Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws once our products are commercialized. In addition, healthcare reform legislation has strengthened these laws and additional laws or requirements may be implemented in the future. For example, the Affordable Care Act, among other things, amended the intent requirement of the federal Anti-Kickback and criminal healthcare fraud statutes. As a result of such amendment, a person or entity no longer needs to have actual knowledge of these statutes or specific intent to violate them in order to have committed a violation. Moreover, the Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
Efforts to ensure that our business arrangements comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our existing or future business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. Any such actions instituted against us could have a significant adverse impact on our business, including the imposition of civil, criminal and administrative penalties, damages, disgorgement, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations. Even if we are successful in defending against such actions, we may nonetheless be subject to substantial costs, reputational harm and adverse effects on our ability to operate our business.
If any of our employees, agents, or the physicians or other providers or entities with whom we expect to do business are found to have violated applicable laws, we may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs, or, if we are not subject to such actions, we may suffer reputational harm for conducting business with persons or entities found, or accused of being, in violation of such laws. Any such events could adversely affect our ability to operate our business and our results of operations.
Inadequate funding for the FDA, the SEC and other government agencies, or a work slowdown or stoppage at those agencies as part of a broader federal government shutdown, could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner, or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities, is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which could adversely affect our business. For example, over the last several years, including December 22, 2018 to January 25, 2019, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA and the SEC, have had to furlough employees and stop critical activities. If a prolonged government shutdown or a series of shutdowns occurs, it could significantly affect the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, future government shutdowns could impact our ability to gain access to the public markets and obtain necessary capital in order to properly capitalize and continue our operations, which could have a material adverse effect on our business and financial condition.
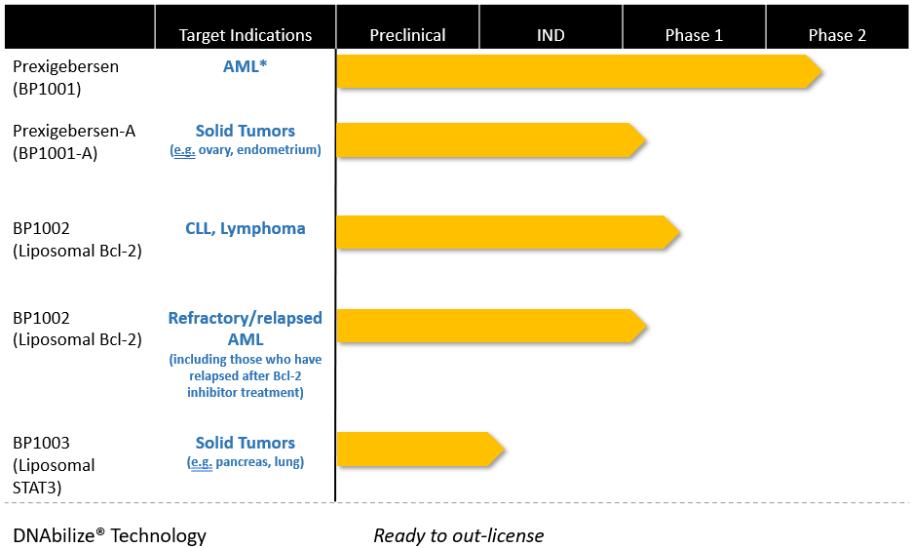
Risks Related to the Development of Our Drug Candidates
We must complete extensive clinical trials to demonstrate the safety and efficacy of our drug candidates. If we are unable to demonstrate the safety and efficacy of our drug candidates, we will not be successful.
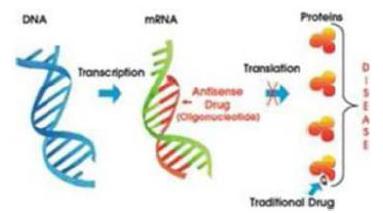
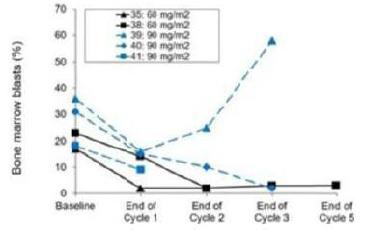
To date, none of our drug candidates have been approved for sale in the U.S. or any foreign country. While antisense therapeutics have been in development for over 20 years, only a limited number of antisense drugs have been successfully developed to date. Further, the development of liposomal antisense therapeutics, which comprise our drug therapeutics technology, has faced