101 to placebo) ratio. Subjects were to have been dosed for 14 consecutive days. Each subject was given a paper diary and instructed to record daily dose administration, concomitant medications, and AEs during the 14-day treatment period.
The safety and tolerability of AV-101 were assessed by evaluating AEs and by physical examinations, vital signs, and clinical laboratory tests (chemistry and hematology assessments) that were performed on Days 1, 7 (±1 day), and 14. Blood sampling for PK was performed on Days 1, 2, 14, and 15. Additionally, ophthalmological examinations were performed at screening and Day 15. Physical examinations, including vital signs, 12-lead electrocardiograms (ECGs), neurocognitive tests, and ataxia tests were performed on Day 1 and Day 14. Before proceeding to the next higher dose, the following criteria were met:
|
·
|
Blinded safety and tolerability data were reviewed and assessed as being satisfactory by the investigator and medical monitor.
|
|
·
|
PK assessments were reviewed by the blinded Cato Research PK specialist to determine if the PK stopping criteria were reached.
|
The doses evaluated in this Phase 1b multi-dose study of AV-101 were based on results obtained in a previously conducted Phase 1a single-dose study of AV-101 in healthy adults. The dose-escalation design was consistent with a standard scheme, and careful monitoring occurred to ensure the safety of all subjects.
The minimum toxic dose was defined as the dose at which the stopping criteria were reached. For this study, the minimum toxic dose was to be (1) the dose at which a drug-related serious adverse event (“SAE”) occurred in an AV-101–treated subject, or (2) the dose at which a severe AE that warranted stopping the study, as determined by the investigator and medical monitor, occurred in an AV-101–treated subject within a cohort. The minimum toxic dose was not reached in this study.
A total of 40 AEs were reported by 24 of 37 (64.9%) subjects receiving AV-101, and 17 AEs were reported by 10 of 13 (76.9%) subject receiving placebo (Table 2). The frequency of AEs was similar among the treatment groups. Thirty-four subjects experienced a total of 57 AEs, with 16 (28.1% of the total AEs) in the 360-mg group, 14 (24.6% of the total AEs) in the 1,040-mg group, 10 (17.5% of the total AEs) in the 1,440-mg group, and 17 (29.8% of the total AEs) in the placebo group. All of the AEs were completely resolved.
Table 2. Summary of Adverse Events, Phase-1b
|
·Dose Cohorts
|
||||
|
·360 mg
AV-101
·(N = 12)
[n (%)]
|
·1,080 mg AV-101
·(N = 13)
[n (%)]
|
·1,440 mg AV-101
·(N = 12)
[n (%)]
|
·Pooled Placebo
·(N = 13)
[n (%)]
|
|
|
● Number of AEs
|
● 16
|
● 14
|
● 10
|
● 17
|
|
● Number of subjects with any AE
|
● 9 (75.0%)
|
● 8 (61.5%)
|
● 7 (58.3%)
|
● 10 (76.9%)
|
|
● Number of SAEs
|
● 0 (0%)
|
● 0 (0%)
|
● 0 (0%)
|
● 0 (0%)
|
|
● Number of AEs resulting in death
|
● 0 (0%)
|
● 0 (0%)
|
● 0 (0%)
|
● 0 (0%)
|
|
Number of AEs leading to discontinuation of study drug
|
● 0 (0%)
|
● 0 (0%)
|
● 0 (0%)
|
● 1 (7.7%)
|
AE = adverse event; SAE = serious adverse event.
The majority of the reported AEs were nervous system disorders (23 subjects, 46% of subjects) and gastrointestinal disorders (7 subjects, 14.0%). The remaining AEs were classified as eye disorders (3 subjects, 6.0%); psychiatric disorders (3 subjects, 6.0%); respiratory, thoracic, and mediastinal disorders (3, 6.0%); skin and subcutaneous tissue disorders (3 subjects, 6.0%); general disorders and administration site conditions (2 subjects, 4.0%); cardiac disorders (1 subject, 2.0%); infections and infestations (1 subject, 2.0%); musculoskeletal and connective tissue disorders (1 subject, 2.0%); and renal disorders (1 subject, 2.0%).
The distribution of AEs by System Organ Class was similar among the cohorts with the exception of headaches and gastrointestinal disorders. Eight of the 18 (44.4%) reported headaches were in the placebo group, 6 (33.3%) were in the 1,080-mg group, 3 (16.7%) were in the 1,440-mg group, and 1 (5.6%) was in the 360-mg group. Three (42.9%) of the 7 reported gastrointestinal disorders were in the 360-mg group, 2 (28.6%) were in the placebo group, 1 (14.3%) was in the 1,080-mg group, and 1 (14.3%) was in the 1,440-mg group.
The determination of the relationship of the AE to the study drug was made when the data were blinded. Ten of the 15 AEs (66.7%) that occurred in the 360-mg AV-101 group, 10 of the 14 AEs (71.4%) that occurred in the 1,040-mg AV-101 group, 7 of the 10 AEs (70.0%) that occurred in the 1,440-mg AV-101 group, and 13 of the 17 AEs (76.5%) that occurred in the placebo group were determined to be possibly related to study drug. One (5.9%) AE in the placebo group was probably related to study drug (rash around neck). Of the 57 reported AEs, 49 (85.9%) were of mild intensity and 8 (14.0%) were of moderate intensity. There were 2 moderate intensity AEs in the 360-mg AV-101 group; 1 was unrelated pain in the right foot, and 1 was a possibly related headache. All other moderate AEs occurred in the placebo group and included nausea or vomiting (2 AEs), headache (2 AEs), and rash around the neck (1 AE). No severe AEs were reported.
Even though these safety studies were not designed to quantitatively assess effects on mood, during the interviews certain subjects who received 360, 1080, and 1440 mg of AV-101, voluntarily acknowledged positive effects on mood. Similar comments were not made by any of the placebo-group subjects.
Phase 1b Pharmacokinetics Summary
Concentration-time data were obtained after dosing of the three cohorts. Plasma concentrations of 4-Cl-KYN (AV-101) and the metabolite, 7-Cl-KYNA, were obtained from subjects that received AV-101. PK parameters were calculated by using WinNonlin Pro Version 5.3. Parameters calculated included Cmax, Tmax, AUC0-t, AUC0-∞, and t1/2.
Plasma concentration-time profiles obtained for 4-Cl-KYN after administration of once-daily oral doses of 360, 1,080, or 1,440 mg AV-101 were consistent with rapid absorption of the oral dose and first-order elimination of both 4-Cl-KYN and 7-Cl-KYNA, with evidence of multicompartment kinetics, particularly for the metabolite 7-Cl-KYNA. Several subjects had plasma concentration-time profiles with a last measurable sample that appeared to be an outlier or suggested multicompartment kinetics, making it challenging to identify a terminal log-linear elimination phase. Particularly for 7-Cl-KYNA, using the last two measurable samples to calculate t1/2 resulted in unrealistic values for some subjects.
Plasma concentration-time profiles for 4-Cl-KYN were more consistently single compartment, but several had a subtle multicompartment appearance. To be consistent in the calculation of t1/2 and to report a meaningful value, the final three samples with measurable concentrations were used to calculate t1/2 for subjects for whom those samples appeared to be log-linear. Otherwise, the last sample was essentially treated as an outlier, and the prior samples in the log-linear phase were used to calculate t1/2 (these samples had a higher coefficient of determination value than the last three samples). In addition, the AUC0-∞ values reported are calculated using the predicted last value rather than observed.
An absolute bioavailability evaluation is not possible from the data; however, an estimate of exposure can be done by comparing the AUC at the same doses. The mean AUC0-∞values in the Phase 1b study were higher at all three doses than seen in Phase 1a study, suggesting similar or even higher bioavailability than that in the Phase 1a study, i.e ≥ 31%.
In conclusion, the PK of AV-101 was fully characterized across the range of doses in this study. Plasma concentration-time profiles obtained for 4-Cl-KYN (AV-101) and 7-Cl-KYNA after administration of a single and multiple, once daily oral doses of 360, 1,080, or 1,440 mg were consistent with rapid absorption of the oral dose and first-order elimination of both analytes, with evidence of multi-compartment kinetics, particularly for the metabolite 7-Cl-KYNA.
Phase 1 - Overall Safety Conclusions
The Phase 1a and Phase 1b safety data indicate that AV-101 was safe and well tolerated at all three doses tested. Overall, 57 AEs were reported by 34 subjects, with 17 AEs (29.8%) occurring in the placebo group. There was a higher rate of AEs reported from subjects that received placebo than from subjects that received AV-101. A total of 40 AEs were reported by 24 of 37 (64.9%) subjects receiving AV-101, and 17 AEs were reported by 10 of 13 (76.9%) subject receiving placebo. Additionally, 49 of the 57 total AEs (85.9%) were considered mild, and the remaining 8 AEs (14.0%) were considered moderate. Of these AEs, headache (nervous system disorder) was the most commonly reported preferred term. All of the AEs were completely resolved.
Overall, the safety results indicate AV-101 is safe and well tolerated in healthy subjects. Subjects receiving AV-101 reported a lower percentage of AEs relative to subjects receiving placebo. Moreover, there were no AEs reported by subjects that received AV-101 that were graded as probably related to study drug. The type and distribution of AEs reported by subjects in this study was considered to be typical for a study in healthy volunteers.
A total of 40 AEs were reported by 24 of 37 (64.9%) subjects receiving AV-101, and 17 AEs were reported by 10 of 13 (76.9%) subject receiving placebo (Table 2). The frequency of AEs was similar among the treatment groups. Thirty-four subjects experienced a total of 57 AEs, with 16 (28.1% of the total AEs) in the 360-mg group, 14 (24.6% of the total AEs) in the 1,040-mg group, 10 (17.5% of the total AEs) in the 1,440-mg group, and 17 (29.8% of the total AEs) in the placebo group. All of the AEs were completely resolved.
Although the Phase 1 safety and pharmacokinetic studies were not designed to measure or evaluate the potential antidepressant effects of AV-101, approximately 9% (5/57) of the healthy volunteer subjects receiving AV-101 and 0% of the 31 subjects receiving placebo reported “feelings of wellbeing” (coded as euphoric mood), similar to the rapid-onset antidepressant effects reported in the literature with ketamine. Table 3 lists the percent of adverse events reported by the subjects in each of the two Phase 1 studies and the number of events reported as euphoric mood per number of subjects on placebo and AV-101.
Table 1. Reports of Well-Being (coded as Euphoric Mood) in Phase 1 Clinical Studies
|
Placebo
% of Adverse Events/N
|
AV-101
% of Adverse Events/N
|
||
|
Phase 1a
|
|||
|
· Nonserious Adverse Events
|
22% (4/18)
|
28% (5/18)
|
|
|
· Feelings of Well-being (coded as euphoric mood)
|
0% (0/18)
|
11% (2/18)
|
|
|
Phase 1b
|
|||
|
· Nonserious Adverse Events
|
77% (10/13)
|
65% (24/37)
|
|
|
· Feelings of Well-being (coded as euphoric mood)
|
0% (0/13)
|
8% (3/37)
|
|
|
Phase 1a and 1b (combined)
|
|||
|
· Feelings of Well-being (coded as euphoric mood)
|
0% (0/31)
|
9% (5/55)
|
|
N = number of subjects
The five events of feeling of well-being (coded as euphoric mood) occurred in one subject each at 360 (7%, 1 of 15 subjects) and 1,080 mg (6%, 1 of 16 subjects), and three subjects at 1,440 mg (20%, 3 of 15 subjects) in the Phase 1a and Phase 1b clinical studies. Four of the five subjects reporting well-being/euphoric mood did not have any other adverse experiences, and one subject (1,080 mg) also reported a mild headache. These results suggest a dose response and that AV-101 at the higher doses may lead to an increased positive mood.
Stem Cell Technology
Overview
We believe better cells lead to better medicines™ and that the key to making better cells is precisely controlling the differentiation of human pluripotent stem cells (“hPSCs”), which are the building blocks of all cells of the human body. Our stem cell technology platform is based on proprietary and licensed technologies for controlling the differentiation of hPSCs and producing the multiple types of mature, non-transformed, functional, adult human cells that we use, or plan to use, to reproduce complex human biology and disease and assess, in vitro, the potential therapeutic benefits and safety risks of new chemical entities (“NCEs”).
We have used our hPSC-derived cardiomyocytes (human heart cells we refer to as VSTA-CMs™) to design and develop CardioSafe 3D™, our novel, customized in vitro bioassay system for predicting potential cardiotoxicity of NCEs, including drug rescue NCEs. We believe CardioSafe 3D is more comprehensive and clinically predictive than the hERG assay, currently the only in vitro cardiac safety assay required by FDA guidelines. We use our stem cell-derived hepatocytes (human liver cells we refer to as VSTA-heps™) as the foundation of LiverSafe 3D™, our second novel, customized bioassay system for predicting potential liver toxicity of new drug candidates, including potential drug metabolism issues and adverse drug-drug interactions. VSTA-heps are highly-functional, non-transformed, and have the majority of the functional properties of mature human hepatocytes. We believe our VSTA-heps have more functionally useful life-span in culture, and overcome numerous problems related to commercially-available primary (cadaver) hepatocytes currently used in FDA-required in vitro hepatocyte assays for drug metabolism. These commercially-available primary hepatocytes are generally in limited supply and the health status and genetic differences of the donor are unknown. We believe our VSTA-CMs, VSTA-heps, CardioSafe 3D and LiverSafe 3D offer a new paradigm for evaluating and predicting potential heart and liver toxicity of NCEs, including drug rescue NCEs, early in development, long before costly, high risk human clinical trials.
Scientific Background
Stem cells are the building blocks of all cells of the human body. They have the potential to develop into many different mature cell types. Stem cells are defined by a minimum of two key characteristics: (i) their capacity to self-renew, or divide in a way that results in more stem cells; and (ii) their capacity to differentiate, or turn into mature, specialized cells that make up tissues and organs. There are many different types of stem cells that come from different places in the body or are formed at different times throughout our lives, including pluripotent stem cells and adult or tissue-specific stem cells, which are limited to differentiating into the specific cell types of the tissues in which they reside. We focus exclusively on human pluripotent stem cells.
Human pluripotent stem cells (“hPSCs”) can be differentiated into all of the more than 200 types of cells in the human body, can be expanded readily, and have diverse medical research, drug discovery, drug rescue, drug development and therapeutic applications. We believe hPSCs can be used to develop numerous cell types, tissues and customized assays that can mimic complex human biology, including heart and liver biology for drug rescue.
Human pluripotent stem cells are either embryonic stem cells (hESCs) or induced pluripotent stem cells (“iPSCs”). Both hESCs and iPSCs have the capacity to be maintained and expanded in an undifferentiated state indefinitely. We believe these features make them highly useful research and development tools and as a source of normal, functionally mature cell populations. We use multiple types of these mature cells as the foundation of to design and develop novel, customized bioassay systems to test the safety and efficacy of NCEs in vitro. These cells also have potential for diverse regenerative medicine applications.
Human Embryonic Stem Cells
According to the NIH, hESCs are derived from excess embryos that develop from eggs that have been fertilized in an in vitro fertilization (“IVF”) clinic and then donated for research purposes with the informed consent of the parental donors after a successful IVF procedure. Human embryonic stem cells are not derived from eggs fertilized in a woman’s body. Human ESCs are isolated when the embryo is approximately 100 cells, well before organs, tissues or nerves have developed.
Human ESCs have the potential to both self-renew and differentiate. They undergo increasingly tissue-restrictive developmental decisions during their differentiation. These “fate decisions” commit the hESCs to becoming only a certain type of mature, functional cells and ultimately tissues. At one of the first fate decision points, hESCs differentiate into epiblasts. Although epiblasts cannot self-renew, they can differentiate into the major tissues of the body. This epiblast stage can be used, for example, as the starting population of cells that develop into millions of blood, heart, muscle, liver and insulin-producing pancreatic beta-islet cells, as well as neurons. In the next step, the presence or absence of certain growth factors, together with the differentiation signals resulting from the physical attributes of the cell culture techniques, induce the epiblasts to differentiate into neuroectoderm or mesendoderm cells. Neuroectoderm cells are committed to developing into cells of the skin and nervous systems. Mesendoderm cells are precursor cells that differentiate into mesoderm and endoderm. Mesoderm cells develop into muscle, bone and blood, among other cell types. Endoderm cells develop into the internal organs such as the heart, liver, pancreas and intestines, among other cell types.
Induced Pluripotent Stem Cells
It is also possible to obtain hPSC lines from individuals without the use of embryos. Induced PSCs are adult cells, typically human skin or fat cells that have been genetically reprogrammed to behave like hESCs by being forced to express genes necessary for maintaining the pluripotential properties of hESCs. Although researchers are exploring non-viral methods, most early iPSCs were produced by using various viruses to express three or four genes required for the immature pluripotential property similar to hESCs. It is not yet precisely known, however, how each gene actually functions to induce cellular pluripotency, nor whether each of the three or four genes is essential for this reprogramming. Although hESCs and iPSCs are believed to be similar in many respects, including their pluripotential ability to form all cells in the body and to self-renew, scientists do not yet know whether they differ in clinically significant ways or have the same ability to self-renew.
We believe the biology and differentiation capabilities of hESCs and iPSCs are likely to be comparable for most if not all purposes. There are, however, specific situations in which we may prefer to use one or the other type of hPSC. For example, we may prefer to use iPSCs for potential drug discovery applications based on the relative ease of generating iPSCs from:
|
·
|
individuals with specific inheritable diseases and conditions that predispose the individual to respond differently to drugs; or
|
|
·
|
individuals with specific variations in genes that directly affect drug levels in the body or alter the manner or efficiency of their metabolism, breakdown and/or elimination of drugs.
|
Because they can significantly affect the therapeutic and/or toxic effects of drugs, these genetic variations have an impact on drug discovery and development. We believe iPSC technologies may allow the rapid and efficient generation of hPSCs from individuals with specific genetic variations. These hPSCs might then be used to produce cells to model specific diseases and genetic conditions for drug discovery and drug rescue purposes.
Proprietary Stem Cell Differentiation Protocols
Proprietary differentiation protocols cover key conditions involved in the differentiation of hPSCs into multiple types of mature human cells. The human cells generated by following these proprietary differentiation protocols are integral to our stem cell technology platform. We believe they support more clinically-predictive in vitro bioassay systems than animal testing or cellular assays currently used in drug discovery and development. Our strategic technology licenses from National Jewish Health in Denver, the Icahn School of Medicine at Mount Sinai in New York and the University Health Network in Toronto relate, among other things, to proprietary stem cell differentiation protocols involving precisely-coordinated temporal and quantitative conditions and interaction of biological molecules.
We believe our bioassay systems will allow us to assess the toxicity profile of NCEs for a wide range of diseases and conditions with greater speed and precision than nonclinical surrogate safety models most often currently used in drug development.
3D “Micro-Organ” Culture Systems
In addition to standard two-dimensional (“2D”) cultures which work well for some cell types and cellular assays, the proprietary hPSC technologies underlying our stem cell technology platform enable us to grow large numbers of normal, non-transformed, mature human cells to produce novel in vitro 3D “micro-organ” culture systems. For example, for CardioSafe 3D, we grow large numbers of normal, non-transformed, human heart cells in vitro in 3D micro-organ culture systems. The 3D micro-organ cultures induce the cells to grow, mature, and develop 3D cell networks and tissue structures. We believe these 3D cell networks and structures more accurately reflect the structures and biology inside the human body than traditional flat, 2D, single cell layers grown on plastic, that are widely used by pharmaceutical companies today. We believe that the more representative human biology afforded by the 3D system will yield responses to drug candidates that are more predictive of human drug responses.
Medicinal Chemistry
Medicinal chemistry involves designing, synthesizing, or modifying a small molecule compound or drug suitable for clinical development. It is a highly interdisciplinary science combining organic chemistry, biochemistry, physical chemistry, computational chemistry, pharmacology, and statistics. The combination of medicinal chemistry with the proprietary and licensed hPSC technologies underlying our stem cell technology platform are core components of our drug rescue business model.
CardioSafe 3D
The limitations of current preclinical drug testing systems used by pharmaceutical companies and others contribute to the high failure rate of NCEs. According to articles published in the Journal of Applied Toxicology, Stem Cell Research and Current Opinion in Cardiology, unexpected cardiotoxicity is one of the top two major safety-related reasons for failure of both drugs and drug candidates. Incorporating novel in vitro assays using hPSC-derived cardiomyocytes (“hPSC-CMs”) early in preclinical development offers the potential to improve clinical predictability, decrease development costs, and avoid adverse patient effects, late-stage clinical termination, and product recall from the market.
With our proprietary stem cell differentiation technology, we produce fully functional, non-transformed hPSC-CMs, which we refer to as VSTA-CMs™, at a level of purity greater than 95% and with normal ratios of all important cardiac cell types. Importantly, our hPSC-CM differentiation protocols do not involve either genetic modification or antibiotic selection. This is important because genetic modification and antibiotic selection can distort the ratio of cardiac cell types and have a direct impact on the ultimate results and clinical predictivity of assays that incorporate hPSC-CMs produced in such a manner. In addition to normal expression all of the key ion channels of the human heart (calcium, potassium and sodium) and various cardiomyocytic markers of the human heart, our VSTA-CMs function reliably in all of our CardioSafe 3D cardiac toxicity assays, screening for both direct cardiomyocyte cytotoxicity and arrhythmogenesis (or development of irregular beating patterns). We believe CardioSafe 3D is sensitive, stable, reproducible and capable of generating data enabling a more accurate prediction of the in vivo cardiac effects of NCEs than is possible with existing preclinical testing systems, particularly the hERG assay.
Limited Clinical Predictivity of the FDA-Required hERG Assay
The hERG assay, which uses either transformed hamster ovary cells or human kidney cells, is currently the only in vitro cardiac safety assay required by FDA Guidelines (“ICH57B”). We believe the clinical predictivity of the hERG assay is limited because it assesses only a single cardiac ion channel - the hERG potassium ion channel. It does not assess any other clinically relevant cardiac ion channels, including calcium, non-hERG potassium and sodium ion channels. Also, importantly, the hERG assay does not assess the normal interaction between these ion channels and their regulators. In addition, the hERG assay does not assess clinically-relevant cardiac biological effects associated with cardiomyocyte viability, including apoptosis and other forms of cytotoxicity, as well as energy, mitochondria and oxidative stress. As a result of its limitations, results of the hERG assay can lead to false negative and false positive predictions regarding the cardiac safety of new drug candidates.
Broad Clinical Predictivity of CardioSafe 3D
As noted above, we have developed and validated two clinically-relevant functional components of our CardioSafe 3D screening system to assess multiple categories of cardiac toxicities, including both direct cardiomyocyte cytotoxicity and arrhythmogenesis (or development of irregular beating patterns). The first functional component of CardioSafe 3D consists of a suite of five fluorescence or luminescence based high-throughput hPSC-CM assays. These five CardioSafe 3D assays measure the following important drug-induced cardiac biological effects:
|
1. cell viability;
|
|
|
2. apoptosis;
|
|
3. mitochondrial membrane depolarization;
|
|
|
4. oxidative stress; and
|
|
5. energy metabolism disruption.
|
These five CardioSafe 3D biological assays were correlated to reported clinical results using reference compounds known to be cardiotoxic in humans versus compounds known to be safe in humans. These reference compounds were representative of eight different drug classes, including:
|
1. ion channel blockers: amiodarone, nifedipine;
|
|
|
2. hERG trafficking blockers: pentamidine, amoxapine;
|
|
|
3. α-1 adrenoreceptors: doxazosin;
|
|
|
4. protein and DNA synthesis inhibitors: emetine;
|
|
|
5. DNA intercalating agents: doxorubicin;
|
|
|
6. antibiotics: ampicillin, cefazolin;
|
|
|
7. NSAID: aspirin; and
|
|
|
8. kinase inhibitors: staurosporine.
|
This suite of five CardioSafe 3D cytotoxicity assays provided measurement of cardiac drug effects with high sensitivity that are consistent with the expected cardiac responses to each of these compounds. Based on our results, we believe CardioSafe 3D provides valuable and far more comprehensive bioanalytical tools for both assessing the effects of pharmaceutical compounds on cardiac cytotoxicity than the hERG assay and can elucidate for us and our medicinal chemistry partner specific mechanisms of cardiac toxicity, thereby laying what we believe is a novel and advantageous foundation for our drug rescue programs.
The other component of our CardioSafe 3D assay system is a sensitive and reliable medium throughput multi-electrode array (“MEA”) assay developed to predict drug-induced alterations of electrophysiological function of the human heart, representing an integrated assessment of not only hERG potassium ion channel activity analogous to the FDA-mandated hERG assay but, in addition, non-hERG potassium channels, and calcium channels and sodium channels, which are well beyond the scope of the hERG assay. Functional electrophysiological assessment is a key component of CardioSafe 3D, and has been validated with reported clinical results involving twelve drugs, each with known toxic or non-toxic cardiac effects in humans. The twelve clinical correlation study compounds are as follows:
|
1.
|
One FDA-approved drug (aspirin) without cardiac liability to serve as a negative control;
|
|
2.
|
Five FDA-approved drugs (astemizole, sotalol, cisapride, terfenadine and sertindole) that were withdrawn from the market due to heart toxicity concerns;
|
|
3.
|
Five FDA-approved drugs (fexofenadine, nifedipine, verapamil, lidocaine and propranolol) that have certain measurable non-toxic cardiac effects consistent with clinical experience with such compounds. Note: fexofenadine is a non-cardiotoxic drug variant of terfenadine; and
|
|
4.
|
One research compound (E-4031) failed in Phase 1 human clinical study before being discontinued due to inducing heart arrhythmias.
|
We have validated that CardioSafe 3D is capable of assessing important electrophysiological activity of drugs or new drug candidates, including spike amplitude, beat period and field potential duration. Our CardioSafe 3D MEA assay, which we refer to as ECG in a test tube™, was reproducible and consistent with the known human cardiac effects of all twelve compounds studied, based on the mechanisms of action and dosage of the compounds. For instance, by using CardioSafe 3D, we were able to distinguish between the arrhythmogenic cardiac effects of terfenadine (Seldane™), withdrawn by the FDA due to cardiotoxicity, and the cardiac effects of the closely structurally-related compound, fexofenadine (Allegra™), a safe variant of terfenadine, which remains on the market. We believe our correlation data demonstrate that CardioSafe 3Dprovides valuable and more comprehensive bioanalytical tools for in vitro cardiac safety screening than the hERG assay. We believe CardioSafe 3D will contribute to our efficient and rapid identification of novel, potentially safer proprietary NCEs in our drug rescue programs.
CardioSafe 3D, Going Far Beyond the hERG Assay
The table below reflects the broad cardiotoxicity screening capabilities CardioSafe 3D, which we believe go far beyond what is possible to assess in vitro using the FDA-required hERG assay:
|
Detects cardiac effects mediated by:
|
hERG assay
|
CardioSafe 3D™
|
|
hERG potassium ion channels
|
ü
|
ü
|
|
Other potassium ion channels
|
ü
|
|
|
Calcium ion channels
|
ü
|
|
|
Sodium ion channels
|
ü
|
|
|
Interactions between ion channels
|
ü
|
|
|
Channel regulatory proteins
|
ü
|
|
|
Cell viability
|
ü
|
|
|
Apoptosis
|
ü
|
|
|
Mitochondria
|
ü
|
|
|
Energy
|
ü
|
|
|
Oxidative Stress
|
ü
|
CardioSafe 3D Assessment of Kinase Inhibitor-Induced Cardiotoxicity
To further evaluate the potential of CardioSafe 3D to predict cardiac toxicity of drug rescue NCEs in vitro, we have assessed cardiac effects induced by small molecule kinase inhibitors (“KIs”), which belong to a new category of drugs that have revolutionized cancer therapy due to decreased systemic toxicity and an increased anti-tumor cell specific effect compared to classic cancer drugs. Since 1998, the FDA has approved numerous small molecule KIs for cancer therapy. However, many of these FDA-approved KIs have been implicated in causing serious adverse cardiac events in patients which were not identified during drug development using traditional preclinical testing systems.
In our KI-induced cardiotoxicity study, we evaluated well-known anti-cancer KIs with CardioSafe 3D, some of which are FDA-approved and have been documented as cardiotoxic. This important validation set of anti-cancer KI compounds is as follows:
|
1.
|
Inhibitors of growth factor receptors: sunitinib, axitinib, imatinib, dasatinib, sorafenib, erlotinib, lapatinib, tyrphostin and AG1478;
|
|
|
2.
|
Inhibitors of the mTOR pathway: everolimus, temsirolimus;
|
|
|
3.
|
Inhibitors of cell cycle regulators: tozasertib, barasertib, alvocidib;
|
|
|
4.
|
Inhibitors of the PI3K pathway : perifosine, LY294002, XL765;
|
|
|
5.
|
Inhibitors of the MEK pathway: PD325901, AZD6264; and
|
|
|
6.
|
Inhibitors of the JAK and other pathways: lestaurtinib.
|
Our validation data indicate that CardioSafe 3D successfully detected cardiotoxicity induced by each of the representative compounds, consistent with adverse cardiac events observed in the clinic. CardioSafe 3D assay system is able to distinguish between cardiotoxic and safe compounds, and even between those compounds which inhibit the same kinase pathways. For instance, both sunitinib and axitinib inhibit VEGFR, PDGFR and c-Kit pathways, whereas our CardioSafe 3D assays indicate that sunitinib is cardiotoxic and axitinib is safe, which is consistent with the reported clinical outcomes.
Importantly, the CardioSafe 3D profile of each KI provided us clues to the potential biological mechanism(s) causing cardiac cytotoxicity. For example, cardiac cytotoxicity induced by perifosine was most potent for producing apoptotic responses, while imatinib was most potent for producing oxidative stress. In addition, no cardiac toxicity or alteration in electrophysiology was detected with drugs that do not have a cardiac liability, emphasizing the specificity of CardioSafe 3D. Having information on the biological pathways associated with the cardiac cytotoxic effects of compounds provides important clues for novel medicinal chemistry approaches and compound modifications for our CardioSafe 3D drug rescue programs.
Another example of the capability of our CardioSafe 3D assay system enabling the sensitive measurement of drug effects that are consistent with reported clinical responses are the results with sunitinib and dasatinib. CardioSafe 3D correctly identified that both compounds would cause QT prolongation, arrhythmia, and/or altered contraction rates, which are consistent with clinical observations.
We believe our CardioSafe 3D correlation data demonstrate that CardioSafe 3D will improve clinical predictivity as a more comprehensive and clinically-relevant in vitro cardiac safety assay system than the hERG assay, helping not only to identify potential cardiac toxicities early in development, but also to discover important potential biological mechanisms of cardiac cytotoxicity. We believe the results of our CardioSafe 3D validation studies indicate that CardioSafe 3D may be effectively used to identify NCEs with reduced cardiotoxicity for our pipeline by providing more accurate and timely indications of alterations in electrophysiological activity, as well as a more clinically relevant assessment of potential cardiac biological effects of drug candidates contributing to cardiac cytotoxicity, than animal models or the hERG assay currently used by pharmaceutical companies. We believe the results of our CardioSafe 3D validation studies support the central premise of our drug rescue business model: by using our hPSC-derived human heart and liver cell bioassay systems at the front end of the drug development process, we have the opportunity to take advantage of substantial prior investment by pharmaceutical companies and others in drug discovery and in vitro efficacy optimization of still-promising drug candidates that have been terminated prior to FDA approval due to unexpected heart or liver toxicity concerns.
LiverSafe 3D
We refer to the highly-functional, non-transformed, mature hPSC-derived hepatocytes we produce as VSTA-heps™. VSTA-heps are the foundation of LiverSafe 3D, a powerful new in vitro hepatotoxicity assay system that we believe goes a step beyond in vitro assays using commercially-available primary (human cadaver cell-based) hepatocytes. By combining the flexibility of an in vitro, non-transformed human hepatocyte-based assay system with genetically-consistent, functionally-reliable, and essentially unlimited production based on hPSCs, VSTA-heps, can be maintained in a healthy state for much longer than the primary hepatocytes used in FDA-required drug metabolism assays, greatly enhancing the reliability and predictability of our hepatotoxicity testing for our drug rescue programs.
Until now, reliable human cell-based hepatotoxicity screening platforms have been difficult to establish for high throughput drug development with currently available primary hepatocyte systems. Commercially available primary (cadaver) hepatocytes are in short supply, are genetically variable, functionally inconsistent, and have a short lifespan in culture, during which they rapidly lose their drug metabolizing capabilities and develop signs of cellular stress. Commercially-available primary hepatocytes also have significant batch-to-batch genetic and functional activity that varies widely batch-to-batch. Primary hepatocytes are derived from individuals with significant genetic differences, unknown differences in health status, and widely different drug exposures, each potentially contributing significant but unquantifiable effects on hepatocyte function, resulting in very large unpredictable ranges of drug metabolism activity. Consequently, it is difficult to maintain reproducible quantitative measurements in drug testing assays using currently available primary (cadaver) hepatocyte assays. This leads to limitations in the quality, reliability, and clinical predictivity of the results and conclusions drawn assays based on primary (cadaver) hepatocytes.
We believe VSTA-heps overcome the foregoing limitations of primary hepatocytes. Our VSTA-heps are derived from the same hPSC line, are genetically identical, normal, non-transformed (that is, not tumor-derived) human cells capable of being produced in essentially unlimited supply. Importantly, VSTA-heps can be indefinitely produced and, we believe, frozen for storage into large, uniform, quality-controlled cell banks.
The table below reflects important characteristics of VSTA-heps compared to commercially-available primary hepatocytes used in FDA-required drug metabolism studies:
|
Characteristics of in vitro hepatocyte assays:
|
Primary hepatocytes
|
VSTA-heps™
|
|
Human cells
|
ü
|
ü
|
|
Liver enzyme activity
|
ü
|
ü
|
|
Within batch reproducibility
|
ü
|
ü
|
|
Batch-to-batch reproducibility
|
ü
|
|
|
Long term culture
|
ü
|
|
|
Maintenance of function in culture
|
ü
|
|
|
Parental cells can be expanded into large batches
|
ü
|
|
|
Uniform genetic background between batches
|
ü
|
|
|
Uniform donor health status between batches
|
ü
|
|
|
Gene “reporters” can be genetically inserted
|
ü
|
VSTA-heps and CYP3A4 Enzyme Expression for Drug Metabolism
In the past, the challenge to using hPSC-derived hepatocytes has been differentiating the stem cells into mature hepatocytes that express a full complement of functional drug metabolizing enzymes, nuclear receptors, and transporters at least as well as primary hepatocytes. While many groups have taken on this challenge in recent years, published reports indicate that current hPSC differentiation protocols yield immature hepatocytes, especially with respect to extremely low expression of certain key adult drug metabolizing enzymes, such as CYP3A4. CYP3A4 is a critical liver enzyme responsible for metabolizing approximately one-third of the FDA-approved drugs currently available on the market. It is an important and well-accepted functional gene found almost exclusively in mature, adult hepatocytes. CYP3A4 is the key functional marker that we have used to optimize our VSTA-hep differentiation cultures for LiverSafe 3D. We believe our optimized LiverSafe 3D assay system enables us to generate more mature hPSC-derived hepatocytes than are currently available from others in the field and that our LiverSafe 3D system provides the unique ability to specifically select for mature CYP3A4-expressing human hepatocytes.
We developed LiverSafe 3D using hPSC differentiation protocols adapted from the laboratory of our co-founder, Dr. Gordon Keller, and our proprietary hPSC cell line, 3A4BLA. This 3A4BLA cell line is a hESC line that contains a humanized BLA “reporter” that is placed in the CYP3A4 gene in a manner resulting in the expression of BLA only in cells that also express CYP3A4. This allows us to visualize by fluorescence cells that express CYP3A4 based on expression of the BLA reporter. By producing a cell line capable of tracking CYP3A4 expression, we have been able to optimize our hPSC differentiation protocols to increase expression of mature hepatocyte markers and drug metabolizing enzymes and to enrich for CYP3A4-expressing cells by cell sorting. However, even in the absence of cell sorting, our LiverSafe 3D hepatocyte populations contain greater than 80% albumin-positive cells and greater than 40% CYP3A4-positive cells, with CYP3A4 mRNA expression reaching levels nearly 60-fold higher than side-by-side 38-week human fetal liver controls. Our VSTA-heps secrete urea and albumin, functional markers of hepatocytes, at levels that exceed commercially-available primary (cadaver) hepatocytes. They also store both glycogen and lipids, which are additional characteristics required of functional, mature adult hepatocytes. Importantly, expression of fetal liver markers decreases over the time course of maturation of our VSTA-heps. This transition to a more mature state with decreased fetal gene expression is expected and essential for the production of adult functional hepatocytes, but it has rarely been reported by others in publications describing their hPSC-derived hepatocytes. With the addition of cell sorting, our VSTA-heps can be highly enriched for CYP3A4-BLA-positive cells, with CYP3A4 message in the positive cell population reaching greater than 30% that of an adult human liver pool control. To our knowledge, this level of CYP3A4 expression exceeds levels reported by others in the literature.
The most important capabilities of LiverSafe 3D relate to “Phase I” and “Phase II” drug metabolism, which are functional characteristics of mature adult hepatocytes. We have validated these capabilities of LiverSafe 3D by demonstrating its ability to metabolize known substrates, such as testosterone, and its ability to respond properly to known inducers of Phase I-mediated CYP3A4 metabolism, such as rifampicin. Moreover, our VSTA-heps demonstrate Phase II-mediated testosterone metabolism levels that exceed commercially-available primary hepatocytes. These functional characteristics of mature adult hepatocytes are critical to the development of a reliable and clinically predictive hepatotoxicity screening platform for our drug rescue programs. We are currently focused on expanding our panel of validation assays and compounds to include more P450 substrates, inducers, and inhibitors, as well as adapting the cellular toxicity assays that have been developed for our CardioSafe 3D assay system to our LiverSafe 3D assay system and to apply specific functional screening, such as albumin and urea secretion assays.
We believe LiverSafe 3D with VSTA-heps offers the capability of producing a genetically-identical, renewable, and reproducible hepatotoxicity assay system for drug rescue and development that provides advantages over in vitro assays using commercially-available primary hepatocytes. In addition, it offers the ability to produce hepatocyte assays that contain common genetic variations in drug metabolizing genes that are expressed in subsets of individuals, and therefore drug development. We have demonstrated that our VSTA-heps, even in the absence of cell sorting, secrete adult hepatocyte levels of albumin and urea and contain greater than 40% CYP3A4-positive cells, historically difficult to achieve in hPSC differentiation cultures. The proprietary 3A4BLA cell line component of LiverSafe 3D allows us the unique opportunity to enrich CYP3A4-positive cells, resulting in CYP3A4 expression reaching greater than 30% of an adult human liver pool, and to the best of our knowledge, a level higher than described in current literature. Most importantly, for drug rescue and development purposes, our VSTA-heps are the foundation of LiverSafe 3D and metabolize known substrates and respond to known inducers in a manner expected only of mature adult hepatocytes, paving the way for our final validation of LiverSafe 3D system as a novel, clinically-relevant hepatotoxicity assay system that can improve clinical predictivity, decrease the cost of drug development, reduce reliance on live animal studies, and improve drug safety.
Using Stem Cell Technology to Produce and Develop Drug Rescue NCEs
We believe using CardioSafe 3D and LiverSafe 3D for our drug rescue programs is the highest-value near term commercial application of the human cells we produce and the novel, customized bioassay systems we have designed and developed. Our drug rescue activities are focused on producing for our internal pipeline proprietary, safer variants of still-promising NCEs previously discovered, optimized and tested for efficacy by pharmaceutical companies and others but terminated before FDA approval due to unexpected heart toxicity or liver toxicity. Our drug rescue strategy involves using CardioSafe 3D and LiverSafe 3D to assess the toxicity that caused certain NCEs available in the public to be terminated, and use that biological insight to produce and develop a new, potentially safer, and proprietary NCEs for our pipeline. We believe the pre-existing public domain knowledge base supporting the therapeutic and commercial potential of NCEs we target for our drug rescue programs will provide us with a valuable head start as we launch each of our drug rescue programs. Leveraging the substantial prior investments by global pharmaceutical companies and others in discovery, optimization and efficacy validation of the NCEs we identify in the public domain is an essential component of our drug rescue strategy.
Our current drug rescue emphasis is on NCEs discontinued prior to FDA market approval due to unexpected cardiac safety concerns. By using CardioSafe 3D to enhance our understanding of the cardiac liability profile of NCEs, biological insight not previously available when the NCEs were originally discovered, optimized for efficacy and developed, we believe we can demonstrate preclincial proof-of-concept (POC) as to the efficacy and safety of new, safer drug rescue NCEs in standard in vitro and in vivo models, as well as in CardioSafe 3D, earlier in development and with substantially less investment in discovery and preclinical development than was required of pharmaceutical companies and others prior to their decision to terminate the original NCE.
We have assessed, and established a CardioSafe 3D cardiotoxicity profile for several drug rescue candidates.
We are now preparing to commence our initial CardioSafe 3D drug rescue program. Our goal in each drug rescue program will be to produce a proprietary drug rescue NCE and establish its preclinical POC, using standard preclinical in vitro and in vivo efficacy and safety models, as well as CardioSafe 3D. In this context, POC means that the lead drug rescue NCE, as compared to the original, previously-terminated NCE, demonstrates both (i) equal or superior efficacy in the same, or a similar, in vitro and in vivo preclinical efficacy models used by the initial developer of the previously-terminated NCE before it was terminated for safety reasons, and (ii) significant reduction of concentration dependent cardiotoxicity in CardioSafe 3D.
We believe our focus on producing proprietary drug rescue NCEs for our internal pipeline based on previously terminated NCEs with therapeutic and commercial potential established by others, and our ability to build on that valuable head start with our novel biological and electrophysiological insight regarding cardiac effects of NCEs that we can generate with CardioSafe 3D, will help us and our medicinal chemistry partner produce and optimize drug rescue NCEs without incurring many of the high costs and risks typically inherent in new drug discovery and preclinical development. Although we plan to continue to identify NCEs for our drug rescue programs in the public domain, we may also seek to acquire rights to previously terminated, but still-promising, NCEs not available to us in the public domain by entering into contractual arrangements with third-parties.
Strategic Development and Commercialization of Drug Rescue NCEs
As a result of research and development productivity issues and diminishing product pipelines, as well as generic competition for established products that are no longer patent protected, we believe there is and will continue to be a critical need among pharmaceutical companies to acquire or in-license the new, safer drug rescue NCEs we are focused on producing and developing, including companies that originally discovered, developed and ultimately discontinued the previously-terminated NCEs we select for drug rescue.
Once we optimize a patentable drug rescue NCE, we intend to develop it internally to establish preclinical POC in established in vitro and in vivo efficacy and safety models, as well as in CardioSafe 3D. After we establish preclinical POC of a patentable drug rescue NCE, we will decide between continuing to develop it internally and out-licensing it to a pharmaceutical company. If we license it to the pharmaceutical company, it will be responsible for all subsequent development, manufacturing, regulatory approval, marketing and sale of the drug rescue NCE and we will generate revenue through payments to us from the license upon signing the license agreement, achievement of development and regulatory milestones, and, if approved and marketed, upon commercial sales, although no assurances can be given that we will seek and complete a partnership, or that the terms of such a beneficial arrangement will be available or offered to us.
Regenerative Medicine and Drug Discovery
Although we believe the best and most valuable near term commercial application of our stem cell technology platform is for small molecule drug rescue, we also believe stem cell technology-based drug discovery and regenerative medicine has the potential to transform healthcare in the U.S. over the next decade by providing new approaches for treating the fundamental mechanisms of disease. We currently intend to explore opportunities to leverage our stem cell technology platform, our expertise in human biology, differentiation of human pluripotent stem cells to develop functional adult human cells and tissues involved in human disease, including blood, bone, cartilage, heart and liver cells, and our expertise in designing and developing novel, customized biological assay systems with the cells we produce, for regenerative medicine purposes, with emphasis on developing novel human disease models for discovery of small molecule drugs with regenerative and therapeutic potential. Among our key objectives will be to assess our regenerative medicine opportunities through exploratory nonclinical POC studies.
Strategic Transactions and Relationships
Strategic collaborations are an important cornerstone of our corporate development strategy. We believe that our strategic outsourcing and sponsorship of application-focused research gives us flexible access to medicinal chemistry, hPSC research and development, manufacturing, clinical development and regulatory expertise at a lower overall cost than developing and maintaining such expertise internally. In particular, we collaborate with the types of third parties identified below for the following functions:
|
·
|
academic research institutions, such as the University Health Network for hPSC technology research and development;
|
|
·
|
contract medicinal chemistry companies, such as Synterys, Inc., to design, produce and analyze drug rescue NCEs; and
|
|
·
|
contract clinical development and regulatory organizations (“CROs”), such as Cato Research, Ltd., for regulatory expertise and clinical development support.
|
Cato Research
Cato Research is a CRO with international resources dedicated to helping biotechnology and pharmaceutical companies navigate the regulatory approval process in order to bring new biologics, drugs and medical devices to markets throughout the world. Cato Research is our CRO for development of AV-101. Cato Research has in-house capabilities to assist its sponsors with aspects of the drug development process including regulatory strategy, nonclinical and toxicology development, clinical development, data processing, data management, statistical analysis, regulatory applications, including INDs and NDAs, chemistry, manufacturing, and control programs, cGCP, cGLP and cGMP audit and compliance activities, and due diligence review of emerging technologies. Cato Research’s senior management team, including co-founders Allen Cato, M.D., Ph.D. and Lynda Sutton, has over 25 years of experience interacting with the FDA and international regulatory agencies and a successful track record of product approvals. Based on our long-term working relationship with Cato Research in connection with the development of AV-101, should we elect to advance development of Drug Rescue Variants internally, as we have done with AV-101, rather than license or sell them to pharmaceutical companies or others, we believe our long term strategic relationship with Cato Research provides us with real time access to the global connections, insight and knowledge necessary to effectively plan, execute and manage successful nonclinical and clinical development programs throughout the world without incurring the substantial expenses typically associated with establishing and maintaining a wide range of drug development capabilities in-house.
Cato BioVentures
Cato Holding Company, doing business as Cato BioVentures, is the venture capital affiliate of Cato Research. Through strategic CRO service agreements with Cato Research, Cato BioVentures invests in therapeutics and medical devices, as well as platform technologies such as our stem cell technology platform, which its principals believe, based on their experience as management of Cato Research, are capable of transforming the traditional drug development process and the research and development productivity of the biotechnology and pharmaceutical industries.
As a result of the access Cato Research has to potential drug rescue NCEs from its biotechnology and pharmaceutical industry network, as well as Cato BioVentures’ strategic long term equity interest in the Company, we believe that our relationships with Cato BioVentures and Cato Research may provide us with unique opportunities relating to our drug rescue efforts that will permit us to leverage both their industry connections and the CRO resources of Cato Research, either on a contract research basis or in exchange for economic participation rights, should we develop drug rescue NCEs s internally rather than out-license them to strategic partners.
University Health Network, McEwen Centre for Regenerative Medicine
University Health Network (“UHN”) in Ontario, Canada is a major landmark in Canada’s healthcare system. UHN is one of the world’s largest research hospitals, with major research in transplantation, cardiology, neurosciences, oncology, surgical innovation, infectious diseases and genomic medicine.
The McEwen Centre for Regenerative Medicine (“McEwen Centre”) is a world-renowned center for stem cell biology and regenerative medicine and a stem cell research facility affiliated with UHN. Dr. Gordon Keller, our co-founder and Chairman of our Scientific Advisory Board, is Director of the McEwen Centre. Dr. Keller’s lab is considered one of the leaders in successfully applying principles from the study of developmental biology of many animal systems to the differentiation of pluripotent stem cell systems, resulting in reproducible, high-yield production of human heart, liver, blood and vascular cells. The results and procedures developed in Dr. Keller’s lab are often quoted and used by academic scientists worldwide.
In September 2007, we entered into a long-term sponsored stem cell research and development collaboration with UHN. In December 2010, we extended the collaboration to September 2017. The primary goal of this ten-year collaboration is to leverage the stem cell research, technology and expertise of Dr. Gordon Keller to develop and commercialize industry-leading human pluripotent stem cell differentiation technology and bioassay systems for drug rescue and development and regenerative cell therapy applications. This sponsored research collaboration builds on our existing strategic licenses from National Jewish Health and the Icahn School of Medicine at Mount Sinai to certain pluripotent stem cell technologies developed by Dr. Keller, and is directed to diverse human pluripotent stem cell-based research projects, including, as expanded and amended, strategic projects related to drug rescue and regenerative medicine. See “Sponsored Research Collaborations and Intellectual Property Rights – University Health Network, McEwen Centre for Regenerative Medicine, Toronto, Ontario”, “Intellectual Property – National Jewish Health Exclusive Licenses” and “Intellectual Property – Icahn School of Medicine at Mount Sinai Exclusive Licenses.”
Cardiac Safety Research Consortium
We have joined the Cardiac Safety Research Consortium (“CSRC”) as an Associate Member. The CSRC, which is sponsored in part by the FDA, was launched in 2006 through an FDA Critical Path Initiative Memorandum of Understanding with Duke University to support research into the evaluation of cardiac safety of medical products. CSRC supports research by engaging stakeholders from industry, academia, and government to share data and expertise regarding several areas of cardiac safety evaluation, including novel stem cell-based approaches, from preclinical through post-market periods.
Cardiac Safety Technical Committee of the Health and Environmental Sciences Institute – FDA’s CIPA Initiative
We have also joined the Cardiac Safety Technical Committee, Cardiac Stem Cell Working Group, and Proarrhythmia Working Group of the Health and Environmental Sciences Institute (“HESI”) to help advance, among other goals, the FDA’s Comprehensive In Vitro Proarrhythmia Assay (“CIPA”) initiative, which is focused on developing innovative preclinical systems for cardiac safety assessment during drug development. HESI is a global branch of the International Life Sciences Institute (“ILSI”), whose members include most of the world’s largest pharmaceutical and biotechnology companies.
The goal of the FDA’s CIPA initiative is to develop a new paradigm for cardiac safety evaluation of new drugs that provides a more comprehensive assessment of proarrhythmic potential by (i) evaluating effects of multiple cardiac ionic currents beyond hERG and ICH S7B Guidelines (inward and outward currents), (ii) providing more complete, accurate assessment of proarrhythmic effects on human cardiac electrophysiology, and (iii) focusing on Torsades de Pointes proarrhythmia rather than surrogate QT prolongation alone.
Centre for Commercialization of Regenerative Medicine
The Toronto-based Centre for Commercialization of Regenerative Medicine (“CCRM”) is a not-for-profit, public-private consortium funded by the Government of Canada, six Ontario-based institutional partners and more than 20 companies representing the key sectors of the regenerative medicine industry. CCRM supports the development of foundational technologies that accelerate the commercialization of stem cell- and biomaterials-based products and therapies.
We are a member of the CCRM’s Industry Consortium. Other members of CCRM’s Industry Consortium include Pfizer and GE Healthcare. The industry leaders that comprise the CCRM consortium benefit from proprietary access to certain licensing opportunities, academic rates on fee-for-service contracts at CCRM and opportunities to participate in large collaborative projects, among other advantages. Our CCRM membership reflects our strong association with CCRM and its core programs and objectives, both directly and through our strategic relationships with Dr. Gordon Keller and UHN. We believe our long-term sponsored research agreement with Dr. Keller, UHN and UHN’s McEwen Centre offers unique opportunities for expanding the commercial applications of our stem cell technology platform by building multi-party collaborations with CCRM and members of its Industry Consortium. We believe these collaborations have the potential to transform medicine and accelerate significant advances in human health and wellness that stem cell technologies and regenerative medicine promise.
United States National Institutes of Health
Since our inception in 1998, the U.S. National Institutes of Health (“NIH”) has awarded us $11.3 million in non-dilutive research and development grants, including $2.3 million to support research and development of our stem cell technology and $8.8 million for nonclinical and Phase 1 clinical development of AV-101.
United States National Institute of Mental Health
The U.S. National Institute of Mental Health (“NIMH”), part of the NIH, is the largest scientific organization in the world dedicated to mental health research. NIMH is one of 27 Institutes and Centers of the NIH, the world’s leading biomedical research organization. The mission of NIMH is to transform the understanding and treatment of mental illnesses through basic and clinical research, paving the way for prevention, recovery and cure. In February 2015, we entered into a Cooperative Research and Development Agreement (“CRADA”) with the NIH providing for the initial AV-101 Phase 2 efficacy and safety in Major Derpessive Disorder. The study will be fully funded by the NIH and will be conducted at the NIMH by Dr. Carlos Zarate. Dr. Zarate is the NIMH’s Chief of Experimental Therapeutics & Pathophysiology Branch and Section on Neurobiology and Treatment of Mood and Anxiety Disorders.
Synterys, Inc.
We have entered into a strategic medicinal chemistry collaboration agreement with Synterys, Inc., a medicinal chemistry and collaborative drug discovery company. We believe this important collaboration will further our drug rescue initiatives with the support of Synterys’ medicinal chemistry expertise. In addition to providing flexible, real-time contract medicinal chemistry services in support of our drug rescue programs, we anticipate potential collaborative opportunities with Synterys wherein we may jointly identify and develop drug rescue NCEs.
Intellectual Property
AV-101
We have developed a portfolio of intellectual property assets around AV-101 that involves both patents and trade secrets. We obtained a multi-patent license from the University of Maryland to certain pharmaceutical formulations of AV-101 and related compounds when we acquired the original licensee, Artemis Neuroscience, Inc. The license included a composition and therapeutic method patent relevant to AV-101, which was originally issued to Merrell Pharmaceuticals and expired in 2011. In early 2013, we filed a provisional application based on discoveries related to specific dosages and dosage ranges of AV-101, as well as to methods of treating depression and hyperalgesia pain. This corresponding PCT application (WO2014/116739) has recently been filed as a national phase application in the US and other countries. This PCT patent application also presents method-of-use claims associated with such dosages. And it includes other claims based on discoveries related to novel clinical activities that are not limited by dose range. In the course of our research and development prior to our Phase 1 clinical studies, our CROs developed two novel methods of synthesizing AV-101, based on extensive research into a range of synthetic routes. As a result, in 2013, we filed two additional provisional applications to these commercially useful production methods, both of which are also now pending as PCT patent applications (WO2014/152752 and WO2014/152835). One of these two PCT patent applications includes pharmaceutical composition claims to a sulfated derivative of 4-chlorokynurenine. A fourth patent application related to additional and expanded clinical uses of AV-101 was recently filed in the U.S. as a provisional application. We plan to pursue national phase applications broadly for all four cases in appropriate global markets.
In addition, among the key components of our commercial protection strategy with respect to AV-101 is the New Drug Product Exclusivity provided by the FDA under section 505(c)(3)(E) and 505(j)(5)(F) of the Federal Food, Drug, and Cosmetic Act (“FDCA”). The FDA’s New Drug Product Exclusivity is available for NCEs such as AV-101, which, by definition, are innovative and have not been approved previously by the FDA, either alone or in combination. The FDA’s New Drug Product Exclusivity protection provides the holder of an FDA-approved new drug application (“NDA”) up to five (5) years of protection from new competition in the U.S. marketplace for the innovation represented by its approved new drug product. This protection precludes FDA approval of certain generic drug applications under section 505(b)(2) of the FDCA, as well certain abbreviated new drug applications (“ANDAs”), during the up to five-year exclusivity period, except that such applications may be submitted after four years if they contain a certification of patent invalidity or non-infringement. We will pursue similar types of regulatory exclusivity in other countries.
Our license agreement related to AV-101 with the University of Maryland, mentioned above, requires us to make royalty payments on net sales of products covered by the licensed patent rights, which have expired. There are no up-front license, milestone or maintenance fees under the agreement. The agreement provides that these royalty obligations will remain in force until 10 years after the first commercial sale of the first product even if the licensed patent rights have expired. However, the U.S. Supreme Court’s recent decision in Kimble v. Marvel Entertainment, LLC determined that patent license royalties that extend beyond a patent’s expiration are not enforceable. Management will be reviewing the impact of this court decision on our license. This agreement may also be terminated earlier at the election of the licensor upon our failure to pay any monies due, our failure to provide updates and reports to the licensor, our failure to provide the necessary financial and other resources required to develop the products, or our failure to cure within 90 days any breach of any provision of the agreement. We may also terminate the agreement at any time upon 90 days’ written notice so long as we make all payments due through the effective date of termination.
Stem Cell Technology
We have established intellectual property rights to stem cell technology through a combination of exclusive and non-exclusive patent and technology licenses, our own patent properties, and trade secrets. To our knowledge, we are the first stem cell company focused on stem cell technology-based drug rescue. We have assembled an intellectual property portfolio around the use of pluripotent stem cell technologies in drug discovery and development and with specific application to drug rescue. The differentiation protocols we have licensed licenses direct the differentiation of pluripotent stem cells through:
|
·
|
a combination of growth factors (molecules that stimulate the growth of cells);
|
|
·
|
the experimentally controlled regulation of developmental genes, which is critical for determining what differentiation path a human cell will take; and
|
|
·
|
precise selection of immature cell populations for further growth and development.
|
By influencing key branch points in the cellular differentiation process, our pluripotent stem cell technologies can produce fully differentiated, non-transformed, highly functional human cells in vitro in an efficient, highly pure and reproducible process.
Licenses
National Jewish Health (“NJH”) Exclusive License
We have an exclusive patent license from NJH. The remaining unexpired patent property is a US patent that contains claims covering composition of matter relating to specific immature multipotent cell populations and applications of such cells for ES Cell-derived immature multipotent precursors of all the cells of the mesoderm and endoderm lineages. Among other cell types, this covers cells of the heart, liver, pancreas, blood, connective tissues, vascular system, gut and lung cells.
This license agreement requires us to pay NJH a percentage of our total revenues for sales of products made under the license in each calendar year, together with minimum annual payments. Additionally, this license agreement requires us to make certain royalty payments on sublicensing of such technology. However, the license agreement also includes anti-stacking provisions which reduce our payment obligations by a percentage of any royalty payments and fees paid to third parties, who have licensed necessary intellectual property to us. This agreement remains in force for the life of the patents so long as neither party elects to terminate the agreement upon the other party’s uncured breach or default of an obligation under the agreement. We also have the right to terminate the agreement at any time without cause.
Icahn School of Medicine at Mount Sinai School (MSSM) Exclusive License
We have an exclusive, field restricted, license to three U.S. patents, three U.S. patent applications, and several foreign counterparts filed by MSSM. Foreign patents have been issued and maintained in Canada, Australia and Singapore. These patent properties have claims covering composition of matter relating to specific populations of cells and precursors, methods to produce such cells, and applications of such cells, including:
|
•
|
the use of certain growth factors to generate mesoderm (that is, the precursors capable of developing into cells of the heart, blood system, connective tissues, and vascular system) from hESCs;
|
|
•
|
the use of certain growth factors to generate endoderm (that is, the precursors capable of developing into cells of the liver, pancreas, lungs, gut, intestines, thymus, thyroid gland, bladder, and parts of the auditory system) from hESCs; and
|
|
•
|
applications of cells derived from mesoderm and endoderm precursors, especially those relating to drug discovery and testing for applications in the field of in vitro drug discovery and development applications.
|
This license agreement requires us to pay annual license fees, patent prosecution and maintenance fees and royalty payments that vary based on product sales and services that are covered by the MSSM patent rights, as well fees for sublicensing. Any drug candidates that we develop, including any drug rescue NCEs, will only require royalty payments to the extent, if any, they require the practice of the licensed technology. To the extent we incur royalty payment obligations from other business activities, the royalty payments are subject to anti-stacking provisions which reduce our payments by a percentage of any royalty payments or fees paid to third parties who have licensed necessary intellectual property to us. The license agreement will remain in force for the life of the patents so long as neither party terminates the agreement for cause (i) due to a material breach or default in performance of any provision of the agreement that is not cured within 60 days or (ii) in the case of failure to pay amounts due within 30 days.
Drug rescue technologies
We have filed two U.S. patent applications on liver stem cells and their applications in drug development relating to toxicity testing; one patent has issued and a second patent application is pending. Of the related international filings, European, Canadian and Korean patents were issued. The European patent has been registered in 11 European countries. We have filed a U.S. patent application, with foreign counterpart filing in Canada and Europe, directed to methods for producing human pluripotent stem cell-derived endocrine cells of the pancreas, with a specific focus on beta-islet cells, the cells that produce insulin, and their uses in diabetes drug discovery and screening.
Our patent portfolio currently related to the generation of human heart and liver cells for use in connection with our drug rescue activities include:
|
Territory
|
Patent No.
|
General Subject Matter
|
Expiration
|
||||
|
US
|
7,763,466
|
Method to produce endoderm cells
|
May 2025
|
||||
|
US
|
7,955,849
|
Method of enriching population of mesoderm cells
|
May 2023
|
||||
|
US
|
8,143,009
|
Toxicity typing using liver stem cells
|
June 2023
|
||||
Trade Secrets
We rely, in part, on trade secrets for protection of some of our intellectual property. We attempt to protect trade secrets by entering into confidentiality agreements with third parties, employees and consultants. Our employees and consultants also sign agreements requiring that they assign to us their interests in patents and copyrights arising from their work for us.
Our Patents
We have filed two U.S. patent applications on liver stem cells and their applications in drug development relating to toxicity testing; one patent has issued and a second patent application is pending. Of the related international filings, European, Canadian and Korean patents were issued. The European patent has been registered in 11 European countries. We have filed a U.S. patent application, with foreign counterpart filing in Canada and Europe, directed to methods for producing human pluripotent stem cell-derived endocrine cells of the pancreas, with a specific focus on beta-islet cells, the cells that produce insulin, and their uses in diabetes drug discovery and screening.
The material patents currently related to the generation of human heart and liver cells for use in connection with our drug rescue activities are set forth below:
|
Territory
|
Patent No.
|
General Subject Matter
|
Expiration
|
||||
|
US
|
7,763,466
|
Method to produce endoderm cells
|
May 2025
|
||||
|
US
|
7,955,849
|
Method of enriching population of mesoderm cells
|
May 2023
|
||||
|
US
|
8,143,009
|
Toxicity typing using liver stem cells
|
June 2023
|
||||
With respect to AV-101, we have filed four new patent applications, as noted above.
Trade Secrets
We rely, in part, on trade secrets for protection of some of our intellectual property. We attempt to protect trade secrets by entering into confidentiality agreements with third parties, employees and consultants. Our employees and consultants also sign agreements requiring that they assign to us their interests in patents and copyrights arising from their work for us.
Sponsored Research Collaborations and Intellectual Property Rights
University Health Network, McEwen Centre for Regenerative Medicine, Toronto, Ontario
Our strategic relationship with our co-founder, Dr. Gordon Keller, Director of the UHN’s McEwen Centre, is focused on, among other things, developing improved methods for differentiation of cardiomyocytes (heart cells) from hPSCs, and their uses in bioassay systems for drug discovery and drug development, including drug rescue. Pursuant to our sponsored research collaboration agreement with UHN, we have acquired exclusive worldwide rights to patent applications in the U.S. and foreign countries on multiple inventions arising from studies we have sponsored, under pre-negotiated license terms. Such pre-negotiated terms provide for royalty payments based on product sales that incorporate the licensed technology and milestone payments based on the achievement of certain events. Any drug rescue NCEs that we develop will not incorporate the licensed technology and, therefore, will not require any royalty payments. To the extent we incur royalty payment obligations from other business activities, the royalty payments will be subject to anti-stacking provisions, which reduce our payments by a percentage of any royalty payments paid to third parties who have licensed necessary intellectual property to us. These licenses will remain in force for so long as we have an obligation to make royalty or milestone payments to UHN, but may be terminated earlier upon mutual consent, by us at any time, or by UHN for our breach of any material provision of the license agreement that is not cured within 90 days.
The sponsored research collaboration agreement (“SRCA”) with UHN, as amended, has a term of ten years, ending on September 18, 2017. We are currently in discussions with Dr. Keller and UHN regarding the scope of potential new sponsored research projects under the SRCA. The ten-year term of the agreement is subject to renewal upon mutual agreement of the parties. The agreement may be terminated earlier upon a material breach by either party that is not cured within 30 days. UHN may elect to terminate the agreement if we become insolvent or if any license granted pursuant to the agreement is prematurely terminated. We have the option to terminate the agreement if Dr. Keller stops conducting his research or ceases to work for UHN.
UHN Licenses for Stem Cell Culture Technology
In October 2011, we licensed stem cell culture technology from UHN’s McEwen Centre pursuant to the SCRA. This exclusive license conveyed rights to a patent application entitled “Methods for enriching pluripotent stem cell derived cardiomyocyte progenitor cells and cardiomyocyte (heart) cells based on SIRPA expression” covered by U.S. Provisional 61/377,665 and PCT patent application WO/2012/024782, and any related patent application claiming priority from these. This technology involves a heretofore unknown cell surface protein, SIRPA (signal-regulatory protein alpha) that is expressed by early immature precursors for cardiomyocytes. Antibodies specific to SIRPA allow the identification and enrichment of these early cardiomyocyte precursors, which we believe will provide benefits in terms of purity, functionality and reproducibility for not only CardioSafe 3D in vitro safety assays for drug screening and development, but also potentially for production of cardiomyocytes for cell therapy and regenerative medicine applications.
In April 2012, we licensed stem cell culture technology from UHN’s McEwen Centre pursuant to the SCRA. The licensed technology may be used to develop hematopoietic precursor stem cells from human pluripotent stem cells, with the goal of developing drug discovery screening and regenerative medicine applications for human blood system disorders. This technology is covered by U.S. Provisional US61/562,094 and PCT patent application WO/2013/075222, and any related patent application claiming priority from these. We believe this stem cell technology dramatically advances our ability to produce and purify this important blood stem cell precursor for both in vitro drug discovery screening and potential regenerative medicine applications. In addition to defining new cell culture methods for our use, the technology describes the surface characteristics of stem cell-derived adult hematopoietic stem cells. Most groups study embryonic blood development from stem cells, but we are able to not only purify the stem cell-derived precursor of all adult hematopoietic cells, but also pinpoint the precise timing when adult blood cell differentiation takes place in these cultures. We believe these early cells have the potential to be the precursors of the ultimate adult, bone marrow-repopulating hematopoietic stem cells to repopulate the blood and immune system when transplanted into patients prepared for bone marrow transplantation. These cells have important potential therapeutic applications for the restoration of healthy blood and immune systems in individuals undergoing transplantation therapies for cancer, organ grafts, HIV infections or for acquired or genetic blood and immune deficiencies.
In December 2014, we licensed stem cell culture technology from UHN’s McEwen Centre pursuant to the SCRA. This exclusive license conveyed rights to a patent application entitled “Methods for generating hepatocytes and cholangiocytes from pluripotent stem cells” covered by PCT patent application WO/2014/124527, and any future patent application claiming priority from it. The licensed technology describes advanced methods for the production of mature hepatocytes and cholangiocytes, the primary cell types of the liver. The liver plays an important role in many bodily functions including protein production, blood clotting, as well as glucose, iron and lipid metabolism. Hepatocytes are the major cells responsible for metabolizing drugs, drug-drug interactions, and are the target for a variety of liver diseases including drug-induced liver failure, Cirrhosis, and viral infections. Cholangiocytes are the precursors for the biliary system found in the liver, i.e. bile ducts and gallbladder. The biliary system is a significant target for many conditions, including drug toxicities, cholecystitis, and liver-related abnormal function associated with the cystic fibrosis mutation. The licensed technology now enables us to more efficiently produce, human hepatocytes and cholangiocytes with more adult-like functions for in vitro drug discovery and LiverSafe 3D toxicity assays to support our drug rescue programs, as well as the therapeutic potential for cell-based therapies.
In December 2014, we licensed stem cell culture technology from UHN’s McEwen Centre pursuant to the SCRA. This exclusive license conveyed rights to patent application entitled “Methods and Compositions for Generating Epicardium Cells” covered by PCT patent application WO/2015/035506 application, and any future patent application claiming priority from it. The epicardium is the outer cell layer on top of the heart muscle (cardiomyocytes), and is essential for proper development of the heart and plays an important role in cardiac recovery during disease. The epicardium plays a critical role in the differentiation, expansion, and maturation of cardiomyocytes during development, or during cardiac repair responses. This technology will be important to developing the next generation of engineered cardiac tissue, or their function in cell therapy approaches.
In December 2014, we licensed stem cell culture technology from UHN’s McEwen Centre pursuant to the SCRA. This exclusive license conveyed rights to patent application entitled “Methods and compositions for generating chondrocyte lineage cells and/or cartilage like tissue” covered by PCT patent application WO/2014/161075 application, and any future patent application claiming priority from it. There are two type of chondrocytes, “articular” and “growth plate”. Articular chondrocytes are responsible for cartilage that lines our joints, whereas growth plate chondrocytes are involved with new bone formation. Osteoarthritis is debilitating joint diseases resulting from the degeneration of articular cartilage leading to inappropriate bone development (spurs) in the joint. These technologies will allow us to develop in vitro assays to study the process of the degeneration of articular cartilage, and provides novel tools for testing drugs that have the potential to reduce this degeneration. These cells also provide the necessary cells for developing cell therapy approaches for treating osteoarthritis.
Competition
The biopharmaceuticals industry is highly competitive. There are many public and private biopharmaceutical companies, universities, governmental agencies and other research organizations actively engaged in the research and development of products that may be similar to our product candidates or address similar markets. It is probable that the number of companies seeking to develop products and therapies similar to our products will increase.
Currently, there are no FDA-approved therapies for MDD with the mechanism of action of AV-101. However, products approved for other indications, for example, the anesthetic ketamine, are being or may be used off-label for treatment of MDD, as well as other CNS indications for which AV-101 may have therapeutic potential. Additionally, other treatment options, such psychotherapy and electroconvulsive therapy, are sometimes used instead of and before antidepressant medications to treat patients with MDD.
In the field of new generation antidepressants focused on modulation of the NMDAR, our principal competitor is Naurex, Inc., which is developing GLYX-13 and NRX-1074 for treatment-resistant MDD. Although each of these drug candidates is a peptide and may not be orally active (GLYX-13 is only administered intravenously and, we believe, NRX-1074 has not yet been administered orally to human subjects), both are new generation NMDA modulators focused on the GyB site of the NMDA receptor.
Many of our potential competitors, alone or with their strategic partners, have substantially greater financial, technical and human resources than we do and significantly greater experience in the discovery and development of product candidates, obtaining FDA and other regulatory approvals of treatments and the commercialization of those treatments. We believe that a range of pharmaceutical and biotechnology companies have programs to develop small molecule drug candidates for the treatment of depression, including MDD, epilepsy, neuropathic pain, Parkinson’s disease and other neurological conditions and diseases, including, but not limited to, Abbott Laboratories, Actavis, Astra Zeneca, Eli Lilly, GlaxoSmithKline, Johnson & Johnson, Lundbeck, Merck, Novartis, Otsuka, Pfizer, Roche, Sumitomo Dainippon, and Takeda. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. We expect that AV-101 will have to compete with a variety of therapeutic products and procedures.
We believe that our human pluripotent stem cell (hPSC) technology platform, the hPSC-derived human cells we produce, and the customized human cell-based assay systems we have formulated and developed are capable of being competitive in the diverse and growing global stem cell and regenerative medicine markets, including markets involving the sale of hPSC-derived cells to third-parties for their in vitro drug discovery and safety testing, contract predictive toxicology drug screening services for third parties, internal drug discovery, drug development and drug rescue of new , and regenerative medicine, including in vivo cell therapy research and development. A representative list of such biopharmaceutical companies pursuing one or more of these potential applications of adult and/or hPSCl technology includes the following: Acea Biosciences, Advanced Cell Technology, Athersys, BioCardia, BioTime, Cellectis Bioresearch, Cellerant Therapeutics, Cytori Therapeutics, Fujifilm Holdings, HemoGenix, International Stem Cell, NeoStem, Neuralstem, Organovo Holdings, PluriStem Therapeutics, Stem Cells, and Stemina BioMarker Discovery. Pharmaceutical companies and other established corporations such as Bristol-Myers Squibb, GE Healthcare Life Sciences, GlaxoSmithKline, Life Technologies, Novartis, Pfizer, Roche Holdings and others have been and are expected to continue pursuing internally various stem cell-related research and development programs. Many of the foregoing companies have greater resources and capital availability and as a result, may be more successful in their research and development programs than us. We anticipate that acceptance and use of hPSC technology for drug development and regenerative medicine will continue to occur and increase at pharmaceutical and biotechnology companies in the future.
Government Regulation
Government authorities in the United States at the federal, state and local level and in other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of drug products. Generally, before a new drug can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific to each regulatory authority, submitted for review and approved by the regulatory authority.
U.S. Drug Development
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and its implementing regulations. Drugs are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include, among other actions, the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls or withdrawals from the market, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
Our product candidates must be approved by the FDA through the NDA process before they may be legally marketed in the United States. The process required by the FDA before a drug may be marketed in the United States generally involves the following:
|
·
|
Completion of extensive non-clinical, sometimes referred to as non-clinical laboratory tests, non-clinical animal studies and formulation studies in accordance with applicable regulations, including the FDA’s current Good Laboratory Practice, or GLP, regulations;
|
|
·
|
Submission to the FDA of an IND application, which must become effective before human clinical trials may begin;
|
|
·
|
Approval by an independent institutional review board, or IRB, or ethics committee at each clinical trial site before each trial may be initiated;
|
|
·
|
Performance of adequate and well-controlled human clinical trials in accordance with applicable IND and other clinical trial-related regulations, sometimes referred to as good clinical practices, or GCPs, to establish the safety and efficacy of the proposed drug for each proposed indication;
|
|
·
|
Submission to the FDA of an NDA, for a new drug;
|
|
·
|
A determination by the FDA within 60 days of its receipt of an NDA to file the NDA for review;
|
|
·
|
Satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities where the drug is produced to assess compliance with cGMP requirements to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity;
|
|
·
|
Potential FDA audit of the non-clinical and/or clinical trial sites that generated the data in support of the NDA; and
|
|
·
|
FDA review and approval of the NDA, including consideration of the views of any FDA advisory committee, prior to any commercial marketing or sale of the drug in the United States.
|
The non-clinical and clinical testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all. Non-clinical tests include laboratory evaluation of product chemistry, formulation, stability and toxicity, as well as animal studies to assess the characteristics and potential safety and efficacy of the product.
The data required to support an NDA is generated in two distinct development stages: non-clinical and clinical. For new chemical entities, the non-clinical development stage generally involves synthesizing the active component, developing the formulation and determining the manufacturing process, as well as carrying out non-human toxicology, pharmacology and drug metabolism studies in the laboratory, which support subsequent clinical testing. The conduct of the non-clinical tests must comply with federal regulations, including GLPs. The sponsor must submit the results of the non-clinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. An IND is a request for authorization from the FDA to administer an investigational drug product to humans. Some non-clinical testing may continue even after the IND is submitted, but an IND must become effective before human clinical trials may begin. The central focus of an IND submission is on the general investigational plan and the protocol(s) for human trials. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions regarding the proposed clinical trials, including subjects will be exposed to unreasonable health risks, and places the IND on clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a drug candidate at any time before or during clinical trials due to safety concerns or non-compliance. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that could cause the trial to be suspended or terminated.
The clinical stage of development involves the administration of the drug candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control, in accordance with GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety and assess efficacy. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. There are also requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries.
A sponsor who wishes to conduct a clinical trial outside the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical trial is not conducted under an IND, the sponsor may submit data from the clinical trial to the FDA in support of an NDA so long as the clinical trial is conducted in compliance with an international guideline for the ethical conduct of clinical research known as the Declaration of Helsinki and/or the laws and regulations of the country or countries in which the clinical trial is performed, whichever provides the greater protection to the participants in the clinical trial.
Clinical Trials
Clinical trials are generally conducted in three sequential phases that may overlap, known as Phase 1, Phase 2 and Phase 3 clinical trials.
|
·
|
Phase 1 clinical trials generally involve a small number of healthy volunteers who are initially exposed to a single dose and then multiple doses of the product candidate. The primary purpose of these clinical trials is to assess the metabolism, pharmacologic action, side effect tolerability and safety of the drug.
|
|
·
|
Phase 2 clinical trials typically involve studies in disease-affected patients to determine the dose required to produce the desired benefits. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected, as well as identification of possible adverse effects and safety risks and preliminary evaluation of efficacy.
|
|
·
|
Phase 3 clinical trials generally involve large numbers of patients at multiple sites (from several hundred to several thousand subjects) and are designed to provide the data necessary to demonstrate the effectiveness of the product for its intended use, its safety in use, and to establish the overall benefit/risk relationship of the product and provide an adequate basis for product approval. Phase 3 clinical trials may include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to mimic the actual use of a product during marketing.
|
Post-approval trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected suspected adverse events, finding from other studies, or any finding from animal or in vitro testing that suggests a significant risk for human subjects. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA, the IRB, or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether or not a trial may move forward at designated check points based on access to certain data from the trial. Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, we must develop methods for testing the identity, strength, quality and purity of the final drug product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
NDA and FDA review process
The results of non-clinical studies and of the clinical trials, together with other detailed information, including extensive manufacturing information and information on the composition of the drug and proposed labeling, are submitted to the FDA in the form of an NDA requesting approval to market the drug for one or more specified indications. The FDA reviews an NDA to determine, among other things, whether a drug is safe and effective for its intended use and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. FDA approval of an NDA must be obtained before a drug may be offered for sale in the United States.
In addition, under the Pediatric Research Equity Act, or PREA, an NDA or supplement to an NDA must contain data to assess the safety and efficacy of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of pediatric data or full or partial waivers.
Under the Prescription Drug User Fee Act, or PDUFA, as amended, each NDA must be accompanied by a user fee. The FDA adjusts the PDUFA user fees on an annual basis. According to the FDA’s fee schedule, effective through December 31, 2014, the user fee for an application requiring clinical data, such as an NDA, is $2.2 million. PDUFA also imposes an annual product fee for human drugs of $0.1 million and an annual establishment fee of $0.6 million on facilities used to manufacture prescription drugs. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on NDAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA for filing. The FDA must make a decision on accepting an NDA for filing within 60 days of receipt. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the goals and policies agreed to by the FDA under PDUFA, the FDA has 10 months from the filing date in which to complete its initial review of a standard NDA and respond to the applicant, and six months from the filing date for a priority NDA. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs, and the review process is often significantly extended by FDA requests for additional information or clarification.
After the NDA submission is accepted for filing, the FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. Before approving an NDA, the FDA will conduct a pre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with cGMPs. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. In addition, before approving an NDA, the FDA may also audit data from clinical trials to ensure compliance with GCP requirements. Additionally, the FDA may refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. The FDA will likely re-analyze the clinical trial data, which could result in extensive discussions between the FDA and the applicant during the review process. The review and evaluation of an NDA by the FDA is extensive and time consuming and may take longer than originally planned to complete, and we may not receive a timely approval, if at all.
After the FDA evaluates an NDA, it may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter usually describes all of the specific deficiencies in the NDA identified by the FDA. The Complete Response Letter may require additional clinical data and/or an additional pivotal Phase 3 clinical trial(s), and/or other significant and time-consuming requirements related to clinical trials, non-clinical studies or manufacturing. If a Complete Response Letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, or withdraw the application. Even if such data and information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data.
There is no assurance that the FDA will ultimately approve a drug product for marketing in the United States and we may encounter significant difficulties or costs during the review process. If a product receives marketing approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling or may condition the approval of the NDA on other changes to the proposed labeling, development of adequate controls and specifications, or a commitment to conduct post-marketing testing or clinical trials and surveillance to monitor the effects of approved products. For example, the FDA may require Phase 4 testing which involves clinical trials designed to further assess a drug’s safety and efficacy and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized. The FDA may also place other conditions on approvals including the requirement for a risk evaluation and mitigation strategy, or REMS, to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA must submit a proposed REMS. The FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Product approvals may be withdrawn for non-compliance with regulatory requirements or if problems occur following initial marketing.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug product available in the United States for this type of disease or condition will be recovered from sales of the product. Orphan product designation must be requested before submitting an NDA. After the FDA grants orphan product designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. Competitors, however, may receive approval of different products for the indication for which the orphan product has exclusivity or obtain approval for the same product but for a different indication than that for which the orphan product has exclusivity. Orphan product exclusivity also could block the approval of one of our products for seven years if a competitor obtains approval of the same product as defined by the FDA or if our product candidate is determined to be contained within the competitor’s product for the same indication or disease. If a drug designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan product exclusivity. Orphan drug status in the European Union has similar, but not identical, benefits.
Expedited Development and Review Programs
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drugs that meet certain criteria. Specifically, new drugs are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new drug or biologic may request the FDA to designate the drug as a Fast Track product at any time during the clinical development of the product. Unique to a Fast Track product, the FDA may review sections of the marketing application on a rolling basis before the complete NDA is submitted, if the sponsor provides a schedule for the submission of the sections of the application, the FDA agrees to accept sections of the application and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the application.
Any product submitted to the FDA for marketing, including under a Fast Track program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or offers a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug designated for priority review in an effort to facilitate the review. A product may also be eligible for accelerated approval. Drugs studied for their safety and efficacy in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical trials establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. If the FDA concludes that a drug shown to be effective can be safely used only if distribution or use is restricted, it will require such post-marketing restrictions, as it deems necessary to assure safe use of the drug, such as:
|
·
|
distribution restricted to certain facilities or physicians with special training or experience; or
|
|
·
|
distribution conditioned on the performance of specified medical procedures.
|
The limitations imposed would be commensurate with the specific safety concerns presented by the drug. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product. Additionally, a drug may be eligible for designation as a breakthrough therapy if the drug is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more indications. The benefits of breakthrough therapy designation includes the same benefits as fast track designation, plus intensive guidance from FDA to ensure an efficient drug development program. Fast Track designation, priority review, accelerated approval and breakthrough designation do not change the standards for approval, but may expedite the development or approval process.
Pediatric Trials
The Food and Drug Administration Safety and Innovation Act, or FDASIA, which was signed into law on July 9, 2012, amended the FDCA to require that a sponsor who is planning to submit a marketing application for a drug that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration submit an initial Pediatric Study Plan, or PSP, within sixty days of an end-of-Phase 2 meeting or as may be agreed between the sponsor and FDA. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. FDA and the sponsor must reach agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from non-clinical studies, early phase clinical trials, and/or other clinical development programs.
Post-Marketing Requirements
Following approval of a new product, a pharmaceutical company and the approved product are subject to continuing regulation by the FDA, including, among other things, monitoring and recordkeeping activities, reporting to the applicable regulatory authorities of adverse experiences with the product, providing the regulatory authorities with updated safety and efficacy information, product sampling and distribution requirements, and complying with promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, restrictions on promoting drugs for uses or in patient populations that are not described in the drug’s approved labeling (known as “off-label use”), limitations on industry-sponsored scientific and educational activities, and requirements for promotional activities involving the Internet. Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such off-label uses. Prescription drug promotional materials must be submitted to the FDA in conjunction with their first use. Further, if there are any modifications to the drug, including changes in indications, labeling, or manufacturing processes or facilities, the applicant may be required to submit and obtain FDA approval of a new NDA or NDA supplement, which may require the applicant to develop additional data or conduct additional non-clinical studies and clinical trials. As with new NDAs, the review process is often significantly extended by FDA requests for additional information or clarification. Any distribution of prescription drug products and pharmaceutical samples must comply with the U.S. Prescription Drug Marketing Act, or the PDMA, a part of the FDCA.
In the United States, once a product is approved, its manufacture is subject to comprehensive and continuing regulation by the FDA. The FDA regulations require that products be manufactured in specific approved facilities and in accordance with cGMP. We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our products in accordance with cGMP regulations. NDA holders using contract manufacturers, laboratories or packagers are responsible for the selection and monitoring of qualified firms, and, in certain circumstances, qualified suppliers to these firms. These manufacturers must comply with cGMP regulations that require among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation and the obligation to investigate and correct any deviations from cGMP. Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance. The discovery of violative conditions, including failure to conform to cGMP, could result in enforcement actions that interrupt the operation of any such facilities or the ability to distribute products manufactured, processed or tested by them. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved NDA, including, among other things, recall or withdrawal of the product from the market.
Discovery of previously unknown problems with a product or the failure to comply with applicable FDA requirements can have negative consequences, including adverse publicity, judicial or administrative enforcement, warning letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties, among others. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
Other Regulatory Matters
Manufacturing, sales, promotion and other activities following product approval are also subject to regulation by numerous regulatory authorities in addition to the FDA, including, in the United States, the Centers for Medicare & Medicaid Services, other divisions of the Department of Health and Human Services, the United States Department of Justice, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency and state and local governments. In the United States, sales, marketing and scientific/educational programs must also comply with state and federal fraud and abuse laws. These laws include the federal Anti-Kickback Statute, which makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, receive, offer, or pay any remuneration that is intended to induce the referral of business, including the purchase, order, or prescription of a particular drug, for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. Violations of this law are punishable by up to five years in prison, criminal fines, administrative civil money penalties, and exclusion from participation in federal healthcare programs. In addition, the Patient Protection and Affordable Health Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively the ACA, among other things, amends the intent requirement of the federal Anti-Kickback Statute and criminal healthcare fraud statutes created by the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA. A person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it. Moreover, the ACA provides that the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
Although we would not submit claims directly to payors, drug manufacturers can be held liable under the federal False Claims Act, which prohibits anyone from knowingly presenting, or causing to be presented, for payment to federal programs (including Medicare and Medicaid) claims for items or services, including drugs, that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. The government may deem manufacturers to have “caused” the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers or promoting a product off-label. In addition, our future activities relating to the reporting of wholesaler or estimated retail prices for our products, the reporting of prices used to calculate Medicaid rebate information and other information affecting federal, state, and third-party reimbursement for our products, and the sale and marketing of our products, are subject to scrutiny under this law. Penalties for a False Claims Act violation include three times the actual damages sustained by the government, plus mandatory civil penalties of between $5,500 and $11,000 for each separate false claim, the potential for exclusion from participation in federal healthcare programs, and, although the federal False Claims Act is a civil statute, conduct that results in a False Claims Act violation may also implicate various federal criminal statutes. If the government were to allege that we were, or convict us of, violating these false claims laws, we could be subject to a substantial fine and may suffer a decline in our stock price. In addition, private individuals have the ability to bring actions under the federal False Claims Act and certain states have enacted laws modeled after the federal False Claims Act.
Pricing and rebate programs must comply with the Medicaid rebate requirements of the U.S. Omnibus Budget Reconciliation Act of 1990 and more recent requirements in ACA. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. The handling of any controlled substances must comply with the U.S. Controlled Substances Act and Controlled Substances Import and Export Act. Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, sales, promotion and other activities are also potentially subject to federal and state consumer protection and unfair competition laws.
The distribution of pharmaceutical products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
The failure to comply with any of these laws or regulatory requirements subjects firms to possible legal or regulatory action. Depending on the circumstances, failure to meet applicable regulatory requirements can result in criminal prosecution, fines or other penalties, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of product approvals, or refusal to allow a firm to enter into supply contracts, including government contracts. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. Prohibitions or restrictions on sales or withdrawal of future products marketed by us could materially affect our business in an adverse way.
Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of our products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business.
U.S. Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of the FDA approval of our drug candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The U.S. PTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restoration of patent term for one of our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA.
Marketing exclusivity provisions under the FDCA can also delay the submission or the approval of certain marketing applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to obtain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another drug based on the same active moiety, regardless of whether the drug is intended for the same indication as the original innovator drug or for another indication, where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder. The FDCA also provides three years of marketing exclusivity for an NDA, or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the modification for which the drug received approval on the basis of the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the active agent for the original indication or condition of use. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the non-clinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and efficacy. Orphan drug exclusivity, as described above, may offer a seven-year period of marketing exclusivity, except in certain circumstances. Pediatric exclusivity is another type of regulatory market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric trial in accordance with an FDA-issued “Written Request” for such a trial.
European Union Drug Development
In the European Union, our future products may also be subject to extensive regulatory requirements. As in the United States, medicinal products can only be marketed if a marketing authorization from the competent regulatory agencies has been obtained.
Similar to the United States, the various phases of non-clinical and clinical research in the European Union are subject to significant regulatory controls. Although the EU Clinical Trials Directive 2001/20/EC has sought to harmonize the EU clinical trials regulatory framework, setting out common rules for the control and authorization of clinical trials in the EU, the EU Member States have transposed and applied the provisions of the Directive differently. This has led to significant variations in the member state regimes. Under the current regime, before a clinical trial can be initiated it must be approved in each of the EU countries where the trial is to be conducted by two distinct bodies: the National Competent Authority, or NCA, and one or more Ethics Committees, or ECs. Under the current regime all suspected unexpected serious adverse reactions to the investigated drug that occur during the clinical trial have to be reported to the NCA and ECs of the Member State where they occurred.
The EU clinical trials legislation is currently undergoing a revision process mainly aimed at harmonizing and streamlining the clinical trials authorization process, simplifying adverse event reporting procedures, improving the supervision of clinical trials, and increasing their transparency.
European Union Drug Review and Approval
In the European Economic Area, or EEA, (which is comprised of the 27 Member States of the European Union (excluding Croatia) plus Norway, Iceland and Liechtenstein), medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. There are two types of marketing authorizations:
The Community MA is issued by the European Commission through the Centralized Procedure, based on the opinion of the Committee for Medicinal Products for Human Use, or CHMP, of the European Medicines Agency, or EMA, and is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, and medicinal products containing a new active substance indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the EU.
National MAs, which are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in another Member States through the Mutual Recognition Procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure. Under the Decentralized Procedure an identical dossier is submitted to the competent authorities of each of the Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member State, or RMS. The competent authority of the RMS prepares a draft assessment report, a draft summary of the product characteristics, or SPC, and a draft of the labeling and package leaflet, which are sent to the other Member States (referred to as the Member States Concerned) for their approval. If the Member States Concerned raise no objections, based on a potential serious risk to public health, to the assessment, SPC, labeling, or packaging proposed by the RMS, the product is subsequently granted a national MA in all the Member States (i.e., in the RMS and the Member States Concerned).
Under the above-described procedures, before granting the MA, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
European Union New Chemical Entity Exclusivity
In the European Union, new chemical entities, sometimes referred to as new active substances, qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity. This data exclusivity, if granted, prevents regulatory authorities in the European Union from referencing the innovator’s data to assess a generic application for eight years, after which generic marketing authorization can be submitted, and the innovator’s data may be referenced, but not approved for two years. The overall ten-year period will be extended to a maximum of 11 years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies.
European Union Orphan Designation and Exclusivity
In the European Union, the EMA’s Committee for Orphan Medicinal Products grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than 5 in 10,000 persons in the European Union Community and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or the product would be a significant benefit to those affected). Additionally, designation is granted for products intended for the diagnosis, prevention, or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in the European Union would be sufficient to justify the necessary investment in developing the medicinal product.
In the European Union, orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity is granted following medicinal product approval. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
Rest of the World Regulation
For other countries outside of the European Union and the United States, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, the clinical trials must be conducted in accordance with cGCP requirements and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Reimbursement
Sales of our products will depend, in part, on the extent to which our products will be covered by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. In the United States no uniform policy of coverage and reimbursement for drug products exists. Accordingly, decisions regarding the extent of coverage and amount of reimbursement to be provided for any of our products will be made on a payor by payor basis. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our product candidates to each payor separately, with no assurance that coverage and adequate reimbursement will be obtained.
Third-party payors are increasingly reducing reimbursements for medical products and services. Additionally, the containment of healthcare costs has become a priority of federal and state governments, and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Decreases in third-party reimbursement for our product candidate or a decision by a third-party payor to not cover our product candidate could reduce physician usage of the product candidate and have a material adverse effect on our sales, results of operations and financial condition.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the MMA, established the Medicare Part D program to provide a voluntary prescription drug benefit to Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities that provide coverage of outpatient prescription drugs. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
The American Recovery and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments for the same illness. The plan for the research was published in 2012 by the Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures will be made to Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payors, it is not clear what effect, if any, the research will have on the sales of our product candidate, if any such product or the condition that it is intended to treat is the subject of a trial. It is also possible that comparative effectiveness research demonstrating benefits in a competitor’s product could adversely affect the sales of our product candidate. If third-party payors do not consider our products to be cost-effective compared to other available therapies, they may not cover our products after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The ACA is expected to have a significant impact on the health care industry. The ACA is expected to expand coverage for the uninsured while at the same time containing overall healthcare costs. With regard to pharmaceutical products, among other things, the ACA is expected to expand and increase industry rebates for drugs covered under Medicaid programs and make changes to the coverage requirements under the Medicare Part D program. We cannot predict the full impact of the ACA on our business as many of the ACA reforms require the promulgation of detailed regulations implementing the statutory provisions that has not yet occurred. For example, the ACA imposed new reporting requirements on drug manufacturers for payments made to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests that are not timely, accurately and completely reported in an annual submission. Drug manufacturers were required to begin collecting data on August 1, 2013 and were required to submit reports to CMS by March 31, 2014 (and by the 90th day of each subsequent calendar year). In addition, many states have adopted laws similar to the federal laws discussed above. Some of these state prohibitions apply to the referral of patients for healthcare services reimbursed by any insurer, not just federal healthcare programs such as Medicare and Medicaid. There has also been a recent trend of increased federal and state regulation of payments made to physicians. Certain states mandate implementation of compliance programs, impose restrictions on drug manufacturers’ marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians. In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. On August 2, 2011, the Budget Control Act of 2011 among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, started in April 2013. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or the ATRA, which delayed for another two months the budget cuts mandated by these sequestration provisions of the Budget Control Act of 2011. The ATRA, among other things, also reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. We expect that additional federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, and in turn could significantly reduce the projected value of certain development projects and reduce our profitability.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the European Union do not follow price structures of the United States and generally prices tend to be significantly lower.
Stem Cell Technology - United States
With respect to our stem cell research and development in the U.S., the U.S. government has established requirements and procedures relating to the isolation and derivation of certain stem cell lines and the availability of federal funds for research and development programs involving those lines. All of the stem cell lines that we are using were either isolated under procedures that meet U.S. government requirements and are approved for funding from the U.S. government, or were isolated under procedures that meet U.S. government requirements.
All procedures we use to obtain clinical samples, and the procedures we use to isolate hESCs, are consistent with the informed consent and ethical guidelines promulgated by either the U.S. National Academy of Science, the International Society of Stem Cell Research (“ISSCR”), or the NIH. These procedures and documentation have been reviewed by an external Stem Cell Research Oversight Committee, and all cell lines we use have been approved under one or more of these guidelines.
The U.S. government and its agencies on July 7, 2009 published guidelines for the ethical derivation of hESCs required for receiving federal funding for hESC research. Should we seek further NIH funding for our stem cell research and development, our request would involve the use of hESC lines that meet the NIH guidelines for NIH funding. In the U.S., the President’s Council on Bioethics monitors stem cell research, and may make recommendations from time to time that could place restrictions on the scope of research using human embryonic or fetal tissue. Although numerous states in the U.S. are considering, or have in place, legislation relating to stem cell research, including California whose voters approved Proposition 71 to provide up to $3 billion of state funding for stem cell research in California, it is not yet clear what affect, if any, state actions may have on our ability to commercialize stem cell technologies.
Stem Cell Technology - Canada
In Canada, stem cell research and development is governed by two policy documents and by one legislative statute: the Guidelines for Human Pluripotent Stem Cell Research (the Guidelines) issued by the Canadian Institutes of Health Research; the Tri-Council Statement: Ethical Conduct for Research Involving Humans (“TCPS”); and the Assisted Human Reproduction Act (Act). The Guidelines and the TCPS govern stem cell research conducted by, or under the auspices of, institutions funded by the federal government. Should we seek funding from Canadian government agencies or should we conduct research under the auspices of an institution so funded, we may have to ensure the compliance of such research with the ethical rules prescribed by the Guidelines and the TCPS.
The Act subjects all research conducted in Canada involving the human embryo, including hESC derivation (but not the stem cells once derived), to a licensing process overseen by a federal licensing agency. However, as of the date of this prospectus, the provisions of the Act regarding the licensing of hESC derivation were not in force
We are not currently conducting stem cell research in Canada. We have, however, sponsored pluripotent stem cell research in Canada by Dr. Gordon Keller at UHN’s McEwen Centre. We anticipate conducting additional hPSC research (with both hESCs and hiPSCs), in collaboration with Dr. Keller and his research team, at UHN’s McEwen Centre during 2015 and beyond. Should the provisions of the Act come into force, we may have to apply for a license for all hESC research we may sponsor or conduct in Canada and ensure compliance of such research with the provisions of the Act.
Subsidiaries and Inter-Corporate Relationships
VistaGen Therapeutics. Inc., a California corporation, is our wholly-owned subsidiary and has the following two wholly-owned subsidiaries: VistaStem Canada Inc., a corporation incorporated pursuant to the laws of the Province of Ontario, intended to facilitate our stem cell-based research and development and drug rescue activities in Canada should we elect to expand our U.S. operations into Canada; and Artemis Neuroscience, Inc., a corporation incorporated pursuant to the laws of the State of Maryland. The operations of VistaGen Therapeutics, Inc., a California corporation, and each of its two wholly owned subsidiaries are managed by our senior management team based in South San Francisco, California.
Employees
As of July 15, 2015, we employed nine full-time employees, three of whom have doctorate degrees. Six full-time employees work in research and development and laboratory support services and three full-time employees work in general and administrative roles. Staffing for all other functional areas is achieved through strategic relationships with service providers and consultants, each of whom provides services on a real-time, as-needed basis, including human resources and payroll, information technology, facilities, legal, stock plan administration, investor relations and website maintenance, regulatory affairs, and FDA program management.
We have never had a work stoppage, and none of our employees is represented by a labor organization or under any collective bargaining agreement. We consider our employee relations to be is good.
Facilities
We lease our office and laboratory space, which consists of approximately 10,000 square feet located in South San Francisco, California. Our lease expires on July 31, 2017.
Legal Proceedings
As of the date of this prospectus, we were not party to any legal matters or claims. In the future, we may become party to legal matters and claims arising in the ordinary course of business, the resolution of which we do not anticipate would have a material adverse impact on our results of operations.
Environmental Regulation
Our business does not require us to comply with any particular unique environmental regulations.
MARKET PRICE OF COMMON STOCK AND OTHER STOCKHOLDER MATTERS
Market Information
On June 21, 2011, our common stock began trading on the OTC Marketplace (“OTCQB”), under the symbol “VSTA”. There was no established trading market for our common stock prior to that date.
Shown below is the range of high and low sales prices for our common stock for the periods indicated as reported by the OTCQB. The market quotations reflect inter-dealer prices, without retail mark-up, mark-down or commissions and may not necessarily represent actual transactions. Effective August 14, 2014, we consummated a 1-for-20 reverse split of our authorized, and issued and outstanding shares of common stock (the Stock Consolidation). Each reference to the price per share of common stock in the table below is on a post-Stock Consolidation basis, and reflects the 1-for-20 adjustment as a result of the Stock Consolidation.
|
High
|
Low
|
|||||||
|
Year Ending March 31, 2015
|
||||||||
|
First quarter ending June 30, 2015
|
$ | 16.50 | $ | 8.00 | ||||
|
Year Ending March 31, 2015
|
||||||||
|
First quarter ending June 30, 2014
|
$ | 14.80 | $ | 5.60 | ||||
|
Second quarter ending September 30, 2014
|
$ | 15.00 | $ | 7.99 | ||||
|
Third quarter ending December 31, 2014
|
$ | 10.50 | $ | 8.00 | ||||
|
Fourth quarter ending March 31, 2015
|
$ | 12.00 | $ | 3.16 | ||||
|
Year Ending March 31, 2014
|
||||||||
|
First quarter ending June 30, 2013
|
$ | 18.00 | $ | 12.00 | ||||
|
Second quarter ending September 30, 2013
|
$ | 17.80 | $ | 11.00 | ||||
|
Third quarter ending December 31, 2013
|
$ | 12.20 | $ | 5.20 | ||||
|
Fourth quarter ending March 31, 2014
|
$ | 10.00 | $ | 5.60 | ||||
On July 17, 2015 the closing price of our common stock on the OTCQB was $10.00 per share.
As of July 17, 2015, we had 1,594,461 shares of common stock outstanding and approximately 300 stockholders of record. On the same date, two stockholders held all 500,000 outstanding restricted shares of our Series A Preferred Stock, which shares are convertible into 750,000 shares of common stock, and 59 stockholders held 3,136,352 outstanding shares of Series B 10% Convertible Preferred Stock, which shares are convertible into 3,136,652 shares of common stock.
Dividend Policy
We have never paid or declared any cash dividends on our common stock, and we do not anticipate paying any cash dividends on our common stock in the foreseeable future. Covenants in certain of our debt agreements prohibit us from paying dividends while the debt remains outstanding. Our Series B Preferred accrues dividends at a rate of 10% per annum, which dividends are payable solely in unregistered shares of our common stock at the time the Series B Preferred is converted into common stock.
Transfer Agent
Our Transfer Agent for our Common Stock is ComputerShare. The transfer agent’s address is 480 Washington Boulevard, 29th Floor, Jersey City, NJ 07310.
SELECTED CONSOLIDATED FINANCIAL DATA
A smaller reporting company is not required to provide the information required by this item.
MANAGEMENT’S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Cautionary Note Regarding Forward-Looking Statements
This prospectus includes forward-looking statements. All statements contained in this prospectus other than statements of historical fact, including statements regarding our future results of operations and financial position, our business strategy and plans, and our objectives for future operations, are forward- looking statements. The words “believe,” “may,” “estimate,” “continue,” “anticipate,” “intend,” “expect” and similar expressions are intended to identify forward-looking statements. We have based these forward- looking statements largely on our current expectations and projections about future events and trends that we believe may affect our financial condition, results of operations, business strategy, short-term and long-term business operations and objectives, and financial needs. These forward-looking statements are subject to a number of risks, uncertainties and assumptions. Our business is subject to significant risks including, but not limited to, our ability to obtain additional financing, the results of our research and development efforts, the results of non-clinical and clinical testing, the effect of regulation by the United States Food and Drug Administration (“FDA”) and other agencies, the impact of competitive products, product development, commercialization and technological difficulties, the effect of our accounting policies, and other risks as detailed in the section entitled “Risk Factors” in this prospectus. Further, even if our product candidates appear promising at various stages of development, our share price may decrease such that we are unable to raise additional capital without significant dilution or other terms that may be unacceptable to our management, Board of Directors and stockholders.
Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties and assumptions, the future events and trends discussed in this prospectus may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements.
You should not rely upon forward-looking statements as predictions of future events. The events and circumstances reflected in the forward-looking statements may not be achieved or occur. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. We are under no duty to update any of these forward-looking statements after the date of this prospectus or to conform these statements to actual results or revised expectations. If we do update one or more forward-looking statements, no inference should be drawn that we will make additional updates with respect to those or other forward-looking statements.
Business Overview
We are a clinical-stage biopharmaceutical company committed to developing and commercializing innovative product candidates for patients with depression, other diseases and various disorders related to the central nervous system (“CNS”), as well as cancer.
More than one billion people worldwide suffer from CNS disorders. Recently, the economic burden of these disorders was estimated at $2.0 trillion in the U.S. and European Union alone, a figure that is expected to triple by 2030. The World Health Organization estimates that 350 million people worldwide are affected by depression. According to the U.S. National Institutes of Health (“NIH”), major depression is one of the most common mental disorders in the U.S.. In 2012, the NIH estimated 16 million adults aged 18 or older in the U.S. had at least one major depressive episode. This represented 6.9 percent of all U.S. adults.
Our lead product candidate, AV-101, is an orally available small molecule prodrug in Phase 2 clinical development for Major Depressive Disorder (“MDD”). AV-101’s mechanism of action (“MOA”), as an N-methyl-D-aspartate receptor (“NMDAR”) antagonist binding selectively at the glycine-binding (“GlyB”) co-agonist site of the NMDAR, is fundamentally different from all antidepressants by the U.S. Food and Drug Administration (“FDA”). In four preclinical studies utilizing well-validated animal models of depression, AV-101 was shown to induce fast-acting, dose-dependent, persistent and statistically significant antidepressant-like responses, following a single treatment, which was equivalent to responses seen with a control single sub-anesthetic dose of ketamine (sometimes used by clinicians off-label to treat suicidal behavior). In the same studies, fluoxetine (Prozac) did not induce rapid onset antidepressant-like responses. Preclinical studies also support the hypothesis that AV-101 has potential to treat several additional CNS disorders, including chronic neuropathic pain, epilepsy and neurodegenerative diseases, such as Parkinson’s disease and Huntington’s disease where modulation of the NMDAR may have therapeutic benefit.
Following two successful randomized, double-blind, placebo-controlled Phase 1 safety studies funded by the NIH, in February 2015, we entered into a Cooperative Research and Development Agreement (“CRADA”) with the U.S. National Institute of Mental Health (“NIMH”), part of the NIH. Under the CRADA, we will collaborate with the NIMH on the initial Phase 2 clinical study of AV-101 in subjects with treatment-resistant MDD. Pursuant to the CRADA, the study will be conducted at the NIMH and be fully funded by the NIMH. It is contemplated that this clinical study will begin in Fall 2015 under the direction of Dr. Carlos Zarate, Jr., the NIMH’s Chief of Experimental Therapeutics & Pathophysiology Branch and of the Section on Neurobiology and Treatment of Mood and Anxiety Disorders.
In addition to developing AV-101 for MDD and other CNS indications, we apply our stem cell technology for drug rescue programs intended to identify and develop proprietary new chemical entities (“NCEs”) for our internal drug candidate pipeline. Drug rescue involves (1) using our customized invitro bioassay systems to predict potential heart and liver toxicity of NCEs, (2) leveraging prior investments by pharmaceutical companies and others related to screening large-scale compound libraries, optimizing and testing for efficacy NCEs that were terminated before FDA approval due to heart or liver toxicity and are now available in the public domain, and (3) applying modern medicinal chemistry to produce safer NCEs for our internal development pipeline. Our CardioSafe 3D™ bioassay system uses our human pluripotent stem cell (“hPSC”)-derived cardiomyocytes, or human heart cells. We believe CardioSafe 3D is more comprehensive and clinically predictive than the hERG assay, which is currently the only in vitro cardiac safety assay required by FDA guidelines. We use our hPSC-derived hepatocytes, or human liver cells, in our LiverSafe 3D™ bioassay system to predict potential liver toxicity of NCEs, including potential drug metabolism issues and adverse drug-drug interactions. CardioSafe 3D and LiverSafe 3D offer a new paradigm for evaluating and predicting potential heart and liver toxicity of NCEs, including drug rescue NCEs, early in the development process, long before costly, high risk animal studies and human clinical trials. We intend to develop internally for our pipeline each lead drug rescue NCEs we produce
The Merger
VistaGen Therapeutics, Inc., a California corporation incorporated on May 26, 1998 (“VistaGen California”), is our wholly-owned subsidiary. Excaliber Enterprises, Ltd. (“Excaliber”), a publicly-held company (formerly OTCBB: EXCA) was incorporated under the laws of the State of Nevada on October 6, 2005. Pursuant to a strategic merger transaction on May 11, 2011, Excaliber acquired all outstanding shares of VistaGen California in exchange for 341,823 shares of our common stock and assumed all of VistaGen California’s pre-Merger obligations (the “Merger”). Shortly after the Merger, Excaliber’s name was changed to “VistaGen Therapeutics, Inc.” (a Nevada corporation).
VistaGen California, as the accounting acquirer in the Merger, recorded the Merger as the issuance of common stock for the net monetary assets of Excaliber, accompanied by a recapitalization. The accounting treatment for the Merger was identical to that resulting from a reverse acquisition, except that we recorded no goodwill or other intangible assets. A total of 78,450 shares of our common stock, representing the shares held by stockholders of Excaliber immediately prior to the Merger and effected for a post-Merger two-for-one (2:1) stock split, have been reflected as outstanding for all periods presented in the Consolidated Financial Statements of the Company included elsewhere in this prospectus. Additionally, the Consolidated Balance Sheets reflect the $0.001 par value of Excaliber’s common stock.
The Consolidated Financial Statements included elsewhere in this prospectus represent the activity of VistaGen California from May 26, 1998, and the consolidated activity of VistaGen California and Excaliber (now VistaGen Therapeutics, Inc., a Nevada corporation), from May 11, 2011 (the date of the Merger). The Consolidated Financial Statements also include the accounts of VistaGen California’s two inactive wholly-owned subsidiaries, Artemis Neuroscience, Inc., a Maryland corporation (Artemis), and VistaStem Canada, Inc., a corporation organized under the laws of Ontario, Canada (“VistaStem Canada”).
Financial Operations Overview
Net Loss
We have not yet achieved revenue-generating status from any of our potential products. Since inception, we have devoted substantially all of our time and efforts to development of AV-101 through its Phase 1 clinical trials and to hPSC research and bioassay development, small molecule drug development, and creating, protecting and patenting intellectual property in support of our drug rescue model, with the corollary initiatives of recruiting personnel and raising working capital. As of March 31, 2015, we had an accumulated deficit of approximately $84.5 million. Our net loss for the years ended March 31, 2015 and 2014 was $13.9 million and $3.0 million, respectively. We expect such losses to continue for the foreseeable future as we engage in Phase 2 clinical trials of AV-101 and its further development and expand our drug rescue activities and the capabilities of our stem cell technology.
Summary of Fiscal Year 2015
Throughout our fiscal years ended March 31, 2015 and 2014, with very limited resources, our scientific personnel have successfully continued to expand the drug rescue capabilities of our novel, customized bioassay systems, CardioSafe 3D™ and LiverSafe 3D™, and advance our internal review and assessment in CardioSafe 3D of multiple prospective drug rescue opportunities. Additionally, as indicated previously, we have entered into a CRADA with the NIH providing for an NIH-sponsored Phase 2 clinical study of AV-101 in Major Depressive Disorder beginning in 2015.
Throughout both fiscal 2015 and 2014, our executive management has been significantly focused on providing sufficient operating capital to continue to advance our AV-101 initiatives, and drug development and research objectives while meeting our continuing operational needs.
To meet our working capital needs during fiscal 2015, between late-March 2014 and March 31, 2015, we entered into securities purchase agreements with accredited investors pursuant to which we sold units to such accredited investors, in private placement transactions, for aggregate cash proceeds of $3,113,500, consisting of (i) convertible promissory notes in the aggregate face amount of $3,113,500 which matured between March 31, 2015 and April 30, 2015, or were automatically convertible into securities we might issue upon the consummation of a Qualified Financing, as defined therein, (ii) an aggregate of 282,850 restricted shares of our common stock; and (iii) warrants exercisable through December 31, 2016 to purchase an aggregate of 282,850 restricted shares of our common stock at an exercise price of $10.00 per share. Through May 2015, we sold an additional $280,000 of such units to accredited investors in private placement transactions. Between May 26, 2015 and the date of the filing of the registration statement, of which this prospectus forms a part, we received gross process of $1,112,500 from the sale of shares of our Series B Preferred and Warrants, as more fully described under “Description of Securities to be Registered” on page 105 of this prospectus.
Given our working capital constraints during fiscal 2015 and the latter portion of fiscal 2014, we attempted to minimize cash commitments and expenditures for both internal and external research and development and general and administrative services to the greatest extent possible. As a consequence, during our fiscal year ended March 31, 2015, Shawn Singh, our CEO, voluntarily elected to receive cash compensation equal to only 24% of his contractual salary. In addition, during fiscal 2015, each of Ralph Snodgrass, PhD, our President and CSO, and Jerrold Dotson, our CFO, voluntarily elected to receive only 52% and 62% of his contractual salary, respectively. Such unpaid compensation has been accrued for accounting purposes, but remains unpaid at the date of this prospectus.
The following table summarizes the results of our operations for the fiscal years ended March 31, 2015 and 2014 (amounts in $000):
|
Fiscal Years Ended March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Operating expenses:
|
||||||||
|
Research and development
|
$
|
2,433
|
$
|
2,481
|
||||
|
General and administrative
|
4,344
|
2,548
|
||||||
|
Total operating expenses
|
6,777
|
5,029
|
||||||
|
Loss from operations
|
(6,777
|
)
|
(5,029
|
)
|
||||
|
Other expenses, net:
|
||||||||
|
Interest expense, net
|
(4,549
|
)
|
(1,503
|
)
|
||||
|
Change in warrant liabilities
|
(35
|
)
|
3,567
|
|||||
|
Loss on extinguishment of debt
|
(2,388
|
)
|
-
|
|||||
|
Other expense
|
(135
|
)
|
-
|
|||||
|
Loss before income taxes
|
(13,884
|
)
|
(2,965
|
)
|
||||
|
Income taxes
|
(2
|
)
|
(3
|
)
|
||||
|
Net loss
|
$
|
(13,886
|
)
|
$
|
(2,968
|
)
|
||
Revenue
We reported no revenue for the fiscal years ended March 31, 2015 or 2014. We have successfully completed our Phase 1 clinical development of AV-101, our orally available, new generation prodrug candidate in clinical development for the treatment of Major Depressive Disorder (“MDD”), with additional potential as a new therapy for neuropathic pain, epilepsy, Parkinson’s disease and Huntington’s disease. Additionally, as indicated previously, we have entered into a CRADA with the NIH providing for an NIH-sponsored Phase 2 clinical study of AV-101 in Major Depressive Disorder beginning in 2015. We presently have no revenue generating arrangements.
Research and Development Expense
Research and development expense represented approximately 36% and 49% of our operating expenses for the fiscal years ended March 31, 2015 and 2014, respectively. Research and development costs are expensed as incurred. Research and development expense consists of both internal and external expenses incurred in drug development activities, costs associated with the development of AV-101, sponsored stem cell research and costs related to the licensing, application and prosecution of our intellectual property. These expenses primarily consist of the following:
|
·
|
salaries and benefits, including stock-based compensation costs, travel and related expense for personnel associated with internal research and development activities;
|
|
·
|
fees to contract research organizations and other professional service providers for services related to the conduct and analysis of clinical trials and other drug development activities;
|
|
·
|
fees to third parties for access to licensed technology and costs associated with securing and maintaining patents related to our internally generated inventions:
|
|
·
|
laboratory supplies and materials;
|
|
·
|
leasing and depreciation of laboratory equipment; and
|
|
·
|
allocated costs of facilities and infrastructure.
|
General and Administrative Expense
General and administrative expense consists primarily of salaries and benefits expense, including stock-based compensation expense, for personnel in executive, finance and accounting, and other support functions. Other costs include professional fees for legal, investor relations and accounting services and other strategic consulting and public company expenses as well as facility costs not otherwise included in research and development expense.
Other Expenses, Net
In both fiscal 2015 and 2014, we incurred interest expense, including significant amounts of non-cash discount amortization attributable to certain notes, on the outstanding balances of our Senior Secured Convertible Promissory Notes issued to Platinum between October 2012 and July 2013, on subordinated convertible promissory notes issued between March 2013 and March 2015 as components of our Unit Private Placements and on unsecured promissory notes issued to various contract research organizations, technology licensors and other professional service providers since fiscal 2011. In fiscal 2015 and 2014, we recorded non-cash expense and income, respectively, related to changes in the fair values of the warrants issued or issuable to Platinum in connection with the various Senior Secured Convertible Promissory Notes we issued to Platinum between October 2012 and July 2013. In fiscal 2015, we incurred non-cash losses on extinguishment of debt resulting from settlements or modifications of indebtedness to Platinum, to various holders of promissory notes issued in connection with our 2013 Unit Private Placement and scheduled to mature on July 30, 2014, and to a technology licensor and a professional service provider. Additionally, in fiscal 2015 we incurred non-cash expense related to the settlement of a note receivable we accepted in fiscal 2012.
Critical Accounting Policies and Estimates
We consider certain accounting policies related to revenue recognition, impairment of long-lived assets, research and development, stock-based compensation, warrant liability and income taxes to be critical accounting policies that require the use of significant judgments and estimates relating to matters that are inherently uncertain and may result in materially different results under different assumptions and conditions. The preparation of financial statements in conformity with United States generally accepted accounting principles (GAAP) requires us to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes to the consolidated financial statements. These estimates include useful lives for property and equipment and related depreciation calculations, and assumptions for valuing options, warrants and other stock-based compensation. Our actual results could differ from these estimates.
Revenue Recognition
Although we do not currently have any such arrangements, we have historically generated revenue principally from collaborative research and development arrangements, technology access fees and government grants. We recognize revenue under the provisions of the SEC issued Staff Accounting Bulletin 104, Topic 13, Revenue Recognition Revised and Updated (SAB 104) and Accounting Standards Codification (ASC) 605-25, Revenue Arrangements-Multiple Element Arrangements (ASC 605-25). Revenue for arrangements not having multiple deliverables, as outlined in ASC 605-25, is recognized once costs are incurred and collectability is reasonably assured.
Revenue arrangements with multiple components are divided into separate units of accounting if certain criteria are met, including whether the delivered component has stand-alone value to the customer. Consideration received is allocated among the separate units of accounting based on their respective selling prices. The selling price for each unit is based on vendor-specific objective evidence, or VSOE, if available, third party evidence if VSOE is not available, or estimated selling price if neither VSOE nor third party evidence is available. The applicable revenue recognition criteria are then applied to each of the units.
We recognize revenue when the four basic criteria of revenue recognition are met: (i) a contractual agreement exists; (ii) the transfer of technology has been completed or services have been rendered; (iii) the fee is fixed or determinable; and (iv) collectability is reasonably assured. For each source of revenue, we comply with the above revenue recognition criteria in the following manner:
|
·
|
Collaborative arrangements typically consist of non-refundable and/or exclusive technology access fees, cost reimbursements for specific research and development spending, and various milestone and future product royalty payments. If the delivered technology does not have stand-alone value, the amount of revenue allocable to the delivered technology is deferred. Non-refundable upfront fees with stand-alone value that are not dependent on future performance under these agreements are recognized as revenue when received, and are deferred if we have continuing performance obligations and have no objective and reliable evidence of the fair value of those obligations. We recognize non-refundable upfront technology access fees under agreements in which we have a continuing performance obligation ratably, on a straight-line basis, over the period in which we are obligated to provide services. Cost reimbursements for research and development spending are recognized when the related costs are incurred and when collectability is reasonably assured. Payments received related to substantive, performance-based “at-risk” milestones are recognized as revenue upon achievement of the milestone event specified in the underlying contracts, which represent the culmination of the earnings process. Amounts received in advance are recorded as deferred revenue until the technology is transferred, costs are incurred, or a milestone is reached.
|
|
·
|
Technology license agreements typically consist of non-refundable upfront license fees, annual minimum access fees and/or royalty payments. Non-refundable upfront license fees and annual minimum payments received with separable stand-alone values are recognized when the technology is transferred or accessed, provided that the technology transferred or accessed is not dependent on the outcome of the continuing research and development efforts. Otherwise, revenue is recognized over the period of our continuing involvement.
|
|
·
|
Government grant awards, which support our research efforts on specific projects, generally provide for reimbursement of approved costs as defined in the terms of grant awards. We recognize grant revenue when associated project costs are incurred.
|
Impairment of Long-Lived Assets
In accordance with ASC 360-10, Property, Plant & Equipment -Overall, we review for impairment whenever events or changes in circumstances indicate that the carrying amount of property and equipment may not be recoverable. Determination of recoverability is based on an estimate of undiscounted future cash flows resulting from the use of the asset and its eventual disposition. In the event that such cash flows are not expected to be sufficient to recover the carrying amount of the assets, we write down the assets to their estimated fair values and recognize the loss in the Consolidated Statements of Operations and Comprehensive Loss.
Research and Development Expenses
Research and development expenses are composed of both internal and external costs. Internal costs include salaries and employment-related expenses of scientific personnel and direct project costs. External research and development expenses consist primarily of costs associated with clinical and non-clinical development of AV-101, our prodrug candidate entering late-stage clinical development for Major Depressive Disorder, sponsored stem cell research and development costs, and costs related to the application and prosecution of patents related to our stem cell technology platform and AV-101. All such costs are charged to expense as incurred.
Stock-Based Compensation
We recognize compensation cost for all stock-based awards to employees based on the grant date fair value of the award. We record non-cash, stock-based compensation expense over the period during which the employee is required to perform services in exchange for the award, which generally represents the scheduled vesting period. We have granted no restricted stock awards nor do we have any awards with market or performance conditions. For equity awards to non-employees, we re-measure the fair value of the awards as they vest and the resulting value is recognized as an expense during the period over which the services are performed.
We use the Black-Scholes option pricing model to estimate the fair value of stock-based awards as of the grant date. The Black-Scholes model is complex and dependent upon key data input estimates. The primary data inputs with the greatest degree of judgment are the expected term of the stock options and the estimated volatility of our stock price. The Black-Scholes model is highly sensitive to changes in these two inputs. The expected term of the options represents the period of time that options granted are expected to be outstanding. We use the simplified method to estimate the expected term as an input into the Black-Scholes option pricing model. We determine expected volatility using the historical method, which, because of the limited period during which our stock has been publicly traded, is based on the historical daily trading data of the common stock of a peer group of public companies over the expected term of the option.
Warrant Liability
We have issued to Platinum Long Term Growth VII, LLC, our largest investor (“Platinum”), warrants to purchase a substantial number of unregistered shares of our common stock and, subject to Platinum’s exercise of its rights to exchange shares of our Series A Preferred Stock that it holds, we are obligated to issue to Platinum an additional warrant to purchase unregistered shares of common stock (collectively, the “Platinum Warrants”). The Platinum Warrants contain an exercise price adjustment feature that will reduce the exercise price of the warrants in the event we subsequently issue equity instruments at a price lower than the exercise price of the Platinum Warrants. We account for the Platinum Warrants as non-cash liabilities and estimate their fair value at the end of each financial reporting period and record the change in the fair value as non-cash expense or non-cash income. The key component in determining the fair value of the Platinum Warrants and the related liability is the market price of our common stock, which is subject to significant fluctuation and is not under our control. The resulting change in the fair value of the warrant liability on our net income or loss is therefore also subject to significant fluctuation and will continue to be so until all of the Platinum Warrants are issued and exercised, amended or expire. Assuming all other fair value inputs remain generally constant, we will record an increase in the warrant liability and non-cash losses when our stock price increases and a decrease in the warrant liability and non-cash gains when our stock price decreases.
Notwithstanding the foregoing, and as described in Note 16, Subsequent Events, to the Consolidated Financial Statements included in this prospectus, on May 12, 2015, we entered into an agreement with Platinum pursuant to which Platinum agreed to amend the Platinum Warrants to (A) fix the exercise price thereof at $7.00 per share, (B) eliminate the exercise price reset features and (C) fix the number of shares of our common stock issuable thereunder. This agreement and the related amendments to the Platinum Warrants will result in the elimination of the warrant liability with respect to the Platinum Warrants during the quarter ending June 30, 2015, which is the first quarter of our fiscal year ending March 31, 2016.
We account for income taxes using the asset and liability approach for financial reporting purposes. We recognize deferred tax assets and liabilities for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. Valuation allowances are established, when necessary, to reduce the deferred tax assets to an amount expected to be realized.
Recent Accounting Pronouncements
See Note 3 to the consolidated financial statements included in Item 8 in this prospectus for information on recent accounting pronouncements.
Results of Operations
Comparison of Years Ended March 31, 2015 and 2014
Revenue
We reported no revenue for the fiscal years ended March 31, 2015 or 2014. We have successfully completed our Phase 1 development of AV-101. Additionally, as indicated previously, we have entered into a CRADA with the NIH providing for an NIH-sponsored Phase 2 clinical study of AV-101 in Major Depressive Disorder beginning in the second half of 2015.
Research and Development Expense
Research and development expense decreased by 2% in fiscal 2015 compared to fiscal 2014. The following table compares the primary components of research and development expense between the periods (in $000):
|
Fiscal Years Ended March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Salaries and benefits
|
$
|
889
|
$
|
902
|
||||
|
Stock-based compensation
|
849
|
453
|
||||||
|
UHN research under SRCA
|
-
|
160
|
||||||
|
Consulting services
|
109
|
53
|
||||||
|
Technology licenses and royalties
|
217
|
484
|
||||||
|
Project-related third-party research and supplies:
|
||||||||
|
AV-101
|
51
|
51
|
||||||
|
All other including CardioSafe and LiverSafe
|
54
|
145
|
||||||
|
105
|
196
|
|||||||
|
Rent
|
220
|
185
|
||||||
|
Depreciation
|
44
|
44
|
||||||
|
All other
|
-
|
4
|
||||||
|
Total Research and Development Expense
|
$
|
2,433
|
$
|
2,481
|
||||
To conserve cash resources, during fiscal 2015 and 2014, Ralph Snodgrass, PhD, our Chief Scientific Officer (CSO), has accepted a voluntary cash pay reduction to substantially less than his contractual pay rate. For fiscal 2015, the CSO’s actual cash pay represented approximately 52% of his contractual pay rate. In fiscal 2014, the CSO voluntarily agreed to accept a cash pay rate of approximately 82% of his contractual rate, not all of which amount was paid. In fiscal 2015, we have accrued the difference between the CSO’s contractual pay rate and his actual cash pay, $147,700, for future payment. In fiscal 2014, we accrued the difference between the CSO’s reduced pay rate and his actual cash pay, $100,400, for future payment. Such accrued amounts for both fiscal 2014 and fiscal 2015 remain unpaid. Pay rates for other scientific personnel remained constant between years. One member of our scientific staff voluntarily resigned at the end of September 2014 and another member has voluntarily reduced her work hours and pay since September 2014.
In January 2015, we granted five-year fully vested warrants to purchase an aggregate of 115,000 restricted shares of our common stock at an exercise price of $8.00 per share to our CSO and other scientific consultants and service providers, recognizing approximately $528,000 in stock-based compensation expense. Stock based compensation expense for fiscal 2015 and fiscal 2014 also reflects approximately $176,000 and $297,000, respectively, related to the ratable four-year amortization of option grants made to scientific staff and consultants in October 2013 and March 2014 and earlier. The ratable amortization of stock compensation expense related to certain options granted in October 2012 with a two-year vesting period ceased when the options became fully-vested in October 2014. An additional component of stock compensation expense is the amortization attributable to grants of warrants made to our CSO in March 2014 and March 2013, amounting to $145,000 in fiscal 2015 and $156,000 in fiscal 2014. The warrants are being amortized over a two-year vesting period, but are subject to certain vesting acceleration events. No further expense will be recognized with respect to the warrants granted in March 2013.
Our most recent sponsored research project budget under the collaboration agreement with Dr. Gordon Keller’s laboratory at UHN ended on September 30, 2013, and we have incurred no sponsored research expense under the agreement since that date. We are engaged in discussions with Dr. Keller and UHN regarding the scope of subsequent sponsored research projects and budget under the agreement, but have not yet finalized such project definitions and budgets.
Consulting services reflects fees paid or accrued for scientific services rendered to us by third-parties, primarily by members of our scientific advisory board.
Stem cell technology license expense reflects both recurring annual fees as well as costs for patent prosecution and protection that we are required to fund under the terms of certain of our license agreements. We recognize the latter costs as they are invoiced to us by the licensors and they do not occur ratably throughout the year or between years. Certain of our technology licensors invoiced us for significant legal fees for patent protection and prosecution during fiscal 2014.
AV-101 expenses in both fiscal years 2015 and 2014 primarily reflect the costs associated with monitoring for and responding to potential feedback related to the Phase 1 clinical trial and preparing other reports required under the terms of our prior NIH grant, primarily through our contract research collaborator, Cato Research Ltd.
The increase in rent expense versus FY 2014 reflects the full-year impact of rental costs related to our relocation to our current facilities in late-July 2013.
General and Administrative Expense
General and administrative expense increased by 70% in fiscal 2015 compared to fiscal 2014. The following table compares the primary components of general and administrative expense between the periods (in $000):
|
Fiscal Years Ended March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Salaries and benefits
|
$
|
714
|
$
|
675
|
||||
|
Stock-based compensation
|
1,611
|
684
|
||||||
|
Consulting Services
|
112
|
94
|
||||||
|
Legal, accounting and other professional fees
|
1,197
|
340
|
||||||
|
Investor relations
|
132
|
120
|
||||||
|
Insurance
|
136
|
130
|
||||||
|
Travel and entertainment
|
71
|
18
|
||||||
|
Rent and utilities
|
155
|
139
|
||||||
|
Warrant modification expense
|
98
|
205
|
||||||
|
All other expenses
|
118
|
143
|
||||||
|
Total General and Administrative Expense
|
$
|
4,344
|
$
|
2,548
|
||||
To conserve cash resources, during fiscal 2015 and 2014, both Shawn Singh, our Chief Executive Officer (CEO), and Jerrold Dotson, our Chief Financial Officer (CFO), have accepted voluntary cash pay reductions to substantially less than their contractual pay rates. For fiscal 2015, the CEO’s and CFO’s actual cash pay represented approximately 24% and 62%, respectively, of contractual rates. In fiscal 2014, the CEO and CFO voluntarily agreed to accept cash pay rates of approximately 72% and 80%, respectively, of their contractual rates, not all of which amounts were paid. In fiscal 2015, we have accrued the difference between the CEO’s and CFO’s contractual pay rate and his actual cash pay, $264,700 and $96,100, respectively, for future payment. In fiscal 2014, we accrued the difference between the CEO’s and CFO’s reduced pay rates and his actual cash pay, $125,000 and $56,700, respectively, for future payment. Such accrued amounts for both fiscal 2014 and fiscal 2015 remain unpaid and owing. Offsetting the impact of the accrual to contractual pay rates for the CEO and CFO for fiscal 2015 is the annual impact of the voluntary resignations of two administrative employees in August and November 2013 who have not been replaced. Pay rates for other administrative employees remained stable between the periods presented.
In January 2015, we granted five-year fully vested warrants to purchase an aggregate of 271,715 restricted shares of our common stock at an exercise price of $8.00 per share to our CEO and CFO, independent members of our Board of Directors and other consultants and service providers, recognizing approximately $1,229,000 in stock-based compensation expense. Stock based compensation expense for fiscal 2015 and fiscal 2014 also reflects approximately $99,000 and $385,000, respectively, related to the ratable four-year amortization of option grants made to employees and consultants in October 2013 and March 2014 and earlier. The ratable amortization of stock compensation expense related to certain options granted in October 2012 with a two-year vesting period ceased when the options became fully-vested in October 2014. An additional component of stock compensation expense is the amortization attributable to grants of warrants made to our CEO, CFO and independent members of our Board of Directors in March 2014 and March 2013, amounting to $283,000 in fiscal 2015 and $299,000 in fiscal 2014. The warrants are being amortized over a two-year vesting period, but are subject to certain vesting acceleration events. No further expense will be recognized with respect to the warrants granted in March 2013.
Consulting services primarily reflects fees paid or accrued for the services of the independent members of our Board of Directors.
The increase in legal, accounting and other professional fees results primarily from the impact of (i) two consulting agreements for strategic advisory and business development services pursuant to which we issued an aggregate of 55,000 restricted shares of our common stock valued at $469,000 at the date of issuance and paid $100,000 as cash compensation for such professional services during fiscal 2015; (ii) direct legal fees aggregating $150,000 related to services provided with respect to our prospective public offering of our equity securities and a proposed private offering of our equity securities; (iii) the expensing of approximately $102,000 of investment banker, banker’s counsel, accounting and other fees and costs related to the cancellation of our prospective public offering; (iv) legal and other costs related to the 1:20 reverse split of our common stock in August 2014 and legal and filing fees for our private placement Unit financing offerings; and (v) costs related to temporary employee fees for part-time administrative services.
Outsourced investor relations service expenses are essentially flat between periods; we have conducted no special awareness or other initiatives during either fiscal 2015 or 2014. Travel expenses related to meetings with potential investors in our attempted registered public offering account for the increase compared to fiscal 2014.
The fiscal 2015 increase in rent and utilities expense reflects the full-year impact of increased costs related to our relocation to expanded facilities in late-July 2013.
Warrant modification expense in fiscal 2015 reflects the extension by one year of the term of outstanding warrants otherwise scheduled to expire during calendar 2015, as approved by our Board of Directors in January 2015. Warrant modification expense in fiscal 2014 reflects the impact of October 2013 and December 2013 reductions in the exercise price of certain outstanding warrants, generally from $35.00 per share or $30.00 per share, to $10.00 per share, and in limited cases, the extension of the term of certain outstanding warrants, and from which we used the proceeds of the warrant exercises as a source of short-term working capital.
The fiscal 2015 decrease in other expenses is attributable to one-time relocation costs incurred in fiscal 2014 in connection with our relocation to expanded facilities in late-July 2013.
Other Expenses, Net
In both fiscal 2015 and 2014, other expenses, net includes interest expense, including non-cash discount amortization, on our outstanding promissory notes, as well as the non-cash impact of changes in the fair value of the warrant liabilities related to warrants issued or issuable to Platinum between October 2012 and July 2013. In fiscal 2015, other expenses, net also includes the non-cash loss on extinguishment of debt resulting from the modification of indebtedness to Platinum, holders of convertible promissory notes originally scheduled to mature on July 30, 2014, and to a technology licensor and a professional service provider. Additionally, in fiscal 2015 we incurred non-cash expense related to the settlement of a note receivable we accepted in fiscal 2012.
The following table compares the primary components of net interest expense between the periods (in $000):
|
Fiscal Years Ended March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Interest expense on promissory notes
|
$
|
1,238
|
$
|
907
|
||||
|
Amortization of discount on promissory notes
|
3,372
|
640
|
||||||
|
Other interest expense, including on capital leases and premium financing
|
7
|
15
|
||||||
|
4,617
|
1,562
|
|||||||
|
Effect of foreign currency fluctuations on notes payable
|
(63
|
)
|
(49
|
)
|
||||
|
Interest income
|
(5
|
)
|
(10
|
)
|
||||
|
Interest expense, net
|
$
|
4,549
|
$
|
1,503
|
||||
The increase in interest expense between the periods is primarily attributable to the accrued interest recorded for the issuances between August 2013 and March 2015 of an aggregate of approximately $4.1 million of unsecured 10% convertible promissory notes pursuant to the 2013 Unit Private Placement and the 2014 Unit Private Placement. As a result of the significant inception-date discounts recorded in connection with the Unit Notes, approximately $2.7 million in fiscal 2015; the relatively short period between issuance and maturity over which the discount on the Unit Notes must be amortized, generally less than 12 months; and the accelerating amount of discount amortization recorded using the effective interest rate method as the notes approach maturity, discount amortization expense increased by approximately $2.7 million between the periods shown in the preceding table.
Under the terms of the October 2012 Note Exchange and Purchase Agreement we entered with Platinum, we issued Senior Secured Convertible Promissory Notes and a related Exchange Warrant and Investment Warrants between October 2012 and March 2013. We issued a similar senior secured promissory note and related warrant to Platinum in July 2013. Upon Platinum’s exchange of the shares of our Series A preferred stock it holds into shares of our common stock, we will also be required to issue a Series A Exchange Warrant to Platinum. We determined that certain exercise price and share adjustment features contained in the various warrants require us to treat the warrants as liabilities. Accordingly, we recorded a non-cash warrant liability at its estimated fair value as of the date the warrant was issued or the contract executed. During fiscal 2015, we recognized a non-cash loss of $34,600 related to the net increase in the estimated fair value of these non-cash liabilities since March 31, 2014, which resulted primarily from the change in the market price of our common stock in relation to the exercise price of the warrants and an additional year elapsed in the remaining term for all but the Series A Exchange Warrant. During fiscal 2014, we recognized a non-cash gain of $3,556,900 related to the net decrease in the estimated fair value of the warrant liabilities since March 31, 2013, which resulted primarily from the decrease in the market price of our common stock in relation to the exercise price of the warrants.
As described more fully in Note 8, Convertible Promissory Notes and Other Notes Payable, and Note 9, Capital Stock, in the Consolidated Financial Statements included in Item 8 of this prospectus, effective May 31, 2014, we entered into agreements with substantially all holders of our 2013 Unit Notes and 2013 Unit Warrants to amend certain terms of the notes and the warrants. We treated the amendments as an extinguishment of debt for accounting purposes. Accordingly, since the fair value of the amended notes and warrants exceeded the carrying amount of the original notes, we recognized non-cash losses on the extinguishment of debt in the aggregate amount of $526,200 attributable to the amendments. We recognized an additional $241,800 as a non-cash loss on extinguishment of debt as a result of the promissory note, shares of our common stock and warrants issued to Icahn School of Medicine at Mount Sinai in settlement of stem cell technology license maintenance fees and reimbursable patent prosecution costs, as described more completely in Note 9, Capital Stock, to the Consolidated Financial Statements included in Item 8 of this prospectus. We recognized a further $16,700 non-cash loss on extinguishment of debt as a result of the shares of our unregistered common stock issued to a professional services provider in settlement of fees for prior services rendered, as also described more completely in Note 9, Capital Stock, to the Consolidated Financial Statements included in Item 8 of this prospectus.
As described more completely in Note 8, Convertible Promissory Notes and Other Notes Payable, to the Consolidated Financial Statements included in this prospectus, in July 2014, we entered into an agreement with Platinum, as further amended in September 2014, pursuant to which Platinum agreed to convert into our unregistered equity securities all Senior Notes and accrued but unpaid interest thereon held by Platinum upon our consummation prior to October 31, 2014 (the “Closing Date”) of either (i) a Private Financing or a Public Offering, each as defined in the agreement. Upon consummation of a Private Financing, the Senior Notes would have converted into that number of unregistered shares of our common stock equal to the Outstanding Balance on the Closing Date, divided by $10.00 per share. Upon consummation of a Public Offering, the Senior Notes would have converted into shares of a newly created Series B Convertible Preferred Stock with an aggregate liquidation preference equal to the Outstanding Balance on the Closing Date. Prior to the agreement, the Senior Notes were convertible, at Platinum’s option, at any time prior to maturity at a conversion price of $10.00 per share. The modification of the conversion feature in the Senior Notes was treated as an extinguishment of the debt for accounting purposes. Accordingly, since the fair value of the amended Senior Notes substantially exceeded the carrying amount of the original notes, we recognized a non-cash loss on the extinguishment of debt in the aggregate amount of $1,603,400 attributable to the amendment.
As described in Note 9, Capital Stock, to the Consolidated Financial Statements included in this prospectus, in October 2014, we accepted a cash payment of $60,000 as settlement in full for a promissory note issued to us in May 2011 for the purchase of shares of our common stock. At the time of the payment, the principal and accrued interest due to us under the note receivable was $194,900, resulting in a recognized loss of $134,900 related to the settlement.
Liquidity and Capital Resources
Since our inception in May 1998 through March 31, 2015, we have financed our operations and technology acquisitions primarily through the issuance and sale of equity and debt securities, including secured and unsecured convertible promissory notes and short-term promissory notes, for cash proceeds of approximately $29.0 million. In addition, we have obtained an aggregate of approximately $16.4 million in government research grant awards, strategic collaboration payments and other revenues. Additionally, we have issued equity securities with an approximate value at issuance of $13.5 million in non-cash settlements of certain liabilities, including liabilities for professional services rendered to us or as compensation for such services.
Between late-March 2014 and March 31, 2015, we entered into securities purchase agreements with accredited investors, including Platinum, pursuant to which we sold units to such accredited investors, in private placement transactions (“2014 Units” or “2014 Unit Private Placement”), for aggregate cash proceeds of $3,113,500, consisting of (i) 2014 Unit Notes in the aggregate face amount of $3,113,500 which matured between March 31, 2015 and April 30, 2015, or were automatically convertible into securities we might issue upon the consummation of a Qualified Financing, as defined, (ii) an aggregate of 282,850 restricted shares of our common stock (“2014 Unit Stock”); and (iii) warrants exercisable through December 31, 2016 to purchase an aggregate of 282,850 restricted shares of our common stock at an exercise price of $10.00 per share (“2014 Unit Warrants”). We currently anticipate that we will need to obtain approximately $7.0 million in financing over the course of the next twelve months to execute our business plan.
At March 31, 2015, we did not have sufficient cash and cash equivalents to enable us to fund our planned operations, including expected cash expenditures of approximately $7.0 million through the next twelve months. To meet our operational needs and fund our working capital requirements after March 31, 2015, we extended the 2014 Unit Private Placement and, through May 14, 2015, we entered into additional securities purchase agreements with accredited investors pursuant to which we sold to such accredited investors 2014 Units, for aggregate cash proceeds of $280,000, consisting of: (i) 10% convertible promissory notes maturing between April 30, 2015 and May 15, 2015, in the aggregate face amount of $280,000, (ii) an aggregate of 33,000 shares of our restricted common stock, and (iii) warrants exercisable through December 31, 2016 to purchase an aggregate of 24,250 restricted shares of our common stock at an exercise price of $10.00 per share.
Effective May 12, 2015, we entered into an agreement with Platinum (“Platinum Agreement”), pursuant to which Platinum:
|
·
|
Converted the approximately $4.5 million outstanding balance (principal and accrued but unpaid interest) of the Senior Notes we issued to Platinum into 641,335 shares of our newly-created Series B 10% Convertible Preferred Stock (“Series B Preferred”), thereby cancelling approximately $4.5 million of our outstanding indebtedness;
|
|
·
|
Released all of its security interests in our assets and those of our subsidiaries by terminating the Amended and Restated Security Agreement, IP Security Agreement and Negative Covenant, each dated October 11, 2012 between us and Platinum;
|
|
·
|
Converted the approximately $1.3 million outstanding balance (principal and accrued but unpaid interest) of the convertible promissory Notes we issued to Platinum in our 2014 Unit Private Placement (“2014 Unit Notes”) into 240,305 shares of Series B Preferred and five-year warrants to purchase 240,305 shares of our common stock at a fixed exercise price of $7.00 per share (“Series B Warrants”), thereby cancelling approximately $1.3 million of our outstanding indebtedness;
|
|
·
|
Purchased approximately $1.5 million (including accrued but unpaid interest thereon) of outstanding 2014 Unit Notes we issued to various investors from the respective holders thereof (“Investor 2014 Unit Notes”) and converted the entire outstanding balance of the Investor 2014 Unit Notes into 265,699 shares of Series B Preferred and Series B Warrants to purchase 265,699 shares of our common stock, thereby cancelling approximately $1.5 million of our outstanding indebtedness;
|
|
·
|
Signed and delivered a Securities Purchase Agreement (“SPA”) to purchase, for $1.0 million, a total of 142,857 shares of Series B Preferred and a Series B Warrant to purchase 142,857 shares of our common stock, on or before June 11, 2015 (a portion of which purchase has been consummated);
|
|
·
|
Amended the Platinum Warrants (all warrants previously issued by us to Platinum in connection with the Senior Notes) and the Series A Exchange Warrant to:
|
|
·
|
fix the exercise price of Platinum Warrants and the Series A Exchange Warrant at $7.00 per share;
|
|
·
|
eliminate the exercise price reset features and fix the number of shares of our common stock issuable under the Platinum Warrants; and
|
|
·
|
eliminate the cashless exercise provisions from the Platinum Warrants and the Series A Exchange Warrant; and
|
|
·
|
Agreed to refrain from the sale of any shares of our common stock held by Platinum or its affiliates until the earlier to occur of an effective registration statement relating to resale of certain specified shares of common stock under the Securities Act of 1933, as amended, or the closing price of our common stock is at least $15.00 per share.
|
As additional consideration for the agreements of Platinum under the Platinum Agreement, we issued to Platinum 400,000 shares of Series B Preferred and Warrants to purchase 1.2 million shares of our common stock and exchanged 30,000 shares of our common stock currently beneficially owned or controlled by Platinum for 30,000 shares of Series B Preferred.
Effective May 20, 2015, holders of the remaining approximately $1.8 million outstanding balance (principal and accrued but unpaid interest) of unsecured 2014 Unit Notes converted such notes into 327,016 shares of Series B Preferred and Series B Warrants to purchase 327,016 shares of our common stock, thereby cancelling an additional approximately $1.8 million of our outstanding indebtedness.
Between May 26, 2015 and July 17,2015, we sold to accredited investors an aggregate of $1,112,500 of units in our Series B Preferred Unit offering, which units consist of Series B Preferred and Series B Warrants (together Series B Preferred Units), including $500,000 to Platinum. We have issued 160,363 shares of Series B Preferred and Series B Warrants to purchase 160,363 shares of our common stock and we have received an aggregate of $557,500 in cash proceeds from the sale of the Series B Preferred Units. As described more completely in Note 16, Subsequent Events, to the Consolidated Financial Statements included in this prospectus, each share of Series B Preferred is convertible into one share of our common stock and the Series B Warrants are exercisable at a fixed price of $7.00 per share for a term of five years following issuance.
Additionally, through July 17, 2015, holders of certain of our promissory notes outstanding at March 31, 2015 and thereafter, including Morrison & Foerster, University Health Network, Cato Research Ltd., McCarthy Tetrault, and certain other service providers converted notes payable or accounts payable having an aggregate balance of approximately $6.6 million (principal and accrued but unpaid interest and certain strategic adjustments) into 946,934 shares of Series B Preferred.
Since March 31, 2015, we have eliminated approximately $15.8 million of promissory notes, other debt and certain adjustments thereto that was either already due and payable or would have otherwise matured prior to March 31, 2016, through conversion into our Series B Preferred and, with respect to a portion of the indebtedness converted, warrants to purchase common stock. Together with the cash proceeds from our Series B Preferred Unit Offering, our working capital position has improved significantly since March 31, 2015. We will, however, need to raise additional capital to fund our operations and execute our business plan over the next year and thereafter.
We believe that our participation in potential strategic collaborations, including potential transactions involving AV-101 such as our February 2015 CRADA with the NIH for an NIH-funded and sponsored Phase 2 study of AV-101 in MDD, may provide resources to support a portion of our future cash needs and working capital requirements, however, no assurances can be provided. When and as necessary, we will seek to raise a material amount of financing through a combination of additional private placements and/or registered public offerings of our securities, which may include both debt and equity securities, stem cell technology-based research and development collaborations, stem cell technology and drug candidate license fees, and government grant awards and collaborations. Our future working capital requirements will depend on many factors, including, without limitation, the scope and nature of strategic opportunities related to our success in clinical trials of and further developing AV-101 as a treatment for MDD and/or other conditions; our stem cell technology platform, including drug rescue and cell therapy research and development efforts and the success of such programs, our ability to obtain government grant awards and our ability to enter into strategic collaborations with institutions on terms acceptable to us. To further advance the clinical development of AV-101 and potential drug rescue applications of our stem cell technology platform, as well as support our operating activities, we plan to continue to carefully manage our routine operating costs, including salaries and benefits, regulatory consulting, contract research and development, legal, accounting, public company compliance and other professional services and working capital costs.
Notwithstanding the foregoing, substantial additional financing may not be available to us on a timely basis, on acceptable terms, or at all. If we are unable to obtain substantial additional financing on a timely basis in the near term, our business, financial condition, and results of operations may be harmed, the price of our stock may decline, we may be required to reduce, defer, or discontinue certain of our research and development activities and we may not be able to continue as a going concern.
The following table summarizes changes in cash and cash equivalents for the periods stated (in thousands):
|
Fiscal Years Ended
|
||||||||
|
March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Net cash used in operating activities
|
$
|
(2,769
|
)
|
$
|
(2,126
|
)
|
||
|
Net cash used in investing activities
|
-
|
(10
|
)
|
|||||
|
Net cash provided by financing activities
|
2,839
|
1,498
|
||||||
|
Net increase (decrease) in cash and cash equivalents
|
70
|
(638
|
)
|
|||||
|
Cash and cash equivalents at beginning of period
|
-
|
638
|
||||||
|
Cash and cash equivalents at end of period
|
$
|
70
|
$
|
-
|
||||
Off-Balance Sheet Arrangements
Other than contractual obligations incurred in the normal course of business, we do not have any off-balance sheet financing arrangements or liabilities, guarantee contracts, retained or contingent interests in transferred assets or any obligation arising out of a material variable interest in an unconsolidated entity. VistaGen California has two inactive, wholly-owned subsidiaries, Artemis Neuroscience, Inc., a Maryland corporation, and VistaStem Canada, Inc., an Ontario corporation.
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
DIRECTORS, EXECUTIVE OFFICERS, PROMOTERS AND CONTROL PERSONS
Our senior management is composed of individuals with significant management experience. Our directors and executive officers are as follows:
|
Name
|
Age
|
Position
|
||||
|
Shawn K. Singh, J.D.
|
52
|
Chief Executive Officer and Director
|
||||
|
H. Ralph Snodgrass, Ph.D.
|
65
|
Founder, President, Chief Scientific Officer and Director
|
||||
|
Jerrold D. Dotson
|
62
|
Vice President, Chief Financial Officer and Secretary
|
||||
|
Jon S. Saxe (1)
|
79
|
Director
|
||||
|
Brian J. Underdown, PhD. (2)
|
74
|
Director
|
||||
|
(1)
|
Chairman of the audit committee and member of the compensation committee and corporate governance and nominating committee
|
|
(2)
|
Member of the audit committee and chairman of the compensation committee and corporate governance and nominating committee
|
Executive Officers
Shawn K. Singh, J.D. has served as our Chief Executive Officer since August 2009; he joined our Board of Directors in 2000 and served on our management team (part-time) from late-2003, following our acquisition of Artemis Neuroscience, of which he was President, to August 2009. Mr. Singh has over 20 years of experience working with biotechnology, medical device and pharmaceutical companies, both private and public. From February 2001 to August 2009, Mr. Singh served as Managing Principal of Cato BioVentures, a life science venture capital firm, and as Chief Business Officer and General Counsel of Cato Research Ltd, a profitable global contract research organization (CRO) affiliated with Cato BioVentures. Mr. Singh served as President (part-time) of Echo Therapeutics (NASDAQ: ECTE), a medical device company, from September 2007 to June 2009, and as a member of its Board of Directors from September 2007 through December 2011. He also served as Chief Executive Officer (part-time) of Hemodynamic Therapeutics, a private biopharmaceutical company affiliated with Cato BioVentures, from November 2004 to August 2009. From late-2000 to February 2001, Mr. Singh served as Managing Director of Start-Up Law, a management consulting firm serving biotechnology companies. Mr. Singh also served as Chief Business Officer of SciClone Pharmaceuticals (NASDAQ: SCLN), a US-based, China-focused specialty pharmaceutical company with a substantial revenue-generating and profitable commercial business and a marketed product portfolio of differentiated therapies for oncology, infectious diseases and cardiovascular disorders, from late-1993 to late-2000, and as a corporate finance associate of Morrison & Foerster LLP, an international law firm, from 1991 to late-1993. Mr. Singh currently serves as a member of the Board of Directors of Armour Therapeutics, a private biotechnology company focused on prostate cancer. Mr. Singh earned a B.A. degree, with honors, from the University of California, Berkeley, and a J.D. degree from the University of Maryland School of Law. Mr. Singh is a member of the State Bar of California.
We selected Mr. Singh to serve on our Board of Directors due to his substantial practical experience and expertise in senior leadership roles with multiple private and public biotechnology, pharmaceutical and medical device companies, and his extensive experience in corporate finance, venture capital, corporate governance and strategic partnering.
H. Ralph Snodgrass, Ph.D. co-founded VistaGen with Dr. Gordon Keller in 1998 and served as our Chief Executive Officer until August 2009. Dr. Snodgrass has served as our President and Chief Scientific Officer since August 2009. He has served as a member of our Board of Directors since 1998. Prior to founding VistaGen, Dr. Snodgrass served as a key member of the executive management team which lead Progenitor, Inc., a biotechnology company focused on developmental biology, through its initial public offering, and was its Chief Scientific Officer from June 1994 to May 1998, and its Executive Director from July 1993 to May 1994. He received his Ph.D. in immunology from the University of Pennsylvania, and has 20 years of experience in senior biotechnology management and over 10 years research experience as a professor at the Lineberger Comprehensive Cancer Center, University of North Carolina Chapel Hill School of Medicine, and as a member of the Institute for Immunology, Basel, Switzerland. Dr. Snodgrass is a past Board Member of the Emerging Company Section of the Biotechnology Industry Organization (BIO), and past member of the International Society Stem Cell Research Industry Committee. Dr. Snodgrass has published more than 50 scientific papers, is the inventor on more than 17 patents and a number of patent applications, is, or has been, the principal investigator on U.S. federal and private foundation sponsored research grants with budgets totaling more than $14.5 million and is recognized as an expert in stem cell biology with more than 28 years’ experience in the uses of stem cells as biological tools for research, drug discovery and development.
We selected Dr. Snodgrass to serve on our Board of Directors due to his expertise in biotechnology focused on developmental biology, including stem cell biology, his extensive senior management experience leading biotechnology companies at all stages of development, as well as his reputation and standing in the fields of biotechnology and stem cell research, allow him to bring to us and the Board of Directors a unique understanding of the challenges and opportunities associated with pluripotent stem cell biology, as well as credibility in the markets in which we operate.
Jerrold D. Dotson, CPA has served as our Chief Financial Officer since September 2011, as our Corporate Secretary since October 2013 and as a Vice President since February 2014. Mr. Dotson served as Corporate Controller for Discovery Foods Company, a privately held Asian frozen foods company from January 2009 to September 2011. From February 2007 through September 2008, Mr. Dotson served as Vice President, Finance and Administration (principal financial and accounting officer) for Calypte Biomedical Corporation (OTCBB: CBMC), a publicly held biotechnology company. Mr. Dotson served as Calypte’s Corporate Secretary from 2001 through September 2008. He also served as Calypte’s Director of Finance from January 2000 through July 2005 and was a financial consultant to Calypte from August 2005 through January 2007. Prior to joining Calypte, from 1988 through 1999, Mr. Dotson worked in various financial management positions, including Chief Financial Officer, for California & Hawaiian Sugar Company, a privately held company. Mr. Dotson is licensed as a CPA in California and received his BS degree in Business Administration with a concentration in accounting from Abilene Christian College.
Directors
Jon S. Saxe, J.D. has served as Chairman of our Board of Directors since 2000. He also serves as the Chairman of our Audit Committee. Mr. Saxe is the retired President and was a director of PDL BioPharma from 1989 to 2008. From 1989 to 1993, he was President, Chief Executive Officer and a director of Synergen, Inc. (acquired by Amgen). Mr. Saxe served as Vice President, Licensing & Corporate Development for Hoffmann-Roche from 1984 through 1989, and Head of Patent Law for Hoffmann-Roche from 1978 through 1989. Mr. Saxe currently is a director of SciClone Pharmaceuticals, Inc. (NASDAQ: SCLN) and Durect Corporation (NASDAQ: DRRX), and five private life science companies, Arbor Vita Corporation, Arcuo Medical, LLC, Armetheon, Inc., Lumos Pharma, Inc. and Trellis Bioscience. Mr. Saxe also has served as a director of other biotechnology and pharmaceutical companies, including ID Biomedical (acquired by GlaxoSmithKline), Sciele Pharmaceuticals, Inc. (acquired by Shionogi), Amalyte (acquired by Kemin Industries), Cell Pathways (acquired by OSI Pharmaceuticals), and other companies, both public and private. Mr. Saxe has a B.S.Ch.E. from Carnegie-Mellon University, a J.D. degree from George Washington University and an LL.M. degree from New York University.
We selected Mr. Saxe to serve as Chairman of our Board of Directors due to numerous years of experience as a senior executive with major biopharmaceutical and biotechnology companies, including Protein Design Labs, Inc., Synergen, Inc. and Hoffmann-Roche, Inc., as well as his extensive experience serving as a director of numerous private and public biotechnology and pharmaceutical companies, serving as Chairman, and Chair and member of audit, compensation and governance committees of both private and public companies. Mr. Saxe provides us and our Board of Directors with highly valuable insight and perspective into the biotechnology and pharmaceutical industries, as well as the strategic opportunities and challenges that we face.
Brian J. Underdown, Ph.D. has served as a member of our Board of Directors since November 2009. Dr. Underdown has served as Managing Director of Lumira Capital Corp. since September 1997, having started in the venture capital industry in 1997 with MDS Capital Corporation (MDSCC). His investment focus has been on therapeutics in both new and established companies in both Canada and the United States. Prior to joining MDSCC, Dr. Underdown held a number of senior management positions in the biopharmaceutical industry and at universities. Dr. Underdown’s past and current board positions include: ID Biomedical, Trillium Therapeutics, Cytochroma Inc., Argos Therapeutics, Locus Therapeutics, Nysa Membrane Technologies, Ception Therapeutics and Transmolecular Therapeutics. He has served on a number of Boards and advisory bodies of government sponsored research organizations including CANVAC, the Canadian National Centre of Excellence in Vaccines, Ontario Genomics Institute, Allergen, the Canadian National Centre of Excellence in Allergy and Asthma. Dr. Underdown obtained his Ph.D. in immunology from McGill University and undertook post-doctoral studies at Washington University School of Medicine.
We selected Dr. Underdown to serve on our Board of Directors due to his extensive background working in the biotechnology and pharmaceutical industries, as a director of numerous private and public companies, as well as his venture capital experience funding and advising start-up and established companies focused on therapeutics.
Election of Executive Officers
Our executive officers are elected by, and serve at the discretion of, our Board of Directors. Each of our executive officers devotes his full time to our affairs. There are no family relationships among any of our directors or executive officers.
Board Composition
Our amended and restated bylaws provide that the authorized number of directors of the Company shall be not less than one nor more than seven, with the exact number of directors currently fixed at seven. The exact number may be amended only by the vote or written consent of a majority of the outstanding shares of our voting stock. Our Board of Directors currently consists of four members. Accordingly, there are currently three vacancies on our Board of Directors. Our Board of Directors anticipates filling each of such vacancies as soon as practicable. All actions of the Board of Directors require the approval of a majority of the directors in attendance at a meeting at which a quorum is present.
Board Committees
Our Board of Directors has established an audit committee, a compensation committee and a corporate governance and nominating committee. The composition and responsibilities of each committee are described below. Members serve on these committees until their resignation or until otherwise determined by our Board of Directors. Our independent directors, Mr. Saxe and Dr. Underdown, are each members of the audit committee. Mr. Saxe and Dr. Underdown also currently serve as members of the compensation committee and the corporate governance and nominating committee.
Audit Committee
Our audit committee is comprised of Mr. Saxe and Dr. Underdown. Mr. Saxe is the chairman of our audit committee and is our audit committee financial expert, as that term is defined under SEC rules implementing Section 407 of the Sarbanes Oxley Act of 2002, and possesses the requisite financial sophistication, as defined under applicable rules. The audit committee operates under a written charter. Our audit committee charter is available on our website. Under its charter, our audit committee is primarily responsible for, among other things,
|
·
|
overseeing our accounting and financial reporting process;
|
|
·
|
selecting, retaining and replacing our independent auditors and evaluating their qualifications, independence and performance;
|
|
·
|
reviewing and approving scope of the annual audit and audit fees;
|
|
·
|
monitoring rotation of partners of independent auditors on engagement team as required by law;
|
|
·
|
discussing with management and independent auditors the results of annual audit and review of quarterly financial statements;
|
|
·
|
reviewing adequacy and effectiveness of internal control policies and procedures;
|
|
·
|
approving retention of independent auditors to perform any proposed permissible non-audit services;
|
|
·
|
overseeing internal audit functions and annually reviewing audit committee charter and committee performance; and
|
|
·
|
preparing the audit committee report that the SEC requires in our annual proxy statement.
|
Compensation Committee
Our compensation committee is comprised of Mr. Saxe and Dr. Underdown, who serves as the committee chairman. Our compensation committee charter is available on our website. Under its charter, the compensation committee is primarily responsible for, among other things,
|
·
|
Reviewing and approving our compensation programs and arrangements applicable to our executive officers (as defined in Rule I 6a-I (f) of the Exchange Act), including all employment-related agreements or arrangements under which compensatory benefits are awarded or paid to, or earned or received by, our executive officers, including, without limitation, employment, severance, change of control and similar agreements or arrangements;
|
|
·
|
Determining the objectives of our executive officer compensation programs;
|
|
·
|
Ensuring corporate performance measures and goals regarding executive officer compensation are set and determining the extent to which they are achieved and any related compensation earned;
|
|
·
|
Establishing goals and objectives relevant to CEO compensation, evaluating CEO performance in light of such goals and objectives, and determining CEO compensation based on the evaluation; and
|
|
·
|
Endeavoring to ensure that our executive compensation programs are effective in attracting and retaining key employees and reinforcing business strategies and objectives for enhancing stockholder value, monitoring the administration of incentive-compensation plans and equity-based incentive plans as in effect and as adopted from time to time by the board.
|
|
·
|
Reviewing and approving any new equity compensation plan or any material change to an existing plan.
|
|
·
|
Reviewing and approving any stock option award or any other type of award as may be required for complying with any tax, securities, or other regulatory requirement, or otherwise determined to be appropriate or desirable by the committee or board.
|
Corporate Governance and Nominating Committee
Our corporate governance and nominating committee is comprised of Mr. Saxe and Dr. Underdown, who serves as the committee chairman. Our corporate governance and nominating committee charter is available on our website. Under its charter, the corporate governance and nominating committee is primarily responsible for, among other things:
|
·
|
Monitoring the size and composition of the board;
|
|
·
|
Making recommendations to the board with respect to the nominations or elections of our directors;
|
|
·
|
Reviewing the adequacy of our corporate governance policies and procedures and our Code of Business Conduct and Ethics, and recommending any proposed changes to the board for approval; and
|
|
·
|
Considering any requests for waivers from our Code of Business Conduct and Ethics and ensure that we disclose such waivers as may be required by the exchange on which we are listed, if any, and rules and regulations of the SEC.
|
All potential candidates for director nominees, including candidates recommended by our stockholders, are reviewed in the context of the current composition of the Board, our operating requirements and the long-term interests of our stockholders. In conducting this assessment, the Committee considers such factors as it deems appropriate given our current needs and those of our Board, to maintain a balance of expertise, experience and capability. The Corporate Governance and Nominating Committee reviews directors’ overall service during their term, including the number of meetings attended, their level of participation and quality of performance. The Committee also determines whether the nominee would be independent, which determination is based upon applicable NASDAQ or other exchange listing standards, applicable SEC rules and regulations and the advice of counsel, if necessary.
The Corporate Governance and Nominating Committee will consider director candidates recommended by stockholders in the same manner as it considers recommendations from current directors or other sources. Stockholders who wish to recommend individuals for consideration by the Corporate Governance and Nominating Committee to become nominees for election to the Board may do so by delivering a written recommendation to the Company Secretary at the following address: 343 Allerton Avenue, South San Francisco, CA 94080 at least 60 days prior, but no more than 90 days prior, to the anniversary date of the last annual meeting of stockholders. Submissions should include the full name, address and age of the proposed nominee, a description of the proposed nominee’s business experience for at least the previous five years, complete biographical information, a description of the proposed nominee’s qualifications as a director, and the number of shares of our stock beneficially owned by the proposed nominee. The nominating stockholder must also provide his or her name and address of record and the number of shares of our stock that he or she owns beneficially or of record.
The Corporate Governance and Nominating Committee has not established specific minimum qualifications for recommended nominees or specific qualities or skills for one or more of our directors to possess, other than as are necessary to meet any requirements under rules and regulations (including any stock exchange rules) applicable to the Company. The Corporate Governance and Nominating Committee uses a subjective process for identifying and evaluating nominees for director, based on the information available to, and the subjective judgments of, the members of the Committee and our then current needs for the Board as a whole. Although it does not have a formal policy regarding the consideration of diversity, the Corporate Governance and Nominating Committee considers the needs for the Board as a whole when identifying and evaluating nominees and, among other things, considers diversity in background, age, experience, qualifications, attributes and skills in identifying nominees.
The Corporate Governance and Nominating Committee’s process for identification and evaluation of director candidates is generally as follows:
(a) In the event of a vacancy or the establishment of a new directorship on the Board, candidate(s) for director nominee(s) shall be presented to the full Board for consideration and approval upon the recommendation of no less than a majority of the independent members of the Board (as independence is defined under any stock exchange rules that may be applicable to the Company at such time).
(b) We believe that the continuing service of qualified incumbents promotes stability and continuity in the boardroom, contributing to the Board's ability to work as a collective body, while giving us the benefit of the familiarity and insight into our affairs that our directors have accumulated during their tenure. Accordingly, the process for identifying nominees reflects our practice of re-nominating incumbent directors who continue to satisfy the criteria for membership on the Board, whom the independent members of the Board believe continue to make important contributions to the Board and who consent to continue their service on the Board. Consistent with this policy, in considering candidates for election at annual meetings of stockholders, the independent members of the Board will first determine the incumbent directors whose terms expire at the upcoming meeting and who wish to continue their service on the Board.
(c) The independent members of the Board will evaluate the qualifications and performance of the incumbent directors that desire to continue their service. In particular, as to each such incumbent director, the independent members of the Board will (i) consider if the director continues to satisfy the minimum qualifications for director candidates adopted by the independent members of the Board, (ii) review any assessments of the performance of the director during the preceding term made by the Board, and (iii) determine whether there exist any special, countervailing considerations against re-nomination of the director.
(d) If the independent members of the Board determine that an incumbent director consenting to re-nomination continues to be qualified and has satisfactorily performed his or her duties as director during the preceding term, and there exist no reasons, including considerations relating to the composition and functional needs of the Board as a whole, why in the view of the independent members of the Board the incumbent should not be re-nominated, the independent members of the Board will, absent special circumstances, propose the incumbent director for reelection.
(e) The process by the independent members of the Board for identifying and evaluating nominees for director, including nominees recommended by a stockholder, involves (with or without the assistance of a retained search firm):
|
·
|
compiling names of potentially eligible candidates;
|
|
·
|
conducting background and reference checks;
|
|
·
|
conducting interviews with candidates and/or others;
|
|
·
|
meeting to consider and approve final candidates; and, as appropriate,
|
|
·
|
preparing and presenting to the full Board an analysis with regard to particular recommended candidates.
|
During the search process, the independent directors shall endeavor to identify director nominees who have the highest personal and professional integrity, have demonstrated exceptional ability and judgment, and, together with other director nominees and current Board members, shall effectively serve the long-term interests of our stockholders and contribute to our overall corporate goals.
(f) In considering potential new directors, the independent members of the Board will review individuals from various disciplines and backgrounds. Among the qualifications to be considered in the selection of candidates are:
|
·
|
personal and professional integrity;
|
|
·
|
broad experience in business, finance or administration;
|
|
·
|
familiarity with our industry; and
|
|
·
|
prominence and reputation.
|
Code of Business Conduct and Ethics
We have adopted a Code of Business Conduct and Ethics applicable to our employees, officers and directors. Our Code of Business Conduct and Ethics is available on our website at www.vistagen.com. We intend to disclose any future amendments to certain provisions of our Code of Business Conduct and Ethics, or waivers of these provisions, on our website or in filings with the SEC under the Exchange Act.
Board Attendance at Board of Directors, Committee and Stockholder Meetings
Our Board of Directors met two times and acted by unanimous written consent six times during the fiscal year ended March 31, 2015. Our Audit Committee met four times and our Compensation Committee requested action by the entire Board of Directors for grants of warrants and the modification of certain warrants during the same period. Each director serving during fiscal 2015 attended all of the meetings of the Board and the committees of the Board upon which such director served.
We do not have a formal policy regarding attendance by members of the Board at our annual meeting of stockholders, but directors are encouraged to attend. We did not hold an annual meeting of stockholders during our fiscal year ended March 31, 2015.
Compensation Committee Interlocks and Insider Participation
Our Compensation Committee consists of Dr. Underdown and Mr. Saxe, each of whom is a non-employee director. Neither member of the Compensation Committee has a relationship that would constitute an interlocking relationship with executive officers or directors of another entity.
Section 16 Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our officers, directors and persons who beneficially own more than ten percent of our common stock (collectively, Reporting Persons) to file reports of ownership on Form 3 and changes in ownership on Form 4 or Form 5 with the Commission. The Reporting Persons are also required by SEC rules to furnish us with copies of all reports that they file pursuant to Section 16(a). We believe that during our fiscal year ended March 31, 2015, all of the Reporting Persons complied with all applicable reporting requirements.
EXECUTIVE COMPENSATION
Our Compensation Objectives
Our compensation practices are designed to attract key employees and to retain, motivate and reward our executive officers for their performance and contribution to our long-term success. Our Board of Directors, through the Compensation Committee, seeks to compensate our executive officers by combining short and long-term cash and equity incentives. It also seeks to reward the achievement of corporate and individual performance objectives, and to align executive officers’ incentives with shareholder value creation. When possible, the Compensation Committee seeks to tie individual goals to the area of the executive officer’s primary responsibility. These goals may include the achievement of specific financial or business development goals. Also, when possible and appropriate taking into account the Company’s financial condition and other related facts and circumstances, the Compensation Committee seeks to set performance goals that reach across all business areas and include achievements in finance/business development and corporate development.
The Compensation Committee makes decisions regarding salaries, annual bonuses, if any, and equity incentive compensation for our executive officers, approves corporate goals and objectives relevant to the compensation of the Chief Executive Officer and our other executive officers. The Compensation Committee solicits input from our Chief Executive Officer regarding the performance of our other executive officers. Finally, the Compensation Committee also administers our incentive compensation and benefit plans.
Although we have no formal policy for a specific allocation between current and long-term compensation, or cash and non-cash compensation, when possible and appropriate taking into account the Company’s financial condition and other related facts and circumstances, we seek to implement a pay mix for our officers with a relatively equal balance of both, providing a competitive salary with a significant portion of compensation awarded on both corporate and personal performance.
Compensation Components
As a general rule, and when possible and appropriate taking into account the Company’s financial condition and other related facts and circumstances, our compensation consists primarily of three elements: base salary, annual bonus and long-term equity incentives. We describe each element of compensation in more detail below.
Base Salary
Base salaries for our executive officers are established based on the scope of their responsibilities and their prior relevant experience, taking into account competitive market compensation paid by other companies in our industry for similar positions and the overall market demand for such executives at the time of hire. An executive officer’s base salary is also determined by reviewing the executive officer’s other compensation to ensure that the executive officer’s total compensation is in line with our overall compensation philosophy.
Base salaries are reviewed annually and increased for merit reasons, based on the executive officers’ success in meeting or exceeding individual objectives. Additionally, we adjust base salaries as warranted throughout the year for promotions or other changes in the scope or breadth of an executive officer’s role or responsibilities. As indicated in the Summary Compensation Table following, to conserve our cash resources during fiscal 2015 and 2014 the cash amounts of annual base salary that we paid to our executives was significantly less than their stated annual base salary rates.
Annual Bonus
The Compensation Committee assesses the level of the executive officer’s achievement of meeting individual goals, as well as that executive officer’s contribution towards our corporate-wide goals. The amount of the cash bonus depends on the level of achievement of the individual performance goals, with a target bonus generally set as a percentage of base salary and based on the achievement of pre-determined milestones. To conserve our cash resources, our management team did not seek, and our Compensation Committee did not award, cash bonuses to executive officers during fiscal 2014 or 2015.
Long-Term Equity Incentives
The Compensation Committee believes that to attract and retain management, key employees and non-management directors the compensation paid to these persons should include, in addition to base salary and potential annual cash incentives, equity based compensation that is competitive with peer companies. The Compensation Committee determines the amount and terms of equity based compensation granted under our stock option plans or pursuant to other awards made to our executives and key employees.
Summary Compensation Table
The following table shows information regarding the compensation of our Named Executive Officers (NEO’s) for services performed in the fiscal years ended March 31, 2015 and 2014.
|
Name and Principal Position
|
Fiscal
Year
|
Salary
($)
|
Bonus
($)
|
Option and Warrant
Awards (10)
($)
|
All Other Compensation
($)
|
Total
($)
|
|||||||||||||||
|
Shawn K. Singh, J.D. (1)
|
2015
|
347,500
|
(4)
|
-
|
688,050
|
(11)
|
-
|
1,035,550
|
|||||||||||||
|
Chief Executive Officer
|
2014
|
250,000
|
(5)
|
-
|
159,802
|
(12)
|
-
|
409,802
|
|||||||||||||
|
H. Ralph Snodgrass, Ph.D. (2)
|
2015
|
305,000
|
(6)
|
-
|
458,700
|
(11)
|
-
|
763,700
|
|||||||||||||
|
President, Chief Scientific Officer
|
2014
|
250,000
|
(7)
|
-
|
102,353
|
(12)
|
-
|
352,353
|
|||||||||||||
|
Jerrold D. Dotson (3)
|
2015
|
250,000
|
(8)
|
-
|
229,350
|
(11)
|
-
|
479,350
|
|||||||||||||
|
Vice President, Chief Financial Officer, Secretary
|
2013
|
200,000
|
(9)
|
-
|
36,846
|
(12)
|
-
|
236,846
|
|||||||||||||
|
(1)
|
Mr. Singh became VistaGen California’s Chief Executive Officer on August 20, 2009 and our Chief Executive Officer in May 2011, in connection with the Merger. In our fiscal years ended March 31, 2015 and 2014, Mr. Singh’s annual base cash salary, pursuant to his January 2010 employment agreement, was contractually set at $347,500. However, to conserve cash for our operations during those years, Mr. Singh voluntarily agreed to receive cash payments of less than his contractual base cash salary. Further, in fiscal 2014 Mr. Singh voluntarily reduced his base cash salary to the amount indicated above. The figures reported above reflect the amount of Mr. Singh’s salary that we expensed for accounting purposes in our financial statements included in this prospectus for the respective fiscal years. As discussed in notes (4) and (5) below, only $82,813 and $125,000 was actually paid in cash to Mr. Singh in our fiscal years ended March 31, 2015 and 2014, respectively. The difference between the amounts expensed for accounting purposes and the amounts actually paid to Mr. Singh has been accrued for payment in the future. Additionally, pursuant to his employment agreement, Mr. Singh is eligible to receive an annual cash incentive bonus of up to fifty percent (50%) of his base cash salary. Again, to conserve cash for our operations during our fiscal years ended March 31, 2015 and 2014, Mr. Singh voluntarily refrained from receiving any cash bonus.
|
|
(2)
|
Through August 20, 2009, Dr. Snodgrass served as VistaGen California’s President and Chief Executive Officer, at which time he became its President and Chief Scientific Officer. He became our President and Chief Scientific Officer in May 2011, in connection with the Merger. In our fiscal years ended March 31, 2015 and 2014, Dr. Snodgrass’ annual base cash salary, pursuant to his January 2010 employment agreement, was contractually set at $305,000. However, to conserve cash for our operations during those years, Dr. Snodgrass voluntarily agreed to receive cash payments of less than his contractual base cash salary. Further, in fiscal 2014 Dr. Snodgrass voluntarily reduced his base cash salary to the amount indicated. The figures reported above reflect the amount of Dr. Snodgrass’ salary that we expensed for accounting purposes in our financial statements included in this prospectus for the respective fiscal years. As discussed in notes (6) and (7) below, only $157,292 and $149,606 was actually paid in cash to Dr. Snodgrass in our fiscal years ended March 31, 2015 and 2014, respectively. The difference between the amounts expensed for accounting purposes and the amounts actually paid to Dr. Snodgrass has been accrued for payment in the future. Additionally, pursuant to his employment agreement, Dr. Snodgrass is eligible to receive an annual cash incentive bonus of up to fifty percent (50%) of his base cash salary. Again, to conserve cash for our operations during our fiscal years ended March 31, 2015 and 2014, Dr. Snodgrass voluntarily refrained from receiving any cash bonus.
|
|
(3)
|
Mr. Dotson served as Chief Financial Officer on a part-time contract basis from September 19, 2011 through August 2012, at which time he became our full-time employee. In our fiscal years ended March 31, 2015 and 2014, Mr. Dotson’s annual base cash salary was $250,000 and $200,000, respectively. However, to conserve cash for our operations during those years, Mr. Dotson voluntarily agreed to receive cash payments of less than his base cash salary. The figures reported above reflect the amount of Mr. Dotson’s salary that we expensed for accounting purposes in our financial statements included in this prospectus for the respective fiscal years. As discussed in notes (8) and (9) below, only $153,917 and $143,333 was actually paid in cash to Mr. Dotson in our fiscal years ended March 31, 2015 and 2014, respectively. The difference between the amounts expensed for accounting purposes and the amounts actually paid to Mr. Dotson has been accrued for payment in the future. To conserve cash for our operations, Mr. Dotson did not receive a cash bonus in either of our fiscal years ended March 31, 2015 or 2014.
|
|
(4)
|
Mr. Singh received only $82,813 in cash compensation in our fiscal year ended March 31, 2015. The remaining balance has been accrued for future payment and remains unpaid at the date of this prospectus.
|
|
(5)
|
Mr. Singh received only $125,000 in cash compensation in our fiscal year ended March 31, 2014. At March 31, 2014, the remaining balance was accrued for future payment and remained outstanding at March 31, 2015 and through the date of this prospectus.
|
|
(6)
|
Dr. Snodgrass received only $157,292 in cash compensation in our fiscal year ended March 31, 2015 and the remaining balance has been accrued for future payment and remains unpaid at the date of this prospectus.
|
|
(7)
|
Dr. Snodgrass received only $149,606 in cash compensation in our fiscal year ended March 31, 2014. At March 31, 2014, the remaining balance was accrued for future payment and remained outstanding at March 31, 2015 and through the date of this prospectus.
|
|
(8)
|
Mr. Dotson received only $153,917 in cash compensation in our fiscal year ended March 31, 2015 and the remaining balance has been accrued for future payment and remains unpaid at the date of this prospectus.
|
|
(9)
|
Mr. Dotson received only $143,333 in cash compensation in our fiscal year ended March 31, 2014. At March 31, 2014, the remaining balance was accrued for future payment and remained outstanding at March 31, 2015 and through the date of this prospectus.
|
|
(10)
|
The amounts in the Option and Warrant Awards column represent the aggregate grant date fair value of options or warrants to purchase restricted shares of our common stock awarded to Mr. Singh, Dr. Snodgrass and Mr. Dotson, or the effect of modifications to prior grants of options or warrants occurring during the fiscal year presented, computed in accordance with the Financial Accounting Standards Board’s Accounting Standards Codification Topic 718, Compensation – Stock Compensation ("ASC 718”). The amounts in this column do not represent any cash payments actually received by Mr. Singh, Dr. Snodgrass or Mr. Dotson with respect to any of such options or warrants to purchase restricted shares of our common stock awarded to them or modified during the periods presented. To date, Mr. Singh, Dr. Snodgrass and Mr. Dotson have not exercised any of such options or warrants to purchase common stock, and there can be no assurance that any of them will ever realize any of the ASC 718 grant date fair value amounts presented in the Option and Warrant Awards column.
|
|
(11)
|
We used the Black Scholes Option Pricing Model and the following assumptions for determining the grant date fair value of the warrants to purchase shares of our common stock granted in January 2015.
|
|
Market price per share
|
$
|
8.00
|
|||
|
Exercise price per share
|
$
|
10.00
|
|||
|
Risk-free interest rate
|
1.45
|
%
|
|||
|
Expected Term (years)
|
5.0
|
||||
|
Volatility
|
75.86
|
%
|
|||
|
Dividend rate
|
0.0
|
%
|
|||
|
Grant date fair value per share
|
$
|
4.59
|
|
Mr. Singh, Dr. Snodgrass and Mr. Dotson were granted warrants to purchase 150,000, 100,000 and 50,000 restricted shares of our common stock, respectively
|
|
|
(12)
|
The table below provides information regarding the option and warrant awards and modifications we granted to Mr. Singh, Dr. Snodgrass and Mr. Dotson during fiscal 2014 and the assumptions used in the Black Scholes Option Pricing Model to determine the grant date fair values of the respective awards and modifications.
|
|
Option
Grant
10/27/2013
|
Warrant
Grant
3/19/2014
|
Option Modification
12/20/2013
|
Warrant Modification
12/20/2013
|
Option/Warrant Exchange (a)
3/19/2014
|
Total
|
|||||||||||||||||||||||||||||||
|
Singh
|
$
|
-
|
$
|
-
|
$
|
134,436
|
$
|
25,366
|
$
|
-
|
$
|
159,802
|
||||||||||||||||||||||||
|
Snodgrass
|
-
|
14,560
|
56,835
|
-
|
30,958
|
102,353
|
||||||||||||||||||||||||||||||
|
Dotson
|
6,380
|
29,120
|
1,346
|
-
|
-
|
36,846
|
||||||||||||||||||||||||||||||
|
$
|
6,380
|
$
|
43,680
|
$
|
192,617
|
$
|
25,366
|
$
|
30,958
|
$
|
299,001
|
|||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||
|
Before
|
After
|
Before
|
After
|
Before
|
After
|
|||||||||||||||||||||||||||||||
|
Market price per share
|
$
|
8.00
|
$
|
9.20
|
$
|
8.00
|
$
|
8.00
|
$
|
8.00
|
$
|
8.00
|
$
|
9.20
|
$
|
9.20
|
||||||||||||||||||||
|
Exercise price per share
|
$
|
8.00
|
$
|
10.00
|
$
|
15.00 to $42.00
|
$
|
0.50
|
$
|
30.00 to $35.00
|
$
|
0.50
|
$
|
10.00
|
$
|
10.00
|
||||||||||||||||||||
|
Risk-free interest rate
|
1.675
|
%
|
1.750
|
%
|
0.7% to 2.68%
|
0.12% to 2.68%
|
0.07% to 1.18%
|
0.75% to 1.18%
|
0.106
|
%
|
1.750
|
%
|
||||||||||||||||||||||||
|
Volatility
|
99.53
|
%
|
80.57
|
%
|
68.8% to 97.6%
|
68.8% to 97.6%
|
68.76% to 78.21%
|
76.51% to 78.21%
|
68.96
|
%
|
80.57
|
%
|
||||||||||||||||||||||||
|
Expected term (years)
|
6.25
|
5.00
|
0.25 to 8.86
|
0.87 to 8.86
|
0.03 to 3.96
|
3.03 to 3.96
|
0.63
|
5.00
|
||||||||||||||||||||||||||||
|
Dividend rate
|
0
|
%
|
0
|
%
|
0
|
%
|
0
|
%
|
0
|
%
|
0
|
%
|
0
|
%
|
0
|
%
|
||||||||||||||||||||
|
Fair value per share
|
$
|
6.38
|
$
|
5.82
|
$
|
0.00 to $6.49
|
$
|
1.40 to $6.76
|
$
|
0.00 to $2.29
|
$
|
3.55 to $4.20
|
$
|
1.70
|
$
|
5.82
|
||||||||||||||||||||
|
Aggregate shares
|
1,000
|
7,500
|
116,125
|
116,125
|
8,303
|
8,303
|
7,500
|
7,500
|
||||||||||||||||||||||||||||
|
(a)
|
On March 19, 2014, the Board and Dr. Snodgrass agreed to cancel a fully-vested option to purchase 7,500 shares of our restricted common stock at a price of $10.00 per share and expiring on November 4, 2014 in exchange for the grant of a five-year warrant to purchase 7,500 shares of our restricted common stock at a price of $10.00 per share. Shares subject to the cancelled option grant were returned to the 2008 Stock Incentive Plan for potential future grants. The cancellation of the option and grant of the warrant was accounted for as a modification of an award under ASC 718 and, accordingly, the difference in the fair value of the two instruments at the modification date was recorded in stock compensation expense and is the amount reported in the table above.
|
None of the NEOs is entitled to perquisites or other personal benefits which, in the aggregate, are worth over $50,000 or over 10% of their base salary.
Benefit Plans
401(k) Plan
We maintain, through a registered agent, a retirement and deferred savings plan for our officers and employees. This plan is intended to qualify as a tax-qualified plan under Section 401(k) of the Internal Revenue Code of 1986, as amended. The retirement and deferred savings plan provides that each participant may contribute a portion of his or her pre-tax compensation, subject to statutory limits. Under the plan, each employee is fully vested in his or her deferred salary contributions. Employee contributions are held and invested by the plan’s trustee. The retirement and deferred savings plan also permits us to make discretionary contributions subject to established limits and a vesting schedule. To date, we have not made any discretionary contributions to the retirement and deferred savings plan on behalf of participating employees.
The following table provides information regarding each unexercised stock option and warrant to purchase restricted shares of our common stock held by each of the named executive officers as of March 31, 2015.
|
Stock Options
|
|||||||||||||
|
Name
|
Number of Securities Underlying Unexercised Options
(#) Exercisable
|
Number of Securities
Underlying Unexercised Options
(#) Unexercisable
|
Option
Exercise
Price
($)
|
Option
Expiration
Date
|
|||||||||
|
Shawn K. Singh, J.D.
|
1,000
|
-
|
16.00
|
12/21/2016
|
|||||||||
|
2,000
|
-
|
14.40
|
5/17/2017
|
||||||||||
|
1,000
|
-
|
10.00
|
1/17/2018
|
||||||||||
|
1,000
|
-
|
10.00
|
1/17/2018
|
||||||||||
|
3,000
|
-
|
10.00
|
3/24/2019
|
||||||||||
|
1,125
|
-
|
10.00
|
6/17/2019
|
||||||||||
|
50,000
|
-
|
10.00
|
11/4/2019
|
||||||||||
|
21,250
|
-
|
10.00
|
12/30/2019
|
||||||||||
|
4,896
|
104
|
10.00
|
4/26/2021
|
||||||||||
|
4,017
|
-
|
10.00
|
12/31/2016
|
||||||||||
|
1,786
|
-
|
10.00
|
12/31/2016
|
||||||||||
|
2,500
|
-
|
10.00
|
12/6/2017
|
||||||||||
|
5,000
|
-
|
20.00
|
7/30/2016
|
||||||||||
|
53,250
|
18,750
|
(1)
|
12.80
|
3/3/2023
|
|||||||||
|
150,000
|
-
|
(3)
|
10.00
|
1/11/2020
|
|||||||||
|
Total:
|
301,824
|
18,854
|
|||||||||||
|
H. Ralph Snodgrass, Ph.D.
|
2,500
|
-
|
10.00
|
3/24/2019
|
|||||||||
|
1,250
|
-
|
10.00
|
6/17/2014
|
||||||||||
|
319
|
-
|
17.60
|
12/20/2016
|
||||||||||
|
12,500
|
-
|
10.00
|
12/30/2019
|
||||||||||
|
4,896
|
104
|
10.00
|
4/25/2021
|
||||||||||
|
37,500
|
12,500
|
(1)
|
12.80
|
3/3/2023
|
|||||||||
|
1,250
|
1,250
|
(2)
|
10.00
|
3/19/2024
|
|||||||||
|
3,750
|
3,750
|
(2)
|
10.00
|
3/19/2024
|
|||||||||
|
100,000
|
-
|
(3)
|
10.00
|
1/11/2020
|
|||||||||
|
Total:
|
163,965
|
17,604
|
|||||||||||
|
Jerrold D. Dotson
|
5,001
|
-
|
10.00
|
10/30/2022
|
|||||||||
|
708
|
292
|
8.00
|
10/27/2023
|
||||||||||
|
7,500
|
2,500
|
(1)
|
12.80
|
3/3/2023
|
|||||||||
|
2,500
|
2,500
|
(2)
|
10.00
|
3/19/2024
|
|||||||||
|
50,000
|
-
|
(3)
|
10.00
|
1/11/2020
|
|||||||||
|
Total:
|
65,709
|
5,292
|
|||||||||||
|
(1)
|
Represents warrant to purchase restricted shares of our common stock granted on March 3, 2013 at the market price of our common stock on the grant date. At March 31, 2015, the warrant is exercisable for 75% of the shares and became exercisable for the remaining 25% of the shares on April 1, 2015.
|
|
(2)
|
Represents warrant to purchase restricted shares of our common stock granted on March 19, 2014 when the market price of our common stock was $9.20 per share. The warrant became exercisable for 50% of the shares on April 1, 2014, and became exercisable for an additional 25% of the shares on April 1, 2015. It becomes exercisable for the remaining 25% of the shares on April 1, 2016, provided that the warrant will become fully vested upon a change in control of the Company, as defined, or upon the consummation by the Company and a third party of a license or sale transaction involving at least one new Drug Rescue Variant.
|
|
(3)
|
Represents warrant to purchase restricted shares of our common stock granted as fully-exercisable on January 11, 2015 when the market price of our common stock was $8.00 per share.
|
Employment or Severance Agreements
We have employment agreements with Mr. Singh and Dr. Snodgrass.
Singh Agreement
We entered into an employment agreement with Mr. Singh on April 28, 2010. Under the agreement, as amended on May 9, 2011, Mr. Singh’s base salary is $347,500 per year. However, to conserve cash for our operations, Mr. Singh has not received his full base salary in any fiscal year since he entered into his agreement in 2010. In our fiscal year ended March 31, 2015, Mr. Singh received only $82,813 in cash. In our fiscal year ended March 31, 2014, Mr. Singh voluntarily reduced his base salary to $250,000, but only received $125,000 in cash. Although, under his agreement, Mr. Singh is eligible to receive an annual incentive cash bonus of up to 50% of his base salary, he has foregone any such cash bonus payment to conserve cash for our operations. Payment of his annual incentive bonus is at the discretion of our Board of Directors. In the event we terminate Mr. Singh’s employment without cause, he is entitled to receive severance in an amount equal to:
|
·
|
twelve months of his then-current base salary payable in the form of salary continuation;
|
|
·
|
a pro-rated portion of the incentive cash bonus that the Board of Directors determines in good faith that Mr. Singh earned prior to his termination; and
|
|
·
|
such amounts required to reimburse him for Consolidated Omnibus Budget Reconciliation Act (COBRA) payments for continuation of his medical health benefits for such twelve-month period.
|
In addition, in the event Mr. Singh terminates his employment with good reason following a change of control, he is entitled to twelve months of his then-current base salary payable in the form of salary continuation.
In December 2006, we accepted a full-recourse promissory note in the amount of $103,411 from Mr. Singh in payment of the exercise price for options and warrants to purchase an aggregate of 6,320 shares of our common stock. On May 11, 2011, in connection with the Merger, the $128,168 outstanding balance of the principal and accrued interest on this note was cancelled in accordance with Mr. Singh's employment agreement and was treated as additional compensation. In accordance with his employment agreement, Mr. Singh is entitled to an income tax gross-up payment on the compensation related to the note cancellation. At March 31, 2015 and 2014, we had accrued $101,936 as an estimate of the gross-up amount. However, as a result of Mr. Singh’s forbearance, we have not yet paid such amount to Mr. Singh to conserve capital for our operations. See Note 14 to our audited consolidated financial statements which are included in this prospectus.
Snodgrass Agreement
We entered into an employment agreement with Dr. Snodgrass on April 28, 2010. Under the agreement, as amended on May 9, 2011, Dr. Snodgrass’s base salary is $305,000 per year. However, to conserve cash for our operations, Dr. Snodgrass has not received his full base salary in any year since he entered into his agreement in 2010. In our fiscal year ended March 31, 2015, Dr. Snodgrass received only $157,292 in cash. In our fiscal year ended March 31, 2014, Dr. Snodgrass voluntarily reduced his annual salary to $250,000, but received only $149,606 in cash. Dr. Snodgrass is eligible to receive an annual incentive cash bonus of up to 50% of his base salary, but he has foregone any such cash bonus payment to conserve cash for our operations. Payment of his annual incentive bonus is at the discretion of the Board of Directors. In the event we terminate Dr. Snodgrass’s employment without cause, he is entitled to receive severance in an amount equal to
|
·
|
twelve months of his then-current base salary payable in the form of salary continuation;
|
|
·
|
a pro-rated portion of the incentive bonus that the Board of Directors determines in good faith that Dr. Snodgrass earned prior to his termination; and
|
|
·
|
such amounts required to reimburse him for COBRA payments for continuation of his medical health benefits for such twelve-month period.
|
In addition, in the event Dr. Snodgrass terminates his employment with good reason, he is entitled to twelve months of his then-current base salary payable in the form of salary continuation.
Change of Control Provisions
Pursuant to each of their respective employment agreements, Dr. Snodgrass is entitled to severance if he terminates his employment at any time for “good reason” (as defined below), while Mr. Singh is entitled to severance if he terminates his employment for good reason after a change of control. Under their respective agreements, “good reason” means any of the following events, if the event is effected by us without the executive’s consent (subject to our right to cure):
|
·
|
a material reduction in the executive’s responsibility; or
|
|
·
|
a material reduction in the executive’s base salary except for reductions that are comparable to reductions generally applicable to similarly situated executives of VistaGen.
|
Furthermore, pursuant to their respective employment agreements and their stock option award agreements as amended, in the event we terminate the executive without cause within twelve months of a change of control, the executive’s remaining unvested option shares become fully vested and exercisable. Upon a change of control in which the successor corporation does not assume the executive’s stock options, the stock options granted to the executive become fully vested and exercisable.
Pursuant to their respective employment agreements, a change of control occurs when: (i) any “person” as such term is used in Sections 13(d) and 14(d) of the Exchange Act (other than VistaGen, a subsidiary, an affiliate, or a VistaGen employee benefit plan, including any trustee of such plan acting as trustee) becoming the “beneficial owner” (as defined in Rule 13d-3 under the Exchange), directly or indirectly, of securities of VistaGen representing 50% or more of the combined voting power of VistaGen’s then outstanding securities; (ii) a sale of substantially all of VistaGen’s assets; or (iii) any merger or reorganization of VistaGen whether or not another entity is the survivor, pursuant to which the holders of all the shares of capital stock of VistaGen outstanding prior to the transaction hold, as a group, fewer than 50% of the shares of capital stock of VistaGen outstanding after the transaction.
In the event that following termination of employment amounts are payable to an executive pursuant to his employment agreement, the executive’s eligibility for severance is conditioned on executive having first signed a release agreement.
Pursuant to their respective employment agreements, the estimated amount that could be paid by us assuming that a change of control occurred on the last business day of our current fiscal year, is $347,500 for Mr. Singh and $305,000 for Dr. Snodgrass, excluding the imputed value of accelerated vesting of incentive stock options, if any.
DIRECTOR COMPENSATION
We do not have a formal compensation plan for our non-employee directors. We adopted a director compensation policy for our independent directors, as independence is defined by the NASDAQ Stock Market, which became effective for our fiscal year beginning April 1, 2014. Under the independent director compensation policy, our independent directors are entitled to receive a $25,000 annual cash retainer. For service on a committee of the board, an independent director is entitled to receive an additional annual cash retainer as follows: $7,500 for audit and compensation committee members and $5,000 for nominating and governance committee members. In lieu of the annual cash retainer for committee participation, each independent director serving as a chair of a board committee shall receive the following annual cash retainer: $15,000 for audit and compensation committee chairs and $10,000 for the nominating and governance committee chairs. We did not pay our independent directors any cash compensation during our fiscal year ended March 31, 2015.
Under our director compensation policy, each independent director will also receive an annual grant of an option or warrant to purchase a minimum of 1,250 shares of our common stock, which will vest monthly over a one-year period from the date of grant. In January 2015, we granted fully-vested warrants to purchase 20,000 shares of our restricted stock at an exercise price of $10.00 per share to each of our independent directors. We expect to make future grants on the same date as our annual meeting. Prorated grants will be made for partial years of service.
The following table sets forth a summary of the compensation earned by our non-employee directors in our fiscal year ended March 31, 2015.
|
Fees Earned or
Paid in Cash (1)
|
Option and Warrant
Awards (2)
|
Other
Compensation
|
Total
|
|||||||||||||
|
Name
|
($)
|
($)
|
($)
|
($)
|
||||||||||||
|
Jon S. Saxe (3)
|
$
|
55,000
|
$
|
91,734
|
(5)
|
-
|
$
|
146,734
|
||||||||
|
Brian J. Underdown, Ph.D. (4)
|
$
|
57,500
|
$
|
91,734
|
(5)
|
-
|
$
|
149,234
|
||||||||
|
(1)
|
The amounts shown represent fees earned for service on our Board of Directors, and Audit Committee, Compensation Committee and Corporate Governance and Nominating Committee during the fiscal year ended March 31, 2015 which we have accrued but have not paid to the director during that period or through the date of this prospectus.
|
|
|
(2)
|
The amounts in this column represent the aggregate grant date fair value of warrants to purchase restricted shares of our common stock awarded to Mr. Saxe and Dr. Underdown occurring during the fiscal year ended March 31, 2015, computed in accordance with the Financial Accounting Standards Board’s Accounting Standards Codification Topic 718, Compensation – Stock Compensation ("ASC 718”). The amounts in this column do not represent any cash payment actually received by Mr. Saxe or Dr. Underdown with respect to any of such warrants to purchase restricted shares of our common stock awarded to them during the fiscal year ended March 31, 2015. To date, Mr. Saxe and Dr. Underdown have not exercised such warrants to purchase common stock, and there can be no assurance that either of them will ever realize any of the ASC 718 grant date fair value amounts presented in the Option and Warrant Awards column.
|
|
|
(3)
|
Mr. Saxe has served as the Chairman of our Board of Directors, the Chairman of our Audit Committee and a member of our Compensation Committee and Corporate Governance and Nominating Committee throughout our fiscal year ended March 31, 2015. At March 31, 2015, Mr. Saxe holds: (i) 1,875 restricted shares of our common stock; (ii) options to purchase 12,500 restricted shares of our common stock, of which options to purchase 12,448 restricted shares are vested; and (iii) warrants to purchase 33,250 restricted shares of our common stock, of which 29,750 are exercisable and of which an additional 2,687 shares became exercisable on April 1, 2015.
|
|
|
(4)
|
Dr. Underdown has served as a member of our Board of Directors, as the Chairman of our Compensation Committee and Corporate Governance and Nominating Committee and as a member of our Audit Committee throughout our fiscal year ended March 31, 2015. At March 31, 2015, Dr. Underdown holds: (i) options to purchase 9,250 restricted shares of our common stock, of which options to purchase 9,198 restricted shares are vested; and (ii) warrants to purchase 32,500 restricted shares of our common stock, of which 29,375 are exercisable and of which an additional 2,500 shares became exercisable on April 1, 2015.
|
|
|
(5)
|
The table below provides information regarding the warrant awards we granted to Mr. Saxe and Dr. Underdown during fiscal 2015 and the assumptions used in the Black Scholes Option Pricing Model to determine the grant date fair values of the awards as reported in the table above.
|
|
|
Market price per share
|
$
|
8.00
|
||
|
Exercise price per share
|
$
|
10.00
|
||
|
Risk-free interest rate
|
1.450
|
%
|
||
|
Volatility
|
75.86
|
%
|
||
|
Expected term (years)
|
5.00
|
|||
|
Dividend rate
|
0
|
%
|
||
|
Fair value per share
|
$
|
4.59
|
||
|
Aggregate shares
|
40,000
|
Director Independence
Our securities are not currently listed on a national securities exchange or on any inter-dealer quotation system that has a requirement that directors be independent, or that a majority of our directors be independent. However, we evaluate independence by the standards for director independence established by applicable laws, rules, and listing standards, including, without limitation, the standards for independent directors established by the SEC, the New York Stock Exchange, Inc. and the NASDAQ Stock Market.
Subject to some exceptions, these standards generally provide that a director will not be independent if (a) the director is, or in the past three years has been, an employee of ours; (b) a member of the director’s immediate family is, or in the past three years has been, an executive officer of ours; (c) the director or a member of the director’s immediate family has received more than $120,000 per year in direct compensation from us other than for service as a director (or for a family member, as a non-executive employee); (d) the director or a member of the director’s immediate family is, or in the past three years has been, employed in a professional capacity by our independent public accountants, or has worked for such firm in any capacity on our audit; (e) the director or a member of the director’s immediate family is, or in the past three years has been, employed as an executive officer of a company where one of our executive officers serves on the compensation committee; or (f) the director or a member of the director’s immediate family is an executive officer of a company that makes payments to, or receives payments from, us in an amount which, in any twelve-month period during the past three years, exceeds the greater of $1,000,000 or two percent of that other company’s consolidated gross revenues.
Our Board of Directors has undertaken a review of its composition, the composition of its committees and the independence of each director. Based upon information requested from and provided by each director concerning his background, employment and affiliations, including family relationships, our Board of Directors has determined that Mr. Saxe and Dr. Underdown are “independent” as that term is defined under the applicable rules and regulations of the SEC. Our Board of Directors has also determined that Mr. Saxe and Dr. Underdown, who comprise our audit committee, compensation committee, corporate governance and nominating committee, satisfy the independence standards for those committees established by applicable SEC rules. In making these determinations, our Board of Directors considered the current and prior relationships that each non-employee director has with the Company and all other facts and circumstances that our Board of Directors deemed relevant.
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
Sales of Securities to Cato Holding Company
Cato Holding Company (CHC), doing business as Cato BioVentures (CBV), the parent of Cato Research Ltd. (CRL), is one of our largest institutional stockholders at March 31, 2015, holding common stock and warrants to purchase our common stock. Shawn Singh, our Chief Executive Officer and member of our Board of Directors, served as Managing Principal of CBV and as an officer of CRL until August 2009. On October 10, 2012, we issued to CHC an unsecured promissory note in the principal amount of $310,443 (the 2012 CHC Note) and a five-year warrant to purchase 12,500 restricted shares of the Company’s common stock at a price of $30.00 per share (the CHC Warrant). Additionally, on October 10, 2012, we issued to CRL: (i) an unsecured promissory note in the initial principal amount of $1,009,000, which is payable solely in restricted shares of our common stock and which accrues interest at the rate of 7.5% per annum, compounded monthly (the CRL Note), as payment in full for all contract research and development services and regulatory advice rendered to us by CRL through December 31, 2012 with respect to the preclinical and clinical development of AV-101, and (ii) a five-year warrant to purchase, at a price of $20.00 per share, 50,450 restricted shares of our common stock. The 2012 CHC Note and the CRL Note mature on March 31, 2016. Total interest expense on notes payable to CHC and CRL was $174,800 and $167,900 for the fiscal years ended March 31, 2015 and 2014, respectively..
Contract Research and Development Agreement with Cato Research Ltd.
During fiscal year 2007, we entered into a contract research organization arrangement with CRL related to the development of AV-101, under which we incurred expenses of $38,100 and $52,500 for the fiscal years ended March 31, 2015 and 2014, respectively.
Note Receivable from Shawn K. Singh, JD and Advances to us by Mr. Singh
Upon the approval of its Board of Directors, in December 2006, VistaGen California accepted a full-recourse promissory note in the amount of $103,400 from Mr. Shawn Singh in payment of the exercise price for options and warrants to purchase an aggregate of 6,320 restricted shares of VistaGen California’s common stock. The note accrued interest at a rate of 4.90% per annum and was due and payable no later than the earlier of (i) December 1, 2016 or (ii) ten days prior to our becoming subject to the requirements of the Exchange Act. On May 11, 2011, in connection with the Merger, the $128,200 outstanding balance of principal and accrued interest on this note was cancelled in accordance with Mr. Singh's employment agreement and recorded as additional compensation. In accordance with his employment agreement, Mr. Singh is also entitled to receive an income tax gross-up on the compensation related to the note cancellation. At March 31, 2015 and 2014, we had accrued $101,900 as an estimate of the gross-up amount, but we had not yet paid any of that amount to Mr. Singh. As a result of Mr. Singh’s forbearance, it remains unpaid through the date of this prospectus.
Between September 2013 and December 2013, Mr. Singh provided short-term cash advances aggregating $64,000 to meet our short-term working capital requirements. In lieu of cash repayment of the entire amount of the advances, in December 2013, Mr. Singh elected to invest $50,000 of the balance due him in the 2013 Unit Private Placement. At March 31, 2015, we have completely repaid the remaining balance of the advances and the $50,000 promissory note issued in connection with his investment in the 2013 Unit Private Placement to Mr. Singh.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth certain information with respect to the beneficial ownership of our common stock as of July 17, 2015 for:
|
·
|
each stockholder known by us to be the beneficial owner of more than 5% of our common stock;
|
|
·
|
each of our directors;
|
|
·
|
each of our named executive officers; and
|
|
·
|
all of our directors and executive officers as a group.
|
Applicable percentage ownership is based on 1,594,461 shares of capital stock outstanding at July 17, 2015. In computing the number of shares of common stock beneficially owned by a person, we deemed to be outstanding all shares of common stock subject to options or warrants held by that person or entity that are currently exercisable or that will become exercisable within 60 days of July 17, 2015 and all shares of common stock issuable pursuant to promissory notes and related accrued interest convertible into shares of common stock at July 17, 2015. In computing the percentage of shares beneficially owned, we deemed to be outstanding all shares of common stock subject to options or warrants held by that person or entity that are currently exercisable or that will become exercisable within 60 days of July 17, 2015 and all shares of common stock issuable pursuant to promissory notes and related accrued interest convertible into shares of common stock at July 17, 2015. Unless otherwise noted below, the address of each beneficial owner listed in the table is c/o VistaGen Therapeutics, Inc., 343 Allerton Avenue, South San Francisco, California 94080.
|
Name and address of beneficial owner
|
Number of shares beneficially owned
|
Percent
of shares beneficially
owned (1)
|
||||
|
Executive officers and directors:
|
||||||
|
Shawn K. Singh, JD (2)
|
341,287
|
17.82
|
%
|
|||
|
H. Ralph Snodgrass, PhD (3)
|
239,293
|
13.49
|
%
|
|||
|
Jerrold D. Dotson (4)
|
70,344
|
4.23
|
%
|
|||
|
Jon S. Saxe (5)
|
46,563
|
2.84
|
%
|
|||
|
Brian J. Underdown, PhD (6)
|
41,125
|
2.51
|
%
|
|||
|
5% Stockholders:
|
||||||
|
Cato BioVentures (7)
|
238,734
|
14.32
|
%
|
|||
|
Platinum Long Term Growth Fund VII/Montsant Partners, LLC (8)
|
164,164
|
9.99
|
%
|
|||
|
Michael Goldberg (9)
|
172,894
|
9.99
|
||||
|
University Health Network (10)
|
150,678
|
8.93
|
%
|
|||
|
Brio Capital Master Fund Ltd. (11)
|
128,594
|
7.64
|
%
|
|||
|
Lincoln Park Capital Fund LLC (12)
|
153,053
|
8.97
|
%
|
|||
|
Morrison & Foerster LLP (13)
|
120,448
|
7.06
|
%
|
|||
|
All executive officers and directors as a group (5 persons) (14)
|
738,613
|
32.82
|
%
|
|||
|
(1)
|
Assumes 1,594,461 shares of common stock are issued and outstanding as of July 17, 2015.
|
|
(2)
|
Includes options to purchase 85,375 restricted shares of common stock exercisable within 60 days of July 17, 2015 and warrants to purchase 235,303 restricted shares of common stock exercisable within 60 days of July 17, 2015.
|
|
(3)
|
Includes options to purchase 21,569 restricted shares of common stock exercisable within 60 days of July 17, 2015 and warrants to purchase 157,500 restricted shares of common stock exercisable within 60 days of July 17, 2015.
|
|
(4)
|
Includes options to purchase 6,594 restricted shares of common stock exercisable within 60 days of July 17, 2015, including options to purchase 676 shares of common stock held by Mr. Dotson’s wife, and warrants to purchase 63,750 restricted shares of common stock exercisable within 60 days of July 17, 2015.
|
|
(5)
|
Includes options to purchase 12,250 restricted shares of common stock exercisable within 60 days of July 17, 2015 and warrants to purchase 32,438 restricted shares of common stock exercisable within 60 days of July 17, 2015.
|
|
(6)
|
Includes options to purchase 9,250 restricted shares of common stock exercisable within 60 days of July 17, 2015 and warrants to purchase 31,875 restricted shares of common stock exercisable within 60 days of July 17, 2015.
|
|
(7)
|
Based upon information contained in Form 4 filed on January 9, 2012, as updated to give effect to transactions through July 17, 2015 as recorded on our books. Includes currently exercisable warrants to purchase 73,191 shares of restricted common stock. The reported number of shares beneficially owned by Cato Holding Company, dba Cato BioVentures, excludes 328,571 restricted shares of our Series B Preferred stock currently exchangeable for 328,571 restricted shares of our common stock. Pursuant to the terms of the Certificate of Designation of the Relative Rights and Preferences of the Series B 10% Convertible Preferred Stock, there is a limitation on conversion of the Series B Preferred such that the number of shares of common stock that Cato may beneficially acquire upon such conversion is limited to the extent necessary to ensure that, following such conversion, the total number of shares of common stock then beneficially owned by Cato does not exceed 9.99% of the total number of then issued and outstanding shares of our common stock without providing us with 61 days’ prior notice thereof. Including the shares otherwise excluded due to the beneficial ownership restrictions noted above, Cato beneficially owns 567,305 shares or 28.42% of our common stock. Lynda Sutton has voting and investment authority over the shares held by Cato Holding Company. The primary business address of Cato BioVentures is 4364 South Alston Avenue, Durham, North Carolina 27713.
|
|
(8)
|
Based upon information contained in Schedule 13G/A filed on February 18, 2015 by Platinum Long Term Growth Fund VII (“Platinum”) and adjusted to give effect to the transactions consummated between Platinum, Montsant Partners, LLC (“Montsant”), a Platinum affiliate, and us pursuant to the Agreement between the Company and Platinum effective May 12, 2015 (“Agreement”) including Montsant’s investment of an aggregate of $500,000 in our Series B Preferred Unit offering pursuant to the Agreement through July 17, 2015. The number of beneficially owned shares reported includes 115,570 restricted shares of common stock owned by Montsant and 28,241 restricted shares of common stock and a warrant to purchase 20,172 restricted shares of common stock that may currently be acquired by Montsant upon exchange of 18,827 restricted shares of our Series A Preferred Stock (“Series A Preferred”).
The reported number of shares beneficially owned excludes 609,259 restricted shares of common stock and a warrant to purchase 435,186 restricted shares of common stock that may currently be acquired by Montsant upon exchange of 406,173 restricted shares of our Series A Preferred. Pursuant to the October 11, 2012 Note Exchange and Purchase Agreement by and between us and Platinum, there is a limitation on exchange such that the number of shares of our common stock that may be acquired by Platinum or its affiliates upon exchange of the Series A Preferred is limited to the extent necessary to ensure that, following such exchange, the total number of shares of our common stock then beneficially owned by Platinum or its affiliates does not exceed 9.99% of the total number of our then issued and outstanding shares of common stock without providing us with 61 days’ prior notice thereof.
Further, the reported number of shares beneficially owned by Montsant also excludes 3,253,410 shares of common stock pursuant to its ownership of 1,512,022 shares of our Series B 10% Convertible Preferred Stock (“Series B Preferred”), immediately convertible into a like number of shares of our restricted common stock, and warrants to purchase 1,741,388 shares of our restricted common stock. Pursuant to the terms of the Certificate of Designation of the Relative Rights and Preferences of the Series B 10% Convertible Preferred Stock and of the respective common stock warrant agreements, there is a limitation on conversion of the Series B Preferred and exercise of the warrants such that the number of shares of common stock that Montsant may beneficially acquire upon such conversion or exercise is limited to the extent necessary to ensure that, following such conversion or exercise, the total number of shares of common stock then beneficially owned by Platinum or Montsant does not exceed 9.99% of the total number of then issued and outstanding shares of our common stock without providing us with 61 days’ prior notice thereof.
Additionally, the reported number of shares beneficially owned excludes currently exercisable warrants owned by Platinum to purchase 320,124 shares of our restricted common stock. Pursuant to the terms of the respective common stock warrant agreements, there is a limitation on the exercise of the warrants such that the number of shares of common stock that Platinum may beneficially acquire upon such exercise is limited to the extent necessary to ensure that, following such exercise, the total number of shares of common stock then beneficially owned by Platinum or Montsant does not exceed 9.99% of the total number of then issued and outstanding shares of our common stock without providing us with 61 days’ prior notice thereof.
Including the shares otherwise excluded due to the beneficial ownership restrictions noted above, Platinum and Montsant beneficially own 4,782,142 shares or 76.38% of our common stock. The primary business address of Platinum Long Term Growth Fund VII and Montsant Partners, LLC is c/o Platinum Partners, 250 West 55th Street, 14th Floor, New York, NY 10019. Mark Nordlicht has voting and investment control over the shares held by Platinum and Montsant.
|
|
(9)
|
Platinum has transferred to Michael Goldberg (“Goldberg”) certain of the equity securities initially issued by us to Platinum. The conversion or exercise restrictions in those securities initially applicable to Platinum remain applicable to Goldberg. The number of shares reported as beneficially owned by Goldberg includes 37,508 restricted shares of common stock and 78,975 restricted shares of common stock and a currently exercisable warrant to purchase 56,411 restricted shares of common stock that may currently be acquired by Goldberg upon exchange of 52,650 restricted shares of our Series A Preferred.
The reported number of shares beneficially owned both before and after the Offering excludes 33,525 restricted shares of common stock and a warrant to purchase 23,946 restricted shares of common stock that may currently be acquired by Goldberg upon exchange of 22,350 restricted shares of our Series A Preferred. Pursuant to the October 11, 2012 Note Exchange and Purchase Agreement by and between us and Platinum, there is a limitation on exchange such that the number of shares of our common stock that may be acquired by Goldberg upon exchange of the Series A Preferred is limited to the extent necessary to ensure that, following such exchange, the total number of shares of our common stock then beneficially owned by Goldberg does not exceed 9.99% of the total number of our then issued and outstanding shares of common stock without providing us with 61 days’ prior notice thereof.
Further, the reported number of shares beneficially owned by Goldberg before the Offering also excludes 193,355 shares of common stock pursuant to his ownership of 147,028 shares of our Series B Preferred, immediately convertible into a like number of shares of our restricted common stock, and warrants to purchase 46,327 shares of our restricted common stock. Pursuant to the terms of the Certificate of Designation of the Relative Rights and Preferences of the Series B 10% Convertible Preferred Stock and of the respective common stock warrant agreements, there is a limitation on conversion of the Series B Preferred and exercise of the warrants such that the number of shares of common stock that Goldberg may beneficially acquire upon such conversion or exercise is limited to the extent necessary to ensure that, following such conversion or exercise, the total number of shares of common stock then beneficially owned by Goldberg does not exceed 9.99% of the total number of then issued and outstanding shares of our common stock without providing us with 61 days’ prior notice thereof.
Additionally, the reported number of shares beneficially owned by Goldberg excludes currently exercisable warrants to purchase 66,251 shares of our restricted common stock. Pursuant to the terms of the respective common stock warrant agreements, there is a limitation on the exercise of the warrants such that the number of shares of common stock that Goldberg may beneficially acquire upon such exercise is limited to the extent necessary to ensure that, following such exercise, the total number of shares of common stock then beneficially owned by Goldberg does not exceed 9.99% of the total number of issued and outstanding shares of our common stock without providing us with 61 days’ prior notice thereof.
Including the shares otherwise excluded due to the beneficial ownership restrictions noted above, Goldberg beneficially owns 489,971 shares or 23.94% of our common stock.
|
|
(10)
|
Includes 93,775 restricted shares of our Series B Preferred currently exchangeable for 93,775 restricted shares of our common stock. The primary business address of University Health Network is 101 College Street, Suite 150, Toronto, Ontario Canada M5G 1L7. Christopher Paige, Ph.D. has voting and investment authority over the shares held by University Health Network.
|
|
(11)
|
Includes currently exercisable warrants to purchase 61,378 restricted shares of common stock and 27,107 restricted shares of our Series B Preferred currently exchangeable for 27,107 restricted shares of our common stock. The primary business address of Brio Capital Master Fund Ltd. is 100 Merrick Road, Suite 401W, Rockville Centre, NY 11570. Shaye Hirsch has voting and investment control over the shares held by Brio Capital Master Fund Ltd.
|
|
(12)
|
Includes currently exercisable warrants to purchase 72,740 restricted shares of common stock and 38,347 restricted shares of our Series B Preferred currently exchangeable for 38,347 restricted shares of our common stock. The primary business address of Lincoln Park Capital Fund LLC is 440 N. Wells Street, Chicago, IL 60654. Joshua Scheinfeld has voting and investment control over the shares held by Lincoln Park Capital Fund LLC.
|
|
(13)
|
Includes currently exercisable warrants to purchase 110,448 restricted shares of common stock. The reported number of shares beneficially owned excludes 257,143 restricted shares of our Series B Preferred stock currently exchangeable for 257,143 restricted shares of our common stock. Pursuant to the terms of the Certificate of Designation of Rights and Preferences of the Series B Preferred, there is a limitation on conversion of the Series B Preferred such that the number of shares of common stock that Morrison & Foerster may beneficially acquire upon such conversion is limited to the extent necessary to ensure that, following such conversion, the total number of shares of common stock then beneficially owned by Morrison & Foerster does not exceed 9.99% of the total number of issued and outstanding shares of our common stock without providing us with 61 days’ prior notice thereof. Effective on June 12, 2015, Morrison & Foerster provided us with such 61 day advance notice. Including the shares otherwise excluded due to the beneficial ownership restrictions noted above, Morrison & Foerster beneficially owns 377,591 shares or 19.24% of our common stock. The primary business address of Morrison & Foerster is 555 Market Street, San Francisco, California 94105. Mark Blumenthal has voting and investment control over the shares held by Morrison & Foerster.
|
|
(14)
|
Includes options to purchase an aggregate of 135,038 restricted shares of common stock exercisable within 60 days of July 17, 2015 and warrants to purchase an aggregate of 520,866 restricted shares of common stock exercisable within 60 days of July 17, 2015.
|
Equity Grants
As of March 31, 2015, options to purchase a total of 207,638 restricted shares of our common stock are outstanding at a weighted average exercise price of $10.09 per share, of which 199,013 options are vested and exercisable at a weighted average exercise price of $10.09 per share and 8,625 are unvested and not exercisable at a weighted average exercise price of $10.22 per share. These options were issued under our 2008 Plan and our 1999 Plan, each as described below. At March 31, 2015, an additional 40,491 shares remain available for future equity grants under our 2008 Plan.
|
Plan category
|
Number of securities
to be issued upon
exercise of
outstanding options,
warrants and rights
(a)
|
Weighted-average
exercise price of
outstanding
options, warrants
and rights
(b)
|
Number of securities
remaining available for future issuance under equity compensation plans
(excluding securities
reflected in column (a))
(c)
|
|||||||||
|
Equity compensation plans approved by security holders
|
194,509
|
$
|
9.99
|
40,491
|
||||||||
|
Equity compensation plans not approved by security holders
|
13,129
|
11.67
|
--
|
|||||||||
|
Total
|
207,638
|
$
|
10.09
|
40,491
|
||||||||
2008 Stock Incentive Plan
Shareholders of VistaGen California adopted our 2008 Plan on December 19, 2008 and we assumed the plan in connection with the Merger. The maximum number of shares of common stock that may be granted pursuant to the 2008 Plan is 250,000. In all cases, the maximum number of shares of common stock under the 2008 Plan will be subject to adjustments for stock splits, stock dividends or other similar changes in our common stock or our capital structure. Notwithstanding the foregoing, the maximum number of shares of common stock available for grant of options intended to qualify as “incentive stock options” under the provisions of Section 422 of the Internal Revenue Code of 1986 (the “Code”), is 250,000.
Our 2008 Plan provides for the grant of stock options, restricted shares of common stock, stock appreciation rights and dividend equivalent rights, collectively referred to as “awards”. Stock options granted under the 2008 Plan may be either incentive stock options under the provisions of Section 422 of the Code, or non-qualified stock options. We may grant incentive stock options only to employees of VistaGen or any parent or subsidiary of VistaGen. Awards other than incentive stock options may be granted to employees, directors and consultants.
Our Board of Directors or the Compensation Committee of the Board of Directors, referred to as the “Administrator”, administers our 2008 Plan, including selecting the award recipients, determining the number of shares to be subject to each award, the exercise or purchase price of each award and the vesting and exercise periods of each award.
The exercise price of all incentive stock options granted under our 2008 Plan must be at least equal to 100% of the fair market value of the shares on the date of grant. If, however, incentive stock options are granted to an employee who owns stock possessing more than 10% of the voting power of all classes of our stock or the stock of any of our subsidiaries, the exercise price of any incentive stock option granted may not be less than 110% of the fair market value on the grant date. The maximum term of incentive stock options granted to employees who own stock possessing more than 10% of the voting power of all classes of our stock or the stock of any of our subsidiaries may not exceed five years. The maximum term of an incentive stock option granted to any other participant may not exceed ten years. The Administrator determines the term and exercise or purchase price of all other awards granted under our 2008 Plan.
Under the 2008 Plan, incentive stock options may not be sold, pledged, assigned, hypothecated, transferred or disposed of in any manner other than by will or by the laws of descent or distribution and may be exercised, during the lifetime of the participant, only by the participant. Other awards shall be transferable:
|
·
|
by will and by the laws of descent and distribution; and
|
|
·
|
during the lifetime of the participant, to the extent and in the manner authorized by the Administrator by gift or pursuant to a domestic relations order to members of the participant’s immediate family.
|
The 2008 Plan permits the designation of beneficiaries by holders of awards, including incentive stock options.
In the event of termination of a participant’s service for any reason other than disability or death, such participant may, but only during the period specified in the award agreement of not less than 30 days (generally 90 days) commencing on the date of termination (but in no event later than the expiration date of the term of such award as set forth in the award agreement), exercise the portion of the participant’s award that was vested at the date of such termination or such other portion of the participant’s award as may be determined by the Administrator. The participant’s award agreement may provide that upon the termination of the participant’s service for cause, the participant’s right to exercise the award shall terminate concurrently with the termination of the participant’s service. In the event of a participant’s change of status from employee to consultant, an employee’s incentive stock option shall convert automatically into a non-qualified stock option on the day three months and one day following such change in status. To the extent that the participant’s award was unvested at the date of termination, or if the participant does not exercise the vested portion of the participant’s award within the period specified in the award agreement of not less than 30 days commencing on the date of termination, the award shall terminate. If termination was caused by death or disability, any options which have become exercisable prior to the time of termination, will remain exercisable for twelve months from the date of termination (unless a shorter or longer period of time is determined by the Administrator).
Following the date that the exemption from application of Section 162(m) of the Code ceases to apply to awards, the maximum number of shares with respect to which options and stock appreciation rights may be granted to any participant in any calendar year will be 125,000 shares of common stock. In connection with a participant’s commencement of service with us, a participant may be granted options and stock appreciation rights for up to an additional 25,000 shares that will not count against the foregoing limitation. In addition, following the date that the exemption from application of Section 162(m) of the Code ceases to apply to awards, for awards of restricted stock and restricted shares of common stock that are intended to be “performance-based compensation” (within the meaning of Section 162(m)), the maximum number of shares with respect to which such awards may be granted to any participant in any calendar year will be 125,000 shares of common stock. The limits described in this paragraph are subject to adjustment in the event of any change in our capital structure as described below.
The terms and conditions of awards are determined by the Administrator, including the vesting schedule and any forfeiture provisions. Awards under the plan may vest upon the passage of time or upon the attainment of certain performance criteria. Although we do not currently have any awards outstanding that vest upon the attainment of performance criteria, the Administrator may establish criteria based on any one of, or combination of, the following:
|
·
|
increase in share price;
|
|
·
|
earnings per share;
|
|
·
|
total shareholder return;
|
|
·
|
operating margin;
|
|
·
|
gross margin;
|
|
·
|
return on equity;
|
|
·
|
return on assets;
|
|
·
|
return on investment;
|
|
·
|
operating income;
|
|
·
|
net operating income;
|
|
·
|
pre-tax profit;
|
|
·
|
cash flow;
|
|
·
|
revenue;
|
|
·
|
expenses;
|
|
·
|
earnings before interest, taxes and depreciation;
|
|
·
|
economic value added; and
|
|
·
|
market share.
|
Subject to any required action by our stockholders, the number of shares of common stock covered by outstanding awards, the number of shares of common stock that have been authorized for issuance under the 2008 Plan, the exercise or purchase price of each outstanding award, the maximum number of shares of common stock that may be granted subject to awards to any participant in a calendar year, and the like, shall be proportionally adjusted by the Administrator in the event of any increase or decrease in the number of issued shares of common stock resulting from certain changes in our capital structure as described in the 2008 Plan.
Effective upon the consummation of a Corporate Transaction (as defined below), all outstanding awards under the 2008 Plan will terminate unless the acquirer assumes or replaces such awards. The Administrator has the authority, exercisable either in advance of any actual or anticipated Corporate Transaction or Change in Control (as defined below) or at the time of an actual Corporate Transaction or Change in Control and exercisable at the time of the grant of an award under the 2008 Plan or any time while an award remains outstanding, to provide for the full or partial automatic vesting and exercisability of one or more outstanding unvested awards under the 2008 Plan and the release from restrictions on transfer and repurchase or forfeiture rights of such awards in connection with a Corporate Transaction or Change in Control, on such terms and conditions as the Administrator may specify. The Administrator also has the authority to condition any such award vesting and exercisability or release from such limitations upon the subsequent termination of the service of the grantee within a specified period following the effective date of the Corporate Transaction or Change in Control. The Administrator may provide that any awards so vested or released from such limitations in connection with a Change in Control, shall remain fully exercisable until the expiration or sooner termination of the award.
Under our 2008 Plan, a Corporate Transaction is generally defined as:
|
·
|
an acquisition of securities possessing more than fifty percent (50%) of the total combined voting power of our outstanding securities but excluding any such transaction or series of related transactions that the Administrator determines shall not be a Corporate Transaction;
|
|
·
|
a reverse merger in which we remain the surviving entity but: (i) the shares of common stock outstanding immediately prior to such merger are converted or exchanged by virtue of the merger into other property, whether in the form of securities, cash or otherwise; or (ii) in which securities possessing more than fifty percent (50%) of the total combined voting power of our outstanding securities are transferred to a person or persons different from those who held such securities immediately prior to such merger;
|
|
·
|
a sale, transfer or other disposition of all or substantially all of the assets of our Corporation;
|
|
·
|
a merger or consolidation in which our Corporation is not the surviving entity; or
|
|
·
|
a complete liquidation or dissolution.
|
Under our 2008 Plan, a Change in Control is generally defined as: (i) the acquisition of more than 50% of the total combined voting power of our stock by any individual or entity which a majority of our Board of Directors (who have served on our board for at least 12 months) do not recommend our shareholders accept; (ii) or a change in the composition of our Board of Directors over a period of 12 months or less.
Unless terminated sooner, our 2008 Plan will automatically terminate in 2017. Our Board of Directors may at any time amend, suspend or terminate our 2008 Plan. To the extent necessary to comply with applicable provisions of U.S. federal securities laws, state corporate and securities laws, the Internal Revenue Code, the rules of any applicable stock exchange or national market system, and the rules of any non-U.S. jurisdiction applicable to awards granted to residents therein, we shall obtain shareholder approval of any such amendment to the 2008 Stock Plan in such a manner and to such a degree as required.
As of March 31, 2015, we have options to purchase an aggregate of 194,509 restricted shares of our common stock outstanding under our 2008 Plan.
1999 Stock Incentive Plan
VistaGen California’s Board of Directors adopted the 1999 Plan on December 6, 1999. The 1999 Plan terminated under its own terms in December 2009, and as a result, no awards may currently be granted under the 1999 Plan. However, the options and awards that have been granted pursuant to the 1999 Plan prior to its expiration remain operative.
The 1999 Plan permitted VistaGen California to make grants of incentive stock options, non-qualified stock options and restricted stock awards. VistaGen California initially reserved 22,500 restricted shares of its common stock for the issuance of awards under the 1999 Plan, which number was subject to adjustment in the event of a stock split, stock dividend or other change in capitalization. Prior to the 1999 Plan’s expiration, shares that were forfeited or cancelled from awards under the 1999 Plan were generally available for future awards.
The 1999 Plan could be administered by either VistaGen California’s Board of Directors or a committee designated by its Board of Directors. VistaGen California’s Board of Directors designated its Compensation Committee as the committee with full power and authority to select the participants to whom awards were granted, to make any combination of awards to participants, to accelerate the exercisability or vesting of any award and to determine the specific terms and conditions of each award, subject to the provisions of the 1999 Plan. All directors, executive officers, and certain other key persons (including employees, consultants and advisors) of VistaGen California were eligible to participate in the 1999 Plan.
The exercise price of incentive stock options awarded under the 1999 Plan could not be less than the fair market value of the common stock on the date of the option grant and could not be less than 110% of the fair market value of the common stock to persons owning stock representing more than 10% of the voting power of all classes of our stock. The exercise price of non-qualified stock options could not be less than 85% of the fair market value of the common stock. The term of each option granted under the 1999 Plan could not exceed ten years (or five years, in the case of an incentive stock option granted to a 10% shareholder) from the date of grant. VistaGen California’s Compensation Committee determined at what time or times each option might be exercised (provided that in no event could it exceed ten years from the date of grant) and, subject to the provisions of the 1999 Plan, the period of time, if any, after retirement, death, disability or other termination of employment during which options could be exercised.
The 1999 Plan also permitted the issuance of restricted stock awards. Restricted stock awards issued by VistaGen California were shares of common stock that vest in accordance with terms and conditions established by VistaGen California’s Compensation Committee. The Compensation Committee could impose conditions to vesting that it determined to be appropriate. Shares of restricted stock that did not vest were subject to our right of repurchase or forfeiture. VistaGen California’s Compensation Committee determined the number of shares of restricted stock granted to any employee. Our 1999 Plan also gave VistaGen California’s Compensation Committee discretion to grant stock awards free of any restrictions.
Unless the Compensation Committee provided otherwise, the 1999 Plan did not generally allow for the transfer of incentive stock options and other awards and only the recipient of an award could exercise an award during his or her lifetime. Non-qualified stock options were transferable only to the extent provided in the award agreement, in a manner consistent with the applicable law, and by will and by the laws of descent and distribution. In the event of a change in control of the Company, as defined in the 1999 Plan, the outstanding options will automatically vest unless our Board of Directors and the Board of Directors of the surviving or acquiring entity make appropriate provisions for the continuation or assumption of any outstanding awards under the 1999 Plan.
As of March 31, 2015, we have options outstanding under the 1999 Plan to purchase an aggregate of 13,129 restricted shares of our common stock.
DESCRIPTION OF SECURITIES TO BE REGISTERED
On May 7, 2015, we filed the Certificate of Designation of the Relative Rights and Preferences of the Series B 10% Preferred Stock of VistaGen Therapeutics, Inc. (the “Certificate of Designation”) with the Nevada Secretary of State in order to designate 4.0 million shares of the Company’s preferred stock as Series B 10% Convertible Preferred Stock (“Series B Preferred”). Each share of the Company’s Series B Preferred is convertible, at the option of the holder thereof (“Voluntary Conversion”), into one (1) share of Common Stock at a fixed conversion price of $7.00 per share, subject to adjustment only for customary stock dividends, reclassifications, splits and similar transactions (“Fixed Conversion Price”). All shares of Series B Preferred are also convertible automatically into Common Stock (“Automatic Conversion”) upon the closing or effective date of any of the following transactions or events: (i) a strategic transaction involving AV-101, the Company’s orally-available new prodrug candidate in clinical development for Major Depressive Disorder and other diseases and disorders of the central nervous system, with an initial up front cash payment to the Company of at least $10.0 million; (ii) a registered public offering of Common Stock with aggregate gross proceeds to the Company of at least $10.0 million; or (iii) for 20 consecutive trading days the Company’s Common Stock trades at least 20,000 shares per day with a daily closing price of at least $12.00 per share; provided, however, that Automatic Conversion and Voluntary Conversion (collectively, “Conversion”) are subject to customary beneficial ownership blockers. Prior to Conversion, shares of Series B Preferred will accrue dividends, payable only in unregistered shares of Common Stock, at a rate of 10% per annum (the “Accrued Dividend”). The Accrued Dividend will be payable only on the date of Conversion solely in that number of shares of Common Stock equal to the Accrued Dividend, divided by the Fixed Conversion Price.
Effective May 12, 2015, the Company and Platinum Long Term Growth VII, LLC (“Platinum”) entered into an agreement (the “Platinum Agreement”), under which Platinum has:
|
·
|
Converted all of the approximately $4.5 million outstanding balance (principal and accrued but unpaid interest) of its Senior Secured Convertible Promissory Notes issued by the Company (the “Senior Secured Notes”), into 641,335 shares of Series B Preferred, thereby cancelling approximately $4.5 million of the Company’s indebtedness;
|
|
·
|
Released all of its security interests in the assets of the Company and its subsidiaries by terminating the Amended and Restated Security Agreement, IP Security Agreement and Negative Covenant, each by and between the Company and Platinum and dated October 11, 2012;
|
|
·
|
Converted all of the approximately $1.3 million outstanding balance (principal and accrued but unpaid interest) of its Subordinated Convertible Promissory Notes issued by the Company (“Platinum Subordinated Notes”) into 240,305 shares of Series B Preferred and five-year warrants to purchase 240,305 shares of Common Stock at a fixed exercise price of $7.00 per share, subject to adjustment only for customary stock dividends, reclassifications, splits and similar transactions (“Warrants”), thereby cancelling approximately $1.3 million of the Company’s indebtedness;
|
|
·
|
Purchased approximately $1.5 million (including accrued but unpaid interest thereon) of outstanding Subordinated Notes issued by the Company from the respective holders thereof (“Investor Subordinated Notes”) and converted the entire outstanding balance of the Investor Subordinated Notes into 265,699 shares of Series B Preferred and 265,699 Warrants, thereby cancelling approximately $1.5 million of the Company’s indebtedness;
|
|
·
|
Signed and delivered a Securities Purchase Agreement (“SPA”) to purchase, for $1.0 million, a total of 142,857 shares of Series B Preferred and 142,857 Warrants on or before June 11, 2015. A form of the SPA is attached to this Current Report on Form 8-K as Exhibit 10.3;
|
|
·
|
Amended all warrants previously issued by the Company to Platinum in connection with the Senior Secured Notes, and warrants that still may be issued pursuant to the Note Exchange and Purchase Agreement, by and between the Company and Platinum, dated October 11, 2012 (the “NEPA”), if any (collectively, the “Exchange Warrants”) to:
|
|
o
|
fix the exercise price thereof at $7.00 per share;
|
|
o
|
fix the number of shares of the Company’s Common Stock issuable thereunder; and
|
|
o
|
eliminate the cashless exercise provisions from all Exchange Warrants; and
|
|
·
|
Agreed to lock-up (refrain from) the potential sale of any shares of common stock held by Platinum or its affiliates until the earlier to occur of an effective registration statement relating to resale of Conversion Shares under the Securities Act of 1933, as amended, or the closing price of the Company’s Common Stock is at least $15.00 per share.
|
As additional consideration for the agreements of Platinum under the Platinum Agreement, the Company issued to Platinum 400,000 shares of Series B Preferred and Warrants to purchase 1.2 million shares of Common Stock and exchanged 30,000 shares of Common Stock currently beneficially owned or controlled by Platinum for 30,000 shares of Series B Preferred.
On June 12, 2015, the Company, and certain of the Company’s note holders and strategic partners agreed to convert an aggregate total of approximately $5.3 million of the Company’s outstanding debt obligations into 750,918 shares of Series B Preferred (the “Debt-to-Equity Conversions”).
In connection with the foregoing Debt-to-Equity Conversions, effective as of June 12, 2015, the Company entered into a letter agreement with Morrison & Foerster LLP (“MF”) (the “Letter Agreement”) with regards to (i) certain promissory notes issued by the Company to MF, each due and payable on or before March 31, 2016, one in the principal amount of approximately $917,000 (“Note A”) and another in the principal amount of approximately $1.4 million (“Note B”), and (ii) certain warrants held by MF to purchase an aggregate of 110,448 shares of the Company’s common stock, par value $0.001 per share at exercise prices of $20.00 per share $40.00 per share (the “MF Warrants”). Subject to certain conditions set forth in the Letter Agreement, each of which has now been satisfied by the Company, MF agreed to convert the principal amount and all accrued but unpaid interest of Note B into 257,143 shares of Series B Preferred and to withhold any and all action to collect amounts due under Note A, and certain amounts owed by the Company to MF in connection with services previously rendered by MF. Additionally, concurrently with the conversion of Note B into shares of Series B Preferred, the Company agreed to amend the MF Warrants to extend the expiration date of each warrant to September 15, 2019, and to set the exercise price of each MF Warrant to $20.00 per share.
This Registration Statement registers for resale shares of Common Stock issuable as Conversion Shares, issuable upon conversion of the shares of Series B Preferred, and Warrant Shares issuable upon exercise of the Warrants issued to Investors and Note Holders in connection with the Series B Offering and Debt-to-Equity Conversions.
SELLING STOCKHOLDERS
The Company, each of the Investors participating in the Series B Offering and the Note Holders participating in the Note Conversion signed a Securities Purchase Agreement (“SPA”), wherein we agreed to register the Warrant Shares issuable upon exercise of the Warrants, and the Conversion Shares issuable upon conversion of the shares of Series B Preferred (collectively, the “Registrable Securities”), under the Securities Act. Accordingly, we filed a Registration Statement on Form S-1, of which this prospectus forms a part, with respect to the resale of the Registrable Securities from time to time. In addition, we agreed in the SPA to use our best efforts to cause the registration statement to be declared effective under the Securities Act by August 30, 2015, and to use our best efforts to keep the registration statement effective until the Registrable Securities are sold or may be sold without registration or prospectus delivery requirements under the Securities Act, subject to certain restrictions.
Selling Stockholders Table
We filed a Registration Statement on Form S-1 with the SEC, of which this prospectus forms a part, with respect to the resale of the Registrable Securities from time to time under Rule 415 of the Securities Act. The Registrable Securities were registered to permit secondary public trading thereof. Subject to the restrictions described in this prospectus, the Selling Stockholders may offer the Registrable Securities for resale from time to time. In addition, subject to the restrictions described in this prospectus, the Selling Stockholders may sell, transfer or otherwise dispose of all or a portion of any Registrable Securities held in transactions exempt from the registration requirements of the Securities Act. See “Plan of Distribution” below for more information.
The table below presents information as of July 17, 2015, regarding the Selling Stockholders and the Registrable Securities the Selling Stockholders (and their donees, pledgees, assignees, transferees and other successors in interest) may offer and sell from time to time under this prospectus. More specifically, the following table sets forth as to the Selling Stockholders:
|
·
|
the number of shares of our Common Stock beneficially owned by each Selling Stockholders prior to the offering for resale of any of the shares of our Common Stock being registered by the registration statement of which this prospectus is a part;
|
|
·
|
the number of shares of our Common Stock that may be offered for resale for the Selling Stockholders’ account under this prospectus; and
|
|
·
|
the number and percent of shares of our Common Stock to be held by the Selling Stockholders after the offering of the resale securities, assuming all of the resale shares of Common Stock are sold by the Selling Stockholders and that the Selling Stockholders do not acquire any other shares of our Common Stock prior to their assumed sale of all of the resale shares.
|
The table is prepared based on information supplied to us by the Selling Stockholders. Although we have assumed for purposes of the table below that the Selling Stockholders will sell all Registrable Securities offered by this prospectus, because the Selling Stockholders may offer from time to time all or some of the Registrable Securities, no assurances can be given as to the actual number of securities that will be resold by the Selling Stockholders or that will be held by the Selling Stockholders after completion of the resales. In addition, the Selling Stockholders may have sold, transferred or otherwise disposed of the Registrable Securities in transactions exempt from the registration requirements of the Securities Act since the date the Selling Stockholders provided the information regarding their securities holdings. Information covering the Selling Stockholders may change from time to time and changed information will be presented in a supplement to this prospectus if and when necessary and required.
Except as described above, there are currently no agreements, arrangements or understandings with respect to the resale of any of the securities covered by this prospectus.
The applicable percentages of ownership are based on an aggregate of 1,594,461 shares of our Common Stock issued and outstanding on July 17, 2015. The number of shares beneficially owned by the Selling Stockholders is determined under rules promulgated by the SEC.
|
Shares Beneficially Owned
Prior to |
Maximum Number of
Shares Being Offered
Pursuant
to this |
Shares Beneficially
Owned After
Offering (25)
|
||||||||||||||
| Name of Selling Stockholder (1) |
Offering (25)
|
Prospectus
|
Number
|
Percent*
|
||||||||||||
|
Montsant Partners LLC (2)
|
164,164 | 1,623,410 | 164,164 | 9.99 | % | |||||||||||
|
Cato Holding Company (3)
|
238,734 | 328,571 | 238,734 | 14.32 | % | |||||||||||
|
Morrison & Foerster LLP (4)
|
377,571 | 257,143 | 120,448 | 7.06 | % | |||||||||||
|
Michael Goldberg (5)
|
172,894 | 193,355 | 172,894 | 9.99 | % | |||||||||||
|
University Health Network (6)
|
150,658 | 93,775 | 56,903 | 3.57 | % | |||||||||||
|
Chardan Capital Markets (7)
|
75,000 | 75,000 | - | * | ||||||||||||
|
Calm Seas Capital (8)
|
82,944 | 71,429 | 11,515 | * | ||||||||||||
|
McCarthy Tetrault LLP (9)
|
64,230 | 59,230 | 5,000 | * | ||||||||||||
|
Brio Capital Master Fund Ltd. (10)
|
128,594 | 54,214 | 74,380 | 4.57 | % | |||||||||||
|
Matthew A. Kreling
|
68,965 | 43,752 | 25,213 | 1.57 | % | |||||||||||
|
Icahn School of Medicine at Mount Sinai (11)
|
73,000 | 43,000 | 30,000 | 1.86 | % | |||||||||||
|
Praim S. Singh & Marlene Singh Trustees, Singh Family Trust 4/25/1997
|
79,560 | 40,760 | 38,800 | 2.42 | % | |||||||||||
|
Plazacorp Investments Limited (12)
|
50,924 | 35,924 | 15,000 | * | ||||||||||||
|
Excell Advisors LLC (13)
|
67,342 | 34,842 | 32,500 | 2.02 | % | |||||||||||
|
Desjardins Securities, Inc. (14)
|
34,098 | 32,143 | 1,955 | * | ||||||||||||
|
John Doherty & Jennifer Doherty, Trustees, The Doherty Revocable Trust dated 6/1/2011
|
61,879 | 30,476 | 31,403 | 1.96 | % | |||||||||||
|
MAZ Partners, LP (15)
|
30,000 | 30,000 | - | * | ||||||||||||
|
Yaakov Yitzchak Bodner
|
36,928 | 29,428 | 7,500 | * | ||||||||||||
|
Daniel Zucker
|
43,852 | 28,852 | 15,000 | * | ||||||||||||
|
Daniel W. Rumsey (16)
|
54,584 | 25,000 | 29,584 | 1.84 | % | |||||||||||
|
Thomas T. Tavares
|
64,582 | 24,446 | 40,136 | 2.50 | % | |||||||||||
|
Nathan Steinmetz
|
35,988 | 23,288 | 12,700 | * | ||||||||||||
|
Michael C. Phillips
|
62,947 | 21,860 | 41,087 | 2.56 | % | |||||||||||
|
Akiva Horowitz
|
21,430 | 21,430 | - | * | ||||||||||||
|
Burr Pilger Mayer, Inc. (17)
|
21,429 | 21,429 | - | * | ||||||||||||
|
Tracy K & Valerie M Price TR Price Family Trust 8/15/2007
|
58,154 | 20,590 | 37,564 | 2.34 | % | |||||||||||
|
Wayne Lindholm
|
37,988 | 20,488 | 17,500 | 1.09 | % | |||||||||||
|
Jeffrey M. Quick
|
20,000 | 20,000 | - | * | ||||||||||||
|
Alexander I. Elperin Living Trust dated August 1, 2012
|
23,890 | 18,890 | 5,000 | * | ||||||||||||
|
Mordechai Schechter
|
38,682 | 18,682 | 20,000 | 1.25 | % | |||||||||||
|
Eva Levi
|
18,636 | 18,636 | - | * | ||||||||||||
|
GMP Securities ITF Robert J. Halpern
|
25,402 | 17,902 | 7,500 | * | ||||||||||||
|
Thomas W. Hanson
|
37,862 | 17,862 | 20,000 | 1.25 | % | |||||||||||
|
National Jewish Health (18)
|
19,700 | 17,857 | 1,843 | * | ||||||||||||
|
MicroConstants, Inc. (19)
|
17,857 | 17,857 | - | * | ||||||||||||
|
M Capital Partners LLC (20)
|
27,144 | 17,144 | 10,000 | * | ||||||||||||
|
Reber-Burness Trust (21)
|
15,000 | 15,000 | - | * | ||||||||||||
|
GGG Investments, LLC (22)
|
15,000 | 15,000 | - | * | ||||||||||||
|
Michael W. Dinnen
|
14,286 | 14,286 | - | * | ||||||||||||
|
Marvin Mermelstein
|
14,286 | 14,286 | - | * | ||||||||||||
|
Jacob J. Tepper
|
14,286 | 14,286 | - | * | ||||||||||||
|
Jeff & Jenifer Culbertson JT
|
21,518 | 14,074 | 7,444 | * | ||||||||||||
|
Luke Matthys & Marilyn Good JT
|
25,674 | 10,394 | 15,280 | * | ||||||||||||
|
Moshe Glassman
|
10,002 | 10,002 | - | * | ||||||||||||
|
Joe Gerard
|
12,706 | 8,956 | 2,500 | * | ||||||||||||
|
IBH Capital LLC (23)
|
7,144 | 7,144 | - | * | ||||||||||||
|
Harry Klein
|
7,144 | 7,144 | - | * | ||||||||||||
|
Eli Scharf
|
7,144 | 7,144 | - | * | ||||||||||||
|
Fell 1996 Trust (24)
|
14,820 | 6,284 | 8,536 | * | ||||||||||||
|
Jeffrey A. Lindeman Trust UAD 09/23/10, Jeffrey A. Lindeman and Mona L. Lindeman Ttees
|
5,000 | 5,000 | - | * | ||||||||||||
|
Daniel J. Turner
|
10,336 | 4,190 | 5,146 | * | ||||||||||||
|
I M Marks and Joan M Marks Family Trust
|
7,019 | 4,072 | 2,947 | * | ||||||||||||
|
Bernhard A. Votteri MD, Trustee, Bernhard A. Votteri Inc. Profit Sharing Trust 08/01/78
|
9,610 | 3,956 | 5,654 | * | ||||||||||||
|
Lawrence C. Gotelli ,Jr.
|
40,633 | 2,090 | 38,543 | 2.39 | % | |||||||||||
|
Christopher A. Andreozzi
|
6,683 | 1,868 | 4,815 | * | ||||||||||||
|
Reid Adler
|
22,000 | 9,500 | 12,500 | * | ||||||||||||
|
(1)
|
Except as otherwise indicated in the following footnotes, the Selling Stockholder listed has voting and investment control over the securities beneficially owned, including the Registrable Securities..
|
|
|
(2)
|
Montsant Partners, LLC (“Montsant”) is an affiliate of Platinum Long Term Growth Fund VII (“Platinum”), to whom Platinum has previously transferred certain of the equity securities initially issued by us to Platinum. The number of beneficially owned shares reported both before and after the Offering includes 115,570 restricted shares of common stock owned by Montsant and 25,241 restricted shares of common stock and a warrant to purchase 20,172 restricted shares of common stock that may currently be acquired by Montsant upon exchange of 18,827 restricted shares of our Series A Preferred Stock (“Series A Preferred”).
The reported number of shares beneficially owned both before and after the Offering excludes 609,259 restricted shares of common stock and a warrant to purchase 435,186 restricted shares of common stock that may currently be acquired by Montsant upon exchange of 406,173 restricted shares of our Series A Preferred. Pursuant to the October 11, 2012 Note Exchange and Purchase Agreement by and between us and Platinum, there is a limitation on exchange such that the number of shares of our common stock that may be acquired by Platinum or its affiliates upon exchange of the Series A Preferred is limited to the extent necessary to ensure that, following such exchange, the total number of shares of our common stock then beneficially owned by Platinum or its affiliates does not exceed 9.99% of the total number of our then issued and outstanding shares of common stock without providing us with 61 days’ prior notice thereof.
Further, the reported number of shares beneficially owned by Montsant before the Offering also excludes 3,253,410 Registrable Shares pursuant to its ownership of 1,512,022 shares of our Series B 10% Convertible Preferred Stock (“Series B Preferred”), immediately convertible into a like number of shares of our restricted common stock, of which 430,000 shares are excluded from Registrable Securities herein, and warrants to purchase 1,741,388 shares of our restricted common stock, of which 1,200,000 shares are excluded from Registrable Securities herein. Pursuant to the terms of the Certificate of Designation of the Relative Rights and Preferences of the Series B 10% Convertible Preferred Stock and of the respective common stock warrant agreements, there is a limitation on conversion of the Series B Preferred and exercise of the warrants such that the number of shares of common stock that Montsant may beneficially acquire upon such conversion or exercise is limited to the extent necessary to ensure that, following such conversion or exercise, the total number of shares of common stock then beneficially owned by Platinum or Montsant does not exceed 9.99% of the total number of then issued and outstanding shares of our common stock without providing us with 61 days’ prior notice thereof.
Additionally, the reported number of shares beneficially owned both before and after the Offering excludes currently exercisable warrants owned by Platinum to purchase 320,124 shares of our restricted common stock. Pursuant to the terms of the respective common stock warrant agreements, there is a limitation on the exercise of the warrants such that the number of shares of common stock that Platinum may beneficially acquire upon such exercise is limited to the extent necessary to ensure that, following such exercise, the total number of shares of common stock then beneficially owned by Platinum or Montsant does not exceed 9.99% of the total number of then issued and outstanding shares of our common stock without providing us with 61 days’ prior notice thereof.
Mark Nordlicht has voting and investment control over the shares held by Platinum and Montsant.
|
|
|
(3)
|
The reported number of shares beneficially owned by Cato Holding Company dba Cato Bio Ventures prior to the Offering excludes 328,571 Registrable Shares, currently exchangeable for 328,571 restricted shares of our common stock. Pursuant to the terms of the Certificate of Designation of the Relative Rights and Preferences of the Series B 10% Convertible Preferred Stock, there is a limitation on conversion of the Series B Preferred such that the number of shares of common stock that Cato may beneficially acquire upon such conversion is limited to the extent necessary to ensure that, following such conversion, the total number of shares of common stock then beneficially owned by Cato does not exceed 9.99% of the total number of then issued and outstanding shares of our common stock without providing us with 61 days’ prior notice thereof. Lynda Sutton has voting and investment authority over the shares held by Cato Holding Company.
|
|
|
(4)
|
Mark J. Blumenthal has voting and investment authority over the shares held by Morrison & Foerster LLP.
|
|
|
(5)
|
Platinum has previously transferred to Michael Goldberg (“Goldberg”) certain of the equity securities initially issued by us to Platinum. The conversion or exercise restrictions in those securities initially applicable to Platinum remain applicable to Goldberg. The number of beneficially owned shares reported both before and after the Offering includes 37,508 restricted shares of common stock owned by Goldberg and 78,975 restricted shares of common stock and a currently exercisable warrant to purchase 56,411 restricted shares of common stock that may currently be acquired by Goldberg upon exchange of 52,650 restricted shares of our Series A Preferred.
The reported number of shares beneficially owned both before and after the Offering excludes 33,525 restricted shares of common stock and a warrant to purchase 23,946 restricted shares of common stock that may currently be acquired by Goldberg upon exchange of 22,350 restricted shares of our Series A Preferred. Pursuant to the October 11, 2012 Note Exchange and Purchase Agreement by and between us and Platinum, there is a limitation on exchange such that the number of shares of our common stock that may be acquired by Goldberg upon exchange of the Series A Preferred is limited to the extent necessary to ensure that, following such exchange, the total number of shares of our common stock then beneficially owned by Goldberg does not exceed 9.99% of the total number of our then issued and outstanding shares of common stock without providing us with 61 days’ prior notice thereof.
Further, the reported number of shares beneficially owned by Goldberg before the Offering also excludes 193,355 Registrable Shares pursuant to his ownership of 147,028 shares of our Series B Preferred, immediately convertible into a like number of shares of our restricted common stock, and warrants to purchase 46,327 shares of our restricted common stock. Pursuant to the terms of the Certificate of Designation of the Relative Rights and Preferences of the Series B 10% Convertible Preferred Stock and of the respective common stock warrant agreements, there is a limitation on conversion of the Series B Preferred and exercise of the warrants such that the number of shares of common stock that Goldberg may beneficially acquire upon such conversion or exercise is limited to the extent necessary to ensure that, following such conversion or exercise, the total number of shares of common stock then beneficially owned by Goldberg does not exceed 9.99% of the total number of then issued and outstanding shares of our common stock without providing us with 61 days’ prior notice thereof.
Additionally, the reported number of shares beneficially owned by Goldberg both before and after the Offering excludes currently exercisable warrants to purchase 66,251 shares of our restricted common stock. Pursuant to the terms of the respective common stock warrant agreements, there is a limitation on the exercise of the warrants such that the number of shares of common stock that Goldberg may beneficially acquire upon such exercise is limited to the extent necessary to ensure that, following such exercise, the total number of shares of common stock then beneficially owned by Goldberg does not exceed 9.99% of the total number of then issued and outstanding shares of our common stock without providing us with 61 days’ prior notice thereof.
|
|
|
(6)
|
Christopher Paige, Ph.D. has voting and investment authority over the shares held by University Health Network.
|
|
|
(7)
|
Chardan Capital Markets (“Chardan”) is a broker dealer and has received its Registrable Shares from us for services rendered and to be rendered to us pursuant to our financial advisory agreement. Steven Urbach has voting and investment authority over the shares held by Chardan.
|
|
|
(8)
|
Michael McCarthy has voting and investment authority over the shares held by Calm Seas Capital.
|
|
|
(9)
|
David E. Woollcombe has voting and investment authority over the shares held by McCarthy Tetrault LLP.
|
|
|
(10)
|
Shaye Hirsch has voting and investment authority over the shares held by Brio Capital Master Fund Ltd.
|
|
|
(11)
|
Robert Hellauer has voting and investment authority over the shares held by Icahn School of Medicine at Mount Sinai.
|
|
|
(12)
|
Anthony Heller has voting and investment authority over the shares held by Plazacorp Investments Limited.
|
|
|
(13)
|
Benjamin Mayer has voting and investment authority over the shares held by Excell Advisors LLC.
|
|
|
(14)
|
Desjardins Securities, Inc. (“Desjardins”) is a broker dealer and has received its Registrable Shares in exchange for the cancellation of certain promissory notes previously issued as payment of certain investment advisory services rendered to us. Jean-Yves Bourgeois has voting and investment authority over the shares held by Desjardins Securities, Inc.
|
|
|
(15)
|
Walter Schenker has voting and investment authority over the shares held by MAZ Partners, LP.
|
|
|
(16)
|
The reported number of shares beneficially owned both before and after the Offering includes 16,667 restricted shares of our common stock owned by Disclosure Law Group, of which Daniel W. Rumsey is the managing partner.
|
|
|
(17)
|
Curtis A. Burr has voting and investment authority over the shares held by Burr Pilger Mayer, Inc.
|
|
|
(18)
|
Christine K. Forkner has voting and investment authority over the shares held by National Jewish Health.
|
|
|
(19)
|
Gilbert Lam has voting and investment authority over the shares held by MicroConstants, Inc.
|
|
|
(20)
|
Benjamin Mayer has voting and investment authority over the shares held by M Capital Partners LLC.
|
|
|
(21)
|
James A. Burness has voting and investment authority over the shares held by Reber-Burness Trust.
|
|
|
(22)
|
Giulio Staiano has voting and investment authority over the shares held by GGG Investments, LLC.
|
|
|
(23)
|
Pincus Kievman has voting and investment authority over the shares held by IBH Capital LLC.
|
|
|
(24)
|
Thomas Fell has voting and investment authority over the shares held by Fell 1996 Trust.
|
|
|
(25)
|
Beneficial ownership assumes the conversion of and/ or exercise of any securities held by the Selling Stockholder that are currently convertible or exercisable or that may become convertible or exercisable within 60 days of July 17, 2015, not otherwise subject to conversion or exercise restrictions.
|
|
|
*
|
Less than 1%.
|
RELATIONSHIPS BETWEEN THE ISSUER AND THE SELLING SECURITY HOLDERS
Other than as disclosed herein, none of the Selling Stockholders has at any time during the past three years acted as one of our employees, officers or directors or had a material relationship with us.
Cato Holding Company, doing business as Cato BioVentures (“CBV”), is the parent of Cato Research Ltd., which has served as our contract research organization for the development and regulatory oversight of AV-101 since 2007. Shawn Singh, our Chief Executive Officer and member of our Board of Directors (“Mr. Singh”), served as Managing Principal of CBV and as an officer of CRL until August 2009.
Morrison & Foerster LLP (“Morrison & Foerster”) has previously provided certain legal services to us, and received its Registrable Securities in exchange for the cancellation of a promissory note previously issued in partial payment of those services.
University Health Network (“UHN”) provides stem cell research and development collaboration services to us through a Sponsored Research Collaboration Agreement (“SRCA”) and is one of our intellectual property licensors. UHN received its Registrable Securities in exchange for the cancellation of a promissory note previously issued as payment for services under the SRCA.
Icahn School of Medicine at Mount Sinai (“ISMMS”) is one of our intellectual property licensors and received its Registrable Securities in exchange for the cancellation of a promissory note previously issued as payment for certain amounts due under our license agreement with ISMMS.
Daniel W. Rumsey, managing partner of Disclosure Law Group, has served as our legal counsel through Disclosure Law Group since late 2011.
Praim S. Singh and Marlene Singh, trustees of the Singh Family Trust 4/25/1997 are the parents of Mr. Singh. Tracy K Price, trustee of the Price Family Trust 8/15/2007 is the brother of Mr. Singh.
Chardan Capital Markets (“Chardan”) provides financial advisory services us. We issued to Chardan its Registrable Securities as compensation for such advisory services.
Desjardins Securities, Inc. (“Desjardins”) previously provided certain investment advisory services to us, and received its Registrable Securities in exchange for the cancellation of a promissory note previously issued as payment of those services and certain current amounts payable by us to Desjardins.
PLAN OF DISTRIBUTION
Each Selling Stockholder and any of their pledgees, assignees and successors-in-interest may, from time to time, resell any or all of their shares of Common Stock covered hereby on the OTCQB or any other stock exchange, market or trading facility on which the shares are traded or in private transactions. These resales may be at fixed or negotiated prices. A Selling Stockholder may use any one or more of the following methods when reselling shares:
|
·
|
on any national securities exchange, market or quotation service on which our Common Stock may be listed or quoted at the time of sale;
|
|
·
|
in transactions other than on these exchanges or systems or in the over-the-counter market;
|
|
·
|
in ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers;
|
|
·
|
in block trades in which the broker-dealer will attempt to sell the securities as agent, but may position and resell a portion of the block as principal to facilitate the transaction;
|
|
·
|
in purchases by a broker-dealer as principal and resale by the broker-dealer for its account;
|
|
·
|
in an exchange distribution in accordance with the rules of the applicable exchange;
|
|
·
|
in privately negotiated transactions;
|
|
·
|
in put or call option transactions;
|
|
·
|
in transactions involving short sales through broker-dealers;
|
|
·
|
in transactions wherein the Selling Stockholder sells securities short themselves and delivers the securities to close out short positions;
|
|
·
|
through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise;
|
|
·
|
in transactions that may involve crosses or block transactions;
|
|
·
|
in transactions where broker-dealers may agree with the Selling Stockholders to sell a specified number of securities at a stipulated price per security;
|
|
·
|
a combination of any such methods of sale; or
|
|
·
|
in any other method permitted by applicable law.
|
The Selling Stockholders may also resell shares under Rule 144 under the Securities Act, if available, rather than under this prospectus.
Broker-dealers engaged by the Selling Stockholders may arrange for other brokers-dealers to participate in sales. Broker-dealers may receive commissions or discounts from the Selling Stockholders (or, if any broker-dealer acts as agent for the purchaser of shares, from the purchaser) in amounts to be negotiated, but, except as set forth in a supplement to this prospectus, in the case of an agency transaction not in excess of a customary brokerage commission in compliance with Rule 2440 of the Financial Industry Regulatory Authority, Inc.; and in the case of a principal transaction a markup or markdown in compliance with IM-2440 of the Financial Industry Regulatory Authority, Inc.
In connection with the sale of the Common Stock or interests therein, the Selling Stockholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the Common Stock in the course of hedging the positions they assume. The Selling Stockholders may also sell shares of the Common Stock short and deliver these securities to close out their short positions, or loan or pledge the Common Stock to broker-dealers that in turn may resell these securities. The Selling Stockholders may also enter into option or other transactions with broker-dealers or other financial institutions or create one or more derivative securities which require the delivery to such broker-dealer or other financial institution of shares offered by this prospectus, which shares such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
The Selling Stockholders and any broker-dealers or agents that are involved in reselling the shares may be deemed to be “underwriters” within the meaning of the Securities Act of 1933, as amended, in connection with such resales. In such event, any commissions received by such broker-dealers or agents and any profit on the resale of the shares purchased by them may be deemed to be underwriting commissions or discounts under the Securities Act. Each Selling Stockholder has informed the Company that it does not have any written or oral agreement or understanding, directly or indirectly, with any person to distribute their shares of Common Stock.
The Company is required to pay certain fees and expenses incurred by the Company incident to the registration of the shares. The Company has agreed to indemnify the Selling Stockholders against certain losses, claims, damages and liabilities, including liabilities under the Securities Act.
Because Selling Stockholders may be deemed to be “underwriters” within the meaning of the Securities Act, they will be subject to the prospectus delivery requirements of the Securities Act, including Rule 172 thereunder. The Selling Stockholders have advised us that there is no underwriter or coordinating broker acting in connection with the proposed resale of shares by the Selling Stockholders.
We agreed to keep this prospectus effective until the earlier of (i) the date on which the shares may be resold by the Selling Stockholders without registration and without regard to any volume or manner-of-sale limitations by reason of Rule 144, without the requirement for the Company to be in compliance with the current public information under Rule 144 under the Securities Act, or any other rule of similar effect (assuming that the shares were at no time held by any affiliate of ours, and all warrants are exercised by “cashless exercise” as provided in each of the warrants) or (ii) all of the shares have been resold pursuant to this prospectus or Rule 144 under the Securities Act, or any other rule of similar effect. The resale of shares will be only through registered or licensed brokers or dealers if required under applicable state securities laws. In addition, in certain states, resale of the shares of Common Stock covered hereby may not be consummated unless they have been registered or qualified for resale in the applicable state or an exemption from the registration or qualification requirement is available and is complied with.
Under applicable rules and regulations under the Exchange Act, any person engaged in the distribution of the shares may not simultaneously engage in market making activities with respect to the Common Stock for the applicable restricted period, as defined in Regulation M, prior to the commencement of the distribution. In addition, the Selling Stockholders will be subject to applicable provisions of the Exchange Act, and the rules and regulations thereunder, including Regulation M, which may limit the timing of purchases and resales of shares of the Common Stock by the Selling Stockholders or any other person under this prospectus. We will make copies of this prospectus available to the Selling Stockholders and have informed them of the need to deliver a copy of this prospectus to each purchaser at or prior to the time of the sale (including by compliance with Rule 172 under the Securities Act).
The financial statements as of March 31, 2015 and 2014, and for the years then ended, included in this prospectus, have been audited by OUM & Co. LLP, an independent registered public accounting firm, as stated in their report appearing herein. Such financial statements have been so included in reliance upon the report of such firm given upon their authority as experts in accounting and auditing.
LEGAL MATTERS
The Disclosure Law Group, San Diego, California, will pass upon the validity of the shares of common stock offered hereby. A partner of the Disclosure Law Group beneficially owns 16,667 restricted shares of our common stock, 25,000 shares of our Series B Preferred stock (included as Registrable Securities in the Selling Stockholder Table) and holds warrants to purchase 12,917 restricted shares of our common stock.
INTERESTS OF NAMED EXPERTS AND COUNSEL
No expert or counsel named in this prospectus as having prepared or certified any part of this prospectus or having given an opinion upon the validity of the securities being registered or upon other legal matters in connection with the resale of shares of common stock being offered by the Selling Stockholders under this prospectus was employed on a contingency basis or had, or is to receive, in connection with the resale of shares of common stock being offered by us under this prospectus, a substantial interest, directly or indirectly, in the Registrant or any of its parents or subsidiaries. Nor was any such person connected with the Registrant or any of its parents, subsidiaries as a promoter, managing or principal underwriter, voting trustee, director, officer, or employee.
WHERE YOU CAN FIND MORE INFORMATION
We are a public company and file annual, quarterly and special reports, proxy statements and other information with the SEC. You may read and copy any document we file at the SEC’s public reference room at 100 F Street, NE, Washington, D.C. 20549. You can request copies of these documents by writing to the SEC and paying a fee for the copying cost. Please call the SEC at 1-800-SEC-0330 for more information about the operation of the public reference room. Our SEC filings are also available, at no charge, to the public at the SEC’s website at http://www.sec.gov.
We have filed with the SEC a Registration Statement on Form S-1 under the Securities Act with respect to the shares of common stock being offered by the Selling Stockholders under this prospectus. This prospectus is part of that registration statement. This prospectus does not contain all of the information set forth in the registration statement or the exhibits to the registration statement. For further information with respect to us and the shares the Selling Stockholders are offering pursuant to this prospectus, you should refer to the registration statement and its exhibits. Statements contained in this prospectus as to the contents of any contract, agreement or other document referred to are not necessarily complete, and you should refer to the copy of that contract or other documents filed as an exhibit to the registration statement. You may read or obtain a copy of the registration statement at the SEC’s public reference facilities and Internet site referred to above.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
|
Page
|
|
|
F-2
|
|
|
F-3
|
|
|
F-4
|
|
|
F-5
|
|
|
F-6
|
|
|
F-7
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
VistaGen Therapeutics, Inc.
We have audited the accompanying consolidated balance sheets of VistaGen Therapeutics, Inc. as of March 31, 2015 and 2014 and the related consolidated statements of operations and comprehensive loss, cash flows, preferred stock, and stockholders’ deficit for the years then ended. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of VistaGen Therapeutics, Inc. at March 31, 2015 and 2014, and the consolidated results of its operations and its cash flows for the years then ended, in conformity with U.S. generally accepted accounting principles.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the consolidated financial statements, the Company has not yet generated sustainable revenues, has suffered recurring losses and negative cash flows from operations and has a stockholders’ deficit, all of which raise substantial doubt about its ability to continue as a going concern. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ OUM & Co. LLP
San Francisco, California
June 29, 2015
VISTAGEN THERAPEUTICS, INC.
CONSOLIDATED BALANCE SHEETS
(Amounts in dollars, except share amounts)
|
VISTAGEN THERAPEUTICS
|
||||||||
|
Consolidated Balance Sheets
|
||||||||
|
Amounts in Dollars
|
||||||||
|
March 31,
|
March 31,
|
|||||||
|
2015
|
2014
|
|||||||
|
ASSETS
|
||||||||
|
Current assets:
|
||||||||
|
Cash and cash equivalents
|
$ | 70,000 | $ | - | ||||
|
Prepaid expenses and other current assets
|
35,700 | 40,500 | ||||||
|
Total current assets
|
105,700 | 40,500 | ||||||
|
Property and equipment, net
|
117,100 | 176,300 | ||||||
|
Security deposits and other assets
|
46,900 | 46,900 | ||||||
|
Total assets
|
$ | 269,700 | $ | 263,700 | ||||
|
LIABILITIES AND STOCKHOLDERS’ DEFICIT
|
||||||||
|
Current liabilities:
|
||||||||
|
Accounts payable
|
$ | 2,251,100 | $ | 2,443,900 | ||||
|
Accrued expenses
|
1,206,500 | 625,600 | ||||||
|
Advance from officer
|
- | 3,600 | ||||||
|
Current maturities of senior secured convertible promissory notes and accrued interest
|
4,146,100 | - | ||||||
|
Current portion of notes payable, net of discount of $474,500 at March 31, 2014 and accrued interest
|
4,117,000 | 1,442,300 | ||||||
|
Current portion of notes payable to related parties, net of discount of $54,500 at March 31, 2015
|
||||||||
|
and accrued interest
|
1,508,800 | 290,400 | ||||||
|
Convertible promissory notes and accrued interest, net of discount of $180,000 at March 31, 2015 and
|
||||||||
|
$697,400 at March 31, 2014, respectively
|
4,157,600 | 396,000 | ||||||
|
Capital lease obligations
|
1,000 | 3,900 | ||||||
|
Total current liabilities
|
17,388,100 | 5,205,700 | ||||||
|
Non-current liabilities:
|
||||||||
|
Senior secured convertible promissory notes, net of discount of $0 at March 31, 2015 and
|
||||||||
|
$2,085,900 at March 31, 2014, respectively, and accrued interest
|
296,200 | 1,929,800 | ||||||
|
Notes payable, net of discount of $0 at March 31, 2015 and $848,100 at March 31, 2014, and
|
||||||||
|
and accrued interest
|
35,600 | 1,797,600 | ||||||
|
Notes payable to related parties, net of discount of $103,200 at March 31, 2014 and accrued interest
|
- | 1,057,100 | ||||||
|
Warrant liability
|
3,008,500 | 2,973,900 | ||||||
|
Deferred rent liability
|
83,000 | 97,400 | ||||||
|
Capital lease obligations
|
1,100 | 2,100 | ||||||
|
Total non-current liabilities
|
3,424,400 | 7,857,900 | ||||||
|
Total liabilities
|
20,812,500 | 13,063,600 | ||||||
|
Commitments and contingencies
|
||||||||
|
Stockholders’ deficit:
|
||||||||
|
Preferred stock, $0.001 par value; 10,000,000 shares, including 500,000 Series A shares, authorized
|
||||||||
|
at March 31, 2015 and March 31, 2014, respectively; 500,000 Series A shares issued and
|
||||||||
|
outstanding at March 31, 2015 and March 31, 2014, respectively
|
500 | 500 | ||||||
|
Common stock, $0.001 par value; 10,000,000 shares authorized at March 31, 2015 and March 31, 2014,
|
||||||||
|
respectively; 1,677,110 shares and 1,310,093 shares issued at March 31, 2015 and March 31,
|
||||||||
|
2014, respectively
|
1,700 | 1,300 | ||||||
|
Additional paid-in capital
|
67,945,800 | 62,001,400 | ||||||
|
Treasury stock, at cost, 135,665 shares of common stock held at March 31, 2015 and March 31, 2014,
|
||||||||
|
respectively
|
(3,968,100 | ) | (3,968,100 | ) | ||||
|
Note receivable from sale of common stock
|
- | (198,100 | ) | |||||
|
Accumulated deficit
|
(84,522,700 | ) | (70,636,900 | ) | ||||
|
Total stockholders’ deficit
|
(20,542,800 | ) | (12,799,900 | ) | ||||
|
Total liabilities and stockholders’ deficit
|
$ | 269,700 | $ | 263,700 | ||||
See accompanying notes to consolidated financial statements.
VISTAGEN THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(Amounts in dollars, except share amounts)
|
Fiscal Years Ended
|
||||||||
|
March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Operating expenses:
|
||||||||
|
Research and development
|
$ | 2,432,700 | $ | 2,480,600 | ||||
|
General and administrative
|
4,344,400 | 2,548,300 | ||||||
|
Total operating expenses
|
6,777,100 | 5,028,900 | ||||||
|
Loss from operations
|
(6,777,100 | ) | (5,028,900 | ) | ||||
|
Other expenses, net:
|
||||||||
|
Interest expense, net
|
(4,548,700 | ) | (1,503,000 | ) | ||||
|
Change in warrant liability
|
(34,600 | ) | 3,566,900 | |||||
|
Loss on extinguishment of debt
|
(2,388,000 | ) | - | |||||
|
Other expense
|
(135,000 | ) | - | |||||
|
Loss before income taxes
|
(13,883,400 | ) | (2,965,000 | ) | ||||
|
Income taxes
|
(2,400 | ) | (2,700 | ) | ||||
|
Net loss and comprehensive loss
|
$ | (13,885,800 | ) | $ | (2,967,700 | ) | ||
|
Basic net loss per common share
|
$ | (10.53 | ) | $ | (2.70 | ) | ||
|
Diluted net loss per common share
|
$ | (10.61 | ) | $ | (3.81 | ) | ||
|
Weighted average shares used in computing
|
||||||||
|
Basic net loss per common share
|
1,318,797 | 1,098,742 | ||||||
|
Diluted net loss per common share
|
1,318,797 | 1,099,216 | ||||||
See accompanying notes to consolidated financial statements.
VISTAGEN THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(Amounts in dollars)
|
Fiscal Years Ended March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Cash flows from operating activities:
|
||||||||
|
Net loss
|
$ | (13,885,800 | ) | $ | (2,967,700 | ) | ||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||
|
Depreciation and amortization
|
59,100 | 54,600 | ||||||
|
Amortization of discounts on convertible and promissory notes
|
3,372,000 | 640,000 | ||||||
|
Change in warrant liability
|
34,600 | (3,566,900 | ) | |||||
|
Stock-based compensation
|
2,460,100 | 1,137,300 | ||||||
|
Expense related to modification of warrants
|
98,400 | 204,300 | ||||||
|
Non-cash rent and relocation expense
|
(14,400 | ) | 56,800 | |||||
|
Interest income on note receivable for stock purchase
|
2,800 | (1,200 | ) | |||||
|
Loss on settlement of note receivable for stock purchase
|
134,900 | - | ||||||
|
Fair value of common stock granted for services
|
469,000 | - | ||||||
|
Fair value of warrants granted for services and interest
|
44,500 | 60,700 | ||||||
|
Gain on currency fluctuation
|
(63,600 | ) | (48,600 | ) | ||||
|
Loss on extinguishment of debt
|
2,388,000 | - | ||||||
|
Changes in operating assets and liabilities:
|
||||||||
|
Prepaid expenses and other current assets
|
107,400 | 92,700 | ||||||
|
Security deposits and other assets
|
- | (17,900 | ) | |||||
|
Accounts payable and accrued expenses, including accrued interest
|
2,024,100 | 2,229,900 | ||||||
|
Net cash used in operating activities
|
(2,768,900 | ) | (2,126,000 | ) | ||||
|
Cash flows from investing activities:
|
||||||||
|
Purchases of equipment, net
|
- | (9,600 | ) | |||||
|
Net cash used in investing activities
|
- | (9,600 | ) | |||||
|
Cash flows from financing activities:
|
||||||||
|
Net proceeds from issuance of common stock and warrants, including Units
|
3,146,600 | 1,075,500 | ||||||
|
Proceeds from exercise of modified warrants
|
- | 264,200 | ||||||
|
Proceeds from sale of note and warrant to Platinum
|
- | 250,000 | ||||||
|
Advance from officer
|
- | 64,000 | ||||||
|
Repayment of capital lease obligations
|
(3,900 | ) | (7,600 | ) | ||||
|
Repayment of notes
|
(303,800 | ) | (148,600 | ) | ||||
|
Net cash provided by financing activities
|
2,838,900 | 1,497,500 | ||||||
|
Net increase (decrease) in cash and cash equivalents
|
70,000 | (638,100 | ) | |||||
|
Cash and cash equivalents at beginning of period
|
- | 638,100 | ||||||
|
Cash and cash equivalents at end of period
|
$ | 70,000 | $ | - | ||||
|
Supplemental disclosure of cash flow activities:
|
||||||||
|
Cash paid for interest
|
$ | 35,700 | $ | 21,000 | ||||
|
Cash paid for income taxes
|
$ | 2,400 | $ | 2,700 | ||||
|
Supplemental disclosure of noncash activities:
|
||||||||
|
Insurance premiums settled by issuing note payable
|
$ | 105,300 | $ | 98,300 | ||||
|
Accounts payable settled by issuance of stock or notes payable and stock
|
$ | 438,400 | $ | - | ||||
|
Recognition of warrant liability upon issuance to Platinum of July 2013
|
||||||||
|
Senior Secured Convertible Note
|
$ | - | $ | 146,800 | ||||
See accompanying notes to consolidated financial statements.
VISTAGEN THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ DEFICIT
Fiscal Years Ended march 31, 2015 and 2014
(Amounts in dollars, except share amounts)
|
Series A Preferred Stock
|
Common Stock
|
Additional
Paid-in |
Treasury
|
Note
Receivable |
Accumulated
|
Total
Stockholders’ |
||||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Stock
|
Stock
|
Deficit
|
Deficit
|
||||||||||||||||||||||||||||
|
Balances at March 31, 2013
|
500,000 | $ | 500 | 1,174,092 | $ | 1,200 | $ | 59,288,300 | $ | (3,968,100 | ) | $ | (209,100 | ) | $ | (67,669,200 | ) | $ | (12,556,400 | ) | ||||||||||||||||
|
Share-based compensation expense
|
- | - | - | - | 1,137,300 | - | - | - | 1,137,300 | |||||||||||||||||||||||||||
|
Proceeds from sale of common stock for cash, including exercises of warrants under Discount Warrant Exercise Program
|
- | - | 32,751 | - | 335,900 | - | - | - | 335,900 | |||||||||||||||||||||||||||
|
Beneficial conversion feature on note issued to Platinum in July 2013
|
- | - | - | - | 100,700 | - | - | - | 100,700 | |||||||||||||||||||||||||||
|
Payment on note receivable from sale of stock
|
- | - | - | - | - | - | 11,000 | - | 11,000 | |||||||||||||||||||||||||||
|
Allocated proceeds from sale of Units for cash under 2013 Unit Private Placement, including beneficial conversion feature
|
- | - | 100,750 | 100 | 838,100 | - | - | - | 838,200 | |||||||||||||||||||||||||||
|
Allocated proceeds from sale of Units for cash under 2014 Unit Private Placement, including beneficial conversion feature
|
- | - | 2,500 | - | 36,000 | 36,000 | ||||||||||||||||||||||||||||||
|
Incremental fair value of warrant modifications
|
- | - | - | - | 204,300 | - | - | - | 204,300 | |||||||||||||||||||||||||||
|
Fair value of warrants issued to Morrison & Foerster, Cato Research Ltd. and University Health Network in connection with accrued interest on underlying notes
|
- | - | - | - | 60,800 | - | - | - | 60,800 | |||||||||||||||||||||||||||
|
Net loss for fiscal year 2014
|
- | - | - | - | - | - | - | (2,967,700 | ) | (2,967,700 | ) | |||||||||||||||||||||||||
|
Balances at March 31, 2014
|
500,000 | $ | 500 | 1,310,093 | $ | 1,300 | $ | 62,001,400 | $ | (3,968,100 | ) | $ | (198,100 | ) | $ | (70,636,900 | ) | $ | (12,799,900 | ) | ||||||||||||||||
|
Allocated proceeds from sale of Units for cash under 2014 Unit Private Placement, including beneficial conversion feature
|
- | - | 280,350 | 300 | 2,746,800 | - | - | - | 2,747,100 | |||||||||||||||||||||||||||
|
Share-based compensation expense
|
- | - | - | 2,460,100 | - | - | - | 2,460,100 | ||||||||||||||||||||||||||||
|
Payment on and settlement of note receivable from sale of stock
|
- | - | - | - | - | - | 198,100 | - | 198,100 | |||||||||||||||||||||||||||
|
Incremental fair value of modified warrants
|
- | - | - | - | 98,400 | - | - | - | 98,400 | |||||||||||||||||||||||||||
|
Fair Value of common stock issued for services
|
- | - | 71,667 | 100 | 635,600 | - | - | - | 635,700 | |||||||||||||||||||||||||||
|
Fair value of common stock and warrants issued in settlement of technology license expenses
|
- | - | 15,000 | - | 230,200 | - | - | - | 230,200 | |||||||||||||||||||||||||||
|
Fair value of warrants issued to Morrison & Foerster, Cato Research Ltd. and University Health Network in connection with accrued interest on underlying notes
|
- | - | - | - | 44,400 | - | - | - | 44,400 | |||||||||||||||||||||||||||
|
Effect of amendments of 2013 Unit Notes and warrants, including repurchase of beneficial conversion feature
|
- | - | - | - | 109,300 | - | - | - | 109,300 | |||||||||||||||||||||||||||
|
Effect of amendments of Platinum Senior Secured Promissory Notes, including repurchase of beneficial conversion feature
|
- | - | - | - | (380,400 | ) | - | - | - | (380,400 | ) | |||||||||||||||||||||||||
|
Net loss for fiscal year 2015
|
- | - | - | - | - | - | - | (13,885,800 | ) | (13,885,800 | ) | |||||||||||||||||||||||||
|
Balances at March 31, 2015
|
500,000 | $ | 500 | 1,677,110 | $ | 1,700 | $ | 67,945,800 | $ | (3,968,100 | ) | $ | - | $ | (84,522,700 | ) | $ | (20,542,800 | ) | |||||||||||||||||
See accompanying notes to consolidated financial statements.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Description of Business
VistaGen Therapeutics, Inc., a Nevada corporation, is a clinical-stage biopharmaceutical company committed to developing and commercializing product candidates for patients with depression, other diseases and disorders related to the central nervous system (CNS), and cancer. Our principal executive offices are located at 343 Allerton Avenue, South San Francisco, California 94080, and our telephone number is (650) 577-3600. Our website address is www.vistagen.com. Unless the context otherwise requires, the words “VistaGen Therapeutics, Inc.” “VistaGen,” “we,” “the Company,” “us” and “our” refer to VistaGen Therapeutics, Inc., a Nevada corporation.
VistaGen Therapeutics, Inc., a California corporation incorporated on May 26, 1998 (VistaGen California), is our wholly-owned subsidiary. Pursuant to a strategic merger transaction on May 11, 2011, we acquired all outstanding shares of VistaGen California in exchange for 341,823 shares of our common stock (Merger), and assumed all of VistaGen California’s pre-Merger obligations. The Consolidated Financial Statements in this report also include the accounts of VistaGen California’s two wholly-owned subsidiaries, Artemis Neuroscience, Inc., a Maryland corporation, and VistaStem Canada, Inc., a corporation organized under the laws of Ontario, Canada.
Our lead product candidate, AV-101, is an orally available small molecule prodrug in Phase 2 development for Major Depressive Disorder (MDD). AV-101’s mechanism of action (MOA), as an N-methyl-D-aspartate receptor (NMDAR) antagonist binding selectively at the glycine-binding (GlyB) co-agonist site of the NMDAR, is fundamentally different from all currently-approved antidepressants. In three preclinical studies utilizing well-validated animal models of depression, AV-101 was shown to induce fast-acting, dose-dependent, persistent and statistically significant antidepressant-like responses, following a single treatment, which were equivalent to response seen with a single sub-anesthetic dose of ketamine. In the same studies, fluoxetine did not induce rapid onset antidepressant-like responses. Preclinical studies also support the hypothesis that AV-101 has potential to treat several additional CNS disorders, including chronic neuropathic pain, epilepsy and neurodegenerative diseases, such as Parkinson’s disease and Huntington’s disease where modulation of the NMDAR may have therapeutic benefit.
Following our two successful randomized, double-blind, placebo-controlled Phase 1 safety studies funded by the U.S. National Institutes of Health (NIH), in February 2015, we entered a Cooperative Research and Development Agreement (CRADA) with the U.S. National Institute of Mental Health (NIMH), part of the NIH. Under this agreement, we will collaborate with the NIH on a Phase 2 clinical study of AV-101 in subjects with treatment resistant Major Depressive Disorder. Pursuant to the CRADA, this study will be conducted and fully-funded by the NIMH. It is contemplated that this clinical study will begin this year under the direction of Dr. Carlos Zarate, Jr., the NIMH’s Chief of Experimental Therapeutics & Pathophysiology Branch and of the Section on Neurobiology and Treatment of Mood and Anxiety Disorders.
In addition to developing AV-101 for MDD and other CNS indications, we are using our stem cell technology platform for drug rescue –to identify and develop proprietary new chemical entities (NCEs) for our internal drug candidate pipeline by leveraging our in vitro bioassay systems, prior investment by pharmaceutical companies and others to discover, optimize and test for efficacy NCEs terminated before FDA approval due to unexpected toxicity and medicinal chemistry. Our CardioSafe 3D™ bioassay system uses our human pluripotent stem cell (hPSC)-derived cardiomyocytes, or heart cells. Our LiverSafe 3D™bioassay system uses our stem cell-derived hepatocytes, or liver cells. We believe CardioSafe 3D and LiverSafe 3D offer a new paradigm for evaluating and predicting potential heart and liver toxicity of NCEs, including potential drug rescue NCEs, early in development, long before costly, high risk animal studies and human clinical trials. We intend to develop each optimized drug rescue NCE internally to establish in vitro and in vivo preclinical proof-of-concept (POC), as to both efficacy and safety, using both established in vitro and in vivo models, as well as in CardioSafe 3D and, when available, LiverSafe 3D.
Although we have previously generated approximately $16.4 million of revenue from grant awards and collaborations, we currently have no commercially available, revenue-generating products and, since inception, we have devoted substantially all of our time and efforts to development of AV-101 for CNS indications and our human pluripotent stem cell technology research and development programs, including, customized bioassay system development, creating, protecting and patenting intellectual property, recruiting personnel and raising working capital.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
2. Basis of Presentation and Going Concern
Effective August 14, 2014, we consummated a 1-for-20 reverse split of our authorized, and issued and outstanding shares of common stock (the Stock Consolidation). Each reference to shares of common stock or the price per share of common stock in these financial statements is post-Stock Consolidation, and reflects the 1-for-20 adjustment as a result of the Stock Consolidation. See Note 8, Capital Stock, for more information regarding the Stock Consolidation.
The accompanying Consolidated Financial Statements have been prepared assuming that we will continue as a going concern. As a developing-technology company having not yet developed commercial products or achieved sustainable revenues, we have experienced recurring losses and negative cash flows from operations resulting in a deficit of $84.5 million accumulated from inception through March 31, 2015. We expect losses and negative cash flows from operations to continue for the foreseeable future as we engage in further potential development of AV-101 and launch and execute our drug rescue programs and pursue potential drug development and regenerative medicine opportunities.
Since our inception in May 1998 through March 31, 2015, we have financed our operations and technology acquisitions primarily through the issuance and sale of equity and debt securities, including convertible promissory notes and short-term promissory notes, for cash proceeds of approximately $29.0 million, as well as from an aggregate of approximately $16.4 million of government research grant awards, strategic collaboration payments and other revenues. Additionally, we have issued equity securities with an approximate value at issuance of $13.5 million in non-cash settlements of certain liabilities, including liabilities for professional services rendered to us or as compensation for such services.
Between late-March 2014 and March 31, 2015, we entered into securities purchase agreements with accredited investors and institutions, including Platinum, pursuant to which we sold units to such accredited investors, in private placement transactions (2014 Units or 2014 Unit Private Placement), for aggregate cash proceeds of approximately $3.1 million, consisting of (i) 2014 Unit Notes in the aggregate face amount of approximately $3.1 million which matured between March 31, 2015 and April 30, 2015, or were automatically convertible into securities we might issue upon the consummation of a Qualified Financing, as defined, (ii) an aggregate of 282,850 restricted shares of our common stock (2014 Unit Stock); and (iii) warrants exercisable through December 31, 2016 to purchase an aggregate of 282,850 restricted shares of our common stock at an exercise price of $10.00 per share (2014 Unit Warrants). At March 31, 2015, we did not have sufficient cash and cash equivalents to enable us to fund our planned operations, including expected cash expenditures of approximately $7 million over the next twelve months, including expenditures required to prepare for further clinical trials of AV-101.
As described more completely in Note 16, Subsequent Events, between April 1 and May 14, 2015, we continued the 2014 Unit Private Placement, pursuant to which we sold to accredited investors 2014 Units, for aggregate cash proceeds of $280,000, consisting of: (i) 10% convertible promissory notes maturing between April 30, 2015 and May 15, 2015, in the aggregate face amount of $280,000, (ii) an aggregate of 33,000 shares of our restricted common stock, and (iii) warrants exercisable through December 31, 2016 to purchase an aggregate of 24,250 restricted shares of our common stock at an exercise price of $10.00 per share. As also described more completely in Note 16, Subsequent Events, during May 2015, we entered into an agreement with Platinum (Platinum Agreement), pursuant to which, Platinum has, among other things:
|
·
|
Converted the approximately $4.5 million outstanding balance (principal and accrued but unpaid interest) of the Senior Notes we issued to Platinum into 641,335 shares of our newly-created Series B 10% Convertible Preferred Stock (Series B Preferred) , thereby cancelling approximately $4.5 million of our outstanding indebtedness;
|
|
·
|
Released all of its security interests in our assets and those of our subsidiaries by terminating the Amended and Restated Security Agreement, IP Security Agreement and Negative Covenant, which we had entered into with Platinum in October 2012; and
|
|
·
|
Converted the approximately $1.3 million outstanding balance (principal and accrued but unpaid interest) of the convertible promissory notes we issued to Platinum in the 2014 Unit Private Placement (2014 Unit Notes) into 240,305 shares of Series B Preferred and five-year warrants to purchase 240,305 shares of our common stock at a fixed exercise price of $7.00 per share (Series B Warrants), thereby cancelling approximately $1.3 million of our outstanding indebtedness; and
|
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
·
|
Purchased approximately $1.5 million (principal and accrued but unpaid interest) of outstanding 2014 Unit Notes we issued to various other investors from the respective holders thereof (Investor 2014 Unit Notes ) and converted the entire outstanding balance of the Investor 2014 Unit Notes into 265,699 shares of Series B Preferred and Series B Warrants to purchase 265,699 shares of our common stock, thereby cancelling approximately $1.5 million of our outstanding indebtedness; and
|
|
·
|
Entered into a Securities Purchase Agreement (SPA) to purchase, for $1.0 million, a total of 142,857 shares of Series B Preferred and a Series B Warrant (Series B Preferred Unit) to purchase 142,857 shares of our common stock, on or before June 11, 2015 (a portion of which purchase was consummated on June 19, 2015).
|
As further described in Note 16, Subsequent Events, effective May 20, 2015, holders of the remaining $1.8 million outstanding balance (principal and accrued but unpaid interest) of 2014 Unit Notes converted such notes into 327,016 shares of Series B Preferred and Series B Warrants to purchase 327,016 shares of our common stock, thereby cancelling an additional approximately $1.8 million of our outstanding indebtedness. Between May 26, 2015 and June 25, 2015, we sold to accredited investors and institutions an aggregate of $557,500 of units in our Series B Preferred Unit offering, which units consist of Series B Preferred and Series B Warrants (together Series B Preferred Units), including $100,000 to Platinum. We issued 79,646 shares of Series B Preferred and Series B Warrants to purchase 79,646 shares of our common stock. We have received an aggregate of $557,500 in cash proceeds from the sale of the Series B Preferred Units.
Additionally, as further described in Note 16, Subsequent Events, holders of certain of our promissory notes outstanding at March 31, 2015 and thereafter, including Morrison & Foerster, Cato Research Ltd., University Health Network, and McCarthy Tetrault, and certain other service providers converted notes payable or accounts payable having an aggregate outstanding balance of approximately $5.8 million (principal and accrued but unpaid interest and certain strategic adjustments) into 831,577 shares of Series B Preferred stock.
Since March 31, 2015, we have eliminated approximately $14.9 million of promissory notes, other debt and certain adjustments thereto that was either already due and payable or would have otherwise matured prior to March 31, 2016, through conversion into our Series B Preferred stock and, with respect to a portion of the indebtedness converted, warrants to purchase common stock. Together with the cash proceeds from our Series B Preferred Unit Offering, our working capital position has improved significantly since March 31, 2015. We will, however, need to raise additional capital to fund our operations and execute our business plan over the next year and thereafter.
We believe that our participation in potential strategic collaborations, including potential transactions involving AV-101 such as our February 2015 Cooperative Research and Development Agreement with the U.S. National Institutes of Health (NIH) for an NIH-funded and sponsored Phase 2 study of AV-101 in major depressive disorder, may provide resources to support a portion of our future cash needs and working capital requirements. When and as necessary, we will seek to raise a material amount of financing through a combination of additional private placements and/or registered public offerings of our securities, which may include both debt and equity securities, stem cell technology-based research and development collaborations, stem cell technology and drug candidate license fees, and government grant awards and collaborations. Our future working capital requirements will depend on many factors, including, without limitation, the scope and nature of strategic opportunities related to our success in clinical trials of and further developing AV-101 as a treatment for major depressive disorder and/or other conditions; our stem cell technology platform, including drug rescue and cell therapy research and development efforts and the success of such programs, our ability to obtain government grant awards and our ability to enter into strategic collaborations with institutions on terms acceptable to us. To further advance the clinical development of AV-101 and potential drug rescue applications of our stem cell technology platform, as well as support our operating activities, we plan to continue to carefully manage our routine operating costs, including salaries and benefits, regulatory and public company consulting, contract research and development, legal, accounting and other professional services and working capital costs.
Notwithstanding the foregoing, substantial additional financing may not be available to us on a timely basis, on acceptable terms, or at all. If we are unable to obtain substantial additional financing on a timely basis in the near term, our business, financial condition, and results of operations may be harmed, the price of our stock may decline, we may be required to reduce, defer, or discontinue certain of our research and development activities and we may not be able to continue as a going concern. These Consolidated Financial Statements do not include any adjustments that might result from the outcome of this uncertainty.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
3. Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles (“U.S. GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant estimates include, but are not limited to, those relating to stock-based compensation, revenue recognition, and the assumptions used to value warrants, warrant modifications and warrant liabilities.
Principles of Consolidation
The accompanying consolidated financial statements include the Company’s accounts, and the accounts of VistaGen California’s wholly-owned inactive subsidiaries, Artemis Neurosciences and VistaStem Canada.
Cash and Cash Equivalents
Cash and cash equivalents are considered to be highly liquid investments with maturities of three months or less at the date of purchase.
Property and Equipment
Property and equipment is stated at cost. Repairs and maintenance costs are expensed in the period incurred. Depreciation is calculated using the straight-line method over the estimated useful lives of the assets. The estimated useful lives of property and equipment range from five to seven years.
Impairment or Disposal of Long-Lived Assets
We evaluate our long-lived assets, primarily property and equipment, for impairment whenever events or changes in circumstances indicate that their carrying value may not be recoverable from the estimated future cash flows expected to result from their use or eventual disposition. If the estimates of future undiscounted net cash flows are insufficient to recover the carrying value of the assets, we record an impairment loss in the amount by which the carrying value of the assets exceeds their fair value. If the assets are determined to be recoverable, but the useful lives are shorter than originally estimated, we depreciate or amortize the net book value of the assets over the newly determined remaining useful lives. We have not recorded any impairment charges to date.
Revenue Recognition
Although we do not currently have any such arrangements, we have historically generated revenue principally from collaborative research and development arrangements, technology transfer agreements, including strategic licenses, and government grants. Revenue arrangements with multiple components are divided into separate units of accounting if certain criteria are met, including whether the delivered component has stand-alone value to the customer. Consideration received is allocated among the separate units of accounting based on their respective selling prices. The selling price for each unit is based on vendor-specific objective evidence, or VSOE, if available, third party evidence if VSOE is not available, or estimated selling price if neither VSOE nor third party evidence is available. The applicable revenue recognition criteria are then applied to each of the units.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
We recognize revenue when the four basic criteria of revenue recognition are met: (i) a contractual agreement exists; (ii) the transfer of technology has been completed or services have been rendered; (iii) the fee is fixed or determinable; and (iv) collectability is reasonably assured. For each source of revenue, we comply with the above revenue recognition criteria in the following manner:
|
·
|
Collaborative arrangements typically consist of non-refundable and/or exclusive up front technology access fees, cost reimbursements for specific research and development spending, and various milestone and future product royalty payments. If the delivered technology does not have stand-alone value, the amount of revenue allocable to the delivered technology is deferred. Non-refundable upfront fees with stand-alone value that are not dependent on future performance under these agreements are recognized as revenue when received, and are deferred if we have continuing performance obligations and have no objective and reliable evidence of the fair value of those obligations. We recognize non-refundable upfront technology access fees under agreements in which we have a continuing performance obligation ratably, on a straight-line basis, over the period during which we are obligated to provide services. Cost reimbursements for research and development spending are recognized when the related costs are incurred and when collectability is reasonably assured. Payments received related to substantive, performance-based “at-risk” milestones are recognized as revenue upon achievement of the milestone event specified in the underlying contracts, which represent the culmination of the earnings process. Amounts received in advance are recorded as deferred revenue until the technology is transferred, costs are incurred, or a milestone is reached.
|
|
·
|
Technology license agreements typically consist of non-refundable upfront license fees, annual minimum access fees, development and/or regulatory milestone payments and/or royalty payments. Non-refundable upfront license fees and annual minimum payments received with separable stand-alone values are recognized when the technology is transferred or accessed, provided that the technology transferred or accessed is not dependent on the outcome of the continuing research and development efforts. Otherwise, revenue is recognized over the period of our continuing involvement, and, in the case of development and/or regulatory milestone payments, when the applicable event triggering such a payment has occurred.
|
|
·
|
Government grants, which support our research efforts on specific projects, generally provide for reimbursement of approved costs as defined in the terms of grant awards. Grant revenue is recognized when associated project costs are incurred.
|
Research and Development Expenses
Research and development expenses are composed of both internal and external costs. Internal costs include salaries and employment-related expenses of scientific personnel and direct project costs. External research and development expenses consist primarily of costs associated with clinical and non-clinical development of AV-101, our prodrug candidate entering late-stage clinical development for Major Depressive Disorder, sponsored stem cell research and development costs, and costs related to the application and prosecution of patents related to our stem cell technology platform and AV-101. All such costs are charged to expense as incurred.
Stock-Based Compensation
We recognize compensation cost for all stock-based awards to employees based on the grant date fair value of the award. We record non-cash, stock-based compensation expense over the period during which the employee is required to perform services in exchange for the award, which generally represents the scheduled vesting period. We have granted no restricted stock awards nor do we have any awards with market or performance conditions. For equity awards to non-employees, we re-measure the fair value of the awards as they vest and the resulting value is recognized as an expense during the period over which the services are performed.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Income Taxes
We account for income taxes using the asset and liability approach for financial reporting purposes. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. Valuation allowances are established, when necessary, to reduce the deferred tax assets to an amount expected to be realized.
Concentrations of Credit Risk
Financial instruments, which potentially subject us to concentrations of credit risk, consist principally of cash and cash equivalents. Our investment policies limit any such investments to short-term, low-risk investments. We deposit cash and cash equivalents with quality financial institutions and are insured to the maximum of federal limitations. Balances in these accounts may exceed federally insured limits at times.
Warrant Liability
We have issued to Platinum Long Term Growth VII, LLC, our largest investor (Platinum), warrants to purchase a substantial number of unregistered shares of our common stock and, subject to Platinum’s exercise of its rights to exchange shares of our Series A Preferred Stock that it holds, we are obligated to issue to Platinum an additional warrant to purchase unregistered shares of common stock (collectively, the Platinum Warrants). The Platinum Warrants contain an exercise price adjustment feature that will lower the exercise price of the warrants in the event we subsequently issue equity instruments at a price lower than the exercise price of the Platinum Warrants. We account for the Platinum Warrants as non-cash liabilities and estimate their fair value as described in Note 4, Fair Value Measurements, Note 8, Convertible Promissory Notes and Other Notes Payable, and Note 9, Capital Stock. We compute the fair value of the warrant liability at each reporting period and record the change in the fair value as non-cash expense or non-cash income. The key component in determining the fair value of the Platinum Warrants and the related liability is the market price of our common stock, which is subject to significant fluctuation and is not under our control. The resulting change in the fair value of the warrant liability on our net loss is therefore also subject to significant fluctuation and will continue to be so until all of the Platinum Warrants are issued and exercised, amended or expire. Assuming all other fair value inputs remain generally constant, we will record an increase in the warrant liability and non-cash losses when our stock price increases and a decrease in the warrant liability and non-cash gains when our stock price decreases. As described in Note 16, Subsequent Events, during May 2015, we entered into an agreement with Platinum pursuant to which Platinum agreed to amend the Platinum Warrants to fix the exercise price thereof at $7.00 per share and eliminate the exercise price reset features and fix the number of shares of our common stock issuable thereunder. This amendment will result in the elimination of the warrant liability with respect to the Platinum Warrants during the first quarter of our fiscal year ended March 31, 2016.
Comprehensive Loss
We have no components of other comprehensive loss other than net loss, and accordingly our comprehensive loss is equivalent to our net loss for the periods presented.
Loss per Common Share
Basic income (loss) per share of common stock excludes the effect of dilution and is computed by dividing net income (loss) by the weighted-average number of shares of common stock outstanding for the period. Diluted income (loss) per share of common stock reflects the potential dilution that could occur if securities or other contracts to issue shares of common stock were exercised or converted into shares of common stock. In calculating diluted net income (loss) per share, we adjust the numerator for the change in the fair value of the warrant liability attributable to the outstanding Platinum Warrants, only if dilutive, and increase the denominator to include the number of potentially dilutive common shares assumed to be outstanding during the period using the treasury stock method. As a result of our net loss for both periods presented, potentially dilutive securities were excluded from the computation of diluted loss per share, as their effect would be antidilutive.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Basic and diluted net loss attributable to common stockholders per share was computed as follows:
|
Fiscal Years Ended March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Numerator:
|
||||||||
|
Net loss attributable to common stockholders for basic earnings per share
|
$ | (13,885,800 | ) | $ | (2,967,700 | ) | ||
|
less: change in fair value of warrant liability attributable to Exchange, Investment and Bridge Warrants issued to Platinum
|
(105,200 | ) | (1,219,500 | ) | ||||
|
Net loss for diluted earnings per share attributable to common stockholders
|
$ | (13,991,000 | ) | $ | (4,187,200 | ) | ||
|
Denominator:
|
||||||||
|
Weighted average basic common shares outstanding
|
1,318,797 | 1,098,742 | ||||||
|
Assumed conversion of dilutive securities:
|
||||||||
|
Warrants to purchase common stock
|
- | 474 | ||||||
|
Potentially dilutive common shares assumed converted
|
- | 474 | ||||||
|
Denominator for diluted earnings per share - adjusted weighted average shares
|
1,318,797 | 1,099,216 | ||||||
|
Basic net loss attributable to common stockholders per common share
|
$ | (10.53 | ) | $ | (2.70 | ) | ||
|
Diluted net loss attributable to common stockholders per common share
|
$ | (10.61 | ) | $ | (3.81 | ) | ||
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Potentially dilutive securities excluded in determining diluted net loss per common share for the fiscal years ended March 31, 2015 and 2014 are as follows:
|
Fiscal Years Ended March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Series A preferred stock issued and outstanding (1)
|
750,000 | 750,000 | ||||||
|
Warrant shares issuable to Platinum upon exercise of common stock warrants by Platinum
|
||||||||
|
upon exchange of Series A Preferred under the terms of the October 11, 2012 Note
|
||||||||
|
Exchange and Purchase Agreement
|
375,000 | 375,000 | ||||||
|
Outstanding options under the 2008 and 1999 Stock Incentive Plans
|
207,638 | 212,486 | ||||||
|
Outstanding warrants to purchase common stock
|
1,544,474 | 854,782 | ||||||
|
10% convertible Exchange Note and Investment Notes issued to Platinum in October 2012,
|
||||||||
|
February 2013 and March 2013, including accrued interest through March 31, 2015
|
||||||||
|
and 2014, respectively (2)
|
414,615 | 374,798 | ||||||
|
10% convertible note issued to Platinum on July 26, 2013, including accrued interest
|
||||||||
|
through March 31, 2015 and 2014, respectively
|
29,620 | 26,776 | ||||||
|
10% convertible notes issued as a component of Unit Private Placements, including accrued interest through March 31, 2014
|
||||||||
|
accrued interest through March 31, 2015 and 2014, respectively (3)
|
433,758 | 109,341 | ||||||
|
Total
|
3,755,105 | 2,703,183 | ||||||
|
____________
|
||||||||
|
(1) Assumes exchange under the terms of the October 11, 2012 Note Exchange and Purchase Agreement with Platinum
|
||||||||
|
(2) Assumes conversion under the terms of the October 11, 2012 Note Exchange and Purchase Agreement with Platinum and the terms of the individual notes
|
||||||||
|
(3) Excludes effect of conversion premium upon conversion into securities which may be issued in a Qualified Financing, as defined in the notes
|
||||||||
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) No. 2014-09, Revenue from Contracts with Customers (Topic 606).which supersedes the revenue recognition requirements in ASC 605, Revenue Recognition., The amendment in this ASU provides guidance on revenue recognition to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The core principle of this update provides guidance to identify the performance obligations under the contract(s) with a customer and how to allocate the transaction price to the performance obligations in the contract. It further provides guidance to recognize revenue when (or as) the entity satisfies a performance obligation. The ASU is effective for public entities for annual and interim periods beginning after December 15, 2016 (the first quarter of our fiscal year ending March 31, 2018). In April 2015, the FASB proposed to defer for one year the effective date of the new revenue standard, with an option that would permit companies to adopt the standard as early as the original effective date. Early adoption prior to the original effective date is not permitted. We have not determined the potential effects of adopting this ASU on our consolidated financial statements.
In June 2014, the FASB issued ASU No. 2014-10, Development Stage Entities (Topic 915): Elimination of Certain Financial Reporting Requirements, Including an Amendment to Variable Interest Entities Guidance in Topic 810, Consolidation. The amendments in this ASU remove all incremental financial reporting requirements for development stage entities. Among other changes, this ASU no longer requires development stage entities to present inception-to-date information about income statement line items, cash flows, and equity transactions. The presentation and disclosure requirements in Topic 915 will no longer be required for the first annual period beginning after December 15, 2014, with early adoption permitted. We have adopted ASU 2014-10 effective with our fiscal year beginning April 1, 2014 and, accordingly, have eliminated inception-to-date information in the accompanying Consolidated Statements of Operations and Comprehensive Loss and Consolidated Statements of Cash Flows.
In August 2014, the FASB issued ASU No. 2014-15, Presentation of Financial Statements—Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. The amendments in this ASU define when and how an entity is required to disclose going concern uncertainties, which must be evaluated each interim and annual period. Specifically, the ASU requires management to determine whether substantial doubt exists regarding the entity’s going concern presumption. Substantial doubt about an entity’s ability to continue as a going concern exists when relevant conditions and events, considered in the aggregate, indicate that it is probable (as defined under ASC 450, Contingencies) that the entity will be unable to meet its obligations as they become due within one year after the date that the financial statements are issued or are available to be issued. If substantial doubt exists, certain disclosures are required, the extent of which depends on an evaluation of management’s plans (if any) to mitigate the going concern uncertainty. This evaluation should include consideration of conditions and events that are either known or are reasonably knowable at the date the financial statements are issued or are available to be issued, as well as whether it is probable that management's plans to address the substantial doubt will be implemented and, if so, whether it is probable that the plans will alleviate the substantial doubt. ASU 2014-15 is effective for annual periods ending after December 15, 2016, and interim and annual periods thereafter. Early application is permitted. In their opinion on our audited financial statements for our fiscal year ended March 31, 2015, our auditors indicated that there was substantial doubt about our ability to continue as a going concern. Although we have not yet adopted ASU 2014-15, we have indicated in Note 2, Basis of Presentation and Going Concern, steps we have taken to eliminate certain of our indebtedness and raise additional financing that is expected to permit us to continue our operations for at least one year. Upon our adoption of ASU 2014-15, assuming conditions at such time indicate there is substantial doubt about our ability to continue as a going concern, or that such doubt has been alleviated, we will conform our disclosure to comply with the guidance contained in ASU 2014-15.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
4. Fair Value Measurements
We follow the principles of fair value accounting as they relate to its financial assets and financial liabilities. Fair value is defined as the estimated exit price received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date, rather than an entry price that represents the purchase price of an asset or liability. Where available, fair value is based on observable market prices or parameters, or derived from such prices or parameters. Where observable prices or inputs are not available, valuation models are applied. These valuation techniques involve some level of management estimation and judgment, the degree of which is dependent on several factors, including the instrument’s complexity. The required fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels is described as follows:
|
·
|
Level 1 — Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs.
|
|
·
|
Level 2 — Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
|
|
·
|
Level 3 — Unobservable inputs (i.e., inputs that reflect the reporting entity’s own assumptions about the assumptions that market participants would use in estimating the fair value of an asset or liability) are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs.
|
A financial instrument’s categorization within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement. Where quoted prices are available in an active market, securities are classified as Level 1 of the valuation hierarchy. If quoted market prices are not available for the specific financial instrument, then the Company estimates fair value by using pricing models, quoted prices of financial instruments with similar characteristics or discounted cash flows. In certain cases where there is limited activity or less transparency around inputs to valuation, financial assets or liabilities are classified as Level 3 within the valuation hierarchy.
We do not use derivative instruments for hedging of market risks or for trading or speculative purposes. In conjunction with the Senior Secured Convertible Promissory Notes issued to Platinum between October 2012 and July 2013 and the related Platinum Warrants (see Note 8, Convertible Promissory Notes and Other Notes Payable), and the contingently issuable Series A Exchange Warrant (see Note 9, Capital Stock), we determined that the warrants included certain exercise price adjustment features requiring the warrants to be treated as liabilities, which were recorded at their issuance-date estimated fair values. We determined the fair value of the warrant liabilities using a Monte Carlo simulation model with Level 3 inputs. Inputs used to determine fair value include the remaining contractual term of the notes, risk-free interest rates, expected volatility of the price of the underlying common stock, and the probability of a financing transaction that would trigger a reset in the warrant exercise price, and, in the case of the Series A Exchange Warrant, the probability of Platinum’s exchange of the shares of Series A Preferred it holds into shares of common stock. Changes in the fair value of these warrant liabilities have been recognized as non-cash income or expense in the Consolidated Statements of Operations and Comprehensive Loss for the fiscal years ended March 31, 2015 and 2014.
The fair value hierarchy for liabilities measured at fair value on a recurring basis is as follows:
|
Fair Value Measurements at Reporting Date Using
|
||||||||||||||||
|
Total
Carrying |
Quoted Prices in
Active Markets for |
Significant Other
Observable Inputs |
Significant Unobservable
Inputs |
|||||||||||||
|
|
Value
|
(Level 1)
|
(Level 2)
|
(Level 3)
|
||||||||||||
|
March 31, 2015:
|
||||||||||||||||
|
Warrant liability
|
$ | 3,008,500 | $ | - | $ | - | $ | 3,008,500 | ||||||||
|
March 31, 2014:
|
||||||||||||||||
|
Warrant liability
|
$ | 2,973,900 | $ | - | $ | - | $ | 2,973,900 | ||||||||
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
During the fiscal years ended March 31, 2015 and 2014, there were no significant changes to the valuation models used for purposes of determining the fair value of the Level 3 warrant liability.
The changes in Level 3 liabilities measured at fair value on a recurring basis are as follows:
|
Fair Value Measurements
Using Significant |
||||
|
(Level 3)
|
||||
|
Warrant Liability
|
||||
|
Balance at March 31, 2013
|
$ | 6,394,000 | ||
|
Recognition of warrant liability upon issuance of Senior Secured Convertible
|
||||
|
Promissory Note and warrant to Platinum on July 26, 2013
|
146,800 | |||
|
Mark to market gain included in net loss
|
(3,566,900 | ) | ||
|
Balance at March 31, 2014
|
2,973,900 | |||
|
Mark to market loss included in net loss
|
34,600 | |||
|
Balance at March 31, 2015
|
$ | 3,008,500 | ||
As described in Note 16, Subsequent Events, during May 2015, we entered into an agreement with Platinum pursuant to which Platinum agreed to amend the Platinum Warrants and the Series A Exchange Warrant to fix the exercise price thereof at $7.00 per share and eliminate the exercise price reset features and fix the number of shares of our common stock issuable thereunder. This amendment will result in the elimination of the warrant liability with respect to these warrants during the first quarter of our fiscal year ended March 31, 2016.
No assets or other liabilities were measured on a recurring basis at fair value at March 31, 2015 or 2014.
5. Prepaid Expenses and Other Current Assets
Prepaid expenses and other current assets consist of the following:
|
March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Insurance
|
$ | 27,300 | $ | 21,800 | ||||
|
Legal fees
|
3,400 | 3,400 | ||||||
|
Interest receivable on note receivable from sale
|
||||||||
|
of common stock
|
- | 2,800 | ||||||
|
Technology license fees and all other
|
5,000 | 12,500 | ||||||
| $ | 35,700 | $ | 40,500 | |||||
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
6. Property and Equipment
Property and equipment consists of the following:
|
March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Laboratory equipment
|
$ | 653,600 | $ | 653,600 | ||||
|
Tenant improvements
|
26,900 | 26,900 | ||||||
|
Computers and network equipment
|
32,200 | 32,200 | ||||||
|
Office furniture and equipment
|
69,500 | 69,500 | ||||||
| 782,200 | 782,200 | |||||||
|
Accumulated depreciation and amortization
|
(665,100 | ) | (605,900 | ) | ||||
|
Property and equipment, net
|
$ | 117,100 | $ | 176,300 | ||||
In connection with the issuance of Senior Secured Convertible Promissory Notes to Platinum beginning in October 2012, we entered into a Security Agreement with Platinum under which the repayment of all amounts due under the terms of the various Senior Secured Convertible Promissory Notes is secured by all of our assets, including our tangible and intangible personal property, licenses, patent licenses, trademarks and trademark licenses (see Note 8, Convertible Promissory Notes and Other Notes Payable). As described in Note 16, Subsequent Events, during May 2015, we entered into an agreement with Platinum pursuant to which Platinum converted all of the Senior Secured Convertible Promissory Notes it held into shares of our newly created Series B Preferred stock and terminated its security interests in our assets.
7. Accrued Expenses
Accrued expenses consist of:
|
March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Accrued professional services
|
$ | 213,800 | $ | 135,700 | ||||
|
Accrued compensation
|
990,700 | 489,900 | ||||||
|
All other
|
2,000 | - | ||||||
| $ | 1,206,500 | $ | 625,600 | |||||
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
8. Convertible Promissory Notes and Other Notes Payable
The following table summarizes the components of the Company’s convertible promissory notes and other notes payable:
|
March 31, 2015
|
March 31, 2014
|
|||||||||||||||||||||||
|
|
Principal
|
Accrued
|
Principal
|
Accrued
|
||||||||||||||||||||
|
Balance
|
Interest
|
Total
|
Balance
|
Interest
|
Total
|
|||||||||||||||||||
|
Senior Secured 10% Convertible Promissory Notes issued to Platinum:
|
||||||||||||||||||||||||
|
Exchange Note issued on October 11, 2012
|
$ | 1,272,600 | $ | 360,200 | $ | 1,632,800 | $ | 1,272,600 | $ | 203,400 | $ | 1,476,000 | ||||||||||||
|
Investment Note issued on October 11, 2012
|
500,000 | 141,500 | 641,500 | 500,000 | 79,900 | 579,900 | ||||||||||||||||||
|
Investment Note issued on October 19, 2012
|
500,000 | 140,100 | 640,100 | 500,000 | 78,600 | 578,600 | ||||||||||||||||||
|
Investment Note issued on February 22, 2013
|
250,000 | 59,100 | 309,100 | 250,000 | 29,400 | 279,400 | ||||||||||||||||||
|
Investment Note issued on March 12, 2013
|
750,000 | 172,600 | 922,600 | 750,000 | 84,100 | 834,100 | ||||||||||||||||||
| 3,272,600 | 873,500 | 4,146,100 | 3,272,600 | 475,400 | 3,748,000 | |||||||||||||||||||
|
Convertible promissory note issued on July 26, 2013
|
250,000 | 46,200 | 296,200 | 250,000 | 17,700 | 267,700 | ||||||||||||||||||
|
Total Senior notes
|
3,522,600 | 919,700 | 4,442,300 | 3,522,600 | 493,100 | 4,015,700 | ||||||||||||||||||
|
Aggregate note discount
|
- | - | - | (2,085,900 | ) | - | (2,085,900 | ) | ||||||||||||||||
|
Net Senior notes
|
3,522,600 | 919,700 | 4,442,300 | 1,436,700 | 493,100 | 1,929,800 | ||||||||||||||||||
|
less: current portion
|
(3,272,600 | ) | (873,500 | ) | (4,146,100 | ) | - | - | - | |||||||||||||||
|
Senior notes - non-current portion and discount
|
$ | 250,000 | $ | 46,200 | $ | 296,200 | $ | 1,436,700 | $ | 493,100 | $ | 1,929,800 | ||||||||||||
|
10% Convertible Promissory Notes (Unit Notes)
|
||||||||||||||||||||||||
|
2013 Unit Notes, due 7/31/14
|
$ | - | $ | - | $ | - | $ | 1,007,500 | $ | 35,700 | $ | 1,043,200 | ||||||||||||
|
2014 Unit Notes, including amended notes, due 3/31/15
|
4,066,900 | 270,700 | 4,337,600 | 50,000 | 200 | 50,200 | ||||||||||||||||||
| 4,066,900 | 270,700 | 4,337,600 | 1,057,500 | 35,900 | 1,093,400 | |||||||||||||||||||
|
Note discounts
|
(180,000 | ) | - | (180,000 | ) | (697,400 | ) | - | (697,400 | ) | ||||||||||||||
|
Net convertible notes (all current)
|
$ | 3,886,900 | $ | 270,700 | $ | 4,157,600 | $ | 360,100 | $ | 35,900 | $ | 396,000 | ||||||||||||
|
Notes Payable to unrelated parties:
|
||||||||||||||||||||||||
|
7.5% Notes payable to service providers for
|
||||||||||||||||||||||||
|
accounts payable converted to notes payable:
|
||||||||||||||||||||||||
|
Burr, Pilger, Mayer
|
$ | 90,400 | $ | 13,100 | $ | 103,500 | $ | 90,400 | $ | 6,800 | $ | 97,200 | ||||||||||||
|
Desjardins
|
156,300 | 24,100 | 180,400 | 178,600 | 14,100 | 192,700 | ||||||||||||||||||
|
McCarthy Tetrault
|
319,700 | 46,000 | 365,700 | 360,900 | 24,800 | 385,700 | ||||||||||||||||||
|
August 2012 Morrison & Foerster Note A
|
918,200 | 193,200 | 1,111,400 | 918,200 | 87,900 | 1,006,100 | ||||||||||||||||||
|
August 2012 Morrison & Foerster Note B (1)
|
1,379,400 | 333,100 | 1,712,500 | 1,379,400 | 195,200 | 1,574,600 | ||||||||||||||||||
|
University Health Network (1)
|
549,500 | 101,800 | 651,300 | 549,500 | 60,600 | 610,100 | ||||||||||||||||||
| 3,413,500 | 711,300 | 4,124,800 | 3,477,000 | 389,400 | 3,866,400 | |||||||||||||||||||
|
Note discount
|
(474,500 | ) | - | (474,500 | ) | (848,100 | ) | - | (848,100 | ) | ||||||||||||||
| 2,939,000 | 711,300 | 3,650,300 | 2,628,900 | 389,400 | 3,018,300 | |||||||||||||||||||
|
less: current portion
|
(3,413,500 | ) | (711,300 | ) | (4,124,800 | ) | (1,130,100 | ) | (133,600 | ) | (1,263,700 | ) | ||||||||||||
|
non-current portion and discount
|
$ | (474,500 | ) | $ | - | $ | (474,500 | ) | $ | 1,498,800 | $ | 255,800 | $ | 1,754,600 | ||||||||||
|
5.75% and 10.25% Notes payable to insurance
|
||||||||||||||||||||||||
|
premium financing company (current)
|
$ | 5,800 | $ | - | $ | 5,800 | $ | 4,900 | $ | - | $ | 4,900 | ||||||||||||
|
10% Notes payable to vendors for accounts
|
||||||||||||||||||||||||
|
payable converted to notes payable
|
$ | 378,300 | $ | 51,500 | $ | 429,800 | $ | 119,400 | $ | 34,700 | $ | 154,100 | ||||||||||||
|
less: current portion
|
(378,300 | ) | (51,500 | ) | (429,800 | ) | (119,400 | ) | (34,700 | ) | (154,100 | ) | ||||||||||||
|
non-current portion
|
$ | - | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||||
|
7.0% Note payable (August 2012)
|
$ | 58,800 | $ | 7,900 | $ | 66,700 | $ | 58,800 | $ | 3,800 | $ | 62,600 | ||||||||||||
|
less: current portion
|
(23,200 | ) | (7,900 | ) | (31,100 | ) | (15,800 | ) | (3,800 | ) | (19,600 | ) | ||||||||||||
|
7.0% Notes payable - non-current portion
|
$ | 35,600 | $ | - | $ | 35,600 | $ | 43,000 | $ | - | $ | 43,000 | ||||||||||||
|
Total notes payable to unrelated parties
|
$ | 3,856,400 | $ | 770,700 | $ | 4,627,100 | $ | 3,660,100 | $ | 427,900 | $ | 4,088,000 | ||||||||||||
|
less: current portion
|
(3,820,800 | ) | (770,700 | ) | (4,591,500 | ) | (1,270,200 | ) | (172,100 | ) | (1,442,300 | ) | ||||||||||||
|
non-current portion
|
35,600 | - | 35,600 | 2,389,900 | 255,800 | 2,645,700 | ||||||||||||||||||
|
less: discount (current at March 31, 2015)
|
- | - | - | (848,100 | ) | - | (848,100 | ) | ||||||||||||||||
|
Net non-current portion
|
$ | 35,600 | $ | - | $ | 35,600 | $ | 1,541,800 | $ | 255,800 | $ | 1,797,600 | ||||||||||||
|
Notes payable to related parties:
|
||||||||||||||||||||||||
|
October 2012 7.5% Note to Cato Holding Co.
|
$ | 293,600 | $ | 55,900 | $ | 349,500 | $ | 293,600 | $ | 30,800 | $ | 324,400 | ||||||||||||
|
October 2012 7.5% Note to Cato Research Ltd. (1)
|
1,009,000 | 204,800 | 1,213,800 | 1,009,000 | 117,300 | 1,126,300 | ||||||||||||||||||
| 1,302,600 | 260,700 | 1,563,300 | 1,302,600 | 148,100 | 1,450,700 | |||||||||||||||||||
|
Note discount
|
(54,500 | ) | - | (54,500 | ) | (103,200 | ) | - | (103,200 | ) | ||||||||||||||
|
Total notes payable to related parties
|
1,248,100 | 260,700 | 1,508,800 | 1,199,400 | 148,100 | 1,347,500 | ||||||||||||||||||
|
less: current portion
|
(1,248,100 | ) | (260,700 | ) | (1,508,800 | ) | (259,600 | ) | (30,800 | ) | (290,400 | ) | ||||||||||||
|
non-current portion and discount
|
$ | - | $ | - | $ | - | $ | 939,800 | $ | 117,300 | $ | 1,057,100 | ||||||||||||
|
____________
|
||||||||||||||||||||||||
|
(1) Note and interest payable solely in restricted shares of the Company's common stock.
|
||||||||||||||||||||||||
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Significant changes in our convertible promissory notes and other promissory notes during the fiscal years ended March 31, 2015 and 2014 are described below:
10% Convertible Notes Issued in Connection with 2014 Unit Private Placement
As described more completely in the section entitled 2014 Unit Private Placement in Note 9, Capital Stock, between late March 2014 and March 31, 2015, we issued to accredited investors 10% convertible notes (the 2014Unit Notes) in the aggregate face amount of $3,113,500, including an aggregate face amount of $1,250,000 of such notes issued to Platinum and 2014 Unit Notes in the aggregate principal amount of $50,000 issued prior to March 31, 2014, in connection with our private placement offering of Units. (See Note 16, Subsequent Events, for information regarding additional notes issued in connection with the 2014 Unit Private Placement after March 31, 2015.) The 2014 Unit Notes mature on March 31, 2015 (Maturity) and the outstanding principal of the 2014 Unit Notes and their related accrued interest (the Outstanding Balance) is convertible into shares of our common stock at a conversion price of $10.00 per share at or prior to Maturity, at the option of the investor. In addition, upon our consummation of either (i) an equity or equity-based public financing registered with the SEC, or (ii) an equity or equity-based private placement, or series of private placements, not registered with the SEC, in either case resulting in gross cash proceeds to us of at least $10.0 million prior to Maturity (a Qualified Financing), the Outstanding Balance of the 2014 Unit Notes will automatically convert into securities substantially similar to those sold in the Qualified Financing, based on the following formula: (the Outstanding Balance as of the closing of the Qualified Financing) x 1.25 / (the per security price of the securities sold in the Qualified Financing). Under certain circumstances, the holders of the 2014 Unit Notes may request payment in cash in lieu of automatic conversion into the securities of the Qualified Financing. See Note 16, Subsequent Events, regarding the conversion of the Outstanding Balance of all of the 2014 Unit Notes pursuant to a private placement financing in May 2015.
We allocated the proceeds from the sale of the units to the 2014 Unit Notes, the common stock and the warrants comprising the units based on the relative fair value of the individual securities in the unit on the date of the unit sale. Based on the short-duration of the 2014 Unit Notes and their other terms, we determined that the fair value of the 2014 Unit Notes at the date of issuance was equal to their face value. Accordingly, we recorded an initial discount attributable to each 2014 Unit Note for an amount representing the difference between the face value of the 2014 Unit Note and its allocated relative value. Additionally, the 2014 Unit Notes contain an embedded conversion feature, most of which had an intrinsic value at the issuance date, which value we treated as an additional discount attributable to such 2014 Unit Notes, subject to limitations on the absolute amount of discount attributable to each 2014 Unit Note. We recorded a corresponding credit to additional paid-in capital, an equity account, attributable to the beneficial conversion feature. We amortize the aggregate discount attributable to the 2014 Unit Notes using the effective interest method over the respective term of each 2014 Unit Note. Because the discount on a 2014 Unit Note may be as great as 99% of its initial face value, and because we must amortize such discount over the period from issuance to maturity, which has generally been less than one year, or in the case of such notes issued after December 31, 2014, less than one calendar quarter, the calculated effective interest rate may be very high. Based on the amounts of their respective discounts and the term between issuance and maturity, the effective interest rates attributable to the 2014 Unit Notes range from approximately 38% to over 10,000%, with the weighted average effective interest rate in excess of 3,000%. During November 2014, we repaid the $10,000 face amount of a 2014 Unit Note issued in October 2014.
Senior Secured Convertible Promissory Notes issued to Platinum
In July and August 2012, we issued to Platinum senior secured convertible promissory notes in the aggregate principal amount of $1,250,000. Each note accrued interest at the rate of 10% per annum and was due and payable on July 2, 2015. The notes were each mandatorily convertible into securities that we might have issued in an equity, equity-based, or debt financing, or series of financings, subsequent to the issuance of the notes resulting in gross proceeds to us of at least $3,000,000, excluding any additional investment by Platinum.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
On October 11, 2012, we entered into a Note Exchange and Purchase Agreement (NEPA Agreement) with Platinum in which the senior secured notes issued in July 2012 and August 2012 and the related accrued interest, were consolidated into and exchanged for a single senior secured convertible note in the amount of $1,272,577 (the Exchange Note) and Platinum agreed to purchase four additional 10% senior secured convertible promissory notes in the aggregate principal amount of $2.0 million (the Investment Notes), issuable over four separate $500,000 tranches between October 2012 and December 2012. The NEPA Agreement was subsequently amended as to timing of subsequent note purchases, but between October 2012 and March 2013, we issued and Platinum purchased an aggregate of $2.0 million of Investment Notes. We also entered into a Security Agreement with Platinum to secure repayment of all obligations due and payable under the terms of the Exchange Note and the Investment Notes.
The Exchange Note and each Investment Note (together, SeniorNotes) accrue interest at a rate of 10% per annum and, subject to certain limitations and exceptions set forth in the Senior Notes, unless voluntarily converted earlier by Platinum, will be due and payable in restricted shares of our common stock on October 11, 2015, or three years following the date of issuance, as determined by the terms of the Investment Notes. Subject to certain terms and conditions, at maturity, all principal and accrued interest under the Senior Notes will be repaid through our issuance of restricted shares of our common stock to Platinum. Subject to certain potential adjustments set forth in the Senior Notes, the number of restricted shares of common stock issuable as payment in full for each of the Notes at maturity will be calculated by dividing the outstanding Senior Note balance plus accrued interest by $10.00 per share. Prior to maturity, the outstanding principal and any accrued interest on the Exchange Note and each of the Investment Notes is convertible, in whole or in part, at Platinum’s option into shares of our common stock at a conversion price of $10.00 per share, subject to certain adjustments. Refer to Note 16, Subsequent Events, for information regarding the conversion of the Senior Notes and accrued interest into shares of Series B Preferred stock during May 2015.
As additional consideration for the purchase of the Investment Notes, we issued to Platinum warrants to purchase an aggregate of 100,000 shares of our common stock, issuable in separate tranches together with each Investment Note. We issued four warrants to Platinum between October 2012 and March 2013 (each an Investment Warrant) to purchase an aggregate of 100,000 shares of our restricted common stock. Additionally, we issued Platinum a warrant to purchase 63,629 shares of our common stock in connection with the issuance of the Exchange Note (Exchange Warrant). At issuance, the Exchange Warrant and each Investment Warrant had a term of five-years and an exercise price of $30.00 per share, subject to certain adjustments. Effective on May 24, 2013, we entered into an Amendment and Waiver agreement with Platinum pursuant to which we agreed to reduce the exercise price of the Exchange Warrant and the Investment Warrants from $30.00 per share to $10.00 per share in consideration for Platinum’s agreement to waive its rights for any increase in the number of shares of common stock issuable under the adjustment provisions of the Exchange Warrant and the Investment Warrants that would otherwise occur from certain issuances and prospective issuances of our securities, including issuances pursuant to the prospective Autilion Financing and other private placement transactions, at a price of less than $30.00 per share.
On July 26, 2013, we issued an additional senior secured convertible promissory note in the principal amount of $250,000 to Platinum (July 2013 Note). The July 2013 Note matures on July 26, 2016 and is otherwise in the same form as the earlier Investment Notes, bearing interest at 10% per annum and being payable in restricted shares of our common stock and convertible, in whole or in part, at Platinum’s option into shares of our restricted common stock at a conversion price of $10.00 per share, subject to certain adjustments. The conversion feature in the July 2013 Note constituted a beneficial conversion feature at the date of issuance. As additional consideration for the purchase of the July 2013 Note, we also issued to Platinum a five-year warrant to purchase 12,500 shares of our restricted common stock at an exercise price of $10.00 per share (July 2013 Warrant). The fair value of the July 2013 Warrant was estimated to be $11.74 per share, or $146,800, at its issuance date using a Monte Carlo simulation model and the following assumptions: market price of common stock: $15.00 per share; exercise price: $10.00 per share; risk-free interest rate: 1.36%; volatility: 96.9%; term: 5.0 years; and dividend rate 0.0%. We recorded the fair value of the July 2013 Warrant at the date of issuance as a liability and as a corresponding discount to the July 2013 Note.
The conversion option embedded in the July 2013 Note resulted in a beneficial conversion feature having intrinsic value of $100,700 at issuance. We recorded the issuance-date intrinsic value of the beneficial conversion feature as an additional component of the discount attributable to the July 2013 Note. The aggregate discount attributable to the July 2013 Note was $247,500, resulting in an issuance date carrying value of $2,500. As with the Investment Notes, we amortize the aggregate discount attributable to each note using the effective interest method over the respective term of each note. Considering the amount of the discount and the term of the note, the effective interest rate of the July 2013 Note was determined to be 159%.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Subject to limited exceptions, the Exchange Warrant, each of the Investment Warrants and the July 2013 Warrant include certain exercise price reset and anti-dilution protection features in the event that we issue other securities during the five-year term of the warrants at a price less than $10.00 per share, as amended for the Exchange Warrant and the Investment Warrants. As a result of these provisions, the Exchange Warrant, the Investment Warrants and the July 2013 Warrant do not meet the criteria set forth in ASC 815, Derivatives and Hedging, to be treated as equity instruments. Consequently, we initially recorded the Exchange Warrant, each of the Investment Warrants and the July 2013 Warrant as liabilities at their fair value and adjust them at the end of each reporting period. (See Note 4, Fair Value Measurements.)
Note Conversion and Warrant Amendment Agreement with Platinum
On July 18, 2014, we entered into an Amended and Restated Note Conversion Agreement and Warrant Amendment with Platinum (Amendment), pursuant to which Platinum agreed to convert into our unregistered equity securities all Senior Secured Convertible Promissory Notes (Senior Notes) held by Platinum, including accrued but unpaid interest thereon (Outstanding Balance), in the aggregate amount of approximately $4.2 million at the date of the agreement, upon our consummation on or before August 31, 2014 (Closing Date), of either (i) a private equity financing resulting in aggregate gross proceeds of at least $36.0 million (Private Financing), or (ii) a public offering of our equity securities registered with the SEC resulting in gross proceeds of at least $10.0 million (Public Offering) (the Private Financing and Public Offering are referred to in this discussion as a Platinum Qualified Financing). In August and September 2014, we amended the Amendment to extend the Closing Date until October 31, 2014. Upon consummation of a Private Financing, the Senior Notes would have converted into that number of unregistered shares of our common stock equal to the Outstanding Balance on the Closing Date, divided by $10.00 per share. Upon consummation of a Public Offering, the Senior Notes would have converted into shares of a new class of Series B Convertible Preferred Stock (Series B Preferred) at the lower of $10.00 per share or the lowest per-share price in the Public Offering.
Additionally, pursuant to the terms and conditions of the Amendment, in the event we had consummated a Platinum Qualified Financing on or before the Closing Date, the exercise price of the Platinum Warrants we have issued to Platinum in connection with the Senior Notes, and warrants that we may still issue pursuant to the Note Exchange and Purchase Agreement between us and Platinum, dated October 11, 2012 (NEPA), if any, would have been fixed at the lower of $10.00 per share or the purchase price of common stock sold in the Platinum Qualified Financing. Finally, the anti-dilutive provisions contained in the Platinum Warrants, other than typical adjustments for stock splits, combinations and dividends, would have been terminated as of the Closing Date.
Through March 31, 2015, we did not request Platinum to extend the Closing Date of the Amendment or enter into any other agreement with Platinum regarding the conversion of the Senior Notes, fixing the exercise price of the Platinum Warrants or terminating the anti-dilution provisions of the Platinum Warrants. Refer to Note 16, Subsequent Events, regarding the conversion of the Senior Notes and accrued interest into shares of Series B Preferred stock and modifications made to the exercise price and anti-dilution provisions of the Platinum Warrants in May 2015.
At the execution of the Amendment, we determined that the Amendment resulted in a modification of the Senior Notes that should be accounted for as an extinguishment of debt. Considering, among other factors, the cash flows and conversion features of the Senior Notes as modified by the Amendment, market interest rates for debt of similar quality and the relative probabilities of conversion of the Senior Notes into either shares of our common stock or Series B Preferred upon consummation of a Qualified Financing, we determined that the fair values of the Senior Notes at July 18, 2014, aggregating $6,475,000, represented a substantial premium over their aggregate $4,138,700 face values plus accrued interest. In accordance with the provisions of ASC 470-20, Debt with Conversion and Other Options, we recognized the premium in excess of the face value and accrued interest, $2,336,300, as a non-cash component of loss on extinguishment of debt in the accompanying Condensed Consolidated Statements of Operations and Comprehensive (Loss) Income with a credit to additional paid-in capital, an equity account. Consequently, we recorded the liability for the Senior Notes at their face values plus accrued interest. We recognized the difference between the pre-modification carrying values of the notes and their face values, an aggregate of $1,983,700, as an additional non-cash charge to loss on extinguishment of debt in the accompanying Condensed Consolidated Statement of Operations and Comprehensive (Loss) Income. Certain of the Senior Notes contained a beneficial conversion feature at the time they were originally issued. We have accounted for the repurchase of the beneficial conversion feature at the time of the modification, an aggregate of $2,716,600, as a reduction to the loss on extinguishment of debt in the accompanying Condensed Consolidated Statements of Operations and Comprehensive (Loss) Income, with a corresponding reduction to additional paid-in capital. The net amount of the loss on extinguishment of debt related to the amendment of the Senior Notes recognized in the accompanying Condensed Consolidated Statements of Operations and Comprehensive (Loss) Income for the nine months ended December 31, 2014 is $1,603,400.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
At the date of its issuance, we recorded the fair value of the Exchange Warrant as a liability and as a corresponding charge to loss on early extinguishment of debt. We recorded the fair value of each Investment Warrant and the July 2013 Warrant at the date of issuance as a liability and as a corresponding discount to the related note. Subject to limitations of the absolute amount of discount attributable to each Investment Note and the July 2013 Note, we treated the issuance-date intrinsic value of the beneficial conversion feature embedded in each Investment Note and the July 2013 Note as an additional component of the discount attributable to such note and recorded a corresponding discount attributable to the beneficial conversion feature for each note. Prior to the Amendment, at which time the remaining unamortized discount was written off in our determination of the loss on extinguishment of debt, we had amortized the aggregate discount attributable to each of the Investment Notes and the July 2013 Note using the effective interest method over the respective term of each note. The effective interest rate of the Exchange Note was 10.0% at inception and the effective interest rate at inception for each of the Investment Notes and the July 2013 Note was 159.1% .
We re-measure the fair value of the Exchange Warrant, the Investment Warrants and the July 2013 Warrant (collectively, the Platinum Warrants) at each quarterly reporting period. As of March 31, 2015 and 2014, we measured the fair value of such warrants at an aggregate of $810,100 and $915,300, respectively. The aggregate decrease in fair value between March 31, 2014 and March 31, 2015 of $105,200 and the aggregate decrease in fair value between March 31, 2013 and March 31, 2014 of $1,219,500 is reflected in the Change in Warrant Liability in the accompanying Consolidated Statement of Operations and Comprehensive Income for the years ended March 31, 2015 and 2014, respectively.
10% Convertible Notes Issued in Connection with 2013 Unit Private Placement
As described more completely in the section entitled 2013 Unit Private Placement in Note 9, Capital Stock, between August 2013 and March 2014, we issued to accredited investors 10% convertible promissory notes (the “2013Unit Notes”) in an aggregate face amount of $1,007,500 in connection with our private placement of Units. The maturity date of the 2013 Unit Notes was July 30, 2014 and each 2013 Unit Note and its related accrued interest was convertible into shares of our common stock at a fixed conversion price of $10.00 per share at or prior to maturity, at the option of the accredited investor.
We allocated the proceeds from the sale of the units to the 2013 Unit Notes, the common stock and the warrants comprising the Units based on the relative fair value of the individual securities in each Unit on the dates of the Unit sales. Based on the short-duration of the 2013 Unit Notes and their other terms, we determined that the fair value of the 2013 Unit Notes at the date of issuance was equal to their face value. Accordingly, we recorded an initial discount attributable to each 2013 Unit Note for an amount representing the difference between the face value of the 2013 Unit Note and its relative value. Additionally, the 2013 Unit Notes contain an embedded conversion feature, certain of which had an intrinsic value at the issuance date, which value we treated as an additional discount attributable to such 2013 Unit Notes, subject to limitations on the absolute amount of discount attributable to each 2013 Unit Note. We recorded a corresponding credit to additional paid-in capital, an equity account in the Consolidated Balance Sheet, attributable to the beneficial conversion feature. We amortized the aggregate discount attributable to each of the 2013 Unit Notes using the effective interest method over the respective term of each 2013 Unit Note. Based on their respective discounts, the weighted average effective interest rate attributable to the 2013 Unit Notes at issuance was 464.1%.
Amendment of 2013 Unit Notes and Warrants
Effective May 31, 2014, we entered into note and warrant amendment agreements with substantially all holders of our 2013 Unit Notes and 2013 Unit Warrants, each of whom agreed to (i) modify certain terms of their 2013 Unit Note to conform to the corresponding terms of the 2014 Unit Notes, including an extension of the maturity date of their 2013 Unit Note from July 30, 2014 to March 31, 2015, as well as adoption of the automatic conversion and 25% conversion premium features related to consummation of a Qualified Financing, as described previously (Amended 2013 Unit Notes), and (ii) modify certain terms of their 2013 Unit Warrants, including the exercise price and expiration date, to conform to the corresponding terms of the 2014 Unit Warrants (Amended 2013 Unit Warrants). Holders of 2013 Unit Notes having an aggregate initial face amount of $895,000 agreed to such amendments. The maturity date of 2013 Unit Notes payable to holders who did not agree to amend their 2013 Unit Note and 2013 Unit Warrant remained July 30, 2014 and the $20.00 per share exercise price and July 30, 2016 expiration date of the 2013 Unit Warrants held by such holders remains unchanged. Between April 1, 2014 and August 15, 2014, we repaid 2013 Unit Notes having an initial face value of $ 112,500 and since the later date, no un-amended 2013 Unit Notes remain outstanding.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
We determined that the modification of the 2013 Unit Notes and the 2013 Unit Warrants should be accounted for as an extinguishment of debt. Considering the cash flows and the non-contingent and contingent beneficial conversion features of the Amended 2013 Notes and other factors, including market interest rates for unsecured debt of similar quality and the probability of their conversion to securities in a Qualified Financing, we determined that the fair values of the Amended 2013 Unit Notes, aggregating $1,394,000, represented a substantial premium over their aggregate $943,400 face values. In accordance with the provisions of ASC 470-20, Debt with Conversion and Other Options, we recognized the premium in excess of the face value, $450,600, as a credit to additional paid-in capital, an equity account. Consequently, we recorded the liability for the Amended 2013 Unit Notes at their face values. We recognized the difference between the pre-modification carrying values of the notes and their fair values, an aggregate of $867,500, as a non-cash charge to loss on extinguishment of debt in the accompanying Condensed Consolidated Statement of Operations and Comprehensive Loss. As described in greater detail in Note 9, Capital Stock, we determined the incremental fair value of the Amended 2013 Unit Warrants, which are treated as equity instruments, to be $272,900. We recognized this incremental fair value as an additional component of loss on extinguishment of debt in the accompanying Condensed Consolidated Statements of Operations and Comprehensive Loss and as a credit to additional paid-in capital. Certain of the 2013 Unit Notes contained a beneficial conversion feature when they were originally issued. We have accounted for the repurchase of the beneficial conversion feature at the time of the modification, an aggregate of $614,200, as a reduction to the loss on extinguishment of debt in the accompanying Condensed Consolidated Statements of Operations and Comprehensive Loss, with a corresponding reduction to additional paid-in capital. The net amount of the loss on extinguishment of debt related to the Amended 2013 Unit Notes and Amended 2013 Unit Warrants recognized in the accompanying Condensed Consolidated Statements of Operations and Comprehensive Loss is $526,200. Since the Amended 2013 Unit Notes have the same features and maturity as the 2014 Unit Notes, the two sets of notes are aggregated in the summary table above.
Issuance of Securities in Satisfaction of Technology License and Maintenance Fees and Patent Expenses
In April 2014, we entered into an agreement with Icahn School of Medicine at Mount Sinai (Icahn School), one of our long-term stem cell technology licensors, pursuant to which we issued (i) a 10% promissory note in the face amount of $300,000 due on the earlier of December 31, 2014, or the completion of a qualified financing, as defined, (ii) 15,000 restricted shares of our common stock and (iii) a warrant exercisable through March 31, 2019 to purchase 15,000 restricted shares of our common stock at an exercise price of $10.00 per share to Icahn School in satisfaction of $288,400 of stem cell technology license maintenance fees and reimbursable patent prosecution costs (the Agreement). Based on the short-duration of the note, its interest rate and other terms, we determined that the fair value of the note at the date of issuance was equal to its face value. We determined the fair value of stock to be $141,000, based on the $9.40 per share quoted market price of our common stock on the date of the agreement. We calculated the fair value of the warrant to be $5.95 per share, or $89,200, using the Black Scholes Option Pricing Model and the following assumptions: market price per share: $9.40; exercise price per share: $10.00; risk-free interest rate: 1.59%; contractual term: 5.0 years; volatility: 80.3%; expected dividend rate: 0%. We recognized a loss on extinguishment of debt in the amount of $241,800 related to this settlement in the accompanying Statement of Operations and Comprehensive Loss. Under the terms of the Agreement, an additional $35,800 of license maintenance fees and reimbursable patent prosecution costs was added to the principal amount of the promissory note through December 31, 2014. We made a payment of $10,000 on the note in January 2015 and it remained outstanding at March 31, 2015.
Notes Payable to Morrison & Foerster
On May 5, 2011, we amended a previously outstanding note (the Original Note) issued to Morrison & Foerster LLP (Morrison & Foerster), then our general corporate and intellectual property counsel, in payment of legal services (the Amended Note). Under the terms of Amended Note, the principal balance of the Original Note was increased to $2,200,000, interest accrued at the rate of 7.5% per annum, and we were required to make certain payments to Morrison & Foerster.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
On August 31, 2012, we restructured the Amended Note (the Restructuring Agreement). Pursuant to the Restructuring Agreement, we issued to Morrison & Foerster two new unsecured promissory notes to replace the Amended Note, one in the principal amount of $1,000,000 (Replacement Note A) and the other in the principal amount of $1,379,400 (Replacement Note B) (together, the Replacement Notes); amended an outstanding warrant to purchase restricted shares of our common stock (the Amended M&F Warrant); and issued a new warrant exercisable at $20.00 per share through September 15, 2017 to purchase restricted shares of our common stock (the New M&F Warrant). Under the terms of the Restructuring Agreement, the Amended Note was cancelled and all of our past due payment obligations under the Amended Note were satisfied. Pursuant to the terms of the Amended Note, we made a payment of $155,000 to Morrison & Foerster on August 31, 2012 and issued the Replacement Notes, each dated as of August 31, 2012. Both Replacement Notes accrue interest at the rate of 7.5% per annum and are due and payable on March 31, 2016. Replacement Note A required monthly payments of $15,000 per month through March 31, 2013, and requires $25,000 per month thereafter until maturity. Between May 2013 and March 31, 2015, we have made no payments on Replacement Note A. In accordance with the terms of the Replacement Notes, the applicable penalty interest rate of 10.0% per annum has become effective and we have accrued interest on the Replacement Notes at that rate since May 2013. Payment of the principal and interest on Replacement Note B will be made solely in restricted shares of our common stock pursuant to Morrison & Foerster’s cancellation from time to time of all or a portion of the principal and interest balance due on Replacement Note B in connection with its concurrent exercise of the New M&F Warrant, at an exercise price of $20.00 per share; provided, however, that Morrison & Foerster shall have the option to require payment of Replacement Note B in cash upon the occurrence of a change in control of the Company or an event of default, and only in such circumstances. As indicated in Note 16, Subsequent Events, in May 2015, Replacement Note B in the aggregate amount (principal and accrued but unpaid interest) of approximately $1.8 million was converted into 257,143 shares of our newly created Series B Preferred, eliminating approximately $1.8 million of our indebtedness. Additionally, Morrison & Foerster agreed to standstill and forbear until December 31, 2016 with respect to further payment requirements on Replacement Note A and accounts payable outstanding at the time of the agreement.
The New M&F Warrant is exercisable for the number of restricted shares of our common stock equal to the principal and accrued interest due under the terms of Replacement Note B divided by the warrant exercise price of $20.00 per share. At the August 31, 2012 date of grant, the New M&F Warrant was exercisable to purchase 68,969 restricted shares of our common stock. The New M&F Warrant effectively permits exercise only by the cancellation in whole or in part of our indebtedness under either of the Replacement Notes. Through March 31, 2015, we have adjusted the New M&F Warrant to increase the number of restricted shares available for purchase by 16,658 shares, based on interest accrued on Replacement Note B through that date. We have recorded the fair value of the additional warrant shares as a charge to interest expense and a corresponding credit to additional paid-in capital.
Note Payable to Cato Research Ltd.
On October 10, 2012, we issued to Cato Research Ltd (CRL) a contract research and development partner and a related party: (i) an unsecured promissory note in the initial principal amount of $1,009,000, which is payable solely in restricted shares of our common stock and which accrues interest at the rate of 7.5% per annum, compounded monthly (the CRL Note), as payment in full for all contract research and development services and regulatory advice (CRO Services) rendered by CRL to us and our affiliates through December 31, 2012 with respect to the non-clinical and clinical development of AV-101, and (ii) a five-year warrant to purchase, at a price of $20.00 per share, 50,450 restricted shares of our common stock, the amount equal to the sum of the principal amount of the CRL Note, plus all accrued interest thereon, divided by $20.00 per share (the CRL Warrant). The CRL Note is due and payable on March 31, 2016 and is payable solely by CRL's cancellation from time to time of all or a portion of the principal and interest balance due on the CRL Note in connection with its concurrent exercise of the CRL Warrant, provided, however, that CRL will have the option to require payment of the CRL Note in cash upon the occurrence of a change in control of the Company or an event of default, and only in such circumstances. Through March 31, 2015, we have adjusted the CRL Warrant to increase the number of restricted shares available for purchase by 10,241 shares, based on interest accrued on the CRL Note through that date. We have recorded the fair value of the additional shares as a charge to interest expense and a corresponding credit to additional paid-in capital.
As disclosed in Note 16, Subsequent Events, in May 2015, the CRL Note and certain accounts payable to CRL for CRO Services related to AV-101 in the aggregate amount (principal and accrued but unpaid interest, plus certain strategic adjustments) of approximately $1.9 million were converted into 278,006 shares of our Series B Preferred, eliminating approximately $1.9 million of our indebtedness.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note Payable to Cato Holding Company
On October 10, 2012, we exchanged a previously outstanding note issued to Cato Holding Company (CHC) for a new unsecured promissory note in the principal amount of $310,400 (the 2012 CHC Note) and a five-year warrant to purchase 12,500 restricted shares of the Company’s common stock at a price of $30.00 per share (the CHC Warrant). The 2012 CHC Note accrues interest at a rate of 7.5% per annum and is due and payable in monthly installments of $10,000, beginning November 1, 2012 and continuing until the outstanding balance is paid in full. Between December 2012 and March 31, 2015, we have made no payments on the 2012 CHC Note.
As disclosed in Note 16, Subsequent Events, in May 2015, the 2012 CHC Note in the aggregate amount (principal and accrued but unpaid interest) of approximately $354,000 was converted into 50,565 shares of our Series B Preferred, eliminating approximately $354,000 of our indebtedness.
Note Payable to University Health Network
On October 10, 2012, we issued to the University Health Network (UHN): (i) an unsecured promissory note in the principal amount of $549,500, which is payable solely in restricted shares of our common stock and which accrues interest at the rate of 7.5% per annum, as payment in full for all sponsored stem cell research and development activities by UHN and Gordon Keller, Ph.D. under the SCRA through September 30, 2012 (the UHN Note), and (ii) a five-year warrant to purchase, at a price of $20.00 per share, 27,475 restricted shares of our common stock, the amount equal to the sum of the principal amount of the UHN Note, plus all accrued interest thereon, divided by $20.00 per share (the UHN Warrant). The UHN Note is due and payable on March 31, 2016 and is payable solely by UHN's cancellation from time to time of all or a portion of the principal and interest balance due on the UHN Note in connection with its concurrent exercise of the UHN Warrant, provided, however, that UHN will have the option to require payment of the UHN Note in cash upon the occurrence of a change in control of the Company or an event of default, and only in such circumstances. Through March 31, 2015, we have adjusted the UHN Warrant to increase the number of restricted shares available for purchase by 5,095 shares, based on interest accrued on the UHN Note through that date. We have recorded the fair value of the additional shares as a charge to interest expense and a corresponding credit to additional paid-in capital.
As disclosed in Note 16, Subsequent Events, in May 2015, the UHN Note in the aggregate amount (principal and accrued but unpaid interest) of approximately $656,400 was converted into 93,775 shares of our Series B Preferred, eliminating approximately $656,400 of our indebtedness.
Notes Payable for Cancellation of Amounts Payable for Services and Royalties
On February 25, 2011, we issued to Burr, Pilger, and Mayer, LLC (BPM) an unsecured promissory note in the principal amount of $98,674 for amounts payable in connection with valuation services provided to us by BPM. The BPM note bears interest at the rate of 7.5% per annum and has payment terms of $1,000 per month, beginning March 1, 2011 and continuing until all principal and interest is paid in full. In addition, a payment of $25,000 shall be due upon the sale of the Company or upon our completing a financing transaction of at least $5.0 million during any three-month period, with the payment increasing to $50,000 (or the amount then owed under the note, if less) upon the Company completing a financing of over $10.0 million. We made no payments on the BPM note during the fiscal year ended March 31, 2015.
On April 29, 2011, we issued to Desjardins Securities, Inc. (Desjardins) an unsecured promissory note in the principal amount of CDN $236,000 for amounts payable for legal fees incurred by Desjardins in connection with investment banking services provided to us by Desjardins. The Desjardins note bears interest at 7.5% and was due, along with all accrued but unpaid interest on the earliest of (i) June 30, 2014, (ii) the consummation of a Change of Control, as defined in the Desjardins note, and (iii) any failure to pay principal or interest when due. We were required to make payments of CDN $4,000 per month beginning May 31, 2011, increasing to CDN $6,000 per month beginning on January 31, 2012. Beginning on January 1, 2012, we are also required to make payments equal to one-half of one percent (0.5%) of the net proceeds of all private or public equity financings closed during the term of the note. The note payable to Desjardins was due on June 30, 2014. We made no payments on the Desjardins note during the fiscal year ended March 31, 2015.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
On May 5, 2011, we issued to McCarthy Tetrault LLP (McCarthy) an unsecured promissory note in the principal amount of CDN $502,797 for amounts payable in connection with Canadian legal services provided to us. The McCarthy note bears interest at 7.5% and was due, along with all accrued but unpaid interest on the earliest of (i) June 30, 2014, (ii) the consummation of a Change of Control, as defined in the McCarthy note, and (iii) any failure to pay principal or interest when due. We were required to make payments of CDN $10,000 per month beginning May 31, 2011, which payment amounts increased to CDN $15,000 per month on January 31, 2012. Beginning on January 1, 2012, we are also required to make payments equal to one percent (1%) of the net proceeds of all private or public equity financings closed during the term of the note. The note payable to McCarthy was due on June 14, 2014. On June 11, 2014, we and McCarthy agreed to extend the maturity date of the McCarthy note from June 14, 2014 to the earlier of (i) September 30, 2014, (ii) consummation of a financing in which we received gross cash proceeds of at least $15.0 million, or (iii) consummation of a change of control of the Company, as defined in the McCarthy note. McCarthy also agreed to forbear with respect to the requirement that we make monthly payments on the note from the date of the agreement until maturity and granted us a waiver with respect to previously missed monthly payments. We made no payments on the McCarthy note during the fiscal year ended March 31, 2015. As disclosed in Note 16, Subsequent Events, in May 2015, the McCarthy note in the aggregate amount (principal and accrued but unpaid interest) of approximately $414,600 was converted into 59.230 shares of our Series B Preferred, eliminating approximately $414,600 of our indebtedness.
On August 30, 2012, we issued a promissory note in the principal amount of $60,000 and 750 restricted shares of our common stock valued at a market price of $18.80 per share to Progressive Medical Research in settlement of past due obligations for clinical research services in the amount of $79,900. Under the terms of the settlement, we also agreed to make monthly cash payments of $5,000 in August 2012 through December 2012. The promissory note bears interest at 7% per annum and requires payments of $1,000 per month beginning January 15, 2013 until all principal and interest is paid in full. The note requires payment in full upon the sale of all or substantially all of our assets or upon our completion of a financing transaction, or series of transactions, resulting in gross proceeds to us of at least $4.0 million in any three-month period, excluding proceeds from stock option or warrant exercises. We made no payments on this note during the fiscal year ended March 31, 2015.
On October 12, 2009, VistaGen California issued a promissory note payable to the Regents of the University of California (“UC”) with a principal balance of $90,000 in exchange for the cancellation of certain amounts payable under a research collaboration agreement (the UC Note 1).. On February 25, 2010, VistaGen California issued a promissory note payable to UC having a principal balance of $170,000 in exchange for the cancellation of the remaining $60,000 principal balance of UC Note 1 and certain amounts payable under a research collaboration agreement (UC Note 2). UC Note 2 was payable in monthly principal installments of $15,000 through May 31, 2010, with the remaining $125,000 plus all accrued and unpaid interest due on or before June 30, 2010. Between June 2010 and December 2010, VistaGen California amended UC Note 2 multiple times, ultimately resulting in a decrease in the monthly payment amount to $5,000, with payments continuing until the outstanding balance of principal and interest is paid in full. We made no payments on UC Note 2 during the fiscal year ended March 31, 2015.
On March 1, 2010, VistaGen California issued a 10% promissory note with a principal balance of $75,000 to National Jewish Health in exchange for the cancellation of certain amounts payable for accrued royalties. The principal balance plus all accrued and unpaid interest was initially due on or before December 31, 2010 (March 2010 Note). On December 28, 2010, VistaGen California amended the March 2010 Note and extended its maturity date to the first to occur of April 30, 2011 or 30 days following the closing of a financing with gross proceeds of $5,000,000 or more. We made no payments on this note during the fiscal year ended March 31, 2015.
On August 13, 2010, VistaGen California issued a 10% promissory note with a principal balance of $41,000 to MicroConstants, Inc. in exchange for the cancellation of certain amounts payable for services rendered. Under the terms of this note, VistaGen California is to make payments of $1,000 per month with any unpaid principal or accrued interest due and payable upon the first to occur of (i) August 1, 2013, (ii) the issuance and sale of equity securities whereby the Company raises at least $5,000,000 or (iii) the sale or acquisition of all or substantially all of our stock or assets. We made no payments on this note during the fiscal year ended March 31, 2015.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
9. Capital Stock
Reverse Split (Stock Consolidation) of our Common Stock
As indicated in Note 2, Basis of Presentation and Going Concern, we consummated the Stock Consolidation, a 1-for-20 reverse split of our authorized, and issued and outstanding shares of common stock, effective on August 14, 2014. The par value of our common stock remained unchanged at $0.001 per share following the Stock Consolidation. The Stock Consolidation was approved by the Financial Industry Regulatory Authority (FINRA) on August 13, 2014, and became effective on the OTCQB at the opening of trading on August 14, 2014. Each reference to shares of common stock or the price per share of common stock in these financial statements is post-Stock Consolidation, and reflects the 1-for-20 adjustment as a result of the Stock Consolidation.
Series A Preferred Stock
In December 2011, our Board of Directors authorized the creation of a series of up to 500,000 shares of Series A Preferred, par value $0.001 (Series A Preferred). Each restricted share of Series A Preferred was initially convertible at the option of the holder into one-half of one restricted share of our common stock. The Series A Preferred ranks prior to the common stock for purposes of liquidation preference.
The Series A Preferred has no separate dividend rights, however, whenever the Board of Directors declares a dividend on the common stock, each holder of record of a share of Series A Preferred shall be entitled to receive an amount equal to such dividend declared on one share of common stock multiplied by the number of shares of common stock into which such share of Series A Preferred could be converted on the Record Date.
Except with respect to transactions upon which the Series A Preferred shall be entitled to vote separately as a class, the Series A Preferred has no voting rights. The restricted common stock into which the Series A Preferred is convertible shall, upon issuance, have all of the same voting rights as other issued and outstanding shares of our common stock.
In the event of the liquidation, dissolution or winding up of the affairs of the Company, after payment or provision for payment of our debts and other liabilities, the holders of Series A Preferred then outstanding shall be entitled to receive an amount per share of Series A Preferred calculated by taking the total amount available for distribution to holders of all of our outstanding common stock before deduction of any preference payments for the Series A Preferred, divided by the total of (x), all of the then outstanding shares of our common stock, plus (y) all of the shares of our common stock into which all of the outstanding shares of the Series A Preferred can be converted before any payment shall be made or any assets distributed to the holders of the common stock or any other junior stock.
At March 31, 2015 and 2014, there were 500,000 restricted shares of Series A Preferred outstanding, all issued to Platinum or its affiliates. Platinum acquired the Series A Preferred pursuant to the transactions described below. In October 2012, Platinum’s exchange rights with respect to the Series A Preferred were modified as described in the section entitled Modification of Series A Preferred Exchange Rights and Deemed Dividend, below.
|
·
|
December 2011 Common Stock Exchange Agreement with Platinum
On December 22, 2011, we entered into a Common Stock Exchange Agreement (the Exchange Agreement") with Platinum, pursuant to which Platinum converted 24,200 restricted shares of our common stock into 45,980 restricted shares of Series A Preferred (the Exchange). Each restricted share of Series A Preferred issued to Platinum was initially convertible into ten restricted shares of our common stock. At the time of the Exchange, we determined the fair value of the common stock subject to the Exchange to be $31.00 per share and have reflected the 24,200 restricted common shares as treasury stock on that basis in the accompanying Consolidated Balance Sheet at March 31, 2015 and 2014.
|
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
·
|
December 2011 Note and Warrant Exchange Agreement with Platinum
On December 29, 2011, we entered into a Note and Warrant Exchange Agreement with Platinum pursuant to which a promissory note in the face amount of $4,000,000 plus accrued interest and all outstanding warrants issued to Platinum to purchase an aggregate of 79,993 restricted shares of our common stock were cancelled in exchange for 391,075 restricted shares of Series A Preferred. Each share of Series A Preferred was initially convertible into one-half of one share of our common stock. We issued 231,090 restricted shares of Series A Preferred to Platinum in connection with the note cancellation based on the sum of the $4,000,000 principal balance of the note plus accrued but unpaid interest through May 11, 2011 adjusted for a 125% conversion premium, net of the $1,719,800 aggregate exercise price of the 79,993 outstanding warrants held by Platinum, and a contractual conversion basis of $35.00 per common share, all adjusted for the initial 1:10 Series A Preferred to common exchange ratio. An additional 159,985 restricted shares of Series A Preferred were issued to Platinum in connection with the warrant exercise and exchange to acquire the common shares issued upon the warrant exercise.
|
|
·
|
2012 Exchange Agreement with Platinum
On June 29, 2012, we entered into an Exchange Agreement (the 2012 Platinum Exchange Agreement) with Platinum pursuant to which we issued Platinum 62,945 restricted shares of Series A Preferred in exchange for 31,473 restricted shares of our common stock then owned by Platinum, in consideration for Platinum’s agreement to purchase from us a senior secured convertible promissory note in the face amount of $500,000. We estimated the fair value of the Series A Preferred shares tendered to Platinum under the terms of the 2012 Platinum Exchange Agreement at $736,400 ($23.40 per share on a common share equivalent basis). The common shares exchanged for shares of Series A Preferred are treated as treasury stock on that basis in the accompanying Consolidated Balance Sheet at March 31, 2015 and 2014.
|
Modification of Series A Preferred Exchange Right
Pursuant to the October 2012 Agreement described more completely in Note 8, Convertible Promissory Notes and Other Notes Payable, Platinum’s exchange rights in the Series A Preferred were modified such that Platinum now has the right and option to exchange the 500,000 restricted shares of our Series A Preferred that it holds for (i) a total of 750,000 restricted shares of our common stock, and (ii) a five-year warrant to purchase 350,000 restricted shares of our common stock, originally at an exercise price of $30.00 per share (Series A Exchange Warrant). The Series A Exchange Warrant, when issued, will have the same features, including exercise price and anti-dilution provisions as the Exchange Warrant and the Investment Warrants issued to Platinum between October 2012 and July 2013 (collectively, the Platinum Warrants) and accordingly, has been treated as a component of Warrant Liability in our Consolidated Balance Sheets since the effective date of the October 2012 Agreement. Effective on May 24, 2013, we entered into an Amendment and Waiver Agreement (the Amendment and Waiver) with Platinum pursuant to which we agreed to reduce the exercise price of the Platinum Warrants and the Series A Exchange Warrant from $30.00 per share to $10.00 per share in consideration for Platinum’s agreement to waive certain of its rights under the anti-dilution provisions of the Platinum Warrants and the Series A Exchange Warrant. See the section entitled Modification of Platinum Warrants, later in this note, for a more complete description of the Amendment and Waiver.
Similarly to the Platinum Warrants, we remeasure the fair value of the Series A Exchange Warrant at each quarterly reporting period and reflect the change in its fair value as a component of the Change in Warrant Liability in the Consolidated Statement of Operations. The fair value of the Series A Exchange Warrant was determined to be $2,198,400 and $2,058,600 as of March 31, 2015 and 2014, respectively, and the $139,800 increase in fair value since March 31, 2014 is reflected as a component of the Change in Warrant Liability in the accompanying Consolidated Statement of Operations and Comprehensive Loss for the fiscal year ended March 31, 2015.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
As described in Note 16, Subsequent Events, during May 2015, we entered into an agreement with Platinum pursuant to which Platinum agreed to amend the Platinum Warrants and the Series A Exchange Warrant to fix the exercise price thereof at $7.00 per share and eliminate the exercise price reset features and fix the number of shares of our common stock issuable thereunder. This amendment will result in the elimination of the warrant liability with respect to these warrants during the first quarter of our fiscal year ended March 31, 2016.
Creation of Series B Preferred Stock
On July 17, 2014, our Board of Directors authorized the creation of a class of Series B Preferred Stock (Series B Preferred) to provide for the potential conversion of the Senior Secured Convertible Promissory Notes held by Platinum totaling approximately $4.3 million in principal and accrued interest at December 31, 2014 (Outstanding Balance) into Series B Preferred. At March 31, 2015, we had not yet filed a certificate of designation with the Nevada Secretary of State to amend our Articles of Incorporation to formally establish the Series B Preferred. Refer to Note 16, Subsequent Events, for information regarding the filing of a Certificate of Designation for the Series B Preferred with the Nevada Secretary of State and the issuance of Series B Preferred in May and June 2015.
2014 Unit Private Placement
Between late-March 2014 and March 31, 2015, we entered into securities purchase agreements with accredited investors, including Platinum, pursuant to which we sold units to such accredited investors in private placement transactions (2014 Units), for aggregate cash proceeds of $3,113,500, consisting of (i) 2014 Unit Notes in the aggregate face amount of $3,113,500 which are due on March 31, 2015 or automatically convertible into securities we may issue upon the consummation of a Qualified Financing, defined as (a) an equity-based public financing registered with the SEC, or (b) a private equity-based financing or series of private equity-based financings, in either case in which we receive at least $10 million in gross cash proceeds prior to March 31, 2015; (ii) an aggregate of 282,850 restricted shares of our common stock (2014 Unit Stock); and (iii) warrants exercisable through December 31, 2016 to purchase an aggregate of 282,850 restricted shares of our common stock at an exercise price of $10.00 per share (2014 Unit Warrants). We sold $1,250,000 of such Units to Platinum, issuing 2014 Unit Notes in the face amount of $1,250,000; 125,000 restricted shares of 2014 Unit Stock and 2014 Unit Warrants to purchase 125,000 shares of our common stock to Platinum. The Outstanding Balance of each 2014 Unit Notes is convertible into shares of our common stock at a conversion price of $10.00 per share at or prior to maturity, at the option of each investor. In addition, however, the Outstanding Balance is automatically convertible into securities substantially similar to those we may issue in a Qualified Financing at an amount determined by multiplying the Outstanding Balance by 1.25, and dividing the resulting number by the price per share of securities offered in the Qualified Financing. Under certain circumstances, the holders of the 2014 Unit Notes may request payment in cash in lieu of automatic conversion into the securities of the Qualified Financing. We sold $50,000 of 2014 Units prior to March 31, 2014, which Units are reflected in the figures above. See Note 16, Subsequent Events, for information regarding additional sales of 2014 Unit Notes in April and May 2015 and the conversion of the 2014 Unit Notes pursuant to a private placement financing in May 2015.
We allocated the proceeds from the sale of the 2014 Units to the various securities based on their relative fair values on the dates of the sales. As described in Note 8, Convertible PromissoryNotes and Other Notes Payable, based on the short-term nature of the Unit Notes, we determined that fair value of the 2014 Unit Notes was equal to their face value. We determined the fair value of the 2014 Unit Stock based on the quoted market price of our common stock on the date of the 2014 Unit sale. We calculated the fair value of the 2014 Unit Warrants using the Black Scholes Option Pricing Model and the weighted average assumptions indicated in the table below. The table below also presents the aggregate allocation of the 2014 Unit sales proceeds based on the relative fair values of the 2014 Unit Stock, 2014 Unit Warrants and 2014 Unit Notes as of their respective 2014 Unit sales dates.
|
Unit Warrants
|
||||||||||||
|
Weighted Average Issuance Date Valuation Assumptions
|
Per Share
Fair |
Aggregate
Fair Value |
Aggregate
Proceeds |
Aggregate Allocation of Proceeds
Based on Relative Fair Value of: |
||||||||
|
Warrant
|
Risk free
|
|||||||||||
|
Shares
|
Market
|
Exercise
|
Term
|
Interest
|
Dividend
|
Value of
|
of Unit
|
of Unit
|
Unit
|
|||
|
Issued
|
Price
|
Price
|
(Years)
|
Rate
|
Volatility
|
Rate
|
Warrant
|
Warrants
|
Sales
|
Unit Stock
|
Warrant
|
Unit Note
|
|
282,850
|
$9.28
|
$10.00
|
2.17
|
0.62%
|
72.36%
|
0.0%
|
$3.63
|
$1,027,000
|
$3,133,500
|
$1,122,400
|
$454,200
|
$1,556,900
|
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
2013 Unit Private Placement
Between August 2013 and March 2014, we entered into securities purchase agreements with accredited investors pursuant to which we sold units to such accredited investors in private placement transactions (2013 Units), for aggregate cash proceeds of $1,007,500, including $50,000 in lieu of repayment of previous advances to us made by one of our executive officers. The 2013 Units consisted of (i) 10% convertible promissory notes in the aggregate face amount of $1,007,500 maturing on July 30, 2014 (2013 Unit Notes) (ii) an aggregate 100,750 restricted shares of our common stock (2013 Unit Stock); and (iii) warrants exercisable through July 30, 2016 to purchase an aggregate of 100,750 restricted shares of our common stock at an exercise price of $20.00 per share (2013 Unit Warrants). The 2013 Unit Notes and related accrued interest were convertible into restricted shares of our common stock at a conversion price of $10.00 per share at or prior to maturity, at the option of each investor.
We allocated the proceeds from the sale of the 2013 Units to the various securities in each 2013 Unit based on their relative fair value on the dates of the sales. As described in Note 8, Convertible PromissoryNotes and Other Notes Payable, based on the short-term nature of the 2013 Unit Notes, we determined that fair value of the 2013 Unit Notes was equal to their face value. We determined the fair value of the 2013 Unit Stock based on the quoted market price of our common stock on the date of the 2013 Unit sale. We calculated the fair value of the 2013 Unit Warrants using the Black Scholes Option Pricing Model and the weighted average assumptions indicated in the table below. The table below also presents the aggregate allocation of the 2013 Unit sales proceeds based on the relative fair values of the 2013 Unit Stock, 2013 Unit Warrants and 2013 Unit Notes at the respective 2013 Unit sales date.
|
2013Unit Warrants
|
||||||||||||
|
Weighted Average Issuance Date Valuation Assumptions
|
Per Share
Fair |
Aggregate
Fair Value |
Aggregate
Proceeds |
Aggregate Allocation of Proceeds
Based on Relative Fair Value of: |
||||||||
|
Warrant
|
Risk free
|
|||||||||||
|
Shares
|
Market
|
Exercise
|
Term
|
Interest
|
Dividend
|
Value of
|
of Unit
|
of Unit
|
Unit
|
|||
|
Issued
|
Price
|
Price
|
(Years)
|
Rate
|
Volatility
|
Rate
|
Warrant
|
Warrants
|
Sales
|
Unit Stock
|
Warrant
|
Unit Note
|
|
100,750
|
$9.01
|
$20.00
|
2.68
|
0.58%
|
76.29%
|
0.0%
|
$2.53
|
$254,700
|
$1,007,500
|
$415,000
|
$111,400
|
$481,100
|
Amendment of 2013 Unit Notes and 2013 Unit Warrants
As indicated in Note 8, Convertible Promissory Notes and Other Notes Payable, effective May 31, 2014, we entered into note and warrant amendment agreements with substantially all holders of 2013 Unit Notes and 2013 Unit Warrants to (i) modify certain terms of their 2013 Unit Notes, including the maturity date and certain conversion features, to conform to the corresponding terms of the 2014 Unit Notes and (ii) to modify certain terms of the 2013 Unit Warrants, including the exercise price and expiration date, to conform to the corresponding terms of the 2014 Unit Warrants. Holders of 2013 Unit Notes having an aggregate initial face amount of $895,000 and warrants to purchase an aggregate of 93,250 restricted shares of our common stock agreed to the amendments.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
We calculated the fair value of the modified 2013 Unit Warrants immediately before and after the modifications and determined that the fair value of the warrants increased by an aggregate of $272,900, which we treated as a component of loss on extinguishment of debt in the accompanying Condensed Consolidated Statements of Operations and Comprehensive Loss with a corresponding credit to additional paid-in capital, an equity account. The warrants subject to the exercise price modifications were valued using the Black-Scholes Option Pricing Model and the following assumptions:
|
Assumption:
|
Pre-modification
|
Post-modification
|
||||||
|
Market price per share
|
$
|
12.60
|
$
|
12.60
|
||||
|
Exercise price per share
|
$
|
20.00
|
$
|
10.00
|
||||
|
Risk-free interest rate
|
0.44%
|
0.62%
|
||||||
|
Remaining contractual term in years
|
2.17
|
2.59
|
||||||
|
Volatility
|
75.6%
|
76.6%
|
||||||
|
Dividend rate
|
0.0%
|
0.0%
|
||||||
|
Fair Value per share
|
$
|
3.73
|
$
|
6.65
|
||||
Issuance of Securities in Satisfaction of Technology License and Maintenance Fees and Patent Expenses
In April 2014, we entered into an agreement with Icahn School of Medicine at Mount Sinai (Icahn School), one of our long-term stem cell technology licensors, pursuant to which we issued (i) a 10% promissory note in the face amount of $300,000 due on the earlier of December 31, 2014, or the completion of a qualified financing, as defined, (ii) 15,000 restricted shares of our common stock and (iii) a warrant exercisable through March 31, 2019 to purchase 15,000 restricted shares of our common stock at an exercise price of $10.00 per share to Icahn School in satisfaction of $288,400 of stem cell technology license maintenance fees and reimbursable patent prosecution costs (the Icahn School Agreement). Based on the short-duration of the note, its interest rate and other terms, we determined that the fair value of the note at the date of issuance was equal to its face value. We determined the fair value of stock to be $141,000, based on the $9.40 per share quoted market price of our common stock on the date of the agreement. We calculated the fair value of the warrant to be $5.95 per share, or $89,200, using the Black Scholes Option Pricing Model and the following assumptions: market price per share: $9.40; exercise price per share: $10.00; risk-free interest rate: 1.59%; contractual term: 5.0 years; volatility: 80.3%; expected dividend rate: 0%. We recognized a loss on extinguishment of debt in the amount of $241,800 related to this settlement in the accompanying Statement of Operations and Comprehensive Loss. Under the terms of the Icahn School Agreement, an additional $35,800 of license maintenance fees and reimbursable patent prosecution costs were added to the principal amount of the promissory note through March 31, 2015. The note remains outstanding at March 31, 2015.
Issuance of Common Stock to Consultants
In May 2014, we entered into a consulting agreement for strategic advisory and business development services pursuant to which we issued 10,000 restricted shares of our common stock as partial compensation for such professional services. We determined the fair value of stock to be $134,000, based on the $13.40 per share quoted market price of our common stock on the date of the agreement. Additionally, under the terms of the agreement, we paid an aggregate of $80,000 between May 2014 and December 31, 2014 as additional compensation for professional services rendered by the consultant. Effective January 12, 2015, we entered into a new consulting agreement with this consultant for similar services through December 31, 2015 pursuant to which we have issued 20,000 restricted shares of our common stock valued at $160,000, based on the $8.00 per share quoted market price of our common stock on the date of the agreement, and made cash payments of $20,000 as compensation for such professional services.
In March 2015, we entered into a consulting agreement with another consultant for additional advisory and business development services pursuant to which we issued 25,000 restricted shares of our common stock as compensation for such professional services. We determined the fair value of stock to be $175,000, based on the $7.50 per share quoted market price of our common stock on the date of the agreement.
In March 2015, we issued 16,667 shares of our common stock valued at $166,700 to our legal counsel in settlement of direct legal fees related to services provided with respect to our prospective public offering of our equity securities in the fall of 2014 and the Autilion Financing. We recognized a loss of $16,700 with respect to this settlement, which is included in Loss on Extinguishment of Debt in the accompanying Statement of Operations and Comprehensive Loss for the year ended March 31, 2015.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Warrants to Purchase Common Stock
Warrant Grants and Exercises
In January 2015, when the market price of our common stock was $8.00 per share, our Board of Directors (Board) authorized the grant of fully-vested five-year warrants to purchase an aggregate of 381,000 restricted shares of our common stock at an exercise price of $10.00 per share, including an aggregate of 340,000 such shares to company officers and independent members of the Board. The Board also granted one-year warrants to purchase 5,715 restricted shares of our common stock at an exercise price of $10.00 per share to consultants whose warrants had expired at December 31, 2014. Additionally, the Board extended by one year the expiration date of outstanding warrants to purchase 90,675 shares of our restricted common stock otherwise expiring during calendar 2015 and reduced the exercise price to $15.00 per share for such of those extended term warrants having exercise prices in excess of that amount.
We valued the new warrant grants at $1,756,900 using the Black Scholes Option Pricing Model and the following assumptions: market price per share: $8.00; exercise price per share: $10.00; risk-free interest rate: 1.45% for five-year warrants and 0.24% for one-year warrants; contractual term: 5 years or 1 year; volatility: 75.86% for five-year warrants and 69.74% for one-year warrants; expected dividend rate: 0%..
We calculated the fair value of the modified warrants immediately before and after the modifications and determined that the fair value of the warrants increased by $98,400, which is reflected in general and administrative expense in the accompanying Consolidated Statements of Operations and Comprehensive Loss for the fiscal year ended March 31, 2015. The warrants subject to the exercise price modifications and term extensions were valued using the Black-Scholes Option Pricing Model and the following assumptions:
|
Assumption:
|
Pre-modification
|
Post-modification
|
||||||
|
Market price per share at modification date
|
$
|
8.00
|
$
|
8.00
|
||||
|
Exercise price per share (weighted average)
|
$
|
23.13
|
$
|
13.00
|
||||
|
Risk-free interest rate (weighted average)
|
0.04%
|
0.31%
|
||||||
|
Contractual term in years (weighted average)
|
0.24
|
1.24
|
||||||
|
Volatility (weighted average)
|
69.7%
|
69.8%
|
||||||
|
Dividend rate
|
0.0%
|
0.0%
|
||||||
|
Weighted Average Fair Value per share
|
$
|
0.22
|
$
|
1.31
|
||||
On March 19, 2014, we granted five-year warrants to purchase an aggregate of 20,750 restricted shares of our unregistered common stock at an exercise price of $10.00 per share to the independent members of our Board and certain of our officers. The warrants became exercisable for 50% of the shares on April 1, 2014, and become exercisable for an additional 25% of the shares on April 1, 2015 and 25% of the shares on April 1, 2016, provided that the warrant will become fully vested upon a change in control of the Company, as defined, or the consummation between us and a third party of a license or sale transaction involving at least one new drug rescue variant. We valued the warrants at $120,800 using the Black Scholes Option Pricing Model and the following assumptions: market price per share: $9.20; exercise price per share: $10.00; risk-free interest rate: 1.75%; contractual term: 5 years; volatility: 80.57%; expected dividend rate: 0%. We recognized stock compensation expense of $54,100 related to the grants in the fourth quarter of the fiscal year ended March 31, 2014 and $27,000 during the fiscal year ended March 31, 2015..
In October 2013, we issued new warrants to purchase an aggregate of 11,875 shares of our restricted common stock to certain former warrant holders whose warrants to purchase an equivalent number of shares of our restricted common stock at an exercise price of $30.00 per share had recently expired. We calculated the fair value of the new warrants as $0.63 per share, using the Black-Scholes Option Pricing Model and the following assumptions. market price per share: $10.00; exercise price per share: $30.00; risk-free interest rate: 0.20%; contractual term: 1.32 years; volatility: 73.5%; and expected dividend rate: 0%. We recorded the aggregate fair value of $7,400 for the new warrants in general and administrative expense in the accompanying Consolidated Statements of Operations and Comprehensive Loss for the fiscal year ended March 31, 2014.
In June 2013 and October 2013, our Chief Executive Officer partially exercised an outstanding warrant to purchase 2,500 and 500 restricted shares of the Company’s common stock, respectively, at an exercise price of $12.80 per share, and we received cash proceeds of $32,000 and $6,400, respectively, from the exercises.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Modification of Warrants Held by Platinum
As indicated earlier in this note, effective on May 24, 2013, we entered into an Amendment and Waiver Agreement (Amendment and Waiver) with Platinum pursuant to which we agreed to reduce the exercise price of the Exchange Warrant and the Investment Warrants issued to Platinum between October 2012 and March 2013 (collectively, the “Warrants”) from $30.00 per share to $10.00 per share in consideration for Platinum’s agreement to waive its rights for any increase in the number of shares of common stock issuable under the adjustment provisions of the Exchange Warrant and the Investment Warrants that would otherwise occur from (i) our sale of shares of our common stock at a price of $10.00 per share in connection with the Autilion Financing; (ii) the March 2013 grant of warrants to certain of our officers and independent directors to purchase an aggregate of 150,000 restricted shares of common stock at an exercise price of $12.80 per share; and (iii) our issuance of restricted shares of our common stock resulting in gross proceeds not to exceed $1.5 million in connection with the exercise by warrant holders, by no later than June 30, 2013, subsequently extended to July 30, 2013, of previously outstanding warrants for which we may reduce the exercise price to not less than $10.00 per share. (See “Other Warrant Modifications and Exercises” below.)
As described in Note 4, Fair Value Measurements and in Note 8, Convertible Promissory Notes and Other Notes Payable, we re-measure the fair value of the Platinum Warrants at the end of each quarterly reporting period. The fair value re-measurement at June 30, 2013 incorporated the modification of the exercise price resulting from the Amendment and Waiver and the corresponding adjustment was reflected as a component of the Warrant Liability at that date. We also re-measure at the end of each reporting period the fair value of the Series A Exchange Warrant which is contingently issuable to Platinum upon the exchange of its shares of our Series A Preferred Stock into shares of our restricted common stock. At March 31, 2015 and 2014, we determined the fair values of the Platinum Warrants and the Series A Preferred Exchange Warrant to be a weighted average of $ 5.09.and $5.40 per share, respectively, or an aggregate of $3,008,500 and 2,973,900, which amounts are reflected as Warrant Liability in the accompanying Consolidated Balance Sheets at March 31, 2015 and 2014, respectively. We determined the fair value of the warrants at March 31, 2015 and 2014 using the assumptions indicated in the table below.
|
March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Market price of common stock
|
$ | 10.00 | $ | 9.20 | ||||
|
Exercise price per share
|
$ | 10.00 | $ | 9.80 to $10.00 | ||||
|
Risk-free interest rate
|
0.74% to 1.37%
|
1.73% | ||||||
|
Volatility
|
73.3% to 75.9%
|
75% | ||||||
|
Term (years)
|
2.5 to 5.0
|
3.5 to 5.0
|
||||||
|
Dividend rate
|
0% | 0% | ||||||
|
Probability of Series A Preferred exchange
|
95% | 95% | ||||||
|
Fair value per share
|
$ | 4.45 to $6.17 | $ | 5.20 to $5.80 | ||||
Other Warrant Modifications and Exercises
During the months of June and July 2013, we offered certain long-term warrant holders the opportunity to exercise warrants having an exercise price of $30.00 per share to purchase shares of our restricted common stock at a reduced exercise price of $10.00 per share through July 30, 2013. Warrant holders exercised warrants to purchase an aggregate of 26,419 restricted shares of our common stock and we received cash proceeds of $264,200. In addition, certain warrant holders exercised modified warrants to purchase 832 restricted shares of our common stock in lieu of our payment in satisfaction of amounts due for professional services in the aggregate amount of $8,300. We calculated the fair value of the warrants exercised immediately before and after the modifications and determined that the fair value of the warrants exercised decreased.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
In October 2013, we modified certain outstanding warrants held by our long-term investors and consultants to purchase an aggregate of 64,639 restricted shares of our common stock to reduce the exercise price of the warrants to $10.00 per share and, for warrants scheduled to expire on December 31, 2013, extend the exercise term of the warrants until January 31, 2015, generally without modifying the exercise price. We calculated the fair value of the warrants immediately before and after the modifications and determined that the fair value of the warrants increased by $77,800, which is reflected in general and administrative expense in the accompanying Consolidated Statements of Operations and Comprehensive Loss for the fiscal year ended March 31, 2014. The warrants subject to the exercise price modifications and term extensions were valued using the Black-Scholes Option Pricing Model and the following assumptions:
|
Assumption:
|
Pre-modification
|
Post-modification
|
||||||
|
Market price per share at modification date
|
$
|
10.00
|
$
|
10.00
|
||||
|
Exercise price per share (weighted average)
|
$
|
31.97
|
$
|
24.68
|
||||
|
Risk-free interest rate (weighted average)
|
0.33%
|
0.44%
|
||||||
|
Contractual term in years (weighted average)
|
1.40
|
2.10
|
||||||
|
Volatility (weighted average)
|
74.4%
|
75.8%
|
||||||
|
Dividend rate
|
0.0%
|
0.0%
|
||||||
|
Weighted Average Fair Value per share
|
$
|
1.08
|
$
|
2.29
|
||||
In December 2013, we modified additional outstanding warrants held by certain of our long-term investors, consultants, and members of management and our Board of Directors to purchase an aggregate of 63,013 restricted shares of our common stock to reduce the exercise price of the warrants to $10.00 per share and, in limited cases, extend the exercise term of the warrants. We calculated the fair value of the warrants immediately before and after the modifications and determined that the fair value of the warrants increased by $344,000, which is reflected in general and administrative expense in the accompanying Consolidated Statements of Operations and Comprehensive Loss for the fiscal year ended March 31, 2014. The warrants subject to the exercise price modifications and term extensions were valued using the Black-Scholes Option Pricing Model and the following assumptions:
|
Assumption:
|
Pre-modification
|
Post-modification
|
||||||
|
Market price per share at modification date
|
$
|
8.00
|
$
|
8.00
|
||||
|
Exercise price per share (weighted average)
|
$
|
33.49
|
$
|
10.00
|
||||
|
Risk-free interest rate (weighted average)
|
0.51%
|
0.57%
|
||||||
|
Contractual term in years (weighted average)
|
2.06
|
2.34
|
||||||
|
Volatility (weighted average)
|
73.6%
|
74.4%
|
||||||
|
Dividend rate
|
0.0%
|
0.0%
|
||||||
|
Weighted Average Fair Value per share
|
$
|
0.91
|
$
|
2.85
|
||||
In making its fair value determinations for both warrant modifications and new grants using the Black Scholes Option Pricing Model, we utilize the following principles in selecting its input assumptions. The market price per share is based on the quoted market price of our common stock on the OTC Markets on the date of the modification or grant. Because of our relatively short history as a public company, we estimate stock price volatility based on the historical volatilities of a peer group of public companies over the contractual or remaining contractual term of the warrant. The contractual term of the warrant is determined based on the grant or modification date and the latest date on which the warrant can be exercised under its terms or under the terms of the discounted exercise price offer. The risk-free rate of interest is based on the quoted constant maturity rate for U.S. Treasury Bills on the date of the grant or modification for the term most closely corresponding with the contractual term or remaining term of the warrant. The dividend rate is zero as we have not paid and do not expect to pay dividends in the near future.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Warrants Outstanding
The following table summarizes outstanding warrants to purchase restricted shares of our common stock as of March 31, 2015. The weighted average exercise price of outstanding warrants at March 31, 2015 was $13.00 per share.
|
Exercise Price per Share
|
Expiration Date
|
Shares Subject
to Purchase at |
|||||
| $ | 10.00 |
1/31/2016 to 1/1/2020
|
1,064,683 | ||||
| $ | 12.80 |
3/3/2023
|
147,000 | ||||
| $ | 15.00 |
1/31/2016 to 6/11/2016
|
54,477 | ||||
| $ | 20.00 |
7/30/2016 to 9/30/2017
|
186,388 | ||||
| $ | 30.00 |
2/13/2016 to 3/4/2018
|
69,426 | ||||
| $ | 40.00 |
9/15/2017
|
21,250 | ||||
| $ | 60.00 |
2/13/2016
|
1,250 | ||||
| 1,544,474 | |||||||
Note Receivable from Sale of Common Stock
In May 2011, the Company accepted a $500,000 short-term note from an investor in payment for shares of the Company’s common stock sold to the investor in a private placement transaction. On October 2, 2014 we received a cash payment of $60,000 from the maker of the note. We have considered that payment to be in full satisfaction of the outstanding principal balance of the note and related accrued interest, aggregating $194,900, at the date of the payment and recognized a loss of $134,900 on the settlement of the note, which is reflected as a component of Other expenses, net in the accompanying Statement of Operations and Comprehensive Loss.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Reserved Shares
At March 31, 2015, the Company has reserved shares of its common stock for future issuance as follows:
|
Upon exchange of all shares of Series A Preferred Stock currently issued and outstanding (1)
|
750,000 | |||
|
Warrant shares issuable to Platinum upon exercise of common stock warrant upon exchange of Series A preferred stock under the terms of the October 11, 2012 Note Purchase and Exchange Agreement
|
375,000 | |||
|
110% of shares issuable upon conversion of 10% senior secured convertible notes issued to Platinum in October 2012, February 2013, March 2013 and July 2013, including interest accrued through maturity (2)
|
563,871 | |||
|
Pursuant to warrants to purchase common stock:
|
||||
|
Subject to outstanding warrants
|
1,544,474 | |||
|
Issuable pursuant to accrued interest through maturity on outstanding promissory notes issued to Morrison & Foerster, Cato Research Ltd., and University Health Network
|
33,612 | |||
| 1,578,086 | ||||
|
Pursuant to stock incentive plans:
|
||||
|
Subject to outstanding options under the 2008 and 1999 Stock Incentive Plans
|
207,638 | |||
|
Available for future grants under the 2008 Stock Incentive Plan
|
40,491 | |||
| 248,129 | ||||
|
For additional issuances under the 2014 Private Placement of Units and upon conversion of notes and accrued interest pursuant to the 2013 Private Placement of Units and 2014 Private Placement of Units
|
807,800 | |||
|
Total
|
4,322,886 |
|
____________
|
||||
|
(1) assumes exchange under the terms of the October 11, 2012 Note Exchange and Purchase Agreement with Platinum
|
||||
|
(2) assumes conversion under the terms of the October 11, 2012 Note Exchange and Purchase Agreement with Platinum and the terms of the individual notes
|
||||
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
10. Research and Development Expenses
The Company recorded research and development expenses of approximately $4.3 million and $2.5 million in the fiscal years ended March 31, 2015 and 2014, respectively. Research and development expense is composed primarily of employee compensation expenses, including stock–based compensation, and direct project expenses, including costs incurred by third-party research collaborators, some of which may be reimbursed under the terms of grant or collaboration agreements.
11. Income Taxes
The provision for income taxes for the periods presented in the Consolidated Statements of Operations and Comprehensive Income represents minimum California franchise taxes. Income tax expense differed from the amounts computed by applying the U.S. federal income tax rate of 34% to pretax losses as a result of the following:
|
Fiscal Years Ended March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Computed expected tax benefit
|
(34.0 | ) % | (34.0 | ) % | ||||
|
Tax effect of Warrant Liability mark to market
|
(0.1 | ) % | 41.5 | % | ||||
|
Other losses not benefitted
|
34.0 | % | (7.5 | ) % | ||||
|
Other
|
0.1 | % | 0.1 | % | ||||
|
Income tax expense
|
0.0 | % | 0.1 | % | ||||
Deferred income taxes reflect the net tax effect of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets are as follows (in thousands):
|
March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Deferred tax assets:
|
||||||||
|
Net operating loss carryovers
|
$ | 23,054 | $ | 19,733 | ||||
|
Basis differences in fixed assets
|
24 | 37 | ||||||
|
Accruals and reserves
|
2,694 | 1,383 | ||||||
|
Total deferred tax assets
|
25,772 | 21,153 | ||||||
|
Valuation allowance
|
(25,772 | ) | (21,153 | ) | ||||
|
Net deferred tax assets
|
$ | - | $ | - | ||||
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Accordingly, the deferred tax assets have been fully offset by a valuation allowance. The valuation allowance increased by $4,619,000 and $2,126,000 during the fiscal years ended March 31, 2015 and 2014, respectively. When realized, deferred tax assets related to employee stock options will be credited to additional paid-in capital.
As of March 31, 2015, we had U.S. federal net operating loss carryforwards of $58.7 million, which will expire in fiscal years 2020 through 2035. As of March 31, 2015, we had state net operating loss carryforwards of $53.1 million, which will expire in fiscal years 2016 through 2035.
U.S. federal and state tax laws include substantial restrictions on the utilization of net operating loss carryforwards in the event of an ownership change of a corporation. We have not performed a change in ownership analysis since our inception in 1998 and accordingly some or all of our net operating loss carryforwards may not be available to offset future taxable income, if any.
The Company files income tax returns in the U.S. federal and Canadian jurisdictions and California and Maryland state jurisdictions. The Company is subject to U.S. federal and state income tax examinations by tax authorities for tax years 2000 through 2015 due to net operating losses that are being carried forward for tax purposes.
The Company does not have any uncertain tax positions or unrecognized tax benefits at March 31, 2015 and 2014. The Company’s policy is to recognize interest and penalties related to income taxes as components of interest expense and other expense, respectively.
12. Licensing and Collaborative Agreements
University Health Network
On September 17, 2007, we entered into a Sponsored Research Collaboration Agreement (SRCA) with University Health Network (UHN) to develop certain stem cell technologies for drug discovery, development and rescue technologies. The SRCA was amended on April 19, 2010 to extend the term to five years and give us various options to extend the term for an additional three years. On December 15, 2010, we entered into a second amendment with UHN to expand the scope of work to include induced pluripotent stem cell technology and to further expand the scope of research and term extension options. On April 25, 2011, we and UHN amended the SRCA a third time to expand the scope to include therapeutic and stem cell therapy applications of induced pluripotent cells and to extend the date during which we may elect to fund additional projects to April 30, 2012. On October 24, 2011, we and UHN amended the SRCA a fourth time to identify five key programs to further support our core drug rescue initiatives and potential cell therapy applications. In October 2012, we issued a promissory note in the principal amount of $549,500 and a warrant to UHN as payment in full for services rendered under the fourth amendment. We also entered into Amendment No. 5 to the SRCA establishing the sponsored research projects and the sponsored research budgets under the SRCA from October 1, 2012 to September 30, 2013. During our fiscal year ended March 31, 2015, our financial condition precluded further sponsored research activities with UHN.
Concurrent with the execution of the fourth amendment to the SRCA, we also entered into a License Agreement with UHN under the terms of which UHN granted us exclusive rights to the use of a novel molecule that can be employed in the identification and isolation of mature and immature human cardiomyocytes from pluripotent stem cells, as well as methods for the production of cardiomyocytes from pluripotent stem cells that express this marker. In consideration for the grant of the license, we have agreed to make payments to UHN totaling $3.9 million, if, and when, we achieves certain commercial milestones set forth in the License Agreement, and to pay UHN royalties based on our receipt of revenue attributable to the licensed patents.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. National Institutes of Health
During fiscal years 2006 through 2008, the U.S. National Institutes of Health ("NIH") awarded VistaGen California a $4.2 million grant to support preclinical development of AV-101, our lead drug candidate for treatment of neuropathic pain and other neurodegenerative diseases such as Huntington’s and Parkinson’s diseases. In June 2009, the NIH awarded VistaGen California a $4.2 million grant to support the Phase I clinical development of AV-101, which amount was subsequently increased to a total of $4.6 million in July 2010. The grant expired in the ordinary course on June 30, 2012 and all funds had been expended. In February 2015, we entered into a Cooperative Research and Development Agreement with the National Institute of Mental Health to collaborate on an NIH-sponsored Phase 2 clinical study of the efficacy and safety of AV-101 in subjects with MDD. The study is expected to commence late in the third quarter of 2015 and be completed in 2016.
Cato Research Ltd.
We have built a strategic development relationship with Cato Research Ltd. (“CRL”), a global contract research and development organization, or CRO, and an affiliate of one of the Company’s largest institutional stockholders. CRL has provided us with access to essential CRO services and regulatory expertise supporting our AV-101 preclinical and clinical development programs and other projects. We recorded research and development expenses for CRO services provided by CRL in the amounts of $38,100 and $52,500 for the fiscal years ended March 31, 2015 and 2014, respectively. In October 2012, we issued an unsecured promissory note in the principal amount of $1,009,000, and a warrant exercisable for 50,450 shares of our common stock, as payment in full of all amounts owed to CRL for CRO services rendered to us through December 31, 2012.
13. Stock Option Plans and 401(k) Plan
We have the following share-based compensation plans.
2008 Stock Incentive Plan
Our 2008 Stock Incentive Plan (the “2008 Plan”) was adopted by the shareholders of VistaGen California on December 19, 2008 and assumed by the Company in connection with the Merger. The maximum number of shares of our common stock that may be granted pursuant to the 2008 Plan is 250,000 shares, subject to adjustments for stock splits, stock dividends or other similar changes in the common stock or capital structure.
1999 Stock Incentive Plan
Our 1999 Stock Incentive Plan (the “1999 Plan”) was adopted by the shareholders of VistaGen California on December 6, 1999 and assumed by the Company in connection with the Merger. We initially reserved 45,000 shares for the issuance of awards under the 1999 Plan. The 1999 Plan has terminated under its own terms and, as a result, no awards may currently be granted under the 1999 Plan. The unexpired options and awards that have already been granted pursuant to the 1999 Plan remain operative.
Description of the 2008 Plan
Under the terms of the 2008 Plan, the Compensation Committee of our Board of Directors may grant shares, options or similar rights having either a fixed or variable price related to the fair market value of the shares and with an exercise or conversion privilege related to the passage of time, the occurrence of one or more events, or the satisfaction of performance criteria or other conditions, or any other security with the value derived from the value of the shares. Such awards include stock options, restricted stock, restricted stock units, stock appreciation rights and dividend equivalent rights.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The Compensation Committee may grant nonstatutory stock options under the 2008 Plan at a price of not less than 100% of the fair market value of our common stock on the date the option is granted. Incentive stock options under the 2008 Plan may be granted at a price of not less than 100% of the fair market value of our common stock on the date the option is granted. Incentive stock options granted to employees who, on the date of grant, own stock representing more than 10% of the voting power of all of our classes of stock are granted at an exercise price of not less than 110% of the fair market value of our common stock and the maximum term of such incentive stock options may not exceed five years. The maximum term of an incentive stock option granted to any other participant may not exceed ten years. The Compensation Committee determines the term and exercise or purchase price of all other awards granted under the 2008 Plan. The Compensation Committee also determines the terms and conditions of awards, including the vesting schedule and any forfeiture provisions. Awards under the 2008 Plan may vest upon the passage of time or upon the attainment of certain performance criteria established by the Compensation Committee. We currently have no performance-based awards outstanding.
Unless terminated sooner, the 2008 Plan will automatically terminate in 2017. The Board of Directors may at any time amend, suspend or terminate our 2008 Plan.
We did not grant any stock options during fiscal 2015. During the third quarter of fiscal 2014, when the quoted market price of our common stock was $8.00 per share, we reduced the exercise price of an aggregate of 196,213 outstanding options to purchase shares of its common stock at exercise prices between $15.00 per share and $59.80 per share held by certain employees, including the Company’s officers and directors, and by certain consultants to $8.00 per share or $10.00 per share. These reductions in exercise price were accounted for as a modification of the options and resulted in a charge of $252,000.
The following table summarizes share-based compensation expense, including share-based expense related to the March 2015 and March 2014 grants of warrants to certain of our officers and to our independent directors as described in Note 9, Capital Stock, included in the accompanying Consolidated Statement of Operations and Comprehensive Loss for the years ended March 31, 2015 and 2014.
|
Fiscal Years Ended
|
||||||||
|
March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Research and development expense:
|
||||||||
|
Stock option grants
|
$ | 176,200 | $ | 296,900 | ||||
|
Fully-vested warrants granted to officer and
|
||||||||
|
consultants in January 2015
|
527,500 | - | ||||||
|
Warrants granted to officer in March 2014 and 2013
|
145,100 | 156,500 | ||||||
| 848,800 | 453,400 | |||||||
|
General and administrative expense:
|
||||||||
|
Stock option grants
|
98,800 | 385,100 | ||||||
|
Fully-vested warrants granted to officers, directors
|
||||||||
|
and consultants in January 2015
|
1,229,400 | - | ||||||
|
Warrants granted to officers and directors in March
|
||||||||
|
2014 and 2013
|
283,100 | 298,800 | ||||||
| 1,611,300 | 683,900 | |||||||
|
Total stock-based compensation expense
|
$ | 2,460,100 | $ | 1,137,300 | ||||
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
We used the Black-Scholes option valuation model with the following assumptions to determine share-based compensation expense related to option grants during the fiscal years ended March 31, 2015 and 2014:
| Fiscal Years Ended March 31, | ||||
|
2015
|
2014
|
|||
|
Exercise price
|
not applicable
|
$8.00 to $16.40
|
||
|
Market price on date of grant
|
not applicable
|
$8.00 to $16.40
|
||
|
Risk-free interest rate
|
not applicable
|
1.08% to 2.53%
|
||
|
Expected term (years)
|
not applicable
|
6.25 to 10.0
|
||
|
Volatility
|
not applicable
|
87.9% to 103.2%
|
||
|
Expected dividend yield
|
not applicable
|
0%
|
||
|
Fair value per share at grant date
|
not applicable
|
$6.38 to $13.63
|
||
The expected term of options represents the period that our share-based compensation awards are expected to be outstanding. We have calculated the weighted-average expected term of the options using the simplified method as prescribed by Securities and Exchange Commission Staff Accounting Bulletins No. 107 and No. 110 (“SAB No. 107 and 110”). The utilization of SAB No. 107 and 110 was based on the lack of relevant historical data due to our limited historical experience as a publicly traded company as well as the lack of liquidity resulting from the limited number of freely-tradable shares of our common stock. Limited historical experience and lack of liquidity in our stock also resulted in our decision to utilize the historical volatilities of a peer group of public companies’ stock over the expected term of the option in determining our expected volatility assumptions. The risk-free interest rate for periods related to the expected life of the options is based on the U.S. Treasury yield curve in effect at the time of grant. The expected dividend yield is zero, as we have not paid any dividends and do not anticipate paying dividends in the near future. We calculated the forfeiture rate based on an analysis of historical data, as it reasonably approximates the currently anticipated rate of forfeitures for granted and outstanding options that have not vested.
The following table summarizes activity for the fiscal years ended March 31, 2015 and 2014 under our stock option plans:
|
Fiscal Years Ended March 31,
|
||||||||||||||||
|
2015
|
2014
|
|||||||||||||||
|
Weighted
|
Weighted
|
|||||||||||||||
|
Average
|
Average
|
|||||||||||||||
|
Number of
|
Exercise
|
Number of
|
Exercise
|
|||||||||||||
|
Shares
|
Price
|
Shares
|
Price
|
|||||||||||||
|
Options outstanding at beginning of period
|
212,486 | $ | 10.09 | 245,653 | $ | 26.43 | ||||||||||
|
Options granted
|
- | $ | - | 19,050 | $ | 10.89 | ||||||||||
|
Options exercised
|
- | $ | - | - | $ | - | ||||||||||
|
Options forfeited
|
(2,001 | ) | $ | 9.25 | (3,954 | ) | $ | 27.22 | ||||||||
|
Options expired
|
(2,847 | ) | $ | 10.56 | (48,263 | ) | $ | 23.94 | ||||||||
|
Options outstanding at end of period
|
207,638 | $ | 10.09 | 212,486 | $ | 10.09 | ||||||||||
|
Options exercisable at end of period
|
199,013 | $ | 10.09 | 182,775 | $ | 10.06 | ||||||||||
|
Weighted average grant-date fair value of
|
||||||||||||||||
|
options granted during the period
|
$ | - | $ | 8.36 | ||||||||||||
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The following table summarizes information on stock options outstanding and exercisable under our stock option plans as of March 31, 2015:
|
Options Outstanding
|
Options Exercisable
|
|||||||||||||||||||||
|
Weighted
|
||||||||||||||||||||||
|
Average
|
Weighted
|
Weighted
|
||||||||||||||||||||
|
Remaining
|
Average
|
Average
|
||||||||||||||||||||
|
Exercise
|
Number
|
Years until
|
Exercise
|
Number
|
Exercise
|
|||||||||||||||||
|
Price
|
Outstanding
|
Expiration
|
Price
|
Exercisable
|
Price
|
|||||||||||||||||
| $ | 8.00 | 49,590 | 7.53 | $ | 8.00 | 46,466 | $ | 8.00 | ||||||||||||||
| $ | 10.00 | 147,939 | 4.88 | $ | 10.00 | 142,751 | $ | 10.00 | ||||||||||||||
| $ | 14.40 to $36.00 | 10,109 | 4.64 | $ | 21.69 | 9,796 | $ | 21.23 | ||||||||||||||
| 207,638 | 5.51 | $ | 10.09 | 199,013 | $ | 10.09 | ||||||||||||||||
At March 31, 2015, there were 40,491 shares of our common stock remaining available for grant under the 2008 Plan. There were no option exercises during the years ended March 31, 2015 or 2014.
Aggregate intrinsic value is the sum of the amounts by which the fair value of the underlying common stock exceeded the exercise price of the option (in-the-money-options). Based on the $10.00 per share quoted market price of our common stock on March 31, 2015, the aggregate intrinsic value of outstanding options at that date was $99,200, of which $92,900 related to exercisable options.
As of March 31, 2015, there was approximately $71,700 of unrecognized compensation cost related to non-vested share-based compensation awards from the 2008 Plan, which is expected to be recognized through May 2016. Additionally, at March 31, 2015 there was approximately $27,000 of unrecognized compensation cost related to unvested warrant grants to independent directors and officers, which is expected to be recognized through March 2016 absent any conditions which would accelerate the vesting of the awards and corresponding expense recognition.
401(k) Plan
Through a third-party agent, we maintain a retirement and deferred savings plan for our employees. This plan is intended to qualify as a tax-qualified plan under Section 401(k) of the Internal Revenue Code. The retirement and deferred savings plan provides that each participant may contribute a portion of his or her pre-tax compensation, subject to statutory limits. Under the plan, each employee is fully vested in his or her deferred salary contributions. Employee contributions are held and invested by the plan’s trustee. The retirement and deferred savings plan also permits us to make discretionary contributions, subject to established limits and a vesting schedule. To date, we have not made any discretionary contributions to the retirement and deferred savings plan on behalf of participating employees.
14. Related Party Transactions
Cato Holding Company (CHC), doing business as Cato BioVentures (CBV), the parent of CRL, is one of our largest institutional stockholders at March 31, 2015, holding common stock and warrants to purchase our common stock. Shawn Singh, our Chief Executive Officer and member of our Board of Directors, served as Managing Principal of CBV and as an officer of CRL until August 2009. On October 10, 2012, we issued to CHC an unsecured promissory note in the principal amount of $310,400 (the 2012 CHC Note) and a five-year warrant to purchase 12,500 restricted shares of our common stock at a price of $30.00 per share (the CHC Warrant). Additionally, on October 10, 2012, we issued to CRL: (i) an unsecured promissory note in the initial principal amount of $1,009,000, which is payable solely in restricted shares of our common stock and which accrues interest at the rate of 7.5% per annum, compounded monthly (the CRL Note), as payment in full for all contract research and development services and regulatory advice rendered to us by CRL through December 31, 2012 with respect to the preclinical and clinical development of AV-101, and (ii) a five-year warrant to purchase, at a price of $20.00 per share, 50,450 restricted shares of our common stock. The 2012 CHC Note and the CRL Note mature on March 31, 2016.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
During fiscal year 2007, VistaGen California entered into a contract research organization arrangement with CRL related to the development of AV-101, under which we incurred expenses of $38,100 and $52,500 for the fiscal years ended March 31, 2015 and 2014, respectively. Total interest expense on notes payable to CHC and CRL was $174,800 and $167,900 for the fiscal years ended March 31, 2015 and 2014, respectively.
Upon the approval of its Board of Directors, in December 2006, VistaGen California accepted a full-recourse promissory note in the amount of $103,400 from Mr. Shawn Singh in payment of the exercise price for options and warrants to purchase an aggregate of 6,320 restricted shares of VistaGen California’s common stock. The note accrued interest at a rate of 4.90% per annum and was due and payable no later than the earlier of (i) December 1, 2016 or (ii) ten days prior to VistaGen California becoming subject to the requirements of the Securities Exchange Act of 1934, as amended (Exchange Act). On May 11, 2011, in connection with the Merger, the $128,200 outstanding balance of principal and accrued interest on this note was cancelled in accordance with Mr. Singh's employment agreement and recorded as additional compensation. In accordance with his employment agreement, Mr. Singh is also entitled to receive an income tax gross-up on the compensation related to the note cancellation. At March 31, 2015 and 2014, we had accrued $101,900 as an estimate of the gross-up amount, but we had not yet paid any of that amount to Mr. Singh.
Between September and December 2013, Mr. Singh provided short-term cash advances aggregating $64,000 to meet our short-term working capital requirements. In lieu of cash repayment of the advances, in December 2013, Mr. Singh elected to invest $50,000 of the balance due him in the 2013 Unit Private Placement. At March 31, 2015, we have completely repaid the balance of the advances and the $50,000 promissory note issued in connection with his investment in the 2013 Unit Private Placement to Mr. Singh.
15. Commitments, Contingencies, Guarantees and Indemnifications
From time to time, we may become involved in claims and other legal matters arising in the ordinary course of business. Management is not currently aware of any claims made or other legal matters that will have a material adverse effect on our consolidated financial position, results of operations or its cash flows.
We indemnify our officers and directors for certain events or occurrences while the officer or director is or was serving at our request in such capacity. The term of the indemnification period is for the officer’s or director’s lifetime. We will indemnify the officers or directors against any and all expenses incurred by the officers or directors because of their status as one of our directors or executive officers to the fullest extent permitted by Nevada law. We have never incurred costs to defend lawsuits or settle claims related to these indemnification agreements. We have a director and officer insurance policy which limits our exposure and may enable us to recover a portion of any future amounts paid. We believe the fair value of these indemnification agreements is minimal. Accordingly, there are no liabilities recorded for these agreements at March 31, 2015 or 2014.
In the normal course of business, we provide indemnifications of varying scopes under agreements with other companies, typically clinical research organizations, investigators, clinical sites, suppliers and others. Pursuant to these agreements, we generally indemnify, hold harmless, and agree to reimburse the indemnified parties for losses suffered or incurred by the indemnified parties in connection with the use or testing of our product candidates or with any U.S. patents or any copyright or other intellectual property infringement claims by any third party with respect to our product candidates. The terms of these indemnification agreements are generally perpetual. The potential future payments we could be required to make under these indemnification agreements is unlimited. We maintain liability insurance coverage that limits our exposure. We believe the fair value of these indemnification agreements is minimal. Accordingly, we have not recorded any liabilities for these agreements as of March 31, 2015 or 2014.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Leases
As of March 31, 2015 and 2014, the following assets are under capital lease obligations and included in property and equipment:
|
March 31,
|
||||||||
|
2015
|
2014
|
|||||||
|
Laboratory equipment
|
$ | - | $ | 19,000 | ||||
|
Office equipment
|
4,500 | 4,500 | ||||||
| 4,500 | 23,500 | |||||||
|
Accumulated depreciation
|
(2,500 | ) | (11,100 | ) | ||||
|
Net book value
|
$ | 2,000 | $ | 12,400 | ||||
Amortization expense for assets recorded under capital leases is included in depreciation expense. Future minimum payments, by year and in the aggregate, required under capital leases are as follows:
|
Fiscal Years Ending March 31,
|
Capital
Leases |
|||
|
2016
|
$ | 1,200 | ||
|
2017
|
1,200 | |||
|
2018
|
100 | |||
|
Future minimum lease payments
|
2,500 | |||
|
Less imputed interest included in minimum lease payments
|
(400 | ) | ||
|
Present value of minimum lease payments
|
2,100 | |||
|
Less current portion
|
(1,000 | ) | ||
|
Non-current capital lease obligation
|
$ | 1,100 | ||
At March 31, 2015, future minimum payments under operating leases relate to our facility lease in South San Francisco, California through July 31, 2017 and are as follows:
|
Fiscal Years Ending March 31,
|
Amount
|
|||
|
2016
|
264,000 | |||
|
2017
|
277,100 | |||
|
2018
|
93,800 | |||
| $ | 634,900 | |||
We incurred total facility rent expense for the fiscal years ended March 31, 2015 and 2014 of $337,000 and $284,100 , respectively.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Long-Term Debt Repayment
At March 31, 2015, assuming that all outstanding convertible notes are converted into shares of common stock in accordance with their respective conversion provisions and that Replacement Note B issued to Morrison & Foerster, the CRL Note and the UHN Note, each as described further in Note 8, Convertible Promissory Notes and Other Notes Payable, are repaid through the issuance of restricted common stock upon the exercise of the warrants associated with such notes, future minimum principal payments related to long-term debt were as follows:
|
Fiscal Years Ending March 31,
|
Amount
|
|||
|
2016
|
$ | 2,185,700 | ||
|
2017
|
9,800 | |||
|
2018
|
10,500 | |||
|
2019
|
11,300 | |||
|
Thereafter through June 2019
|
4,000 | |||
| $ | 2,221,300 | |||
16. Subsequent Events
We have evaluated subsequent events through the date of this report and have identified the following material events and transactions that occurred after March 31, 2015.
2014 Unit Private Placement
From April 1, 2015 through May 14, 2015, we entered into securities purchase agreements with accredited investors pursuant to which we sold to such accredited investors Units, for aggregate cash proceeds of $280,000, consisting of (i) 10% convertible notes in the aggregate face amount of $280,000 due between April 30, 2015 and May 15, 2015 or automatically convertible into securities we may issue upon the consummation of a Qualified Financing, as defined in the note (ii) an aggregate of 33,000 restricted shares of our common stock; and (iii) warrants exercisable through December 31, 2016 to purchase an aggregate of 24,250 restricted shares of our common stock at an exercise price of $10.00 per share.
Creation of Series B Preferred Stock
On May 7, 2015, we filed a Certificate of Designation of the Relative Rights and Preferences of the Series B 10% Preferred Stock of VistaGen Therapeutics, Inc. (Certificate of Designation) with the Nevada Secretary of State to designate 4.0 million shares of our authorized preferred stock as Series B 10% Convertible Preferred Stock (Series B Preferred).
Each share of Series B Preferred is convertible, at the option of the holder (Voluntary Conversion), into one (1) share of our common stock at a fixed conversion price of $7.00 per share (Conversion Price). The Conversion Price is subject to adjustment only for customary stock dividends, reclassifications, splits and similar transactions set forth in the Certificate of Designation. All outstanding shares of Series B Preferred are also convertible automatically into shares of our common stock (“Automatic Conversion”) upon the closing or effective date of any of the following transactions or events: (i) a strategic transaction involving AV-101 with an initial up-front cash payment to us of at least $10.0 million; (ii) a registered public offering of our common stock with aggregate gross proceeds to us of at least $10.0 million; or (iii) for 20 consecutive trading days, our common stock trades at least 20,000 shares per day with a daily closing price of at least $12.00 per share; provided, however, that Automatic Conversion and Voluntary Conversion (collectively, Conversion) are subject to certain beneficial ownership blockers as set forth in the Certificate of Designation.
Prior to Conversion, shares of Series B Preferred will accrue dividends, payable only in shares of our common stock, at a rate of 10% per annum (Accrued Dividend). The Accrued Dividend will be payable on the date of either a Voluntary Conversion or Automatic Conversion solely in that number of shares of common stock equal to the Accrued Dividend, divided by the Conversion Price.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Agreement with Platinum
On May 5, 2015, we entered into an Agreement with Platinum, which, as modified by an Acknowledgement and Agreement, became effective on May 12, 2015 (Platinum Agreement). Under the Platinum Agreement, Platinum:
|
·
|
Converted all of the approximately $4.5 million outstanding balance (principal and accrued but unpaid interest) of the Senior Notes we issued to Platinum into 641,335 shares of Series B Preferred, thereby cancelling approximately $4.5 million of our outstanding indebtedness;
|
|
·
|
Released all of its security interests in our assets and those of our subsidiaries by terminating the Amended and Restated Security Agreement, IP Security Agreement and Negative Covenant, each dated October 11, 2012 between us and Platinum; and
|
|
·
|
Converted all of the approximately $1.3 million outstanding balance (principal and accrued but unpaid interest) of the 2014 Unit Notes that we issued to Platinum into 240,305 shares of Series B Preferred and five-year warrants to purchase 240,305 shares of our common stock at a fixed exercise price of $7.00 per share (Series B Warrants), thereby cancelling approximately $1.3 million of our outstanding indebtedness; and
|
|
·
|
Purchased approximately $1.5 million (including accrued but unpaid interest thereon) of outstanding 2014 Unit Notes we issued to various investors from the respective holders thereof (Investor 2014 Unit Notes ) and converted the entire outstanding balance of the Investor 2014 Unit Notes into 265,699 shares of Series B Preferred and Series B Warrants to purchase 265,699 shares of our common stock, thereby cancelling approximately $1.5 million of our outstanding indebtedness; and
|
|
·
|
Entered into a Securities Purchase Agreement (SPA) to purchase, for $1.0 million, a total of 142,857 shares of Series B Preferred and a Series B Warrant to purchase 142,857 shares of our common stock, on or before June 11, 2015 (a portion of which purchase was completed on June 19, 2015); and
|
|
·
|
Amended the Platinum Warrants (all warrants previously issued by us to Platinum in connection with the Senior Notes) and the Series A Exchange Warrant to:
|
|
o
|
fix the exercise price thereof at $7.00 per share;
|
|
o
|
eliminate the exercise price reset features and fix the number of shares of our common stock issuable thereunder; and
|
|
o
|
eliminate the cashless exercise provisions from the Platinum Warrants and the Series A Exchange Warrant; and
|
|
·
|
Agreed to refrain from the sale of any shares of our common stock held by Platinum or its affiliates until the earlier to occur of an effective registration statement relating to resale of certain specified shares of common stock under the Securities Act of 1933, as amended, or the closing price of our common stock is at least $15.00 per share.
|
As additional consideration for the agreements of Platinum under the Platinum Agreement, we issued to Platinum 400,000 shares of Series B Preferred and Series B Warrants to purchase 1.2 million shares of our common stock, and exchanged 30,000 shares of our common stock currently beneficially owned or controlled by Platinum for 30,000 shares of Series B Preferred.
Conversion of 2014 Unit Notes
Effective May 20, 2015, holders of the remaining approximately $1.8 million outstanding balance (principal and accrued but unpaid interest) of 2014 Unit Notes converted such notes into 327,016 shares of Series B Preferred and Series B Warrants to purchase 327,016 shares of our common stock, thereby cancelling an additional approximately $1.8 million of our outstanding indebtedness.
VISTAGEN THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Sale of Series B Preferred Units
Between May 26, 2015 and June 25, 2015, we sold to accredited investors and institutions an aggregate of $557,500 of units in our Series B Preferred Unit offering, which units consist of Series B Preferred and Series B Warrants (together Series B Preferred Units), including $100,000 to Platinum. We issued 79,646 shares of Series B Preferred and Series B Warrants to purchase 79,646 shares of our common stock. We have received an aggregate of $557,500 in cash proceeds from the sale of the Series B Preferred Units.
Conversion of Notes and Accounts Payable
During May and June 2015, holders of certain of our promissory notes outstanding at March 31, 2015 and thereafter, including Morrison & Foerster, Cato Research Ltd., University Health Network, and McCarthy Tetrault, and certain other service providers converted notes payable or accounts payable having an aggregate outstanding balance of approximately $5.8 million (principal and accrued but unpaid interest and certain adjustments thereto) into 831,577 shares of Series B Preferred, thereby cancelling an additional approximately $5.8 million of our outstanding debt.
17. Supplemental Financial Information
The following table presents the unaudited statements of operations data for each of the eight quarters in the period ended March 31, 2015. The information has been presented on the same basis as the audited financial statements and all necessary adjustments, consisting only of normal recurring adjustments, have been included in the amounts below to present fairly the unaudited quarterly results when read in conjunction with the audited financial statements and related notes. The operating results for any quarter should not be relied upon as necessarily indicative of results for any future period.
Quarterly Results of Operations (Unaudited)
(in thousands, except share and per share amounts)
|
Three Months Ended
|
Total
|
|||||||||||||||||||
|
June 30, 2014
|
September 30, 2014
|
December 31, 2014
|
March 31, 2015
|
Fiscal Year 2015
|
||||||||||||||||
|
Revenues:
|
$ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||
|
Operating expenses:
|
||||||||||||||||||||
|
Research and development
|
474 | 558 | 445 | 956 | 2,433 | |||||||||||||||
|
General and administrative
|
797 | 556 | 671 | 2,320 | 4,344 | |||||||||||||||
|
Total operating expenses
|
1,271 | 1,114 | 1,116 | 3,276 | 6,777 | |||||||||||||||
|
Loss from operations
|
(1,271 | ) | (1,114 | ) | (1,116 | ) | (3,276 | ) | (6,777 | ) | ||||||||||
|
Other expenses, net:
|
||||||||||||||||||||
|
Interest expense, net
|
(785 | ) | (606 | ) | (792 | ) | (2,366 | ) | (4,549 | ) | ||||||||||
|
Change in warrant liabilities
|
(1,727 | ) | 1,302 | 953 | (563 | ) | (35 | ) | ||||||||||||
|
Loss on extinguishment of debt
|
(768 | ) | (1,603 | ) | - | (17 | ) | (2,388 | ) | |||||||||||
|
Other expense, net
|
- | - | (135 | ) | - | (135 | ) | |||||||||||||
|
Loss before income taxes
|
(4,551 | ) | (2,021 | ) | (1,090 | ) | (6,222 | ) | (13,884 | ) | ||||||||||
|
Income taxes
|
(2 | ) | - | - | - | (2 | ) | |||||||||||||
|
Net loss
|
$ | (4,553 | ) | $ | (2,021 | ) | $ | (1,090 | ) | $ | (6,222 | ) | $ | (13,886 | ) | |||||
|
Basic net (loss) per common share
|
$ | (3.70 | ) | $ | (1.58 | ) | $ | (0.84 | ) | $ | (4.24 | ) | $ | (10.53 | ) | |||||
|
Diluted net loss per common share
|
$ | (3.70 | ) | $ | (1.90 | ) | $ | (1.08 | ) | $ | (4.24 | ) | $ | (10.61 | ) | |||||
|
Weighted average shares used in computing:
|
||||||||||||||||||||
|
Basic net (loss) per common share
|
1,229,488 | 1,279,251 | 1,302,300 | 1,466,386 | 1,318,797 | |||||||||||||||
|
Diluted net loss per common share
|
1,229,488 | 1,299,099 | 1,302,300 | 1,466,386 | 1,318,797 | |||||||||||||||
|
Three Months Ended
|
Total
|
|||||||||||||||||||
|
June 30, 2013
|
September 30, 2013
|
December 31, 2013
|
March 31, 2014
|
Fiscal Year 2014
|
||||||||||||||||
|
Revenues:
|
$ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||
|
Operating expenses:
|
||||||||||||||||||||
|
Research and development
|
695 | 669 | 551 | 566 | 2,481 | |||||||||||||||
|
General and administrative
|
605 | 546 | 897 | 500 | 2,548 | |||||||||||||||
|
Total operating expenses
|
1,300 | 1,215 | 1,448 | 1,066 | 5,029 | |||||||||||||||
|
Loss from operations
|
(1,300 | ) | (1,215 | ) | (1,448 | ) | (1,066 | ) | (5,029 | ) | ||||||||||
|
Other expenses, net:
|
||||||||||||||||||||
|
Interest expense, net
|
(316 | ) | (323 | ) | (361 | ) | (503 | ) | (1,503 | ) | ||||||||||
|
Change in warrant liabilities
|
1,805 | 79 | 1,940 | (257 | ) | 3,567 | ||||||||||||||
|
Income (loss) before income taxes
|
189 | (1,459 | ) | 131 | (1,826 | ) | (2,965 | ) | ||||||||||||
|
Income taxes
|
(3 | ) | - | - | - | (3 | ) | |||||||||||||
|
Net income (loss)
|
186 | $ | (1,459 | ) | $ | 131 | $ | (1,826 | ) | $ | (2,968 | ) | ||||||||
|
Basic net income (loss) per common share
|
$ | 0.18 | $ | (1.35 | ) | $ | 0.12 | $ | (1.57 | ) | $ | (2.70 | ) | |||||||
|
Diluted net loss per common share
|
$ | (0.44 | ) | $ | (1.37 | ) | $ | (0.44 | ) | $ | (1.57 | ) | $ | (3.81 | ) | |||||
|
Weighted average shares used in computing:
|
||||||||||||||||||||
|
Basic net income (loss) per common share
|
1,042,081 | 1,081,529 | 1,110,529 | 1,162,636 | 1,098,742 | |||||||||||||||
|
Diluted net loss per common share
|
1,061,544 | 1,128,152 | 1,110,529 | 1,162,636 | 1,099,216 | |||||||||||||||
3,623,341 Shares of Common Stock
VISTAGEN THERAPEUTICS, INC.
Prospectus
Until ______ __, 2015, all dealers that buy, sell or trade the common stock may be required to deliver a prospectus, regardless of whether they are participating in this offering by the Selling Stockholders. This is in addition to the dealers’ obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution
The following table presents the costs and expenses in connection with the issuance and distribution of the securities to be registered. No underwriting discounts and commissions shall be payable by us in connection with the resale of common stock being registered. Except as otherwise noted, we will pay all of these amounts. All amounts are estimates except the SEC registration fee.
|
SEC Filing fees
|
$
|
4,210
|
||
|
Legal fees and expenses
|
30,000
|
|||
|
Accounting fees and expenses
|
10,000
|
|||
|
Miscellaneous expenses
|
6,000
|
|||
|
Total
|
$
|
50,210
|
Item 14. Indemnification of Directors and Officers
Limitations of liability and indemnification
Our amended and restated bylaws provide that we will indemnify our directors, officers and employees to the fullest extent permitted by the Nevada Revised Statutes (“NRS”).
If the NRS are amended to authorize corporate action further eliminating or limiting the personal liability of a director, then the liability of our directors will be eliminated or limited to the fullest extent permitted by the NRS, as so amended. Our articles of incorporation do not eliminate a director’s duty of care and, in appropriate circumstances, equitable remedies, such as injunctive or other forms of non-monetary relief, will remain available under the NRS. This provision also does not affect a director’s responsibilities under any other laws, such as the federal securities laws or other state or federal laws. Under our bylaws, we are empowered to enter into indemnification agreements with our directors, officers and employees to purchase insurance on behalf of any person whom we are required or permitted to indemnify.
In addition to the indemnification required in our bylaws, we have entered into indemnification agreements with each of the individuals serving on our board of directors. These agreements provide for the indemnification of our directors to the fullest extent permitted by law. We believe that these bylaw provisions and indemnification agreements are necessary to attract and retain qualified persons as directors, officers and employees. We also maintain directors’ and officers’ liability insurance.
The limitation of liability and indemnification provisions in our bylaws may discourage shareholders from bringing a lawsuit against our directors and officers for breach of their fiduciary duties. They may also reduce the likelihood of derivative litigation against directors and officers, even though an action, if successful, might benefit us and our stockholders. Further, a stockholder’s investment may be adversely affected to the extent that we pay the costs of settlement and damage awards against directors and officers pursuant to these indemnification provisions.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers and certain employees pursuant to the foregoing provisions, or otherwise, we have been advised that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act, and is, therefore, unenforceable.
There is no pending litigation or proceeding naming any of our directors or officers as to which indemnification is being sought, nor are we aware of any pending or threatened litigation that may result in claims for indemnification
Item 15. Recent Sales of Unregistered Securities
During the three years preceding the date of this prospectus, we issued the following securities in private placement transactions with accredited investors which were not registered under the Securities Act of 1933, as amended (Securities Act) and that have not been previously reported in a Quarterly Report on Form 10-Q or a Current Report on Form 8-K:
Between March 10, 2015 and May 14, 2015, we entered into securities purchase agreements with accredited investors pursuant to which we sold Units consisting of an aggregate of (i) a 10% convertible subordinated notes in the face amount of $530,000 maturing between March 31, 2015 and May 15, 2015 (“2014 Unit Notes”); (ii) 59,250 shares of our restricted common stock; and (iii) warrants exercisable through December 31, 2016 to purchase 50,500 shares of our restricted common stock at an exercise price of $10.00 per share (Unit Warrant). We received cash proceeds of $530,000 which we used for general corporate purposes. The Unit Notes and related accrued interest are convertible into shares of our restricted common stock at a conversion price of $10.00 per share at or prior to maturity at the option of the investor. The Units were offered and sold in a transaction exempt from registration under the Securities Act, in reliance on Section 4(2) thereof and Rule 506 of Regulation D thereunder.
Between May 26, 2015 and July 17, 2015, we sold to accredited investors an aggregate of $1,112,500 of units in our Series B Preferred Unit offering, which units consist of Series B Preferred and warrants to purchase shares of our common stock (together Series B Preferred Units), including $500,000 from Platinum. We issued 160,363 shares of Series B Preferred and warrants to purchase 160,363 shares of our common stock. We have received an aggregate of $1,112,500 in cash proceeds from the sale of the Series B Preferred Units. The Units were offered and sold in a transaction exempt from registration under the Securities Act, in reliance on Section 4(2) thereof and Rule 506 of Regulation D thereunder.
Between July 6, 2015 and July 17, 2015, two professional service providers and a technology licensor holding outstanding promissory notes and unpaid trade receivables from us agreed to convert such debt obligations in the aggregate amount of approximately $177,500 into an aggregate of 25,357 shares of our Series B Preferred. We entered into Securities Purchase Agreements in the form attached to our Current Report on Form 8-K filed on May 13, 2015 (“Securities Purchase Agreement”) with these entities. The shares of Series B Preferred issued pursuant to the Securities Purchase Agreement were offered and sold in transactions exempt from registration under the Securities Act of 1933, as amended, in reliance on 3(a)(9) thereof and Rule 506 of Regulation D thereunder. Each recipient of shares of Series B Preferred represented that it is an "accredited investor" as defined in Regulation D.
Securities Issued for Professional Services
On March 4, 2015, we issued to an accredited investor 25,000 restricted shares of our common stock as compensation for consulting services. Similarly, on March 31, 2015, we issued to an accredited investor 16,667 restricted shares of our common stock in payment of an outstanding balance for professional services. On June 16, 2015, we issued 25,000 restricted shares of our common stock each to two accredited investors as compensation for consulting services. Additionally, between June 10, 2015 and July 13, 2015, we issued an aggregate of 125,000 shares of our Series B Preferred to five professional service providers as compensation for services rendered or to be rendered to us. The securities were issued in private placement transactions exempt from registration under the Securities Act, in reliance on Section 4(2) thereof and Rule 506 of Regulation D thereunder.
Item 16. Exhibits and Financial Statement Schedules
(a) Exhibits. The exhibits are incorporated by reference to the Exhibit Index attached hereto and a part hereof by reference.
(b) Financial Statements. See page F-1 for an index of the financial statements and financial statement schedules included in the Registration Statement.
Item 17. Undertakings
(a) The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement.
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(b) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
SIGNATURES
Pursuant to the requirements of the Securities Act of l933, the registrant has duly caused this Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of South San Francisco, California on the 21st day of July 2015
|
|
VistaGen Therapeutics, Inc.
By: /s/ Shawn K. Singh, JD
Shawn K. Singh, JD
Chief Executive Officer
|
|
|
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints each of Shawn K. Singh, J.D. and Jerrold D. Dotson his true and lawful attorney-in-fact and agent, with full power of substitution, for him and in his name, place and stead, in any and all capacities, to sign any and all amendments to this Registration Statement on Form S-1, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or his substitutes or substitute, may lawfully do or cause to be done by virtue hereof.
IN WITNESS WHEREOF, each of the undersigned has executed this Power of Attorney as of the date indicated opposite his name.
Pursuant to the requirements of the Securities Exchange Act of 1934, Registration Statement has been signed by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
|
Signature
|
|
Title
|
Date
|
|
|
/s/ Shawn K. Singh
Shawn K. Singh, JD
|
|
Chief Executive Officer, and Director
(Principal Executive Officer)
|
July 21, 2015
|
|
|
/s/ Jerrold D. Dotson
Jerrold D. Dotson
|
Vice President and Chief Financial Officer
(Principal Financial and Accounting Officer)
|
July 21, 2015
|
||
|
/s/ H. Ralph Snodgrass
H. Ralph Snodgrass, Ph.D
|
|
President, Chief Scientific Officer and Director
|
July21, 2015
|
|
|
/s/ Jon S. Saxe
Jon S. Saxe
|
|
Chairman of the Board of Directors
|
July 21, 2015
|
|
|
/s/ Brian J. Underdown
Brian J. Underdown, Ph. D
|
|
Director
|
July 21, 2015
|
|
|
Exhibit No.
|
Description*
|
|
|
2.1 *
|
Agreement and Plan of Merger by and among Excaliber Enterprises, Ltd., VistaGen Therapeutics, Inc. and Excaliber Merger Subsidiary, Inc.
|
|
|
3.1 *
|
Articles of Incorporation, dated October 6, 2005.
|
|
|
3.2
|
Certificate of Amendment filed with the Nevada Secretary of State on December 6, 2011, incorporated by reference from Exhibit 3.3 to the Company's Annual Report on Form 10-K, filed July 2, 2012.
|
|
|
3.3
|
Amended and Restated Bylaws as of February 5, 2014, incorporated by reference from the Company’s Report on Form 8-K filed on February 7, 2014.
|
|
|
3.4
|
Articles of Merger filed with the Nevada Secretary of State on May 24, 2011, incorporated by reference from Exhibit 3.1 to the Company's Current Report on Form 8-K filed on May 31, 2011.
|
|
|
3.5
|
Certificate of Designations Series A Preferred, incorporated by reference from Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on December 23, 2011.
|
|
|
3.6
|
Certificate of Change filed with the Nevada Secretary of State on August 11, 2014 incorporated by reference from Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on August 14, 2014.
|
|
|
3.7
|
Certificate of Designation of the Relative Rights and Preferences of the Series B 10% Convertible Preferred Stock of VistaGen Therapeutics, Inc., filed with the Nevada Secretary of State on May 7, 2015, incorporated by reference from Exhibit 3.1 to the Company’s Current Report on Form 8-K filed on May 13, 2015.
|
|
| 5.1 | Opinion of Disclosure Law Group (to be filed by amendment). | |
|
10.1 *
|
1999 Stock Incentive Plan.
|
|
|
10.2 *
|
Form of Option Agreement under VistaGen’s 1999 Stock Incentive Plan.
|
|
|
10.5 *
|
2008 Stock Incentive Plan.
|
|
|
10.6 *
|
Form of Option Agreement under VistaGen’s 2008 Stock Incentive Plan.
|
|
|
10.20 *
|
Strategic Development Services Agreement, dated February 26, 2007, by and between VistaGen and Cato Research Ltd.
|
|
|
10.21 *
|
License Agreement by and between National Jewish Medical and Research Center and VistaGen, dated July 12, 1999, as amended by that certain Amendment to License Agreement dated January 25, 2001, as amended by that certain Second Amendment to License Agreement dated November 6, 2002, as amended by that certain Third Amendment to License Agreement dated March 1, 2003, and as amended by that certain Fourth Amendment to License Agreement dated April 15, 2010.
|
|
|
10.22 *
|
License Agreement by and between Mount Sinai School of Medicine of New York University and the Company, dated October 1, 2004.
|
|
|
10.24 *
|
Sponsored Research Collaboration Agreement, dated September 18, 2007, between VistaGen and University Health Network, as amended by that certain Amendment No. 1 and Amendment No. 2, dated April 19, 2010 and December 15, 2010, respectively.
|
|
|
10.26 *
|
License Agreement, dated October 24, 2001, by and between the University of Maryland, Baltimore, Cornell Research Foundation and Artemis Neuroscience, Inc.
|
|
|
10.40 *
|
Employment Agreement, by and between, VistaGen and Shawn K. Singh, dated April 28, 2010, as amended May 9, 2011.
|
|
|
10.41 *
|
Employment Agreement, by and between, VistaGen and H. Ralph Snodgrass, PhD, dated April 28, 2010, as amended May 9, 2011.
|
|
|
10.46
|
Notice of Award by National Institutes of Health, Small Business Innovation Research Program, to VistaGen Therapeutics, Inc. for project, Clinical Development of 4-CI-KYN to Treat Pain dated June 22, 2009, with revisions dated July 19, 2010 and August 9, 2011, incorporated by reference from Exhibit 10.46 to the Company’s Current Report on Form 8-K/A filed on December 20, 2011.
|
|
|
10.47
|
Notice of Grant Award by California Institute of Regenerative Medicine and VistaGen Therapeutics, Inc. for Project: Development of an hES Cell-Based Assay System for Hepatocyte Differentiation Studies and Predictive Toxicology Drug Screening, dated April 1, 2009, incorporated by reference from Exhibit 10.47 to the Company’s Current Report on Form 8-K/A filed on December 20, 2011.
|
|
|
10.48
|
Amendment No. 4, dated October 24, 2011, to Sponsored Research Collaboration Agreement between VistaGen and University Health Network, incorporated by reference from Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on November 30, 2011.
|
|
|
10.49
|
License Agreement No. 1, dated as of October 24, 2011 between University Health Network and VistaGen Therapeutics, Inc., incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on November 30, 2011.
|
|
|
10.50
|
Strategic Medicinal Chemistry Services Agreement, dated as of December 6, 2011, between Synterys, Inc. and VistaGen Therapeutics, Inc., incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on December 7, 2011.
|
|
|
10.51
|
Common Stock Exchange Agreement, dated as of December 22, 2011 between Platinum Long Term Growth VII, LLC and VistaGen Therapeutics, Inc., incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on December 23, 2011.
|
|
|
10.52
|
Note and Warrant Exchange Agreement, dated as of December 28, 2011 between Platinum Long Term Growth VII, LLC and VistaGen Therapeutics, Inc., incorporated by reference from Exhibit 10.1 to the Current Report on Form 8-K filed on January 4, 2012.
|
|
|
10.55
|
Form of Warrant to Purchase Common Stock, dated as of February 28, 2012, incorporated by reference from Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on March 2, 2012.
|
|
|
10.57
|
License Agreement No. 2, dated as of March 19, 2012 between University Health Network and VistaGen Therapeutics, Inc., incorporated by reference from Exhibit 10.57 to the Company’s Annual Report on Form 10-K filed on July 2, 2012.
|
|
|
10.58
|
Exchange Agreement dated as of June 29, 2012 between Platinum Long Term Growth VII, LLC and VistaGen Therapeutics. Inc., incorporated by reference from Exhibit 10.58 to the Company’s Annual Report on Form 10-K filed on July 2, 2012.
|
|
|
10.63
|
Unsecured Promissory Note in the face amount of $1,000,000 issued to Morrison & Foerster LLP on August 31, 2012 (Replacement Note A), incorporated by reference from Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on September 6, 2012.
|
|
|
10.64
|
Unsecured Promissory Note in the face amount of $1,379,376 issued to Morrison & Foerster LLP on August 31, 2012 (Replacement Note B), incorporated by reference from Exhibit 10.4 to the Company’s Current Report on Form 8-K filed on September 6, 2012.
|
|
|
10.65
|
Stock Purchase Warrant issued to Morrison & Foerster LLP on August 31, 2012 to purchase 1,379,376 shares of the Company’s common stock (New Morrison & Foerster Warrant), incorporated by reference from Exhibit 10.5 to the Company’s Current Report on Form 8-K filed on September 6, 2012.
|
|
|
10.66
|
Warrant to Purchase Common Stock issued to Morrison & Foerster LLP on August 31, 2012 to purchase 425,000 shares of the Company’s common stock (Amended Morrison & Foerster Warrant), incorporated by reference from Exhibit 10.6 to the Company’s Current Report on Form 8-K filed on September 6, 2012.
|
|
|
10.67
|
Note Exchange and Purchase Agreement dated as of October 11, 2012 by and between VistaGen Therapeutics, Inc. and Platinum Long Term Growth VII, LLP, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on October 16, 2012.
|
|
|
10.69
|
Form of Warrant to Purchase Shares of Common Stock issued to Platinum Long Term Growth VII, LLP under the Note Exchange and Purchase Agreement, incorporated by reference from Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on October 16, 2012.
|
|
10.73
|
Amendment to Note Exchange and Purchase Agreement as of November 14, 2012 between VistaGen Therapeutics Inc. and Platinum Long Term Growth VII, LLP, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on November 20, 2012.
|
|
|
10.75
|
Amendment No. 2 to Note Exchange and Purchase Agreement as of January 31, 2013 between VistaGen Therapeutics Inc. and Platinum Long Term Growth VII, LLP, incorporated by reference from Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q filed on February 14, 2013.
|
|
|
10.76
|
Amendment No. 3 to Note Exchange and Purchase Agreement as of February 22, 2013 between VistaGen Therapeutics Inc. and Platinum Long Term Growth VII, LLP, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on February 28, 2013.
|
|
|
10.77
|
Form of Warrant to Purchase Common Stock issued to independent members of the Company’s Board of Directors and its executive officers on March 3, 2013, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on March 6, 2013.
|
|
|
10.80
|
Note Conversion Agreement as of April 4, 2013 between VistaGen Therapeutics Inc. and Platinum Long Term Growth VII, LLP, incorporated by reference from Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on April 10, 2013.
|
|
|
10.81
|
Assignment and Assumption Agreement between Autilion AG and Bergamo Acquisition Corp. PTE LTD dated April 12, 2013, incorporated by reference from Exhibit 10.81 to the Company’s Annual Report on Form 10-K filed July 18, 2013.
|
|
|
10.82
|
Amendment No. 1 to Securities Purchase Agreement dated April 30, 2013 between VistaGen Therapeutics, Inc. and Bergamo Acquisition Corp. PTE LTD, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on May 1, 2013.
|
|
|
10.83
|
Lease between Bayside Area Development, LLC and VistaGen Therapeutics, Inc. (California) dated April 24, 2013, incorporated by reference from Exhibit 10.83 to the Company’s Annual Report on Form 10-K filed July 18, 2013.
|
|
|
10.84
|
Indemnification Agreement effective May 20, 2013 between the Company and Jon S. Saxe, incorporated by reference from Exhibit 10.84 to the Company's Annual Report on Form 10-K filed on July 18, 2013.
|
|
|
10.85
|
Indemnification Agreement effective May 20, 2013 between the Company and Shawn K. Singh, incorporated by reference from Exhibit 10.85 to the Company’s Annual Report on Form 10-K filed on July 18, 2013.
|
|
|
10.86
|
Indemnification Agreement effective May 20, 2013 between the Company and H. Ralph Snodgrass, incorporated by reference from Exhibit 10.86 to the Company’s Annual Report on Form 10-K filed on July 18, 2013.
|
|
|
10.87
|
Indemnification Agreement effective May 20, 2013 between the Company and Brian J. Underdown, incorporated by reference from Exhibit 10.87 to the Company’s Annual Report on Form 10-K filed on July 18, 2013.
|
|
|
10.88
|
Indemnification Agreement effective May 20, 2013 between the Company and Jerrold D. Dotson, incorporated by reference from Exhibit 10.88 to the Company’s Annual Report on Form 10-K filed on July 18, 2013.
|
|
|
10.89
|
Amendment and Waiver effective May 24, 2013 between the Company and Platinum Long Term Growth VII, LLC, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on June 3, 2013.
|
|
|
10.92
|
Common Stock Warrant, dated July 26, 2013 issued to Platinum Long Term Growth VII, LLP, incorporated by reference from Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on August 2, 2013.
|
|
10.93
|
Form of Subscription Agreement between the Company and investors in the Fall 2013 Unit Private Placement, incorporated by reference from Exhibit 10.93 to the Company’s Annual Report on Form 10-K filed on June 24, 2014.
|
|
|
10.94
|
Form of Convertible Promissory Note between the Company and investors in the Fall 2013 Unit Private Placement, incorporated by reference from Exhibit 10.94 to the Company’s Annual Report on Form 10-K filed on June 24, 2014.
|
|
|
10.95
|
Form of Common Stock Purchase Warrant between the Company and investors in the Fall 2013 Unit Private Placement, incorporated by reference from Exhibit 10.95 to the Company’s Annual Report on Form 10-K filed on June 24, 2014.
|
|
|
10.96
|
Form of Amendment to Convertible Promissory Note and Warrant between the Company and investors in the Fall 2013 Unit Private Placement, effective May 31, 2014, incorporated by reference from Exhibit 10.96 to the Company’s Annual Report on Form 10-K filed on June 24, 2014.
|
|
|
10.97
|
Form of Unit Subscription Agreement between the Company and investors in the Spring 2014 Unit Private Placement dated April 1, 2014, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on April 8, 2014.
|
|
|
10.98
|
Form of Subordinate Convertible Promissory Note between the Company and investors in the Spring 2014 Unit Private Placement dated April 1, 2014, incorporated by reference from Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on April 8, 2014.
|
|
|
10.99
|
Form of Common Stock Purchase Warrant between the Company and investors in the Spring 2014 Unit Private Placement dated April 1, 2014, incorporated by reference from Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on April 8, 2014.
|
|
|
10.100
|
Common Stock Purchase Warrant between the Company and Platinum Long Term Growth Fund VII dated May 14, 2014, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on May 19, 2014.
|
|
|
10.103
|
Amendment No. 3 to Sponsored Research Collaboration Agreement, dated April 25, 2011, by and between VistaGen and University Health Network, incorporated by reference from Exhibit 10.103 to the Company’s Annual Report on Form 10-K filed on June 24, 2014.
|
|
|
10.104
|
Amendment No. 5 to Sponsored Research Collaboration Agreement, dated October 10, 2012, by and between VistaGen and University Health Network, incorporated by reference from Exhibit 10.104 to the Company’s Annual Report on Form 10-K filed on June 24, 2014.
|
|
|
10.105
|
Amended and Restated Note Conversion Agreement and Warrant Amendment, by and between VistaGen Therapeutics, Inc. and Platinum Long Term Growth VII, LLC, dated July 18, 2014, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on July 22, 2014.
|
|
|
10.106
|
Amendment No. 1 to Amended and Restated Note Conversion Agreement and Warrant Amendment, by and between VistaGen Therapeutics, Inc. and Platinum Long Term Growth VII, LLC, dated September 2, 2014, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on September 4, 2014.
|
|
|
10.107
|
Amendment No. 2 to Amended and Restated Note Conversion Agreement and Warrant Amendment, by and between VistaGen Therapeutics, Inc. and Platinum Long Term Growth VII, LLC, dated September 30, 2014, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on October 3, 2014.
|
|
|
10.108
|
Agreement, by and between VistaGen Therapeutics, Inc. and Platinum Long Term Growth VII, LLC, dated May 5, 2015, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on May 13, 2015.
|
|
|
10.109
|
Acknowledgement and Agreement, by and between VistaGen Therapeutics, Inc. and Platinum Long Term Growth VII, LLC, dated May 12, 2015, incorporated by reference from Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on May 13, 2015.
|
|
|
10.110
|
Form of Securities Purchase Agreement by and between VistaGen Therapeutics, Inc. and Platinum Long Term Growth VII, LLC, dated May 12, 2015, incorporated by reference from Exhibit 10.3 to the Company’s Current Report on Form 8-K filed on May 13, 2015.
|
|
|
21.1*
|
List of Subsidiaries.
|
|
| 23.1 |
Consent of Disclosure Law Group (included in Exhibit 5.1).
|
|
| 23.2 | Consent of OUM & Co., LLP, independent registered public accounting firm (filed herewith). | |
|
24.1
|
Power of Attorney (included on signature page to this registration statement).
|
|
|
101.INS
|
XBRL Instance Document
|
|
|
101.SCH
|
XBRL Taxonomy Schema
|
|
|
101.CAL
|
XBRL Taxonomy Extension Calculation Linkbase
|
|
|
101.DEF
|
XBRL Taxonomy Extension Definition Linkbase
|
|
|
101.LAB
|
XBRL Taxonomy Extension Label Linkbase
|
|
|
101.PRE
|
XBRL Taxonomy Extension Presentation Linkbase
|
| ___________ |
|
* Incorporated by reference from the like-numbered exhibit filed with our Current Report on Form 8-K on May 16, 2011.