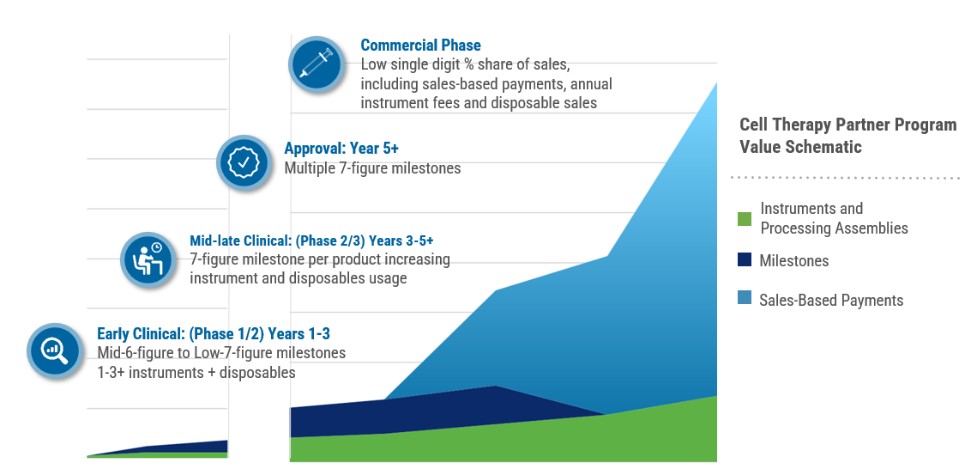
| • | Recurring revenue model provides high visibility, with drivers of potential long-term upside. Our business model enables us to generate revenue from five sources: sales of instruments, disposables and consumables to new customers; additional sales of instruments, disposables and consumables to our existing installed base; annual instrument license fees from cell therapy customers; potential precommercial milestones under SPL partnerships; and potential commercial sales-based payments under SPL partnerships. We generate recurring revenue from our ExPERT instrumentation licenses, as well as disposables and consumables (or buffer) sales, which provides high visibility into future near-term revenue. In addition to recurring revenue, we have the potential to receive meaningful precommercial and commercial payments under SPL partnerships as our customers achieve success in advancing programs through the clinical stage and into the commercial stage. In aggregate, given our SPLs entered into to-date, we have the potential to receive over $1.95 billion in precommercial milestone payments, if all of the programs were to be granted regulatory approvals. |
| • | Leadership team and workforce with deep domain knowledge. Our management team combines strong and broad subject matter expertise with a demonstrated history of commercial and operational execution. Moreover, our workforce has deep domain knowledge across a range of scientific, engineering, regulatory and business disciplines. We have supplemented our diverse technical experience by assembling a deep operational team with expertise in manufacturing, legal, sales, marketing, customer service and finance. We believe the team we have assembled with talent from multiple disciplines and a science- and customer-focused culture represents a significant competitive advantage for us. As of December 31, 2023, of our 143 full-time employees, 81 have advanced degrees including 29 with Ph.D. degrees. |
Our Technology Platform
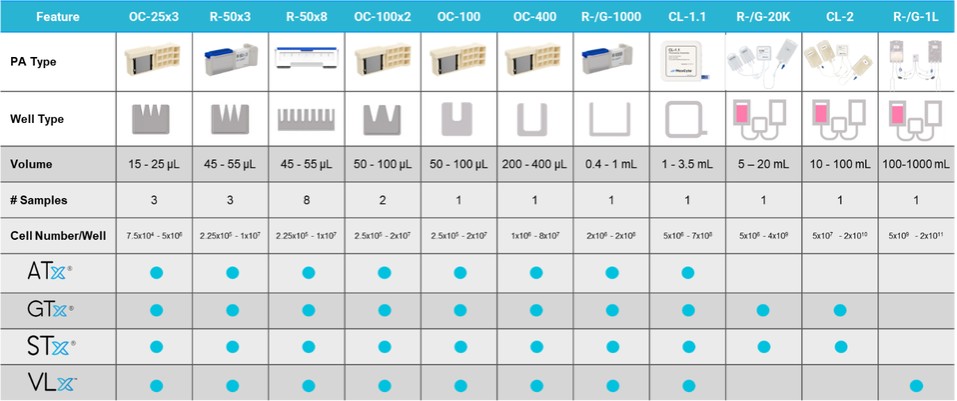
The foundation of our technology platform is our proprietary and patented Flow Electroporation technology, which we have developed and optimized for more than 20 years. Electroporation, or electro-permeabilization, leverages the fundamental properties of cell membranes, the ability to create reversible permeability in the presence of an electric charge, as a universal method to introduce foreign molecules, or transfect, eukaryotic cells, which are cells with a cell membrane and nucleus. Electroporation can be applied to almost any eukaryotic cell type to deliver a broad range of molecules, including DNA, human messenger RNA (“mRNA”), small interfering RNA (“siRNA”), and proteins. Our proprietary Flow Electroporation platform is fully scalable and can support small-scale research and development through large-scale cell engineering for development of commercial therapeutics.

Our technology platform is marketed under the ExPERT brand, the elements of which are depicted in the graphic above. The value of our ExPERT brand starts with Efficiency—with high delivery Efficiency, users can achieve Potency; with high Potency, users improve their chances of therapeutic Efficacy; and if this can be repeated, Reproducibly from patient to patient, users can have a successful Therapy. By delivering high efficiency at any scale, the ExPERT platform is designed to improve our customers’ ability to achieve the required therapeutic index, enabling accelerated, cost-efficient translation of complex cellular therapies from research to clinical development.
10