000103526712/312022FYFALSEhttp://fasb.org/us-gaap/2022#NonoperatingIncomeExpensehttp://fasb.org/us-gaap/2022#NonoperatingIncomeExpensehttp://fasb.org/us-gaap/2022#NonoperatingIncomeExpensehttp://fasb.org/us-gaap/2022#RevenueFromContractWithCustomerExcludingAssessedTaxhttp://fasb.org/us-gaap/2022#RevenueFromContractWithCustomerExcludingAssessedTaxhttp://fasb.org/us-gaap/2022#RevenueFromContractWithCustomerExcludingAssessedTaxhttp://fasb.org/us-gaap/2022#RevenueFromContractWithCustomerExcludingAssessedTaxhttp://fasb.org/us-gaap/2022#RevenueFromContractWithCustomerExcludingAssessedTaxhttp://fasb.org/us-gaap/2022#RevenueFromContractWithCustomerExcludingAssessedTaxhttp://www.intuitivesurgical.com/20221231#IntangibleAndOtherAssetshttp://www.intuitivesurgical.com/20221231#IntangibleAndOtherAssetshttp://fasb.org/us-gaap/2022#OtherLiabilitiesCurrenthttp://fasb.org/us-gaap/2022#OtherLiabilitiesCurrenthttp://fasb.org/us-gaap/2022#LiabilitiesOtherThanLongtermDebtNoncurrenthttp://fasb.org/us-gaap/2022#LiabilitiesOtherThanLongtermDebtNoncurrenthttp://fasb.org/us-gaap/2022#Liabilitieshttp://fasb.org/us-gaap/2022#Liabilities12.50002.083312.50002.083314.58332.083325.00002.083333.33332.777825.000033.333300010352672022-01-012022-12-3100010352672022-06-30iso4217:USD00010352672023-02-07xbrli:shares0001035267isrg:SystemsMember2022-01-012022-12-3100010352672022-12-3100010352672021-12-31iso4217:USDxbrli:shares0001035267us-gaap:ProductMember2022-01-012022-12-310001035267us-gaap:ProductMember2021-01-012021-12-310001035267us-gaap:ProductMember2020-01-012020-12-310001035267us-gaap:ServiceMember2022-01-012022-12-310001035267us-gaap:ServiceMember2021-01-012021-12-310001035267us-gaap:ServiceMember2020-01-012020-12-3100010352672021-01-012021-12-3100010352672020-01-012020-12-310001035267us-gaap:CorporateJointVentureMember2022-01-012022-12-310001035267us-gaap:CorporateJointVentureMember2021-01-012021-12-310001035267us-gaap:CorporateJointVentureMember2020-01-012020-12-310001035267us-gaap:CommonStockMember2019-12-310001035267us-gaap:AdditionalPaidInCapitalMember2019-12-310001035267us-gaap:RetainedEarningsMember2019-12-310001035267us-gaap:AccumulatedOtherComprehensiveIncomeMember2019-12-310001035267us-gaap:ParentMember2019-12-310001035267us-gaap:NoncontrollingInterestMember2019-12-3100010352672019-12-310001035267srt:CumulativeEffectPeriodOfAdoptionAdjustmentMemberus-gaap:RetainedEarningsMember2019-12-310001035267srt:CumulativeEffectPeriodOfAdoptionAdjustmentMemberus-gaap:ParentMember2019-12-310001035267srt:CumulativeEffectPeriodOfAdoptionAdjustmentMember2019-12-310001035267us-gaap:CommonStockMember2020-01-012020-12-310001035267us-gaap:AdditionalPaidInCapitalMember2020-01-012020-12-310001035267us-gaap:ParentMember2020-01-012020-12-310001035267us-gaap:RetainedEarningsMember2020-01-012020-12-310001035267us-gaap:AccumulatedOtherComprehensiveIncomeMember2020-01-012020-12-310001035267us-gaap:NoncontrollingInterestMember2020-01-012020-12-310001035267us-gaap:CommonStockMember2020-12-310001035267us-gaap:AdditionalPaidInCapitalMember2020-12-310001035267us-gaap:RetainedEarningsMember2020-12-310001035267us-gaap:AccumulatedOtherComprehensiveIncomeMember2020-12-310001035267us-gaap:ParentMember2020-12-310001035267us-gaap:NoncontrollingInterestMember2020-12-3100010352672020-12-310001035267us-gaap:CommonStockMember2021-01-012021-12-310001035267us-gaap:AdditionalPaidInCapitalMember2021-01-012021-12-310001035267us-gaap:ParentMember2021-01-012021-12-310001035267us-gaap:RetainedEarningsMember2021-01-012021-12-310001035267us-gaap:AccumulatedOtherComprehensiveIncomeMember2021-01-012021-12-310001035267us-gaap:NoncontrollingInterestMember2021-01-012021-12-310001035267us-gaap:CommonStockMember2021-12-310001035267us-gaap:AdditionalPaidInCapitalMember2021-12-310001035267us-gaap:RetainedEarningsMember2021-12-310001035267us-gaap:AccumulatedOtherComprehensiveIncomeMember2021-12-310001035267us-gaap:ParentMember2021-12-310001035267us-gaap:NoncontrollingInterestMember2021-12-310001035267us-gaap:CommonStockMember2022-01-012022-12-310001035267us-gaap:AdditionalPaidInCapitalMember2022-01-012022-12-310001035267us-gaap:ParentMember2022-01-012022-12-310001035267us-gaap:RetainedEarningsMember2022-01-012022-12-310001035267us-gaap:AccumulatedOtherComprehensiveIncomeMember2022-01-012022-12-310001035267us-gaap:NoncontrollingInterestMember2022-01-012022-12-310001035267us-gaap:CommonStockMember2022-12-310001035267us-gaap:AdditionalPaidInCapitalMember2022-12-310001035267us-gaap:RetainedEarningsMember2022-12-310001035267us-gaap:AccumulatedOtherComprehensiveIncomeMember2022-12-310001035267us-gaap:ParentMember2022-12-310001035267us-gaap:NoncontrollingInterestMember2022-12-3100010352672021-09-272021-09-27xbrli:pure00010352672021-09-270001035267us-gaap:AccountsReceivableMemberus-gaap:GeographicDistributionDomesticMemberus-gaap:GeographicConcentrationRiskMember2022-01-012022-12-310001035267us-gaap:AccountsReceivableMemberus-gaap:GeographicDistributionDomesticMemberus-gaap:GeographicConcentrationRiskMember2021-01-012021-12-310001035267us-gaap:GeographicDistributionDomesticMemberus-gaap:GeographicConcentrationRiskMemberisrg:TotalRevenueMember2022-01-012022-12-310001035267us-gaap:GeographicDistributionDomesticMemberus-gaap:GeographicConcentrationRiskMemberisrg:TotalRevenueMember2021-01-012021-12-310001035267us-gaap:GeographicDistributionDomesticMemberus-gaap:GeographicConcentrationRiskMemberisrg:TotalRevenueMember2020-01-012020-12-310001035267us-gaap:GeographicDistributionForeignMemberus-gaap:GeographicConcentrationRiskMemberisrg:TotalRevenueMember2022-01-012022-12-310001035267us-gaap:GeographicDistributionForeignMemberus-gaap:GeographicConcentrationRiskMemberisrg:TotalRevenueMember2021-01-012021-12-310001035267us-gaap:GeographicDistributionForeignMemberus-gaap:GeographicConcentrationRiskMemberisrg:TotalRevenueMember2020-01-012020-12-310001035267us-gaap:BuildingMember2022-01-012022-12-310001035267us-gaap:BuildingImprovementsMember2022-01-012022-12-310001035267isrg:EquipmentAndFurnitureMember2022-01-012022-12-310001035267us-gaap:OtherCapitalizedPropertyPlantAndEquipmentMembersrt:MinimumMember2022-01-012022-12-310001035267srt:MaximumMemberus-gaap:OtherCapitalizedPropertyPlantAndEquipmentMember2022-01-012022-12-310001035267us-gaap:ComputerEquipmentMember2022-01-012022-12-310001035267isrg:EnterpriseWideSoftwareMember2022-01-012022-12-310001035267isrg:PurchasedSoftwareMember2022-01-012022-12-310001035267srt:MaximumMemberus-gaap:SoftwareAndSoftwareDevelopmentCostsMember2022-01-012022-12-31isrg:segment0001035267srt:MinimumMember2022-01-012022-12-310001035267srt:MaximumMember2022-01-012022-12-310001035267srt:MinimumMember2022-12-310001035267srt:MaximumMember2022-12-310001035267isrg:HighCreditRatingMember2022-12-310001035267isrg:ModerateCreditRatingMember2022-12-310001035267isrg:LowCreditRatingMember2022-12-310001035267us-gaap:CashMember2022-12-310001035267us-gaap:MoneyMarketFundsMemberus-gaap:FairValueInputsLevel1Member2022-12-310001035267us-gaap:USTreasurySecuritiesMemberus-gaap:FairValueInputsLevel1Member2022-12-310001035267us-gaap:FairValueInputsLevel1Member2022-12-310001035267us-gaap:CommercialPaperMemberus-gaap:FairValueInputsLevel2Member2022-12-310001035267us-gaap:FairValueInputsLevel2Memberus-gaap:CorporateDebtSecuritiesMember2022-12-310001035267us-gaap:FairValueInputsLevel2Memberus-gaap:USGovernmentAgenciesDebtSecuritiesMember2022-12-310001035267us-gaap:MunicipalNotesMemberus-gaap:FairValueInputsLevel2Member2022-12-310001035267us-gaap:FairValueInputsLevel2Member2022-12-310001035267us-gaap:CashMember2021-12-310001035267us-gaap:MoneyMarketFundsMemberus-gaap:FairValueInputsLevel1Member2021-12-310001035267us-gaap:USTreasurySecuritiesMemberus-gaap:FairValueInputsLevel1Member2021-12-310001035267us-gaap:FairValueInputsLevel1Member2021-12-310001035267us-gaap:CommercialPaperMemberus-gaap:FairValueInputsLevel2Member2021-12-310001035267us-gaap:FairValueInputsLevel2Memberus-gaap:CorporateDebtSecuritiesMember2021-12-310001035267us-gaap:FairValueInputsLevel2Memberus-gaap:USGovernmentAgenciesDebtSecuritiesMember2021-12-310001035267us-gaap:MunicipalNotesMemberus-gaap:FairValueInputsLevel2Member2021-12-310001035267us-gaap:FairValueInputsLevel2Member2021-12-310001035267us-gaap:USTreasurySecuritiesMember2022-12-310001035267us-gaap:CorporateNoteSecuritiesMember2022-12-310001035267us-gaap:USTreasuryAndGovernmentMember2022-12-310001035267us-gaap:MunicipalNotesMember2022-12-310001035267us-gaap:USTreasurySecuritiesMember2021-12-310001035267us-gaap:CommercialPaperMember2021-12-310001035267us-gaap:CorporateNoteSecuritiesMember2021-12-310001035267us-gaap:USTreasuryAndGovernmentMember2021-12-310001035267us-gaap:MunicipalNotesMember2021-12-310001035267us-gaap:FairValueInputsLevel1Member2022-01-012022-12-310001035267us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:FairValueInputsLevel1Member2022-12-310001035267isrg:IntangibleAndOtherAssetsNetMemberus-gaap:FairValueInputsLevel1Member2022-12-310001035267us-gaap:FairValueInputsLevel2Member2022-01-012022-12-310001035267us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:FairValueInputsLevel2Member2022-12-310001035267us-gaap:FairValueInputsLevel2Memberisrg:IntangibleAndOtherAssetsNetMember2022-12-310001035267us-gaap:FairValueInputsLevel1Memberisrg:BroncusHoldingCorporationMember2022-01-012022-12-310001035267us-gaap:NondesignatedMemberus-gaap:ForeignExchangeForwardMember2022-01-012022-12-310001035267us-gaap:NondesignatedMemberus-gaap:ForeignExchangeForwardMember2021-01-012021-12-310001035267us-gaap:NondesignatedMemberus-gaap:ForeignExchangeForwardMember2020-01-012020-12-310001035267us-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMember2022-12-310001035267us-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMember2021-12-310001035267us-gaap:NondesignatedMemberus-gaap:ForeignExchangeForwardMember2022-12-310001035267us-gaap:NondesignatedMemberus-gaap:ForeignExchangeForwardMember2021-12-310001035267us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMember2022-12-310001035267us-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMember2021-12-310001035267us-gaap:NondesignatedMemberus-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:ForeignExchangeForwardMember2022-12-310001035267us-gaap:NondesignatedMemberus-gaap:PrepaidExpensesAndOtherCurrentAssetsMemberus-gaap:ForeignExchangeForwardMember2021-12-310001035267us-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherLiabilitiesMember2022-12-310001035267us-gaap:DesignatedAsHedgingInstrumentMemberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherLiabilitiesMember2021-12-310001035267us-gaap:NondesignatedMemberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherLiabilitiesMember2022-12-310001035267us-gaap:NondesignatedMemberus-gaap:ForeignExchangeForwardMemberus-gaap:OtherLiabilitiesMember2021-12-310001035267us-gaap:GeographicDistributionDomesticMemberisrg:InstrumentsandAccessoriesMember2022-01-012022-12-310001035267us-gaap:GeographicDistributionDomesticMemberisrg:InstrumentsandAccessoriesMember2021-01-012021-12-310001035267us-gaap:GeographicDistributionDomesticMemberisrg:InstrumentsandAccessoriesMember2020-01-012020-12-310001035267us-gaap:GeographicDistributionDomesticMemberisrg:SystemsMember2022-01-012022-12-310001035267us-gaap:GeographicDistributionDomesticMemberisrg:SystemsMember2021-01-012021-12-310001035267us-gaap:GeographicDistributionDomesticMemberisrg:SystemsMember2020-01-012020-12-310001035267us-gaap:ServiceMemberus-gaap:GeographicDistributionDomesticMember2022-01-012022-12-310001035267us-gaap:ServiceMemberus-gaap:GeographicDistributionDomesticMember2021-01-012021-12-310001035267us-gaap:ServiceMemberus-gaap:GeographicDistributionDomesticMember2020-01-012020-12-310001035267us-gaap:GeographicDistributionDomesticMember2022-01-012022-12-310001035267us-gaap:GeographicDistributionDomesticMember2021-01-012021-12-310001035267us-gaap:GeographicDistributionDomesticMember2020-01-012020-12-310001035267us-gaap:GeographicDistributionForeignMemberisrg:InstrumentsandAccessoriesMember2022-01-012022-12-310001035267us-gaap:GeographicDistributionForeignMemberisrg:InstrumentsandAccessoriesMember2021-01-012021-12-310001035267us-gaap:GeographicDistributionForeignMemberisrg:InstrumentsandAccessoriesMember2020-01-012020-12-310001035267isrg:SystemsMemberus-gaap:GeographicDistributionForeignMember2022-01-012022-12-310001035267isrg:SystemsMemberus-gaap:GeographicDistributionForeignMember2021-01-012021-12-310001035267isrg:SystemsMemberus-gaap:GeographicDistributionForeignMember2020-01-012020-12-310001035267us-gaap:ServiceMemberus-gaap:GeographicDistributionForeignMember2022-01-012022-12-310001035267us-gaap:ServiceMemberus-gaap:GeographicDistributionForeignMember2021-01-012021-12-310001035267us-gaap:ServiceMemberus-gaap:GeographicDistributionForeignMember2020-01-012020-12-310001035267us-gaap:GeographicDistributionForeignMember2022-01-012022-12-310001035267us-gaap:GeographicDistributionForeignMember2021-01-012021-12-310001035267us-gaap:GeographicDistributionForeignMember2020-01-012020-12-310001035267isrg:InstrumentsandAccessoriesMember2022-01-012022-12-310001035267isrg:InstrumentsandAccessoriesMember2021-01-012021-12-310001035267isrg:InstrumentsandAccessoriesMember2020-01-012020-12-310001035267isrg:SystemsMember2021-01-012021-12-310001035267isrg:SystemsMember2020-01-012020-12-3100010352672023-01-012022-12-310001035267isrg:VariableLeaseRevenueUsageBasedArrangementsMember2022-01-012022-12-310001035267isrg:VariableLeaseRevenueUsageBasedArrangementsMember2021-01-012021-12-310001035267isrg:VariableLeaseRevenueUsageBasedArrangementsMember2020-01-012020-12-310001035267us-gaap:PrepaidExpensesAndOtherCurrentAssetsMember2022-12-310001035267us-gaap:PrepaidExpensesAndOtherCurrentAssetsMember2021-12-310001035267us-gaap:OtherNoncurrentAssetsMember2022-12-310001035267us-gaap:OtherNoncurrentAssetsMember2021-12-310001035267us-gaap:TechnologyBasedIntangibleAssetsMember2022-12-310001035267us-gaap:TechnologyBasedIntangibleAssetsMember2021-12-310001035267us-gaap:DistributionRightsMember2022-12-310001035267us-gaap:DistributionRightsMember2021-12-310001035267us-gaap:CustomerRelationshipsMember2022-12-310001035267us-gaap:CustomerRelationshipsMember2021-12-310001035267isrg:RexMedicalLPMember2022-10-192022-10-190001035267us-gaap:CommonStockMember2022-07-010001035267isrg:AugustAcceleratedShareRepurchaseProgramMember2022-08-310001035267isrg:AugustAcceleratedShareRepurchaseProgramMember2022-08-012022-08-310001035267isrg:AugustAcceleratedShareRepurchaseProgramMember2022-09-012022-09-300001035267isrg:AugustAcceleratedShareRepurchaseProgramMember2022-08-012022-09-300001035267isrg:OctoberAcceleratedShareRepurchaseProgramMember2022-10-310001035267isrg:OctoberAcceleratedShareRepurchaseProgramMember2022-10-012022-10-310001035267isrg:OctoberAcceleratedShareRepurchaseProgramMember2022-12-012022-12-310001035267isrg:OctoberAcceleratedShareRepurchaseProgramMember2022-10-012022-12-310001035267us-gaap:CommonStockMemberisrg:CommonStockRepurchaseProgramMember2022-01-012022-12-310001035267us-gaap:CommonStockMemberisrg:CommonStockRepurchaseProgramMember2021-01-012021-12-310001035267us-gaap:CommonStockMemberisrg:CommonStockRepurchaseProgramMember2020-01-012020-12-310001035267us-gaap:AccumulatedNetGainLossFromDesignatedOrQualifyingCashFlowHedgesMember2021-12-310001035267us-gaap:AccumulatedNetUnrealizedInvestmentGainLossMember2021-12-310001035267us-gaap:AccumulatedTranslationAdjustmentMember2021-12-310001035267us-gaap:AccumulatedDefinedBenefitPlansAdjustmentMember2021-12-310001035267us-gaap:AccumulatedNetGainLossFromDesignatedOrQualifyingCashFlowHedgesMember2022-01-012022-12-310001035267us-gaap:AccumulatedNetUnrealizedInvestmentGainLossMember2022-01-012022-12-310001035267us-gaap:AccumulatedTranslationAdjustmentMember2022-01-012022-12-310001035267us-gaap:AccumulatedDefinedBenefitPlansAdjustmentMember2022-01-012022-12-310001035267us-gaap:AccumulatedNetGainLossFromDesignatedOrQualifyingCashFlowHedgesMember2022-12-310001035267us-gaap:AccumulatedNetUnrealizedInvestmentGainLossMember2022-12-310001035267us-gaap:AccumulatedTranslationAdjustmentMember2022-12-310001035267us-gaap:AccumulatedDefinedBenefitPlansAdjustmentMember2022-12-310001035267us-gaap:AccumulatedNetGainLossFromDesignatedOrQualifyingCashFlowHedgesMember2020-12-310001035267us-gaap:AccumulatedNetUnrealizedInvestmentGainLossMember2020-12-310001035267us-gaap:AccumulatedTranslationAdjustmentMember2020-12-310001035267us-gaap:AccumulatedDefinedBenefitPlansAdjustmentMember2020-12-310001035267us-gaap:AccumulatedNetGainLossFromDesignatedOrQualifyingCashFlowHedgesMember2021-01-012021-12-310001035267us-gaap:AccumulatedNetUnrealizedInvestmentGainLossMember2021-01-012021-12-310001035267us-gaap:AccumulatedTranslationAdjustmentMember2021-01-012021-12-310001035267us-gaap:AccumulatedDefinedBenefitPlansAdjustmentMember2021-01-012021-12-310001035267isrg:AmendedAndRestatedTwentyTenStockIncentivePlanMember2010-04-012021-12-310001035267isrg:AmendedAndRestatedTwentyTenStockIncentivePlanMember2022-01-012022-12-310001035267isrg:AmendedAndRestatedTwentyTenStockIncentivePlanMember2022-03-310001035267isrg:AmendedAndRestatedTwentyTenStockIncentivePlanMember2019-04-300001035267isrg:AmendedAndRestatedTwentyTenStockIncentivePlanMember2022-12-310001035267isrg:AmendedAndRestatedTwentyTenStockIncentivePlanMemberus-gaap:RestrictedStockUnitsRSUMember2022-12-310001035267isrg:AmendedAndRestatedTwentyZeroNineStockIncentivePlanMember2022-01-012022-12-310001035267isrg:AmendedAndRestatedTwentyZeroNineStockIncentivePlanMember2018-03-310001035267isrg:AmendedAndRestatedTwentyZeroNineStockIncentivePlanMember2015-04-300001035267isrg:EmployeesMemberus-gaap:RestrictedStockUnitsRSUMember2022-01-012022-12-310001035267isrg:AnnualGrantOptionsMemberisrg:AmendedAndRestatedTwentyTenStockIncentivePlanMemberisrg:AugustGrantMember2022-01-012022-12-310001035267isrg:TwoThousandNonEmployeeDirectorsStockOptionPlanMember2009-10-310001035267isrg:TwoThousandNonEmployeeDirectorsStockOptionPlanMember2022-01-012022-12-310001035267isrg:InitialGrantOptionsMemberisrg:TwoThousandNonEmployeeDirectorsStockOptionPlanMember2019-01-012019-12-310001035267isrg:AnnualGrantOptionsMemberisrg:TwoThousandNonEmployeeDirectorsStockOptionPlanMember2019-01-012019-12-310001035267us-gaap:EmployeeStockMemberisrg:TwoThousandEmployeeStockPurchasePlanMember2022-01-012022-12-310001035267us-gaap:EmployeeStockMemberisrg:TwoThousandEmployeeStockPurchasePlanMember2022-12-31isrg:period0001035267us-gaap:EmployeeStockMemberisrg:TwoThousandEmployeeStockPurchasePlanMember2020-03-310001035267us-gaap:EmployeeStockMemberisrg:TwoThousandEmployeeStockPurchasePlanMember2017-04-300001035267us-gaap:EmployeeStockMember2022-12-310001035267us-gaap:RestrictedStockUnitsRSUMember2021-12-310001035267us-gaap:RestrictedStockUnitsRSUMember2022-01-012022-12-310001035267us-gaap:RestrictedStockUnitsRSUMember2022-12-310001035267us-gaap:RestrictedStockUnitsRSUMember2021-01-012021-12-310001035267us-gaap:RestrictedStockUnitsRSUMember2020-01-012020-12-310001035267isrg:RangeOneMember2022-01-012022-12-310001035267isrg:RangeOneMember2022-12-310001035267isrg:RangeTwoMember2022-01-012022-12-310001035267isrg:RangeTwoMember2022-12-310001035267isrg:RangeThreeMember2022-01-012022-12-310001035267isrg:RangeThreeMember2022-12-310001035267isrg:RangeFourMember2022-01-012022-12-310001035267isrg:RangeFourMember2022-12-310001035267isrg:RangeFiveMember2022-01-012022-12-310001035267isrg:RangeFiveMember2022-12-310001035267isrg:RangeSixMember2022-01-012022-12-310001035267isrg:RangeSixMember2022-12-310001035267isrg:RangeSevenMember2022-01-012022-12-310001035267isrg:RangeSevenMember2022-12-310001035267isrg:RangeEightMember2022-01-012022-12-310001035267isrg:RangeEightMember2022-12-310001035267isrg:RangeNineMember2022-01-012022-12-310001035267isrg:RangeNineMember2022-12-310001035267isrg:RangeTenMember2022-01-012022-12-310001035267isrg:RangeTenMember2022-12-310001035267isrg:PerformanceShareUnitsPSUsMember2021-12-310001035267isrg:PerformanceShareUnitsPSUsMember2022-12-310001035267isrg:PerformanceShareUnitsPSUsMember2022-01-012022-12-310001035267us-gaap:EmployeeStockMember2022-01-012022-12-310001035267us-gaap:EmployeeStockMember2021-01-012021-12-310001035267us-gaap:EmployeeStockMember2020-01-012020-12-310001035267isrg:CostOfSalesProductMemberus-gaap:CostOfSalesMember2022-01-012022-12-310001035267isrg:CostOfSalesProductMemberus-gaap:CostOfSalesMember2021-01-012021-12-310001035267isrg:CostOfSalesProductMemberus-gaap:CostOfSalesMember2020-01-012020-12-310001035267isrg:CostOfSalesServiceMemberus-gaap:CostOfSalesMember2022-01-012022-12-310001035267isrg:CostOfSalesServiceMemberus-gaap:CostOfSalesMember2021-01-012021-12-310001035267isrg:CostOfSalesServiceMemberus-gaap:CostOfSalesMember2020-01-012020-12-310001035267us-gaap:CostOfSalesMember2022-01-012022-12-310001035267us-gaap:CostOfSalesMember2021-01-012021-12-310001035267us-gaap:CostOfSalesMember2020-01-012020-12-310001035267isrg:SellingGeneralAndAdministrativeMember2022-01-012022-12-310001035267isrg:SellingGeneralAndAdministrativeMember2021-01-012021-12-310001035267isrg:SellingGeneralAndAdministrativeMember2020-01-012020-12-310001035267isrg:ResearchAndDevelopmentMember2022-01-012022-12-310001035267isrg:ResearchAndDevelopmentMember2021-01-012021-12-310001035267isrg:ResearchAndDevelopmentMember2020-01-012020-12-310001035267us-gaap:StockOptionMember2022-01-012022-12-310001035267us-gaap:StockOptionMember2021-01-012021-12-310001035267us-gaap:StockOptionMember2020-01-012020-12-310001035267isrg:PerformanceShareUnitsPSUsMember2021-01-012021-12-310001035267isrg:PerformanceShareUnitsPSUsMember2020-01-012020-12-310001035267isrg:NonvestedStockOptionMember2022-12-310001035267isrg:NonvestedStockOptionMember2022-01-012022-12-310001035267isrg:AnnualGrantOptionsMember2022-01-012022-12-310001035267isrg:AnnualGrantOptionsMemberisrg:FebruaryGrantMember2022-01-012022-12-310001035267isrg:AnnualGrantOptionsMemberisrg:AugustGrantMember2022-01-012022-12-310001035267isrg:NewHireOptionsMember2022-01-012022-12-310001035267isrg:InitialGrantOptionsMemberisrg:TwoThousandNonEmployeeDirectorsStockOptionPlanMember2000-03-012015-12-310001035267isrg:InitialRSUgrantsMembersrt:DirectorMember2021-01-012021-12-310001035267isrg:DomesticAndForeignMember2022-12-310001035267stpr:CAus-gaap:StateAndLocalJurisdictionMemberus-gaap:ResearchMember2022-12-310001035267us-gaap:AllowanceForCreditLossMember2021-12-310001035267us-gaap:AllowanceForCreditLossMember2022-01-012022-12-310001035267us-gaap:AllowanceForCreditLossMember2022-12-310001035267us-gaap:AllowanceForCreditLossMember2020-12-310001035267us-gaap:AllowanceForCreditLossMember2021-01-012021-12-310001035267us-gaap:AllowanceForCreditLossMember2019-12-310001035267us-gaap:AllowanceForCreditLossMember2020-01-012020-12-31
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
(MARK ONE) | | | | | | | | |
| ☒ | | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2022
OR | | | | | | | | |
| ☐ | | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number 000-30713
Intuitive Surgical, Inc.
(Exact name of Registrant as specified in its Charter) | | | | | | | | |
| Delaware | | 77-0416458 |
| (State or Other Jurisdiction of Incorporation or Organization) | | (I.R.S. Employer Identification Number) |
1020 Kifer Road
Sunnyvale, California 94086
(Address of principal executive offices) (Zip Code)
(408) 523-2100
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act: | | | | | | | | |
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
| Common Stock, par value $0.001 per share | ISRG | The Nasdaq Global Select Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | | | | | | | | | |
| Large accelerated filer | ☒ | | Accelerated filer | ☐ |
| Non-accelerated filer | ☐ | | Smaller reporting company | ☐ |
| | | Emerging growth company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☒
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the voting and non-voting common equity held by non-affiliates on June 30, 2022, based upon the closing price of Common Stock on such date as reported on The Nasdaq Global Select Market, was approximately $71.4 billion. Shares of voting stock held by each officer and director have been excluded in that such persons may be deemed to be affiliates. This assumption regarding affiliate status is not necessarily a conclusive determination for other purposes.
The number of outstanding shares of the registrant’s common stock as of February 7, 2023, was 350,389,679.
DOCUMENTS INCORPORATED BY REFERENCE
Part III incorporates information by reference to the definitive proxy statement for the Company’s Annual Meeting of Stockholders to be held on or about April 27, 2023, to be filed within 120 days of the registrant’s fiscal year ended December 31, 2022.
INTUITIVE SURGICAL, INC.
INDEX
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This report contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Forward-looking statements relate to expectations concerning matters that are not historical facts. Statements using words such as “estimates,” “projects,” “believes,” “anticipates,” “plans,” “expects,” “intends,” “may,” “will,” “could,” “should,” “would,” “targeted,” and similar words and expressions are intended to identify forward-looking statements. These forward-looking statements include, but are not limited to, statements related to the expected impacts of the COVID-19 pandemic on our business, financial condition, and results of operations, future results of operations, future financial condition, our financing plans and future capital requirements, our potential tax assets or liabilities, and statements based on current expectations, estimates, forecasts, and projections about the economies and markets in which we operate and our beliefs and assumptions regarding these economies and markets. These forward-looking statements are necessarily estimates reflecting the judgment of our management and involve a number of risks and uncertainties that could cause actual results to differ materially from those suggested by the forward-looking statements. These forward-looking statements should be considered in light of various important factors, including, but not limited to, the following: the overall macroeconomic environment, which impacts customer spending and our costs, including increased inflation and interest rates; the conflict in Ukraine; disruption to our supply chain, including increased difficulties in obtaining a sufficient supply of materials in the semiconductor and other markets; the risk that the COVID-19 pandemic could lead to material delays and cancellations of, or reduced demand for, procedures; curtailed or delayed capital spending by hospitals; closures of our facilities; delays in surgeon training; delays in gathering clinical evidence; delays in obtaining new product approvals, clearances, or certifications from the U.S. Food and Drug Administration (“FDA”), comparable regulatory authorities, or notified bodies; diversion of resources to respond to COVID-19 outbreaks; the impact of global and regional economic and credit market conditions on healthcare spending; the risk of our inability to comply with complex FDA and other regulations, which may result in significant enforcement actions; regulatory approvals, clearances, certifications, and restrictions or any dispute that may occur with any regulatory body; guidelines and recommendations in the healthcare and patient communities; healthcare reform legislation in the U.S. and its impact on hospital spending, reimbursement, and fees levied on certain medical device revenues; changes in hospital admissions and actions by payers to limit or manage surgical procedures; the timing and success of product development and market acceptance of developed products; the results of any collaborations, in-licensing arrangements, joint ventures, strategic alliances, or partnerships, including the joint venture with Shanghai Fosun Pharmaceutical (Group) Co., Ltd.; our completion of and ability to successfully integrate acquisitions, including Orpheus Medical; procedure counts; intellectual property positions and litigation; competition in the medical device industry and in the specific markets of surgery in which we operate; risks associated with our operations and any expansion outside of the United States; unanticipated manufacturing disruptions or the inability to meet demand for products; our reliance on sole and single source suppliers; the results of legal proceedings to which we are or may become a party, including, but not limited to, product liability claims; adverse publicity regarding us and the safety of our products and adequacy of training; the impact of changes to tax legislation, guidance, and interpretations; changes in tariffs, trade barriers, and regulatory requirements; and other risks and uncertainties, including those listed under the caption “Risk Factors.” Readers are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date of this report and which are based on current expectations and are subject to risks, uncertainties, and assumptions that are difficult to predict. Our actual results may differ materially and adversely from those expressed in any forward-looking statement, and we undertake no obligation to publicly update or release any revisions to these forward-looking statements, except as required by law. Additional risks are described throughout this report, particularly in Part I, “Item 1A. Risk Factors,” and include, but are not limited to, those summarized on the following pages.
RISKS RELATING TO OUR BUSINESS
•Macroeconomic conditions could have a materially adverse impact on our business, financial condition, or results of operations.
•Our reliance on sole and single source suppliers and ability to purchase at acceptable prices a sufficient supply of materials, parts, and components could harm our ability to meet product demand in a timely manner or within budget.
•Public health crises or epidemic diseases, or the perception of their effects, could materially adversely affect our business, financial condition, or results of operations.
•We are subject to litigation, investigations, and other legal proceedings relating to our products, our customers, our competitors, and government regulators that could materially adversely affect our financial condition, divert management’s attention, and harm our business.
•Because our markets are highly competitive, customers may choose to purchase our competitors’ products or services or may not accept robotic-assisted medical procedures, which could result in reduced revenue and loss of market share.
•If our products do not achieve and maintain market acceptance, we will not be able to generate the revenue necessary to support our business.
•If hospitals are unable to obtain coverage and reimbursement for procedures using our products, if reimbursement is insufficient to cover the costs of purchasing our products, or if limitations are imposed by governments on the amount hospitals can charge for certain procedures, we may be unable to generate sufficient sales to support our business.
•If our products contain defects or encounter performance problems, we may have to recall our products and, as a result, incur additional unforeseen costs, and our reputation may suffer.
•We could be subject to significant, uninsured losses, which may have a material adverse impact on our business, financial condition, or results of operations.
•If we lose key personnel or are unable to attract and retain other personnel, our ability to compete will be harmed, and increases in labor costs could materially adversely impact our business, financial condition, or results of operations.
•Negative publicity, whether accurate or inaccurate, concerning our products or our company could reduce market acceptance of our products and could result in decreased product demand and reduced revenues.
•We experience long and variable capital sales cycles and seasonality in our business, which may cause fluctuations in our financial results.
•New product developments and introductions may adversely affect our business, financial condition, or results of operations.
•We are subject to a variety of risks due to our operations outside of the U.S.
•We may incur losses associated with currency fluctuations and may not be able to effectively hedge our exposure.
•Our customers may use remanufactured and/or unauthorized third-party instruments and accessories, which could result in reduced revenue and negatively impact our reputation.
•Information technology system failures, cyberattacks, or deficiencies in our cybersecurity could harm our business, customer relations, financial condition, or results of operations.
•Our business is subject to complex and evolving laws and regulations regarding privacy, data protection, and other matters relating to information collection.
•If we fail to successfully acquire or integrate new businesses, products, and technology, we may not realize expected benefits, or our business may be harmed.
•If we do not successfully manage our collaboration arrangements, licensing arrangements, joint ventures, strategic alliances, or partnerships with third parties, we may not realize the expected benefits from such alliances, which may have a material adverse effect on our business, financial condition, or results of operations.
•We expect gross profit margins to vary over time, and changes in our gross profit margins could adversely affect our business, financial condition, or results of operations.
•We utilize distributors for a portion of our sales and service of our products in certain countries, which subjects us to a number of risks that could harm our business, financial condition, or results of operations.
•We offer alternative capital acquisition approaches and, as a result, we are exposed to the credit risk of some of our customers and the risk of losses of revenue, which could result in material losses.
•We are exposed to credit risk and fluctuations in the market value of our investments.
•The ongoing armed conflict between Russia and Ukraine could adversely affect our business, financial condition, or results of operations.
•We may encounter manufacturing problems or delays that could result in lost revenue.
•Disruptions at the FDA and other government agencies or notified bodies caused by funding shortages or global health concerns could hinder their ability to hire, retain, or deploy key leadership and other personnel, or otherwise prevent products from being developed, cleared, certified, approved, or commercialized in a timely manner or at all, which may adversely affect our business.
•We are subject to risks associated with real estate construction and development.
•Continued consolidation in the healthcare industry could have an adverse effect on our business, financial condition, or results of operations.
•Climate change and natural disasters or other events beyond our control could disrupt our business and result in loss of revenue or higher expenses.
•Changes in our effective tax rate may adversely affect our business, financial condition, or results of operations.
•We use estimates, make judgments, and apply certain methods in determining our financial results and in measuring the progress of our business. As these estimates, judgments, and methods change, our results of operations and our assessment of the progress of our business could vary.
RISKS RELATING TO OUR REGULATORY ENVIRONMENT
•Complying with FDA and foreign regulations is a complex process, and our failure to fully comply could subject us to significant enforcement actions.
•Our products are subject to a lengthy and uncertain domestic regulatory review process. If we do not obtain and maintain the necessary domestic regulatory authorizations, we will not be able to sell our products in the U.S.
•Our products may cause or contribute to adverse medical events or be subject to failures or malfunctions that we are required to report to the FDA and foreign regulatory authorities and, if we fail to do so, we would be subject to sanctions that could harm our reputation, business, financial condition, or results of operations.
•If our manufacturing facilities do not continue to meet federal, state, or other manufacturing standards, we may be required to temporarily cease all or part of our manufacturing operations, import/export of our products, and/or recall some products, which would result in significant product delivery delays and lost revenue.
•Our products are subject to international regulatory processes and approval or certification requirements. If we do not obtain and maintain the necessary international regulatory approvals or certifications, we will not be able to sell our products in other countries.
•Changes in healthcare legislation and policy may have a material adverse effect on our business, financial condition, or results of operations.
•We are subject to federal, state, and foreign laws governing our business practices, which, if violated, could result in substantial penalties. Additionally, challenges to, or investigation into, our practices could cause adverse publicity and be costly to respond to and, thus, could harm our business.
•If hospitals and other surgery facilities do not continue to meet federal, state, or other regulatory standards, they may be required to temporarily cease all or part of their system utilization.
RISKS RELATING TO OUR INTELLECTUAL PROPERTY
•If we are unable to fully protect and successfully defend our intellectual property from use by third parties, our ability to compete in the market may be harmed.
•Others may be successful in asserting that our products infringe their intellectual property rights, which may cause us to pay substantial damages and/or enjoin us from commercializing our products.
•Our products rely on licenses from third parties, which may not be available to us on commercially reasonable terms or at all. If we lose access to these technologies, our revenues could decline.
GENERAL RISK FACTORS
•Our future operating results may be below securities analysts’ or investors’ expectations, which could cause our stock price to decline.
•Our stock price has been, and will likely continue to be, volatile.
•Changes to financial accounting standards may affect our reported results of operations.
The summary of material risk factors described above should be read together with the text of the full risk factors below in the section entitled “Item 1A. Risk Factors” and the other information set forth in this Annual Report on Form 10-K, including our Consolidated Financial Statements and the related notes, as well as other documents that we file with the U.S. Securities and Exchange Commission. The risks summarized above or described in full below are not the only risks that we face. Additional risks and uncertainties not precisely known to us or that we currently deem to be immaterial may also materially adversely affect our business, financial condition, or results of operations.
PART I
ITEM 1. BUSINESS
In this report, “Intuitive Surgical,” “Intuitive,” the “Company,” “we,” “us,” and “our” refer to Intuitive Surgical, Inc. and its wholly and majority-owned subsidiaries. Intuitive®, Intuitive Surgical®, da Vinci®, da Vinci S®, da Vinci Si®, da Vinci X®, da Vinci Xi®, da Vinci SP®, EndoWrist®, Firefly®, Ion®, Iris®, OnSite®, SimNow®, SureForm®, and SynchroSeal® are trademarks or registered trademarks of the Company.
Company Background
As part of Intuitive’s mission, we believe minimally invasive care is life-enhancing care. Through ingenuity and intelligent technology, we expand the potential of physicians to heal without constraints. We envision a future of care that is less invasive and profoundly better, where diseases are identified earlier and treated quickly so patients can get back to what matters most.
Intuitive is committed to advancing minimally invasive care through a comprehensive ecosystem of products and services. This connected ecosystem includes systems, instruments and accessories, learning, and services connected by a digital portfolio that enables actionable digital insights across the care continuum and provides enhanced capabilities, intraoperative guidance, decision support, and a personalized learning journey, all with the goal to help improve outcomes and efficiency.
Intuitive brings nearly three decades of experience and technical innovation to our robotic-assisted surgical solutions. While surgery and acute interventions have improved significantly in the past decades, there remains a significant need for better outcomes and decreased variability of these outcomes across care teams. The current healthcare environment continues to stress critical resources, including the professionals who staff care teams: surgeons, anesthesiologists, nurses, and other staff. At the same time, governments strain to cover the healthcare needs of their populations and demand lower total costs per patient to treat disease. In the face of these challenges, we believe scientific and technological advances in biology, computing, imaging, algorithms, and robotics may offer new methods to solve continued and difficult problems.
We address our customers’ needs by sharing their goals reflected in the quadruple aim. First, we focus on improving patient outcomes through an ecosystem of advanced robotic systems, instruments and accessories, progressive technology learning pathways, and comprehensive support and program assistance services. Second, we seek to improve the patient experience by minimizing disruption to lives and creating greater predictability for the treatment experience. Third, we seek to improve care team satisfaction by creating products and services that are dependable, smart, and optimized for the care environment in which they are used. Finally, we seek to lower the total cost to treat per patient episode when compared with existing treatment alternatives, providing a return on investment for hospitals and healthcare systems and value for payers.
Products
Systems
Advanced robotic systems provide precise, powerful systems with high-performance vision, extending the care team’s capabilities to enhance minimally invasive care. These systems include the da Vinci Surgical System, which was designed to enable complex surgery using a minimally invasive approach, and the Ion endoluminal system, which extends our commercial offerings beyond surgery into diagnostic procedures, enabling minimally invasive biopsies in the lung.
Da Vinci Surgical Systems
By striving to find less invasive ways to enter the body, provide clearer views of anatomy and more precise tissue interactions, and help hone surgical skills, Intuitive launched its first da Vinci Surgical System in 1999. In 2000, the FDA cleared da Vinci for general laparoscopic surgery.
There are several models of the da Vinci Surgical System: our fourth generation da Vinci X, da Vinci Xi, and da Vinci SP Surgical Systems, our third generation da Vinci Si Surgical System, our second generation da Vinci S Surgical System, and our first generation da Vinci standard Surgical System. The da Vinci surgical systems are designed to enable surgeons to perform a wide range of surgical procedures within our targeted general surgery, urologic, gynecologic, cardiothoracic, and head and neck specialties. To date, surgeons have used the da Vinci Surgical System to perform dozens of different types of surgical procedures. Da Vinci systems offer surgeons three-dimensional, high definition (“3DHD”) vision, a magnified view, and robotic and computer assistance. They use specialized instrumentation, including a miniaturized surgical camera (endoscope) and wristed instruments (e.g., scissors, scalpels, and forceps) that are designed to help with precise dissection and reconstruction deep inside the body.
Our da Vinci surgical systems are comprised of the following components:
Surgeon’s Console. The da Vinci Surgical System allows surgeons to operate while comfortably seated at an ergonomic console viewing a 3DHD image of the surgical field. The surgeon’s fingers grasp instrument controls below the display
with the surgeon’s hands naturally positioned relative to his or her eyes. Using electronic hardware, software, algorithms, and mechanics, our technology translates the surgeon’s hand movements into precise and corresponding real-time micro movements of the da Vinci instruments positioned inside the patient. On most of our current systems (da Vinci X, da Vinci Xi, da Vinci SP, and da Vinci Si), a second surgeon’s console may be used in two ways: to provide assistance to the primary surgeon during surgery or to act as an active aid during surgeon-proctor training sessions. With the da Vinci X, da Vinci Xi, da Vinci SP, and da Vinci Si, a surgeon sitting at a second console can view the same surgery as the primary surgeon and can be passed control of some or all of the da Vinci instruments during the surgery. In addition, surgeons can control 3D virtual pointers to augment the dual-surgeon experience. The da Vinci Surgical System is designed to allow surgeons to operate while seated, which may be clinically advantageous because of reduced surgeon fatigue. The da Vinci Surgical System’s design provides natural hand-eye alignment at the surgeon’s console. Because the da Vinci Surgical System’s robotic arms hold the camera and instruments steady, there is less surgeon and assistant fatigue.
Patient-Side Cart. The patient-side cart holds electromechanical arms that manipulate the instruments inside the patient. Up to four arms attached to the cart can be positioned, as appropriate, and then locked into place. At least two arms hold surgical instruments, one representing the surgeon’s left hand and one representing the surgeon’s right hand. A third arm positions the endoscope, allowing the surgeon to easily move, zoom, and rotate the field of vision. A fourth instrument arm extends surgical capabilities by enabling the surgeon to add a third instrument to perform additional tasks. The fourth instrument arm is a standard, integrated feature on the da Vinci X, da Vinci Xi, and da Vinci Si Surgical Systems. Our da Vinci SP Surgical System includes a single arm with three multi-jointed, wristed instruments and the first da Vinci fully wristed, 3DHD camera. The instruments and the camera all emerge through a single cannula and are triangulated around the target anatomy to avoid external instrument collisions that can occur in narrow surgical workspaces.
3DHD Vision System. Our vision system includes a 3DHD endoscope with two independent vision channels linked to two separate color monitors through sophisticated image processing electronics and software. The resulting 3DHD image has high resolution, high contrast, low flicker, and low cross fading. A digital zoom feature in the 3DHD vision system allows surgeons to magnify the surgical field of view without adjusting the endoscope position and, thereby, reduces interference between the endoscope and instruments. The 3DHD vision system is a standard, integrated feature on the da Vinci X, da Vinci Xi, da Vinci SP, da Vinci Si, and da Vinci S Surgical Systems.
Firefly Fluorescence Imaging (“Firefly”). Firefly is a standard feature of the da Vinci X, da Vinci Xi, and da Vinci SP Surgical Systems and is available as an upgrade on our da Vinci Si Surgical System. This imaging capability combines an injectable fluorescent dye with a specialized da Vinci camera head, endoscope, and laser-based illuminator to allow surgeons to identify vasculature, tissue perfusion, or biliary ducts in three dimensions beneath tissue surfaces in real-time. The most common procedural categories for the use of Firefly are urology, gynecology, and general surgery.
Da Vinci Xi Integrated Table Motion. Integrated Table Motion coordinates the movements of the da Vinci robotic arms with an advanced operating room (“OR”) table, the TS 7000dV OR Table sold by HillromTM, to enable managing the patient’s position in real-time while the da Vinci robotic arms remain docked. This gives OR teams the capability to improve the positioning of the operating table during da Vinci Surgical System procedures. Integrated Table Motion enables the patient to be dynamically positioned during the procedure. It enables surgeons to extend reach, facilitate access, and choose the angle of approach to target anatomy, as well as reposition the table during the procedure to enhance anesthesiologists’ management of the patient.
Ion Endoluminal System
In 2019, the FDA cleared our Ion endoluminal system, which is a flexible, robotic-assisted, catheter-based platform that utilizes instruments and accessories for which the first cleared indication is minimally invasive biopsies in the lung. Our Ion system extends our commercial offering beyond surgery into diagnostic, endoluminal procedures. The system features an ultra-thin, ultra-maneuverable catheter that can articulate 180 degrees in all directions and allows navigation far into the peripheral lung and provides the stability necessary for precision in a biopsy. Many suspicious lesions found in the lung may be small and difficult to access, which can make diagnosis challenging, and Ion helps physicians obtain tissue samples from deep within the lung, which could help enable earlier diagnosis.
Instruments and Accessories
We offer a comprehensive suite of stapling, energy, and core instrumentation for our surgical systems. Our technology is designed to transform the surgeon’s natural hand movements outside of the body into corresponding micro-movements inside the patient’s body and suture with precision, just as they can in open surgery. With our technology, a surgeon can also use “motion scaling,” a feature that translates, for example, a three-millimeter hand movement outside the patient’s body into a one-millimeter instrument movement in the surgical field inside the patient’s body. Motion scaling is designed to allow precision and control for delicate tasks. In addition, our technology filters the tremor inherent in a surgeon’s hands.
Da Vinci Instruments. Most of the various instruments that we manufacture incorporate EndoWrist technology with wristed joints for natural dexterity and tips customized for various surgical procedures. Da Vinci instruments are offered in a variety of diameters, of which 8mm and 12mm diameter sizes are the most commonly sold. Various da Vinci instrument tips include forceps, scissors, electrocautery tools, scalpels, and other surgical tools that are familiar to the surgeon from open surgery and conventional minimally invasive surgery (“MIS”). A variety of instruments may be selected and used interchangeably during a surgery. Most instruments are sterilizable at the hospital, while others are provided sterile, and most are reusable for a defined number of procedures. A programmed memory chip inside each instrument performs several functions that help determine how the da Vinci system and instruments work together. In addition, the chip generally will not allow the instrument to be used for more than the prescribed number of procedures to help ensure that its performance meets specifications during each procedure.
In 2020, we announced our “Extended Use Program,” which consists of select da Vinci Xi and da Vinci X instruments possessing 12 to 18 uses (“Extended Use Instruments”) compared to the previously 10 uses. These Extended Use Instruments represent some of our higher volume instruments but exclude stapling, monopolar, and advanced energy instruments. Instruments included in the program are used across a number of da Vinci surgeries. Their increased uses are the result of continuous, significant investments in the design and production capabilities of our instruments, resulting in improved quality and durability. Extended Use Instruments were introduced in the U.S. and Europe in the fourth quarter of 2020 and were launched in most other countries around the world during the first half of 2021, except China due to regulatory timelines. We believe that, as of the end of 2021, in the U.S. and Europe, full cutover to Extended Use Instruments had occurred, as customers had substantially utilized all of their remaining 10 use instruments.
Da Vinci Stapling. The EndoWrist and SureForm Staplers are wristed, stapling instruments intended for resection, transection, and creation of anastomoses. These instruments enable operators to precisely position and fire the stapler. We have various clearances for five staplers that can be used with the da Vinci X and da Vinci Xi Surgical Systems: the EndoWrist Stapler 30 and 45 and the SureForm Stapler 30, 45, and 60, where the numeric designation indicates the length of the staple line. The EndoWrist Stapler 30 is intended to deliver particular utility with fine tissue interaction in lobectomy and other thoracic procedures. The EndoWrist Stapler 45 is used in general surgery, gynecologic, thoracic, and urologic procedures. The SureForm Staplers 30, 45, and 60 are single-use, fully wristed, stapling instruments intended to be used in general surgery, thoracic, gynecologic, urologic, and pediatric surgery procedures. The SureForm Stapler 30 may deliver particular utility in thoracic procedures. The SureForm 45 may deliver particular utility in thoracic and colorectal procedures where maneuverability and visualization are limited. The SureForm Stapler 60 is intended to deliver particular utility in bariatric procedures. We also have various clearances for five stapler reloads: gray (2.0 mm), white (2.5 mm), blue (3.5 mm), green (4.3 mm), and black (4.6 mm). Not all reloads are available for use on all staplers. Not all staplers or reloads are available in all countries.
Da Vinci Energy. The EndoWrist One Vessel Sealer is a wristed, single-use instrument intended for bipolar coagulation and mechanical transection of vessels up to 7mm in diameter and tissue bundles that fit in the jaws of the instrument. This instrument enables surgeons to fully control vessel sealing, while providing the benefits of robotic-assisted surgery. This instrument is designed to enhance surgical efficiency and autonomy in a variety of general surgery and gynecologic procedures. The da Vinci Vessel Sealer Extend is our newest instrument in the Vessel Sealing family of products. The da Vinci Vessel Sealer Extend is a single-use, fully wristed bipolar electrosurgical instrument compatible with our fourth-generation multiport systems. It is intended for grasping and blunt dissection of tissue and for bipolar coagulation and mechanical transection of vessels up to 7mm in diameter and tissue bundles that fit in the jaws of the instrument.
The E-100 generator is Intuitive’s first generator and is offered as an upgrade to power our da Vinci Vessel Sealer Extend and SynchroSeal instruments. SynchroSeal enables a surgeon to perform rapid, one-step sealing and transection with a single pedal press. SynchroSeal uses advanced bipolar energy from its raised cut electrode to transect tissue and then cool down quickly.
Accessory Products. We sell various accessory products, which are used in conjunction with the da Vinci Surgical Systems as surgical procedures are performed. Accessory products include sterile drapes used to help ensure a sterile field during surgery, vision products, such as replacement 3D stereo endoscopes, camera heads, and light guides, and other items that facilitate use of the da Vinci Surgical Systems.
Learning
Intuitive provides progressive learning pathways to support the safe and effective use of our technology. They are structured and measured training pathways for surgeons, physicians, and care teams. These pathways leverage both learning engagements and learning technologies. Learning engagement touchpoints vary by specific pathway, skill level, and interest, while learning technologies enable and provide training directly to the customer. The portfolio of learning offerings includes role-specific Training Pathways, Learning Engagements, and Learning Technology.
Training Pathways. Intuitive Training Pathways are progressive learning journeys that help our customers achieve proficiency using Intuitive technology. There are pathways for surgeons and physicians, residents and fellows, OR care teams, patient side assists, and robotic coordinators, as well as recommendations for executives.
Learning Engagements. Intuitive Learning Engagements are touchpoints that support customers throughout their learning journeys. They vary by pathway, skill level, and focus area. Engagements include case observations, online education, in-service training, simulation/skills training, OR care team training, technology training, reprocessing training, proctoring, advanced training, and curriculum development support. Many of these programs take place at established Intuitive training centers and include instruction by expert surgeons and physicians.
Learning Technology. Learning Technologies include solutions that provide education and training directly to the customer as well as the enabling technologies that make provision possible. Intuitive’s enabling technologies include Telepresence and the Procedure Analytics Platform. Specific technology solutions include Intuitive Learning, SimNow, customized training models, remote case observations, and remote proctoring. Two of the technology solutions most heavily utilized by customers are Intuitive Learning and SimNow.
Intuitive Learning. Intuitive Learning provides our customers with access to the technology, procedure, and simulation materials essential to their specific learning journeys. Both assignment of learning materials and tracking of learning progress occur seamlessly within the platform. While Intuitive Learning plans guide learners through each step in their pathways, customers are also able to search the platform independently for additional materials that may be relevant to their area of focus. This platform also provides customers with immediate access to their various training certificates.
SimNow. Our cloud-enabled SimNow simulation platform is a practice tool that gives a user the opportunity to practice their skills and gain familiarity with the surgeon console controls and supports the user’s progressive learning pathway. SimNow incorporates 3D, physics-based computer simulation technology to immerse the user within a virtual environment and provides training capabilities that have been used extensively by surgeons. The user navigates through the environment and completes exercises by controlling virtual instruments from the surgeon console. Upon completion of a skills exercise, the skills simulator provides a quantitative assessment of user performance based on a variety of task-specific metrics. The SimNow online connection drives real-time simulation performance tracking for surgeons and administrators through an online dashboard and supports remote updates of the VR content and 3DHD videos to drive a more interactive and engaging customer experience. SimNow is intended to augment, not replace, existing training programs for the da Vinci X, da Vinci Xi, and da Vinci SP Surgical Systems.
Services
We have a network of field service engineers across the U.S., Europe, and Asia and maintain relationships with various distributors around the globe. This infrastructure of service and support specialists offers a full complement of services for our customers, including installation, repair, maintenance, 24/7 technical support, and proactive system health monitoring.
Our comprehensive support and program assistance helps ensure customers and care teams maximize program performance and protect their investment. Services include readiness support, maintenance support, perioperative consulting, Custom Hospital Analytics, and market consulting optimization.
Readiness and Maintenance Support. Readiness support is operational support to ensure smooth onboarding and adoption of new systems and technology. Maintenance support helps to maximize operational efficiency and reduce unplanned equipment downtime. It includes services care plans, support teams, OnSite monitoring, software upgrades and updates, as well as a customer portal. The service plan portfolio offers flexible service plans to ensure reliability of the systems and instruments and help optimize the robotics program. The support team of expert field service, remote technical support, and customer care agents resolve and prevent any technology issues that could inhibit optimal utilization. OnSite monitoring offers remote service in real-time for pre-operative and intraoperative troubleshooting, as well as proactive monitoring of system performance. Software upgrades and updates enable the latest product innovations, enhancements, and reliability improvements. The customer portal is an online tool that enables customers to access system utilization and program analytics, view orders and maintenance history, and initiate product returns and exchanges to help achieve the operational and financial goals of a robotics program.
Perioperative Consulting. Perioperative consulting is a suite of customized solutions to improve a hospital’s efficiency and performance with Intuitive technologies. New system integration support is available to streamline the start-up process and expedite increased procedure volumes. Overall program assessments help to support efficiency improvements, cost reductions, and system access optimization.
Program Analytics. Our Custom Hospital Analytics program enables the integration of data sources so that individual health institutions can analyze their data in their own environment. Using this data, executives, administrators, care teams, and surgeons can gain alignment around their programs based on their KPIs, determine best practices, assess gaps, and take actionable steps to address any gaps.
Digital Solutions
Integrated digital capabilities provide connected offerings, streamlining performance for hospitals with program-enhancing insights. Secure-by-design, cloud-enabled products analyze and simplify essential data to continuously optimize the use of time, tools, and techniques.
Intuitive Hosted & Managed Services. The vast majority of our systems are network connected and directly communicate with Intuitive to enable proactive monitoring as well as provide software updates and data insights to Intuitive customers.
3D Modeling Services. Iris is our augmented reality imaging product for use in kidney procedures. The service extracts CT scans, runs them through machine-learning algorithms and, after technicians’ revision and radiologists’ review, returns a 3D segmented model of the kidney for use in planning for a procedure and for intraoperative visualization of the area. The tool uses augmented reality to give surgeons an image with details of the kidney anatomy – blood vessels, tumor shape, and size – that they may not be able to see well with other imaging. Intuitive designed this to help with pre-operative planning and intraoperative guidance as well as to be shared as a teaching tool for other physicians and patients. It can also be part of the viewing experience inside of the da Vinci surgeon console to enhance information and let surgeons know where critical anatomy sits as they work through a procedure. We have completed several pilot studies with Iris to obtain customer feedback, which is currently being incorporated to support a future product launch.
My Intuitive. This mobile and web application was developed to be the single point for Intuitive customers to access individual or program-level data from Intuitive. The application also offers comparisons of those insights with anonymized national benchmarks to help drive operational efficiencies and decreased costs. The most recent version enables mobile access to Intuitive’s Learning platform, case reports generated automatically for the surgeon, and an ability for surgeons to publish their practice information online for patients seeking local physicians.
Intuitive Hub. Intuitive Hub is part of our OR informatics platform that integrates multiple applications and data sets to help orchestrate medical procedure workflows. For the care team, Intuitive Hub acts as a point-of-care device that automates tasks, such as video recording and bookmarking, and can be used to facilitate peer-to-peer collaboration utilizing our Telepresence offering. For physicians, Intuitive Hub connects video and other data that can be accessed after a medical procedure to help facilitate personalized learning and increased efficiency.
Business Strategy
We align our goals to those of our customers, often called the Quadruple Aim: enabling physicians and hospitals to improve outcomes for their patients, improve their patient’s and the care team’s experience, and lower the total cost to treat per patient episode. Through the use of smart, connected systems, robotic technologies, advanced imaging, and informatics, our objective is to create value for patients, surgeons, and hospitals, as summarized below.
Patient Value. We believe that the value of a medical procedure to a patient can be defined: Patient Value = Procedure Efficacy / Invasiveness. We define procedure efficacy as a measure of the success of the procedure in helping resolve the underlying disease and invasiveness as a measure of patient pain and disruption of regular activities. When the patient value of a procedure using an Intuitive product is greater than that of alternative treatment options, patients may benefit from seeking out surgeons and hospitals that offer those products, which could potentially result in a local market share shift. Adoption of Intuitive technology occurs procedure by procedure and market by market and is driven by the relative patient value and the total treatment costs of da Vinci procedures as compared to alternative treatment options for the same disease state or condition. We believe that most patients will place a higher value on procedures that are not only more efficacious but also less invasive than alternative treatments. Our goal is to provide products to surgeons who, in turn, provide patients with procedure options that are both highly effective and less invasive than others.
Surgeon Value. We offer physicians and their operating room staff training on the technical use of our products. We provide an ergonomic platform through our da Vinci surgical system for surgeons to perform their procedures. We seek to provide surgeons with reliable and easy-to-use products. For example, the change to cloud-based analytics and routine use of local analytics may help surgeons track their procedures and processes and, with a network-connected smartphone and the My Intuitive app, surgeons can access and explore their procedure data, such as console time and instrument usage, to gain insights into their program.
Hospital Value. We assist hospitals in building value by offering patient value using da Vinci products, thereby increasing surgical revenue and reducing costs through lower complication rates and reduced lengths of patient stay. For example, we believe robotic-assisted surgery with the da Vinci Surgical System is a cost-effective approach to many surgeries as compared to alternative treatment options, as recognized in many published studies. We also offer our Custom Hospital Analytics program, which enables the integration of data sources so that individual health institutions can analyze their data in their own environment. Using this data, administrators, chiefs of surgery, and surgeons can gain
alignment around their programs based on their KPIs, determine best practices, assess gaps, and take actionable steps to address any gaps.
Clinical Applications
We are the beneficiaries of productive collaborations with leading surgeons in exploring and developing new techniques and applications for robotic-assisted surgery with the da Vinci Surgical System and minimally invasive biopsies with the Ion endoluminal system—an important part of our creative process. We primarily focus our development efforts on those procedures in which we believe our products bring the highest patient value, surgeon value, and hospital value. We currently focus on five surgical specialties: general surgery, urologic surgery, gynecologic surgery, cardiothoracic surgery, and head and neck surgery. Key procedures that we are focused on include hernia repair, colon and rectal procedures, cholecystectomy, bariatric surgery, prostatectomy, partial nephrectomy, hysterectomy, sacrocolpopexy, lobectomy, and transoral robotic surgery. We also focus on minimally invasive biopsies in the lung. Representative surgical applications are described below.
General Surgery
Hernia Repair. A hernia occurs when an organ or other tissue squeezes through a weak spot in a surrounding muscle or connective tissue. During a hernia repair surgery, the weakened tissue is secured, and defects are repaired. Common types of hernias are ventral and inguinal. Ventral, or abdominal hernia, may occur through a scar after surgery in the abdomen. Inguinal hernia is a bulge in the groin and is more common in men. Hernia repair can be performed using traditional open surgery or MIS. There is a wide range of complexity in hernia repair surgeries and varying surgeon opinions regarding optimal surgical approach. The benefits of minimally invasive and robotic-assisted hernia repair surgery vary by patient.
Colorectal Surgery. These procedures typically involve benign or cancerous conditions of the lower digestive system, in particular the rectum or colon. Common procedures in this area include hemicolectomy, sigmoidectomy, low anterior resection, and abdominoperineal resection. Surgeons have reported that the use of robotic-assisted surgery with a da Vinci Surgical System and our latest technologies, such as the EndoWrist Stapler and da Vinci Energy, has enabled them to offer MIS approaches to a broader range of colorectal surgery patients.
Cholecystectomy. Cholecystectomy, or the surgical removal of the gall bladder, is a commonly performed general surgery procedure. Cholecystectomy is the primary method for the treatment of gallstones and other gall bladder diseases. Most cholecystectomies are performed using multi-port MIS techniques, although some surgeons choose to perform cholecystectomy using manual single-port instrumentation. Firefly technology can be used to visualize biliary anatomy in three dimensions beneath the tissue surfaces during multi-port da Vinci cholecystectomies.
Bariatric Surgery. A body of literature points to the benefit of surgery to treat patients with morbid obesity and its secondary effects, such as diabetes. Sleeve gastrectomy and Roux-en-Y gastric bypass (“RYGB”) are commonly performed surgical procedures for morbid obesity in the U.S. The body habitus of morbidly obese patients can make laparoscopic surgery physically challenging for the surgeon, and certain surgeons have found value in using the da Vinci Surgical System to improve upon the ergonomics when performing MIS in morbidly obese patients. In addition, RYGB can be a technically challenging procedure due to the suturing, stapling, and tissue (bowel) manipulation that is required. Surgeons using the da Vinci Surgical System have reported a reduction in a critical complication (anastomotic leaks) relative to laparoscopic RYGB. Also, we believe SureForm 60 may have particular utility in bariatric procedures.
Urologic Surgery
Prostatectomy. Radical prostatectomy is the removal of the prostate gland in patients diagnosed with clinically localized prostate cancer. The standard approach to the removal of the prostate was via an open surgical procedure. The conventional laparoscopic approach is an option, but it is difficult and poses challenges to even the most skilled urologist. The da Vinci Surgical System has enabled a large number of surgeons to convert from using an open surgical technique to a minimally invasive technique.
Partial Nephrectomy. Partial nephrectomy is the removal of a small portion of a kidney (typically, an area of the kidney containing a tumor). Partial nephrectomies are most commonly performed in patients diagnosed with clinically localized renal cancer. Excluding robotic-assisted surgery with a da Vinci Surgical System, there are three common surgical approaches to performing partial nephrectomies: open surgical technique, laparoscopy, and hand-assisted laparoscopy, which is a hybrid of the open and laparoscopic techniques. Surgeons have reported that the da Vinci Surgical System’s capabilities may enable a large number of these procedures to be performed through a minimally invasive technique, conferring the benefits of MIS to a broader range of partial nephrectomy patients. Treatment guidelines for patients with localized renal cancer recommend partial nephrectomy due to the benefits that nephron-sparing surgery has in long-term patient outcomes. Published clinical literature has shown that the presence of a da Vinci Surgical System is associated with a higher-proportion of patients receiving a guideline-recommended partial nephrectomy.
Gynecologic Surgery
Hysterectomy. Removal of the uterus is one of the most commonly performed surgeries in gynecology and is performed for a variety of underlying benign and cancerous conditions. Hysterectomies can be performed using open surgery (laparotomy) or MIS techniques, which include vaginal, laparoscopic, and robotic-assisted approaches. Prior to the clearance of the da Vinci Surgical System for use in gynecological procedures in 2005, the majority of hysterectomies performed were open surgeries. We believe that robotic-assisted surgery with the da Vinci Surgical System provides patients the opportunity to receive a minimally invasive treatment as an alternative to an open hysterectomy.
Sacrocolpopexy. The abdominal (open) sacrocolpopexy is one of the operations performed to treat vaginal vault prolapse. Sacrocolpopexy involves suturing a synthetic mesh that connects and supports the vagina to the sacrum (tailbone). A sacrocolpopexy can be performed using a conventional laparoscopic technique; however, it is generally described as difficult and cumbersome to perform. Surgeons have reported that the da Vinci Surgical System’s capabilities may enable a larger number of these procedures to be performed through a minimally invasive technique, conferring the benefits of MIS to a broader range of sacrocolpopexy patients.
Cardiothoracic Surgery
Thoracic Surgery. Conventional approaches to surgical procedures in the thorax include both open and video-assisted thoracoscopic approaches. Procedures performed via these methods include pulmonary wedge resection, pulmonary lobectomy, thymectomy, mediastinal mass excision, and esophagectomy. Many thoracic procedures remain open procedures. Surgeons have reported that the use of robotic-assisted surgery with a da Vinci Surgical System in thoracic surgery has enabled them to offer MIS approaches to a broader range of thoracic surgery patients and improved clinical outcomes compared to open and video-assisted thoracic surgery in published single-center, multi-center, and national database clinical studies. Also, we believe the EndoWrist Stapler 30 and the SureForm Stapler 30 may have particular utility in thoracic procedures.
Head and Neck Surgery
Transoral Surgery. Head and neck cancers are typically treated by either surgical resection or chemo-radiation, or a combination of both. Surgical resection performed by an open approach may require a “jaw-splitting” mandibulotomy. This procedure, while effective in treating cancer, is potentially traumatic and disfiguring to the patient. MIS approaches via the mouth (transoral surgery) are challenged by line-of-sight limitations dictated by conventional endoscopic tools. Chemo-radiation as a primary therapy does allow patients to avoid traumatic surgical incisions; however, the literature suggests that this modality diminishes patients’ ability to speak and swallow normally. Surgeons have reported that da Vinci transoral surgery allows them to operate on tumors occurring in the oropharynx (i.e., tonsil and base of tongue) and larynx via the mouth and to overcome some of the line-of-sight limitations of conventional transoral surgery.
Da Vinci Procedure Mix
Our da Vinci procedure business is broadly split into two categories: (1) cancer procedures and (2) procedures for benign conditions. Cancer and other highly complex procedures tend to be reimbursed at higher rates than less complex procedures for benign conditions. Thus, hospitals are more sensitive to the costs associated with treating less complex, benign conditions. Our strategy is to provide hospitals with attractive clinical and economical solutions across the spectrum of procedure complexity. Our fully featured da Vinci Xi Surgical System with advanced instruments, including the da Vinci Energy and EndoWrist and SureForm Stapler products, and our Integrated Table Motion product, targets the more complex procedure segment. Our da Vinci X Surgical System is targeted toward price-sensitive markets and procedures. Our da Vinci SP Surgical System complements the da Vinci Xi and X Surgical Systems by enabling surgeons to access narrow workspaces.
Clinical Summary
There are over 70 representative clinical uses for da Vinci Surgical Systems. We believe that there are numerous additional applications that can be addressed with the da Vinci Surgical System, and we work closely with our surgeon customers to refine and explore new techniques in which a da Vinci Surgical System may bring value. As of December 31, 2022, we had an installed base of 7,544 da Vinci Surgical Systems, including 4,563 in the U.S., 1,388 in Europe, 1,234 in Asia, and 359 in the rest of the world. We estimate that surgeons using our technology completed approximately 1,875,000 surgical procedures of various types in hospitals throughout the world during the year ended December 31, 2022.
Additionally, over time, we believe that there are numerous additional applications that can be addressed with the Ion endoluminal system. As of December 31, 2022, we had an installed base of 321 Ion systems, 320 of which are located in the U.S. We plan to seek additional clearances for Ion in markets outside of the U.S. (“OUS”) over time.
Sales and Customer Support
Sales Model
We provide our products through direct sales organizations in the U.S., Europe (excluding Spain, Portugal, Italy, Greece, and most Eastern European countries), China, Japan, South Korea, India, Taiwan and, as of June 2022, Canada. We provide products and services in China through our majority-owned joint venture (“Joint Venture”) with Shanghai Fosun Pharmaceutical (Group) Co., Ltd. (“Fosun Pharma”) and its affiliates. See “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” for further details on the Joint Venture. In the remainder of our markets outside of the U.S., we provide our products through distributors. During the years ended December 31, 2022, 2021, and 2020, domestic revenue accounted for 67%, 67%, and 68%, respectively, of total revenue, while revenue from our OUS markets accounted for 33%, 33%, and 32%, respectively, of total revenue.
Our direct sales organization is composed of a capital sales team, responsible for selling systems, and a clinical sales team, responsible for supporting the systems used in procedures performed at our hospital accounts. Our hospital accounts include both individual hospitals and healthcare facilities as well as hospitals and healthcare facilities that are part of an integrated delivery network (“IDN groups”). The initial system sale into an account is a major capital equipment purchase by our customers and typically has a lengthy sales cycle that can be affected by macroeconomic factors, capital spending prioritization, the timing of budgeting cycles, and competitive bidding processes. Capital sales activities include educating surgeons or physicians and hospital staff across multiple specialties on the benefits of robotic-assisted surgery with a da Vinci Surgical System or robotic-assisted bronchoscopy with an Ion endoluminal system, total treatment costs, and the clinical applications that our technology enables. We also train our sales organization to educate hospital management on the potential benefits of adopting our technology, including the clinical benefits of robotic-assisted surgery with a da Vinci Surgical System or robotic-assisted bronchoscopy with an Ion endoluminal system, in support of their quadruple aim objectives.
Our clinical sales team works on site at hospitals, interacting with surgeons or physicians, operating room staff, and hospital administrators to develop and sustain successful robotic-assisted surgery or bronchoscopy programs. They assist the hospital in identifying surgeons or physicians who have an interest in robotic-assisted surgery or bronchoscopy and the potential benefits provided by the da Vinci Surgical System and the Ion endoluminal system. Our clinical sales team provides current clinical information on robotic-assisted surgery or bronchoscopy practices and new product applications to the hospital teams. Our clinical sales team has grown with the expanded installed bases of da Vinci Surgical Systems and Ion endoluminal systems as well as the total number of procedures performed. We expect this organization to continue to grow as our business expands.
Our customers place orders to replenish their supplies of instruments and accessories on a regular basis. Orders received are typically shipped within one business day. New direct customers who purchase a system typically place an initial stocking order of instruments and accessories soon after they receive their system.
Our business is subject to seasonal fluctuations. Historically, our sales of da Vinci Surgical Systems have tended to be heavier in the fourth quarter and lighter in the first quarter, as hospital budgets are reset. In addition, we have historically experienced lower procedure volume in the first and third quarters and higher procedure volume in the second and fourth quarters. More than half of da Vinci procedures performed are for benign conditions. These benign procedures and other short-term elective procedures tend to be more seasonal than cancer procedures and surgeries for other life-threatening conditions. In the U.S., volumes for procedures associated with benign conditions are typically seasonally higher in the fourth quarter when more patients have met annual deductibles and lower in the first quarter when deductibles are reset. Seasonality outside of the U.S. varies and is more pronounced around local holidays and vacation periods. The timing of procedures and changes in procedure volume impact the timing of instruments and accessories and capital purchases. As a result of factors outlined in “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations—COVID-19 Pandemic” below, including the past and potential future recommendations of authorities to defer elective procedures, historical procedure patterns have been and may continue to be disrupted.
Customer Support
We have a network of field service and technical support engineers across the U.S., Europe, and Asia and maintain relationships with various distributors around the globe. This infrastructure of service and support specialists, along with advanced service tools and solutions, offers a full complement of services for our customers, including installation, repair, maintenance, 24/7 technical support, and proactive system health monitoring. We generate service revenue by providing these services to our customers through comprehensive service contracts and time and material programs.
Research and Development
We focus our research and development efforts on innovation and improvement for products and services that align with our mission: We believe that minimally invasive care is life-enhancing care. Through ingenuity and intelligent technology, we
believe that we can expand the potential of physicians to heal without constraints. We employ engineering and research and development staff to focus on delivering future innovations and sustaining improvements that advance our mission. In certain instances, we complement our research and development effort through collaborations with other companies, such as Baxter International Inc.
Manufacturing
We manufacture our systems at our facilities in Sunnyvale, California, Durham, North Carolina and, as of 2022, Peachtree Corners, Georgia. We manufacture our instruments at our facilities in Sunnyvale, California and Mexicali, Mexico. We also have manufacturing at multiple sites in Germany.
We purchase both custom and off-the-shelf components from a large number of suppliers and subject them to stringent quality specifications and processes. Some of the components necessary for the assembly of our products are currently provided to us by sole-sourced suppliers (the only recognized supply source available to us) or single-sourced suppliers (the only approved supply source for us among other sources). We purchase the majority of our components and major assemblies through purchase orders rather than long-term supply agreements and generally do not maintain large volumes of finished goods relative to our anticipated demand.
Competition
We face competition in the forms of existing open surgery, conventional MIS, drug therapies, radiation treatment, and other emerging diagnostic and interventional surgical approaches. Our success depends on continued clinical and technical innovation, quality, and reliability, as well as educating hospitals, surgeons, and patients on the demonstrated results associated with robotic-assisted medical procedures using da Vinci Surgical Systems or Ion endoluminal systems and their value relative to other techniques. We also face competition from several companies that have introduced or are developing new approaches and products for the MIS market. We believe that the entrance or emergence of competition validates MIS and robotic-assisted surgery or robotic-assisted bronchoscopy.
Moreover, as we add new robotically controlled products (e.g., da Vinci Stapling and da Vinci Energy) that compete with product offerings traditionally within the domains of open surgery and/or conventional MIS, we face greater competition from larger and well-established companies, such as Johnson & Johnson and Medtronic plc.
The companies that have introduced products in the field of robotic-assisted medical procedures or have made explicit statements about their efforts to enter the field include, but are not limited to, the following companies: Asensus Surgical, Inc.; avateramedical GmbH; CMR Surgical Ltd.; Johnson & Johnson; Medicaroid Corporation; Medrobotics Corporation; Medtronic plc; meerecompany Inc.; Olympus Corporation; Samsung Electronics Co., Ltd; Shandong Weigao Group Medical Polymer Company Ltd.; Shanghai Microport Medbot (Group) Co., Ltd.; and Titan Medical Inc. Other companies with substantial experience in industrial robotics could potentially expand into the field of medical robotics and become a competitor. In addition, research efforts utilizing computers and robotics for medical procedures are underway at various companies and research institutions. Our revenues may be adversely impacted as our competitors announce their intent to enter our markets and as our customers anticipate the availability of competing products.
Intellectual Property
We place considerable importance on obtaining and maintaining patent, copyright, trademark, and trade secret protection for significant new technologies, products, and processes.
We generally rely upon a combination of intellectual property laws, confidentiality procedures, and contractual provisions to protect our proprietary technology. For example, we have trademarks, both registered and unregistered, that provide distinctive identification of our products in the marketplace. We also have exclusive and non-exclusive patent licenses with various third parties to supplement our own robust patent portfolio.
As of December 31, 2022, we owned more than 4,300 patents granted and still in force and more than 2,100 patents pending worldwide. We intend to continue filing new patent applications in the U.S. and foreign jurisdictions to seek protection for our technology.
Patents are granted for finite terms. Upon expiration, the inventions claimed in a patent enter the public domain.
Government Regulation
Our products and operations are subject to regulation in the U.S. by the FDA and the State of California as well as by other countries and regions in which we market and promote our products. In addition, our products must meet the requirements of a large and growing body of international standards, which govern the design, manufacture, materials content and sourcing, testing, certification, packaging, installation, use, and disposal of our products. We must continually keep abreast of these regulations, standards, and requirements and integrate our compliance into the development and regulatory documentation for
our products. Failure to meet these standards could limit our ability to market our products in those regions that require compliance with such standards. Examples of standards to which we are subject include electrical safety standards, such as those of the International Electrotechnical Commission (e.g., IEC 60601-ss series of standards), and composition standards, such as the Reduction of Hazardous Substances (“RoHS”) and the Waste Electrical and Electronic Equipment (“WEEE”) Directives.
U.S. Regulation
Our products are subject to regulation as medical devices in the United States under the Federal Food, Drug, and Cosmetic Act (“FFDCA”), as implemented and enforced by the FDA. The FDA regulates the development, design, non-clinical and clinical research, manufacturing, safety, efficacy, labeling, packaging, storage, installation, recordkeeping, complaint and adverse event reporting, clearance, approval, certification, promotion, marketing, export, import distribution, and service of medical devices in the U.S. to ensure that medical devices distributed domestically are safe and effective for their intended uses.
Under the FFDCA, medical devices are classified into one of three classes—Class I, Class II, or Class III—depending on the degree of risk associated with each medical device and the extent of control needed to ensure safety and effectiveness. Our current products are Class II medical devices.
Class II medical devices are those that are subject to general controls, and most require premarket demonstration of adherence to certain performance standards or other special controls, as specified by the FDA, and special controls as deemed necessary by the FDA to ensure the safety and effectiveness of the device. These special controls can include performance standards, post-market surveillance, patient registries, and FDA guidance documents.
Manufacturers of most Class II devices are required to submit to the FDA a premarket notification under Section 510(k) of the FFDCA requesting authorization to commercially distribute the device. The FDA’s authorization to commercially distribute a device subject to a 510(k) premarket notification is generally known as 510(k) clearance. Our current products are subject to premarket notification and clearance under section 510(k) of the FFDCA. To obtain 510(k) clearance, we must submit to the FDA a premarket notification submission demonstrating that the proposed device is “substantially equivalent” to a legally marketed predicate device.
The FDA may require additional information, including clinical data, to make a determination regarding substantial equivalence. In addition, the FDA collects user fees for certain medical device submissions and annual fees for medical device establishments.
If the FDA agrees that the device is substantially equivalent to a predicate device, it will grant clearance to commercially market the device in the U.S. The FDA has a statutory 90-day period to respond to a 510(k) submission; however, as a practical matter, clearance often takes longer. The FDA may require further information, including clinical data, to make a determination regarding substantial equivalence. If the FDA determines that the device, or its intended use, is not “substantially equivalent,” the device may be designated as a Class III device. The device sponsor must then fulfill more rigorous PMA requirements or can request a risk-based classification determination for the device in accordance with the de novo classification pathway, which is a route to market for novel medical devices that are low to moderate risk and are not substantially equivalent to a predicate device.
The PMA process is more demanding than the 510(k) premarket notification process. In a PMA application, the manufacturer must demonstrate that the device is safe and effective, and the PMA application must be supported by extensive data, including data from preclinical studies and human clinical trials. The FDA, by statute and regulation, has 180 days to review a PMA application, although the review more often occurs over a significantly longer period of time and can take up to several years. In approving a PMA application or clearing a 510(k) submission, the FDA may also require some additional manufacturing controls, design control activities and approvals, as well as specific post-market surveillance requirements when necessary to protect the public health or to provide additional safety and effectiveness data for the device. In such cases, the manufacturer might be required to follow certain patient groups for a number of years and make periodic reports to the FDA on the clinical status of those patients.
Clinical trials are almost always required to support a PMA and are sometimes required to support a 510(k) submission. All clinical investigations designed to determine the safety and effectiveness of a medical device must be conducted in accordance with the FDA’s investigational device exemption (“IDE”) regulations, which govern investigational device labeling, prohibit the promotion of the investigational device, and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. Regardless of the degree of risk presented by the medical device, clinical studies must be approved by, and conducted under the oversight of, an Institutional Review Board (“IRB”) for each clinical site. During a study, the sponsor is required to comply with the applicable FDA requirements, including, for example, trial monitoring, selecting clinical investigators and providing them with the investigational plan, ensuring IRB review, adverse event reporting, record keeping, and prohibitions on the promotion of investigational devices or on making safety or effectiveness claims for them. The clinical investigators in the clinical study are also subject to the FDA’s regulations and must obtain patient informed
consent, rigorously follow the investigational plan and study protocol, control the disposition of the investigational device, and comply with all reporting and recordkeeping requirements. Additionally, after a trial begins, we, the FDA, or the IRB could suspend or terminate a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits.
Over the last several years, the FDA has proposed reforms to its 510(k) clearance process, and such proposals could include increased requirements for clinical data and a longer review period and make it more difficult for manufacturers to utilize the 510(k) clearance process for their products. For example, in September 2019, the FDA issued revised final guidance describing an optional “safety and performance based” premarket review pathway for manufacturers of “certain, well-understood device types” to demonstrate substantial equivalence under the 510(k) clearance pathway by showing that such device meets objective safety and performance criteria established by the FDA, thereby obviating the need for manufacturers to compare the safety and performance of their medical devices to specific predicate devices in the clearance process. The FDA maintains a list of device types appropriate for the “safety and performance based” pathway and continues to develop product-specific guidance documents that identify the performance criteria for each such device type, as well as the recommended testing methods, where feasible.
After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change or modification in its intended use, will require a new 510(k) clearance or, depending on the modification, PMA approval or de novo classification. The FDA requires each manufacturer to determine whether the proposed change requires submission of a 510(k), de novo classification, or a PMA in the first instance, but the FDA can review any such decision and disagree with a manufacturer’s determination. If the FDA disagrees with a manufacturer’s determination, the FDA can require the manufacturer to cease marketing and/or request the recall of the modified device until 510(k) marketing clearance, approval of a PMA, or issuance of a de novo classification. Also, in these circumstances, the manufacturer may be subject to significant regulatory fines or penalties.
In addition, the FDA may place significant limitations upon the intended use of our products as a condition of granting marketing authorization. Moreover, after a device is placed on the market, numerous FDA and other regulatory requirements continue to apply. These requirements include establishment registration and device listing with the FDA; compliance with medical device reporting regulations, which require that manufacturers report to the FDA if their device caused or contributed, or may have caused or contributed, to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; compliance with corrections and removal reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FFDCA that may present a risk to health; the FDA’s recall authority, whereby the agency can order device manufacturers to recall from the market a product that is in violation of governing laws and regulations; and post-market surveillance activities and regulations, which apply when deemed by the FDA to be necessary to protect the public health or to provide additional safety and effectiveness data for the device. In addition, the FDA and the Federal Trade Commission also regulate the advertising and promotion of our products to ensure that the claims we make are consistent with our regulatory clearances, that there is scientific data to substantiate the claims, and that our advertising is neither false nor misleading. In general, we may not promote or advertise our products for uses not within the scope of our intended use statement in our clearances or make unsupported safety and effectiveness claims.
Our manufacturing processes are required to comply with the Quality System Regulation (“QSR”). The QSR covers, among other things, the methods used in, and the facilities and controls used for, the design, testing, controlling, documenting, manufacture, packaging, labeling, storage, installation, and servicing of all medical devices intended for human use. The QSR also requires maintenance of extensive records, which demonstrate compliance with the FDA regulation, the manufacturer’s own procedures, specifications, and testing, as well as distribution and post-market experience. Compliance with the QSR is necessary for a manufacturer to be able to continue to market cleared or approved product offerings in the U.S. A company’s facilities, records, and manufacturing processes are subject to periodic scheduled or unscheduled inspections by the FDA. Failure to maintain compliance with applicable QSR requirements could result in the shut-down of, or restrictions on, manufacturing operations and the recall or seizure of marketed products. If the FDA determines that a manufacturer has failed to comply with applicable regulatory requirements, it can take a variety of compliance or enforcement actions, which may result in any of the following sanctions:
•warning letters, untitled letters, fines, injunctions, consent decrees, administrative penalties, and civil or criminal penalties;
•recalls, withdrawals, or administrative detention or seizure of our products;
•operating restrictions or partial suspension or total shutdown of production;
•refusing or delaying requests for 510(k) marketing clearance or PMA approvals of new products or modified products;
•withdrawing 510(k) clearances or PMA approvals that have already been granted;
•refusal to grant export approvals for our products; or
•criminal prosecution.
In addition, the discovery of previously unknown problems with any marketed products, including unanticipated adverse events or adverse events of increasing severity or frequency, whether resulting from the use of the device within the scope of its clearance or off-label by a physician in the practice of medicine, could result in restrictions on the device, including the removal of the product from the market or voluntary or mandatory device recalls.
Products manufactured outside of the U.S. by or for us are subject to U.S. Customs and FDA inspection upon entry into the U.S. We must demonstrate compliance of such products with U.S. regulations and carefully document the eventual distribution or re-exportation of such products. Failure to comply with all applicable regulations could prevent us from having access to products or components critical to the manufacture of finished products and lead to shortages and delays.
Data Privacy and Security Laws
Numerous state, federal, and foreign laws, regulations, and standards govern the collection, use, access to, confidentiality, and security of health-related and other personal information and could apply now or in the future to our operations or the operations of our partners. In the U.S., numerous federal and state laws and regulations, including data breach notification laws, health information privacy and security laws, and consumer protection laws and regulations govern the collection, use, disclosure, and protection of health-related and other personal information. In addition, certain foreign laws govern the privacy and security of personal data, including health-related data. Privacy and security laws, regulations, and other obligations are constantly evolving, may conflict with each other to complicate compliance efforts, and can result in investigations, proceedings, or actions that lead to significant civil and/or criminal penalties and restrictions on data processing.
We collect, process, share, disclose, transfer, and otherwise use data, some of which contains personal information about identifiable individuals including, but not limited to, our employees, clinical trial participants, partners, and vendors. Therefore, we are subject to U.S. (federal, state, local) and international laws and regulations, including those in the European Economic Area (“EEA”) and the UK regarding data privacy and security and our use of such data.
We are subject to the European Union General Data Protection Regulation 2016/679 and applicable national supplementing laws (collectively, the “EU GDPR”) and to the United Kingdom General Data Protection Regulation and Data Protection Act 2018 (collectively, the “UK GDPR”) (the EU GDPR and UK GDPR together referred to as the “GDPR”). The GDPR imposes comprehensive data privacy compliance obligations in relation to our collection, processing, sharing, disclosure, transfer, and other use of data relating to an identifiable living individual or “personal data,” including a principle of accountability and the obligation to demonstrate compliance through policies, procedures, training, and audit.
The EU GDPR and UK GDPR also regulate cross-border transfers of personal data out of the EEA and the UK. Recent legal developments in Europe have created complexity and uncertainty regarding such transfers, in particular in relation to transfers to the United States.
Cybersecurity
In the normal course of business, we may collect and store personal information and other sensitive information, including proprietary and confidential business information, trade secrets, intellectual property, patient information, sensitive third-party information, and employee information. To protect this information, our existing cybersecurity policies require continuous monitoring and detection programs, network security precautions, encryption of critical data, and in-depth security assessments of vendors. We maintain various protections designed to safeguard against cyberattacks, including firewalls and virus detection software. We have established and regularly test our disaster recovery plan, and we protect against business interruption by backing up our major systems. In addition, we periodically scan our environment for any vulnerabilities, perform penetration testing, and engage third parties to assess the effectiveness of our data security practices. In addition, we maintain insurance that includes cybersecurity coverage.
Our cybersecurity program is executed by a team of highly skilled cybersecurity professionals, including, but not limited to, risk and threat analysts, penetration testers, security operations center analysts, cyber regulatory analysts, and risk/threat modelers. Team members maintain certification(s) and practical application of skills through organizations, such as ISC2 (Certified Information Security Systems Professional or CISSP), Global Information Assurance (GIAC), EC-Council, the Committee on National Security Systems (CNSS), and the National Security Agency (NSA). The program incorporates industry-standard frameworks, policies, and practices designed to protect the privacy and security of our sensitive information. Our cybersecurity team reports to the Board of Directors on a quarterly basis on information security and cybersecurity matters or more frequently as needed. Our Audit Committee, which is comprised of several members of our Board of Directors, has oversight responsibility for our data security practices, and we believe that the committee has the requisite skills and visibility into the design and operation of our data security practices to fulfill this responsibility effectively. Five members of our Board
of Directors have enhanced information security expertise, including Gary S. Guthart, Ph.D., Joseph C. Beery, Amal M. Johnson, Keith R. Leonard Jr., and Mark J. Rubash.
Despite the implementation of our cybersecurity program, our security measures cannot guarantee that a significant cyberattack will not occur. A successful attack on our information technology systems could have significant consequences for the business. While we devote resources to our security measures to protect our systems and information, these measures cannot provide absolute security. See “Risk Factors – Information technology system failures, cyberattacks, or deficiencies in our cybersecurity could harm our business, customer relations, financial condition, or results of operations” for additional information about the risks to our business associated with a breach or compromise to our information technology systems.
In April 2022, the FDA issued draft guidance on cybersecurity related to quality systems and premarket submission content for medical devices. The 2022 draft guidance for premarket cybersecurity in medical devices would increase the expectations that the FDA has for manufacturers. We are diligently pursuing compliance with this new guidance.
Foreign Regulation
In order for us to market our products in countries outside the United States, we must obtain regulatory approvals or certifications and comply with extensive product and quality system regulations in other countries. These regulations, including the requirements for approvals, clearance, or certifications and the time required for regulatory review, vary from country to country. Some countries have regulatory review processes that are substantially longer than U.S. processes. Failure to obtain regulatory approval or certification in a timely manner and meet all of the local requirements, including language and specific safety standards, in any foreign country in which we plan to market our products could prevent us from marketing products in such countries or subject us to sanctions and fines.
China
China has its own regulatory agency. They require regulatory approvals and compliance with extensive safety and quality system regulations. Failure to obtain regulatory approval or failure to comply with any regulation may negatively impact our ability to generate revenue and harm our business. In addition to product registration approvals, our system sales into China are also dependent on obtaining importation authorizations and provincial approvals, as well as hospitals completing a tender and hospital listing process under the authorization. In October 2018, the China National Health Commission published on its official website the quota for major medical equipment to be imported and sold in China through 2020. After an adjustment notice was published in the third quarter of 2020 (ref. NHC Financial Notice [2020] 315), the government will allow for the total sale of 225 new Endoscopic Surgical Instrument Control Systems (surgical robots) into China, which could include da Vinci Surgical Systems as well as surgical systems introduced by others. Sales of da Vinci Surgical Systems under the quota are uncertain, as they are dependent on hospitals completing a tender process and receiving associated approvals.
Japan
Most medical devices must undergo thorough safety examinations and demonstrate medical efficacy before they receive regulatory approval to be sold in Japan. We obtained approval from the Japanese Ministry of Health, Labor, and Welfare (“MHLW”) for our da Vinci Si Surgical System in October 2012, for our da Vinci Xi Surgical System in March 2015, and for our da Vinci X Surgical System in April 2018. National reimbursement status in Japan was received for prostatectomy procedures in April 2012 and for da Vinci partial nephrectomy procedures in April 2016. An additional 12 da Vinci procedures were granted reimbursement effective April 1, 2018, including gastrectomy, low anterior resection, lobectomy, and hysterectomy, for both malignant and benign conditions. An additional seven da Vinci procedures were granted reimbursement effective April 1, 2020. An additional eight da Vinci procedures were granted reimbursement effective April 1, 2022, including colon resection. In addition, we received higher reimbursement for da Vinci gastrectomy procedures, as compared to open and conventional laparoscopic procedure reimbursements. The additional reimbursed procedures have varying levels of conventional laparoscopic penetration and will generally be reimbursed at rates equal to the conventional laparoscopic procedures. Given the reimbursement level and laparoscopic penetration for these additional procedures, there can be no assurance that the adoption pace for these procedures will be similar to prostatectomy or partial nephrectomy, given their higher reimbursement, or any other da Vinci procedure. If these procedures are not adopted and we are not successful in obtaining adequate procedure reimbursements for additional procedures, then the demand for our products in Japan could be limited. The process of reimbursement for new da Vinci surgical procedures in Japan is led by the surgical societies. The societies submit for reimbursement or incremental reimbursement to the MHLW for their evaluation. The decision to reimburse requires in-country clinical data and is fixed in April of even-numbered years. In September 2022, we received regulatory clearance for the da Vinci SP Surgical System in Japan for the same set of procedures as can be performed on the da Vinci Xi Surgical System in Japan.
European Union
In the European Union (“EU”), all medical devices placed on the EU market must meet the essential requirements, including the requirement that a medical device must be designed and manufactured in such a way that it will not compromise
the clinical condition or safety of patients, or the safety and health of users and others. In addition, the device must achieve the performance intended by the manufacturer and be designed, manufactured, and packaged in a suitable manner.
Until and including May 25, 2021, medical devices were regulated by Council Directive 93/42/EEC (the “EU Medical Devices Directive” or “MDD”), which has been repealed and replaced by Regulation (EU) No 2017/745 (the “EU Medical Devices Regulation” or “MDR”). Our current certificates have been granted under the MDD. However, as of May 26, 2021, some of the MDR requirements apply in place of the corresponding requirements of the MDD with regard to the registration of economic operators and of devices, post-market surveillance, and vigilance. Pursuing marketing of medical devices in the EU requires that our devices be certified under the new regime set forth in the MDR, and we are diligently pursuing our plan to be fully compliant by May 26, 2024. Under recently proposed draft legislation issued by the European Commission, this date would be extended to December 2027 for higher classification devices (Class III and certain Class C IIb implantable devices) and to December 2028 for medium- and lower-risk devices (for the other Class IIb devices, Class IIa, and some Class I devices). We may adjust our transition plans to take this transitional period into account, if said legislative proposal is adopted by the European Parliament and Council.
Medical Devices Directive
Under the EU Medical Devices Directive, all medical devices placed on the market in the EU must meet the essential requirements laid down in Annex I to the EU Medical Devices Directive, including the requirement that a medical device must be designed and manufactured in such a way that it will not compromise the clinical condition or safety of patients or the safety and health of users and others. In addition, the device must achieve the performance intended by the manufacturer and be designed, manufactured, and packaged in a suitable manner. The European Commission has adopted various standards applicable to medical devices. These include standards governing common requirements, such as the sterilization and safety of medical electrical equipment and product standards for certain types of medical devices. There are also harmonized standards relating to design and manufacture. While not mandatory, compliance with these standards is viewed as the easiest way to satisfy the essential requirements as a practical matter, as it creates a rebuttable presumption that the device satisfies the essential requirements.
Except for low-risk medical devices (Class I non-sterile, non-measuring devices), where the manufacturer can self-assess the conformity of its products with the essential requirements (except for any parts that relate to sterility or metrology), a conformity assessment procedure requires the intervention of a notified body. Notified bodies are independent organizations designated by EU member states to assess the conformity of devices before being placed on the market. A notified body would typically audit and examine a product’s technical dossiers and the manufacturer’s quality system (the notified body must presume that quality systems that implement the relevant harmonized standards, which is ISO 13485:2016 for Medical Devices Quality Management Systems, conform to these requirements). If satisfied that the relevant product conforms to the relevant essential requirements, the notified body issues a certificate of conformity, which the manufacturer uses as a basis for its own declaration of conformity. The manufacturer may then apply the Conformité Européenne mark (“CE mark”) to the device, which allows the device to be placed on the market throughout the EU.
Throughout the term of the certificate of conformity, the manufacturer will be subject to periodic surveillance audits to verify continued compliance with the applicable requirements. In particular, there will be a new audit by the notified body before it will renew the relevant certificate(s).
Medical Devices Regulation
On April 5, 2017, the MDR was adopted with the aim of ensuring better protection of public health and patient safety. The MDR establishes a uniform, transparent, predictable, and sustainable regulatory framework across the EU for medical devices and ensures a high level of safety and health while supporting innovation. Unlike directives, regulations are directly applicable in EU member states without the need for member states to implement into national law. This aims at increasing harmonization across the EU.
The MDR became effective on May 26, 2021. Devices lawfully placed on the market pursuant to the MDD prior to May 26, 2021, may generally continue to be made available on the market or put into service until and including May 26, 2025, provided that the requirements of the transitional provisions are fulfilled (referred to as the “sell-off” provision). In particular, the certificate in question must still be valid. However, even in this case, manufacturers must comply with a number of new or reinforced requirements set forth in the MDR, in particular the obligations described below. If it is adopted by the European Parliament and Council, under draft legislation proposed by the European Commission, the sell-off provision would be removed.
The MDR requires that, before placing a device on the market, other than a custom-made device, manufacturers (as well as other economic operators, such as authorized representatives and importers) must register by submitting identification information to the electronic system (EUDAMED), unless they have already registered. The information to be submitted by manufacturers (and authorized representatives) also includes the name, address, and contact details of the person or persons
responsible for regulatory compliance. The new Regulation also requires that, before placing a device on the market, other than a custom-made device, manufacturers must assign a unique identifier to the device and provide it along with other core data to the unique device identifier (“UDI”) database. These new requirements aim at ensuring better identification and traceability of the devices.
All manufacturers placing medical devices on the market in the EU must comply with the EU medical device vigilance system, which has been reinforced by the MDR. Under this system, serious incidents and Field Safety Corrective Actions (“FSCAs”) must be reported to the relevant authorities of the EU member states. These reports will have to be submitted through Eudamed (once functional) and aim to ensure that, in addition to reporting to the relevant authorities of the EU member states, other actors, such as the economic operators in the supply chain, will also be informed. Until Eudamed is fully functional, the corresponding provisions of the MDD continue to apply. Manufacturers are required to take FSCAs, which are defined as any corrective action for technical or medical reasons to prevent or reduce a risk of a serious incident associated with the use of a medical device that is made available on the market.
The advertising and promotion of medical devices are subject to some general principles set forth in EU legislation. According to the MDR, only devices that are CE marked may be marketed and advertised in the EU in accordance with their intended purpose. Directive 2006/114/EC concerning misleading and comparative advertising and Directive 2005/29/EC on unfair commercial practices, while not specific to the advertising of medical devices, also apply to the advertising thereof and contain general rules, for example, requiring that advertisements are evidenced, balanced, and not misleading. Specific requirements are defined at a national level. EU member states’ laws related to the advertising and promotion of medical devices, which vary between jurisdictions, may limit or restrict the advertising and promotion of products to the general public and may impose limitations on promotional activities with healthcare professionals.
Many EU member states have adopted specific anti-gift statutes that further limit commercial practices for medical devices, in particular vis-à-vis healthcare professionals and organizations. Additionally, there has been a recent trend of increased regulation of payments and transfers of value provided to healthcare professionals or entities and many EU member states have adopted national “Sunshine Acts,” which impose reporting and transparency requirements (often on an annual basis), similar to the requirements in the United States, on medical device manufacturers. Certain countries also mandate implementation of commercial compliance programs.
In the EU, regulatory authorities have the power to carry out announced and, if necessary, unannounced inspections of companies, as well as suppliers and/or sub-contractors and, where necessary, the facilities of professional users. Failure to comply with regulatory requirements (as applicable) could require time and resources to respond to the regulatory authorities’ observations and to implement corrective and preventive actions, as appropriate. Regulatory authorities have broad compliance and enforcement powers and, if such issues cannot be resolved to their satisfaction, can take a variety of actions, including untitled or warning letters, fines, consent decrees, injunctions, or civil or criminal penalties.
The aforementioned EU rules are generally applicable in the EEA, which consists of the 27 EU Member States as well as Iceland, Liechtenstein, and Norway.
Brexit
Since January 1, 2021, the Medicines and Healthcare Products Regulatory Agency (“MHRA”) has become the sovereign regulatory authority responsible for the Great Britain (i.e., England, Wales, and Scotland) medical device market. Following the end of the Brexit transitional period on January 1, 2021, new regulations require all medical devices to be registered with the MHRA before being placed on the Great Britain market. From January 1, 2022, non-UK manufacturers were required to appoint a UK Responsible Person for the purposes of registering devices placed on the Great Britain market. Under the terms of the Protocol on Ireland/Northern Ireland, the MDR applies to medical devices placed on the Northern Ireland market in the same way as it applies to medical devices marketed in the EU.
On June 26, 2022, the MHRA published its response to a 10-week consultation on the future regulation of medical devices in the UK. Regulations implementing the new regime were originally scheduled to come into force in July 2023 but have recently been postponed to July 2024. Devices bearing CE marks issued by EU notified bodies under the MDR or MDD are now subject to transitional arrangements. In its consultation response, the MHRA indicated that the future UK regulations will allow devices certified under the MDR to be placed on the market in Great Britain under the CE mark until either the certificate expires or for five years after the new regulations take effect, whichever is sooner. Following these transitional periods, all medical devices will require a UK Conformity Assessed (“UKCA”) mark in order to be placed on the market in Great Britain. Manufacturers may choose to use the UKCA mark on a voluntary basis prior to entry of the new regulations on July 1, 2024. However, from July 2024, products that do not have existing and valid certification under the EU Medical Devices Directive or EU MDR and, therefore, are not subject to the transitional arrangements will be required to carry the UKCA mark if they are to be sold into the market in Great Britain. For products to be sold into the market in Northern Ireland, CE marking will continue to be recognized as a result of the Northern Ireland Protocol implemented following the UK’s exit from the EU. UKCA
marking will not be recognized in the EU. Following the transitional period, compliance with the UK regulations will be a prerequisite to be able to affix the UKCA mark to our products, without which they cannot be sold or marketed in Great Britain.
In addition, the Trade Deal between the UK and the EU generally provides for cooperation and exchange of information between the parties in the areas of product safety and compliance, including market surveillance, enforcement activities and measures, standardization-related activities, exchanges of officials, and coordinated product recalls. As such, processes for compliance and reporting should reflect requirements from regulatory authorities.
Other countries
Regulations in other countries, including the requirements for approvals, certification, or clearance and the time required for regulatory review, vary from country to country. Certain countries, such as South Korea, Brazil, Australia, India, and Canada, have their own regulatory agencies. These countries typically require regulatory approvals and compliance with extensive safety and quality system regulations included in the MDSAP (Medical Device Single Audit Program) that we comply with every year as part of our annual audit program. Failure to obtain regulatory approval in any foreign country in which we plan to market our products, or failure to comply with any regulation in any foreign country in which we market our products may negatively impact our ability to generate revenue and harm our business.
In addition, local regulations may apply, which govern the use of our products and which could have an adverse effect on our product utilization if they are unfavorable. All such regulations are revised from time to time and, in general, are increasing in complexity and in the scope and degree of documentation and testing required. There can be no assurance that the outcomes from such documentation and testing will be acceptable to any particular regulatory agency or will continue to be acceptable over time. There are further regulations governing the importation, marketing, sale, distribution, use, and service as well as the removal and disposal of medical devices in the regions in which we operate and market our products. Failure to comply with any of these regulations could result in sanctions or fines and could prevent us from marketing our products in these regions.
Third-Party Coverage and Payment
Our customers, including physicians, hospitals, and outpatient facilities, typically bill third-party payors for the costs and fees associated with the procedures in which our products are used. In the U.S., in order to receive payment for the procedures performed using our products, our customers must report codes that describe the services or products furnished and determine the medical necessity of the service or whether the service is included in the payors’ policy. In the U.S. and most markets globally where we sell our products, payment for medical services and surgical procedures to hospitals, outpatient facilities, and surgeons (collectively “providers”) is determined by the government, commercial payors (insurers), or a combination of both.
In the U.S., the Centers for Medicare and Medicaid Services (“CMS”) and its fiscal intermediaries (Medicare Administrative Contractors) and state Medicaid programs establish reimbursement policies for medical and surgical services at the state and federal level for the Medicare and Medicaid programs. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own coverage and payment policies but also have their own methods and approval processes. Commercial payors in non-capitated contracts commonly establish payment to providers based on a percentage of the Medicare payment rate.
Physicians and outpatient facilities bill for medical and surgical services by reporting a combination of billing codes. Current Procedural Terminology (“CPT”) codes are created by the American Medical Association (“AMA”) with input from CMS and commercial payors to describe medical and surgical procedures. CPT codes currently exist for minimally invasive surgical procedures, which may involve the da Vinci surgical system, as well as for robotic-assisted bronchoscopy, which may involve the Ion endoluminal system. In general, the majority of payors, including Medicare, consider robotic assistance as a tool used to perform the procedure and do not pay providers more for a procedure that involves robotic assistance using the da Vinci, Ion, or any other robotic system. Because there is often no separate payment for the use of our products, the additional cost associated with the use of our products can affect the profit margin of the hospital or surgery center where the procedure is performed. If hospitals do not obtain sufficient payment from third-party payors for procedures performed with our products, or if governmental and private payors’ policies do not cover surgical procedures performed using our products, we may not be able to generate the revenue necessary to support our business.
Hospitals bill for inpatient services by reporting ICD-10-PCS codes. CMS is primarily responsible for overseeing changes and modifications to ICD-10-PCS codes. Medicare payments to hospitals for services provided during an inpatient stay are based on the Inpatient Prospective Payment System (“IPPS”). Under the IPPS, each patient discharge is categorized into a Medicare Severity Adjusted Diagnosis-Related Group (“MS-DRG” or “DRG”). Each DRG has an assigned payment weight based on the average resources used for Medicare patients in that DRG, taking into account the patient’s principal diagnosis, surgical procedures, age, discharge status, and additional or secondary diagnoses, among other things. The DRG is a single, bundled payment intended to cover all costs associated with the inpatient admission.
The use of robotic technology does not influence the MS-DRG assignment or payment for an inpatient admission related to a surgical procedure. CMS annually updates hospital inpatient and outpatient payments based on hospitals’ charge data. Hospital inpatient and outpatient payments are also adjusted based on whether the hospital is a teaching hospital, its geographic location, and any failures to meet certain quality metrics, among other factors.
Commercial payors commonly establish inpatient facility payment for providers using published Medicare DRG rates as a benchmark. Commercial payment to providers varies depending on the procedure performed, geographic location, contractual allowances, and other factors.
Medicare and commercial payor payments to facilities for medical and surgical services may not always fully reimburse providers for all costs associated with furnishing these procedures. If payment is insufficient for procedures involving our technology, hospitals and physicians may decide not to use our products.
In countries outside of the U.S., payment for surgical services to physicians and facilities differs considerably and varies by country. In some markets, there is a single public payor who provides a global annual budget to hospitals to provide all care to the population served in a designated geographic area. In other markets, private insurance can be purchased or is provided by employers to supplement public health insurance. In some countries, patients may be permitted to pay directly for surgical services; however, such “co-pay” practices are not common (or allowed) in many countries. Further, in many global markets, access to procedures and technology is governed or heavily influenced by Health Technology Assessment (“HTA”) organizations, which conduct periodic and extensive evidence-based reviews of the clinical value and cost effectiveness of a new technology. To effectively conduct our business, we may need to seek OUS reimbursement approvals, and we do not know if these required approvals will be obtained in a timely manner or at all. In addition, in some markets, HTA organizations may publish reports with mixed conclusions about the clinical and economic value of our products to the population. Such reviews could negatively impact hospital adoption of our technology.
Healthcare Reform
In the U.S., there have been, and continue to be, a number of legislative initiatives to contain healthcare costs. In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the “ACA”), was enacted. The ACA made changes that have significantly impacted healthcare providers, insurers, pharmaceutical companies, and medical device manufacturers. The ACA contained a number of provisions designed to generate the revenues necessary to fund health insurance coverage expansion and appropriated funding to research the comparative effectiveness of healthcare treatments and strategies. To date, this research has had a negligible effect on Medicare coverage and reimbursement decisions as well as influence on other third-party payor coverage and reimbursement policies.
Since its enactment, there have been judicial, executive, and Congressional challenges to certain aspects of the ACA. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA brought by several states without specifically ruling on the constitutionality of the ACA. Thus, the ACA remained in effect in its current form.
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. These changes included an aggregate reduction in Medicare payments, which went into effect on April 1, 2013, and will remain in effect through 2031, unless additional Congressional action is taken, with the exception of a temporary suspension due to the COVID-19 pandemic from May 1, 2020, through March 31, 2022. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers, and cancer treatment centers. The Medicare Access and CHIP Reauthorization Act of 2015, enacted on April 16, 2015 (“MACRA”), repealed the formula by which Medicare made annual payment adjustments to physicians and replaced the former formula with fixed annual updates and a new system of incentive payments that began in 2019 and are based on various performance measures and physicians’ participation in alternative payment models, such as accountable care organizations. Individual states in the U.S. have also become increasingly aggressive in passing legislation and implementing regulations designed to control product pricing, including price or patient reimbursement constraints and discounts, and require marketing cost disclosure and transparency measures.
In the U.S. and abroad, reimbursement is dynamic and subject to change annually by public and private payors. Congress and government agencies may also intervene and pass legislation that is intended to reduce healthcare spending, which could impact market access. Such legislative interventions can vacillate significantly based on government leadership. Other federal or state healthcare reform measures that may be adopted in the future could have a material adverse effect on our business. Any regulatory or legislative developments in domestic or foreign markets that eliminate or reduce reimbursement rates for procedures performed with our products could harm our ability to sell our products or cause downward pressure on the prices of our products, either of which would adversely affect our business, financial condition, and results of business operations.
For instance, in December 2021, the EU Regulation No 2021/2282 on HTA, amending Directive 2011/24/EU, was adopted. This regulation, which entered into force in January 2022, intends to boost cooperation among EU member states in assessing health technologies, including certain medical devices, and provides the basis for cooperation at the EU level for joint
clinical assessments in these areas. The regulation foresees a three-year transitional period and will permit EU member states to use common HTA tools, methodologies, and procedures across the EU, working together in four main areas, including joint clinical assessment of the innovative health technologies with the most potential impact for patients, joint scientific consultations whereby developers can seek advice from HTA authorities, identification of emerging health technologies to identify promising technologies early, and continuing voluntary cooperation in other areas. Individual EU member states will continue to be responsible for assessing non-clinical (e.g., economic, social, ethical, etc.) aspects of health technology and making decisions on pricing and reimbursement.
Human Capital
The future success of our company depends on our ability to attract, retain, and further develop top talent. We enable this by continuously striving to make Intuitive an inclusive, diverse, and safe workplace with opportunities for our employees to grow and develop in their careers. These objectives are supported through strong compensation, benefits, programs that encourage employee health and wellness, and connections between our employees and the communities in which they live and work.
As of December 31, 2022, we had approximately 12,120 full-time employees, 1,651 of whom were engaged directly in research and development, 4,843 in manufacturing operations, 3,834 in commercial and service operations, and 1,792 in administrative activities. During 2022, the number of employees increased by approximately 2,327. Our employees are based in 29 different countries around the world. Our global workforce consists of diverse, highly skilled talent at all levels. During 2022, our turnover rate was approximately 10.9%.
Inclusion and Diversity
Intuitive’s inclusion and diversity (“I&D”) vision is to empower our employees and customers from every background to fully contribute toward our mission to expand the potential of physicians to heal without constraints. We want to build an environment where every individual can belong and flourish – in our company and the communities we serve.
We believe that everyone should feel included and fairly treated, and we embrace the unique qualities that make people who they are. This includes all genders and gender identities, races, ethnicities, ages, national origins, native languages, disabilities, sexual orientations, body sizes, military backgrounds, socioeconomic backgrounds, religions, and family structures. We believe in seeking the different to propel innovation and creativity forward.
We have a four-part strategy to guide our I&D progress: ensuring an inclusive experience, where employees from all backgrounds feel welcome, supported, and valued; building a diverse workforce to fuel innovation and better mirror the patients we serve; continuously investing in and enhancing the fairness of our people practices and sharing progress; and strengthening industry engagement through collaboration with the healthcare community, diversity-focused organizations, and shareholders to drive positive change. Employee Resource Groups (“ERGs”) have been one key area of I&D focus and growth, providing support and community for traditionally marginalized groups, including women, people of color, members of the lesbian, gay, bisexual, transgender, queer, and/or questioning (“LGBTQ+”) community, military veterans, and employees with disabilities. Details of our employee workforce composition, including a link to our Employer Information Report (“EEO-1”) submission to the U.S. Equal Employment Opportunity Commission (“EEOC”), are available on our website. Although we reference the availability of our EEO-1 on our website in this Annual Report on Form 10-K, our EEO-1 and any other materials on our website are not incorporated by reference into this Annual Report on Form 10-K or any of our other filings under the Securities Act of 1933, as amended, or the Exchange Act. While matters discussed in such EEO-1 and other website materials may be significant, any significance should not be read as necessarily rising to the level of materiality used for the purposes of our compliance with the U.S. federal securities laws, even if we use the word “material” or “materiality” in such materials.
From a governance perspective, maintaining a mix of backgrounds and experience in our Board composition is essential to understanding and reflecting the needs of our diverse stakeholders. Currently, four of our 11 Board members self-identify as women, and three of our 11 board members self-identify as individuals from underrepresented communities (defined as an individual who self-identifies as Black, African American, Hispanic, Latino, Asian, Pacific Islander, Native American, Native Hawaiian, or Alaska Native, or who self-identifies as LGBTQ+).
Health, Safety, and Wellness
The health, safety, and wellness of our employees is a priority in which we continue to invest and expand. We provide our employees and their families with access to a variety of innovative, flexible, and convenient health and wellness programs. Program benefits are intended to provide protection, peace of mind, and security, including workplace health and safety best practices integrated into everyday activities and programs that support employee time away from work, family care, mental health, or financial well-being.
We continue to evolve our programs to respond to the best interest of our changing workforce, as well as the communities in which we operate, in compliance with government regulations. Each Intuitive location manages overall safety with guidance based on regional, country, and local regulations and best practices.
In 2022, we focused on collecting internal and external insights to inform decision-making on work models that would align with how employees will work in the current environment, which has evolved as a result of the COVID-19 pandemic. These efforts will allow for an improved employee experience, regardless of whether an employee is working from home, fully on-site, or in a hybrid fashion. As we move into 2023, our focus will be on the implementation and sustained success of these new work models, including on-site engagement activities that reinforce our differentiating culture and facilitate cross-team networking, collaboration, and innovation.
Compensation and Benefits
We provide compensation and benefits programs to help meet the needs of our employees. In addition to base compensation, these programs, which vary by country and region, include annual bonuses, stock awards, an Employee Stock Purchase Plan, retirement savings plans, healthcare, income protection benefits, paid time off, family leave, family care resources, and flexible work schedules, among many others.
Ensuring fair and equitable pay is integral to our commitment to our employees. Our executive team and Board of Directors strongly support this commitment. We regularly review pay for internal equity and ensure our compensation structure is appropriate, including with regard to race/ethnicity and gender. We also engage outside counsel to ensure compliance with pay equity laws. When we identify any potential differences in pay for whatever reason, we research those differences and act if appropriate. Employees are encouraged to share any pay equity concerns with management, Human Resources, or confidentially through our reporting hotline, including anonymously. Intuitive has a non-retaliation policy for raising any workplace concerns, including around pay.
Talent Development
We value our employees and the passion, commitment, and expertise they contribute to Intuitive. To enhance employee retention and engagement, we offer ongoing learning and leadership training opportunities that support growth.
With a commitment to building strong, inclusive leaders, in 2022, we rolled out an enhancement to our existing manager development program designed to equip our people leaders with fundamental management skills to accelerate leadership success. This program is delivered over a twelve-month period through a mix of in-person, virtual, and on-demand learning. For our individual contributor employee population, we offer a variety of general and targeted development opportunities, including technical skills training, career learning journeys, and networking opportunities. We also provide extra support for Employee Resource Group leaders via a 1:1 coaching program.
We have robust annual global performance and compensation planning processes for reviewing all employees’ performance and pay. As part of this process, Intuitive supports career development and growth through our Talent Action Planning program, which encourages employees to work on targeted talent development actions to enhance on-the-job performance and prepare for future career opportunities with the support of their leaders. To support our people leaders in managing the performance of their teams, we train them on conducting effective career conversations and performance reviews and making fair and equitable compensation recommendations. Compensation guidelines are provided to leaders, which take into consideration market pay data and performance, as well as job experience. In 2022, we added a new focus on clarity and consistency in our internal promotion processes.
Community Programs
We believe that building connections between our employees, their families, and our communities creates a more meaningful, fulfilling, and enjoyable workplace. Through our engagement programs, our employees can pursue their interests and hobbies, connect to volunteering and giving opportunities, and enjoy unique recreational experiences with family members.
The Intuitive Foundation is a nonprofit organization established in 2018 and funded by Intuitive. Since its founding, the Intuitive Foundation has been dedicated to promoting health, advancing education, and reducing human suffering. The Foundation supports outreach programs financially while we provide the volunteers and mentors from within our company. Since its inception, we have contributed $85 million to the Intuitive Foundation to fulfill its mission.
We encourage you to review the “Employee health and safety” and “Global access and engagement outreach initiatives” sections of our 2022 ESG Annual Report (to be made available on our website) for more detailed information regarding our Human Capital programs and initiatives. Although we reference our ESG Annual Report available on our website in this report, this report and any other materials on our corporate website are not incorporated by reference into this Annual Report or any other filing of the Company under the Securities Act of 1933, as amended, or the Exchange Act. While matters discussed in such ESG Annual Report and website materials may be significant, any significance should not be read as necessarily rising to
the level of materiality used for the purposes of our compliance with the U.S. federal securities laws, even if we use the word “material” or “materiality” in such materials.
General
We make our periodic and current reports, including our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and any amendments to those reports, available free of charge on our website as soon as practicable after such material is electronically filed or furnished with the Securities and Exchange Commission (the “SEC”). Our website address is www.intuitive.com, and the reports are filed under “SEC Filings” on the Company — Investor Relations portion of our website. Periodically, we webcast Company announcements, product launch events, and executive presentations, which can be viewed via our Investor Relations page on our website. In addition, we provide notifications of our material news, including SEC filings, investor events, and press releases as part of our Investor Relations page on our website. The contents of our website are not intended to be incorporated by reference into this report or in any other report or document we file, and any references to our website are intended to be inactive textual references only. The SEC maintains an internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC at www.sec.gov. The contents of these websites are not incorporated into this filing. Further, references to the URLs for these websites are intended to be inactive textual references only.
We operate our business as one segment, as defined by U.S. generally accepted accounting principles. Our financial results for the years ended December 31, 2022, 2021, and 2020 are discussed in “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Item 8. Financial Statements and Supplementary Data” of this Annual Report.
Intuitive Surgical, Inc. was founded in 1995. We are a Delaware corporation with our principal executive offices located at 1020 Kifer Road, Sunnyvale, California 94086. Our telephone number is (408) 523-2100, and our website address is www.intuitive.com.
ITEM 1A. RISK FACTORS
You should consider each of the following risk factors, which could materially affect our business, financial condition, or future results of operations. Additional risks and uncertainties not currently known to us or that we currently deem to be immaterial also may materially adversely affect our business, financial condition, or future results of operations. In addition, the global economic environment and additional or unforeseen effects from the COVID-19 pandemic amplify many of these risks.
RISKS RELATING TO OUR BUSINESS
MACROECONOMIC CONDITIONS COULD HAVE A MATERIALLY ADVERSE IMPACT ON OUR BUSINESS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
Macroeconomic conditions, such as high inflation, changes to monetary policy, increasing interest rates, volatile currency exchange rates, credit and sovereign debt concerns in certain European countries, concerns about slowed growth in China and other OUS markets, decreasing consumer confidence and spending, including capital spending, and global or local recessions can adversely impact demand for our products, which could negatively impact our business, financial condition, or results of operations. Recent macroeconomic conditions have been adversely impacted by political instability and military hostilities in multiple geographies (including the conflict between Ukraine and Russia), monetary and financial uncertainties, and the ongoing COVID-19 pandemic. The results of these macroeconomic conditions, and the actions taken by governments, central banks, companies, and consumers in response, have and may continue to result in higher inflation in the U.S. and globally, which is likely, in turn, to lead to an increase in costs and may cause changes in fiscal and monetary policy, including increased interest rates. Other adverse impacts of recent macroeconomic conditions have been and may continue to be supply chain constraints, logistics challenges, and fluctuations in labor availability.
In a higher inflationary environment, we may be unable to raise the prices of our products and services sufficiently to keep up with the rate of inflation. Impacts from inflationary pressures could be more pronounced and materially adversely impact aspects of our business where revenue streams and cost commitments are linked to contractual agreements that extend further into the future, as we may not be able to quickly or easily adjust pricing, reduce costs, or implement countermeasures. A higher inflationary environment can also negatively impact raw material, component, and logistics costs that, in turn, may increase the costs of producing and distributing our products. Recently, the costs of raw materials, transportation, construction, services, and energy necessary for the production and distribution of our products have increased significantly.
Furthermore, hospitals and distributors may choose to postpone or reduce spending due to financial difficulties or difficulties in obtaining credit to finance purchases of our products due to increased interest rates and restraints on credit. Hospitals, in particular, are experiencing and may continue to experience financial and operational pressures as a result of staffing shortages, the supply chain environment, and increased inflation, which could impact their ability to access capital markets and other funding sources, increase the cost of funding, or impede their ability to comply with debt covenants, all of which could impede their ability to provide patient care, defer elective surgeries, and impact their profitability. To the extent that hospitals face financial pressures, reductions in government spending, or higher interest rates, hospitals’ ability or willingness to spend on capital equipment may be adversely impacted, all of which could have a material adverse effect on our business, financial condition, or results of operations.
We are unable to predict the impact of efforts by central banks and federal, state, and local governments to combat elevated levels of inflation. If their efforts to create downward pressure on inflation are too aggressive, they may lead to a recession. Alternatively, if they are insufficient or are not sustained long enough to bring inflation to lower, more acceptable levels, hospitals’ ability or willingness to spend on capital equipment may be impacted for a prolonged period of time. If a recession occurs, economies weaken, or inflationary trends continue, our business and operating results could be materially adversely affected.
In addition, in early 2023, the U.S. Government reached its existing statutory limit on the amount of permissible federal debt, and this limit must be raised in order for the U.S. Government to continue to pay its obligations on a timely basis. If the debt ceiling is not raised, it is unclear how the U.S. Government would prioritize its payments towards its various programs, which could have a significant impact on the overall economy as well as on medical procedures performed.
Also, we have and may continue to experience supply chain constraints due to the current supply chain environment and logistic challenges, including difficulties obtaining a sufficient supply of component materials used in our products. If interest rates continue to rise, access to credit may become more difficult, which may result in the insolvency of key suppliers, including single-source suppliers, which would exacerbate supply chain challenges. Such supply chain constraints could cause us to fail to meet product demand, which could result in deferred or canceled procedures.
OUR RELIANCE ON SOLE AND SINGLE SOURCE SUPPLIERS AND ABILITY TO PURCHASE AT ACCEPTABLE PRICES A SUFFICIENT SUPPLY OF MATERIALS, PARTS, AND COMPONENTS COULD HARM OUR ABILITY TO MEET PRODUCT DEMAND IN A TIMELY MANNER OR WITHIN BUDGET.
Some of the components necessary for the assembly of our products are currently provided to us by sole-sourced suppliers or single-sourced suppliers. We generally purchase components through purchase orders rather than long-term supply agreements and generally do not maintain large volumes of inventory. While alternative suppliers exist and could be identified for single-sourced components, the disruption or termination of the supply of components, or inflationary pressure in our supply chain, could cause a significant increase in the costs of these components, which could affect our operating results. A disruption or termination in the supply of components could also result in our inability to meet demand for our products, which could harm our ability to generate revenues, lead to customer dissatisfaction, and damage our reputation and our brand. Furthermore, if we are required to change the manufacturer of a key component of our products, we may be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. The time and processes associated with the verification of a new manufacturer could delay our ability to manufacture our products on schedule or within budget, which may have a material adverse impact on our business, financial condition, or results of operations.
In addition, our ability to meet customers’ demands depends, in part, on our ability to timely obtain an adequate delivery of quality materials, parts, and components from our suppliers. An information technology systems interruption, including cyberattacks, could adversely affect the ordering, distribution, and manufacturing processes of our suppliers. Difficulties remain in obtaining a sufficient supply of semiconductor and other component materials, and we expect such difficulties to persist in the foreseeable future. Prices of such materials have also increased, and global supply has become significantly constrained due to the increased demand for materials, including semiconductors, to support expansion of server and cloud networks as a greater proportion of the global population worked remotely, the introduction of 5G, and the continued electrification of vehicles. We engage in activities to seek to mitigate such supply disruptions by, for example, increasing our communications with our suppliers and modifying our purchase order coverage and inventory levels. Such global shortages in important components have resulted in, and will continue to cause, inflationary pressure in our supply chain, which would impact our profits and profit margin. If shortages and price increases in important supply-chain materials in the semiconductor or other markets continue, we could also fail to meet product demand, which would adversely impact our business, financial condition, or results of operations.
PUBLIC HEALTH CRISES OR EPIDEMIC DISEASES, OR THE PERCEPTION OF THEIR EFFECTS, COULD MATERIALLY ADVERSELY AFFECT OUR BUSINESS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
Our global operations expose us to risks associated with public health crises and outbreaks of epidemic, pandemic, or contagious diseases, such as the current outbreak of a novel strain of coronavirus (COVID-19). To date, COVID-19 has had, and may continue to have, an adverse impact on our operations, our supply chains and distribution systems, and our expenses, including as a result of preventive and precautionary measures that we, other businesses, and governments have taken and may continue to take. In addition, hospitals are experiencing staffing shortages and supply chain issues that could impact their ability to provide patient care. Due to these impacts and measures, we have experienced and may continue to experience significant and unpredictable reductions in the demand for our products as healthcare customers divert medical resources and priorities towards the treatment of that disease. Also, our customers have delayed, cancelled, or redirected and, in the future, may delay, cancel, or redirect, planned capital expenditures in order to focus resources on COVID-19 or in response to economic disruption related to COVID-19. For example, as a result of the global COVID-19 pandemic, in the first half of 2020, we experienced a significant decline in procedure volume in the U.S. and Western Europe, as healthcare systems diverted resources to meet the increasing demands of managing COVID-19. In addition, U.S. and global public health bodies have, at times, recommended delaying elective surgeries during the COVID-19 pandemic, which may continue to negatively impact the usage of our products and the number of da Vinci procedures performed. Also, as we are conducting IDE studies to support 510(k) submission for da Vinci platforms and for seeking new indications, we may experience delays in obtaining new product approvals, or clearances from the FDA or foreign approvals or certifications from foreign authorities or notified bodies or delays in recruiting patients in our ongoing and planned clinical studies.
As a result of the COVID-19 outbreak, we experienced significant business disruptions, including restrictions on our ability to travel as well as distribute and service our products, temporary closures of our facilities and the facilities of our suppliers and their contract manufacturers, and a reduction in access to our customers due to diverted resources and priorities and the business hours of hospitals, as governments institute prolonged shelter-in-place and/or self-quarantine mandates. For example, our corporate headquarters and many of our operations, including certain of our manufacturing facilities, are located in California, which previously instituted risk reduction orders applicable to our employees in that region, significantly impacting the ability of our employees to get to their places of work to produce products and hampering our products from moving through the supply chain. These unprecedented measures to slow the spread of the virus taken by local governments and healthcare
authorities globally, including the deferral of elective medical procedures and social distancing measures, had, and may continue to have, a negative impact on our operations and financial results. Furthermore, our future ways of working changes, including working from home, fully on-site, or in a hybrid fashion, may present additional risks, uncertainties, and costs that could affect our performance, including increased operational risk, uncertainty regarding office space needs, heightened vulnerability to cyberattacks due to remote work, potential reduced productivity, changes to our company culture, and increased costs to ensure our offices are safe and functional as hybrid offices that enable effective collaboration of both remote and in-person colleagues.
In addition, the COVID-19 pandemic adversely affected and may continue to adversely affect the economies and financial markets of many countries, which may result in a period of regional, national, and global economic slowdown or regional, national, or global recessions that could curtail or delay spending by hospitals and affect demand for our products as well as increased risk of customer defaults or delays in payments. Our customers may terminate or amend their agreements for the purchase, lease, or service of our products due to bankruptcy, lack of liquidity, lack of funding, operational failures, or other reasons. COVID-19 and the current financial, economic, and capital markets environment, and future developments in these and other areas present material uncertainty and risk with respect to our performance, financial condition, volume of business, or results of operations.
Outbreaks of other epidemic, pandemic, or contagious diseases, such as, historically, the Ebola virus, Middle East Respiratory Syndrome, Severe Acute Respiratory Syndrome, or the H1N1 virus, could also divert medical resources and priorities towards the treatment of that disease. An outbreak of other contagious diseases could negatively affect hospital admission rates or disrupt our business similar to the impact of the COVID-19 pandemic highlighted above. Any of these outbreaks could negatively impact the number of procedures performed and have a material adverse effect on our business, financial condition, or results of operations.
WE ARE SUBJECT TO LITIGATION, INVESTIGATIONS, AND OTHER LEGAL PROCEEDINGS RELATING TO OUR PRODUCTS, OUR CUSTOMERS, OUR COMPETITORS, AND GOVERNMENT REGULATORS THAT COULD MATERIALLY ADVERSELY AFFECT OUR FINANCIAL CONDITION, DIVERT MANAGEMENT’S ATTENTION, AND HARM OUR BUSINESS.
We are, and may become, subject to various legal proceedings and claims that arise in or outside the ordinary course of business. Certain current lawsuits and pending proceedings to which we are party, including purported class actions, product liability litigation, and patent litigation, are described in Note 8 to the Consolidated Financial Statements included in Part II, Item 8.
In particular, our business exposes us to significant risks of patent claims, product liability claims, and competition claims (including antitrust claims), many of which are common in the medical device industry. For example, product liability claims have been brought against us by, or on behalf of, individuals alleging that they have sustained personal injuries and/or death as a result of purported product defects, the alleged failure to warn, and/or the alleged inadequate training by us of physicians regarding the use of the da Vinci Surgical System. The individuals who have brought the product liability claims seek recovery for their alleged personal injuries and, in many cases, punitive damages. Current product liability claims have resulted in negative publicity regarding our Company, and these and any other product liability or negligence claims or product recalls also could harm our reputation. Refer to our risk factor titled “Negative Publicity, Whether Accurate or Inaccurate, Concerning Our Products or Our Company Could Reduce Market Acceptance of Our Products and Could Result in Decreased Product Demand and Reduced Revenues” for additional risks related to the potential effects of negative publicity on our business. Also, antitrust claims have been brought against us by third parties looking to compete in the instruments or servicing space and by certain customers.
The outcome of these product liability claims and other legal proceedings cannot be predicted with certainty. We purchase and maintain business insurance for certain liabilities and self-insure our product liability claims through a fronting policy. We cannot determine whether our existing business insurance program would be sufficient to cover the costs or potential losses related to these lawsuits and proceedings or otherwise be excluded under the terms of any insurance policy. Regardless of merit, litigation may be time-consuming and disruptive to our operations and cause significant legal costs (including settlements, judgments, legal fees, and other related defense costs) and diversion of management attention. If we do not prevail in these legal proceedings, we may be faced with significant monetary damages or injunctive relief against us that could have a material adverse effect on our business, financial condition, or results of operations. We could also be subject to governmental investigations in connection with many of these claims.
BECAUSE OUR MARKETS ARE HIGHLY COMPETITIVE, CUSTOMERS MAY CHOOSE TO PURCHASE OUR COMPETITORS’ PRODUCTS OR SERVICES OR MAY NOT ACCEPT ROBOTIC-ASSISTED MEDICAL PROCEDURES, WHICH COULD RESULT IN REDUCED REVENUE AND LOSS OF MARKET SHARE.
Robotic-assisted surgery with a da Vinci Surgical System or robotic-assisted bronchoscopy using an Ion endoluminal system are technologies that compete with established and emerging treatment options in reconstructive medical procedures or
disease management. These competitive treatment options include open surgery, conventional MIS, drug therapies, radiation treatment, and other emerging diagnostic and interventional surgical approaches. Some of these procedures are widely accepted in the medical community and, in many cases, have a long history of use. Technological advances could make such treatments more effective or less expensive than using our products, which could render our products obsolete or unmarketable. Studies could be published that show that other treatment options are more beneficial and/or cost-effective than robotic-assisted medical procedures. We cannot be certain that physicians will use our products to replace or supplement established treatments or that our products will continue to be competitive with current or future technologies.
Additionally, we face or expect to face competition from companies that develop or have developed wristed, robotic-assisted, or computer-assisted medical systems and products. Companies have introduced products in the field of robotic medical procedures or have made explicit statements about their efforts to enter the field including, but not limited to, the following companies: Asensus Surgical, Inc.; avateramedical GmbH; CMR Surgical Ltd.; Johnson & Johnson; Medicaroid Corporation; Medrobotics Corporation; Medtronic plc; meerecompany Inc.; Olympus Corporation; Samsung Electronics Co., Ltd; Shandong Weigao Group Medical Polymer Company Ltd.; Shanghai Microport Medbot (Group) Co., Ltd.; and Titan Medical Inc. Other companies with substantial experience in industrial robotics could potentially expand into the field of medical robotics and become competitors. Our revenues may be reduced due to pricing pressure or eliminated if our competitors develop and market products that are more effective or less expensive than our products. If we are unable to compete successfully, our revenues will suffer, which could have a material adverse effect on our business, financial condition, or result of operations. We may not be able to maintain or improve our competitive position against current or potential competitors, especially those with greater resources.
In addition, third-party service providers that provide services to da Vinci Surgical System and Ion endoluminal system operators may emerge and compete with us on price or offerings. To date, substantially all of our customers have sourced services on their systems from us through service contract commitments or time and materials contracts. Furthermore, there are third-party service providers offering consulting services targeted at analyzing the cost-effectiveness of hospitals’ robotic-assisted medical programs, including procedures performed, placement of systems, and consumption of instruments and accessories. We currently provide similar services and analysis to our customers, but it is difficult to assess the impact that this may have on our business. If we are unable to compete successfully with any third-party service providers, our revenues may suffer.
IF OUR PRODUCTS DO NOT ACHIEVE AND MAINTAIN MARKET ACCEPTANCE, WE WILL NOT BE ABLE TO GENERATE THE REVENUE NECESSARY TO SUPPORT OUR BUSINESS.
The da Vinci Surgical System and our other products represent a fundamentally new way of performing medical procedures. Achieving and maintaining physician, patient, and third-party payor acceptance of robotic-assisted medical procedures as a preferred method of performing these procedures is crucial to our success. If our products fail to achieve or maintain market acceptance, customers will not purchase our products, and we will not be able to generate the revenue necessary to support our business. We believe that physicians’ and third-party payors’ acceptance of the benefits of procedures performed using our products will be essential for acceptance of our products by patients. Physicians will not recommend the use of our products unless we can demonstrate that they produce results comparable or superior to existing techniques. Even if we can prove the effectiveness of our products through clinical studies, physicians may elect not to use our products for any number of other reasons. For example, cardiologists may continue to recommend conventional heart surgery simply because such surgery is already widely accepted. In addition, physicians may be slow to adopt our products because of the perceived liability risks arising from the use of new products and the uncertainty of reimbursement from third-party payors, particularly in light of ongoing healthcare reform initiatives and the evolving U.S. healthcare environment.
We expect that there will continue to be a learning process involved for patient care teams to become proficient in the use of our products. Broad use of our products requires training of patient care teams. Market acceptance could be delayed by the time required to complete this training. We may not be able to rapidly train patient care teams in numbers sufficient to generate adequate demand for our products.
IF HOSPITALS ARE UNABLE TO OBTAIN COVERAGE AND REIMBURSEMENT FOR PROCEDURES USING OUR PRODUCTS, IF REIMBURSEMENT IS INSUFFICIENT TO COVER THE COSTS OF PURCHASING OUR PRODUCTS, OR IF LIMITATIONS ARE IMPOSED BY GOVERNMENTS ON THE AMOUNT HOSPITALS CAN CHARGE FOR CERTAIN PROCEDURES, WE MAY BE UNABLE TO GENERATE SUFFICIENT SALES TO SUPPORT OUR BUSINESS.
In the U.S., hospitals generally bill for the services performed with our products to various third-party payors, such as Medicare, Medicaid, other government programs, and private insurance plans. If hospitals do not obtain sufficient reimbursement from third-party payors for procedures performed with our products, or if government and private payors’ policies do not cover surgical procedures performed using our products, we may not be able to generate the revenues necessary to support our business. In addition, to the extent that there is a shift from an inpatient setting to outpatient settings, we may
experience pricing pressure and a reduction in the number of procedures performed. Our success in OUS markets also depends on the eligibility of our products for coverage and reimbursement through government-sponsored healthcare payment systems and third-party payors. Reimbursement practices vary significantly by country. Many OUS markets have government-managed healthcare systems that control reimbursement for new products and procedures. Other foreign markets have both private insurance systems and government-managed systems that control reimbursement for new products and procedures. Market acceptance of our products may depend on the availability and level of coverage and reimbursement in a country within a particular time. In addition, healthcare cost containment efforts similar to those in the U.S. are prevalent in many of the other countries in which we sell, and intend to sell, our products, and these efforts are expected to continue. Refer to our risk factor titled “Changes in Healthcare Legislation and Policy May Have a Material Adverse Effect on Our Business, Financial Condition, or Results of Operations” for additional risks related to the ability of hospitals to obtain reimbursements.
In China, the Hunan Provincial Healthcare Security Administration implemented significant limits on what hospitals can charge patients for surgeries using robotic surgical technology, including soft tissue surgery and orthopedics. This rule has had and may continue to have a material negative impact on our procedures performed in the Hunan province. In addition to the Hunan province, the Hainan province (an island province of China) recently announced a policy to implement almost identical limits on what hospitals can charge patients for surgeries using robotic surgical technology. We cannot assure you that other provincial healthcare administrations will not impose similar limits.
IF OUR PRODUCTS CONTAIN DEFECTS OR ENCOUNTER PERFORMANCE PROBLEMS, WE MAY HAVE TO RECALL OUR PRODUCTS AND, AS A RESULT, INCUR ADDITIONAL UNFORESEEN COSTS, AND OUR REPUTATION MAY SUFFER.
Our success depends on the quality and reliability of our products. While we subject components sourced and products manufactured to stringent quality specifications and processes, our products incorporate mechanical parts, electrical components, optical components, and computer software, any of which may contain errors or exhibit failures, especially when products are first introduced. Component failures, manufacturing flaws, design defects, or inadequate disclosure of product-related risks with respect to our products could result in an unsafe condition or injury to, or death of, the patient. In addition, new products or enhancements may contain undetected errors or performance problems that, despite testing, are discovered only after commercial shipment. Because our products are designed to be used to perform complex surgical procedures, due to the serious and costly consequences of product failure, we and our customers have an increased sensitivity to such defects. In the past, we have voluntarily recalled certain products. Although our products are subject to stringent quality processes and controls, we cannot provide assurance that our products will not experience component aging, errors, or performance problems. If we experience product flaws or performance problems, any or all of the following could occur:
•delays in product shipments;
•loss of revenue;
•delay in market acceptance;
•diversion of our resources;
•damage to our reputation;
•product recalls, which can include, but not be limited to, product withdrawals from the market, labeling changes, design changes, customer notifications, and notifications to global regulatory bodies;
•regulatory actions;
•increased service or warranty costs; or
•product liability claims.
Costs associated with defects or performance problems of our products could have a material adverse effect on our business, financial condition, or results of operations.
WE COULD BE SUBJECT TO SIGNIFICANT, UNINSURED LOSSES, WHICH MAY HAVE A MATERIAL ADVERSE IMPACT ON OUR BUSINESS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
For certain risks, we do not maintain insurance coverage due to cost and/or availability. For example, we self-insure our product liability risks, and we indemnify our directors and officers for third-party claims and do not carry insurance to cover that indemnity or the related underlying potential losses. Also, we do not carry, among other types of coverage, earthquake insurance. In addition, in the future, we may not continue to maintain certain existing insurance coverage or adequate levels of coverage. Premiums for many types of insurance have increased significantly in recent years and, depending on market conditions and our circumstances, certain types of insurance, such as directors’ and officers’ insurance, may not be available in the future on acceptable terms or at all. Because we retain some portion of our insurable risks and, in some cases, we are entirely self-insured, unforeseen or catastrophic losses in excess of insurance coverage could require us to pay substantial amounts, which may have a material adverse impact on our business, financial condition, or results of operations.
IF WE LOSE KEY PERSONNEL OR ARE UNABLE TO ATTRACT AND RETAIN OTHER PERSONNEL, OUR ABILITY TO COMPETE WILL BE HARMED, AND INCREASES IN LABOR COSTS COULD MATERIALLY ADVERSELY IMPACT OUR BUSINESS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
We are highly dependent on the principal members of our management and scientific staff. For example, our product development plans depend, in part, on our ability to attract and retain software, mechanical, electrical, and robotics engineers. Attracting and retaining qualified personnel will be critical to our success, and competition for qualified personnel is intense. We may not be able to attract and retain personnel on acceptable terms given the constrained labor market and competition for such personnel among technology and healthcare companies. Additionally, as a result of recent declines in our stock price, certain long-term incentive benefits, such as recently issued stock options, may be viewed as having less value and, accordingly, could lead to higher attrition. Moreover, we may encounter higher recruiting expenses, wage rates, and retention benefits, which may result from higher inflationary environments.
Fluctuations in labor availability globally, including labor shortages and staff burnout and attrition, may also impact our ability to hire and retain personnel critical to our manufacturing, logistics, and commercial operations. The extent and duration of the impact of labor market challenges are subject to numerous factors, including the continuing impact of the COVID-19 pandemic, availability of qualified and highly skilled persons in the markets where we operate and unemployment levels within these markets, behavioral changes, such as fully engaging employees and earning loyalty, prevailing wage rates, health and other insurance and benefit costs, inflation, adoption of new or revised employment and labor laws and regulations or government programs, safety levels of our operations, and our reputation within the labor market. The loss of any of our qualified personnel or our inability to attract and retain qualified personnel could harm our business and our ability to compete, and related expenses could adversely affect our results of operations and financial condition.
Moreover, if we fail to attract, motivate, or retain personnel, or relax our standards in order to meet the demands of our growth, our corporate culture, our ability to achieve our strategic objectives, and our compliance with obligations under our internal controls and other requirements may be harmed. We believe that a critical contributor to our success has been our corporate culture, which we believe fosters innovation, teamwork, and a focus on execution, as well as facilitates critical knowledge transfer and knowledge sharing. Many of our employees have worked remotely during the COVID-19 pandemic, which makes it challenging to maintain or enhance our culture. While we are exploring ways to improve the employee experience, regardless of whether an employee is working from home, fully on-site, or in a hybrid fashion, the impact this will have on our corporate culture, innovation, collaboration, and ability to attract and retain talent is uncertain.
NEGATIVE PUBLICITY, WHETHER ACCURATE OR INACCURATE, CONCERNING OUR PRODUCTS OR OUR COMPANY COULD REDUCE MARKET ACCEPTANCE OF OUR PRODUCTS AND COULD RESULT IN DECREASED PRODUCT DEMAND AND REDUCED REVENUES.
There have been reports and articles published questioning patient safety and efficacy associated with robotic-assisted surgery with the da Vinci Surgical System, its cost relative to other disease management methods, and the adequacy of surgeon training. Negative publicity, including statements made by public officials, whether accurate or inaccurate, concerning our products or our Company could reduce market acceptance of our products and could result in decreased product demand and a decline in revenues. In addition, significant negative publicity could result in an increased number of product liability claims, regardless of whether these claims are meritorious. The number of claims could be further increased by plaintiffs’ law firms that use a wide variety of media to advertise their services and solicit clients for product liability cases against us.
WE EXPERIENCE LONG AND VARIABLE CAPITAL SALES CYCLES AND SEASONALITY IN OUR BUSINESS, WHICH MAY CAUSE FLUCTUATIONS IN OUR FINANCIAL RESULTS.
The sales and purchase order cycle of our systems is lengthy, because the systems are major capital items and their purchase generally requires the approval of senior management of hospitals, their parent organizations, purchasing groups, and government bodies, as applicable. In addition, sales to some of our customers are subject to competitive bidding or public tender processes. These approval and bidding processes can be lengthy. As a result, hospitals may delay or accelerate system purchases in conjunction with the timing of their capital budget timelines. Further, IDN groups are creating larger networks of system users with increasing purchasing power and are increasingly evaluating their robotic-assisted surgery programs to optimize the efficiency of surgeries using da Vinci Surgical Systems. Further, the introduction of new products could adversely impact our sales cycle as customers take additional time to assess the benefits and costs of such products. As a result, it is difficult for us to predict the length of capital sales cycles and, therefore, the exact timing of capital sales. Historically, our sales of da Vinci Surgical Systems have tended to be heavier in the fourth quarter and lighter in the first quarter, as hospital budgets are reset.
We have experienced procedure growth for a number of benign conditions, including hysterectomies, sacrocolpopexies, hernia repairs, cholecystectomies, bariatrics, and certain other surgeries. Many of these types of surgeries may be postponed in the short term by patients to avoid vacation periods and for other personal scheduling reasons. Patients may also accelerate procedures to take advantage of insurance funding cut-off dates. Historically, we have experienced lower procedure volume in
the first and third quarters of the year and higher procedure volume in the second and fourth quarters of the year. The timing of procedures and changes in procedure growth directly affect the timing of instruments and accessories and capital purchases by customers.
The above factors may contribute to substantial fluctuations in our quarterly operating results. Because of these fluctuations, it is possible that, in future periods, our operating results will fall below the expectations of securities analysts or investors. If that happens, the market price of our stock would likely decrease. These fluctuations, among other factors, also mean that our operating results in any particular period may not be relied upon as an indication of future performance.
NEW PRODUCT DEVELOPMENTS AND INTRODUCTIONS MAY ADVERSELY AFFECT OUR BUSINESS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
We develop and introduce new products with enhanced features and extended capabilities from time to time. We may introduce new products that target different markets than what our existing products target. The success of new product introductions depends on a number of factors including, but not limited to, timely and successful research and development, regulatory clearances, approvals, or certifications, pricing, competition, market and consumer acceptance, the effective forecasting and management of product demand, inventory levels, the management of manufacturing and supply costs, and the risk that new products may have quality or other defects in the early stages of introduction.
We invest substantially in various research and development projects to expand our product offerings. Our research and development efforts are critical to our success, and our research and development projects may not be successful. We may be unable to develop and market new products successfully, and the products we invest in and develop may not be well-received by customers or meet our expectations. Our research and development investments may not generate significant operating income or contribute to our future operating results for several years, and such contributions may not meet our expectations or even cover the costs of such investments. In addition, the introduction or announcement of new products or product enhancements may shorten the life cycle of our existing products or reduce demand for our current products, thereby offsetting any benefits of successful product introductions and potentially leading to challenges in managing inventory of existing products.
Our products are subject to various regulatory processes, and we must obtain and maintain regulatory approvals and certifications in order to sell our new products. If a potential purchaser believes that we plan to introduce a new product in the near future or is located in a country where a new product that we have introduced has not yet received regulatory clearance or certification, planned purchases may be deferred or delayed. In the past, we have experienced a slowdown in demand for existing products in advance of new product introductions, and we may experience a slowdown in demand in the future as well. It is also possible that a new product introduction could cause downward pressure on the prices of our existing products or require us to change how we sell our products, either of which could have material adverse effects on our revenues.
If we fail to effectively develop new products and manage new product introductions in the future, our business, financial condition, or results of operations could be adversely impacted.
WE ARE SUBJECT TO A VARIETY OF RISKS DUE TO OUR OPERATIONS OUTSIDE OF THE U.S.
We manufacture, perform research and development activities, and distribute our products in OUS markets. Revenue from OUS markets accounted for approximately 33%, 33%, and 32% of our revenue for the years ended December 31, 2022, 2021, and 2020, respectively. Our OUS operations are, and will continue to be, subject to a number of risks including:
•the failure to obtain or maintain the same degree of protection against infringement of our intellectual property rights due to differing intellectual property protection laws in OUS countries from those in the U.S.;
•multiple OUS regulatory requirements that are subject to change and that could impact our ability to manufacture and sell our products;
•changes in tariffs, trade barriers, and regulatory requirements;
•protectionist laws, policies, and business practices that favor local competitors or lead to non-U.S. customers favoring domestic technology solutions, which could slow our growth in OUS markets;
•local or national regulations that make it difficult or impractical to market or use our products;
•U.S. relations with the governments of the other countries in which we operate;
•the inability or regulatory limitations on our ability to move goods across borders;
•the risks associated with foreign currency exchange rate fluctuations;
•the difficulty in establishing, staffing, and managing OUS operations, including differing labor relations;
•the expense of establishing facilities and operations in new foreign markets;
•the building and maintenance of an organization capable of supporting geographically dispersed operations, including appropriate business procedures and controls;
•anti-corruption laws, such as the U.S. Foreign Corrupt Practices Act (“FCPA”), and other local laws prohibiting corrupt payments to governmental officials;
•antitrust and anti-competition laws;
•economic weakness, including inflation, or political instability in particular foreign economies and markets, including exposure to a higher degree of financial risk if we extend credit to customers in these economies; and
•business interruptions due to natural disasters, outbreak of disease, climate change, and other events beyond our control.
We have increased, and will continue to increase, our operations in China. There is inherent risk, based on the complex relationships between China and the U.S., that political, diplomatic, military, or other events could result in business disruptions, including increased regulatory enforcement against companies, tariffs, trade embargoes, or export restrictions. Tariffs increase the cost of our products and the components and raw materials that go into making them. These increased costs adversely impact the gross margin that we earn on our products. Tariffs can also make our products more expensive for customers, which could make our products less competitive and reduce consumer demand. Countries may also adopt other measures, such as controls on imports or exports of goods, technology, or data, which could adversely impact our operations and supply chain and limit our ability to offer our products and services as designed. These measures can require us to take various actions, including changing suppliers and restructuring business relationships. Changing our operations in accordance with new or changed trade restrictions can be expensive, time-consuming, disruptive to our operations and distracting to management. Such restrictions can be announced with little or no advance notice, and we may not be able to effectively mitigate all adverse impacts from such measures. Political uncertainty surrounding trade and other international disputes could also have a negative effect on consumer confidence and spending. Any of these events could reduce customer demand, increase the cost of our products and services, or otherwise have a materially adverse impact on our customers’ and suppliers’ businesses or results of operations.
For example, in 2020, the U.S. government amended the Entity List rules to expand the requirement to obtain a license prior to the export of certain technologies. In addition, in 2020, a new U.S. regulation seeks to prohibit the U.S. government from contracting with companies who use the products or services of certain Chinese companies. We believe that these regulations do not materially adversely impact our business at this time but cannot predict the impact that additional regulatory changes may have on our business in the future. These actions or similar actions may result in policies and regulations in response that could adversely affect our business operations in China or may otherwise limit our ability to offer our products and services in China and other parts of the world.
Following a national referendum and enactment of legislation by the government of the UK, the UK formally withdrew from the EU and ratified a trade and cooperation agreement governing its relationship with the EU. The EU–UK Trade and Cooperation Agreement (the “TCA”) was applied provisionally as of January 1, 2021, and entered into force on May 1, 2021. The TCA does not specifically refer to medical devices. However, as a result of Brexit, the EU Medical Devices Regulation will not be implemented in the UK, and previous legislation that sought to mirror the EU Medical Devices Regulation in the UK law has been revoked. The regulatory regime for medical devices in Great Britain continues to be based on the requirements derived from previous EU legislation, and the UK may choose to retain regulatory flexibility or align with the EU Medical Devices Regulation going forward. On June 26, 2022, the MHRA published its response to a 10-week consultation on the future regulation of medical devices in the UK. Regulations implementing the new regime were originally scheduled to come into force in July 2023 but have recently been postponed to July 2024. Devices issued by EU notified bodies are now subject to transitional arrangements. Following these transitional periods, it is expected that all medical devices will require a UKCA mark, but CE marks issued by EU notified bodies will remain valid until this time. Manufacturers may choose to use the UKCA mark on a voluntary basis until June 30, 2023. However, UKCA marking will not be recognized in the EU. Following the transitional period, compliance with the UK legislation will be a prerequisite to be able to affix the UKCA mark to our products, without which they cannot be sold or marketed in Great Britain. The TCA does provide for cooperation and exchange of information in the area of product safety and compliance, including market surveillance, enforcement activities and measures, standardization-related activities, exchanges of officials, and coordinated product recalls (or other similar actions). For medical devices that are locally manufactured but use components from other countries, the “rules of origin” criteria will need to be reviewed. Depending on which countries products will ultimately be sold in, manufacturers may start seeking alternative sources for components if this would allow them to benefit from no tariffs. The rules for placing medical devices on the Northern Ireland market will differ from those in Great Britain. These developments, or the perception that any related developments could occur, have had and may continue to have a material adverse effect on global economic conditions and financial markets, and our business would likely be impacted and the demand for our products could be depressed.
In addition, the U.S. federal government has made changes to the U.S. trade policy, including entering into a successor to the North American Free Trade Agreement (“NAFTA”), known as the United States-Mexico-Canada Agreement (“USMCA”), effective as of July 1, 2020. In addition, the U.S. federal government has implemented, or is considering the imposition of, tariffs on certain foreign goods. Such tariffs and, if enacted, any further legislation or actions taken by the U.S. federal
government that restrict trade, such as additional tariffs, trade barriers, and other protectionist or retaliatory measures taken by governments in Europe, Asia, and other countries, could adversely impact our ability to sell products and services in our OUS markets. Tariffs could increase the cost of our products and the components and raw materials that go into making them. These increased costs could adversely impact the gross margin that we earn on our products, which could make our products less competitive and reduce consumer demand. Countries may also adopt other protectionist measures that could limit our ability to offer our products and services.
Furthermore, a large portion of our OUS sales are denominated in U.S. dollars. As a result, an increase in the value of the U.S. dollar relative to foreign currencies could make our products less competitive and/or less affordable in OUS markets.
If we are unable to meet and manage these risks, our OUS operations may not be successful, which would limit the growth of our business and could have a material adverse effect on our business, financial condition, or result of operations.
WE MAY INCUR LOSSES ASSOCIATED WITH CURRENCY FLUCTUATIONS AND MAY NOT BE ABLE TO EFFECTIVELY HEDGE OUR EXPOSURE.
Our operating results are subject to volatility due to fluctuations in foreign currency exchange rates. Our primary exposure to fluctuations in foreign currency exchange rates relates to revenue and operating expenses denominated in currencies other than the U.S. dollar. The weakening of foreign currencies relative to the U.S. dollar adversely affects our foreign currency-denominated revenue. Margins on OUS revenue could also be materially adversely affected by foreign currency exchange rate fluctuations, as we may not be able to raise local prices to fully offset the strengthening of the U.S. dollar. Conversely, the strengthening of foreign currencies relative to the U.S. dollar, while generally beneficial to our foreign currency-denominated revenue and earnings, may cause us to reduce pricing on our products in our OUS markets and may cause us to incur losses on our foreign currency hedging instruments, thereby limiting the benefit that strengthened foreign currencies could have on our results of operations.
We attempt to mitigate a portion of these risks through foreign currency hedging, based on our judgment of the appropriate trade-offs among risk, opportunity, and expense. Although we have established a hedging program to partially hedge our exposure to foreign currency exchange rate fluctuations, primarily related to transactions denominated in the Euro, the British Pound, the Japanese Yen, the Korean Won, the New Taiwan Dollar, and the Swiss Franc, and we regularly review our hedging program and make adjustments as necessary, our hedging activities may not offset more than a portion of the adverse financial impact caused by unfavorable movement in foreign currency exchange rates, which could materially adversely affect our financial condition or results of operations. See “Item 7A. Quantitative and Qualitative Disclosures About Market Risk” for additional discussion on the impact of foreign exchange risk.
OUR CUSTOMERS MAY USE REMANUFACTURED AND/OR UNAUTHORIZED THIRD-PARTY INSTRUMENTS AND ACCESSORIES, WHICH COULD RESULT IN REDUCED REVENUE AND NEGATIVELY IMPACT OUR REPUTATION.
A significant portion of our revenue is generated through our sales of instruments and accessories. Third parties have offered and may continue to offer customers counterfeit instruments and accessories and/or instruments and accessories that have been remanufactured and/or are unauthorized, including instruments that have been remanufactured to support the use of some of our limited-use instruments beyond their labeled useful life. As of the filing date, we are unaware that the FDA or any other regulatory agency has granted 510(k) or equivalent clearance for the remanufacturing of any instruments for use with a da Vinci X or da Vinci Xi Surgical System, but we understand that the FDA has granted 510(k) clearance to one company for the remanufacturing of an EndoWrist instrument used with our da Vinci S and da Vinci Si Surgical Systems. While our sales arrangements with customers generally prohibit the use of unauthorized and unapproved instruments and accessories that lack FDA clearance or other applicable regulatory approval or certification with our systems, such activities could potentially result in reduced revenue, increased patient safety risks, and negative publicity for us if these products cause injuries and/or do not function as intended when used with our systems, any of which could have a material adverse effect on our business, financial condition, or results of operations.
INFORMATION TECHNOLOGY SYSTEM FAILURES, CYBERATTACKS, OR DEFICIENCIES IN OUR CYBERSECURITY COULD HARM OUR BUSINESS, CUSTOMER RELATIONS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
Our information technology systems are critical to the success of our products, help us operate effectively and efficiently, interface with customers, maintain our supply chain and manufacturing operations, maintain financial accuracy and efficiency, and help us produce our Consolidated Financial Statements. If we do not allocate and effectively manage the resources necessary to build and sustain the proper information technology infrastructure, we could be subject to transaction errors, processing inefficiencies, the loss of existing customers, difficulty attracting new customers, business operation disruptions, diversion of the attention of management and key information technology resources, security breaches, or the unauthorized access to, loss of, or damage to intellectual property, confidential information, or personal information. Our information
technology systems and those of our third-party service providers, strategic partners, and other contractors or consultants are vulnerable to attack, damage, or interruption from a variety of sources. These sources include computer viruses and malware (e.g., ransomware), malicious code, natural disasters, terrorism, war, telecommunication and electrical failures, hacking, cyberattacks, phishing attacks and other social engineering schemes, employee theft or misuse, human error, fraud, denial or degradation of service attacks, sophisticated nation-state and nation-state-supported actors, or unauthorized access or use by persons inside our organization, or persons with access to systems inside our organization. If our information technology systems do not effectively and securely collect, store, process, and report relevant data for the operation of our business, our ability to effectively plan, forecast, and execute our business plan and comply with applicable laws and regulations could be impaired. Any such impairment could materially and adversely affect our financial condition, results of operations, and the timeliness with which we report our internal and external operating results.
Our business requires us to use and store customer, employee, and business partner personal information. This may include names, addresses, phone numbers, email addresses, contact preferences, tax identification numbers, and payment account information. We require usernames and passwords in order to access our information technology systems. We also use encryption and authentication technologies to secure the transmission and storage of data. These security measures may be compromised as a result of security breaches by unauthorized persons, employee error, malfeasance, faulty password management, or other irregularity and result in persons obtaining unauthorized access to our data or accounts. Third parties may attempt to fraudulently induce employees or customers into disclosing usernames, passwords, or other sensitive information, which may, in turn, be used to access our information technology systems. As a result of the COVID-19 pandemic, we may also face increased cybersecurity risks due to our reliance on internet technology and the number of our employees who are working remotely, which may create additional opportunities for cybercriminals to exploit vulnerabilities.
In addition, unauthorized persons may attempt to hack into our products or systems to obtain personal data relating to patients or employees, our confidential or proprietary information, or confidential information we hold on behalf of third parties. If the unauthorized persons successfully hack into or interfere with our connected products or services, they may create issues with product functionality that could pose a risk of the loss of data, a risk to patient safety, and a risk of product recall or field action, which could adversely impact our business and reputation. We have programs in place to detect, contain, and respond to data security incidents, and we make ongoing improvements to our information-sharing products in order to minimize vulnerabilities, in accordance with industry and regulatory standards. However, because the techniques used to obtain unauthorized access to or steal personal information or intellectual property, or sabotage systems containing personal information or intellectual property, change frequently and may originate from less regulated and remote areas of the world and be difficult to detect, we may not be able to anticipate and prevent these intrusions or mitigate them when and if they occur. Even if identified, we may be unable to adequately investigate or remediate incidents or breaches due to attackers increasingly using tools and techniques that are designed to circumvent controls, avoid detection, and remove or obfuscate forensic evidence.
We also rely on external vendors to supply and/or support certain aspects of our information technology systems. The systems of these external vendors may contain defects in design or manufacture or other problems that could unexpectedly compromise the security of our own information technology systems, and we are dependent on these third parties to deploy appropriate security programs to protect their systems. In addition to potential exposure to data breaches, security and cybersecurity incidents, or other actions that may compromise the security of or interfere with the function of our systems, defects or vulnerabilities in the software or systems of our external vendors may expose failures in our internal controls and risk management processes, which may adversely impact our business, financial condition, or results of operations and may also harm our reputation, brand, and customer relationships.
While we devote significant resources to network security, data encryption, and other security measures to protect our systems and data, these security measures cannot provide absolute security. We and certain of our service providers are, from time to time, subject to cyberattacks and security breaches and incidents. We consider such cyberattacks or security breaches and incidents to be in the ordinary course of business for a company of our size in our industry. While we do not believe that we have experienced any significant system failure, accident, or security breach to date, if such an event were to occur, it could impair our ability to attract and retain customers for our products, impact the price of our stock, materially damage commercial relationships, and expose us to litigation or government investigations, which could result in penalties, fines, or judgments against us. The costs to us to eliminate or alleviate network security problems, bugs, viruses, worms, ransomware and other malicious software programs, and security vulnerabilities could be significant. Our efforts to address these problems may not be successful and could result in unexpected interruptions, delays, cessation of service, and harm to our business operations. Moreover, if a security breach affects our systems or results in the unauthorized release of personal information, our reputation and brand could be materially damaged, and use of our products and services could decrease. We would also be exposed to a risk of loss, litigation and potential liability, and regulatory scrutiny, which could have a material adverse impact on our business, financial condition, or results of operations.
Globally, attacks are expected to continue accelerating in both frequency and sophistication with increasing use of tools and techniques that are designed to circumvent controls, avoid detection, and remove or obfuscate forensic evidence, all of which hinders our ability to identify, investigate, and recover from incidents.
Furthermore, due to the political uncertainty involving Russia and Ukraine, there is also an increased likelihood that the tensions could result in cyberattacks or cybersecurity incidents that could either directly or indirectly impact our operations. Any attempts by cyber-attackers to disrupt our services or information technology systems or the services or information technology systems of our third-party service providers, strategic partners, and other contractors or consultants, if successful, could harm our business, result in the misappropriation of funds, be expensive to remedy, and damage our reputation or brand.
While we maintain cyber insurance coverage that is intended to address data security risks, such insurance coverage may be insufficient to cover all losses or claims that may arise.
OUR BUSINESS IS SUBJECT TO COMPLEX AND EVOLVING LAWS AND REGULATIONS REGARDING PRIVACY, DATA PROTECTION, AND OTHER MATTERS RELATING TO INFORMATION COLLECTION.
There are numerous state, federal, and foreign laws, regulations, decisions, and directives regarding privacy and the collection, storage, transmission, use, processing, disclosure, and protection of different types of personal data and personal information and other customer or other data, the scope of which is continually evolving and subject to differing interpretations. We may be subject to significant consequences, including penalties and fines, for any failure to comply with such laws, regulations, and directives.
For example, the GDPR, which is in effect across the EEA, imposes several stringent requirements for controllers and processors of personal data including, for example, imposing strict standards when obtaining consent from individuals to process their personal data, requiring robust disclosures to individuals, providing individual data rights, imposing short timelines for data breach notifications, limiting retention periods and secondary use of information, imposing certain requirements pertaining to health data as well as pseudonymized (i.e., key-coded) data, regulating cross-border transfers of personal data out of the EEA, as well as additional obligations when we contract third-party processors in connection with the processing of personal data. The GDPR provides that EEA member states may make their own further laws and regulations limiting the processing of genetic, biometric, or health data, which could limit our ability to use and share personal data or could cause our costs to increase and harm our business and financial condition. Failure to comply with the requirements of the GDPR and the applicable national data protection laws of the EEA member states may result in fines of up to 4% of the total worldwide annual turnover of the preceding financial year and other administrative penalties. In addition to fines, a breach of the GDPR may result in regulatory investigations, reputational damage, orders to cease/change our data processing activities, enforcement notices, assessment notices (for a compulsory audit), and/or civil claims (including class actions). Compliance with new data protection rules imposed by GDPR may be onerous and adversely affect our business, financial condition, or results of operations.
Further, since 2021, we have been subject to the UK GDPR, which, together with the amended UK Data Protection Act 2018, retains the GDPR in UK national law. The UK GDPR mirrors the fines under the GDPR, e.g., fines up to 4% of worldwide annual turnover of the preceding financial year. The relationship between the UK and the EU in relation to certain aspects of data protection law remains unclear, and it is unclear how UK data protection laws and regulations will develop in the medium to longer term and how data transfers to and from the UK will be regulated in the long term. These changes may lead to additional costs and increase our overall risk exposure.
In the United States, the Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and regulations implemented thereunder (collectively, “HIPAA”), imposes privacy, security, and breach notification obligations on certain healthcare providers, health plans, and healthcare clearinghouses, known as covered entities, as well as their business associates that perform certain services that involve creating, receiving, maintaining, or transmitting individually identifiable health information for or on behalf of such covered entities and their covered subcontractors. Entities that are found to be in violation of HIPAA, as the result of a breach of unsecured personal information, a complaint about privacy practices, or an audit by the U.S. Department of Health and Human Services (“HHS”), may be subject to significant civil, criminal, and administrative fines and penalties and/or additional reporting and oversight obligations if they are required to enter into a resolution agreement and corrective action plan with HHS to settle allegations of HIPAA non-compliance.
Even when HIPAA does not apply, according to the Federal Trade Commission (the “FTC”), violating consumers’ privacy rights or failing to take appropriate steps to keep consumers’ personal information secure may constitute unfair and/or deceptive acts or practices in violation of Section 5(a) of the Federal Trade Commission Act. The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities.
Further, the California Consumer Privacy Act (the “CCPA”) went into effect in 2020 and gives California residents expanded rights to access and delete their personal information, opt out of certain personal information sharing, and receive detailed information and how their personal information is used. The CCPA imposes compliance burdens on many organizations doing business in California that collect personal information about California residents. The CCPA’s definition of personal information is very broad and specifically includes biometric information. The CCPA allows for significant fines by the state attorney general, as well as a private right of action from individuals in relation to certain security breaches. The enactment of the CCPA is prompting a wave of similar legislative developments in other U.S. states and creating the potential for a patchwork of overlapping but different state laws. These developments are increasing our compliance burden and our risk, including risks of regulatory fines, litigation, and associated reputational harm. Additionally, a new California ballot initiative, the California Privacy Rights Act (the “CPRA”) recently passed in California. The CPRA will substantially expand the requirements of the CCPA and will impose additional data protection obligations on companies doing business in California. The majority of the provisions will go into effect on January 1, 2023, and additional compliance investment and potential business process changes may be required. Similar laws have passed in Virginia, Colorado, Connecticut, and Utah and have been proposed in other states and at the federal level, reflecting a trend toward more stringent privacy legislation in the U.S.
In addition, recent legal developments in Europe have created complexity and compliance uncertainty regarding certain transfers of personal data from the EEA or UK to the United States. For example, on July 16, 2020, the Court of Justice of the European Union (the “CJEU”) invalidated the EU-U.S. Privacy Shield Framework (“Privacy Shield”) under which personal data could be transferred from the EU to U.S. entities who had self-certified under the Privacy Shield scheme. The CJEU further noted that reliance on the standard contractual clauses (a standard form of contract approved by the European Commission as an adequate personal data transfer mechanism and potential alternative to the Privacy Shield) alone may not necessarily be sufficient in all circumstances and that transfers must be assessed on a case-by-case basis. European court and regulatory decisions subsequent to the CJEU decision of July 16, 2020, have taken a restrictive approach to international data transfers.
We rely on a mixture of mechanisms to transfer personal data from our EU business to the U.S. (including having previously relied on Privacy Shield) and are evaluating whether additional mechanisms will be required to establish adequate safeguards for personal data. As supervisory authorities issue further guidance on personal data export mechanisms, including circumstances where the standard contractual clauses cannot be used and/or start taking enforcement action, we could suffer additional costs, complaints, and/or regulatory investigations or fines. Moreover, if we are unable to transfer Personal Information between and among countries and regions in which we operate, it could affect the manner in which we provide our services and could adversely affect our financial results.
In Israel, The Protection of Privacy Law, 5741-1981 (the “Israeli Privacy Law”) regulates the protection of privacy and personal data, along with several other specific regulations enacted thereunder and, in particular, the Privacy Protection Regulations (Data Security), 5777-2017 (together with Israeli Privacy Law, the “Israeli Privacy Law and Regulations”). Under the Israeli Privacy Law and Regulations, organizations are subject to various privacy and data protection requirements, including mandatory registration of databases with the Israeli Registrar of Databases (if certain conditions are met), executing data processing agreements with data recipients, safeguarding the collection and processing of personal data, safeguarding the transfer of personal data (which is specifically subject to the requirements of the Privacy Protection Regulations), personal data breach notification obligations, and other requirements. The Privacy Protection Authority (the “PPA”) is responsible for enforcement of the Israeli Privacy Law and Regulations and periodically publishes opinions and guidelines on privacy matters. In terms of enforcement, failure to comply with the Israeli Privacy Law and Regulations can result in PPA investigations, administrative fines or sanctions, and civil or criminal actions (civil proceedings may include statutory damages without the need to prove actual damages).
Furthermore, any failure, or perceived failure, by us to comply with or make effective modifications to our policies or to comply with any federal, state, or international privacy, data-retention, or data-protection-related laws, regulations, orders, or industry self-regulatory principles could result in proceedings or actions against us by governmental entities or others, a loss of customer confidence, damage to our brand and reputation, and a loss of customers, any of which could have an adverse effect on our business. In addition, various federal, state, and foreign legislative or regulatory bodies may enact new or additional laws and regulations concerning privacy, data-retention, and data-protection issues, including laws or regulations mandating disclosure to domestic or international law enforcement bodies, which could adversely impact our business or our reputation with customers. For example, some countries have adopted laws mandating that some personal information regarding customers in their country be maintained solely in their country. Having to maintain local data centers and redesign product, service, and business operations to limit personal information processing to within individual countries could increase our operating costs significantly.
IF WE FAIL TO SUCCESSFULLY ACQUIRE OR INTEGRATE NEW BUSINESSES, PRODUCTS, AND TECHNOLOGY, WE MAY NOT REALIZE EXPECTED BENEFITS, OR OUR BUSINESS MAY BE HARMED.
We need to grow our businesses in response to changing technologies, customer demands, and competitive pressures. In some circumstances, we may decide to grow our business through the acquisition of complementary businesses, products, or technologies rather than through internal development.
Identifying suitable acquisition candidates can be difficult, time-consuming, and costly, and we may not be able to identify suitable candidates or successfully complete identified acquisitions. In addition, completing an acquisition can divert our management and key personnel from our business operations, which could harm our business and affect our financial results. Even if we complete an acquisition, we may not be able to successfully integrate newly acquired organizations, products, technologies, or employees into our operations or may not fully realize some of the expected synergies. An acquired company may have deficiencies in product quality, regulatory marketing authorizations or certifications, or intellectual property protections, which are not detected during due diligence activities or which are unasserted at the time of acquisition. It may be difficult, expensive, and time-consuming for us to re-establish market access, regulatory compliance, or cure such deficiencies in product quality or intellectual property protection in such cases, which may have a material adverse impact on our business, financial condition, or results of operations.
Integrating an acquisition can also be expensive and time-consuming and may strain our resources. In many instances, integrating a new business will also involve implementing or improving internal controls appropriate for a public company at a business that lacks them. In addition, we may be unable to retain the employees of acquired companies or the acquired company’s customers, suppliers, distributors, or other partners for a variety of reasons, including that these entities may be our competitors or may have close relationships with our competitors. Furthermore, acquired companies may have less mature or less sophisticated information systems, securities practices, or training, which may result in an increased risk of security and cybersecurity incidents when such companies are integrated. In 2020, we acquired Orpheus Medical Ltd. and its wholly owned subsidiaries (“Orpheus Medical”) to deepen and expand our integrated informatics platform. The integration of this acquisition involves complex operations across different geographic locations and new products, distribution networks, and legal jurisdictions. Therefore, we cannot assure you that we can successfully integrate this acquisition or realize the expected benefits from this acquisition. Failure to successfully integrate our acquisitions may have a material adverse impact on our business, financial condition, or results of operations.
IF WE DO NOT SUCCESSFULLY MANAGE OUR COLLABORATION ARRANGEMENTS, LICENSING ARRANGEMENTS, JOINT VENTURES, STRATEGIC ALLIANCES, OR PARTNERSHIPS WITH THIRD PARTIES, WE MAY NOT REALIZE THE EXPECTED BENEFITS FROM SUCH ALLIANCES, WHICH MAY HAVE A MATERIAL ADVERSE EFFECT ON OUR BUSINESS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
From time to time, we enter into collaborations, in-licensing arrangements, joint ventures, strategic alliances, or partnerships to complement or augment our research and development, product development, training, procedure development, and marketing efforts. For example, in 2016, we entered into an agreement to form the Joint Venture. In January 2019, the Joint Venture acquired certain assets related to the da Vinci distribution business of Chindex, a subsidiary of Fosun Pharma, following which the Joint Venture began direct distribution operations for da Vinci products and services in China. There can be no assurance that we and the Joint Venture can successfully complete development of robotic-assisted, catheter-based medical devices, or that we and the Joint Venture will successfully commercialize such products. There can also be no assurance that the Joint Venture will not require additional contributions to fund its business, that the Joint Venture will become profitable, or that the expected benefits of the acquisition of certain assets of Chindex will be realized. Proposing, negotiating, and implementing collaborations, in-licensing agreements, joint ventures, strategic alliances, or partnerships may be a lengthy and complex process. In addition, other companies, including those with substantially greater financial, marketing, sales, technology, or other business resources, may compete with us for these opportunities or arrangements. As a result, we may not identify, secure, or complete any such arrangements in a timely manner, on a cost-effective basis, or on otherwise favorable terms, if it all.
There can be no assurance that we will realize the expected benefits from these alliances. In addition, we may not be in a position to exercise sole decision-making authority regarding any collaboration or other arrangement, which could create the potential risk of creating impasses on decisions, and our alliances may have economic or business interests that are, or that may become, inconsistent with our interests. It is possible that conflicts may arise in these relationships, such as conflicts concerning the achievement of performance milestones or the interpretation of significant terms under any agreement, such as those related to financial obligations, termination rights, or the ownership or control of intellectual property developed during the collaboration. These alliances can be difficult to manage, given the potentially different interests of the parties involved, and we could suffer delays in product development or other operational difficulties.
There can be no assurance that we will realize a return on our strategic investments. Further, if we acquire privately held companies, valuations of such companies are inherently complex due to the lack of readily available market data. If we determine that our investments in privately held companies have experienced a decline in value, we may be required to record impairments, which could be material and have an adverse effect on our results of operations.
These alliances may also involve significant costs and divert the focus and attention of our management and other key personnel. Any of these relationships may require us to incur non-recurring and other charges, increase our near- and long-term expenditures, or disrupt our ordinary business activities. Such arrangements may also expose us to numerous known and unknown risks, including unique risks with respect to the economic, political, and regulatory environment of any foreign entities with whom we partner, including Fosun Pharma. Any of the foregoing may have a material adverse effect on our business, financial condition, or results of operations.
WE EXPECT GROSS PROFIT MARGINS TO VARY OVER TIME, AND CHANGES IN OUR GROSS PROFIT MARGINS COULD ADVERSELY AFFECT OUR BUSINESS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
Our gross profit margins have fluctuated from period to period, and we expect that they will continue to fluctuate in the future. Our gross profit margins may be adversely affected by numerous factors, including:
•changes in customer, geographic, or product mix, including the mix of system models sold or leased;
•changes in the portion of sales involving a trade-in of another system and the amount of trade-in credits given;
•our introduction of new products, which may have lower margins than our existing products;
•our inability to maintain or reduce production costs;
•changes to our pricing strategy;
•competition;
•changes in production volume driven by demand for our products;
•changes in material, labor, or other manufacturing-related costs, including the impact of foreign exchange rate fluctuations for foreign currency-denominated costs;
•changes to U.S. and foreign trade policies, such as the enactment of tariffs on goods imported into the U.S. including, but not limited to, goods imported from Mexico where we manufacture a majority of our instruments that we sell;
•inventory obsolescence, which may result from maintaining significant inventories of raw materials, components, and finished goods;
•product recall charges; and
•market conditions.
If we are unable to offset the unfavorable impact of the factors noted above by increasing the volume of products shipped, reducing product manufacturing costs, or otherwise, our business, financial condition, or results of operations may be adversely affected.
WE UTILIZE DISTRIBUTORS FOR A PORTION OF OUR SALES AND SERVICE OF OUR PRODUCTS IN CERTAIN COUNTRIES, WHICH SUBJECTS US TO A NUMBER OF RISKS THAT COULD HARM OUR BUSINESS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
We have strategic relationships with a number of key distributors for the sale and service of our products in certain countries. If these strategic relationships are terminated and not replaced, our revenues and/or ability to sell or service our products in the markets serviced by these distributors could be adversely affected. In addition, we may be named as a defendant in lawsuits against our distributors related to sales or service of our products performed by them. Refer to our risk factor titled “We are subject to litigation, investigations, and other legal proceedings relating to our products, our customers, our competitors, and government regulators that could materially adversely affect our financial condition, divert management’s attention, and harm our business.” Our distributors may affect our ability to effectively market our products in certain countries or regulatory jurisdictions if a distributor holds the regulatory authorization or certification in such countries or within such regions and causes, by action or inaction, the suspension of such marketing authorization or certification or sanctions for non-compliance. It may be difficult, expensive, and time-consuming for us to re-establish market access or regulatory compliance in such cases.
WE OFFER ALTERNATIVE CAPITAL ACQUISITION APPROACHES AND, AS A RESULT, WE ARE EXPOSED TO THE CREDIT RISK OF SOME OF OUR CUSTOMERS AND THE RISK OF LOSSES OF REVENUE, WHICH COULD RESULT IN MATERIAL LOSSES.
We believe customer financing through leasing is an important consideration for some of our customers and have experienced an increase in demand for customer financing. Lease financing arrangements have the effect of reducing cash flows at lease commencement and, instead, spread them over the life of the lease term, which increases the time taken to recover our product costs and can impact our liquidity. We may experience losses from a customer’s failure to make payments according to the contractual lease terms. Our exposure to the credit risks relating to our lease financing arrangements may increase if our customers are adversely affected by changes in healthcare laws, coverage and reimbursement, economic pressures or uncertainty, or other customer-specific factors.
Although we have programs in place that are designed to monitor and mitigate the associated risks, there can be no assurance that such programs will be effective in reducing credit risks relating to these lease financing arrangements. If the level of credit losses we experience in the future exceeds our expectations, such losses could have a material adverse effect on our financial condition or results of operations.
Certain of our leasing arrangements allow customers to cancel, return, or upgrade the systems leased prior to the end of the lease term without incurring a financial penalty. We also lease our systems to certain qualified customers where the lease payments are based on their usage of the systems. While leases and usage-based arrangements enable our customers to upgrade and get access to new technologies faster, it may also enable competitors to more easily induce customers to switch to a competitor’s system. Furthermore, depending on the timing and terms of the upgrade transaction, the amount of revenue generated on the initial and upgraded lease arrangements may not, in the aggregate, generate the same amount of revenue that a traditional sale and trade-in transaction would. Also, if customers do not perform a sufficient number of procedures on systems leased under usage-based arrangements, or return or terminate leases prematurely, it could have a material adverse effect on our business, financial condition, or result of operations.
WE ARE EXPOSED TO CREDIT RISK AND FLUCTUATIONS IN THE MARKET VALUE OF OUR INVESTMENTS.
Our investment portfolio includes both domestic and international investments. The credit ratings and pricing of our investments can be negatively affected by liquidity concerns, credit deterioration, financial results, economic risk, political risk, or other factors. As a result, the value and liquidity of our cash equivalents and marketable securities could fluctuate substantially. Our other income and expense could also vary materially from expectations depending on gains or losses realized on the sale or exchange of investments, impairment charges resulting from revaluations of debt and equity securities and other investments, changes in interest rates, increases or decreases in cash balances, volatility in foreign exchange rates, and changes in the fair value of derivative instruments. Increased volatility in the financial markets and overall economic uncertainty could increase the risk that actual amounts realized on our investments may differ significantly from the fair values currently assigned to them.
Our Intuitive Ventures fund plans to invest in early-stage companies, which involve substantial risks and uncertainties. These risks and uncertainties include, among other things, uncertainties inherent in research and development; uncertainties regarding the ability of Intuitive Ventures to identify investment candidates; uncertainties regarding the success of Intuitive Ventures’ investments; uncertainties and variables inherent in the operating and financial performance in investments made, including, among other things, competitive developments and general economic, political, business, industry, regulatory and market conditions; future exchange and interest rates; and changes in tax and other laws, regulations, rates and policies.
While we have not realized any significant losses on our cash equivalents, marketable securities, or other investments, future fluctuations in their value could have a material adverse impact on our business, financial condition, or results of operations.
THE ONGOING ARMED CONFLICT BETWEEN RUSSIA AND UKRAINE COULD ADVERSELY AFFECT OUR BUSINESS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
In February 2022, Russian military forces launched a military action in Ukraine, and sustained conflict and disruption in the region has continued. The length, impact, and outcome of this ongoing military conflict is highly unpredictable and could lead to significant market and other disruptions, including significant volatility in commodity prices and supply of energy resources, instability in financial markets, supply chain interruptions, political and social instability, trade disputes or trade barriers, changes in consumer or purchaser preferences, as well as an increase in cyberattacks and espionage.
Russia’s military actions against Ukraine have led to substantial expansion of sanction programs imposed by the United States, the European Union, the United Kingdom, Canada, Switzerland, Japan, and other countries against Russia, Belarus, the
Crimea Region of Ukraine, the so-called Donetsk People’s Republic, and the so-called Luhansk People’s Republic, including, among others:
•blocking sanctions against some of the largest state-owned and private Russian financial institutions (and their subsequent removal from the Society for Worldwide Interbank Financial Telecommunication payment system) and certain Russian businesses, some of which have significant financial and trade ties to the European Union;
•blocking sanctions against Russian and Belarusian individuals, including the Russian President, other politicians, and those with government connections or involved in Russian military activities; and
•blocking of Russia’s foreign currency reserves as well as expansion of sectoral sanctions and export and trade restrictions, limitations on investments and access to capital markets, and bans on various Russian imports.
In retaliation against new international sanctions and as part of measures to stabilize and support the volatile Russian financial and currency markets, the Russian authorities also imposed significant currency control measures aimed at restricting the outflow of foreign currency and capital from Russia, imposed various restrictions on transacting with non-Russian parties, banned exports of various products, and imposed other economic and financial restrictions. The situation is rapidly evolving, and additional sanctions by Russia on the one hand, and by the other countries on the other hand, could adversely affect the global economy, financial markets, energy supply and prices, certain critical materials and metals, supply chains, and global logistics and could adversely affect our business, financial condition, or results of operations.
We are actively monitoring the situation in Ukraine and Russia and assessing its impact on our business, including our business partners and customers. To date, we have not experienced any material interruptions in our infrastructure, supplies, technology systems, or networks needed to support our operations. We have no way to predict the progress or outcome of the military conflict in Ukraine or its impacts in Ukraine, Russia, Belarus, Europe, or the U.S. The extent and duration of the military action, sanctions, other consequences, such as Russia imposing restrictions on transactions or banning the export of energy products, including natural gas, and the resulting market disruptions could be significant and could potentially have substantial impact on the global economy and our business for an unknown period of time. Impacts to our business may include, but are not limited to, procedures performed, demand for our products, and ability to spend on capital equipment and healthcare in general. Any such disruption may also magnify the impact of other risks described.
WE MAY ENCOUNTER MANUFACTURING PROBLEMS OR DELAYS THAT COULD RESULT IN LOST REVENUE.
Manufacturing our products is a complex process. We (or our critical suppliers) may encounter difficulties in scaling up or maintaining production of our products, including:
•problems involving production yields;
•quality control and assurance;
•component supply shortages;
•import or export restrictions on components, materials, or technology;
•shortages of qualified personnel; and
•compliance with state, federal, and foreign regulations.
If demand for our products exceeds our manufacturing capacity, we could develop a substantial backlog of customer orders. If we are unable to develop or maintain larger-scale manufacturing capabilities or build new manufacturing capabilities or facilities on schedule or within budget, our ability to generate revenue and maintain profit margins as expected will be limited and our reputation in the marketplace could be damaged, all of which may have a material adverse impact on our business, financial condition, or results of operations.
DISRUPTIONS AT THE FDA AND OTHER GOVERNMENT AGENCIES OR NOTIFIED BODIES CAUSED BY FUNDING SHORTAGES OR GLOBAL HEALTH CONCERNS COULD HINDER THEIR ABILITY TO HIRE, RETAIN, OR DEPLOY KEY LEADERSHIP AND OTHER PERSONNEL, OR OTHERWISE PREVENT PRODUCTS FROM BEING DEVELOPED, CLEARED, CERTIFIED, APPROVED, OR COMMERCIALIZED IN A TIMELY MANNER OR AT ALL, WHICH MAY ADVERSELY AFFECT OUR BUSINESS.
Hospitals, health systems, and physicians depend on a number of government agencies and services to effectively deliver healthcare to their patients. A prolonged government shutdown could impact inspections, regulatory review and certifications, grants, or approvals or could cause other situations that could impede their ability to effectively deliver healthcare, including attempts to reduce payments and other reimbursements to hospitals by federal healthcare programs. These situations could adversely affect our customers’ ability to perform procedures with our devices and/or their decisions to purchase additional products from us.
In addition, the ability of the FDA, foreign authorities, and notified bodies to review and clear, approve, or certify new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable. Disruptions at the FDA and other agencies or notified bodies, including a prolonged government shutdown, may cause significant regulatory delays and, therefore, delay our efforts to seek clearances, approvals, or certifications from the FDA, foreign authorities, and notified bodies and adversely affect business travel and import and export of products, all of which could have a material adverse effect on our business, financial condition, or results of operations. For example, over the last several years, the U.S. government has shut down several times, and certain regulatory agencies, such as the FDA, have had to furlough critical FDA employees and stop critical activities.
Separately, in response to the global COVID-19 pandemic, the FDA postponed most inspections of domestic and foreign manufacturing facilities at various points. Even though the FDA has since resumed standard inspection operations of domestic facilities where feasible, the FDA has continued to monitor and implement changes to its inspectional activities to ensure the safety of its employees and those of the firms it regulates as it adapts to the evolving COVID-19 pandemic, and any resurgence of the virus or emergence of new variants may lead to further inspectional delays. Regulatory authorities outside the United States have adopted similar restrictions or other policy measures in response to the COVID-19 pandemic. If a prolonged government shutdown occurs, or if global health concerns continue to prevent the FDA, other regulatory authorities, or notified bodies from conducting their regular inspections, reviews, or other regulatory activities, it could significantly impact the ability of the FDA, other regulatory authorities, or notified bodies to timely review and process our regulatory submissions, which could have a material adverse effect on our business.
For instance, in the EU, notified bodies must be officially designated to certify products and services in accordance with the EU Medical Devices Regulation. While several notified bodies have been designated, the COVID-19 pandemic has significantly slowed down their designation process, and the current designated notified bodies are facing a large number of requests with the new regulation, as a consequence of which review times have lengthened. Unless additional transitional measures are implemented, this situation could impact our ability to grow our business in the EU and EEA.
WE ARE SUBJECT TO RISKS ASSOCIATED WITH REAL ESTATE CONSTRUCTION AND DEVELOPMENT.
The development of our facilities is subject to risks relating to our ability to complete our projects on schedule or within budget. Factors that may result in a development project being prevented or delayed from completion or exceeding budget include, but are not limited to (i) construction delays due to labor challenges, poor weather, defects, or cost overruns, which may increase project development costs; (ii) cost escalations associated with materials, including changes in availability, proximity, and cost of materials, such as steel, cement, concrete, aggregates, oil, fuel, and other construction materials, including changes in U.S. trade policies and retaliatory responses from other countries, changes in foreign exchange rates, as well as cost escalations associated with subcontractors and labor; (iii) the discovery of hazardous or toxic substances, or other environmental, culturally-sensitive, or related issues; (iv) an inability to obtain, or a significant delay in obtaining, zoning, construction, occupancy, and other required governmental permits and authorizations; (v) difficulty in complying with local, city, county, and state rules and regulations regarding permitting, zoning, subdivision, utilities, and water quality, as well as federal rules and regulations regarding air and water quality and protection of endangered species and their habitats; (vi) insufficient infrastructure capacity or availability (e.g., water, sewer, and roads) to serve the needs of our projects; (vii) failure to achieve or sustain anticipated occupancy levels; (viii) condemnation of all or parts of development or operating properties, which could adversely affect the value or viability of such projects; and (ix) natural disasters and other extreme weather conditions, including, but not limited to, hurricanes, tornadoes, earthquakes, wildfires, or flooding.
CONTINUED CONSOLIDATION IN THE HEALTHCARE INDUSTRY COULD HAVE AN ADVERSE EFFECT ON OUR BUSINESS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
The healthcare industry has been consolidating, and organizations continue to consolidate purchasing decisions for many of our healthcare provider customers. Numerous initiatives and reforms by legislators, regulators, and third-party payers to curb the rising cost of healthcare have catalyzed a consolidation of aggregate purchasing power within the markets in which we sell our products. As the healthcare industry consolidates, competition to provide products and services is expected to continue to intensify, resulting in pricing pressures and decreased average selling prices. We expect that market demand, government regulation, third-party payor coverage and reimbursement policies, government contracting requirements, and societal pressures will continue to change the worldwide healthcare industry, resulting in further consolidation, which may exert further downward pressure on prices of our products and services and may have a material adverse impact on our business, financial condition, or results of operations.
CLIMATE CHANGE AND NATURAL DISASTERS OR OTHER EVENTS BEYOND OUR CONTROL COULD DISRUPT OUR BUSINESS AND RESULT IN LOSS OF REVENUE OR HIGHER EXPENSES.
Natural disasters, terrorist activities, and other events beyond our control including, but not limited to, internet security threats and violence motivated by political or social causes, could adversely affect our business, financial condition, or results of operations. Moreover, global climate change could result in certain types of natural disasters occurring more frequently or with more intense effects. The impacts of climate change may include physical risks (such as frequency and severity of extreme weather conditions), social and human effects (such as population dislocations or harm to health and well-being), compliance costs, transition risks, shifts in market trends, and other adverse effects. Such impacts may disrupt parties in our supply chain, our customers, and our operations. For example, the March 2011 earthquake and tsunami in Japan, and their aftermath, created economic uncertainty and disrupted economic activities in Japan, including a reduction in hospital spending.
Physical risks associated with climate change are subject to increasing societal, regulatory, and political focus in the U.S. and globally. Shifts in weather patterns caused by climate change are expected to increase the frequency, severity, or duration of certain adverse weather conditions and natural disasters, such as hurricanes, tornadoes, earthquakes, wildfires, droughts, extreme temperatures, or flooding, which could cause more significant business and supply chain interruptions, damage to our products and facilities as well as the infrastructure of hospitals, medical care facilities, and other customers, reduced workforce availability, increased costs of raw materials and components, increased liabilities, and decreased revenues than what we have experienced in the past from such events. The geographic location of our California headquarters and many of our manufacturing facilities, as well as the facilities of certain of our key suppliers and service providers, subject them to earthquake, drought, and wildfire risks. If a major earthquake, wildfire, or other natural disaster were to damage our facilities or the facilities of our suppliers and service providers, or impact the ability of our employees or the employees of our suppliers and service providers to travel to their workplace, we may experience potential impacts ranging from production and shipping delays to lost revenues and increased costs, which could harm our business. Moreover, periods with increased drought and annual periods of wildfire danger may increase the probability of planned power outages in the communities where we work and live. For example, in October 2019, Pacific Gas and Electric, the public electric utility in the Northern California region, used planned power outages to avoid and contain wildfires sparked during strong wind events by downed power lines or equipment failure. If prolonged or frequent, such planned blackouts could impact our operations and the operations of our suppliers and service providers located in the Northern California region. While this danger has a low assessed risk of disrupting normal business operations, it has a potential impact on our employees’ abilities to commute to work or to work from home and stay connected effectively. We do not have multiple-site capacity for all of our operations in the event of a business disruption, and we are predominantly self-insured and may not be able to sufficiently cover losses or additional expenses that we may sustain. Furthermore, the impacts of global climate change on water resources may result in water scarcity, which could impact our ability to access sufficient quantities of water in certain locations and result in increased costs.
In addition, the increasing concern over climate change has resulted and may continue to result in more legal and regulatory requirements designed to mitigate the effects of climate change on the environment, including regulating greenhouse gas emissions, alternative energy policies, and sustainability initiatives. If such laws or regulations are more stringent than current legal or regulatory requirements, we may experience increased compliance burdens and costs to meet the regulatory obligations. Changes in requirements may adversely affect raw material sourcing from suppliers, our manufacturing operations and those of our suppliers, and the distribution of our products. Further, there may be increasing scrutiny and changing expectations from the market and other stakeholders with respect to Environmental, Social, and Governance (ESG) practices. Any such regulatory changes or increased market expectations could also have a significant effect on our operating and financial decisions, including those involving capital expenditures to reduce emissions and comply with other regulatory requirements or stakeholder expectations.
CHANGES IN OUR EFFECTIVE TAX RATE MAY ADVERSELY AFFECT OUR BUSINESS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
We are subject to taxes in the U.S. and other jurisdictions around the world. Tax rates in these jurisdictions may be subject to significant change due to economic and/or political conditions. A number of other factors may also impact our future effective tax rate, including:
•the jurisdictions in which profits are determined to be earned and taxed;
•the resolution of issues arising from tax audits with various tax authorities;
•changes in the valuation of our deferred tax assets and liabilities;
•increases in expenses not deductible for tax purposes, including write-offs of acquired intangibles and impairment of goodwill in connection with acquisitions;
•changes in the availability of tax credits, tax holidays, and tax deductions;
•changes in share-based compensation; and
•changes in tax laws or the interpretation of such tax laws and changes in generally accepted accounting principles.
We are unable to predict what changes to the tax laws of the U.S. and other jurisdictions may be proposed or enacted in the future or what effect such changes would have on our business. Any significant increase in our future effective tax rate could have a material adverse impact on our business, financial condition, or results of operations.
WE USE ESTIMATES, MAKE JUDGMENTS, AND APPLY CERTAIN METHODS IN DETERMINING OUR FINANCIAL RESULTS AND IN MEASURING THE PROGRESS OF OUR BUSINESS. AS THESE ESTIMATES, JUDGMENTS, AND METHODS CHANGE, OUR RESULTS OF OPERATIONS AND OUR ASSESSMENT OF THE PROGRESS OF OUR BUSINESS COULD VARY.
The methods, estimates, and judgments we use in applying our accounting policies have a significant impact on our results of operations. Such methods, estimates, and judgments are, by their nature, subject to substantial risks, uncertainties, and assumptions, and factors may arise over time that may lead us to change our methods, estimates, and judgments. Changes in any of our assumptions may adversely affect our reported financial results.
We utilize methods for determining surgical market sizes, the number and type (cancerous or benign) of certain procedures performed, and the installed base of our systems that involve estimates and judgments, which are, by their nature, subject to substantial risks, uncertainties, and assumptions. Our estimates of surgical market sizes, the number and type of procedures performed, or the installed base of our systems do not have an impact on our results of operations but are used to estimate the progress of our business. Estimates and judgments for determining surgical market sizes, the number and type of procedures, and the installed base of our systems and the accuracy of these estimates may be impacted over time with changes in treatment modalities, hospital reporting behavior, system internet connectivity, distributor reporting behavior, increases in procedures per field employee, and other factors. In addition, from time to time, we may change the method for determining market sizes, the number and type of procedures, and the installed base of our systems, causing variation in our reporting.
RISKS RELATING TO OUR REGULATORY ENVIRONMENT
COMPLYING WITH FDA AND FOREIGN REGULATIONS IS A COMPLEX PROCESS, AND OUR FAILURE TO FULLY COMPLY COULD SUBJECT US TO SIGNIFICANT ENFORCEMENT ACTIONS.
Because our products are commercially distributed, numerous quality and post-market regulatory requirements apply, including the following:
•continued compliance with the FDA’s QSR, which requires manufacturers to follow design, testing, control, documentation, and other quality assurance procedures during the development and manufacturing process;
•labeling regulations;
•the FDA’s general prohibition against false or misleading statements in the labeling or promotion of products for unapproved or “off-label” uses;
•stringent complaint reporting and Medical Device Reporting regulations, which require that manufacturers keep detailed records of investigations or complaints against their devices and report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur;
•adequate use of the corrective and preventive actions process to identify and correct or prevent significant, systemic failures of products or processes or in trends which suggest the same; and
•the reporting of corrections and removals, which requires that manufacturers report to the FDA recalls and field corrective actions taken to reduce a risk to health or to remedy a violation of the FFDCA that may pose a risk to health.
We are subject to inspection and marketing surveillance by the FDA to determine our compliance with regulatory requirements. If the FDA finds that we have failed to comply, it can institute a wide variety of enforcement actions, ranging from inspectional observations (as set forth on FDA Form 483) to a public Warning Letter to more severe civil and criminal sanctions, including the seizure of our products and equipment or ban on the import or export of our products. The FDA has, in the past, issued and could, in the future, issue Warning Letters or other adverse communications to us. If we fail to satisfy or remediate the matters discussed in any such Warning Letters or communications, the FDA could take further enforcement actions, including prohibiting the sale or marketing of the affected product. Our failure to comply with applicable requirements could lead to an enforcement action that may have an adverse effect on our financial condition or results of operations. The receipt of a Warning Letter could place certain limits on the ability to obtain FDA-issued Certificates to Foreign Government (“CFGs”) used for new and re-registration of products in certain other countries.
The FDA also strictly regulates labeling, advertising, promotion, and other activities relating to the marketing of our products. Medical devices may be promoted only for their cleared or approved indications and in accordance with the provisions of the cleared or approved label. It is possible that federal or state enforcement authorities might take action if they consider our promotional or training materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under a variety of statutory authorities, including under the FFDCA, as well as laws prohibiting false claims for reimbursement.
In addition, any modification or change of medical devices cleared for the market requires the manufacturer to make a determination whether the change is significant enough to require new 510(k) clearance. We have created labeling, advertising, and user training for the da Vinci Surgical System to describe specific surgical procedures that we believe are fully within the scope of our existing 510(k) indications for use stated in our 510(k) clearances. Although we have relied on expert in-house and external staff, consultants, and advisors, some of whom were formerly employed by the FDA and are familiar with the FDA perspective, we cannot provide assurance that the FDA would agree that all such specific procedures are within the scope of the existing general clearance or that we have compiled adequate information to support the safety and efficacy of using the da Vinci Surgical System for all such specific procedures. From time to time, we modify our products, including the hardware and software in the da Vinci Surgical System, after we obtain 510(k) clearance from the FDA for the devices in ways that we do not believe require new 510(k) clearance. We cannot provide assurance that the FDA would agree in all cases with our determinations not to seek new 510(k) clearance for any of these changes. If the FDA disagrees with our assessments that a new 510(k) clearance was not required prior to commercializing the devices with these changes or modifications, then the FDA could impose enforcement sanctions and/or require us to obtain 510(k) clearance or other FDA marketing authorization for any modification to our products. We may be prohibited from marketing the modified device until such marketing authorization is granted.
We have a wholly owned manufacturing facility located in Mexicali, Mexico, which manufactures reusable and disposable surgical instruments. This facility is registered with the FDA as well as with Mexican authorities. The facility is operated under U.S. and international quality system regulations, including those applicable to Canada, the EU, and Japan, among others. Our wholly owned manufacturing facility in Mexicali, Mexico has an FDA Establishment Registration but has not been inspected by the FDA to date. If the FDA were to identify non-conformances in our product documentation or quality system compliance, it could hold indefinitely the importation of instruments at the border, which would deprive us of the ability to sell and supply the majority of our customers until the FDA requirements have been satisfied. Similar supply disruptions could occur if key suppliers outside of the U.S. were to encounter non-conformances with their documentation or quality system compliance.
OUR PRODUCTS ARE SUBJECT TO A LENGTHY AND UNCERTAIN DOMESTIC REGULATORY REVIEW PROCESS. IF WE DO NOT OBTAIN AND MAINTAIN THE NECESSARY DOMESTIC REGULATORY AUTHORIZATIONS, WE WILL NOT BE ABLE TO SELL OUR PRODUCTS IN THE U.S.
Our products and operations are subject to extensive regulation in the U.S. by the FDA. The FDA regulates the development and clinical testing, manufacturing, labeling, storage, record keeping, promotion, sales, distribution, and post-market support and medical device reporting in the U.S. to ensure that medical products distributed domestically are safe and effective for their intended uses. In order for us to market products for use in the U.S., we generally must first obtain clearance from the FDA pursuant to Section 510(k) of the FFDCA or approval of the product through the premarket approval (“PMA”) pathway. Clearance under Section 510(k) requires demonstration that a new device is substantially equivalent to another device with 510(k) clearance or grandfathered (“pre-amendment”) status and for which a PMA is not required. If we develop products in the future that are not considered to be substantially equivalent to a device with 510(k) clearance or grandfathered status, we may be required to obtain marketing authorization through the more burdensome PMA process or alternatively through the de novo classification process, which is a path to market for novel devices that are low to moderate risk and for which a predicate device is not available. A PMA is typically a much more complex, lengthy, and burdensome application than a 510(k) or a de novo classification request. To support a PMA, the FDA would likely require that we conduct one or more clinical studies to demonstrate that the device is safe and effective for its intended uses. In some cases, such studies may also be required to support a 510(k) application or a de novo classification request. The FDA may not act favorably or quickly in its review of any marketing application submissions, or we may encounter significant difficulties and costs in our efforts to obtain marketing authorization from the FDA, either of which could delay or preclude the sale of new products in the U.S. In addition, the FDA may place significant limitations upon the intended use of our products as a condition of granting marketing authorization. Product applications can also be denied or withdrawn due to failure to comply with regulatory requirements or the occurrence of unforeseen problems following marketing authorization. Any delays or failure to obtain FDA marketing authorization for new or modified products that we develop, any limitations imposed by the FDA on new product use, or the costs of obtaining FDA clearance or approvals could have a material adverse effect on our business, financial condition, or results of operations.
In addition, the FDA or other regulatory agencies may change their policies, adopt additional regulations, revise existing regulations, or take other actions that may prevent or delay approval or clearance of our products under development or impact our ability to modify our currently approved or cleared products on a timely basis. We may be found non-compliant as a result
of future changes in, or interpretations of, regulations by the FDA or other regulatory agencies. For example, on February 23, 2022, the FDA issued a proposed rule to amend the QSR, which establishes current good manufacturing practice requirements for medical device manufacturers, to align more closely with the International Organization for Standardization (“ISO”) standards. This proposal has not yet been finalized or adopted. Accordingly, it is unclear the extent to which this or any other proposals, if adopted, could impose additional or different regulatory requirements on us that could increase the costs of compliance or otherwise create competition that may negatively affect our business.
Additionally, in September 2019, the FDA issued revised guidance describing an optional “safety and performance based” premarket review pathway for manufacturers of “certain, well-understood device types” to demonstrate substantial equivalence under the 510(k) clearance pathway by showing that such device meets objective safety and performance criteria established by the FDA, thereby obviating the need for manufacturers to compare the safety and performance of their medical devices to specific predicate devices in the clearance process. The FDA maintains a list of device types appropriate for the “safety and performance based” pathway and continues to develop product-specific guidance documents that identify the performance criteria for each such device type, as well as the recommended testing methods, where feasible. The FDA may establish performance criteria for classes of devices for which we or our competitors seek or currently have received clearance, and it is unclear the extent to which such performance standards, if established, could impact our ability to obtain new 510(k) clearances or otherwise create competition that may negatively affect our business.
In order to conduct a clinical investigation involving human subjects for the purpose of demonstrating the safety and effectiveness of a medical device, a company must, among other things, apply for and obtain IRB approval of the proposed investigation. In addition, if the clinical study involves a “significant risk” (as defined by the FDA) to human health, the sponsor of the investigation must also submit and obtain FDA approval of an IDE application. Many of our products to date have been or would be considered significant risk devices requiring IDE approval prior to investigational use. We may not be able to obtain FDA and/or IRB approval to undertake clinical trials in the U.S. for any new devices that we intend to market in the U.S. in the future.
Even if we obtain such approvals, we may not be able to conduct studies that comply with the IDE and other regulations governing clinical investigations or the data from any such trials may not support clearance or approval of the investigational device. Clinical testing is difficult to design and implement, can take many years, can be expensive, and carries uncertain outcomes and, if we fail to complete our planned or ongoing clinical trials or if such clinical trials produce negative or inconclusive results, we may be delayed or prevented from obtaining regulatory clearances or approvals to commercialize our products for new or expanded indications. Additionally, we may experience delays in our ongoing clinical trials for any number of reasons, which could adversely affect the costs, timing, or successful completion of our clinical trials. Moreover, the disruptions caused by the COVID-19 pandemic may increase the likelihood that we encounter such difficulties or delays in initiating, enrolling, conducting, or completing our planned and ongoing clinical trials. If we fail to complete our planned and ongoing clinical trials or if such clinical trials produce negative or inconclusive results, we may be delayed or prevented from obtaining regulatory clearances or approvals to commercialize our products for new or expanded indications, which may limit the market for our products.
Failure to obtain such approvals or to comply with such regulations could have a material adverse effect on our business, financial condition, or results of operations. Certainty that clinical trials will meet desired endpoints, produce meaningful or useful data, and be free of unexpected adverse effects or that the FDA will accept the validity of foreign clinical study data cannot be assured, and such uncertainty could preclude or delay market clearance or authorizations resulting in significant financial costs and reduced revenue.
OUR PRODUCTS MAY CAUSE OR CONTRIBUTE TO ADVERSE MEDICAL EVENTS OR BE SUBJECT TO FAILURES OR MALFUNCTIONS THAT WE ARE REQUIRED TO REPORT TO THE FDA AND FOREIGN REGULATORY AUTHORITIES AND, IF WE FAIL TO DO SO, WE WOULD BE SUBJECT TO SANCTIONS THAT COULD HARM OUR REPUTATION, BUSINESS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
We are subject to the FDA’s medical device reporting regulations and similar foreign regulations, which require us to report to the FDA and foreign regulatory authorities when we receive or become aware of information that reasonably suggests that one or more of our products may have caused or contributed to a death or serious injury or malfunctioned in a way that, if the malfunction were to recur, it could cause or contribute to a death or serious injury. The timing of our obligation to report is triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events of which we become aware within the prescribed timeframe. We may also fail to recognize that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of the product. If we fail to comply with our reporting obligations, the FDA or foreign regulatory authorities could take action, including warning letters, untitled letters, administrative actions, criminal prosecution, imposition of civil monetary penalties, revocation of our device clearance, approval, or certification, seizure of our products or delay in clearance, approval, or certification of future products.
The FDA and foreign regulatory bodies have the authority to require the recall of commercialized products in the event of material deficiencies or defects in the design or manufacture of a product or in the event that a product poses an unacceptable risk to health. The FDA’s authority to require a recall must be based on a finding that there is reasonable probability that the device could cause serious injury or death. We may also choose to voluntarily recall a product if any material deficiency is found. A government‑mandated or voluntary recall by us could occur as a result of an unacceptable risk to health, component failures, malfunctions, manufacturing defects, labeling or design deficiencies, packaging defects, or other deficiencies or failures to comply with applicable regulations. Product defects or other errors may occur in the future.
Depending on the corrective action we take to redress a product’s deficiencies or defects, the FDA or foreign regulatory authorities may require, or we may decide, that we will need to obtain new clearances, approvals, or certifications for the device before we may market or distribute the corrected device. Seeking such clearances, approvals, or certifications may delay our ability to replace the recalled devices in a timely manner. Moreover, if we do not adequately address problems associated with our devices, we may face additional regulatory enforcement actions, including FDA or foreign regulatory authorities warning letters, product seizure, injunctions, administrative penalties, or civil or criminal fines.
Companies are required to maintain certain records of recalls and corrections, even if they are not reportable to the FDA or foreign regulatory authorities. We may initiate voluntary withdrawals or corrections for our products in the future that we determine do not require notification of the FDA or foreign regulatory authorities. If the FDA or foreign regulatory authorities disagree with our determinations, it could require us to report those actions as recalls, and we may be subject to enforcement actions. A future recall announcement could harm our reputation with customers, potentially lead to product liability claims against us, and negatively affect our sales. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, would require the dedication of our time and capital, distract management from operating our business, and may harm our reputation and financial results.
IF OUR MANUFACTURING FACILITIES DO NOT CONTINUE TO MEET FEDERAL, STATE, OR OTHER MANUFACTURING STANDARDS, WE MAY BE REQUIRED TO TEMPORARILY CEASE ALL OR PART OF OUR MANUFACTURING OPERATIONS, IMPORT/EXPORT OF OUR PRODUCTS, AND/OR RECALL SOME PRODUCTS, WHICH WOULD RESULT IN SIGNIFICANT PRODUCT DELIVERY DELAYS AND LOST REVENUE.
Our manufacturing facilities are subject to periodic inspection by regulatory authorities and notified bodies, and our operations will continue to be regulated and inspected by the FDA and other regulatory agencies and notified bodies for compliance with Good Manufacturing Practice requirements contained in the QSR and other regulatory requirements. We are also required to comply with ISO quality system standards as well as EU legislation and norms in order to produce products for sale in the EU. In addition, many countries, such as Canada and Japan, have very specific additional regulatory requirements for quality assurance and manufacturing. If we fail to continue to comply with Good Manufacturing Practice requirements, as well as ISO or other regulatory standards, we may be required to cease all or part of our operations until we comply with these regulations.
We continue to be subject to FDA and certain other inspections by other regulatory authorities and notified bodies at any time. Maintaining such compliance is difficult and costly. We cannot be certain that our facilities will be found to comply with Good Manufacturing Practice requirements or ISO standards and other regulatory requirements in future inspections and audits by regulatory authorities and notified bodies.
We are currently participating in the Medical Device Single Audit Program (“MDSAP”), which allows an MDSAP-recognized auditing organization to conduct a single regulatory audit of a medical device manufacturer that evaluates our quality system to assess compliance with the requirements of multiple regulatory jurisdictions, including the U.S., Japan, Brazil, Australia, and Canada. The information collected in an MDSAP audit is shared and reviewed amongst all the regulatory authorities participating in the MDSAP, who may or may not determine that additional information or auditing is required.
Our Sunnyvale, California facility is licensed by the State of California to manufacture medical devices. We have been subject to periodic inspections by the California Department of Health Services Food and Drug Branch and, if we are unable to maintain this license following any future inspections, we will be unable to manufacture or ship some products, which would have a material adverse effect on our results of operations. In addition, both our Sunnyvale, California and Mexicali, Mexico facilities are subject to periodic inspections by other regulatory bodies, including third-party auditors on behalf of national regulatory authorities. Compliance with multiple regulatory standards is complex, difficult, and costly to maintain, and material deficiencies could result in significant limitations on our ability to manufacture, transport, and sell our products in one or more countries.
OUR PRODUCTS ARE SUBJECT TO INTERNATIONAL REGULATORY PROCESSES AND APPROVAL OR CERTIFICATION REQUIREMENTS. IF WE DO NOT OBTAIN AND MAINTAIN THE NECESSARY INTERNATIONAL REGULATORY APPROVALS OR CERTIFICATIONS, WE WILL NOT BE ABLE TO SELL OUR PRODUCTS IN OTHER COUNTRIES.
To be able to sell our products in other countries, we must obtain regulatory approvals or certifications and comply with the regulations of those countries, which may differ substantially from those of the U.S. These regulations, including the requirements for approvals or certifications and the time required for regulatory review, vary from country to country. Obtaining and maintaining foreign regulatory approvals or certifications is complex, and the time to obtain clearances or certifications in other countries varies; therefore, we cannot be certain that we will receive regulatory approvals or certifications in any other country in which we plan to market our products or obtain such approvals or certifications on a favorable schedule. If we fail to obtain or maintain regulatory approval or certification in any other country in which we plan to market our products, our ability to generate revenue will be harmed. In particular, if the FDA refuses to provide CFGs, our ability to register products or renew such registrations may be delayed or denied.
For instance, one of the most significant moving targets related to the regulatory landscape is in the EU; more specifically, the regulation of medical devices has recently evolved. The EU Medical Devices Regulation, which repeals and replaces the EU Medical Devices Directive, became applicable on May 26, 2021. Devices lawfully placed on the market pursuant to the EU Medical Devices Directive prior to May 26, 2021, may generally continue to be made available on the market or put into service until May 26, 2025, provided that the requirements of the transitional provisions are fulfilled. In particular, the certificate in question must still be valid. However, since May 26, 2021, manufacturers must already comply with a number of new, or reinforced, requirements set forth in the EU Medical Devices Regulation, including registration of economic operators and of devices, post-market surveillance, market surveillance, and vigilance requirements.
Subject to the transitional provisions, in order to sell our products in EU member states, our products must comply with the general safety and performance requirements of the EU Medical Devices Regulation. Compliance with these requirements is a prerequisite to be able to affix the CE mark to our products, without which they cannot be sold or marketed in the EU. All medical devices placed on the market in the EU must meet the general safety and performance requirements laid down in Annex I to the EU Medical Devices Regulation, including the requirement that a medical device must be designed and manufactured in such a way that, during normal conditions of use, it is suitable for its intended purpose. It is the responsibility of the Person Responsible for Regulatory Compliance (“PRRC”) to ensure such requirements are fulfilled and in place in the company. Medical devices must be safe and effective and must not compromise the clinical condition or safety of patients or the safety and health of users and, where applicable, other persons, provided that any risks that may be associated with their use constitute acceptable risks when weighed against the benefits to the patient and are compatible with a high level of protection of health and safety, taking into account the generally acknowledged state of the art. To demonstrate compliance with the general safety and performance requirements, we must undergo a conformity assessment procedure, which varies according to the type of medical device and its (risk) classification and may include a technical documentation assessment and an onsite audit. Except for low-risk medical devices (Class I), where the manufacturer can self-assess the conformity of its products with the general safety and performance requirements (except for any parts that relate to sterility, metrology, or reuse aspects), a conformity assessment procedure requires the intervention of a notified body. The notified body would typically audit and examine the technical file and the quality system for the manufacture, design, and final inspection of our devices. If satisfied that the relevant product conforms to the relevant general safety and performance requirements and we have the organizational structure to support it (i.e., PRRC), the notified body issues a certificate of conformity, which the manufacturer uses as a basis for its own declaration of conformity. The manufacturer may then apply the CE mark to the device, which allows the device to be placed on the market throughout the EU. If we fail to comply with applicable laws and regulations, we would be unable to affix the CE mark to our products, which would prevent us from selling them within the EU or any countries recognizing the CE mark. The aforementioned EU rules are generally applicable in the EEA.
In January 1999, further to their certification by our notified body, Presafe, we affixed the CE mark to our da Vinci Surgical System and EndoWrist instruments, attesting compliance with the former EU Medical Devices Directive, and we have maintained these certifications continuously since that time. Subsequent products and accessories have also received certifications by our notified body in accordance with the former EU Medical Devices Directive. Where required, we are maintaining our certificates granted under the former EU Medical Devices Directive and have either gained, or are working towards, certification under the EU Medical Devices Regulation for all medical devices that we intend to continue to market in the EU and EEA. Should we not gain such certification by the end of the validity of our certificates granted under the former EU Medical Devices Directive, which may not exceed the transitional period currently set forth in the EU Medical Devices Regulation of May 26, 2024, this would prevent us from selling our products in the EU and EEA. However, under recently proposed draft legislation issued by the European Commission, this date could be extended to December 2027 for higher classification devices (Class III and certain Class IIb implantable devices) and to December 2028 for medium- and lower-risk devices (for the other Class IIb devices, Class IIa devices, and some Class I devices).
Further, Switzerland, which is the country from which we import our products into the EU and where our EU regulatory team is based, has not yet entered into a Mutual Recognition Agreement with the EU that covers the EU Medical Device Regulation and allows medical devices to move freely between Switzerland and the EU. Therefore, for future needs, we will adjust the manner in which we bring our products into the EU market. Any such adjustments could cause temporary disruptions in and have adverse financial implications to our business in Europe.
To date, we received approvals from the Japanese Ministry of Health, Labor and Welfare for our da Vinci S, Si, Xi, X, and SP Surgical Systems and various associated instruments and accessories for use in certain da Vinci procedures. We may seek additional approvals for other products and/or indications; however, there can be no assurance that such approvals will be granted. In addition, because not all of our instruments have received product approvals and reimbursement is an additional process to generate market acceptance, it is possible that procedures will be adopted slowly or not at all. Sales of our products depend, in part, on the extent to which the costs of our products are reimbursed by governmental health administration authorities. There are multiple pathways to obtain reimbursement for procedures including those that require in-country clinical data and which are considered for reimbursed status in April of even-numbered years. If we are not successful in obtaining the necessary reimbursement approvals or obtaining approvals for future products and procedures, then the demand for our products could be limited. These limitations could eliminate a significant market opportunity for our products in Japan.
Our capital sales in China are subject to importation authorizations and purchasing tender processes. In October 2018, the China National Health Commission published on its official website the quota for major medical equipment to be imported and sold in China through 2020. After an adjustment notice was published in the third quarter of 2020 (ref. NHC Financial Notice [2020] 315), the government will allow for the total sale of 225 new Endoscopic Surgical Instrument Control Systems (surgical robots) into China, which could include da Vinci Surgical Systems as well as surgical systems introduced by others. Future system sales and our ability to grow future procedure volumes are dependent on the completion of these purchasing tender authorizations. The timing and magnitude of these future authorizations, which may determine our system placements in future years, is not certain, and we expect to continue to experience variability in the timing of capital sales in China.
CHANGES IN HEALTHCARE LEGISLATION AND POLICY MAY HAVE A MATERIAL ADVERSE EFFECT ON OUR BUSINESS, FINANCIAL CONDITION, OR RESULTS OF OPERATIONS.
In the U.S., there have been, and continue to be, a number of legislative initiatives to contain healthcare costs. In March 2010, the ACA was enacted, which made changes that have impacted and are expected to significantly impact the pharmaceutical and medical device industries.
The ACA contained a number of provisions designed to generate the revenues necessary to fund health insurance coverage expansions among other things. This included a number of Medicare payment system reforms, including a national pilot program on payment bundling to encourage hospitals, physicians, and other providers to improve the coordination, quality, and efficiency of certain healthcare services through bundled payment models and appropriated funding for comparative effectiveness research.
Since its enactment, there have been judicial, executive branch, and Congressional challenges to certain aspects of the ACA. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA brought by several states without specifically ruling on the constitutionality of the ACA. Thus, the ACA will remain in effect in its current form.
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. These changes included an aggregate reduction in Medicare payments, which went into effect on April 1, 2013, and will remain in effect through 2031, unless additional Congressional action is taken, with the exception of a temporary suspension due to the COVID-19 pandemic from May 1, 2020, through March 31, 2022. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers, and cancer treatment centers. MACRA repealed the formula by which Medicare made annual payment adjustments to physicians and replaced the former formula with fixed annual updates and a new system of incentive payments that began in 2019 and are based on various performance measures and physicians’ participation in alternative payment models, such as accountable care organizations. Individual states in the U.S. have also become increasingly aggressive in passing legislation and implementing regulations designed to control product pricing, including price or patient reimbursement constraints and discounts, and require marketing cost disclosure and transparency measures.
We expect additional state and federal healthcare reform measures to be adopted in the future that could have a material adverse effect on our industry generally and on our customers. Any changes to, or uncertainty with respect to, future reimbursement rates or changes in hospital admission rates could impact our customers’ demand for our products and services, which, in turn, could have a material adverse effect on our business, financial condition, or results of operations.
Further, the federal, state, and local governments, Medicare, Medicaid, managed-care organizations, and foreign governments have, in the past, considered, are currently considering, and may, in the future, consider healthcare policies and proposals intended to curb rising healthcare costs, including those that could significantly affect both private and public
reimbursement for healthcare services. Future significant changes in the healthcare systems in the U.S. or other countries, including retroactive and prospective rate and coverage criteria changes, competitive bidding or tender processes for certain products and services, and other changes intended to reduce expenditures along with uncertainty about whether and how changes may be implemented, could have a negative impact on the demand for our products. We are unable to predict whether other healthcare policies, including policies stemming from legislation or regulations affecting our business may be proposed or enacted in the future, what effect such policies would have on our business, or what effect ongoing uncertainty about these matters will have on the purchasing decisions of our customers.
For instance, in December 2021, the EU Regulation No. 2021/2282 on Health Technology Assessment (“HTA”), amending Directive 2011/24/EU, was adopted. This regulation, which entered into force in January 2022, intends to boost cooperation among EU member states in assessing health technologies, including certain high-risk medical devices, and provides the basis for cooperation at the EU level for joint clinical assessments in these areas. The regulation foresees a three-year transitional period and will permit EU member states to use common HTA tools, methodologies, and procedures across the EU, working together in four main areas, including joint clinical assessment of the innovative health technologies with the most potential impact for patients, joint scientific consultations whereby developers can seek advice from HTA authorities, identification of emerging health technologies to identify promising technologies early, and continuing voluntary cooperation in other areas. Individual EU member states will continue to be responsible for assessing non-clinical (e.g., economic, social, ethical, etc.) aspects of health technology and making decisions on pricing and reimbursement.
WE ARE SUBJECT TO FEDERAL, STATE, AND FOREIGN LAWS GOVERNING OUR BUSINESS PRACTICES, WHICH, IF VIOLATED, COULD RESULT IN SUBSTANTIAL PENALTIES. ADDITIONALLY, CHALLENGES TO, OR INVESTIGATION INTO, OUR PRACTICES COULD CAUSE ADVERSE PUBLICITY AND BE COSTLY TO RESPOND TO AND, THUS, COULD HARM OUR BUSINESS.
The Dodd-Frank Wall Street Reform and Consumer Protection Act requires us to track and disclose the source of any tantalum, tin, gold, and tungsten used in manufacturing that may originate in the Democratic Republic of the Congo or adjoining regions (so called “conflict minerals”). These metals are central to the technology industry and are present in some of our products as component parts. In most cases, no acceptable alternative material exists that has the necessary properties that our products require. Because it is not possible to determine the source of the metals by analysis, we must obtain a good faith description of the source of the intermediate components and raw materials from parties in our supply chain. The components that incorporate those metals may originate from many sources, and we purchase fabricated products from manufacturers who may have a long and difficult-to-trace supply chain. As the spot price of these materials varies, producers of the metal intermediates can be expected to change the mix of sources used. Accordingly, components and assemblies we buy may have a mix of sources as their origin. We are required to carry out a diligent effort to determine and disclose the source of these materials. There can be no assurance that we can obtain this information accurately or reliably, or at all, from intermediate producers who may be unwilling or unable to provide this information or further identify their sources of supply or to notify us if these sources change. In addition, these metals are subject to price fluctuations and shortages that can affect our ability to obtain the manufactured materials that we rely on at favorable terms or from consistent sources. These changes could have an adverse impact on our ability to manufacture and market our devices and products.
We are also subject to healthcare regulation and enforcement by the federal government and the states and foreign governments where we conduct our business. The healthcare laws and regulations that may affect our ability to operate include the federal Anti-Kickback Statute, which prohibits, among other things, payments or other remuneration that could be considered to induce hospitals, physicians, or other potential purchasers of our products either to refer patients or to purchase, lease, order, or arrange for or recommend the purchase, lease, or order of healthcare products or services for which payment may be made under federal and state healthcare programs, such as Medicare and Medicaid and any other third-party payor programs. Further, a person or entity does not need to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. Similar laws must be complied with in foreign jurisdiction.
The federal civil and criminal false claims laws, including the federal civil False Claims Act, and civil monetary penalties laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other federal healthcare programs that are false or fraudulent. Although we do not submit claims directly to government payors, manufacturers can be held liable under the federal false claim act if they are deemed to “cause” the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers or promoting a product off-label. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
The Health Insurance Portability and Accountability Act of 1996, which created additional federal criminal statutes prohibit, among other things, executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation.
These laws may affect our sales, marketing, and other promotional activities by limiting the kinds of financial arrangements that we may have with hospitals, physicians, or other potential purchasers of our products. They particularly impact how we structure our sales offerings, including discount practices, customer support, speaker, education, and training programs, physician consulting, and other service arrangements. These laws are broadly written, and it is often difficult to determine precisely how these laws will be applied to specific circumstances. Violating anti-kickback laws and false claims laws can result in civil and criminal fines and penalties, which can be substantial and include monetary damages and penalties, imprisonment, and exclusion from government healthcare programs for non-compliance. Even an unsuccessful challenge or investigation into our practices could cause adverse publicity and be costly to defend and, thus, could harm our business, financial condition, or results of operations.
The federal Physicians Payments Sunshine Act imposes reporting and disclosure requirements on certain device manufacturers for any “transfer of value” made or distributed to physicians (including family members), as defined by statute, certain non-physician practitioners, including physician assistants and nurse practitioners, and teaching hospitals. Such information must be made publicly available in a searchable format. In addition, device manufacturers are required to report and disclose any ownership or investment interests held by physicians and their immediate family members, as well as any transfers of value made to such physician owners and investors, during the preceding calendar year. Similar requirements apply in foreign jurisdictions. Failure to submit required information may result in civil monetary penalties for all payments, transfers of value, or ownership or investment interests not reported in an annual submission. Device manufacturers are required to submit reports to CMS by the 90th day of each calendar year.
Many states have similar laws and regulations, such as anti-kickback and false claims laws, which may be broader in scope and may apply regardless of payor, in addition to items and services reimbursed under Medicaid and other state programs. Certain states mandate implementation of commercial compliance programs to ensure compliance with these laws, impose restrictions on device manufacturer marketing practices, and/or require the tracking and reporting of gifts, compensation, and other remuneration to physicians or marketing expenditures and pricing information. The shifting commercial compliance environment and the need to build and maintain robust and expandable systems to comply with multiple jurisdictions with different compliance and/or reporting requirements increases the possibility that a healthcare company may be found out of compliance with one or more of the requirements, subjecting us to significant civil monetary penalties.
Additionally, to the extent that our product is sold in a foreign country, we may be subject to similar foreign laws.
Compliance with complex foreign and U.S. laws and regulations that apply to our OUS operations increases our cost of doing business in foreign jurisdictions and could expose us or our employees to fines and penalties in the U.S. and/or abroad. These numerous, and sometimes conflicting, laws and regulations include U.S. laws, such as the FCPA, and similar laws in other countries, such as the U.K. Bribery Act of 2010. Violations of these laws and regulations could result in fines, criminal sanctions against us, our officers, or our employees, prohibitions on the conduct of our business, and damage to our reputation. Although we have implemented policies and procedures designed to ensure compliance with these laws, there can be no assurance that our employees, contractors, or agents will not violate our policies.
Our operations are subject to certain antitrust and competition laws in the jurisdictions in which we conduct our business, in particular the U.S. and the EU. These laws prohibit, among other things, anticompetitive agreements and practices. If any of our commercial agreements or practices are found to violate or infringe such laws, we may be subject to civil and other penalties. We may also be subject to third-party claims for damages. Further, agreements that infringe upon these antitrust and competition laws may be void and unenforceable, in whole or in part, or require modification in order to be lawful and enforceable. If we are unable to enforce our commercial agreements, whether at all or in material part, our business, financial condition, or results of operations could be adversely affected.
We are also subject to claims, lawsuits, and government investigations involving labor and employment. Such claims, lawsuits, and government investigations are inherently uncertain. Regardless of the outcome, any of these types of legal proceedings can have an adverse impact on us because of legal costs, diversion of management resources, and other factors.
We are also exposed to the risk that our employees, independent contractors, consultants, manufacturers, suppliers, and any other third parties that we may engage in connection with the development and commercialization of our products may engage in fraudulent or illegal activity. Misconduct by these parties could include intentional, reckless, and/or negligent conduct or disclosure of unauthorized activities to us that violates: (i) the laws of the FDA and other similar regulatory authorities, including those laws requiring the reporting of true, complete, and accurate information to such authorities; (ii) manufacturing standards; (iii) data privacy, security, fraud, and abuse laws and regulations; or (iv) laws that require the true, complete, and accurate reporting of financial information or data. Activities subject to these laws could also involve the improper use or misrepresentation of information obtained in the course of clinical trials or the creation of fraudulent data in clinical trials, which could result in regulatory sanctions and cause serious harm to our reputation. It is not always possible to identify and deter misconduct by employees and other third parties, and the precautions we take to detect and prevent this activity may not
be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with such laws or regulations.
Additionally, we are subject to the risk that a person or government could allege fraud or other misconduct, even if none occurred. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business or results of operations, including the imposition of significant civil, criminal, and administrative penalties, damages, monetary fines, disgorgements, possible exclusion from participation in Medicare, Medicaid, other U.S. federal healthcare programs, or healthcare programs in other jurisdictions, integrity oversight and reporting obligations to resolve allegations of non-compliance, imprisonment, other sanctions, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations.
IF HOSPITALS AND OTHER SURGERY FACILITIES DO NOT CONTINUE TO MEET FEDERAL, STATE, OR OTHER REGULATORY STANDARDS, THEY MAY BE REQUIRED TO TEMPORARILY CEASE ALL OR PART OF THEIR SYSTEM UTILIZATION.
Our global customers are subject to periodic inspection by regulatory authorities. Our customers are required to comply with applicable local and international regulations, including with respect to the reprocessing of our instruments and accessories. Hospitals may not follow cleaning and sterilization instructions properly, or equipment used for cleaning and sterilization may malfunction or be used improperly. If our customers deviate from cleaning and sterilization instructions, regulatory authorities may require them to suspend the use of our systems.
RISKS RELATING TO OUR INTELLECTUAL PROPERTY
IF WE ARE UNABLE TO FULLY PROTECT AND SUCCESSFULLY DEFEND OUR INTELLECTUAL PROPERTY FROM USE BY THIRD PARTIES, OUR ABILITY TO COMPETE IN THE MARKET MAY BE HARMED.
Our commercial success depends in part on obtaining patent protection for the proprietary technologies contained in our products and on successfully defending our patents against infringing products and/or services in litigation or administrative proceedings, including patent oppositions, reviews, or reexaminations. We incur substantial costs in obtaining patents and, if necessary, defending our patent rights. We do not know whether we will be successful in obtaining the desired patent protection for our new proprietary technologies or that the protection we do obtain will be found valid and enforceable when challenged. The success of defending our proprietary rights can be highly uncertain, because it involves complex and often evolving legal issues and procedures that are dependent on the particular facts of each case.
In addition to patents, we also rely on other intellectual property rights, such as trade secret, copyright, and trademark laws to protect proprietary technologies. We further utilize nondisclosure agreements and other contractual provisions as well as technical measures to protect our proprietary technologies. Nevertheless, these measures may be inadequate in protecting our technologies. If these measures prove to be inadequate in protecting our technologies, our competitive advantages may be reduced. Moreover, we may not have adequate remedies for potential breaches by employees, consultants, and others who participate in developing our proprietary technologies against their agreements with us regarding intellectual property. As a result, our trade secrets may be lost. Notwithstanding our efforts to protect our intellectual property, our competitors may independently develop similar or alternative technologies or products that are equal to or superior to our technologies without infringing any of our intellectual property, which would harm our ability to compete in the market.
As foreign markets become more significant in revenue for us, our foreign operations and strategic alliances with foreign entities will likely increase. Our exposure to risks associated with these operations requires us to increase our reliance on protecting our intellectual property against infringing products and/or services in markets outside of the U.S. The laws and judicial systems in these countries may introduce yet another level of uncertainty in our effort to obtain the desired protection as well as defending our rights.
OTHERS MAY BE SUCCESSFUL IN ASSERTING THAT OUR PRODUCTS INFRINGE THEIR INTELLECTUAL PROPERTY RIGHTS, WHICH MAY CAUSE US TO PAY SUBSTANTIAL DAMAGES AND/OR ENJOIN US FROM COMMERCIALIZING OUR PRODUCTS.
As we continue to introduce and commercialize new products and technologies, there may be U.S. and foreign patents issued to third parties that relate to our products. Some of these patents may be broad enough to cover one or more aspects of our products. We do not know whether any of these patents, if challenged, would be held valid, enforceable, and infringed. From time to time, we receive, and likely will continue to receive, letters from third parties accusing us of infringing and/or inviting us to license their patents. We may be sued by, or become involved in an administrative proceeding with, one or more of these third parties.
We cannot be certain that a court or administrative body would agree with any arguments or defenses that we may have concerning invalidity, unenforceability, or non-infringement of any third-party patent. In addition, other parties may have filed
or will file patent applications covering products that are similar to or identical to ours. We cannot be certain that patents issuing from our own patent applications covering our products will have a priority date over any patents issuing from applications filed by a third party.
The medical device industry has experienced extensive intellectual property litigation and administrative proceedings. If third parties assert infringement claims or institute administrative proceedings against us, our technical and management personnel will need to spend significant time and effort, and we will incur large expenses in defending against these attacks. We cannot be certain that we will prevail in defending against infringement, validity, or enforceability claims against us. If plaintiffs in patent administrative proceedings are successful, our patent portfolio may be adversely affected. If plaintiffs in any patent action are successful, we may be enjoined from selling or importing our products, we may have to pay substantial damages, including treble damages, or we may be required to obtain a license that requires us to pay substantial royalties or relocate our manufacturing facilities. In addition, any public announcements related to litigation or administrative proceedings initiated or threatened against us could cause our stock price to decline.
OUR PRODUCTS RELY ON LICENSES FROM THIRD PARTIES, WHICH MAY NOT BE AVAILABLE TO US ON COMMERCIALLY REASONABLE TERMS OR AT ALL. IF WE LOSE ACCESS TO THESE TECHNOLOGIES, OUR REVENUES COULD DECLINE.
We rely on technology that we license from others, including technology that is integral to our products. There is no assurance that we can obtain or retain licenses on acceptable terms or at all. The license agreements we have entered into with several industry partners may be terminated for breach. If any of these agreements are terminated, we may be unable to reacquire the necessary license on satisfactory terms or at all. The failure to obtain, retain, or maintain licenses could prevent or delay further development or commercialization of our products, which may have a material adverse effect on our business, financial condition, or results of operations.
GENERAL RISK FACTORS
OUR FUTURE OPERATING RESULTS MAY BE BELOW SECURITIES ANALYSTS’ OR INVESTORS’ EXPECTATIONS, WHICH COULD CAUSE OUR STOCK PRICE TO DECLINE.
Due to the nascent nature of our industry, we have limited insight into trends that may emerge in our market and affect our business. The revenue and income potential of our market are unproven, and we may be unable to maintain or grow our revenue. Our products typically have lengthy sales cycles. In addition, our costs may be higher than we anticipated. If we fail to generate sufficient revenues or our costs are higher than we expect, our results of operations may be materially adversely affected. Further, future revenue from sales of our products is difficult to forecast, because the market for new surgical technologies is still evolving. Our results of operations could be impacted by numerous factors, including:
•the extent to which our products achieve and maintain market acceptance;
•actions relating to regulatory matters;
•product quality and supply problems;
•inflationary pressures on the cost of producing and distributing our products;
•our timing and ability to develop our manufacturing and sales and marketing capabilities;
•demand for our products;
•the size and timing of particular sales and any collection delays related to those sales;
•the progress of surgical training in the use of our products;
•our ability to develop, introduce, and market new or enhanced versions of our products on a timely basis;
•third-party payor reimbursement policies;
•our ability to protect our proprietary rights and defend against third-party challenges;
•our ability to license additional intellectual property rights; and
•the progress and results of any clinical trials.
Our operating results in any particular period will not be a reliable indication of our future performance. It is possible that, in future periods, our operating results will be below the expectations of securities analysts or investors. If this occurs, the price of our common stock and the value of your investment will likely decline.
OUR STOCK PRICE HAS BEEN, AND WILL LIKELY CONTINUE TO BE, VOLATILE.
The market price of our common stock has experienced fluctuations and may fluctuate significantly in the future. For example, during 2020, the adjusted closing price of our common stock reached a high of $272.70 and a low of $122.58; during
2021, it reached a high of $365.42 and a low of $228.30; and, during 2022, it reached a high of $360.00 and a low of $183.06. Our stock price can fluctuate for a number of reasons, including:
•announcements about us or our competitors;
•variations in our operating results and financial guidance;
•our introduction or abandonment of new technologies or products;
•regulatory approvals and enforcement actions;
•changes in our product pricing policies;
•changes in earnings estimates or recommendations by analysts;
•changes in accounting policies;
•economic changes and overall market volatility;
•announcements relating to product quality and the supply chain for our products;
•litigation;
•media coverage, whether accurate or inaccurate, fair or misleading;
•political uncertainties;
•short sales on shares of our common stock or other activities by short sellers; and
•our stock repurchase program.
Future stock repurchase programs will be contingent on a variety of factors, including our financial condition, results of operations, and business requirements. There can be no assurance that we will continue repurchasing our common stock in the future, consistent with historical levels or at all, or that our stock repurchase programs will have a beneficial impact on our stock price.
In addition, stock markets generally have experienced, and in the future may experience, significant price and volume volatility. This volatility has a substantial effect on the market prices of securities of many public companies for reasons frequently unrelated or disproportionate to the operating performance of the specific companies. Further, the securities of many medical device companies, including us, have historically been subject to extensive price and volume fluctuations that may affect the market price of their common stock. If these broad market fluctuations continue, it may have a material adverse impact on the market price of our common stock.
CHANGES TO FINANCIAL ACCOUNTING STANDARDS MAY AFFECT OUR REPORTED RESULTS OF OPERATIONS.
A change in accounting standards can have a significant effect on our reported results and may retroactively affect previously reported results. New accounting pronouncements and varying interpretations of accounting pronouncements have occurred and may occur in the future. Changes to existing standards or the reevaluation of current practices may adversely affect our reported financial results or the way we conduct our business.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
As of December 31, 2022, we own approximately 2.0 million square feet of space on 112 acres of land in Sunnyvale, California, where we house our principal headquarters, research and development, service, and support functions, as well as certain of our manufacturing operations.
Outside of Sunnyvale, California, we own facilities in other U.S. locations that are used for sales, training, manufacturing, engineering, and administrative functions, including approximately 530,000 square feet of space on 60 acres of land in Peachtree Corners, Georgia. We also lease approximately 750,000 square feet of space for certain engineering, warehousing, and support functions at various locations in the U.S. Outside of the U.S., we own properties in Mexicali, Mexico, primarily for manufacturing operations, and Aubonne, Switzerland, primarily for our international headquarters. In China, our Joint Venture leases facilities for research and development, manufacturing, and sales operations. In Germany, we own and lease facilities for manufacturing operations, as we build out operations of our acquisition of certain assets and operations from Schölly Fiberoptic GmbH. In Israel, we lease facilities, including space for the operations of our subsidiary, Orpheus Medical. In addition, we lease various international facilities for sales and other operations.
ITEM 3. LEGAL PROCEEDINGS
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS, AND ISSUER PURCHASES OF EQUITY SECURITIES
All share and per-share information presented have been retroactively adjusted to reflect the three-for-one stock split of our issued and outstanding common stock in October 2021.
COMMON STOCK
Our common stock is traded on The Nasdaq Global Select Market under the symbol “ISRG.”
As of February 7, 2023, there were 130 stockholders of record of our common stock, although there are a significantly larger number of beneficial owners of our common stock.
DIVIDENDS
We have never declared or paid any cash dividends on our common stock. We currently intend to retain earnings for use in the operation and expansion of our business. In addition, we may use a portion of our retained earnings to repurchase shares of our common stock, if appropriate.
SECURITIES AUTHORIZED FOR ISSUANCE UNDER EQUITY COMPENSATION PLANS
Please see Item 12. "Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters" under Part III of this Annual Report on Form 10-K for information on where to find information required by Item 201(d) of Regulation S-K.
RECENT SALES OF UNREGISTERED SECURITIES
None.
ISSUER PURCHASES OF EQUITY SECURITIES
The table below summarizes our stock repurchase activity for the quarter ended December 31, 2022.
| | | | | | | | | | | | | | | | | | | | | | | |
| Fiscal Period | Total Number of
Shares
Repurchased | | Average
Price Paid
Per Share | | Total Number of
Shares Purchased As
Part of a Publicly
Announced Program | | Approximate Dollar
Amount of Shares That
May Yet be Purchased
Under the Program (1) |
October 1 to October 31, 2022 | 3,635,474 | | | $ | 254.46 | | | 3,635,474 | | | $ | 1.6 | billion |
November 1 to November 30, 2022 | — | | | $ | — | | | — | | | $ | 1.6 | billion |
December 1 to December 31, 2022 | 298,017 | | | $ | 254.48 | | | 298,017 | | | $ | 1.5 | billion |
Total during quarter ended December 31, 2022 | 3,933,491 | | | $ | 254.46 | | | 3,933,491 | | | |
(1) Since March 2009, we have had an active stock repurchase program (the “Repurchase Program”). As of December 31, 2022, our Board of Directors (our “Board”) had authorized an aggregate amount of up to $10.0 billion for stock repurchases, of which the most recent authorization occurred in July 2022, when our Board increased the authorized amount available under our Repurchase Program to $3.5 billion. The remaining amount available to repurchase shares under the authorized Repurchase Program as of December 31, 2022, is $1.5 billion. The authorized Repurchase Program does not have an expiration date.
STOCK PERFORMANCE GRAPH
This graph is not “soliciting material” or deemed “filed” with the SEC for purposes of Section 18 of the Exchange Act, or otherwise subject to liabilities under that Section, and shall not be deemed incorporated by reference into any filings of Intuitive Surgical, Inc. under the Securities Act of 1933, as amended, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.
The graph set forth below compares the cumulative total stockholder return on our common stock between December 31, 2017, and December 31, 2022, with the cumulative total return of (i) the Nasdaq Composite Index, (ii) the S&P 500 Healthcare Index, and (iii) the S&P 500 Index over the same period. This graph assumes an investment of $100.00 on December 31, 2017, in our common stock, the Nasdaq Composite Index, the S&P Healthcare Index, and the S&P 500 Index and assumes the re-investment of dividends, if any.
The comparisons shown in the graph below are based on historical data. We caution that the stock price performance shown in the graph below is not necessarily indicative of, nor is it intended to forecast, the potential future performance of our common stock.
| | |
| COMPARISON OF CUMULATIVE TOTAL RETURN AMONG INTUITIVE, NASDAQ COMPOSITE, S&P HEALTHCARE INDEX, AND S&P 500 INDEX |
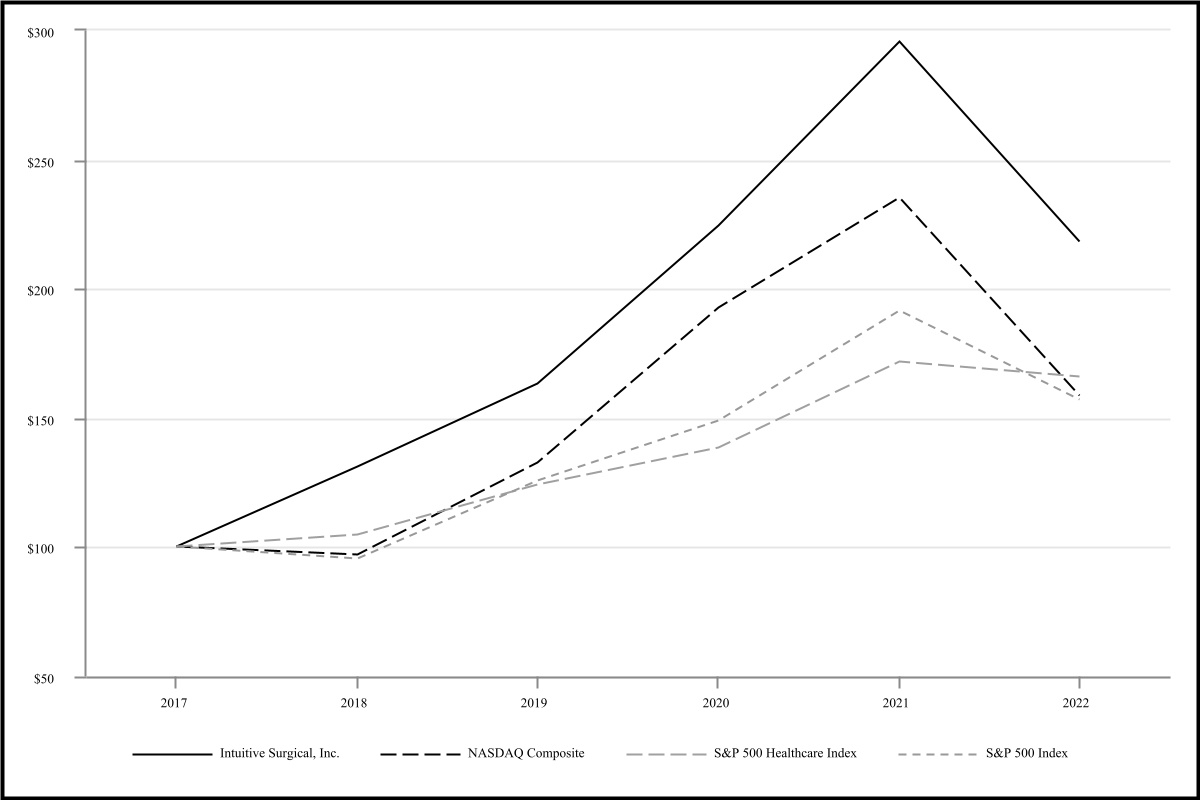
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| December 31, |
| 2017 | | 2018 | | 2019 | | 2020 | | 2021 | | 2022 |
| Intuitive Surgical, Inc. | $ | 100.00 | | | $ | 131.23 | | | $ | 163.08 | | | $ | 224.17 | | | $ | 295.36 | | | $ | 218.13 | |
| Nasdaq Composite | $ | 100.00 | | | $ | 97.16 | | | $ | 132.81 | | | $ | 192.47 | | | $ | 235.15 | | | $ | 158.65 | |
| S&P 500 Healthcare Index | $ | 100.00 | | | $ | 104.69 | | | $ | 124.25 | | | $ | 138.45 | | | $ | 171.90 | | | $ | 165.80 | |
| S&P 500 Index | $ | 100.00 | | | $ | 95.62 | | | $ | 125.72 | | | $ | 148.85 | | | $ | 191.58 | | | $ | 156.88 | |
ITEM 6.
[RESERVED]
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Overview
Open surgery remains the predominant form of surgery and is used in almost every area of the body. However, the large incisions required for open surgery create trauma to patients, typically resulting in longer hospitalization and recovery times, increased hospitalization costs, and additional pain and suffering relative to minimally invasive surgery, where MIS is available. For over three decades, MIS has reduced trauma to patients by allowing selected surgeries to be performed through small ports rather than large incisions. MIS has been widely adopted for certain surgical procedures.
Da Vinci Surgical Systems enable surgeons to extend the benefits of MIS to many patients who would otherwise undergo a more invasive surgery by using computational, robotic, and imaging technologies to overcome many of the limitations of traditional open surgery or conventional MIS. Surgeons using a da Vinci Surgical System operate while seated comfortably at a console viewing a 3D, high-definition image of the surgical field. This immersive console connects surgeons to the surgical field and their instruments. While seated at the console, the surgeon manipulates instrument controls in a natural manner, similar to open surgical technique. Our technology is designed to provide surgeons with a range of articulation of the surgical instruments used in the surgical field analogous to the motions of a human wrist, while filtering out the tremor inherent in a surgeon’s hand. In designing our products, we focus on making our technology easy and safe to use.
Our da Vinci products fall into five broad categories: da Vinci Surgical Systems, da Vinci instruments and accessories, da Vinci Stapling, da Vinci Energy, and da Vinci Vision, including Firefly Fluorescence imaging systems and da Vinci Endoscopes. We also provide a comprehensive suite of systems, learning, and services offerings. Digitally-enabled for nearly three decades, these three offerings aim to decrease variability by providing dependable, consistent functionality and an integrated user experience. Our systems category includes robotic platforms, software, vision, energy, and instruments and accessories. Our learning category includes educational technology, such as simulation and telepresence, as well as technical training programs and personalized peer-to-peer learning opportunities. Our services category assists and optimizes minimally invasive programs through readiness, on-demand support, consultation for minimally invasive program optimization, and hospitals customized analytics. Within our integrated ecosystem, our focus is to decrease variability in surgery by offering actionable insights, with digital solutions, to take action with the potential to improve outcomes, personalize learning, and optimize efficiency. We take a holistic approach, offering intelligent technology and systems designed to work together to make MIS intervention more available and applicable.
We have commercialized the following da Vinci Surgical Systems: the da Vinci standard Surgical System in 1999, the da Vinci S Surgical System in 2006, the da Vinci Si Surgical System in 2009, and the fourth generation da Vinci Xi Surgical System in 2014. We extended our fourth-generation platform by adding the da Vinci X Surgical System, commercialized in 2017, and the da Vinci SP Surgical System, commercialized in 2018. The da Vinci SP Surgical System accesses the body through a single incision, while the other da Vinci Surgical Systems access the body through multiple incisions. All da Vinci systems include a surgeon’s console (or consoles), imaging electronics, a patient-side cart, and computational hardware and software. We are in the early stages of launching our da Vinci SP Surgical System, and we have an installed base of 121 da Vinci SP Surgical Systems as of December 31, 2022. We have received FDA clearance for the da Vinci SP Surgical System for urologic and certain transoral procedures, and we have received regulatory clearance in South Korea, where the da Vinci SP Surgical System may be used for a broad set of procedures. In September 2022, we also received regulatory clearance for the da Vinci SP Surgical System in Japan for the same set of procedures as can be performed on the da Vinci Xi Surgical System in Japan. We plan to seek FDA clearances for additional indications for da Vinci SP over time. We also plan to seek clearances in other OUS markets over time. The success of the da Vinci SP Surgical System is dependent on positive experiences and improved clinical outcomes for the procedures for which it has been cleared as well as securing additional clinical clearances.
We offer approximately 70 different multi-port da Vinci instruments to provide surgeons with flexibility in choosing the types of tools needed to perform a particular surgery. These multi-port instruments are generally robotically controlled and provide end effectors (tips) that are similar to those used in either open or laparoscopic surgery. We offer advanced instrumentation for the da Vinci X and da Vinci Xi platforms, including da Vinci Energy and da Vinci Stapler products, to provide surgeons with sophisticated, computer-aided tools to precisely and efficiently interact with tissue. Da Vinci X and da Vinci Xi Surgical Systems share the same instruments, whereas the da Vinci Si Surgical System uses instruments that are not compatible with da Vinci X or da Vinci Xi systems. We currently offer nine core instruments on our da Vinci SP Surgical System. We plan to expand the SP instrument offering over time.
Training technologies include our Intuitive Simulation products, our Intuitive Telepresence remote case observation and telementoring tools, and our dual console for use in surgeon proctoring and collaborative surgery.
In 2019, the FDA cleared our Ion endoluminal system to enable minimally invasive biopsies in the lung. Our Ion system extends our commercial offering beyond surgery into diagnostic procedures with this first application. Our rollout of the Ion
system is progressing well, and we are continuing to gather additional clinical evidence. We plan to seek additional clearances for the Ion system in OUS markets over time.
The success of new product introductions depends on a number of factors including, but not limited to, pricing, competition, market and consumer acceptance, the effective forecasting and management of product demand, inventory levels, the management of manufacturing and supply costs, and the risk that new products may have quality or other defects in the early stages of introduction.
Macroeconomic Environment
Uncertainty surrounding macroeconomic factors in the U.S. and globally characterized by the supply chain environment, inflationary pressure, rising interest rates, labor shortages, and significant disruption in the commodities’ markets as a result of the Russia and Ukraine conflict may result in a recession, which could have a material adverse effect on our long-term business.
We have experienced increased difficulties in obtaining a sufficient supply of a number of component materials used in our products, such as semiconductor components as well as a range of other materials, including, but not limited to, metals and polymers, as global supply has become significantly constrained due to increased demand for certain materials. Additionally, prices of such materials have increased due to the increased demand and supply shortage. With rising interest rates, access to credit may become more difficult, and any insolvency of key suppliers, including sole-source and single-source suppliers, may exacerbate current supply chain challenges. We are engaged in activities to seek to mitigate supply disruptions, but the global supply chain shortages will remain a challenge for the foreseeable future.
Such global shortages in important components as well as certain logistics challenges have resulted in, and will continue to cause, inflationary cost pressure in our supply chain. To date, these supply chain challenges have not materially impacted our results of operations or ability to deliver products and services to our customers. However, if shortages in important supply chain materials in the semiconductor or other markets or logistics challenges continue, we could fail to meet product demand, which could result in deferred or canceled procedures. If inflationary pressures in logistics or component costs persist, we may not be able to adjust pricing, reduce costs, or implement countermeasures. Additionally, there is uncertainty surrounding the impact of any monetary policy changes taken by the U.S. Federal Reserve and other central banks to address the structural risks associated with inflation.
Fluctuations in labor availability globally, including labor shortages and staff burnout and attrition, could also impact our ability to hire and retain personnel critical to our manufacturing, logistics, and commercial operations. We are also highly dependent on the principal members of our management and scientific staff. The loss of critical members of our team, or our inability to attract and retain qualified personnel, could significantly harm our operations, business, and ability to compete.
The current macroeconomic environment is impacting our customers financially and operationally as well. Hospitals are experiencing staffing shortages and supply chain issues that could affect their ability to provide patient care. Additionally, hospitals are facing significant financial pressure as supply chain constraints and inflation drive up operating costs, rising interest rates make access to credit more expensive, unrealized losses decrease available cash reserves, and fiscal stimulus programs enacted during the COVID-19 pandemic wind down. As a consequence of the financial pressures and decreased profitability, some hospitals have indicated that they are lowering their capital investment plans and tightening their operational budgets. We believe that these factors have contributed to a softening in our U.S. capital pipeline, and we expect that demand for capital, particularly in the U.S., will continue to be impacted while macroeconomic conditions remain challenging. In addition, as competition progresses in various markets, we will likely experience longer selling cycles and pricing pressures. Any or all of these factors could negatively impact the number of da Vinci procedures performed or the number of system placements and have a material adverse effect on our business, financial condition, or results of operations resulting in failure to achieve our anticipated financial results.
COVID-19 Pandemic
Procedures
In 2020, as a result of the outbreak of a novel strain of coronavirus (COVID-19), we saw a substantial reduction in da Vinci procedures. The initial decline in procedures in the first quarter of 2020 was significant, most notably in China and then later in Western Europe and the U.S. as the pandemic spread. The second quarter of 2020 saw a further significant decline in procedures, followed by a period of recovery and new resurgences. In the U.S., procedures initially continued to decline, before starting a recovery as COVID-19 cases dropped and elective procedures were permitted. In China, procedure volumes recovered strongly, while the impact on procedure volumes of other countries varied. The third and fourth quarters of 2020 were characterized by the continued recovery of procedure volumes in the U.S. and China, while the procedure volumes of other countries, such as Japan, continued to vary depending on the spread and/or resurgence of COVID-19.
In 2021, COVID-19 resurgences affected da Vinci procedure volumes at various times throughout the year in most of the markets that we operate in. After each resurgence, as COVID-19 cases and hospitalizations subsided, we saw procedure
volumes recover. In the U.S., the impact of high COVID-related hospitalization rates on procedure volumes was exacerbated by staffing shortages. Although hospitals were better equipped to handle COVID patients as compared to the outset of the pandemic, COVID-19 resurgences challenged hospital resources and negatively impacted da Vinci procedure volumes. In addition, delays in diagnosis and treatment of underlying conditions had a negative impact on da Vinci procedure volumes. Volumes associated with benign procedures were generally impacted to a higher degree when COVID-19 cases and hospitalizations increased, reflecting the deferability of certain elective surgeries.
In early 2022, a resurgence of COVID-19 resulted in a significant increase in infections and hospitalization rates in the U.S. and certain countries in Europe, which, in turn, negatively impacted procedure volumes in January and February. As infections and hospitalizations started to decrease in February in the U.S. and Europe, we saw a recovery of procedure volumes. In March and during the second quarter of 2022, we also saw a resurgence in COVID-19 cases and increased hospitalizations and government interventions impacting parts of Asia, particularly China, which negatively impacted procedure volumes. During the third quarter of 2022, we did not experience significant disruptions from COVID-19. In the fourth quarter of 2022, we saw a resurgence in COVID-19 cases in China, which had a significant negative impact on our procedure volumes in the region.
The depth and extent to which the COVID-19 pandemic will impact individual markets will vary based on the availability of vaccinations, personal protective equipment, intensive care units and operating rooms, and medical staff, as well as government interventions. The impact of COVID-19 on our procedure volumes varies widely by country, region, and type. When COVID-19 infection rates spike in a particular region, procedure volumes have been negatively impacted and the diagnoses of new conditions and their related treatments have been deferred. While there is a backlog of patients, it is unpredictable when those patients will ultimately seek diagnosis and treatment and whether they will be treated through surgery. Based on our experience during the last three years, we do not expect all markets, regions, and procedure types to recover at the same time or at the same pace.
System Demand
As the impact of the COVID-19 pandemic progressed throughout 2020, customers in affected regions deferred decisions to purchase or lease systems into future quarters and, in some cases, indefinitely. In addition, the year-over-year stagnation in procedures during 2020 and, in turn, reduced utilization of our systems had resulted in unused capacity in the existing installed base. On the other hand, throughout 2021, we experienced strong system demand, as utilization levels recovered. In general, we believe that the COVID-19 pandemic had less of an impact on hospital spending capacity and that customers recognize that surgery meets their quadruple aim objectives better than other surgical approaches. This system demand continued during 2022; however, in 2022, our system demand was also impacted by macroeconomic challenges impacting our customers, primarily in the U.S. Refer to the factors outlined in the Macroeconomic Environment section above.
Customer Relief Program
In April 2020, we announced a program to provide financial relief to our customers. The program consisted of three main elements. The first element provided credits against service fees otherwise due in the six-month period from April 1 through September 30, 2020, which generally reflected the underutilization of the system during that period. Those credits were offered to most customers worldwide. The second element of the program deferred certain lease payments, and the third element extended certain payment terms. Service fee credits resulted in an $80 million decrease in service revenue in 2020. While the short-term payment relief offered did not have a material impact on the results of operations, we deferred $15 million of lease billings and extended payment terms associated with $181 million of trade receivables during the program, of which $19 million remained outstanding as of December 31, 2020. All of the trade receivables with extended payment terms were collected as of December 31, 2021. We may be subject to increased credit risks resulting in collection delinquencies and defaults, which could materially impact our bad debt write-offs and provisions for credit losses. Although we have programs in place that are designed to monitor and mitigate the associated risks, there can be no assurance that such programs will be effective in reducing credit risks relating to these lease financing arrangements and extended payment terms. There was no similar customer relief program offered in 2021 or 2022.
General Increase in Risks
The COVID-19 pandemic and local actions, such as “shelter-in-place” orders and restrictions on our ability to travel and access our customers or temporary closures of our facilities, including our training and manufacturing operations, or the facilities of our suppliers and their contract manufacturers, could further significantly impact our sales and our ability to produce and ship our products and supply our customers.
In addition, COVID-19 has contributed to the staffing shortages experienced by hospitals, which impacts hospitals’ ability to provide patient care and, in some cases, results in the deferral of elective surgeries.
Our Response
Our priorities and actions during the COVID-19 pandemic have been and remain as follows. First, we are focused on the health and safety of all those we serve—patients, customers, our communities, and our employees—implementing continuous updates to our health and safety policies and processes. Second, we are supporting our customers according to their priorities—clinical, operational, and economic—and ensuring continuity of supply by working with our suppliers and our distributors. Third, we are securing our workforce economically. We have built a valuable team over the years, and we believe they will be important in a recovery that follows the pandemic. Finally, we will continue to invest in our priority development programs while eliminating avoidable spend.
As COVID-19 vaccination rates increase and the severity of cases declines, we are implementing our return-to-office strategy. We collected internal and external insights to inform decision-making on work models that would align with how employees will work in the current environment, which has evolved as a result of the COVID-19 pandemic. These efforts will allow for an improved employee experience, regardless of whether an employee is working from home, fully on-site, or in a hybrid fashion. Our top priority in this process continues to be the health and safety of our employees.
Business Model
Overview
We generate revenue from the placement of da Vinci Surgical Systems, in sales or sales-type lease arrangements where revenue is recognized up-front or in operating lease and usage-based arrangements where revenue is recognized over time. We earn recurring revenue from the sales of instruments, accessories, and services, as well as the revenue from operating leases. The da Vinci Surgical System generally sells for between $0.5 million and $2.5 million, depending upon the model, configuration, and geography, and represents a significant capital equipment investment for our customers when purchased. Our instruments and accessories have limited lives and will either expire or wear out as they are used in surgery, at which point they need to be replaced. We generally earn between $600 and $3,500 of instruments and accessories revenue per surgical procedure performed, depending on the type and complexity of the specific procedures performed and the number and type of instruments used. In late 2020, we launched our Extended Use Program (refer to further discussion in the section below) in the U.S. and Europe, with the intention to reduce the cost for customers to treat patients, which in turn will reduce our overall instruments and accessories revenue per procedure. We typically enter into service contracts at the time systems are sold or leased at an annual fee between $80,000 and $190,000, depending upon the configuration of the underlying system and the composition of the services offered under the contract. These service contracts have generally been renewed at the end of the initial contractual service periods.
We generate revenue from our Ion endoluminal system in a business model consistent with the da Vinci Surgical System model described above. We generate revenue from the placement of Ion systems, in sales or sales-type lease arrangements where revenue is recognized up-front or in operating lease and usage-based arrangements where revenue is recognized over time. We earn recurring revenue from the sales of instruments, accessories, and services, as well as revenue from operating leases. The average selling price of an Ion system is generally significantly lower than the average selling price of a da Vinci Surgical System. For the years ended December 31, 2022, 2021, and 2020, Ion’s contribution to revenue and gross margin was not significant.
Additionally, as part of our ecosystem of products and services, we provide a portfolio of learning offerings and digital solutions. We do not currently generate material revenue from these offerings.
Extended Use Program
In 2020, we introduced our Extended Use Program in the U.S. and Europe, which consists of select da Vinci Xi and da Vinci X instruments possessing 12 to 18 uses compared to the previously 10 uses. These Extended Use Instruments represent some of our higher volume instruments but exclude stapling, monopolar, and advanced energy instruments. Instruments included in the program are used across a number of da Vinci surgeries. Their increased uses are the result of continuous, significant investments in the design and production capabilities of our instruments, resulting in improved quality and durability. Extended Use Instruments were then launched in most other countries around the world in the first half of 2021, except China due to regulatory timelines. In addition, simultaneous with the regional launches of Extended Use Instruments, we have lowered the price of certain instruments that are most commonly used in lower acuity procedures and/or lower reimbursed procedures within the region. These actions have reduced the cost for customers to treat patients, which in turn has reduced our revenue per procedure. In the U.S. and Europe, during 2021, we saw customers adjust their instrument buying patterns to reduce their inventory levels to reflect the additional uses per instrument. We believe that, as of the end of 2021, in the U.S. and Europe, full cutover to Extended Use Instruments has occurred, as customers have substantially utilized all of their remaining 10 use instruments. The precise impact of these actions on future revenue will be dependent on the future volume and mix of procedures and whether cost elasticity will enable greater penetration into available markets.
Recurring Revenue
Recurring revenue consists of instruments and accessories revenue, service revenue, and operating lease revenue. Recurring revenue increased to $4.9 billion, or 79% of total revenue in 2022, compared to $4.3 billion, or 75% of total revenue in 2021, and $3.4 billion, or 77% of total revenue in 2020.
Instruments and accessories revenue has grown at a faster rate than systems revenue over time. Instruments and accessories revenue increased to $3.52 billion in 2022, compared to $3.10 billion in 2021 and $2.46 billion in 2020. The increase in instruments and accessories revenue largely reflects continued procedure adoption.
Service revenue was $1.02 billion in 2022, compared to $0.92 billion in 2021 and $0.72 billion in 2020. The increase in service revenue was primarily driven by the growth of the base of installed da Vinci Surgical Systems producing service revenue, as well as the effects of the Customer Relief Program in 2020, which resulted in an $80 million decrease in service revenue. The installed base of da Vinci Surgical Systems grew 12% to approximately 7,544 as of December 31, 2022; 12% to approximately 6,730 as of December 31, 2021; and 7% to approximately 5,989 as of December 31, 2020.
We use the installed base, number of placements, and utilization of systems as metrics for financial and operational decision-making and as a means to evaluate period-to-period comparisons. Management believes that the installed base, number of placements, and utilization of systems provide meaningful supplemental information regarding our performance, as management believes that the installed base, number of placements, and utilization of systems are an indicator of the rate of adoption of robotic-assisted surgery or bronchoscopy as well as an indicator of future recurring revenue. Management believes that both it and investors benefit from referring to the installed base, number of placements, and utilization of systems in assessing our performance and when planning, forecasting, and analyzing future periods. The installed base, number of placements, and utilization of systems also facilitate management’s internal comparisons of our historical performance. We believe that the installed base, number of placements, and utilization of systems are useful to investors as metrics because (1) they allow for greater transparency with respect to key metrics used by management in its financial and operational decision-making, and (2) they are used by institutional investors and the analyst community to help them analyze the performance of our business. The vast majority of installed systems are connected via the internet. System logs can also be accessed by field engineers for systems that are not connected to the internet. We utilize this information as well as other information from agreements and discussions with our customers that involve estimates and judgments, which are, by their nature, subject to substantial uncertainties and assumptions. Estimates and judgments for determining the installed base, number of placements, and utilization of systems may be impacted over time by various factors, including system internet connectivity, hospital and distributor reporting behavior, and inherent complexities in new agreements. Such estimates and judgments are also susceptible to technical errors. In addition, the relationship between the installed base, number of placements, and utilization of systems and our revenues may fluctuate from period to period, and growth in the installed base, number of placements, and utilization of systems may not correspond to an increase in revenue. The installed base, number of placements, and utilization of systems are not intended to be considered in isolation or as a substitute for, or superior to, revenue or other financial information prepared and presented in accordance with U.S. generally accepted accounting principles (“GAAP”).
Intuitive System Leasing
Since 2013, we have entered into sales-type and operating lease arrangements directly with certain qualified customers as a way to offer customers flexibility in how they acquire systems and expand their robotic-assisted programs while leveraging our balance sheet. These leases generally have commercially competitive terms as compared to other third-party entities that offer equipment leasing. We have also entered into usage-based arrangements with qualified customers that have committed da Vinci programs where we charge for the system and service as the systems are utilized. We believe that these alternative financing structures have been effective and well-received, and we are willing to expand the proportion of these structures based on customer demand. We include operating and sales-type leases, and systems placed under usage-based arrangements, in our system placement and installed base disclosures. We exclude operating lease-related revenue, usage-based revenue, and Ion system revenue from our da Vinci Surgical System average selling price (“ASP”) computations.
In the years ended December 31, 2022, 2021, and 2020, we placed 591, 668, and 432 da Vinci Surgical Systems, respectively, under lease and usage-based arrangements, of which 492, 517, and 317 systems, respectively, were operating lease and usage-based arrangements. In the years ended December 31, 2022, 2021, and 2020, we placed 112, 57, and 9 Ion systems, respectively, under lease and usage-based arrangements, of which 101, 50, and 9 systems, respectively, were operating lease and usage-based arrangements.
Revenue from operating lease arrangements is generally recognized on a straight-line basis over the lease term or, in the case of usage-based arrangements, as the systems are used. We generally set operating lease and usage-based pricing at a modest premium relative to purchased systems reflecting the time value of money and, in the case of usage-based arrangements, the risk that system utilization may fall short of anticipated levels. Variable lease revenue recognized from usage-based arrangements has been included in our operating lease metrics herein. Operating lease revenue has grown at a faster rate than overall systems revenue and was $377 million, $277 million, and $177 million for the years ended December 31, 2022, 2021,
and 2020, respectively, of which $133 million, $78 million, and $28 million, respectively, was variable lease revenue. As revenue for operating leases and usage-based systems is recognized over time, total systems revenue growth is reduced in a period when the number of operating lease and usage-based placements increases as a proportion of total system placements. Generally, lease transactions generate similar gross margins as our sale transactions. A total of 1,683, 1,294, and 901 da Vinci Surgical Systems were installed at customers under operating lease or usage-based arrangements as of December 31, 2022, 2021, and 2020, respectively. A total of 132, 61, and 11 Ion systems were installed at customers under operating lease or usage-based arrangements as of December 31, 2022, 2021, and 2020, respectively.
Our exposure to the credit risks relating to our lease financing arrangements may increase if our customers are adversely affected by changes in healthcare laws, coverage and reimbursement, economic pressures or uncertainty, or other customer-specific factors. As a result of these macroeconomic factors impacting our customers, we may be exposed to defaults under our lease financing arrangements. Moreover, usage-based arrangements generally contain no minimum payments; therefore, customers may exit such arrangements without paying a financial penalty to us.
For some operating lease arrangements, our customers are provided with the right to purchase the leased system at certain points during and/or at the end of the lease term. Revenue generated from customer purchases of systems under operating lease arrangements (“Lease Buyouts”) was $72 million, $96 million, and $52 million for the years ended December 31, 2022, 2021, and 2020, respectively. We expect that revenue recognized from customer exercises of the buyout options will fluctuate based on the timing of when, and if, customers choose to exercise their buyout options.
Systems Revenue
System placements are driven by procedure growth in most markets. In some markets, system placements are constrained by regulation. In geographies where da Vinci procedure adoption is in an early stage or system placements are constrained by regulation, system sales will precede procedure growth. System placements also vary due to seasonality largely aligned with hospital budgeting cycles. We typically place a higher proportion of annual system placements in the fourth quarter and a lower proportion in the first quarter as customer budgets are reset. Systems revenue is also affected by the proportion of system placements under operating lease and usage-based arrangements, recurring operating lease and usage-based revenue, operating lease buyouts, product mix, ASPs, trade-in activities, and customer mix. Systems revenue declined 1% to $1.68 billion in 2022. Systems revenue grew 44% to $1.69 billion in 2021. Systems revenue declined 12% to $1.18 billion in 2020. Based on the factors outlined in the COVID-19 Pandemic section above, we believe that historical system placement trends may not be a good indicator of future system placements.
Procedure Mix / Products
Our da Vinci Surgical Systems are generally used for soft tissue surgery for areas of the body between the pelvis and the neck, primarily in general surgery, gynecologic surgery, urologic surgery, cardiothoracic surgery, and head and neck surgery. Within these categories, procedures range in complexity from cancer and other highly complex procedures to less complex procedures for benign conditions. Cancer and other highly complex procedures tend to be reimbursed at higher rates than less complex procedures for benign conditions. Thus, hospitals are more sensitive to the costs associated with treating less complex, benign conditions. Our strategy is to provide hospitals with attractive clinical and economical solutions across the spectrum of procedure complexity. Our fully featured da Vinci Xi Surgical System with advanced instruments (including da Vinci Energy and EndoWrist and SureForm Stapler products) and our Integrated Table Motion product targets the more complex procedure segment. Our da Vinci X Surgical System is targeted toward price-sensitive markets and procedures. Our da Vinci SP Surgical System complements the da Vinci Xi and X Surgical Systems by enabling surgeons to access narrow workspaces.
Procedure Seasonality
More than half of da Vinci procedures performed are for benign conditions, most notably hernia repairs, hysterectomies, and cholecystectomies. These benign procedures and other short-term elective procedures tend to be more seasonal than cancer operations and surgeries for other life-threatening conditions. Seasonality in the U.S. for procedures for benign conditions typically results in higher fourth quarter procedure volume when more patients have met annual deductibles and lower first quarter procedure volume when deductibles are reset. Seasonality outside of the U.S. varies and is more pronounced around local holidays and vacation periods. As a result of the factors outlined in the COVID-19 Pandemic section above, including past and potentially future recommendations of authorities to defer elective procedures, historical procedure patterns may be disrupted.
Distribution Channels
We provide our products through direct sales organizations in the U.S., Europe (excluding Spain, Portugal, Italy, Greece, and most Eastern European countries), China (through our Intuitive-Fosun Pharma joint venture), Japan, South Korea, India, Taiwan and, as of June 2022, Canada. In the remainder of our OUS markets, we provide our products through distributors.
Regulatory Activities
Overview
Our products must meet the requirements of a large and growing body of international standards that govern the product safety, efficacy, advertising, labeling, safety reporting design, manufacture, materials content and sourcing, testing, certification, packaging, installation, use, and disposal of our products. Examples of such standards include electrical safety standards, such as those of the International Electrotechnical Commission, and composition standards, such as the Reduction of Hazardous Substances and the Waste Electrical and Electronic Equipment Directives. Failure to meet these standards could limit our ability to market our products in those regions that require compliance with such standards.
Our products and operations are also subject to increasingly stringent medical device, privacy, and other regulations by regional, federal, state, and local authorities. After a device is placed on the market, numerous FDA and other regulatory requirements continue to apply. These requirements include establishment registration and device listing with the FDA and compliance with medical device reporting regulations, which require that manufacturers report to the FDA if their device caused or contributed, or may have caused or contributed, to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur.
We recently revised our medical device reporting policies, which had been developed based on previous feedback from the FDA. These revisions have been made in consultation with the FDA to better align with existing regulations. There has been an increase in medical device reporting filings due to changes in our reportability criteria. In addition, we have been investing in resources and utilizing external experts to strengthen our quality system. These efforts are ongoing.
We also anticipate that timelines for the introduction of new products and/or indications may be extended relative to past experience as a result of these regulations. For example, we have seen elongated regulatory approval timelines in the U.S. and Europe.
Clearances, Approvals, and Certifications
We have generally obtained the regulatory clearances, approvals, and certifications required to market our products associated with our da Vinci Surgical Multiport Systems (Standard, S, Si, Xi, and X systems) for our targeted surgical specialties within the U.S., South Korea, Japan, and the European markets in which we operate. Since 2020, we have obtained regulatory clearances, approvals, and certifications for the following products:
•In September 2022, we obtained regulatory clearance for the da Vinci SP Surgical System in Japan for use in general surgeries, thoracic surgeries (excluding cardiac procedures and intercostal approaches), urologic surgeries, gynecological surgeries, and trans-oral head and neck surgeries. We obtained the initial FDA clearance for our da Vinci SP Surgical System in April 2014 and have since invested in important platform refinements. We also received regulatory clearance in South Korea for our da Vinci SP Surgical System in May 2018.
•In February 2022, we received regulatory clearance in China to market both our 12 mm SureForm 45 Stapler and SureForm 60 Stapler and corresponding reloads.
•In January 2022, we received regulatory clearance in China to market our da Vinci Vessel Sealer Extend with up to 7 mm vascular indications.
•In December 2021, we obtained FDA clearance for our 8 mm SureForm 30 Curved-Tip Stapler and reloads for use in general, thoracic, gynecologic, urologic, and pediatric surgery. The 8 mm SureForm 30 Curved-Tip Stapler is expected to launch in the U.S. in 2023, with other countries to follow. In October 2022, we received regulatory clearance in Japan to market our 8 mm SureForm 30 Curved-Tip and Straight-Tip Stapler instruments and reloads for use in general, thoracic (except for cardiac), gynecologic, and urologic surgery.
•In late 2020 and early 2021, we obtained FDA clearance, European certification, and other regulatory clearances in most of our significant markets to market our Extended Use Instruments.
•In November 2019, we obtained FDA clearance for our SynchroSeal instrument and E-100 generator. Following the FDA clearance, in February 2020, we obtained European certification for both products. In March 2020, we received regulatory clearance in Japan to market both our SynchroSeal instrument and E-100 generator. We received regulatory clearance in South Korea to market our SynchroSeal instrument and E-100 generator in January 2020 and August 2020, respectively.
•In July 2019, we obtained FDA clearance for our SureForm 45 Curved-Tip Stapler and SureForm 45 Gray reload, which round out our SureForm 45 portfolio. We have also obtained European certification for our SureForm 45 Curved-Tip Stapler and SureForm 45 Gray reload. In September 2019, we received regulatory clearance in Japan to market both our SureForm 45 Curved-Tip Stapler and SureForm 45 Gray reload. We received regulatory clearance in South Korea to market our SureForm 45 Curved-Tip Stapler and SureForm 45 Gray reload in June 2021 and July 2021, respectively.
•In June 2019, we obtained European certification for our da Vinci Endoscope Plus for the da Vinci Xi and da Vinci X Surgical Systems in Europe. Following the European certification, in July 2019, we obtained FDA clearance for our da Vinci Endoscope Plus. We have also received regulatory clearances in South Korea and Japan to market our da Vinci Endoscope Plus in December 2019 and May 2020, respectively. In March 2022, we received regulatory clearance in China to market our da Vinci Endoscope Plus.
•In June 2019, we obtained FDA clearance for our da Vinci Handheld Camera and, in February 2020, we obtained European certification.
Refer to the descriptions of our new products that received regulatory clearances, approvals, or certifications in 2022, 2021, and 2020 in the Recent Product Introductions section below.
In October 2018, the China National Health Commission published on its official website the quota for major medical equipment to be sold in China through 2020. After an adjustment notice was published in the third quarter of 2020, the government will now allow for the total sale of 225 new surgical robots into China, which could include da Vinci Surgical Systems as well as surgical systems introduced by others. As of December 31, 2022, we have sold 187 da Vinci Surgical Systems under this quota, and we believe that four system quotas are no longer available; therefore, 34 surgical robots should still be available for sale under this quota. Future sales of da Vinci Surgical Systems under the quota are uncertain, as they are dependent on hospitals completing a tender process and receiving associated approvals. Additionally, any delays in the granting of a new quota in China will constrain our ability to further grow our installed base in China as well as limit our capacity for procedure growth in China.
During the third quarter of 2022, the Hunan Provincial Healthcare Security Administration implemented significant limits on what hospitals can charge patients for surgeries using robotic surgical technology, including soft tissue surgery and orthopedics. This rule has had a material negative impact on our procedures performed in the Hunan province. In addition to the Hunan province, the Hainan province (an island province of China) recently announced a policy to implement almost identical limits on what hospitals can charge patients for surgeries using robotic surgical technology. However, currently, only a small portion of our installed base in China is located in the Hunan and Hainan provinces. Companies providing robotic surgical technology, including our joint venture in China, are meeting with Chinese government healthcare agencies to discuss this development and provide feedback. We cannot assure you that other provincial healthcare administrations will not impose similar limits.
The Japanese Ministry of Health, Labor, and Welfare considers reimbursement for procedures in April of even-numbered years. The process for obtaining reimbursement requires Japanese university hospitals and surgical societies, with our support, to seek reimbursement. There are multiple pathways to obtain reimbursement for procedures, including those that require in-country clinical data/economic data. In April 2012 and April 2016, the MHLW granted reimbursement status for prostatectomy and partial nephrectomy, respectively. Most prostatectomies and partial nephrectomies were open procedures prior to da Vinci reimbursement. Da Vinci procedure reimbursement for prostatectomy and partial nephrectomy procedures are higher than open and conventional laparoscopic procedure reimbursements. An additional 12 da Vinci procedures were granted reimbursement effective April 1, 2018, including gastrectomy, low anterior resection, lobectomy, and hysterectomy, for both malignant and benign conditions, and an additional seven da Vinci procedures were granted reimbursement effective April 1, 2020. An additional eight da Vinci procedures were granted reimbursement effective April 1, 2022, including colon resection. In addition, we received higher reimbursement for da Vinci gastrectomy procedures, as compared to open and conventional laparoscopic procedure reimbursements. The additional reimbursed procedures have varying levels of conventional laparoscopic penetration and will generally be reimbursed at rates equal to the conventional laparoscopic procedures. Given the reimbursement level and laparoscopic penetration for these additional procedures, there can be no assurance that the adoption pace for these procedures will be similar to prostatectomy or partial nephrectomy, given their higher reimbursement, or any other da Vinci procedure.
Recalls and Corrections
Medical device companies have regulatory obligations to correct or remove medical devices in the field that could pose a risk to health. The definition of “recalls and corrections” is expansive and includes repair, replacement, inspections, relabeling, and issuance of new or additional instructions for use or reinforcement of existing instructions for use and training when such actions are taken for specific reasons of safety or compliance. These field actions require stringent documentation, reporting, and monitoring worldwide. There are other actions that a medical device manufacturer may take in the field without reporting including, but not limited to, routine servicing and stock rotations.
As we determine whether a field action is reportable in any regulatory jurisdiction, we prepare and submit notifications to the appropriate regulatory agency for the particular jurisdiction. Regulators can require the expansion, reclassification, or change in scope and language of the field action. In general, upon submitting required notifications to regulators regarding a field action that is a recall or correction, we will notify customers regarding the field action, provide any additional documentation required in their national language, and arrange, as required, the return or replacement of the affected product or a field service visit to perform the correction.
Field actions, as well as certain outcomes from regulatory activities, can result in adverse effects on our business, including damage to our reputation, delays by customers of purchase decisions, reduction or stoppage of the use of installed systems, and reduced revenue as well as increased expenses.
Due to an internally-discovered defect in a component manufactured by a third party and used in certain da Vinci arms, we will be issuing a voluntary field action on or about February 10, 2023. As agreed with the FDA, the field action recommends that customers stop using approximately 109 da Vinci X and Xi systems until we can replace the affected arms, which we anticipate completing during February 2023. The affected part does not have patient contact, and there have been no adverse events reported related to this issue. We do not expect this field action to have a material impact on our business, financial condition, or results of operations.
Procedures
We model patient value as equal to procedure efficacy / invasiveness. In this equation, procedure efficacy is defined as a measure of the success of the surgery in resolving the underlying disease, and invasiveness is defined as a measure of patient pain and disruption of regular activities. When the patient value of a da Vinci procedure is greater than that of alternative treatment options, patients may benefit from seeking out surgeons and hospitals that offer da Vinci Surgery, which could potentially result in a local market share shift. Adoption of da Vinci procedures occurs procedure by procedure and market by market and is driven by the relative patient value and total treatment costs of da Vinci procedures as compared to alternative treatment options for the same disease state or condition.
We use the number and type of procedures as metrics for financial and operational decision-making and as a means to evaluate period-to-period comparisons. Management believes that the number and type of procedures provide meaningful supplemental information regarding our performance, as management believes procedure volume is an indicator of the rate of adoption of robotic-assisted surgery or bronchoscopy as well as an indicator of future revenue (including revenue from usage-based arrangements). Management believes that both it and investors benefit from referring to the number and type of procedures in assessing our performance and when planning, forecasting, and analyzing future periods. The number and type of procedures also facilitate management’s internal comparisons of our historical performance. We believe that the number and type of procedures are useful to investors as metrics, because (1) they allow for greater transparency with respect to key metrics used by management in its financial and operational decision-making, and (2) they are used by institutional investors and the analyst community to help them analyze the performance of our business. The vast majority of our installed systems are connected via the internet. System logs can also be accessed by field engineers for systems that are not connected to the internet. We utilize certain methods that rely on information collected from the installed systems for determining the number and type of procedures performed that involve estimates and judgments, which are, by their nature, subject to substantial uncertainties and assumptions. Estimates and judgments for determining the number and type of procedures may be impacted over time by various factors, including changes in treatment modalities, hospital and distributor reporting behavior, and system internet connectivity. Such estimates and judgments are also susceptible to algorithmic or other technical errors. In addition, the relationship between the number and type of procedures and our revenues may fluctuate from period to period, and procedure volume growth may not correspond to an increase in revenue. The number and type of procedures are not intended to be considered in isolation or as a substitute for, or superior to, revenue or other financial information prepared and presented in accordance with GAAP.
Worldwide Procedures
Our systems and instruments are regulated independently in various countries and regions of the world. The discussion of indications for use and representative or target procedures is intended solely to provide an understanding of the market for our products and is not intended to promote for sale or use any Intuitive product outside of its licensed or cleared labeling and indications for use.
The adoption of robotic-assisted surgery using the da Vinci Surgical System has the potential to grow for those procedures that offer greater patient value than to non-da Vinci alternatives and competitive total economics for healthcare providers. Our da Vinci Surgical Systems are used primarily in general surgery, urologic surgery, gynecologic surgery, cardiothoracic surgery, and head and neck surgery. We focus our organization and investments on developing, marketing, and training products and services for procedures in which da Vinci can bring patient value relative to alternative treatment options and/or economic benefit to healthcare providers. Target procedures in general surgery include hernia repair (both ventral and inguinal), colorectal, cholecystectomy, and bariatric procedures. Target procedures in urology include prostatectomy and partial nephrectomy. Target procedures in gynecology include hysterectomy for both cancer and benign conditions and sacrocolpopexy. In cardiothoracic surgery, target procedures include lobectomy. In head and neck surgery, target procedures include transoral surgery. Not all indications, procedures, or products described may be available in a given country or region or on all generations of da Vinci Surgical Systems. Surgeons and their patients need to consult the product labeling in their specific country and for each product in order to determine the cleared uses, as well as important limitations, restrictions, or contraindications.
Similarly, the adoption of robotic-assisted bronchoscopy using the Ion system has the potential to grow if it can offer greater patient value than non-Ion alternatives and competitive total economics for healthcare providers.
In 2022, approximately 1,875,000 surgical procedures were performed with da Vinci Surgical Systems, compared to approximately 1,594,000 and 1,243,000 surgical procedures performed with da Vinci Surgical Systems in 2021 and 2020, respectively. The increase in our overall procedure volume in 2022 reflects the disruption caused by the COVID-19 pandemic in 2022 and 2021, as noted in the COVID-19 Pandemic section above, and was driven by growth in U.S. general surgery, OUS urology, and OUS general surgery (particularly cancer) procedures.
In 2022, approximately 23,500 biopsy procedures were performed with Ion systems, compared to approximately 7,400 and 1,700 biopsy procedures performed with Ion systems in 2021 and 2020, respectively. The increase in our overall procedure volume in 2022 reflects a larger installed base of approximately 321 systems, an increase of 149% compared to the installed base of approximately 129 systems as of 2021. Currently, the vast majority of Ion biopsy procedures are performed in the U.S.
U.S. da Vinci Procedures
Overall U.S. procedure volume with da Vinci Surgical Systems grew to approximately 1,282,000 in 2022, compared to approximately 1,109,000 in 2021 and approximately 876,000 in 2020. General surgery was our largest and fastest growing U.S. specialty in 2022 with procedure volume that grew to approximately 720,000 in 2022, compared to approximately 588,000 in 2021 and approximately 434,000 in 2020. Gynecology was our second largest U.S. surgical specialty in 2022 with procedure volume that grew to approximately 341,000 in 2022, compared to approximately 316,000 in 2021 and approximately 267,000 in 2020. Urology was our third largest U.S. surgical specialty in 2022 with procedure volume that grew to approximately 162,000 in 2022, compared to approximately 153,000 in 2021 and approximately 134,000 in 2020.
OUS da Vinci Procedures
Overall OUS procedure volume with da Vinci Surgical Systems grew to approximately 593,000 in 2022, compared to approximately 485,000 in 2021 and approximately 367,000 in 2020. Urology was our largest OUS specialty in 2022 with procedure volume that grew to approximately 316,000 in 2022, compared to approximately 264,000 in 2021 and approximately 215,000 in 2020. General surgery was our second largest OUS specialty in 2022 with procedure volume that grew to approximately 133,000 in 2022, compared to approximately 101,000 in 2021 and approximately 68,000 in 2020. Gynecology procedures also contributed to OUS procedure growth.
Recent Business Events and Trends
Procedures
Overall. Total da Vinci procedures performed by our customers grew approximately 18% for the year ended December 31, 2022, compared to approximately 28% for the year ended December 31, 2021. The 2022 and 2021 procedure results (and comparative 2020 procedures results) reflected disruption caused by the COVID-19 pandemic, as noted in the COVID-19 Pandemic section above, which significantly impacted our procedures in certain periods in geographies and markets where there was a resurgence of the virus. The 2022 procedure growth was largely attributable to growth in U.S. general surgery, OUS urology, and OUS general surgery (particularly cancer) procedures. Delays in both the diagnosis and treatments of diseases reflecting disruptions caused by COVID-19 have previously and may continue to impact the number of procedures performed by our customers.
U.S. Procedures. U.S. da Vinci procedures grew approximately 16% for the year ended December 31, 2022, compared to approximately 27% for the year ended December 31, 2021. The 2022 and 2021 procedure results (and comparative 2020 procedures results) reflected disruption caused by the COVID-19 pandemic, as noted in the COVID-19 Pandemic section above, which significantly impacted our procedures. The 2022 U.S. procedure growth was largely attributable to growth in general surgery procedures, most notably hernia repair, cholecystectomy, and bariatric procedures. Growth in the more mature gynecologic and urologic procedure categories was more moderate.
U.S. General Surgery. General surgery procedures in the U.S. grew to approximately 720,000 in 2022, compared to approximately 588,000 in 2021 and approximately 434,000 in 2020. Inguinal and ventral hernia repairs, cholecystectomies, and bariatric procedures contributed the most incremental procedures in 2022 and 2021, while cholecystectomies and bariatric procedures contributed the most incremental procedures in 2020.
We believe that growth in hernia repair using da Vinci reflects improved clinical outcomes within certain patient populations, as well as potential cost benefits relative to certain alternative treatments. We believe that hernia repair procedures represent a significant opportunity with the potential to drive growth in future periods. However, given the differences in surgical complexity associated with the treatment of various hernia patient populations and varying surgeon opinions regarding optimal surgical technique, it is difficult to estimate the timing of and to what extent hernia repair procedure volume will grow in the future. We expect a large portion of hernia repairs will continue to be performed via different modalities of surgery.
Given the already very high level of laparoscopic techniques used in cholecystectomy, it is unclear whether our growth is sustainable and to what extent da Vinci may be adopted.
Bariatric procedures have grown significantly over the last three years. These procedures have been an increased area of focus and may also have benefited from certain patients prioritizing weight loss, as obesity is a significant COVID-19 risk factor. In addition, our SureForm 60mm Stapler provides surgeons with a more optimized robotic tool set for bariatric procedures. However, the diagnoses and treatment pathways for bariatric patients are long, and we cannot provide any assurance that we will continue to see significant growth in bariatric procedures in future periods.
Adoption of da Vinci for colorectal procedures, which includes several underlying procedures, including low anterior resections for rectal cancers and certain colon procedures for benign and cancerous conditions, has been ongoing for several years and is supported by certain technologies, such as the EndoWrist and SureForm Staplers, energy devices, and Integrated Table Motion.
OUS Procedures. OUS da Vinci procedures grew approximately 22% for the year ended December 31, 2022, compared to approximately 32% for the year ended December 31, 2021. The 2022 and 2021 procedure results (and comparative 2020 procedures results) reflected disruption caused by the COVID-19 pandemic, as noted in the COVID-19 Pandemic section above, which significantly impacted our procedures. The 2022 OUS procedure growth was driven by continued growth in urologic procedures, including prostatectomies and partial nephrectomies, and earlier stage growth in general surgery (particularly colorectal), gynecologic, and thoracic procedures. The 2022 OUS procedure growth rate reflects continued da Vinci adoption in European and Asian markets. We saw strong procedure growth in Japan, Germany, and the UK during 2022. Procedure growth in China was negatively impacted by COVID-19 restrictions in effect during 2022, particularly during Q2, and the subsequent effect of lifting those restrictions in Q4, which resulted in a surge of COVID-19 cases. We believe that growth in these global markets is being driven by increased acceptance among surgeons and health systems, supported by expanded global evidence validating the clinical and economic value of da Vinci procedures as well as increased surgeon training.
OUS Urology. Along with general surgery, OUS urology procedures have been a strong contributor to our overall procedure growth. OUS urology procedures grew to approximately 316,000 in 2022, compared to approximately 264,000 in 2021 and approximately 215,000 in 2020. In the U.S., da Vinci is the standard of care for the surgical treatment of prostate cancer, and we believe that the growth is largely aligned with surgical volumes of prostate cancer. For OUS, prostatectomy is at varying states of adoption in different areas of the world but is the largest overall da Vinci procedure. In 2022, we saw more moderate growth in OUS prostatectomy procedures compared to higher growth in 2021, as we are further up the adoption curve.
Kidney cancer procedures have also been a strong contributor to our recent global urology procedure growth. Clinical publications have demonstrated that the use of a da Vinci system increases the likelihood that a patient will receive nephron sparing surgery through a partial nephrectomy, which is typically the surgical society guideline recommended therapy.
OUS General Surgery. OUS general surgery procedures grew to approximately 133,000 in 2022, compared to approximately 101,000 in 2021 and approximately 68,000 in 2020. Colorectal procedures contributed the most incremental procedures in 2022, 2021, and 2020, aided by improved clinical outcomes relative to open and laparoscopic techniques within certain patient populations, along with enabling technologies, such as EndoWrist and SureForm staplers, energy devices, and Integrated Table Motion.
System Demand
We placed 1,264 da Vinci Surgical Systems in 2022, compared to 1,347 systems in 2021. The decrease in systems placed reflects a smaller number of third generation da Vinci systems available for trade-in along with the macroeconomic challenges impacting our customers, primarily in the U.S. As a consequence of the macroeconomic challenges, some hospitals have indicated that they are lowering their capital investment plans and tightening operational budgets. We expect that demand for capital will be impacted while macroeconomic conditions remain challenging.
2022 da Vinci placements declined 6% compared with 2021, and we expect that future placements of da Vinci Surgical Systems will be impacted by a number of factors: supply chain risks; economic and geopolitical factors; inflationary pressures; rising interest rates; hospital staffing shortages; the impact of the current COVID-19 pandemic, as noted in the COVID-19 Pandemic section above; hospital response to the evolving healthcare environment; procedure growth rates; hospital consolidation trends; evolving system utilization and point of care dynamics; capital replacement trends, including a declining number of older generation systems available for trade-in transactions; additional reimbursements in various global markets, including Japan; the timing around governmental tenders and authorizations, including China; the timing of when we receive regulatory clearance in our other OUS markets for our da Vinci Xi Surgical System, da Vinci X Surgical System, and da Vinci SP Surgical System, and related instruments; and market response. Market acceptance of our da Vinci SP Surgical System and the nature and timing of additional da Vinci SP regulatory indications may also impact future system placements.
Demand may also be impacted by competition, including from companies that have introduced products in the field of robotic-assisted medical procedures or have made explicit statements about their efforts to enter the field including, but not limited to, the following companies: Asensus Surgical, Inc.; avateramedical GmbH; CMR Surgical Ltd.; Johnson & Johnson; Medicaroid Corporation; Medrobotics Corporation; Medtronic plc; meerecompany Inc.; Olympus Corporation; Samsung Electronics Co., Ltd; Shandong Weigao Group Medical Polymer Company Ltd.; Shanghai Microport Medbot (Group) Co., Ltd.; and Titan Medical Inc.
Many of the above factors will also impact future demand for our Ion system, as we extend our commercial offering into diagnostics, along with additional factors associated with a new product introduction, including, but not limited to, our ability to optimize manufacturing and our supply chain, competition, clinical data to demonstrate value, and market acceptance.
Recent Product Introductions
SureForm 30 Curved-Tip Stapler and Reloads. In December 2021, we obtained FDA clearance for our 8 mm SureForm 30 Curved-Tip Stapler and reloads (gray, white, and blue) for use in general, thoracic, gynecologic, urologic, and pediatric surgery. We designed this instrument to help surgeons better visualize and reach anatomy through a combination of the 8 mm diameter instrument shaft and jaws, 120-degree cone of wristed articulation, and the curved tip. As it fits through the 8 mm da Vinci surgical system instrument cannula, the stapler allows different angles for surgeons to approach patient anatomy. Consistent with our other SureForm staplers, the 8 mm SureForm 30 Curved-Tip Stapler integrates SmartFire technology, which makes automatic adjustments to the firing process as staples are formed and the transection is made. The technology makes more than 1,000 measurements per second, helping achieve a consistent staple line. We completed initial evaluations of the 8 mm SureForm 30 stapler with certain customers in the U.S. in 2022. The full U.S. product launch will occur in late 2023, with other countries to follow. In October 2022, we received regulatory clearance in Japan to market our 8 mm SureForm 30 Curved-Tip and Straight-Tip Stapler instruments and reloads for use in general, thoracic (except for cardiac), gynecologic, and urologic surgery.
SynchroSeal and E-100 Generator. In November 2019, we obtained FDA clearance for our SynchroSeal instrument and E-100 generator. Following the FDA clearance, in February 2020, we received European certification for both products. In March 2020, we received regulatory clearance in Japan to market both our SynchroSeal instrument and E-100 generator. In August 2020, we received regulatory clearance in South Korea to market our E-100 generator. SynchroSeal is a single-use, bipolar, electrosurgical instrument intended for grasping, dissection, sealing, and transection of tissue. With its wristed articulation, rapid sealing cycle, and refined curved jaw, SynchroSeal offers enhanced versatility to the da Vinci Energy portfolio. The E-100 generator is an electrosurgical generator developed to power two key instruments–Vessel Sealer Extend and SynchroSeal–on the da Vinci X and da Vinci Xi Surgical Systems. The generator delivers high frequency energy for cutting, coagulation, and vessel sealing of tissues.
SureForm 45 Curved-Tip Stapler and Gray Reload. In July 2019, we obtained FDA clearance for the SureForm 45 Curved-Tip Stapler and SureForm 45 Gray reload. We have also obtained European certification for our SureForm 45 Curved-Tip Stapler and SureForm 45 Gray reload. In September 2019, we received regulatory clearance in Japan to market both our SureForm 45 Curved-Tip Stapler and SureForm 45 Gray reload. We received regulatory clearance in South Korea to market our SureForm 45 Curved-Tip Stapler and SureForm 45 Gray reload in June 2021 and July 2021, respectively. SureForm 45 Curved-Tip Stapler is a single-use, fully wristed stapling instrument with a curved tip intended for resection, transection, and/or creation of anastomoses. SureForm 45 Gray reload is a new, single-use cartridge that contains multiple staggered rows of implantable staples and a stainless steel knife. The SureForm 45 Curved-Tip Stapler and Gray reload have particular utility in thoracic procedures and round out our SureForm 45 portfolio. Not all reloads or staplers are available for use on all systems or in all countries.
Da Vinci Endoscope Plus. In June 2019, we obtained European certification for our da Vinci Endoscope Plus, an enhanced 3D endoscope for use with our da Vinci X and Xi Surgical Systems. Following the European certification, in July 2019, we obtained FDA clearance for our da Vinci Endoscope Plus. We have also received regulatory clearances in South Korea and Japan to market our da Vinci Endoscope Plus in December 2019 and May 2020, respectively. In March 2022, we received regulatory clearance in China to market our da Vinci Endoscope Plus. The da Vinci Endoscope Plus leverages new sensor technology to allow for increased sharpness and color accuracy.
Da Vinci Handheld Camera. In June 2019, we obtained FDA clearance for our da Vinci Handheld Camera, a lightweight, 2D camera head, which can be connected to third-party laparoscopes. This allows the laparoscopic image to be displayed on the da Vinci X/Xi vision cart to address aspects of da Vinci procedures that may require the use of a laparoscope, thus eliminating the need for redundant equipment in the operating room and increasing procedure efficiency. In February 2020, we obtained European certification for our da Vinci Handheld Camera. We broadly launched the da Vinci Handheld Camera in our European direct markets and in the U.S. in May 2020 and June 2020, respectively.
Acquisition of Orpheus Medical
In February 2020, we acquired Orpheus Medical Ltd. and its wholly owned subsidiaries to deepen and expand our integrated informatics platform. Orpheus Medical provides hospitals with information technology connectivity, as well as expertise in processing and archiving surgical videos. Orpheus Medical is a wholly owned subsidiary of Intuitive.
Intuitive Ventures
In 2020, we launched Intuitive Ventures, an inaugural $100 million fund focused on investment opportunities in companies that share Intuitive’s commitment to advancing positive outcomes in healthcare. As of December 31, 2022, we have invested $33 million of the $100 million.
2022 Operational and Financial Highlights
•Total revenue increased by 9% to $6.2 billion for the year ended December 31, 2022, compared to $5.7 billion for the year ended December 31, 2021.
•Approximately 1,875,000 da Vinci procedures were performed during the year ended December 31, 2022, an increase of 18% compared to approximately 1,594,000 da Vinci procedures for the year ended December 31, 2021.
•Approximately 23,500 Ion procedures were performed during the year ended December 31, 2022, an increase of 218% compared to approximately 7,400 Ion procedures for the year ended December 31, 2021.
•Instruments and accessories revenue increased by 13% to $3.52 billion for the year ended December 31, 2022, compared to $3.10 billion for the year ended December 31, 2021.
•Systems revenue decreased by 1% to $1.68 billion for the year ended December 31, 2022, compared to $1.69 billion for the year ended December 31, 2021.
•1,264 da Vinci Surgical Systems were placed during the year ended December 31, 2022, a decrease of 6% compared to 1,347 systems during the year ended December 31, 2021.
•As of December 31, 2022, we had a da Vinci Surgical System installed base of approximately 7,544 systems, an increase of 12% compared to the installed base of approximately 6,730 systems as of December 31, 2021.
•Utilization of da Vinci Surgical Systems, measured in terms of procedures per system per year, increased 4% relative to 2021.
•192 Ion systems were placed during the year ended December 31, 2022, an increase of 106% compared to 93 systems during the year ended December 31, 2021.
•As of December 31, 2022, we had an Ion system installed base of approximately 321 systems, an increase of 149% compared to the installed base of approximately 129 systems as of December 31, 2021.
•Gross profit as a percentage of revenue was 67.4% for the year ended December 31, 2022, compared to 69.3% for the year ended December 31, 2021.
•Operating income decreased by 13% to $1.58 billion for the year ended December 31, 2022, compared to $1.82 billion for the year ended December 31, 2021. Operating income included $517 million and $457 million of share-based compensation expense related to employee stock plans and $45.4 million and $37.0 million of intangible asset-related charges for the years ended December 31, 2022, and 2021, respectively.
•As of December 31, 2022, we had $6.74 billion in cash, cash equivalents, and investments. Cash, cash equivalents, and investments decreased by $1.88 billion, compared to $8.62 billion as of December 31, 2021, primarily as a result of cash used for share repurchases of $2.61 billion, capital expenditures, and taxes paid related to net share settlements of equity awards, as well as unrealized losses on interest-bearing debt securities classified as available for sale, partially offset by cash provided by operating activities and proceeds from stock options exercises and employee stock purchases.
Results of Operations
The following discussion should be read in conjunction with our Consolidated Financial Statements and Notes thereto. This section of the Annual Report on Form 10-K generally discusses 2022 and 2021 items and year-to-year comparisons between 2022 and 2021. Discussions of 2020 items and year-to-year comparisons between 2021 and 2020 that are not included in this report on Form 10-K can be found in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in Part II, Item 7 of the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2021.
The following table sets forth, for the years indicated, certain Consolidated Statements of Income information (in millions, except percentages):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2022 | | % of total Revenue | | 2021 | | % of total Revenue | | 2020 | | % of total Revenue |
| Revenue: | | | | | | | | | | | |
| Product | $ | 5,198.0 | | | 84 | % | | $ | 4,793.9 | | | 84 | % | | $ | 3,634.6 | | | 83 | % |
| Service | 1,024.2 | | | 16 | % | | 916.2 | | | 16 | % | | 723.8 | | | 17 | % |
| Total revenue | 6,222.2 | | | 100 | % | | 5,710.1 | | | 100 | % | | 4,358.4 | | | 100 | % |
| Cost of revenue: | | | | | | | | | | | |
| Product | 1,700.3 | | | 28 | % | | 1,464.1 | | | 26 | % | | 1,230.3 | | | 28 | % |
| Service | 325.9 | | | 5 | % | | 287.5 | | | 5 | % | | 266.9 | | | 6 | % |
| Total cost of revenue | 2,026.2 | | | 33 | % | | 1,751.6 | | | 31 | % | | 1,497.2 | | | 34 | % |
| Product gross profit | 3,497.7 | | | 56 | % | | 3,329.8 | | | 58 | % | | 2,404.3 | | | 55 | % |
| Service gross profit | 698.3 | | | 11 | % | | 628.7 | | | 11 | % | | 456.9 | | | 11 | % |
| Gross profit | 4,196.0 | | | 67 | % | | 3,958.5 | | | 69 | % | | 2,861.2 | | | 66 | % |
| Operating expenses: | | | | | | | | | | | |
| Selling, general and administrative | 1,739.9 | | | 28 | % | | 1,466.5 | | | 25 | % | | 1,216.3 | | | 28 | % |
| Research and development | 879.0 | | | 14 | % | | 671.0 | | | 12 | % | | 595.1 | | | 14 | % |
| Total operating expenses | 2,618.9 | | | 42 | % | | 2,137.5 | | | 37 | % | | 1,811.4 | | | 42 | % |
| Income from operations | 1,577.1 | | | 25 | % | | 1,821.0 | | | 32 | % | | 1,049.8 | | | 24 | % |
| Interest and other income, net | 29.7 | | | 1 | % | | 69.3 | | | 1 | % | | 157.2 | | | 4 | % |
| Income before taxes | 1,606.8 | | | 26 | % | | 1,890.3 | | | 33 | % | | 1,207.0 | | | 28 | % |
| Income tax expense | 262.4 | | | 4 | % | | 162.2 | | | 3 | % | | 140.2 | | | 3 | % |
| Net income | 1,344.4 | | | 22 | % | | 1,728.1 | | | 30 | % | | 1,066.8 | | | 24 | % |
| Less: net income attributable to noncontrolling interest in joint venture | 22.1 | | | 1 | % | | 23.5 | | | — | % | | 6.2 | | | — | % |
| Net income attributable to Intuitive Surgical, Inc. | $ | 1,322.3 | | | 21 | % | | $ | 1,704.6 | | | 30 | % | | $ | 1,060.6 | | | 24 | % |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Total Revenue
Total revenue increased by 9% to $6.2 billion for the year ended December 31, 2022, compared to $5.7 billion for the year ended December 31, 2021. Total revenue for the year ended December 31, 2021, increased by 31% compared to $4.4 billion for the year ended December 31, 2020. The increase in total revenue for the year ended December 31, 2022, resulted from 13% higher instruments and accessories revenue, driven by approximately 18% higher da Vinci procedure volume, partially offset by foreign currency impacts and customer buying patterns, and 12% higher service revenue, partially offset by 1% lower systems revenue, driven by 6% lower da Vinci system placements, partially offset by higher operating lease revenue. In conjunction with our 2020 COVID-19 Customer Relief Program implemented in the second quarter of 2020, service revenue in 2020 was reduced by $80 million as a result of service fee credits provided to customers.
Revenue denominated in foreign currencies as a percentage of total revenue was approximately 24%, 23%, and 23% for the years ended December 31, 2022, 2021, and 2020, respectively. We generally sell our products and services in local currencies where we have direct distribution channels. Foreign currency rate fluctuations, as determined by comparing current period revenue in USD to current period revenue in local currency using the same foreign exchange rates as the prior year same period, net of the impacts from foreign currency hedging, had an unfavorable impact on OUS total revenue of $138 million for the year
ended December 31, 2022, as compared to 2021. Foreign currency rate fluctuations, net of the impacts from foreign currency hedging, had a favorable impact on OUS total revenue of $35 million for the year ended December 31, 2021, as compared to 2020.
Revenue generated in the U.S. accounted for 67%, 67%, and 68% of total revenue for the years ended December 31, 2022, 2021, and 2020, respectively. We believe that U.S. revenue has accounted for the large majority of total revenue due to U.S. patients’ ability to choose their provider and method of treatment, reimbursement structures supportive of innovation and MIS, and our initial investments focused on U.S. infrastructure. We have been investing in our business in OUS markets, and our OUS procedures have grown faster in proportion to U.S. procedures. We expect that our OUS procedures and revenue will make up a greater portion of our business in the long term.
The following table summarizes our revenue and system unit placements for the years ended December 31, 2022, 2021, and 2020, respectively (in millions, except percentages and unit placements):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2022 | | 2021 | | 2020 |
| Revenue | | | | | |
| Instruments and accessories | $ | 3,517.9 | | | $ | 3,100.5 | | | $ | 2,455.7 | |
| Systems | 1,680.1 | | | 1,693.4 | | | 1,178.9 | |
| Total product revenue | 5,198.0 | | | 4,793.9 | | | 3,634.6 | |
| Services | 1,024.2 | | | 916.2 | | | 723.8 | |
| Total revenue | $ | 6,222.2 | | | $ | 5,710.1 | | | $ | 4,358.4 | |
| U.S. | $ | 4,157.6 | | | $ | 3,853.2 | | | $ | 2,962.7 | |
| OUS | 2,064.6 | | | 1,856.9 | | | 1,395.7 | |
| Total revenue | $ | 6,222.2 | | | $ | 5,710.1 | | | $ | 4,358.4 | |
| % of Revenue — U.S. | 67% | | 67% | | 68% |
| % of Revenue — OUS | 33% | | 33% | | 32% |
| | | | | |
| Instruments and accessories | $ | 3,517.9 | | | $ | 3,100.5 | | | $ | 2,455.7 | |
| Services | 1,024.2 | | | 916.2 | | | 723.8 | |
| Operating lease revenue | 376.5 | | | 276.9 | | | 176.7 | |
| Total recurring revenue | $ | 4,918.6 | | | $ | 4,293.6 | | | $ | 3,356.2 | |
| % of Total revenue | 79% | | 75% | | 77% |
| | | | | |
| Da Vinci Surgical System Placements by Region | | | | | |
| U.S. unit placements | 692 | | | 865 | | | 600 | |
| OUS unit placements | 572 | | | 482 | | | 336 | |
| Total unit placements* | 1,264 | | | 1,347 | | | 936 | |
| *Systems placed under operating leases (included in total unit placements) | 492 | | | 517 | | | 317 | |
| | | | | |
| Da Vinci Surgical System Placements involving System Trade-ins | | | | | |
| Unit placements involving trade-ins | 345 | | | 510 | | | 447 | |
| Unit placements not involving trade-ins | 919 | | | 837 | | | 489 | |
| | | | | |
Ion System Placements** | 192 | | | 93 | | | 26 | |
| **Systems placed under operating leases (included in total unit placements) | 101 | | | 50 | | | 9 | |
Product Revenue
Product revenue increased by 8% to $5.20 billion for the year ended December 31, 2022, compared to $4.79 billion for the year ended December 31, 2021. Product revenue for the year ended December 31, 2021, increased by 32% compared to $3.63 billion for the year ended December 31, 2020.
Instruments and accessories revenue increased by 13% to $3.52 billion for the year ended December 31, 2022, compared to $3.10 billion for the year ended December 31, 2021. The increase in instruments and accessories revenue was primarily driven by da Vinci procedure growth of approximately 18% and incremental sales of our advanced instruments. The increase was partially offset by foreign currency impacts, customer buying patterns, and hospitals normalizing their purchasing of new Extended Use Instruments as a result of the recent launch of our Extended Use Program. The 2022 U.S. da Vinci procedure growth was approximately 16%, driven by strong growth in general surgery procedures, most notably hernia repair, cholecystectomy, and bariatric procedures, as well as moderate growth in the more mature gynecologic and urologic procedure categories. The 2022 OUS da Vinci procedure growth was approximately 22%, driven by continued growth in urologic procedures, including prostatectomies and partial nephrectomies, and earlier stage growth in general surgery (particularly colorectal), gynecologic, and thoracic procedures. Both growth rates were impacted by the disruption caused by the COVID-19 pandemic, as noted in the COVID-19 Pandemic section above. Geographically, the 2022 OUS da Vinci procedure growth was driven by procedure expansion in a number of markets with particular strength in Japan, Germany, and the UK. We also saw procedure growth in China, despite the procedure growth rate being lower than the prior year as a result of an increase in COVID-19 cases, particularly in the fourth quarter of 2022.
Systems revenue decreased by 1% to $1.68 billion for the year ended December 31, 2022, compared to $1.69 billion for the year ended December 31, 2021. The decrease in systems revenue was primarily driven by lower sales-type lease revenue, fewer da Vinci system placements, lower 2022 da Vinci ASPs, lower lease buyout revenue, and a higher proportion of da Vinci system placements under operating leases, partially offset by higher operating lease revenue and more Ion system placements.
During 2022, 1,264 da Vinci Surgical Systems were placed compared to 1,347 systems during 2021. By geography, 692 systems were placed in the U.S., 280 in Europe, 244 in Asia, and 48 in other markets during 2022, compared to 865 systems placed in the U.S., 232 in Europe, 203 in Asia, and 47 in other markets during 2021. The decrease in system placements was primarily driven by a smaller number of third generation da Vinci systems available for trade-in along with the macroeconomic challenges impacting our customers, primarily in the U.S. Nevertheless, the incremental system placements reflect continued procedure growth and further customer validation that robotic-assisted surgery addresses their quadruple aim objectives. As of December 31, 2022, we had a da Vinci Surgical System installed base of approximately 7,544 systems, compared to an installed base of approximately 6,730 systems as of December 31, 2021.
We placed 591 and 668 da Vinci Surgical Systems under lease or usage-based arrangements, of which 492 and 517 systems were classified as operating leases for the years ended December 31, 2022, and 2021, respectively. Operating lease revenue, including the contribution from Ion systems, was $377 million for the year ended December 31, 2022, compared to $277 million for the year ended December 31, 2021. Da Vinci Surgical Systems placed as operating leases represented 39% of total placements during 2022, compared to 38% during 2021. 1,683 da Vinci Surgical Systems were installed at customers under operating lease or usage-based arrangements as of December 31, 2022, compared to 1,294 systems as of December 31, 2021. Revenue from Lease Buyouts was $72 million for the year ended December 31, 2022, compared to $96 million for the year ended December 31, 2021. We expect revenue from Lease Buyouts to fluctuate from period to period depending on the timing of when, and if, customers choose to exercise the buyout options embedded in their leases.
The da Vinci Surgical System ASP, excluding systems placed under operating lease or usage-based arrangements and Ion systems, was approximately $1.49 million for the year ended December 31, 2022, compared to approximately $1.55 million for the year ended December 31, 2021. The lower 2022 ASP was largely driven by unfavorable foreign currency impacts, unfavorable geographic mix, and higher pricing discounts, partially offset by favorable product mix and fewer trade-ins. ASP fluctuates from period to period based on geographic and product mix, product pricing, systems placed involving trade-ins, and changes in foreign exchange rates.
During 2022, 192 Ion systems were placed compared to 93 systems during 2021. We placed 112 and 57 Ion systems under lease or usage-based arrangements, of which 101 and 50 systems were classified as operating leases for the years ended December 31, 2022, and 2021, respectively. Ion systems placed as operating leases represented 53% of total placements during 2022, compared to 54% during 2021.
Service Revenue
Service revenue increased by 12% to $1.02 billion for the year ended December 31, 2022, compared to $0.92 billion for the year ended December 31, 2021. Service revenue increased by 27% for the year ended December 31, 2021, compared to $0.72 billion for the year ended December 31, 2020. The increase in service revenue in 2022 was primarily driven by a larger installed base of da Vinci Surgical Systems producing service revenue, partially offset by foreign currency impacts. The increase in service revenue in 2021 was primarily driven by a larger installed base of da Vinci Surgical Systems producing service revenue, as well as the effects of the Customer Relief Program in the prior year, which resulted in an $80 million decrease in service revenue in 2020 as a result of service fee credits provided to customers.
Gross Profit
Product gross profit for the year ended December 31, 2022, increased by 5% to $3.50 billion, representing 67.3% of product revenue, compared to $3.33 billion, representing 69.5% of product revenue, for the year ended December 31, 2021. The higher product gross profit for the year ended December 31, 2022, was primarily driven by higher product revenue, partially offset by lower product gross profit margin. The lower product gross profit margin for the year ended December 31, 2022, was primarily driven by higher fixed overhead costs from investments to drive the growth of the business and strengthen our operating capabilities, higher freight and materials costs, unfavorable foreign currency impacts, and lower 2022 da Vinci system ASPs.
Product gross profit for the years ended December 31, 2022, and 2021, included share-based compensation expense of $67.6 million and $68.9 million, respectively, and intangible assets amortization expense of $17.3 million and $17.6 million, respectively.
Service gross profit for the year ended December 31, 2022, increased by 11% to $0.70 billion, representing 68.2% of service revenue, compared to $0.63 billion, representing 68.6% of service revenue, for the year ended December 31, 2021. The higher service gross profit for the year ended December 31, 2022, was primarily driven by higher service revenue, reflecting a larger installed base of da Vinci Surgical Systems, partially offset by a lower service gross profit margin. The lower service gross profit margin for the year ended December 31, 2022, was primarily driven by higher freight and overhead costs, as well as unfavorable foreign currency impacts, partially offset by lower materials costs for repairs.
Service gross profit for the years ended December 31, 2022, and 2021, included share-based compensation expense of $23.6 million and $22.2 million, respectively, and intangible assets amortization expense of $1.9 million and $1.0 million, respectively.
Selling, General and Administrative Expenses
Selling, general and administrative expenses include costs for sales, marketing and administrative personnel, sales and marketing activities, trade show expenses, legal expenses, regulatory fees, and general corporate expenses.
Selling, general and administrative expenses for the year ended December 31, 2022, increased by 19% to $1.74 billion, compared to $1.47 billion for the year ended December 31, 2021. The increase in selling, general and administrative expenses for the year ended December 31, 2022, was primarily driven by higher headcount, resulting in increased fixed and share-based compensation expense, higher variable compensation, increased infrastructure costs to support our growth, and higher litigation charges, partially offset by favorable foreign currency impacts. In addition, there were higher travel, training, and marketing expenses for the year ended December 31, 2022, as compared with the prior year. In the fourth quarter of 2021, we made a charitable contribution of $30 million to the Intuitive Foundation, a not-for-profit organization whose mission is to reduce the global burden of disease and suffering through research, education, and philanthropy aimed at better outcomes for patients around the globe.
Selling, general and administrative expenses for the years ended December 31, 2022, and 2021, included share-based compensation expense of $261 million and $232 million, respectively, and intangible assets amortization expense of $5.9 million and $7.3 million, respectively.
Research and Development Expenses
Research and development costs are expensed as incurred. Research and development expenses include costs associated with the design, development, testing, and significant enhancement of our products.
Research and development expenses for the year ended December 31, 2022, increased by 31% to $0.88 billion, compared to $0.67 billion for the year ended December 31, 2021. The increase in research and development expenses for the year ended December 31, 2022, was primarily driven by higher personnel-related expenses, including share-based compensation expense, project costs incurred to support a broader set of product development initiatives, including future generations of robotics, Ion and SP platform investments, and digital investments, and intangible asset-related charges.
Research and development expenses for the years ended December 31, 2022, and 2021, included share-based compensation expense of $164 million and $134 million, respectively, and intangible asset-related charges of $20.3 million and $11.1 million, respectively.
Research and development expenses fluctuate with project timing. Based upon our broader set of product development initiatives and the stage of the underlying projects, we expect to continue to make substantial investments in research and development and anticipate that research and development expenses will continue to increase in the future.
Interest and Other Income, Net
Interest and other income, net, for the year ended December 31, 2022, decreased by 57% to $30 million, compared to $69 million for the year ended December 31, 2021. Interest and other income, net decreased by 56% for the year ended December 31, 2021, compared to $157 million for the year ended December 31, 2020. The decrease in interest and other income, net, for the year ended December 31, 2022, was primarily driven by unrealized losses on investments resulting from strategic arrangements (compared to unrealized gains on investments resulting from strategic arrangements in 2021) and higher foreign exchange losses, partially offset by higher interest income earned, despite lower cash and investment balances, due to an increase in average interest rates.
We held an equity investment in preferred shares of Broncus Holding Corporation (“Broncus”), which was reflected in our Consolidated Financial Statements on a cost basis. In the first quarter of 2021, we recorded an unrealized gain on our investment in Broncus of approximately $14 million. In September 2021, Broncus completed an initial public offering (“IPO”) of common shares on the Stock Exchange of Hong Kong. Upon completion of the IPO, the preferred shares were converted to common shares in Broncus, and we recognized a net gain on this investment in the third quarter of 2021 of approximately $8 million. We were restricted from selling these shares for a period of six months. For the year ended December 31, 2022, we recognized a loss on this investment of approximately $21 million.
We held an equity investment in common shares of Bolder Surgical Holdings, Inc. (“Bolder”), which was reflected in our Consolidated Financial Statements on a cost basis. During the fourth quarter of 2021, Hologic, Inc., a publicly traded company, completed its acquisition of Bolder. Under the terms of the acquisition agreement, we received cash on the date of closing and recognized a gain on this investment of approximately $10 million.
We held an equity investment in preferred shares of InTouch Technologies, Inc. (“InTouch”), which was reflected in our Consolidated Financial Statements on a cost basis. On July 1, 2020, Teladoc Health, Inc. (“Teladoc”), a publicly traded company, completed its acquisition of InTouch. Based on the terms of the agreement, we received Teladoc shares on the date of closing and recognized a gain on our investment of approximately $45 million. We were restricted from selling these shares for a period of six months. In January 2021, we sold all of our shares in Teladoc and recognized a gain on this investment of approximately $11 million. This gain was offset by a $7.5 million loss recognized upon the settlement of a corresponding derivative collar contract in January 2021.
Additionally, the Company recorded unrealized gains on other strategic investments in 2020 of approximately $22 million.
Income Tax Expense
Income tax expense was $262 million and $162 million for the years ended December 31, 2022, and 2021, respectively. Our effective tax rate for 2022 was approximately 16.3% compared to 8.6% for 2021.
Our effective tax rates for 2022 and 2021 differed from the U.S. federal statutory rate of 21% primarily due to the excess tax benefits associated with employee equity plans, the effect of income earned by certain overseas entities being taxed at rates lower than the federal statutory rate, and the federal research and development credit benefit, partially offset by U.S. tax on foreign earnings and state income taxes (net of federal benefit).
The provision for income taxes for the year ended December 31, 2022, reflected the impact of a change in U.S. tax law effective January 1, 2022, which requires the capitalization and amortization of research and development expenditures incurred after December 31, 2021.
The increase in income tax expense for the year ended December 31, 2022, was primarily due to the impact of the capitalization of research and development and lower excess tax benefits, as discussed below, as well as the fact that income tax expense for the year ended December 31, 2021, included a one-time benefit of $66.4 million from the re-measurement of our Swiss deferred tax assets resulting from the extension of the economic useful life of certain intangible assets. The increase was partially offset by a one-time charge of $13.6 million in 2021, which related to intercompany charges for share-based compensation for relevant periods prior to 2020, triggered by additional Internal Revenue Service (“IRS”) guidance issued in July 2021 associated with a Ninth Circuit Court of Appeals opinion involving an independent third party.
Our provision for income taxes for 2022 and 2021 included excess tax benefits associated with employee equity plans of $99 million and $186 million, respectively, which reduced our effective tax rate by 6.1 and 9.8 percentage points, respectively. The amount of excess tax benefits or deficiencies will fluctuate from period to period based on the price of our stock, the volume of share-based awards settled or vested, and the value assigned to employee equity awards under GAAP, which results in increased income tax expense volatility.
On August 16, 2022, the Inflation Reduction Act (“IRA”) was enacted in the United States. The IRA introduces a 15% alternative minimum tax based on the financial statement income of certain large corporations, effective for tax years beginning after December 31, 2022. The IRA also includes a 1% excise tax on the net fair market value of stock repurchases made after
December 31, 2022. We have considered the applicable tax law changes, and there is no impact on our tax provision for the year ended December 31, 2022. We will continue to evaluate the impact of these tax law changes on future periods.
We file federal, state, and foreign income tax returns in many jurisdictions in the U.S. and abroad. Years prior to 2016 are considered closed for most significant jurisdictions. Certain of our unrecognized tax benefits could change due to activities of various tax authorities, including evolving interpretations of existing tax laws in the jurisdictions we operate, potential assessment of additional tax, possible settlement of audits, or through normal expiration of various statutes of limitations, which could affect our effective tax rate in the period in which they change. Due to the uncertainty related to the timing and potential outcome of audits, we cannot estimate the range of reasonably possible changes in unrecognized tax benefits that may occur in the next 12 months.
We are subject to the examination of our income tax returns by the IRS and other tax authorities. The outcome of these audits cannot be predicted with certainty. Management regularly assesses the likelihood of adverse outcomes resulting from these examinations to determine the adequacy of our provision for income taxes. If any issues addressed in our tax audits are resolved in a manner not consistent with management’s expectations, we could be required to adjust our provision for income taxes in the period such resolution occurs.
Net Income Attributable to Noncontrolling Interest in Joint Venture
The Company’s Joint Venture with Fosun Pharma was established to research, develop, manufacture, and sell robotic-assisted, catheter-based medical devices. The Joint Venture is owned 60% by us and 40% by Fosun Pharma and is located in China. The catheter-based technology will initially target early diagnosis and cost-effective treatment of lung cancer, one of the most commonly diagnosed forms of cancer in the world. Distribution of catheter-based medical devices in China will be conducted by the Joint Venture, while distribution outside of China will be conducted by us.
In January 2019, the Joint Venture acquired certain assets, including distribution rights, customer relationships, and certain personnel, from Chindex and its affiliates, a subsidiary of Fosun Pharma, and began direct operations for da Vinci products and services in China. As of December 31, 2022, the companies have contributed $55 million of up to $100 million required by the joint venture agreement.
Net income attributable to noncontrolling interest in Joint Venture for the year ended December 31, 2022, was $22.1 million, compared to $23.5 million for the year ended December 31, 2021, and $6.2 million for the year ended December 31, 2020. The decrease in net income attributable to noncontrolling interest in Joint Venture for the year ended December 31, 2022, was primarily due to a decrease in sales and an increase in selling, general and administrative expenses in China, partially offset by a decrease in income tax expense in China.
Liquidity and Capital Resources
Sources and Uses of Cash and Cash Equivalents
Our principal source of liquidity is cash provided by operations and by the issuance of common stock through the exercise of stock options and our employee stock purchase program. Cash and cash equivalents plus short- and long-term investments decreased by $1.88 billion to $6.74 billion as of December 31, 2022, from $8.62 billion as of December 31, 2021, primarily from cash used in share repurchases, capital expenditures, and taxes paid related to net share settlements of equity awards, as well as unrealized losses on interest-bearing debt securities classified as available for sale, offset by cash provided by our operations and proceeds from stock option exercises and employee stock purchases. Cash and cash equivalents plus short- and long-term investments increased by $1.75 billion to $8.62 billion as of December 31, 2021, from $6.87 billion as of December 31, 2020, primarily from cash provided by our operations and proceeds from stock option exercises and employee stock purchases, partially offset by capital expenditures and taxes paid related to net share settlements of equity awards.
Our cash requirements depend on numerous factors, including market acceptance of our products, the resources we devote to developing and supporting our products, and other factors. We expect to continue to devote substantial resources to expand procedure adoption and acceptance of our products. We have made substantial investments in our commercial operations, product development activities, facilities, and intellectual property. Based on our business model, we anticipate that we will continue to be able to fund future growth through cash provided by our operations. We believe that our current cash, cash equivalents, and investment balances, together with income to be derived from the sale of our products, will be sufficient to meet our liquidity requirements for the foreseeable future. However, we may experience reduced cash flow from operations as a result of the increasing risk of a recession along with other macroeconomic and geopolitical headwinds.
As of December 31, 2022, $396 million of our cash, cash equivalents, and investments was held by foreign subsidiaries. We intend to repatriate earnings from our Swiss subsidiary and our joint venture in Hong Kong, as needed, since the U.S. and foreign tax implications of such repatriations are not expected to be significant. We will continue to indefinitely reinvest earnings from the rest of our foreign subsidiaries, which are not significant.
See “Item 7A. Quantitative and Qualitative Disclosures About Market Risk” for discussion on the impact of interest rate risk and market risk on our investment portfolio.
Consolidated Cash Flow Data
The following table summarizes our cash flows for the years ended December 31, 2022, 2021, and 2020 (in millions):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2022 | | 2021 | | 2020 |
| | | | | |
| Net cash provided by (used in): | | | | | |
| Operating activities | $ | 1,490.8 | | | $ | 2,089.4 | | | $ | 1,484.8 | |
| Investing activities | 1,370.8 | | | (2,461.5) | | | (940.6) | |
| Financing activities | (2,572.3) | | | 43.0 | | | (85.7) | |
| Effect of exchange rates on cash, cash equivalents, and restricted cash | 5.4 | | | (3.4) | | | (2.6) | |
| Net increase (decrease) in cash, cash equivalents, and restricted cash | $ | 294.7 | | | $ | (332.5) | | | $ | 455.9 | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
Operating Activities
For the year ended December 31, 2022, net cash provided by operating activities of $1.49 billion exceeded our net income of $1.34 billion, primarily due to the following factors:
1.Our net income included non-cash charges of $766 million, consisting primarily of the following significant items: share-based compensation of $513 million; depreciation expense and losses on the disposal of property, plant, and equipment of $338 million; changes in deferred income taxes of $(185) million; and net losses on investments, accretion, and amortization of $49 million.
2.The non-cash charges outlined above were partially offset by changes in operating assets and liabilities that resulted in $619 million of cash used in operating activities during the year ended December 31, 2022. Inventory, including the transfer of equipment from inventory to property, plant, and equipment, increased by $547 million, primarily to address the growth in the business as well as to mitigate risks of disruption that could arise from trade, supply, or other matters. Refer to further details in the supplemental cash flow information in Note 4 to the Consolidated Financial Statements. Accounts receivable increased by $159 million, primarily due to the timing of billings and collections. Prepaid expenses and other assets increased by $129 million, primarily due to an increase in net investments in sales-type leases and an increase in recoverable VAT related to growth in our OUS activities. The unfavorable impact of these items on cash provided by operating activities was partially offset by a $122 million increase in other liabilities, primarily due to additional accruals related to capital expenditures and timing of income tax payments, a $52 million increase in accrued compensation and employee benefits, primarily due to higher headcount and variable compensation, and a $21 million increase in accounts payable, primarily due to the timing of billing and payments.
For the year ended December 31, 2021, net cash provided by operating activities of $2.09 billion exceeded our net income of $1.73 billion, primarily due to the following factors:
1.Our net income included non-cash charges of $729 million, consisting primarily of the following significant items: share-based compensation of $449 million; depreciation expense and losses on the disposal of property, plant, and equipment of $283 million; changes in deferred income taxes of $(63) million; and amortization of intangible assets of $27 million.
2.The non-cash charges outlined above were partially offset by changes in operating assets and liabilities that resulted in $368 million of cash used in operating activities during the year ended December 31, 2021. Inventory, including the transfer of equipment from inventory to property, plant, and equipment, increased by $256 million, primarily to address the growth in the business as well as to mitigate risks of disruption that could arise from trade, supply, or other matters. Refer to further details in the supplemental cash flow information in Note 4 to the Consolidated Financial Statements. Prepaid expenses and other assets increased by $205 million, primarily due to an increase in net investments in sales-type leases, and accounts receivable increased by $142 million, primarily due to the timing of billings and collections. The unfavorable impact of these items on cash used in operating activities was partially offset by a $115 million increase in accrued compensation and employee benefits, primarily due to higher headcount and variable compensation, a $51 million increase in other liabilities, primarily due to additional accruals related to capital expenditures and timing of income tax payments, a $36 million increase in accounts payable, primarily due to the timing of payments and vendor billings, and a $33 million increase in deferred revenue, primarily due to increased volume of sales contracts.
Investing Activities
Net cash provided by investing activities for the year ended December 31, 2022, consisted primarily of proceeds from maturities and sales of investments, net of purchases, of $1.92 billion, partially offset by $532 million paid for the acquisition of property, plant, and equipment.
Net cash used in investing activities for the year ended December 31, 2021, consisted primarily of purchases of investments, net of proceeds from maturities and sales, of $2.10 billion and $340 million paid for the acquisition of property, plant, and equipment.
Net cash used in investing activities for the year ended December 31, 2020, consisted of purchases of investments, net of proceeds from maturities and sales, of $561 million, $342 million paid for the acquisition of property, plant, and equipment, and $38 million paid for the Orpheus Medical acquisition, net of cash acquired.
We invest predominantly in high quality, fixed income securities. Our investment portfolio may, at any time, contain investments in U.S. treasury and U.S. government agency securities, taxable and tax-exempt municipal notes, corporate notes and bonds, commercial paper, non-U.S. government agency securities, cash deposits, and money market funds.
Financing Activities
Net cash used in financing activities for the year ended December 31, 2022, consisted primarily of cash used in the repurchase of approximately 11.2 million shares of our common stock for $2.61 billion, 8.5 million shares of which related to accelerated share buyback programs executed and settled during 2022 and further described in Note 9 to the Consolidated Financial Statements, and taxes paid on behalf of employees related to net share settlements of vested employee equity awards of $194 million, partially offset by proceeds from stock option exercises and employee stock purchases of $234 million.
Net cash provided by financing activities for the year ended December 31, 2021, consisted primarily of proceeds from stock option exercises and employee stock purchases of $277 million, partially offset by taxes paid on behalf of employees related to net share settlements of vested employee equity awards of $212 million, and the payment of deferred purchase consideration of $22 million.
Net cash used in financing activities for the year ended December 31, 2020, consisted primarily of taxes paid on behalf of employees related to net share settlements of vested employee equity awards of $175 million, cash used in the repurchase of approximately 0.7 million shares of our common stock in the open market for $134 million, and the payment of deferred purchase consideration of $85 million, partially offset by proceeds from stock option exercises and employee stock purchases of $309 million.
Capital Expenditures
Our capital expenditures are increasing as we continue to build the Company to supply our customers with highly differentiated products manufactured in highly automated factories that facilitate outstanding performance in product quality, availability, and cost. A significant portion of this investment involves the construction of facilities to expand our manufacturing and commercial capabilities. We have also been vertically integrating key technologies to develop a more robust supply chain and bring important products to market at attractive price points. These investments include increased ownership of our imaging pipelines, investments in strategic instruments and accessories technologies, as well as the development of software and digital products that allow us to serve our customers better. We expect these capital investments to increase significantly in 2023 to a range between $800 million and $1 billion, over half of which will be facilities-related investments. We intend to fund these capital investments with cash generated from operations.
Contractual Obligations and Commercial Commitments
Operating leases. We lease spaces for our operations in the U.S. as well as in Japan, China, Israel, Mexico, Germany, South Korea, the United Kingdom, and other countries. We also lease automobiles for certain sales and field service employees. These leases have varying terms of up to 20 years. Operating lease amounts include future minimum lease payments under all of our non-cancellable operating leases with an initial term in excess of one year. Refer to Note 6 to the Consolidated Financial Statements included in Part II, Item 8 for further details.
Purchase commitments and obligations. Total purchase commitments and obligations as of December 31, 2022, are estimated to be $2.14 billion, of which $1.79 billion is expected to be due within a year. These amounts include an estimate of all open purchase orders and contractual obligations in the ordinary course of business, including commitments with contract manufacturers and suppliers for which we have not received the goods or services, commitments for capital expenditures, including construction-related activities, for which we have not received the goods or services, and commitments for the acquisition and licensing of intellectual property. More than one third of our estimated purchase commitments and obligations are facilities-related. Although open purchase orders are considered enforceable and legally binding, the terms generally allow us the option to cancel, reschedule, or adjust our requirements based on our business needs prior to the delivery of goods or
performance of services. In addition to the above, we have committed to making potential future milestone payments to third parties as part of licensing, collaboration, and development arrangements. Payments under these agreements generally become due and payable only upon achievement of certain developmental, regulatory, and/or commercial milestones. For instances in which the achievement of these milestones is neither probable nor reasonably estimable, such contingencies have not been recorded on our Consolidated Balance Sheets.
2017 Tax Act deemed repatriation tax. As of December 31, 2022, our obligation associated with the deemed repatriation tax is $161 million, of which $40 million is due within a year. Amounts due are expected to be paid in installments in accordance with the 2017 Tax Act.
We are unable to make a reasonably reliable estimate as to when payments may occur for our unrecognized tax benefits.
Off-Balance Sheet Arrangements
As of December 31, 2022, we did not have any significant off-balance sheet arrangements, as defined in Item 303(a)(4)(ii) of Regulation S-K promulgated under the Exchange Act.
Critical Accounting Estimates
Our Consolidated Financial Statements are prepared in conformity with GAAP, which requires us to make judgments, estimates, and assumptions. See “Note 2. Summary of Significant Accounting Policies,” in Notes to the Consolidated Financial Statements, which is included in “Item 8. Financial Statements and Supplementary Data,” for a description of our significant accounting policies and methods used in the preparation of our Consolidated Financial Statements. The methods, estimates, and judgments that we use in applying our accounting policies require us to make difficult and subjective judgments, often as a result of the need to make estimates regarding matters that are inherently uncertain. Our most critical accounting estimates include:
•the valuation and recognition of investments, which impacts our investment portfolio balance when we assess fair value and interest and other income, net, when we record impairments;
•the standalone selling prices used to allocate the contract consideration to the individual performance obligations, which impacts revenue recognition;
•the valuation of inventory, which impacts gross profit margins;
•the valuation of and assessment of the recoverability of intangible assets and goodwill and the estimated useful lives of intangible assets, which primarily impacts gross profit margin or operating expenses when we record asset impairments or accelerate their amortization;
•the recognition and measurement of current and deferred income taxes (including the measurement of uncertain tax positions), which impact our provision for taxes; and
•the estimate of probable loss associated with legal contingencies, which impacts accrued liabilities and operating expenses.
Investments Valuation
Fair Value. Our investments may include, at any time, a diversified portfolio of cash equivalents and short- and long-term investments in a variety of high-quality securities, including money market funds, U.S. treasury and U.S. government agency securities, corporate notes and bonds, commercial paper, non-U.S. government agency securities, and municipal notes, as well as equity investments with and without readily determinable value. The assessment of the fair value of investments can be difficult and subjective. GAAP establishes three levels of inputs that may be used to measure fair value. Each level of input has different levels of subjectivity and difficulty involved in determining fair value. Valuation of Level 1 and 2 instruments generally do not require significant management judgment, and the estimation is not difficult. Level 3 instruments include unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. The determination of fair value for Level 3 instruments requires the most management judgment and subjectivity. There were no Level 3 securities for the periods presented.
After determining the fair value of our available-for-sale instruments, we identify instruments with an amortized cost basis in excess of their estimated fair value. Available-for-sale instruments in an unrealized loss position are written down to fair value through a charge to interest and other income, net in the Consolidated Statements of Income, if we intend to sell the security or it is more likely than not we will be required to sell the security before recovery of its amortized cost basis. For the remaining securities, we assess what amount of the excess, if any, is caused by expected credit losses. Factors considered in determining whether a credit-related loss exists include the financial condition and near-term prospects of the investee, the extent of the loss related to the credit of the issuer, and the expected cash flows from the security. These judgments could prove to be wrong, and companies with relatively high credit ratings and solid financial conditions may not be able to fulfill their obligations.
No significant impairment charges were recorded during the years ended December 31, 2022, 2021, and 2020. As of December 31, 2022, and 2021, net unrealized losses on investments of $154.2 million and $16.0 million, net of tax, respectively, were included in accumulated other comprehensive loss.
Revenue recognition. Our system sale arrangements contain multiple products and services, including system(s), system components, system accessories, instruments, accessories, and services. Other than services, we generally deliver all of the products upfront. Each of these products and services is a distinct performance obligation. System accessories, instruments, accessories, and services are also sold on a standalone basis.
For multiple-element arrangements, revenue is allocated to each performance obligation based on its relative standalone selling price. Standalone selling prices are based on observable prices at which we separately sell the products or services. If a standalone selling price is not directly observable, then we estimate the standalone selling prices considering market conditions and entity-specific factors including, but not limited to, features and functionality of the products and services, geographies, type of customer, and market conditions. We regularly review standalone selling prices and maintain internal controls over establishing and updating these estimates.
Our system sales arrangements generally include a five-year period of service. The first year of service is generally free and included in the system sale arrangement, and the remaining four years are billed at a stated service price. Revenue that is allocated to the service obligation is deferred and recognized ratably over the service period.
Inventory valuation. Inventory is stated at the lower of cost or net realizable value on a first-in, first-out basis. The cost basis of our inventory is reduced for any products that are considered excess or obsolete based on assumptions about future demand and market conditions. If actual future demand or market conditions are less favorable than those projected by management, additional inventory write-downs may be required, which could have a material adverse effect on the results of our operations.
Valuation of intangible assets and goodwill. We allocate the fair value of purchase consideration, including contingent consideration, to assets acquired and liabilities assumed in a business combination based on their estimated fair values at the acquisition date. The excess of the fair value of the purchase consideration over the fair value of assets acquired, liabilities assumed, and any noncontrolling interest is recorded as goodwill. When determining the fair value of assets acquired, liabilities assumed, and any noncontrolling interest, management is required to make certain estimates and assumptions, especially with respect to intangible assets. The estimates and assumptions used in valuing intangible assets include, but are not limited to, the amount and timing of projected future cash flows, the discount rate used to determine the present value of these cash flows, and the determination of the assets’ life cycle. These estimates are inherently uncertain and, therefore, actual results may differ from the estimates made.
Our intangible assets include identifiable intangible assets and goodwill. Identifiable intangible assets include developed technology, patents, distribution rights, customer relationships, licenses, and non-competition arrangements. Currently, all of our identifiable intangible assets have finite lives. Goodwill and intangible assets with indefinite lives are subject to an annual impairment review (or more frequent if impairment indicators arise) by applying a fair value-based test. There have been no such impairments.
Identifiable intangible assets with finite lives are subject to impairment testing and are reviewed for impairment when events or circumstances indicate that the carrying value of an asset is not recoverable and its carrying amount exceeds its fair value. We evaluate the recoverability of the carrying value of these identifiable intangible assets based on estimated undiscounted cash flows to be generated from such assets. If the cash flow estimates or the significant operating assumptions upon which they are based change in the future, we may be required to record additional impairment charges.
The valuation and classification of intangible assets and goodwill and the assignment of useful lives for purposes of amortization involves judgments and the use of estimates. The evaluation of these intangible assets and goodwill for impairment under established accounting principles is required on a recurring basis. Changes in business conditions could potentially require future adjustments to the assumptions made. When we determine that the useful lives of assets are shorter than we had originally estimated, we accelerate the rate of amortization over the assets’ new, shorter useful lives. No impairment charges or accelerated amortization were recorded for the years ended December 31, 2022, 2021, and 2020. A considerable amount of judgment is required in assessing impairment, which includes financial forecasts. If conditions are different from management’s current estimates, material write-downs of long-lived assets may be required, which would adversely affect our operating results.
Accounting for income taxes. Significant management judgment is required in determining our provision for income taxes, deferred tax assets and liabilities, and any valuation allowance recorded against net deferred tax assets in accordance with GAAP. These estimates and judgments occur in the calculation of tax credits, benefits, and deductions and in the calculation of certain tax assets and liabilities, which arise from differences in the timing of recognition of revenue and expense for tax and
financial statement purposes, as well as the interest and penalties related to uncertain tax positions. Significant changes to these estimates may result in an increase or decrease in our tax provision in the current period or subsequent periods.
Also, we must assess the likelihood that we will be able to recover our deferred tax assets. In the event that all or part of our deferred tax assets are not recoverable in the future, we must increase our provision for taxes by recording a valuation allowance to reduce our deferred tax assets to the amount that is more likely than not to be recoverable. In order for our deferred tax assets to be recoverable, we must be able to generate sufficient taxable income in those jurisdictions where the deferred tax assets are located. We consider forecasted income, including income that may be generated as a result of certain tax planning strategies, together with future reversals of existing taxable temporary differences, in determining the need for a valuation allowance. As of December 31, 2022, we believe that it is more likely than not that our deferred tax assets will ultimately be recovered, with the exception of our California deferred tax assets. We believe that, due to the computation of California taxes under the single sales factor, it is more likely than not that our California deferred tax assets will not be realized. Should there be a change in our ability to recover our deferred tax assets, our tax provision would be affected in the period in which such change takes place.
The calculation of our tax liabilities involves dealing with uncertainties in the application of complex tax regulations. We recognize liabilities for uncertain tax positions based on a two-step process. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained on audit, including resolution of related appeals or litigation processes, if any. If we determine that a tax position will more likely than not be sustained on audit, then the second step requires us to estimate and measure the tax benefit as the largest amount that is more than 50% likely to be realized upon ultimate settlement. It is inherently difficult and subjective to estimate such amounts, as we have to determine the probability of various possible outcomes. We re-evaluate these uncertain tax positions on a quarterly basis. This evaluation is based on factors including, but not limited to, changes in facts or circumstances, changes in tax law, effective settlement of audit issues, and new audit activity. Such a change in the recognition or measurement would result in the recognition of a tax benefit or an additional charge to the tax provision.
Accounting for legal contingencies. From time to time, we are involved in a number of legal proceedings involving product liability, intellectual property, shareholder derivative actions, securities class actions, insurance, employee-related, and other matters. We record a liability and related charge to earnings in our Consolidated Financial Statements for legal contingencies when the loss is considered probable and the amount can be reasonably estimated. Our assessment is re-evaluated each accounting period and is based on all available information, including discussion with any outside legal counsel that represents us. If a reasonable estimate of a known or probable loss cannot be made, but a range of probable losses can be estimated, the low-end of the range of losses is recognized if no amount within the range is a better estimate than any other. If a loss is reasonably possible, but not probable, and can be reasonably estimated, the estimated loss or range of loss is disclosed in the Notes to the Consolidated Financial Statements.
When determining the estimated probable loss or range of losses, significant judgment is required to be exercised in order to estimate the amount and timing of the loss to be recorded. Estimates of probable losses resulting from litigation are inherently difficult to make, particularly when the matters are in early procedural stages with incomplete facts and information. The final outcome of legal proceedings is dependent on many variables and is difficult to predict and, therefore, the ultimate cost to entirely resolve such matters may be materially different than the amount of current estimates. Consequently, new information or changes in judgments and estimates could have a material adverse effect on our business, financial condition, and results of operations or cash flows.
RECENT ACCOUNTING PRONOUNCEMENTS
See “Note 2. Summary of Significant Accounting Policies” of the Notes to Consolidated Financial Statements in “Item 8. Financial Statements and Supplementary Data” for additional information regarding recent accounting pronouncements, including the respective expected dates of adoption and estimated effects, if any, on our Consolidated Financial Statements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate and Market Risk
The primary objective of our investment activities is to preserve principal while supporting our liquidity requirements. To achieve this objective, we maintain a diversified portfolio of cash equivalents and short- and long-term investments in a variety of high-quality securities, including money market funds, U.S. treasury and U.S. government agency securities, corporate notes and bonds, commercial paper, non-U.S. government agency securities, and municipal notes. The securities are classified as available-for-sale and consequently are recorded at fair value with unrealized gains or losses reported as a separate component of accumulated other comprehensive loss. The weighted average duration of our portfolio as of December 31, 2022, was approximately 0.8 years. If interest rates rise, the market value of our investments may decline, which could result in a realized loss if we are forced to sell an investment before its scheduled maturity. A hypothetical increase or decrease in interest rates by 25 basis points would have resulted in a decrease or increase in the fair value of our net investment position of approximately $12 million, respectively, as of December 31, 2022. We do not utilize derivative financial instruments to manage our interest rate risks.
Uncertain financial markets could result in a tightening in the credit markets, a reduced level of liquidity in many financial markets, and extreme volatility in fixed income and credit markets. The credit ratings of the securities we have invested in could deteriorate and may have an adverse impact on the carrying value of these investments.
Foreign Exchange Risk
The majority of our revenue, expense, and capital purchasing activities are transacted in U.S. dollars. However, we generally sell our products and services in local currencies where we have direct distribution channels. We operate in a number of markets on a direct sales basis and incur operating expenses in local currencies. We also purchase certain product components from non-U.S. suppliers in local currency. As a result, because a portion of our operations consists of sales activities outside of the U.S., we have foreign exchange exposures to non-U.S. dollar revenues, operating expenses, accounts receivable, accounts payable, and foreign currency bank balances.
For the year ended December 31, 2022, sales denominated in foreign currencies were approximately 24% of total revenue. The objective of our hedging program is to mitigate the impact of changes in currency exchange rates on our net cash flow from foreign currency-denominated sales and expenses. For the year ended December 31, 2022, our revenue would have decreased by approximately $100 million if the U.S. dollar exchange rate strengthened by 10%. We also hedge the net recognized non-functional currency balance sheet exposures with foreign exchange forward contracts to reduce the risk that our earnings and cash flows will be adversely affected by changes in exchange rates. A 10% strengthening of the U.S. dollar exchange rate against all currencies to which we have exposure, after considering foreign currency hedges and offsetting positions as of December 31, 2022, would have resulted in an approximately $2.7 million increase in the carrying amounts of those net assets. Actual gains and losses in the future may differ materially from the hypothetical gains and losses discussed above based on changes in the timing and amount of foreign currency exchange rate movements and our actual exposure and hedging transactions. Bank counterparties to foreign exchange forward contracts expose us to credit-related losses in the event of their nonperformance. To mitigate that risk, we only contract with counterparties that meet certain minimum requirements under our counterparty risk assessment process. We monitor credit ratings and potential downgrades on at least a quarterly basis. Based on our ongoing assessment of counterparty risk, we will adjust our exposure to various counterparties.
Although we sell to distributors outside of the U.S. in U.S. dollars, strengthening of the dollar can impact our distributors’ margins and could impact the end customers’ ability to purchase our products if our distributors seek to recover the impact of the change in the dollar by increasing product and service prices. Less than 10% of our revenue is conducted through distributors outside of the U.S. Strengthening of the dollar relative to non-U.S. currencies could have an adverse impact on our business.
Our operations outside of the U.S. are subject to risks typical of operations outside of the U.S. including, but not limited to, differing economic conditions, changes in the political climate, differing tax structures, other regulations and restrictions, and foreign exchange rate volatility.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Index To Consolidated Financial Statements All other schedules have been omitted, because they are not applicable or the required information is shown in the Consolidated Financial Statements or the Notes thereto.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Intuitive Surgical, Inc.
Opinions on the Financial Statements and Internal Control over Financial Reporting
We have audited the accompanying consolidated balance sheets of Intuitive Surgical, Inc. and its subsidiaries (the “Company”) as of December 31, 2022 and 2021, and the related consolidated statements of income, of comprehensive income, of stockholders’ equity and of cash flows for each of the three years in the period ended December 31, 2022, including the related notes and financial statement schedule listed in the index appearing under Item 15(a)(2) (collectively referred to as the “consolidated financial statements”). We also have audited the Company’s internal control over financial reporting as of December 31, 2022, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2022 and 2021, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2022 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2022, based on criteria established in Internal Control - Integrated Framework (2013) issued by the COSO.
Basis for Opinions
The Company’s management is responsible for these consolidated financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in Management’s Report on Internal Control Over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on the Company’s consolidated financial statements and on the Company’s internal control over financial reporting based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud, and whether effective internal control over financial reporting was maintained in all material respects.
Our audits of the consolidated financial statements included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Critical Audit Matters
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that (i) relates to accounts or disclosures that are material to the consolidated financial statements and (ii) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Determination of Standalone Selling Prices Related to System Sale Arrangements
As described in Notes 2 and 5 to the consolidated financial statements, the Company recognized $1,680.1 million of systems revenue, during the year ended December 31, 2022. The Company’s system sale arrangements include a combination of the following performance obligations: system(s); system components; system accessories; instruments; accessories; and system service. For multiple-element arrangements, revenue is allocated to each performance obligation based on its relative standalone selling price. Standalone selling prices are based on observable prices at which the Company separately sells the products or services. If a standalone selling price is not directly observable, then management estimates the standalone selling price considering market conditions and entity-specific factors including, but not limited to, features and functionality of the products and services, geographies, and type of customer.
The principal considerations for our determination that performing procedures relating to the determination of standalone selling prices related to system sale arrangements is a critical audit matter are the significant judgment by management when determining estimates of standalone selling prices, which in turn led to a high degree of auditor judgment, subjectivity, and effort in performing procedures and evaluating audit evidence relating to the estimates of standalone selling prices used to allocate the transaction price of an arrangement to each distinct performance obligation.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures included testing the effectiveness of controls over the revenue recognition process, including controls over the determination of the estimates of standalone selling prices. These procedures also included, among others, (i) testing management’s process for determining the estimates of standalone selling prices; (ii) evaluating the appropriateness of the overall methodology used by management to develop the estimates, including the appropriateness of the data inputs related to the products and services, geographies, and type of customer used in the methodology; (iii) testing the completeness and accuracy of the data used in the methodology; and (iv) testing the accuracy of management’s calculations of estimated selling prices.
/s/ PricewaterhouseCoopers LLP
San Jose, California
February 10, 2023
We have served as the Company’s auditor since 2014.
INTUITIVE SURGICAL, INC.
CONSOLIDATED BALANCE SHEETS
(IN MILLIONS, EXCEPT PAR VALUE AMOUNTS)
| | | | | | | | | | | |
| December 31, |
| 2022 | | 2021 |
| ASSETS | | | |
| Current assets: | | | |
| Cash and cash equivalents | $ | 1,581.2 | | | $ | 1,290.9 | |
| Short-term investments | 2,536.7 | | | 2,913.1 | |
Accounts receivable, net of allowances of $22.4 and $20.2 as of December 31, 2022, and 2021, respectively | 942.1 | | | 782.7 | |
| Inventory | 893.2 | | | 587.1 | |
| Prepaids and other current assets | 299.8 | | | 271.1 | |
| Total current assets | 6,253.0 | | | 5,844.9 | |
| Property, plant, and equipment, net | 2,374.2 | | | 1,876.4 | |
| Long-term investments | 2,623.6 | | | 4,415.5 | |
| Deferred tax assets | 664.6 | | | 441.4 | |
| Intangible and other assets, net | 710.1 | | | 633.2 | |
| Goodwill | 348.5 | | | 343.6 | |
| Total assets | $ | 12,974.0 | | | $ | 13,555.0 | |
| | | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY | | | |
| Current liabilities: | | | |
| Accounts payable | $ | 147.0 | | | $ | 121.2 | |
| Accrued compensation and employee benefits | 401.6 | | | 350.1 | |
| Deferred revenue | 397.3 | | | 377.2 | |
| Other accrued liabilities | 476.2 | | | 301.3 | |
| Total current liabilities | 1,422.1 | | | 1,149.8 | |
| Other long-term liabilities | 439.3 | | | 453.7 | |
| Total liabilities | 1,861.4 | | | 1,603.5 | |
| Commitments and contingencies (Note 8) | | | |
| Stockholders’ equity: | | | |
Preferred stock, 2.5 shares authorized, $0.001 par value, issuable in series; no shares issued and outstanding as of December 31, 2022, and 2021 | — | | | — | |
Common stock, 600.0 shares authorized, $0.001 par value, 350.0 shares and 357.7 shares issued and outstanding as of December 31, 2022, and 2021, respectively | 0.4 | | | 0.4 | |
| Additional paid-in capital | 7,703.9 | | | 7,164.0 | |
| Retained earnings | 3,500.1 | | | 4,760.9 | |
| Accumulated other comprehensive loss | (162.5) | | | (24.2) | |
| Total Intuitive Surgical, Inc. stockholders’ equity | 11,041.9 | | | 11,901.1 | |
| Noncontrolling interest in joint venture | 70.7 | | | 50.4 | |
| Total stockholders’ equity | 11,112.6 | | | 11,951.5 | |
| Total liabilities and stockholders’ equity | $ | 12,974.0 | | | $ | 13,555.0 | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
The accompanying notes are an integral part of these Consolidated Financial Statements.
INTUITIVE SURGICAL, INC.
CONSOLIDATED STATEMENTS OF INCOME
(IN MILLIONS, EXCEPT PER SHARE AMOUNTS)
22
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| 2022 | | 2021 | | 2020 |
| Revenue: | | | | | |
| Product | $ | 5,198.0 | | | $ | 4,793.9 | | | $ | 3,634.6 | |
| Service | 1,024.2 | | | 916.2 | | | 723.8 | |
| Total revenue | 6,222.2 | | | 5,710.1 | | | 4,358.4 | |
| Cost of revenue: | | | | | |
| Product | 1,700.3 | | | 1,464.1 | | | 1,230.3 | |
| Service | 325.9 | | | 287.5 | | | 266.9 | |
| Total cost of revenue | 2,026.2 | | | 1,751.6 | | | 1,497.2 | |
| Gross profit | 4,196.0 | | | 3,958.5 | | | 2,861.2 | |
| Operating expenses: | | | | | |
| Selling, general and administrative | 1,739.9 | | | 1,466.5 | | | 1,216.3 | |
| Research and development | 879.0 | | | 671.0 | | | 595.1 | |
| Total operating expenses | 2,618.9 | | | 2,137.5 | | | 1,811.4 | |
| Income from operations | 1,577.1 | | | 1,821.0 | | | 1,049.8 | |
| Interest and other income, net | 29.7 | | | 69.3 | | | 157.2 | |
| Income before taxes | 1,606.8 | | | 1,890.3 | | | 1,207.0 | |
| Income tax expense | 262.4 | | | 162.2 | | | 140.2 | |
| Net income | 1,344.4 | | | 1,728.1 | | | 1,066.8 | |
| Less: net income attributable to noncontrolling interest in joint venture | 22.1 | | | 23.5 | | | 6.2 | |
| Net income attributable to Intuitive Surgical, Inc. | $ | 1,322.3 | | | $ | 1,704.6 | | | $ | 1,060.6 | |
| Net income per share attributable to Intuitive Surgical, Inc.: | | | | | |
| Basic | $ | 3.72 | | | $ | 4.79 | | | $ | 3.02 | |
| Diluted | $ | 3.65 | | | $ | 4.66 | | | $ | 2.94 | |
| Shares used in computing net income per share attributable to Intuitive Surgical, Inc.: | | | | | |
| Basic | 355.7 | | | 356.1 | | | 351.1 | |
| Diluted | 362.0 | | | 365.8 | | | 361.0 | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
The accompanying notes are an integral part of these Consolidated Financial Statements.
INTUITIVE SURGICAL, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME
(IN MILLIONS)
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| 2022 | | 2021 | | 2020 |
| Net income | $ | 1,344.4 | | | $ | 1,728.1 | | | $ | 1,066.8 | |
| Other comprehensive income (loss), net of tax: | | | | | |
| Change in foreign currency translation gains (losses) | (0.5) | | | (13.3) | | | 5.2 | |
| Available-for-sale securities (net of tax): | | | | | |
| Change in unrealized gains (losses) | (138.2) | | | (45.5) | | | 13.8 | |
| Less: Reclassification adjustment for gains on securities | — | | | — | | | (4.7) | |
| Net change | (138.2) | | | (45.5) | | | 9.1 | |
| Hedge instruments (net of tax): | | | | | |
| Change in unrealized gains (losses) | (35.0) | | | 12.3 | | | (0.8) | |
| Less: Reclassification adjustment for (gains) losses on hedge instruments | 27.6 | | | (4.9) | | | (2.8) | |
| Net change | (7.4) | | | 7.4 | | | (3.6) | |
| Employee benefit plans (net of tax): | | | | | |
| Change in unrealized gains | 5.8 | | | 0.1 | | | 1.0 | |
| Less: Reclassification adjustment for losses on employee benefit plans | 0.2 | | | 1.5 | | | 1.3 | |
| Net change | 6.0 | | | 1.6 | | | 2.3 | |
| Other comprehensive income (loss), net of tax | (140.1) | | | (49.8) | | | 13.0 | |
| Total comprehensive income | $ | 1,204.3 | | | $ | 1,678.3 | | | $ | 1,079.8 | |
| Less: comprehensive income attributable to noncontrolling interest | $ | 20.3 | | | $ | 22.8 | | | $ | 6.7 | |
| Total comprehensive income attributable to Intuitive Surgical, Inc. | $ | 1,184.0 | | | $ | 1,655.5 | | | $ | 1,073.1 | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
The accompanying notes are an integral part of these Consolidated Financial Statements.
INTUITIVE SURGICAL, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(IN MILLIONS)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Common Stock | | Additional
Paid-In
Capital | | Retained
Earnings | | Accumulated
Other
Comprehensive
(Loss)/Income | | Total Intuitive Surgical, Inc. Stockholders’ Equity | | Noncontrolling
Interest
in
Joint
Venture | | Total Stockholders’ Equity |
| Shares | | Amount | | | | | | |
Balances as of December 31, 2019 | 347.9 | | | $ | 0.3 | | | $ | 5,756.6 | | | $ | 2,494.5 | | | $ | 12.4 | | | $ | 8,263.8 | | | $ | 20.9 | | | $ | 8,284.7 | |
Adoption of new accounting standards (1) | — | | | — | | | — | | | (0.1) | | | — | | | (0.1) | | | — | | | (0.1) | |
Issuance of common stock through employee stock plans | 6.8 | | | 0.1 | | | 308.7 | | | — | | | — | | | 308.8 | | | — | | | 308.8 | |
Shares withheld related to net share settlement of equity awards | (0.9) | | | — | | | (7.9) | | | (167.3) | | | — | | | (175.2) | | | — | | | (175.2) | |
Share-based compensation expense related to employee stock plans | — | | | — | | | 395.4 | | | — | | | — | | | 395.4 | | | — | | | 395.4 | |
Repurchase and retirement of common stock | (0.7) | | | — | | (7.9) | | | (126.4) | | | — | | (134.3) | | | — | | (134.3) | |
| Net income attributable to Intuitive Surgical, Inc. | — | | | — | | | — | | | 1,060.6 | | | — | | | 1,060.6 | | | — | | | 1,060.6 | |
Other comprehensive income (loss) | — | | | — | | | — | | | — | | | 12.5 | | | 12.5 | | | 0.5 | | | 13.0 | |
Net income attributable to noncontrolling interest in joint venture | — | | | — | | | — | | | — | | | — | | | — | | | 6.2 | | | 6.2 | |
Balances as of December 31, 2020 | 353.1 | | | $ | 0.4 | | | $ | 6,444.9 | | | $ | 3,261.3 | | | $ | 24.9 | | | $ | 9,731.5 | | | $ | 27.6 | | | $ | 9,759.1 | |
Issuance of common stock through employee stock plans | 5.4 | | | — | | | 276.5 | | | — | | | — | | | 276.5 | | | — | | | 276.5 | |
Shares withheld related to net share settlement of equity awards | (0.8) | | | — | | | (6.6) | | | (205.0) | | | — | | | (211.6) | | | — | | | (211.6) | |
Share-based compensation expense related to employee stock plans | — | | | — | | | 449.2 | | | — | | | — | | | 449.2 | | | — | | | 449.2 | |
Net income attributable to Intuitive Surgical, Inc. | — | | | — | | | — | | | 1,704.6 | | | — | | | 1,704.6 | | | — | | | 1,704.6 | |
Other comprehensive income | — | | | — | | | — | | | — | | | (49.1) | | | (49.1) | | | (0.7) | | | (49.8) | |
Net income attributable to noncontrolling interest in joint venture | — | | | — | | | — | | | — | | | — | | | — | | | 23.5 | | | 23.5 | |
Balances as of December 31, 2021 | 357.7 | | | $ | 0.4 | | | $ | 7,164.0 | | | $ | 4,760.9 | | | $ | (24.2) | | | $ | 11,901.1 | | | $ | 50.4 | | | $ | 11,951.5 | |
| | | | | | | | | | | | | | | |
Issuance of common stock through employee stock plans | 4.2 | | | — | | | 233.8 | | | — | | | — | | | 233.8 | | | — | | | 233.8 | |
Shares withheld related to net share settlement of equity awards | (0.7) | | | — | | | (7.4) | | | (186.8) | | | — | | | (194.2) | | | — | | | (194.2) | |
Share-based compensation expense related to employee stock plans | — | | | — | | | 524.6 | | | — | | | — | | | 524.6 | | | — | | | 524.6 | |
Repurchase and retirement of common stock | (11.2) | | | — | | | (211.1) | | | (2,396.3) | | | — | | | (2,607.4) | | | — | | | (2,607.4) | |
Net income attributable to Intuitive Surgical, Inc. | — | | | — | | | — | | | 1,322.3 | | | — | | | 1,322.3 | | | — | | | 1,322.3 | |
Other comprehensive income (loss) | — | | | — | | | — | | | — | | | (138.3) | | | (138.3) | | | (1.8) | | | (140.1) | |
| | | | | | | | | | | | | | | |
Net income attributable to noncontrolling interest in joint venture | — | | | — | | | — | | | — | | | — | | | — | | | 22.1 | | | 22.1 | |
Balances as of December 31, 2022 | 350.0 | | | $ | 0.4 | | | $ | 7,703.9 | | | $ | 3,500.1 | | | $ | (162.5) | | | $ | 11,041.9 | | | $ | 70.7 | | | $ | 11,112.6 | |
|
|
(1) Represents the adjustment related to the adoption of Accounting Standards Update (“ASU”) 2016-13, Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments. |
The accompanying notes are an integral part of these Consolidated Financial Statements.
INTUITIVE SURGICAL, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(IN MILLIONS)
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| 2022 | | 2021 | | 2020 |
| Operating activities: | | | | | |
| Net income | $ | 1,344.4 | | | $ | 1,728.1 | | | $ | 1,066.8 | |
| Adjustments to reconcile net income to net cash provided by operating activities: | | | | | |
| Depreciation and loss on disposal of property, plant, and equipment, net | 338.0 | | | 282.8 | | | 226.4 | |
| Amortization of intangible assets | 27.8 | | | 27.4 | | | 49.8 | |
| Gain on sale of business | (3.8) | | | — | | | — | |
| Loss (gain) on investments, accretion of discounts, and amortization of premiums on investments, net | 49.0 | | | 10.6 | | | (55.1) | |
| Deferred income taxes | (185.3) | | | (62.6) | | | 57.6 | |
| Share-based compensation expense | 513.2 | | | 449.2 | | | 395.4 | |
| Amortization of contract acquisition assets | 26.6 | | | 22.0 | | | 17.1 |
| Changes in operating assets and liabilities, net of effects of acquisitions: | | | | | |
| Accounts receivable | (159.3) | | | (142.3) | | | 5.7 | |
| Inventory | (546.6) | | | (256.0) | | | (170.1) | |
| Prepaids and other assets | (129.2) | | | (204.9) | | | (111.8) | |
| Accounts payable | 21.3 | | | 36.0 | | | (32.3) | |
| Accrued compensation and employee benefits | 51.5 | | | 115.1 | | | (16.6) | |
| Deferred revenue | 21.5 | | | 32.6 | | | 15.0 | |
| Other liabilities | 121.7 | | | 51.4 | | | 36.9 | |
| Net cash provided by operating activities | 1,490.8 | | | 2,089.4 | | | 1,484.8 | |
| Investing activities: | | | | | |
| Purchase of investments | (1,399.5) | | | (6,452.0) | | | (4,292.9) | |
| Proceeds from sales of investments | 61.1 | | | 84.9 | | | 800.7 | |
| Proceeds from maturities of investments | 3,254.4 | | | 4,267.8 | | | 2,930.8 | |
| Purchase of property, plant, and equipment | (532.4) | | | (339.5) | | | (341.5) | |
| Acquisition of businesses, net of cash, and intellectual property and other investing activities | (12.8) | | | (22.7) | | | (37.7) | |
| Net cash provided by (used in) investing activities | 1,370.8 | | | (2,461.5) | | | (940.6) | |
| Financing activities: | | | | | |
| Proceeds from issuance of common stock relating to employee stock plans | 233.8 | | | 276.5 | | | 308.8 | |
| Taxes paid related to net share settlement of equity awards | (194.2) | | | (211.6) | | | (175.2) | |
| Repurchase of common stock | (2,607.4) | | | — | | | (134.3) | |
| | | | | |
| Payment of deferred purchase consideration | (4.5) | | | (21.9) | | | (85.0) | |
| Net cash provided by (used in) financing activities | (2,572.3) | | | 43.0 | | | (85.7) | |
| Effect of exchange rate changes on cash, cash equivalents, and restricted cash | 5.4 | | | (3.4) | | | (2.6) | |
| Net increase (decrease) in cash, cash equivalents, and restricted cash | 294.7 | | | (332.5) | | | 455.9 | |
| Cash, cash equivalents, and restricted cash, beginning of year | 1,306.0 | | | 1,638.5 | | | 1,182.6 | |
| Cash, cash equivalents, and restricted cash, end of year | $ | 1,600.7 | | | $ | 1,306.0 | | | $ | 1,638.5 | |
The accompanying notes are an integral part of these Consolidated Financial Statements.
INTUITIVE SURGICAL, INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1. DESCRIPTION OF THE BUSINESS
Intuitive Surgical, Inc. (“Intuitive” or the “Company”) develops, manufactures, and markets the da Vinci® Surgical System and the Ion® endoluminal system. The Company’s products and related services enable physicians and healthcare providers to improve the quality of and access to minimally invasive care. The systems consist of a surgeon console or consoles, a patient-side cart, and a high-performance vision system and use proprietary instruments and accessories.
NOTE 2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The accompanying Consolidated Financial Statements have been prepared in accordance with U.S. generally accepted accounting principles (“GAAP”) and include the accounts of the Company and its wholly and majority-owned subsidiaries. All intercompany balances and transactions have been eliminated in consolidation.
The Consolidated Financial Statements include the results and balances of the Company’s majority-owned joint venture (“Joint Venture”) with Shanghai Fosun Pharmaceutical (Group) Co., Ltd. (“Fosun Pharma”). Chindex Medical Limited (“Chindex”), a subsidiary of Fosun Pharma, has been its distribution partner for da Vinci Surgical Systems in China. The Company holds a controlling financial interest in the Joint Venture, and the noncontrolling interest is reflected as a separate component of the consolidated stockholders’ equity. The noncontrolling interest’s share of the earnings in the Joint Venture is presented separately in the Consolidated Statements of Income and Comprehensive Income for the years ended December 31, 2022, 2021, and 2020.
Common Stock Split
Shares issued pursuant to the three-for-one stock split (the “Stock Split”) of the Company’s issued and outstanding common stock, par value $0.001 per share, were distributed on October 4, 2021, to stockholders of record as of September 27, 2021. All share and per-share information presented in the Consolidated Financial Statements have been retroactively adjusted to reflect the Stock Split.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the Consolidated Financial Statements and accompanying Notes to the Consolidated Financial Statements. The accounting estimates that require management’s most significant, complex, and subjective judgments include the valuation and recognition of investments, the standalone selling prices used to allocate the contract consideration to the individual performance obligations, the valuation of inventory, the valuation of and assessment of the recoverability of intangible assets and goodwill, the recognition and measurement of current and deferred income taxes, including the measurement of uncertain tax positions, and the estimates for legal contingencies. Actual results could differ materially from these estimates.
Concentrations of Credit Risk and Other Risks and Uncertainties
The carrying amounts for financial instruments consisting of cash and cash equivalents, accounts receivable, accounts payable, and accrued liabilities approximate fair value due to their short maturities. Marketable securities and derivative instruments are stated at their estimated fair values, based on quoted market prices for the same or similar instruments. The counterparties to the agreements relating to the Company’s investment securities and derivative instruments consist of various major corporations, financial institutions, municipalities, and government agencies of high credit standing.
The Company’s accounts receivable are primarily derived from billings related to revenue arrangements with customers and distributors located throughout the world. The Company performs credit evaluations of its customers’ financial condition and, generally, requires no collateral from its customers. The Company provides reserves for potential credit losses but has not experienced significant losses to date. As of December 31, 2022, and 2021, 60% and 67%, respectively, of accounts receivable were from domestic customers.
During the years ended December 31, 2022, 2021, and 2020, domestic revenue accounted for 67%, 67%, and 68% of total revenue, respectively, while outside of the U.S. (“OUS”) revenue accounted for 33%, 33%, and 32%, respectively, of total revenue for each of the years then ended.
The Company’s future results of operations and liquidity could be materially adversely affected by macroeconomic factors contributing to delays in payments of outstanding receivables, supply chain disruptions, including materials shortages and inflationary pressure, uncertain or reduced demand, and the impact of any initiatives or programs that the Company may undertake to address financial and operational challenges faced by its customers.
In particular, the Company has experienced increased difficulties in obtaining a sufficient supply of a number of component materials used in its products, such as semiconductor components as well as a range of other materials including, but not limited to, metals and polymers, as global supply has become significantly constrained due to increased demand for certain materials. Additionally, prices of such materials have increased due to the increased demand and supply shortage. With rising interest rates, access to credit may become more difficult, and any insolvency of the Company’s key suppliers, including sole-source and single-source suppliers, may exacerbate current supply chain challenges. The Company is engaged in activities to seek to mitigate supply disruptions by, for example, increasing its communications with its suppliers and modifying its purchase order coverage and inventory levels. However, the global supply chain shortages will remain a challenge for the foreseeable future.
Such global shortages in important components as well as certain logistics challenges have resulted in, and will continue to cause, inflationary cost pressure in the Company’s supply chain. To date, the inflationary cost pressure has been more pronounced in the Company’s logistics costs, but these supply chain challenges have not materially impacted the Company’s results of operations or ability to deliver products and services to its customers. However, if shortages in important supply chain materials in the semiconductor or other markets or logistics challenges continue, the Company could fail to meet product demand, which could result in deferred or canceled procedures. Additionally, if inflationary pressures in logistics or component costs persist, the Company may not be able to quickly or easily adjust pricing, reduce costs, or implement countermeasures. Additionally, there is uncertainty surrounding the impact of any monetary policy changes taken by the U.S. Federal Reserve and other central banks to address the structural risks associated with inflation.
Fluctuations in labor availability globally, including labor shortages and staff burnout and attrition, could also impact the Company’s ability to hire and retain personnel critical to its manufacturing, logistics, and commercial operations. The Company is also highly dependent on the principal members of its management and scientific staff. The loss of critical members of the Company’s team, or its inability to attract and retain qualified personnel, could significantly harm its operations, business, and ability to compete.
Hospitals are also experiencing staffing shortages and supply chain issues that could affect their ability to provide patient care. Additionally, hospitals are facing significant financial pressure as supply chain constraints and inflation drive up operating costs, rising interest rates make access to credit more expensive, unrealized losses decrease available cash reserves, and fiscal stimulus programs enacted during the COVID-19 pandemic wind down. To the extent macroeconomic conditions remain challenging, it is likely that hospitals’ spend on capital equipment will be adversely impacted. In addition, as competition progresses in various markets, longer selling cycles and pricing pressures are likely to result. As of the date of issuance of these Consolidated Financial Statements, the extent to which these macroeconomic factors may materially adversely affect the Company’s financial condition, liquidity, or results of operations is uncertain.
The Company is also subject to additional risks and uncertainties due to the ongoing COVID-19 pandemic. The extent of the impact on the Company’s business is highly uncertain and difficult to predict. The Company’s customers may divert resources to treat COVID-19 patients and defer some elective surgical procedures, both of which may impact the Company’s customers’ ability to meet their obligations, including to the Company. The severity of the impact of the COVID-19 pandemic on the Company’s business will depend on a number of factors, including, but not limited to, the duration and severity of the pandemic and the extent and severity of the impact on the Company’s customers, all of which are uncertain and cannot be predicted.
Customer Relief Program
During the second quarter of 2020, the Company introduced a series of programs to provide financial relief to customers (the “Customer Relief Program”). As part of the Customer Relief Program, the Company provided its customers service fee credits, extended payment terms, and deferred payments related to Intuitive System Leasing arrangements. The Customer Relief Program ended at the end of the third quarter of 2020. There was no similar customer relief program offered in 2021 or 2022.
Service fee credits. As part of the Customer Relief Program, the Company provided service fee credits to customers based on the reduction in the utilization of their systems during the second and third quarters of 2020 relative to a pre-COVID-19 level baseline. The Company reflected the service fee credits as a reduction of service revenue and accounts receivable in the quarter they were earned by its customers. The service fee credit program resulted in an $80 million decrease in service revenue in 2020.
Short-term payment relief. In response to the COVID-19 pandemic, the Company introduced a payment deferral program to provide financial relief to qualified customers. This relief extended payment terms up to 180 days for qualified and creditworthy customers.
The Company also introduced a lease payment deferral program in which creditworthy customers with active Intuitive system leasing arrangements could elect to defer lease payments up to five months that are payable at the end of the lease by extending the lease term. This program did not result in substantial increases in the rights of the lessor or the obligations of the
lessee, and the Company elected to apply the relief provided by the Financial Accounting Standards Board (“FASB”) FAQ on accounting for COVID-19 and market volatility by not applying the lease modification guidance in ASC 842 to the lease arrangements affected by the deferrals and lease extensions.
For operating lease arrangements where the lease term was extended by adding the deferred period to the end of the contract, the Company recalculated the straight-line revenue based on the revised terms, consistent with the treatment accepted by the FASB FAQ on accounting for COVID-19. For its sales-type lease arrangements impacted, the Company accounted for the deferral in the timing of lease payments as if there were no changes in the lease contract, consistent with the treatment accepted by the FASB FAQ on accounting for COVID-19. While the short-term payment relief offered did not have a material impact on the results of operations, the Company deferred $15 million of lease billings and extended payment terms associated with $181 million of billings during the program, of which $19 million remained outstanding as of December 31, 2020. All of the trade receivables with extended payment terms have been collected as of December 31, 2021.
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original maturity from the date of purchase of 90 days or less to be cash equivalents.
Restricted Cash
As of December 31, 2022, and 2021, the Company had $19.5 million and $17.9 million, respectively, of restricted cash primarily associated with its insurance programs. Restricted cash was included in prepaids and other current assets and intangible and other assets, net on the Consolidated Balance Sheets.
Investments
Available-for-sale debt securities. The Company’s investments may consist of money market funds, U.S. treasury and U.S. government agency securities, high-quality corporate notes and bonds, commercial paper, non-U.S. government agency securities, and taxable and tax-exempt municipal notes. The Company has designated all investments as available-for-sale and, therefore, the investments are subject to periodic impairment under the available-for-sale debt security impairment model. Available-for-sale debt securities in an unrealized loss position are written down to fair value through a charge to interest and other income, net, if the Company intends to sell the security or it is more likely than not the Company will be required to sell the security before recovery of its amortized cost basis. The Company evaluates the remaining securities to determine what amount of the excess, if any, is caused by expected credit losses. A decline in fair value attributable to expected credit losses is recorded to interest and other income, net, while any portion of the loss related to non-credit factors is recorded in accumulated other comprehensive income (loss). For securities sold prior to maturity, the cost of the securities sold is based on the specific identification method. Realized gains and losses on the sale of investments are recorded in interest and other income, net in the Consolidated Statements of Income. Investments with remaining maturities at the date of purchase greater than 90 days and remaining maturities as of the reporting period of less than one year are classified as short-term investments. Investments with remaining maturities greater than one year are classified as long-term investments.
Equity investments. The Company holds equity investments with readily determinable fair values and equity investments without readily determinable fair values. The Company recognizes equity investments with readily determinable fair values at the quoted market price with changes in value recorded in interest and other income, net. The Company generally recognizes equity investments that do not have readily determinable fair values at cost minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for the identical or a similar investment of the same issuer.
Fair Value Measurements
The Company measures the fair value of money market funds, certain U.S. treasury securities, and equity investments with readily determinable value based on quoted prices in active markets for identical assets as Level 1 securities. Marketable securities measured at fair value using Level 2 inputs are primarily comprised of commercial paper, corporate notes and bonds, U.S. and non-U.S. government agencies, municipal notes, and equity investments without readily determinable value. The Company reviews trading activity and pricing for these investments as of the measurement date. When sufficient quoted pricing for identical securities is not available, the Company uses market pricing and other observable market inputs for similar securities obtained from various third-party data providers. These inputs either represent quoted prices for similar assets in active markets or have been derived from observable market data. This approach results in the Level 2 classification of these securities within the fair value hierarchy.
Inventory
Inventory is stated at the lower of cost or net realizable value on a first-in, first-out basis. Inventory costs include direct materials, direct labor, and normal manufacturing overhead. The cost basis of the Company’s inventory is reduced for any products that are considered excessive or obsolete based upon assumptions about future demand and market conditions.
Additionally, the cost basis of the Company’s inventory does not include any unallocated fixed overhead costs associated with abnormally low utilization of its factories.
Property, Plant, and Equipment
Property, plant, and equipment are stated at cost, net of accumulated depreciation. Depreciation is computed on a straight-line basis over the estimated useful lives of the assets, generally, as follows:
| | | | | |
| | Useful Lives |
| Building | Up to 30 years |
| Building improvements | Up to 15 years |
| Leasehold improvements | Lesser of useful life or term of lease |
| Equipment and furniture | 5 years |
| Operating lease assets | Greater of lease term or 1 to 5 years |
| Computer and office equipment | 3 years |
| Enterprise-wide software | 5 years |
| Purchased software | Lesser of 3 years or life of license |
Depreciation expense for the years ended December 31, 2022, 2021, and 2020, was $326 million, $280 million, and $221 million, respectively.
Capitalized Software Costs for Internal Use
The Company capitalizes direct costs associated with developing or obtaining internal use software, including enterprise-wide business software, which are incurred during the application development stage. These capitalized costs are recorded as capitalized software within property, plant, and equipment. Costs related to preliminary project activities and post-implementation activities are expensed as incurred. Once the software is ready for its intended use, amounts capitalized are amortized over an estimated useful life of up to 5 years, generally on a straight-line basis.
Implementation Costs in a Cloud Computing Arrangement
The Company capitalizes qualified implementation costs incurred in a hosting arrangement that is a service contract for which it is the customer in accordance with the requirements for capitalizing costs incurred to develop internal-use software. These capitalized implementation costs are recorded within intangible and other assets, net, and are generally amortized over the fixed, non-cancellable term of the associated hosting arrangement on a straight-line basis.
Business Combinations
The Company accounts for business acquisitions in accordance with ASC 805, Business Combinations. This standard requires the acquiring entity in a business combination to recognize the assets acquired, liabilities assumed, and any noncontrolling interest in the acquiree using acquisition-date fair values. Certain provisions of this standard prescribe, among other things, the determination of acquisition-date fair value of consideration paid in a business combination, including contingent consideration. The excess of the acquisition-date fair value of consideration paid over the fair values of the identifiable assets and liabilities is recorded as goodwill. Acquisition-related costs are recognized separately from the business combination and are expensed as incurred. The Company includes the results of operations of the businesses that are acquired as of the acquisition date.
Goodwill and Intangible Assets
Goodwill and intangible assets with indefinite useful lives are not amortized but are tested for impairment at least annually during the fourth quarter, or if circumstances indicate their value may no longer be recoverable. Goodwill represents the excess of the purchase price over the fair value of net identifiable assets and liabilities. The Company continues to operate in one segment, which is considered to be the sole reporting unit and, therefore, goodwill was tested for impairment at the enterprise level.
Intangible assets are carried at cost, net of accumulated amortization. The Company does not have intangible assets with indefinite useful lives other than goodwill. Amortization is recorded on a straight-line basis over the intangible assets’ useful lives, which range from approximately 3 to 9 years.
Impairment of Long-lived Assets
The Company evaluates long-lived assets, which include finite-lived intangible and tangible assets, for impairment whenever events or changes in circumstances indicate that the carrying value of long-lived assets may not be recoverable.
Recoverability is measured by comparing the net book value to the future undiscounted cash flows attributable to such assets. The Company recognizes an impairment charge equal to the amount by which the net book value exceeds its fair value. No material impairment losses were incurred in the periods presented.
Revenue Recognition
The Company’s revenue consists of product revenue, resulting from the sale of systems, system components, and instruments and accessories, and service revenue. The Company accounts for a contract with a customer when there is a legally enforceable contract between the Company and its customer, the rights of the parties are identified, the contract has commercial substance, and the collectability of the contract consideration is probable. The Company’s revenues are measured based on the consideration specified in the contract with each customer, net of any sales incentives and taxes collected from customers that are remitted to government authorities.
The Company’s system sale arrangements generally contain multiple products and services. For these bundled sale arrangements, the Company accounts for individual products and services as separate performance obligations if they are a distinct product or service that is separately identifiable from other items in bundled packages and if a customer can benefit from the product or service on its own or with other resources that are readily available to the customer. The Company’s system sale arrangements include a combination of the following performance obligations: system(s); system components; system accessories; instruments; accessories; and system service. The Company’s system sale arrangements generally include a five-year period of service. The first year of service is generally free and included in the system sale arrangement, and the remaining four years are generally included at a stated service price. The Company considers the service terms in the arrangements that are legally enforceable to be performance obligations. Other than service, the Company generally satisfies all of the performance obligations at a point in time. System components, system accessories, instruments, accessories, and service are also sold on a stand-alone basis.
The Company recognizes revenue as the performance obligations are satisfied by transferring control of the product or service to a customer. The Company generally recognizes revenue for the performance obligations in the following manner:
System sales. For systems (including system components and system accessories) sold directly to end customers, revenue is recognized when the Company transfers control to the customer, which is generally at the point when acceptance occurs that indicates customer acknowledgment of delivery or installation, depending on the terms of the arrangement. For systems sold through distributors, revenue is recognized generally at the time of shipment. The Company’s system arrangements generally do not provide a right of return. The systems are generally covered by a one-year warranty. Warranty costs were not material for the periods presented.
Instruments and accessories. Revenue from sales of instruments and accessories is recognized when control is transferred to the customers, which generally occurs at the time of shipment but also could occur at the time of delivery, depending on the customer arrangement. The Company generally allows its customers in the normal course of business to return unused products for a limited period of time subsequent to the initial purchase and records an allowance against revenue for estimated returns.
Service. Service revenue is recognized over the term of the service period, as the customer benefits from the services throughout the service period. Revenue related to services performed on a time-and-materials basis is recognized when performed.
The Company offers its customers the opportunity to trade in their older systems for a credit towards the purchase of a newer generation system. The Company generally does not provide specified price trade-in rights or upgrade rights at the time of system purchase. Such trade-in or upgrade transactions are separately negotiated based on the circumstances at the time of the trade-in or upgrade, based on the then-fair value of the system, and are generally not based on any pre-existing rights granted by the Company. Accordingly, such trade-ins and upgrades are not considered separate performance obligations in the arrangement for a system sale. Traded-in systems can be reconditioned and resold. The Company accounts for the fair value of the traded-in system in the total consideration in the arrangement by including the net realizable value of the traded-in system less a normal profit margin. The value of the traded-in system is determined as the amount, after reconditioning costs are added, that will allow a normal profit margin on the sale of the reconditioned unit to be generated. When there is no market for the traded-in units, no value is assigned. The assigned value of the traded-in units is reported as a component of inventory until resold or otherwise disposed.
In addition, customers may also have the opportunity to upgrade their systems at a price determined at the time of the upgrade, for example, by adding a second surgeon console for use with the da Vinci Surgical System. Such upgrades are performed by completing component level upgrades at the customer’s site. Upgrade revenue is recognized when the component level upgrades are complete and all revenue recognition criteria are met.
For multiple-element arrangements, revenue is allocated to each performance obligation based on its relative standalone selling price. Standalone selling prices are based on observable prices at which the Company separately sells the products or
services. If a standalone selling price is not directly observable, then the Company estimates the standalone selling price considering market conditions and entity-specific factors including, but not limited to, features and functionality of the products and services, geographies, and type of customer. The Company regularly reviews standalone selling prices and updates these estimates, as necessary.
Assets Recognized from the Costs to Obtain a Contract with a Customer
The Company has determined that certain sales incentives provided to the Company’s sales team are required to be capitalized when the Company expects to generate future economic benefits from the related revenue-generating contracts subsequent to the initial system sales transaction. When determining the economic life of the contract acquisition assets recognized, the Company considers historical service renewal rates, expectations of future customer renewals of service contracts, and other factors that could impact the economic benefits that the Company expects to generate from the relationship with its customers. The costs capitalized as contract acquisition costs included in intangible and other assets, net in the Consolidated Balance Sheets were $72.3 million and $71.8 million as of December 31, 2022, and 2021, respectively. The Company did not incur any impairment losses during the periods presented.
Intuitive System Leasing
The Company enters into lease arrangements with certain qualified customers. Leases have terms that generally range from 24 to 84 months and are usually collateralized by a security interest in the underlying assets. The Company also leases systems to certain qualified customers under usage-based arrangements that have terms of up to 84 months. For these usage-based lease arrangements, the lease fee is generally billed monthly in arrears based on a contractual per-use fee, and usage is generally defined as the number of procedures performed with the system.
Revenue related to multiple-element arrangements are allocated to lease and non-lease elements based on their relative standalone selling prices as prescribed by the Company’s revenue recognition policy. Lease elements generally include a system or system component, while non-lease elements generally include service. For some lease arrangements, customers are provided with the right to purchase the leased system at some point during and/or at the end of the lease term. Except for certain usage-based lease arrangements, lease arrangements generally do not provide rights for the customers to exit or terminate the lease without incurring a penalty. Certain lease arrangements may also include upgrade rights that allow customers to upgrade the leased system to newer technology at some point during the lease term. Generally, these upgrade rights do not specify the terms, including the price or structure of the future upgrade transactions, as those terms are negotiated based on the circumstances at the time of the upgrade, including the then-fair value of the system as well as other factors.
In determining whether a transaction should be classified as a sales-type or operating lease, the Company considers the following terms at lease commencement: (1) whether title of the system transfers automatically or for a nominal fee by the end of the lease term; (2) whether the present value of the minimum lease payments equals or exceeds substantially all of the fair value of the leased system; (3) whether the lease term is for the major part of the remaining economic life of the leased system; (4) whether the lease grants the lessee an option to purchase the leased system that the lessee is reasonably certain to exercise; and (5) whether the underlying system is of such a specialized nature that it is expected to have no alternative use to the Company at the end of the lease term.
The Company generally recognizes revenue from sales-type lease arrangements at the time the system is accepted by the customer, assuming all other revenue recognition criteria have been met. Revenue related to lease elements from sales-type leases is presented as product revenue. Revenue related to lease elements from operating lease arrangements is generally recognized on a straight-line basis over the lease term and is presented as product revenue. Revenue related to lease elements from usage-based arrangements is recognized as the customers utilize the systems and is presented as product revenue.
Other Leasing Arrangements
The Company determines if an arrangement contains a lease at inception. For arrangements where the Company is the lessee, operating leases are included in intangible and other assets, net, other accrued liabilities, and other long-term liabilities on the Consolidated Balance Sheet as of December 31, 2022. The Company currently does not have any finance leases.
Operating lease right-of-use (“ROU”) assets and operating lease liabilities are recognized based on the present value of the future minimum lease payments over the lease term at the commencement date. ROU assets also include any initial direct costs incurred and any lease payments made at or before the lease commencement date, less lease incentives received. The Company uses its incremental borrowing rate based on the information available at the commencement date in determining the lease liabilities, as the Company does not have insight into the inputs necessary to calculate the implicit rate of the leases. Lease terms may include options to extend or terminate when the Company is reasonably certain the option will be exercised. Lease expense is recognized on a straight-line basis over the lease term.
The Company also has lease arrangements with lease and non-lease components. The Company elected the practical expedient not to separate non-lease components from lease components for the Company’s real estate and automobile leases.
Additionally, the Company applied a portfolio approach to effectively account for the operating lease ROU assets and lease liabilities for the Company’s automobile leases. The Company also elected to apply the short-term lease measurement and recognition exemption in which ROU assets and lease liabilities are not recognized for leases with terms of 12 months or less.
Credit Losses
Trade accounts receivable. The allowance for doubtful accounts is based on the Company’s assessment of the collectability of customer accounts. The Company regularly reviews the allowance by considering factors such as historical experience, credit quality, age of the accounts receivable balances, and current economic conditions that may affect a customer’s ability to pay. For the years ended December 31, 2022, and 2021, bad debt expense was not material.
Net investment in sales-type leases. The Company enters into sales-type leases with certain qualified customers to purchase its systems. Sales-type leases have terms that generally range from 24 to 84 months and are usually collateralized by a security interest in the underlying assets. The allowance for loan loss is based on the Company’s assessment of the current expected lifetime loss on lease receivables. The Company regularly reviews the allowance by considering factors such as historical experience, credit quality, age of the lease receivable balances, and current economic conditions that may affect a customer’s ability to pay. Lease receivables are considered past due 90 days after invoice.
The Company manages the credit risk of the net investment in sales-type leases using a number of factors, including, but not limited to, the following: size of operations; profitability, liquidity, and debt ratios; payment history; and past due amounts. The Company also uses credit scores obtained from external providers as a key indicator for the purposes of determining credit quality. The following table summarizes the amortized cost basis by year of origination and by credit quality for the net investment in sales-type leases as of December 31, 2022 (in millions):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 2022 | | 2021 | | 2020 | | 2019 | | 2018 | | | Prior | | Net Investment |
| Credit Rating: | | | | | | | | | | | | | | |
| High | $ | 67.3 | | | $ | 86.7 | | | $ | 42.6 | | | $ | 14.5 | | | $ | 2.2 | | | | $ | 0.4 | | | $ | 213.7 | |
| Moderate | 98.4 | | | 69.6 | | | 34.1 | | | 7.7 | | | 4.1 | | | | 0.3 | | | 214.2 | |
| Low | 2.6 | | | 1.6 | | | 2.9 | | | — | | | — | | | | — | | | 7.1 | |
| Total | $ | 168.3 | | | $ | 157.9 | | | $ | 79.6 | | | $ | 22.2 | | | $ | 6.3 | | | | $ | 0.7 | | | $ | 435.0 | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
For the year ended December 31, 2022, and 2021, credit losses related to the net investment in sales-type leases were not material.
The Company’s exposure to credit losses may increase if its customers are adversely affected by changes in healthcare laws, procedure coverage, and reimbursement, economic pressures or uncertainty associated with local or global economic recessions, disruption associated with the current COVID-19 pandemic, or other customer-specific factors. Although the Company has historically not experienced significant credit losses, it is possible that there could be a material adverse impact from potential adjustments of the carrying amount of trade and lease receivables as hospital cash flows are impacted by their response to the COVID-19 pandemic.
Available-for-sale debt securities. The Company regularly reviews the securities in an unrealized loss position and evaluates the current expected credit loss by considering factors such as historical experience, market data, financial condition and near-term prospects of the investee, the extent of the loss related to the credit of the issuer, and the expected cash flows from the security. The Company segments its portfolio based on the underlying risk profiles of the securities and has a zero-loss expectation for U.S. treasury and U.S. government agency securities. The basis for this assumption is that these securities have consistently high credit ratings by rating agencies, have a long history with no credit losses, are explicitly guaranteed by a sovereign entity, which can print its own currency, and are denominated in a currency that is routinely held by central banks, used in international commerce, and commonly viewed as a reserve currency. Additionally, all of the Company’s investments in corporate debt securities are in securities with high-quality credit ratings, which have historically experienced low rates of default. For the years ended December 31, 2022, and 2021, credit losses related to available-for-sale debt securities were not material.
Allowance for Sales Returns
The allowance for sales returns is based on the Company’s estimates of potential future returns of certain products related to current period product revenue. The Company analyzes historical returns, current economic trends, and changes in customer demand and acceptance of the Company’s products.
Share-Based Compensation
The Company grants long-term equity awards under its stock-based compensation plans to certain employees of the Company. These awards include restricted stock units, stock options, and performance stock units. The Company accounts for share-based compensation plans using the fair value recognition and measurement provisions under GAAP. The Company’s share-based compensation cost is measured at the grant date, based on the fair value of the award, and is recognized as expense on a straight-line basis over the requisite service period. The Company estimates expected forfeitures at the time of grant and revises the estimate, if necessary, in subsequent periods if actual forfeitures differ from those estimated.
Restricted stock units. The fair value of restricted stock units (“RSUs”) is determined based on the closing quoted price of the Company’s common stock on the date of the grant.
Stock options. The Black-Scholes-Merton option-pricing model is used to estimate the fair value of stock options granted and utilizes the following inputs: (1) closing quoted price of the Company’s common stock on the date of grant; (2) expected term; (3) expected volatility; and (4) risk-free interest rate.
Expected Term: The expected term represents the weighted-average period that the stock options are expected to be outstanding prior to being exercised. The Company determines expected term based on historical exercise patterns and its expectation of the time that it will take for employees to exercise options still outstanding.
Expected Volatility: The Company uses market-based implied volatility for purposes of valuing stock options granted. Market-based implied volatility is derived based on actively traded options with expirations greater than one year on the Company’s common stock. The extent to which the Company relies on market-based volatility when valuing options depends, among other things, on the availability of traded options on the Company’s stock and the term of such options. Due to the sufficient volume of the traded options, the Company used 100% market-based implied volatility to value options granted, which the Company believes is more representative of future stock price trends than historical volatility.
Risk-Free Interest Rate: The risk-free interest rate is based on the U.S. treasury yield curve in effect at the time of grant for the expected term of the stock option.
Performance stock units. Performance stock units (“PSUs”) include predefined performance and market conditions. The fair value of performance stock units with performance conditions is based on the closing quoted price of the Company’s common stock on the date of the grant and is recognized on a straight-line basis over the requisite service period. The Company estimates the number of awards with performance conditions that will ultimately vest based on the probability of achievement each quarter to determine the amount of compensation expense to recognize each reporting period. The fair value of performance stock units that include a market condition is determined using a Monte Carlo valuation model and is recognized on a straight-line basis over the requisite service period.
Employee stock purchase plan. The fair value of shares to be issued under the Company’s Employee Stock Purchase Plan (the “ESPP”) is computed using the Black-Scholes-Merton model at the commencement of an offering period in February and August of each year utilizing the following inputs: (1) closing quoted price of the Company’s common stock on the initial date of the offering period; (2) expected term; (3) expected volatility; and (4) risk-free interest rate. Share-based compensation for the ESPP is expensed straight-line over the two-year offering period.
See “Note 10. Share-Based Compensation” for a detailed discussion of the Company’s stock plans and share-based compensation expense.
Computation of Net Income per Share
Basic net income per share attributable to Intuitive Surgical, Inc. is computed using the weighted-average number of shares outstanding during the period. Diluted net income per share attributable to Intuitive Surgical, Inc. is computed using the weighted-average number of the Company’s shares and dilutive potential shares outstanding during the period. Dilutive potential shares primarily consist of employee stock options, restricted stock units, and shares to be purchased by employees under the Company’s employee stock purchase plan.
GAAP requires that employee equity share options, non-vested shares, and similar equity instruments granted by the Company be treated as potential common shares outstanding in computing diluted earnings per share. Diluted shares outstanding include the dilutive effect of equity awards, which is calculated based on the average share price for each fiscal period using the treasury stock method. Under the treasury stock method, the amount the employee must pay for exercising stock options and the amount of compensation cost for future service that the Company has not yet recognized are assumed to be used to repurchase shares.
Research and Development Expenses
Research and development costs are expensed as incurred and include amortization of intangible assets, costs associated with co-development research and development licensing arrangements, costs of prototypes, salaries, benefits and other headcount-related costs, contract and other outside service fees, and facilities and overhead costs.
Foreign Currency and Other Hedging Instruments
For subsidiaries whose local currency is their functional currency, their assets and liabilities are translated into U.S. dollars at exchange rates at the balance sheet date, and revenues and expenses are translated using average exchange rates in effect during the period. Gains and losses from foreign currency translation are included in accumulated other comprehensive income (loss) within stockholders’ equity in the Consolidated Balance Sheets. For all non-functional currency account balances, the re-measurement of such balances to the functional currency results in either a foreign exchange gain or loss, which is recorded to interest and other income, net in the Consolidated Statements of Income in the same accounting period that the re-measurement occurred.
The Company uses derivatives to partially offset its business exposure to foreign currency exchange risk. The terms of the Company’s derivative contracts are generally twelve months or shorter. The Company typically hedges portions of its forecasted foreign currency exposure associated with revenue and expenses. The Company may also enter into foreign currency forward contracts to offset the foreign currency exchange gains and losses generated by the re-measurement of certain assets and liabilities denominated in non-functional currencies. The hedging program is not designated for trading or speculative purposes.
The Company’s accounting policies for these instruments are based on whether the instruments are designated as hedging or non-hedging instruments. The Company records all derivatives on the Consolidated Balance Sheets at fair value. The effective portions of cash flow hedges are recorded in other comprehensive income (loss) (“OCI”) until the hedged item is recognized in earnings. Derivative instruments designated as cash flow hedges are de-designated as hedges when it is probable that the forecasted hedged transaction will not occur in the initially identified time period or within a subsequent two-month time period. Gains and losses in OCI associated with such derivative instruments are reclassified immediately into earnings through interest and other income, net. Any subsequent changes in the fair value of such derivative instruments also are reflected in current earnings. Derivatives that are not designated as hedging instruments and the ineffective portions of cash flow hedges are adjusted to fair value through earnings in interest and other income, net.
Income Taxes
Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the year in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. Valuation allowances are established, when necessary, to reduce deferred tax assets to the amounts that are expected more likely than not to be realized in the future.
The Company recognizes tax benefits from uncertain tax positions only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The tax benefits recognized in the Consolidated Financial Statements from such positions are then measured based on the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement. The Company includes interest and penalty on unrecognized tax benefits as a component of its income tax expense.
The Company recognizes excess tax benefits and tax deficiencies in the provision for income taxes as discrete items in the period when the awards vest or are settled. The Company accounts for Global Intangible Low-Taxed Income (“GILTI”) as period costs when incurred.
Segments
The Company operates in one segment. The chief operating decision maker regularly reviews the operating results of the Company on a consolidated basis as part of making decisions for allocating resources and evaluating performance. As of both December 31, 2022, and 2021, 84% of long-lived assets were in the United States. Revenue from external customers is attributed to individual countries based on customer location.
Legal Contingencies
From time to time, the Company is involved in a number of legal proceedings involving product liability, intellectual property, shareholder derivative actions, securities class actions, and other matters. A liability and related charge are recorded to earnings in the Company’s Consolidated Financial Statements for legal contingencies when the loss is considered probable and the amount can be reasonably estimated. The assessment is re-evaluated each period and is based on all available information,
including discussion with outside legal counsel. If a reasonable estimate of a known or probable loss cannot be made, but a range of probable losses can be estimated, the low-end of the range of losses is recognized if no amount within the range is a better estimate than any other. If a material loss is reasonably possible but not probable and can be reasonably estimated, the estimated loss or range of loss is disclosed in the notes to the Consolidated Financial Statements. The Company expenses legal fees as incurred.
When determining the estimated probable loss or range of losses, significant judgment is required to be exercised in order to estimate the amount and timing of the loss to be recorded. Estimates of probable losses resulting from litigation are inherently difficult to make, particularly when the matters are in early procedural stages with incomplete facts and information. The final outcome of legal proceedings is dependent on many variables that are difficult to predict and, therefore, the ultimate cost to entirely resolve such matters may be materially different than the amount of current estimates. Consequently, new information or changes in judgments and estimates could have a material adverse effect on the Company’s business, financial condition, and results of operations or cash flows.
Recently Adopted Accounting Pronouncements
Business Combinations
In October 2021, the FASB issued ASU No. 2021-08, Business Combinations (Topic 805): Accounting for Contract Assets and Contract Liabilities from Contracts with Customers (“ASU 2021-08”), which creates an exception to the general recognition and measurement principle in ASC 805 by requiring companies to apply ASC 606 to recognize and measure contract assets and contract liabilities from contracts with customers acquired in a business combination. The guidance additionally clarifies that companies should apply the definition of a performance obligation in ASC 606 when recognizing contract liabilities assumed in a business combination. The Company early adopted ASU 2021-08 as of January 1, 2022, on a prospective basis. The impact of the adoption of ASU 2021-08 had an immaterial impact on the Company’s Financial Statements in the year ended December 31, 2022.
Fair Value Measurement of Equity Securities Subject to Contractual Sale Restrictions
In June 2022, the FASB issued ASU No. 2022-03, Fair Value Measurement (Topic 820): Fair Value Measurement of Equity Securities Subject to Contractual Sale Restrictions (“ASU 2022-03”), which applies to all equity securities measured at fair value that are subject to contractual sale restrictions. This change prohibits entities from taking into account contractual restrictions on the sale of equity securities when estimating fair value and introduces required disclosures for such transactions. The Company early adopted ASU 2022-03 as of July 1, 2022, on a prospective basis. There was no impact of the adoption of ASU 2022-03 on the Company’s Financial Statements in the year ended December 31, 2022.
Recently Issued Accounting Pronouncements
Troubled Debt Restructurings and Vintage Disclosures
In March 2022, the FASB issued ASU No. 2022-02, Financial Instruments-Credit Losses (Topic 326): Troubled Debt Restructurings and Vintage Disclosures (“ASU 2022-02”), which eliminates the accounting guidance for troubled debt restructurings by creditors while enhancing disclosure requirements for certain loan refinancings and restructurings by creditors when a borrower is experiencing financial difficulty. Additionally, the standard requires disclosure of current-period gross write-offs by year of origination for financing receivables and net investments in leases within the scope of Subtopic ASC 326-20, Financial Instruments-Credit Losses-Measured at Amortized Cost. The standard will become effective for the Company beginning January 1, 2023, and should be applied prospectively. The adoption of ASU 2022-02 is not expected to have a material impact on the Company’s future Financial Statements.
NOTE 3. FINANCIAL INSTRUMENTS
Cash, Cash Equivalents, and Investments
The following tables summarize the Company’s cash and available-for-sale debt securities’ amortized cost, gross unrealized gains, gross unrealized losses, allowance for credit loss, and fair value by significant investment category reported as cash and cash equivalents, short-term investments, or long-term investments as of December 31, 2022, and 2021 (in millions):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | Reported as: |
| Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Allowance for Credit Loss | | Fair
Value | | Cash and
Cash
Equivalents | | Short-term
Investments | | Long-term
Investments |
| December 31, 2022 | | | | | | | | | | | | | | | |
| Cash | $ | 497.2 | | | $ | — | | | $ | — | | | $ | — | | | $ | 497.2 | | | $ | 497.2 | | | $ | — | | | $ | — | |
| Level 1: | | | | | | | | | | | | | | | |
| Money market funds | 1,084.0 | | | — | | | — | | | — | | | 1,084.0 | | | 1,084.0 | | | — | | | — | |
| U.S. treasuries | 2,715.2 | | | — | | | (96.6) | | | — | | | 2,618.6 | | | — | | | 1,542.4 | | | 1,076.2 | |
| Subtotal | 3,799.2 | | | — | | | (96.6) | | | — | | | 3,702.6 | | | 1,084.0 | | | 1,542.4 | | | 1,076.2 | |
| Level 2: | | | | | | | | | | | | | | | |
| Commercial paper | 20.0 | | | — | | | — | | | — | | | 20.0 | | | — | | | 20.0 | | | — | |
| Corporate debt securities | 2,022.0 | | | — | | | (76.0) | | | (1.1) | | | 1,944.9 | | | — | | | 651.8 | | | 1,293.1 | |
| U.S. government agencies | 447.2 | | | — | | | (19.9) | | | — | | | 427.3 | | | — | | | 247.8 | | | 179.5 | |
| | | | | | | | | | | | | | | |
| Municipal securities | 155.5 | | | — | | | (6.0) | | | — | | | 149.5 | | | — | | | 74.7 | | | 74.8 | |
| Subtotal | 2,644.7 | | | — | | | (101.9) | | | (1.1) | | | 2,541.7 | | | — | | | 994.3 | | | 1,547.4 | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| Total assets measured at fair value | $ | 6,941.1 | | | $ | — | | | $ | (198.5) | | | $ | (1.1) | | | $ | 6,741.5 | | | $ | 1,581.2 | | | $ | 2,536.7 | | | $ | 2,623.6 | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | Reported as: |
| Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Allowance for Credit Loss | | Fair
Value | | Cash and
Cash
Equivalents | | Short-term
Investments | | Long-term
Investments |
| December 31, 2021 | | | | | | | | | | | | | | | |
| Cash | $ | 572.3 | | | $ | — | | | $ | — | | | $ | — | | | $ | 572.3 | | | $ | 572.3 | | | $ | — | | | $ | — | |
| Level 1: | | | | | | | | | | | | | | | |
| Money market funds | 696.6 | | | — | | | — | | | — | | | 696.6 | | | 696.6 | | | — | | | — | |
| U.S. treasuries | 3,429.1 | | | 6.3 | | | (15.4) | | | — | | | 3,420.0 | | | 17.0 | | | 1,100.3 | | | 2,302.7 | |
| Subtotal | 4,125.7 | | | 6.3 | | | (15.4) | | | — | | | 4,116.6 | | | 713.6 | | | 1,100.3 | | | 2,302.7 | |
| Level 2: | | | | | | | | | | | | | | | |
| Commercial paper | 717.7 | | | — | | | — | | | — | | | 717.7 | | | — | | | 717.7 | | | — | |
| Corporate debt securities | 2,485.6 | | | 2.7 | | | (11.9) | | | — | | | 2,476.4 | | | 5.0 | | | 886.7 | | | 1,584.7 | |
| U.S. government agencies | 526.1 | | | 0.2 | | | (2.9) | | | — | | | 523.4 | | | — | | | 137.8 | | | 385.6 | |
| | | | | | | | | | | | | | | |
| Municipal securities | 213.4 | | | 0.7 | | | (1.0) | | | — | | | 213.1 | | | — | | | 70.6 | | | 142.5 | |
| Subtotal | 3,942.8 | | | 3.6 | | | (15.8) | | | — | | | 3,930.6 | | | 5.0 | | | 1,812.8 | | | 2,112.8 | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| Total assets measured at fair value | $ | 8,640.8 | | | $ | 9.9 | | | $ | (31.2) | | | $ | — | | | $ | 8,619.5 | | | $ | 1,290.9 | | | $ | 2,913.1 | | | $ | 4,415.5 | |
The following table summarizes the contractual maturities of the Company’s cash equivalents and available-for-sale debt securities (excluding money market funds) as of December 31, 2022 (in millions):
| | | | | | | | | | | |
| Amortized
Cost | | Fair
Value |
| Mature in less than one year | $ | 2,586.6 | | | $ | 2,536.7 | |
| Mature in one to five years | 2,773.3 | | | 2,623.6 | |
| Total | $ | 5,359.9 | | | $ | 5,160.3 | |
| | | |
| | | |
| | | |
| | | |
Actual maturities may differ from contractual maturities, because certain borrowers have the right to call or prepay certain obligations. Gross realized gains and losses recognized on the sale of investments were immaterial for the years ended December 31, 2022, and 2021.
As of December 31, 2022, and 2021, net unrealized losses on available-for-sale debt securities, net of tax, of $154.2 million and $16.0 million, respectively, were included in accumulated other comprehensive loss in the accompanying Consolidated Balance Sheets.
The following tables present the breakdown of the available-for-sale debt securities with unrealized losses as of December 31, 2022, and 2021 (in millions):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Unrealized losses less
than 12 months | | Unrealized losses 12
months or greater | | Total |
| December 31, 2022 | Fair
Value | | Unrealized
Losses | | Fair
Value | | Unrealized
Losses | | Fair
Value | | Unrealized
Losses |
| U.S. treasuries | $ | 731.7 | | | $ | (26.0) | | | $ | 1,886.9 | | | $ | (70.6) | | | $ | 2,618.6 | | | $ | (96.6) | |
| | | | | | | | | | | |
| Corporate debt securities | 631.4 | | | (17.6) | | | 1,221.9 | | | (58.4) | | | 1,853.3 | | | (76.0) | |
| U.S. government agencies | 102.7 | | | (4.4) | | | 324.6 | | | (15.5) | | | 427.3 | | | (19.9) | |
| Municipal securities | 44.6 | | | (1.1) | | | 104.9 | | | (4.9) | | | 149.5 | | | (6.0) | |
| Total | $ | 1,510.4 | | | $ | (49.1) | | | $ | 3,538.3 | | | $ | (149.4) | | | $ | 5,048.7 | | | $ | (198.5) | |
| | | | | | | | | | | |
| December 31, 2021 | | | | | | | | | | | |
| U.S. treasuries | $ | 2,596.3 | | | $ | (15.4) | | | $ | — | | | $ | — | | | $ | 2,596.3 | | | $ | (15.4) | |
| Commercial paper | 4.0 | | | — | | | — | | | — | | | 4.0 | | | — | |
| Corporate debt securities | 1,687.9 | | | (11.9) | | | — | | | — | | | 1,687.9 | | | (11.9) | |
| U.S. government agencies | 412.5 | | | (2.9) | | | — | | | — | | | 412.5 | | | (2.9) | |
| Municipal securities | 156.0 | | | (1.0) | | | — | | | — | | | 156.0 | | | (1.0) | |
| Total | $ | 4,856.7 | | | $ | (31.2) | | | $ | — | | | $ | — | | | $ | 4,856.7 | | | $ | (31.2) | |
The unrealized losses on the Company’s available-for-sale debt securities were caused by interest rate increases. The contractual terms of those investments do not permit the issuer to settle the securities at a price less than the amortized cost basis of the investments. As of December 31, 2022, the Company does not intend to sell the investments, and it is not more likely than not that the Company will be required to sell the investments before recovery of their amortized cost basis, which may be at maturity. Additional factors considered in determining the treatment of unrealized losses include the financial condition and near-term prospects of the investee, the extent of the loss related to the credit of the issuer, and the expected cash flows from the security.
Equity Investments
The following table is a summary of the activity related to equity investments (in millions):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | Reported as: |
| December 31, 2021 Carrying Value | | Changes in Fair Value (1) | | Purchases / Sales / Other (2) | | December 31, 2022 Carrying Value | | Prepaids and other current assets | | Intangible and other assets, net |
| Equity investments with readily determinable value (Level 1) | $ | 26.9 | | | (21.2) | | | (1.4) | | | $ | 4.3 | | | $ | 4.3 | | | $ | — | |
| Equity investments without readily determinable value (Level 2) | $ | 15.6 | | | 0.2 | | | 43.3 | | | $ | 59.1 | | | $ | — | | | $ | 59.1 | |
| | | | | | | | | | | |
(1) Recorded in interest and other income, net. |
(2) Other includes foreign currency translation gains/(losses). |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
In September 2021, Broncus Holding Corporation (“Broncus”) completed an initial public offering (“IPO”) of common shares on the Stock Exchange of Hong Kong. Upon completion of its IPO, the Company’s preferred shares of Broncus were converted into common shares, which have a readily determinable value (Level 1). The Company was restricted from selling these shares for a period of six months. In 2022, the Company recognized a $21.2 million loss on this investment reflected in changes in fair value for Level 1 equity investments, which was reflected in Interest and other income, net.
The Company recognized a $0.2 million increase in fair value, which was reflected in interest and other income, net, due to changes in observable prices for certain equity investments that lack readily determinable market values (Level 2). There were no decreases in fair value reflected in net income due to impairments.
Foreign Currency Derivatives
The objective of the Company’s hedging program is to mitigate the impact of changes in currency exchange rates on net cash flow from foreign currency-denominated sales, expenses, intercompany balances, and other monetary assets or liabilities denominated in currencies other than the U.S. dollar (“USD”). The terms of the Company’s derivative contracts are generally twelve months or shorter. The derivative assets and liabilities are measured using Level 2 fair value inputs.
Cash Flow Hedges. The Company enters into currency forward contracts as cash flow hedges to hedge certain forecasted revenue transactions denominated in currencies other than the USD, primarily the Euro (“EUR”), the British Pound (“GBP”), the Japanese Yen (“JPY”), the Korean Won (“KRW”), and the New Taiwan Dollar (“TWD”). The Company also enters into currency forward contracts as cash flow hedges to hedge certain forecasted expense transactions denominated in EUR and the Swiss Franc (“CHF”).
For these derivatives, the Company reports the unrealized after-tax gain or loss from the hedge as a component of accumulated other comprehensive loss in stockholders’ equity and reclassifies the amount into earnings in the same period in which the hedged transaction affects earnings. The amounts reclassified to revenue and expenses related to the hedged transactions and the ineffective portions of cash flow hedges were not material for the periods presented.
Other Derivatives Not Designated as Hedging Instruments. Other derivatives not designated as hedging instruments consist primarily of forward contracts that the Company uses to hedge intercompany balances and other monetary assets or liabilities denominated in currencies other than the USD, primarily the EUR, GBP, JPY, KRW, CHF, TWD, Indian Rupee (“INR”), Mexican Peso (“MXN”), Chinese Yuan (“CNY”), and Canadian Dollar (“CAD”).
These derivative instruments are used to hedge against balance sheet foreign currency exposures. The related gains and losses were as follows (in millions):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2022 | | 2021 | | 2020 |
| Recognized gains (losses) in interest and other income, net | $ | 26.9 | | | $ | 15.5 | | | $ | (12.3) | |
| Foreign exchange gains (losses) related to balance sheet re-measurement | $ | (54.2) | | | $ | (16.4) | | | $ | 10.9 | |
The notional amounts for derivative instruments provide one measure of the transaction volume. Total gross notional amounts (in USD) for outstanding derivatives and the aggregate gross fair value at the end of each period were as follows (in millions):
| | | | | | | | | | | | | | | | | | | | | | | |
| Derivatives Designated as Hedging Instruments | | Derivatives Not Designated as Hedging Instruments |
| December 31,
2022 | | December 31,
2021 | | December 31,
2022 | | December 31,
2021 |
| Notional amounts: | | | | | | | |
| Forward contracts | $ | 188.4 | | | $ | 181.2 | | | $ | 496.3 | | | $ | 318.8 | |
| Gross fair value recorded in: | | | | | | | |
| Prepaids and other current assets | $ | 1.8 | | | $ | 5.7 | | | $ | 4.3 | | | $ | 6.9 | |
| Other accrued liabilities | $ | 5.3 | | | $ | 0.5 | | | $ | 4.2 | | | $ | 0.8 | |
NOTE 4. CONSOLIDATED FINANCIAL STATEMENT DETAILS
The following tables provide details of selected consolidated financial statement items (in millions):
| | | | | | | | | | | |
| | December 31, |
| 2022 | | 2021 |
| Accounts receivable, net | | | |
| Trade accounts receivable, net | $ | 864.9 | | | $ | 731.0 | |
| | | |
| | | |
| Unbilled accounts receivable and other | 91.7 | | | 64.8 | |
| Sales returns and allowances | (14.5) | | | (13.1) | |
| Total accounts receivable, net | $ | 942.1 | | | $ | 782.7 | |
| | | | | | | | | | | |
| | December 31, |
| 2022 | | 2021 |
| Inventory | | | |
| Raw materials | $ | 382.9 | | | $ | 214.6 | |
| Work-in-process | 159.9 | | | 96.4 | |
| Finished goods | 350.4 | | | 276.1 | |
| Total inventory | $ | 893.2 | | | $ | 587.1 | |
| | | | | | | | | | | |
| December 31, |
| 2022 | | 2021 |
| Prepaids and other current assets | | | |
| Net investment in sales-type leases – short-term | 131.2 | | | 110.3 | |
| Equity investments | 4.3 | | | 26.9 | |
| Other prepaids and other current assets | 164.3 | | | 133.9 | |
| Total prepaids and other current assets | $ | 299.8 | | | $ | 271.1 | |
| | | | | | | | | | | |
| | December 31, |
| | 2022 | | 2021 |
| Property, plant, and equipment, net | | | |
| Land | $ | 388.6 | | | $ | 367.8 | |
| Building and building/leasehold improvements | 866.5 | | | 812.5 | |
| Machinery and equipment | 566.4 | | | 497.6 | |
| Operating lease assets – Intuitive System Leasing | 806.4 | | | 616.1 | |
| Computer and office equipment | 134.7 | | | 123.7 | |
| Capitalized software | 240.9 | | | 217.6 | |
| Construction-in-process | 608.6 | | | 209.7 | |
| Gross property, plant, and equipment | 3,612.1 | | | 2,845.0 | |
| Less: Accumulated depreciation* | (1,237.9) | | | (968.6) | |
| Total property, plant, and equipment, net | $ | 2,374.2 | | | $ | 1,876.4 | |
| | | |
| *Accumulated depreciation associated with operating lease assets – Intuitive System Leasing | $ | (285.8) | | | $ | (182.1) | |
| | | | | | | | | | | |
| | December 31, |
| 2022 | | 2021 |
| Other accrued liabilities – short-term | | | |
| Income and other taxes payable | $ | 96.1 | | | $ | 54.1 | |
| Accrued construction-related capital expenditures | 50.3 | | | 23.1 | |
| Litigation-related accruals | 23.0 | | | 2.4 | |
| Current portion of deferred and contingent purchase consideration | 7.8 | | | 12.0 | |
| Other accrued liabilities | 299.0 | | | 209.7 | |
| Total other accrued liabilities – short-term | $ | 476.2 | | | $ | 301.3 | |
| | | | | | | | | | | |
| | December 31, |
| | 2022 | | 2021 |
| Other long-term liabilities | | | |
| Income taxes – long-term | $ | 288.0 | | | $ | 316.6 | |
| Deferred revenue – long-term | 41.0 | | | 36.8 | |
| Other long-term liabilities | 110.3 | | | 100.3 | |
| Total other long-term liabilities | $ | 439.3 | | | $ | 453.7 | |
Supplemental Cash Flow Information
The following table provides supplemental cash flow information (in millions):
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| 2022 | | 2021 | | 2020 |
| Income taxes paid | $ | 444.2 | | | $ | 180.0 | | | $ | 34.4 | |
| Supplemental non-cash investing and financing activities: | | | | | |
| Equipment transfers from inventory to property, plant, and equipment | $ | 279.2 | | | $ | 302.4 | | | $ | 186.5 | |
| Acquisition of property, plant, and equipment in accounts payable and accrued liabilities | $ | 73.4 | | | $ | 32.1 | | | $ | 47.3 | |
NOTE 5. REVENUE
The following table presents revenue disaggregated by types and geography (in millions):
| | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| U.S. | | 2022 | | 2021 | | 2020 |
| Instruments and accessories | | $ | 2,507.2 | | | $ | 2,225.1 | | | $ | 1,785.1 | |
| Systems | | 966.0 | | | 1,024.8 | | | 695.0 | |
| Services | | 684.4 | | | 603.3 | | | 482.6 | |
| Total U.S. revenue | | $ | 4,157.6 | | | $ | 3,853.2 | | | $ | 2,962.7 | |
| | | | | | |
OUS | | | | | | |
| Instruments and accessories | | $ | 1,010.7 | | | $ | 875.4 | | | $ | 670.6 | |
| Systems | | 714.1 | | | 668.6 | | | 483.9 | |
| Services | | 339.8 | | | 312.9 | | | 241.2 | |
| Total OUS revenue | | $ | 2,064.6 | | | $ | 1,856.9 | | | $ | 1,395.7 | |
| | | | | | |
| Total | | | | | | |
| Instruments and accessories | | $ | 3,517.9 | | | $ | 3,100.5 | | | $ | 2,455.7 | |
| Systems | | 1,680.1 | | | 1,693.4 | | | 1,178.9 | |
| Services | | 1,024.2 | | | 916.2 | | | 723.8 | |
| Total revenue | | $ | 6,222.2 | | | $ | 5,710.1 | | | $ | 4,358.4 | |
Remaining Performance Obligations
The transaction price allocated to remaining performance obligations relates to amounts allocated to products and services for which revenue has not yet been recognized. A significant portion of these performance obligations relate to service obligations in the Company’s system sale and lease arrangements that will be satisfied and recognized as revenue in future periods. The transaction price allocated to the remaining performance obligations was $1.86 billion as of December 31, 2022. The remaining performance obligations are expected to be satisfied over the term of the system sale, lease, and service arrangements, which are generally up to 5 years.
Contract Assets and Liabilities
The following information summarizes the Company’s contract assets and liabilities (in millions):
| | | | | | | | | | | |
| December 31, |
| 2022 | | 2021 |
| Contract assets | $ | 45.0 | | | $ | 46.9 | |
| Deferred revenue | $ | 438.3 | | | $ | 414.0 | |
The Company invoices its customers based on the billing schedules in its sales arrangements. Payments are generally due 30 to 60 days from the date of invoice. Contract assets for the periods presented primarily represent the difference between the revenue that was recognized based on the relative standalone selling price of the related performance obligations satisfied and the contractual billing terms in the arrangements. Deferred revenue for the periods presented primarily relates to service contracts where the service fees are billed up-front, generally quarterly or annually, prior to those services having been performed. The associated deferred revenue is generally recognized over the term of the service period. The Company did not have any significant impairment losses on its contract assets for the periods presented.
During the year ended December 31, 2022, the Company recognized $380 million of revenue that was included in the deferred revenue balance as of December 31, 2021. During the year ended December 31, 2021, the Company recognized $351 million of revenue that was included in the deferred revenue balance as of December 31, 2020.
Intuitive System Leasing
The following table presents product revenue from Intuitive System Leasing arrangements (in millions):
| | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2022 | | 2021 | | 2020 |
| Sales-type lease revenue | | $ | 156.4 | | | $ | 220.3 | | | $ | 154.4 | |
| Operating lease revenue* | | $ | 376.5 | | | $ | 276.9 | | | $ | 176.7 | |
| | | | | | |
*Variable lease revenue relating to usage-based arrangements included within operating lease revenue | | $ | 133.0 | | | $ | 78.1 | | | $ | 27.5 | |
NOTE 6. LEASES
Lessor Information related to Intuitive System Leasing
Sales-type Leases. Lease receivables relating to sales-type lease arrangements are presented on the Consolidated Balance Sheets as follows (in millions):
| | | | | | | | | | | |
| December 31, |
| 2022 | | 2021 |
| Gross lease receivables | $ | 449.4 | | | $ | 404.0 | |
| Unearned income | (14.4) | | | (11.4) | |
| Subtotal | 435.0 | | | 392.6 | |
| Allowance for credit loss | (3.0) | | | (3.6) | |
| Net investment in sales-type leases | $ | 432.0 | | | $ | 389.0 | |
| | | |
| Reported as: | | | |
| Prepaids and other current assets | $ | 131.2 | | | $ | 110.3 | |
| Intangible and other assets, net | 300.8 | | | 278.7 | |
| Net investment in sales-type leases | $ | 432.0 | | | $ | 389.0 | |
Contractual maturities of gross lease receivables as of December 31, 2022, are as follows (in millions):
| | | | | |
| Fiscal Year | Amount |
| 2023 | $ | 137.5 | |
| 2024 | 126.3 | |
| 2025 | 95.5 | |
| 2026 | 60.3 | |
| 2027 | 26.5 | |
| 2028 and thereafter | 3.3 | |
| Total | $ | 449.4 | |
Operating Leases. The Company’s operating lease terms are generally less than seven years. Future minimum lease payments related to the non-cancellable portion of operating leases (which excludes contingent payments related to usage-based arrangements) as of December 31, 2022, are as follows (in millions):
| | | | | |
| Fiscal Year | Amount |
| 2023 | $ | 300.0 | |
| 2024 | 250.5 | |
| 2025 | 194.4 | |
| 2026 | 130.2 | |
| 2027 | 58.7 | |
| 2028 and thereafter | 20.2 | |
| Total | $ | 954.0 | |
Lessee Information
The Company enters into operating leases for real estate, automobiles, and certain equipment. Operating lease expense was $25.7 million, $20.4 million, and $21.0 million for the years ended December 31, 2022, 2021, and 2020, respectively. For leases with terms of 12 months or less, the related expense was immaterial for each of the years ended December 31, 2022, 2021, and 2020.
Supplemental cash flow information for the years ended December 31, 2022, 2021, and 2020 related to operating leases was as follows (in millions): | | | | | | | | | | | | | | | | | | | | |
| Years Ended December 31, | | | |
| 2022 | | 2021 | | 2020 | | | |
| Cash paid for leases that were included within operating cash outflows | $ | 33.8 | | | $ | 23.2 | | | $ | 11.0 | | | | |
| Right-of-use assets recognized related to new lease obligations | $ | 34.0 | | | $ | 30.6 | | | $ | 9.6 | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Supplemental balance sheet information, as of December 31, 2022, and 2021, related to operating leases was as follows (in millions, except lease term and discount rate): | | | | | | | | | | | | | | |
| December 31, | | | |
| 2022 | | 2021 | | | |
| Intangible and other assets, net (Right-of-use assets) | $ | 82.2 | | | $ | 74.4 | | | | |
| | | | | | |
| Other accrued liabilities | $ | 24.2 | | | $ | 20.4 | | | | |
| Other long-term liabilities | 69.6 | | | 66.6 | | | | |
| Total lease liabilities | $ | 93.8 | | | $ | 87.0 | | | | |
| | | | | | |
| Weighted-average remaining lease term | 4.5 years | | 4.9 years | | | |
| Weighted-average discount rate | 3.0 | % | | 2.5 | % | | | |
As of December 31, 2022, the future payments related to the Company’s operating lease liabilities are scheduled as follows (in millions): | | | | | | | | |
| Fiscal Year | Amount | | | |
| 2023 | $ | 26.0 | | | | |
| 2024 | 21.7 | | | | |
| 2025 | 19.5 | | | | |
| 2026 | 16.6 | | | | |
| 2027 | 8.3 | | | | |
| 2028 and thereafter | 8.9 | | | | |
| Total lease payments | 101.0 | | | | |
| Less: imputed interest | (7.2) | | | | |
| Total operating lease liabilities | $ | 93.8 | | | | |
NOTE 7. GOODWILL AND INTANGIBLE ASSETS
Acquisitions
There were no material acquisitions in 2022 and 2021.
In February 2020, the Company acquired Orpheus Medical Ltd. and its wholly owned subsidiaries (“Orpheus Medical”) to deepen and expand the Company’s integrated informatics platform. Orpheus Medical provides hospitals with information technology connectivity, as well as expertise in processing and archiving surgical videos.
Goodwill
The following table summarizes the changes in the carrying amount of goodwill (in millions):
| | | | | | | | |
| | Amount |
Balance as of December 31, 2020 | | $ | 336.7 | |
| Acquisition activity | | 8.0 | |
| Translation and other | | (1.1) | |
Balance as of December 31, 2021 | | 343.6 | |
| Acquisition activity | | 6.5 | |
| Translation and other | | (1.6) | |
Balance as of December 31, 2022 | | $ | 348.5 | |
The Company completed its annual goodwill impairment test and determined that no impairment existed. As of December 31, 2022, there has been no impairment of goodwill.
Intangible Assets
The following table summarizes the components of gross intangible asset, accumulated amortization, and net intangible asset balances as of December 31, 2022, and 2021 (in millions):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | December 31, 2022 | | December 31, 2021 |
| | Gross Carrying Amount | | Accumulated Amortization | | Net
Carrying
Amount | | Gross Carrying Amount | | Accumulated Amortization | | Net
Carrying
Amount |
| Patents and developed technology | | $ | 199.1 | | | $ | (167.4) | | | $ | 31.7 | | | $ | 219.3 | | | $ | (173.2) | | | $ | 46.1 | |
| Distribution rights and others | | 11.0 | | | (7.4) | | | 3.6 | | | 26.3 | | | (19.4) | | | 6.9 | |
| Customer relationships | | 32.6 | | | (18.1) | | | 14.5 | | | 31.8 | | | (14.3) | | | 17.5 | |
| Total intangible assets | | $ | 242.7 | | | $ | (192.9) | | | $ | 49.8 | | | $ | 277.4 | | | $ | (206.9) | | | $ | 70.5 | |
Amortization expense related to intangible assets was $27.8 million, $27.4 million, and $49.8 million for the years ended December 31, 2022, 2021, and 2020, respectively.
The estimated future amortization expense related to intangible assets as of December 31, 2022, is as follows (in millions):
| | | | | |
| Fiscal Year | Amount |
| 2023 | $ | 19.2 | |
| 2024 | 15.0 | |
| 2025 | 10.3 | |
| 2026 | 3.4 | |
| 2027 | 1.0 | |
| 2028 and thereafter | 0.9 | |
| Total | $ | 49.8 | |
The preceding expected amortization expense is an estimate. Actual amounts of amortization expense may differ from estimated amounts due to additional intangible asset acquisitions, measurement period adjustments to intangible assets, changes in foreign currency exchange rates, impairment of intangible assets, accelerated amortization of intangible assets, and other events.
NOTE 8. COMMITMENTS AND CONTINGENCIES
Commitments
As of December 31, 2022, the Company’s commitments include an estimated amount of approximately $2.14 billion relating to the Company’s open purchase orders and contractual obligations that occur in the ordinary course of business, including commitments with contract manufacturers and suppliers for which the Company has not received the goods or services, commitments for capital expenditures and construction-related activities for which the Company has not received the services, and acquisition and licensing of intellectual property. Although open purchase orders are considered enforceable and legally binding, the terms generally allow the Company the option to cancel, reschedule, and adjust its requirements based on
its business needs prior to the delivery of goods or performance of services. Additionally, the Company has committed to making certain future milestone payments to third parties as part of licensing, collaboration, and development arrangements. Payments under these arrangements generally become due and payable only upon the achievement of certain specified developmental, regulatory, and/or commercial milestones. For instances in which the achievement of these milestones is neither probable nor reasonably estimable, such contingencies are not included in the estimated amount.
Contingencies
From time to time, the Company is involved in a variety of claims, lawsuits, investigations, and proceedings relating to securities laws, product liability, intellectual property, commercial, insurance, contract disputes, employment, and other matters. Certain of these lawsuits and claims are described in further detail below. It is not possible to predict what the outcome of these matters will be, and the Company cannot guarantee that any resolution will be reached on commercially reasonable terms, if at all.
A liability and related charge to earnings are recorded in the Consolidated Financial Statements for legal contingencies when the loss is considered probable and the amount can be reasonably estimated. The assessment is re-evaluated each accounting period and is based on all available information, including the impact of negotiations, settlements, rulings, advice of legal counsel, and other information and events pertaining to each case. Nevertheless, it is possible that additional future legal costs (including settlements, judgments, legal fees, and other related defense costs) could have a material adverse effect on the Company’s business, financial condition, or future results of operations.
The Company incurred $28.1 million of pre-tax litigation charges in 2022. During the years ended 2021 and 2020, pre-tax litigation charges were insignificant.
Product Liability Litigation
The Company is currently named as a defendant in a number of individual product liability lawsuits filed in various state and federal courts. The plaintiffs generally allege that they or a family member underwent surgical procedures that utilized the da Vinci Surgical System and sustained a variety of personal injuries and, in some cases, death as a result of such surgery. Several of the filed cases have trial dates in the next 12 months.
The cases raise a variety of allegations including, to varying degrees, that plaintiffs’ injuries resulted from purported defects in the da Vinci Surgical System and/or failure on the Company’s part to provide adequate training resources to the healthcare professionals who performed plaintiffs’ surgeries. The cases further allege that the Company failed to adequately disclose and/or misrepresented the potential risks and/or benefits of the da Vinci Surgical System. Plaintiffs also assert a variety of causes of action, including, for example, strict liability based on purported design defects, negligence, fraud, breach of express and implied warranties, unjust enrichment, and loss of consortium. Plaintiffs seek recovery for alleged personal injuries and, in many cases, punitive damages. The Company disputes these allegations and is defending against these claims.
The Company’s estimate of the anticipated cost of resolving the pending cases is based on negotiations with attorneys for the claimants. The final outcome of the pending lawsuits and claims, and others that might arise, is dependent on many variables that are difficult to predict, and the ultimate cost associated with these product liability lawsuits and claims may be materially different than the amount of the current estimate and accruals and could have a material adverse effect on the Company’s business, financial condition, or future results of operations. Although there is a reasonable possibility that a loss in excess of the amount recognized exists, the Company is unable to estimate the possible loss or range of loss in excess of the amount recognized at this time.
Patent Litigation
On June 30, 2017, Ethicon LLC, Ethicon Endo-Surgery, Inc., and Ethicon US LLC (collectively, “Ethicon”) filed a complaint for patent infringement against the Company in the U.S. District Court for the District of Delaware. The complaint, which was served on the Company on July 12, 2017, alleges that the Company’s EndoWrist Stapler instruments infringe several of Ethicon’s patents. Ethicon asserts infringement of U.S. Patent Nos. 9,585,658; 8,479,969; 9,113,874; 8,998,058; 8,991,677; 9,084,601; and 8,616,431. A claim construction hearing occurred on October 1, 2018, and the Court issued a scheduling order on December 28, 2018. On March 20, 2019, the Court granted the Company’s Motion to Stay pending an Inter Partes Review to be held at the Patent Trademark and Appeals Board to review patentability of six of the seven patents noted above and vacated the trial date. On August 1, 2019, the Court granted the parties’ joint stipulation to modify the stay in light of Ethicon’s U.S. International Trade Commission (“USITC”) complaint against Intuitive involving U.S. Patent Nos. 8,479,969 and 9,113,874, discussed below. There is currently no trial date scheduled for this matter.
On August 27, 2018, Ethicon filed a second complaint for patent infringement against the Company in the U.S. District Court for the District of Delaware. The complaint alleges that the Company’s SureForm 60 Staplers infringe five of Ethicon’s patents. Ethicon asserts infringement of U.S. Patent Nos. 9,884,369; 7,490,749; 8,602,288; 8,602,287; and 9,326,770. The Company filed an answer denying all claims. On March 19, 2019, Ethicon filed a Motion for Leave to File a First Amended
Complaint, removing allegations related to U.S. Patent No. 9,326,770 and adding allegations related to U.S. Patent Nos. 9,844,379 and 8,479,969. On July 17, 2019, the Court entered an order denying the amendment, without prejudice, and granting the parties’ joint stipulation to stay the case in its entirety in light of the USITC investigation involving U.S. Patent Nos. 9,844,369 and 7,490,749, discussed below. There is currently no trial date scheduled for this matter.
Based on currently available information, the Company is unable to make a reasonable estimate of loss or range of losses, if any, arising from these matters.
On May 30, 2019, Ethicon filed a complaint with the USITC, asserting infringement of U.S. Patent Nos. 9,884,369 (“’369”); 7,490,749 (“’749”); 9,844,379 (“’379”); 9,113,874 (“’874”); and 8,479,969 (“’969”). On June 28, 2019, the USITC voted to institute an investigation (No. 337-TA-1167) with respect to the claims in this complaint. The accused products include the Company’s EndoWrist 30, EndoWrist 45, SureForm 45, and SureForm 60 Staplers, as well as the stapler reload cartridges. In March 2020, Ethicon dismissed its claims concerning the ’749 patent. The evidentiary hearing took place in February 2021. On March 26, 2021, the U.S. Patent Trial and Appeal Board (“PTAB”) issued a Final Written Decision in which it found the claims in the ’379 patent asserted against the Company in this USITC proceeding to be invalid. On June 8, 2021, the Chief Administrative Law Judge issued an Initial Determination concluding that (1) the accused products do not infringe the asserted claims in the ’874 or ’969 patents; (2) the asserted claims in the ’874 and ’969 patents are invalid; (3) the accused SureForm staplers and associated reload cartridges infringe two claims of the ’369 patent; (4) the accused SureForm staplers and associated reload cartridges infringe two claims of the ’379 patent; and (5) the Company was estopped from contending that the asserted claims in the ’379 patent are invalid. Ethicon has not challenged the Initial Determination with regard to the findings that absolve Intuitive of any liability regarding the accused EndoWrist staplers and associated reload cartridges. On October 14, 2021, the USITC issued its Opinion in which it made the following rulings: (1) the USITC absolved Intuitive from any liability regarding the ’874, ’969, and ’369 patents; and (2) the USITC found that, while the SureForm staplers and their associated reload cartridges infringe the asserted claims in the ’379 patent, it has suspended the imposition of any remedial order pending an opinion from the U.S. Court of Appeal for the Federal Circuit of whether the Patent and Trademark Office correctly found the asserted claims in this patent to be invalid. On May 23, 2022, the U.S. Court of Appeal for the Federal Circuit affirmed the earlier PTAB Final Written Decision invalidating the asserted claims in the ’379 patent. A hearing before the U.S. Court of Appeal for the Federal Circuit has been scheduled for March 8, 2023, on Ethicon’s appeal of the USITC’s Opinion. An adverse ruling on Ethicon’s appeal of the USITC’s Opinion could result in a prohibition on importing the accused SureForm products into the U.S. or necessitating workarounds. Based on currently available information, the Company does not believe that any losses arising from this matter would be material.
On October 19, 2022, a jury rendered a verdict against the Company awarding $10 million in damages to Rex Medical, L.P. in a patent infringement lawsuit. The Company intends to appeal the decision and vigorously defend its position. Based on currently available information, the Company does not believe that any losses arising from this matter would be material.
Commercial Litigation
On February 27, 2019, Restore Robotics LLC and Restore Repair LLC (“Restore”) filed a complaint in the Northern District of Florida alleging anti-trust claims against the Company. On May 13, 2019, Restore filed an amended complaint alleging anti-trust claims relating to the da Vinci Surgical System and EndoWrist service, maintenance, and repair processes. On September 16, 2019, the Court partially granted and partially denied the Company’s Motion to Dismiss the amended complaint.
On September 30, 2019, the Company filed an answer denying the anti-trust allegations and filed a counterclaim against Restore. The Company filed amended counterclaims after the Court partially granted and partially denied Restore’s Motion to Dismiss the counterclaim. The amended counterclaims allege that Restore violated the Federal Lanham Act, the Federal Computer Fraud and Abuse Act, and Florida’s Deceptive and Unfair Trade Practices Act and that Restore is also liable to the Company for Unfair Competition and Tortious Interference with Contract. On January 7, 2020, the Court denied Restore’s Motion to Dismiss the amended counterclaims.
On April 11, 2022, the Court granted in part and denied in part the parties’ Motions for Summary Judgment. Shortly thereafter, Restore’s licensor, Rebotix Repair LLC (“Rebotix”) filed a notice in a separate legal action that it had received an email from the U.S. Food and Drug Administration (“FDA”) confirming that “extending the number of uses and modifying [EndoWrist] instrument[s] with a new chip” constitutes “remanufacturing” and required 510(k) clearance, which neither Restore nor Rebotix has obtained. In light of this communication with the FDA, on April 13, 2022, the Company filed a Motion for Reconsideration to the Court’s Summary Judgment Order, which the Court denied on May 31, 2022. This case was scheduled for trial in February 2023. On January 23, 2023, the parties resolved all of their claims against each other and, on January 27, 2023, the case was dismissed with prejudice. The settlement had an immaterial impact on the Company’s Consolidated Financial Statements for the year ended December 31, 2022.
Similar to the claims asserted in the Restore case, on May 10, 2021, Surgical Instrument Service Company, Inc. (“SIS”) filed a complaint in the Northern District of California Court alleging anti-trust claims against the Company relating to
EndoWrist service, maintenance, and repair processes. The Court granted in part and denied in part the Company’s Motion to Dismiss, and discovery has commenced. The Company filed an answer denying the anti-trust allegations and filed counterclaims against SIS. The counterclaims allege that SIS violated the Federal Lanham Act, California’s Unfair Competition Law, and California’s False Advertising Law and that SIS is also liable to the Company for Unfair Competition and Tortious Interference with Contract. Based on currently available information, the Company is unable to make a reasonable estimate of loss or range of losses, if any, arising from this matter.
Three class action complaints were filed against the Company in the Northern District of California Court alleging anti-trust allegations relating to the service and repair of certain instruments manufactured by the Company. A complaint by Larkin Community Hospital was filed on May 20, 2021, a complaint by Franciscan Alliance, Inc. and King County Public Hospital District No. 1 was filed on July 6, 2021, and a complaint by Kaleida Health was filed on July 8, 2021. The Court has consolidated the Franciscan Alliance, Inc. and King County Public Hospital District No. 1 and Kaleida Health cases with the Larkin Community Hospital case, which is now captioned on the Larkin docket as “In Re: da Vinci Surgical Robot Antitrust Litigation.” A Consolidated Amended Class Action Complaint has been filed on behalf of each plaintiff named in the earlier-filed cases. On January 14, 2022, Kaleida Health voluntarily dismissed itself as a party to this case. On January 18, 2022, the Company filed an answer against the plaintiffs in this matter, and discovery has commenced. Based on currently available information, the Company is unable to make a reasonable estimate of loss or range of losses, if any, arising from this matter.
NOTE 9. STOCKHOLDERS’ EQUITY
Stock Repurchase Program
Through December 31, 2022, the Company’s Board of Directors (the “Board”) has authorized an aggregate of $10.0 billion of funding for the Company’s common stock repurchase program (the “Repurchase Program”) since its establishment in March 2009. The most recent authorization occurred in July 2022 when the Board increased the authorized amount available under the Repurchase Program to $3.5 billion, including amounts remaining under previous authorization. As of December 31, 2022, the remaining amount of share repurchases authorized by the Board under the Repurchase Program was approximately $1.5 billion.
In August 2022, the Company entered into an accelerated share repurchase program (the “August ASR Program”) with Goldman, Sachs & Co. (“Goldman”) to repurchase $1.0 billion of the Company’s common stock. The Company made a payment of $1.0 billion to Goldman and received an initial delivery of approximately 3.5 million shares of the Company’s common stock, which were subsequently retired. In September 2022, the August ASR Program was completed, and an additional 1.1 million shares of common stock were received and retired. In total, 4.6 million shares were repurchased at an average price per share of $217.52. The total cost of the August ASR Program was reflected as a reduction to equity in the Consolidated Balance Sheets.
In October 2022, the Company entered into an accelerated share repurchase program (the “October ASR Program”) with Citibank, N.A. (“Citibank”) to repurchase $1.0 billion of the Company’s common stock. The Company made an initial payment of $1.0 billion to Citibank and received an initial delivery of approximately 3.6 million shares of the Company’s common stock, which were subsequently retired. In December 2022, the October ASR Program was completed, and an additional 0.3 million shares of common stock were received and retired. In total, 3.9 million shares were repurchased at an average price per share of $254.48. The total cost of the October ASR Program was reflected as a reduction to equity in the Consolidated Balance Sheets.
The following table summarizes stock repurchase activities (in millions, except per share amounts): | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2022 | | 2021 | | 2020 |
| Shares repurchased | 11.2 | | | — | | | 0.7 | |
| Average price per share | $ | 233.70 | | | $ | — | | | $ | 183.84 | |
| Value of shares repurchased | $ | 2,607.4 | | | $ | — | | | $ | 134.3 | |
The Company uses the par value method of accounting for its stock repurchases. As a result of share repurchase activities during the years ended December 31, 2022, 2021, and 2020, the Company reduced common stock and additional paid-in capital by an aggregate of $211 million, zero, and $8 million, respectively, and charged $2.396 billion, zero, and $126 million, respectively, to retained earnings.
Accumulated Other Comprehensive Income (Loss), Net of Tax, Attributable to Intuitive Surgical, Inc.
The components of accumulated other comprehensive income (loss), net of tax, attributable to Intuitive Surgical, Inc. are as follows (in millions):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, 2022 |
| | Gains (Losses)
on Hedge
Instruments | | Unrealized
Gains (Losses)
on
Available-for-Sale Securities | | Foreign
Currency
Translation
Gains
(Losses) | | Employee Benefit Plans | | Total |
| Beginning balance | $ | 4.5 | | | $ | (16.0) | | | $ | (7.9) | | | $ | (4.8) | | | $ | (24.2) | |
Other comprehensive income (loss) before reclassifications | (35.0) | | | (138.2) | | | 1.3 | | | 5.8 | | | (166.1) | |
Amounts reclassified from accumulated other comprehensive income (loss) | 27.6 | | | — | | | — | | | 0.2 | | | 27.8 | |
| Net current-period other comprehensive income (loss) | (7.4) | | | (138.2) | | | 1.3 | | | 6.0 | | | (138.3) | |
| Ending balance | $ | (2.9) | | | $ | (154.2) | | | $ | (6.6) | | | $ | 1.2 | | | $ | (162.5) | |
| | | | | | | | | |
| | Year Ended December 31, 2021 |
| | Gains (Losses)
on Hedge
Instruments | | Unrealized
Gains (Losses)
on
Available-for-Sale Securities | | Foreign
Currency
Translation
Gains
(Losses) | | Employee Benefit Plans | | Total |
| Beginning balance | $ | (2.9) | | | $ | 29.5 | | | $ | 4.7 | | | $ | (6.4) | | | $ | 24.9 | |
Other comprehensive income (loss) before reclassifications | 12.3 | | | (45.5) | | | (12.6) | | | 0.1 | | | (45.7) | |
Amounts reclassified from accumulated other comprehensive income (loss) | (4.9) | | | — | | | — | | | 1.5 | | | (3.4) | |
| Net current-period other comprehensive income (loss) | 7.4 | | | (45.5) | | | (12.6) | | | 1.6 | | | (49.1) | |
| Ending balance | $ | 4.5 | | | $ | (16.0) | | | $ | (7.9) | | | $ | (4.8) | | | $ | (24.2) | |
The tax impacts for amounts recognized in other comprehensive income before reclassifications were as follows (in millions):
| | | | | | | | | | | | | |
| | Years Ended December 31, | | |
| Available-for-sale securities | 2022 | | 2021 | | |
| Income tax benefit for net losses recorded in other comprehensive income (loss) | $ | 39.1 | | | $ | 14.9 | | | |
The tax impacts for amounts recognized in other comprehensive income (loss) before reclassifications for hedge instruments, foreign currency translation gains (losses), and employee benefit plans in 2022 and 2021 were not material to the Company’s Consolidated Financial Statements. The tax impacts for amounts reclassified from accumulated other comprehensive loss relating to hedge instruments, available-for-sale securities, foreign currency translation gains (losses), and employee benefit plans in 2022 and 2021 were not material to the Company’s Consolidated Financial Statements.
NOTE 10. SHARE-BASED COMPENSATION
Stock Plans
2010 Incentive Award Plan. In April 2010, the Company’s stockholders approved the 2010 Incentive Award Plan (“2010 Plan”). Under this plan, the Company can issue RSUs, nonqualified stock options (“NSOs”), and PSUs to employees, non-employee directors, and consultants. Equity awards granted to employees and non-employee directors include a mix of RSUs, stock options, and, as applicable, PSUs. The 2010 Plan generally permits NSOs to be granted at no less than the fair market value of the common stock on the date of grant. Prior to 2022, NSOs were granted with terms of 10 years from the date of the grant. Beginning in 2022, the Company changed the term of its new NSO grants to 7 years from the date of the grant. The 2010 Plan expires in 2032. In April 2022, the Company’s shareholders approved an amended and restated 2010 Plan to provide for an increase in the number of shares of common stock reserved for issuance thereunder from 103,350,000 to 110,350,000. As of
December 31, 2022, approximately 27.2 million shares were reserved for future issuance under the 2010 Plan. A maximum of approximately 11.8 million of these shares can be awarded as RSUs.
2009 Employment Commencement Incentive Plan. In October 2009, the Board adopted the 2009 Employment Commencement Incentive Plan (“New Hire Plan”). The New Hire Plan provides for the shares to be used exclusively for the grant of RSUs and NSOs to new employees (“New Hire Options”), who were not previously employees or non-employee directors of the Company. The Compensation Committee approves all equity awards under the New Hire Plan, which are granted to newly-hired employees once a month on the fifth business day of each month after their hire. Options are granted at an exercise price not less than the fair market value of the stock on the date of grant and have a term not to exceed 10 years.
In April 2015, the Board of Directors amended and restated the New Hire Plan to provide for an increase in the number of shares of common stock authorized for issuance pursuant to awards granted under the New Hire Plan from 10,395,000 to 13,095,000. The New Hire Plan expired in October 2019 and, therefore, there are no shares reserved for future grants under the New Hire Plan. However, awards granted prior to the plan’s expiration continue to remain outstanding until their original expiration date.
Restricted Stock Units. The RSUs granted to employees vest in one-fourth increments annually over a four-year period. The RSUs granted to existing non-employee directors vest one year from the date of grant or at the next Annual Shareholders Meeting, whichever comes first. New non-employee directors receive pro-rated RSU grants that vest on the same term as the annual RSU grants. The number of shares issued on the date the RSUs vest is net of the minimum statutory tax withholdings, which are paid in cash to the appropriate taxing authorities on behalf of the Company’s employees.
Nonqualified Stock Options. Prior to 2020, annual NSO grants were made to employees on February 15 (or the next business day if the date is not a business day) and on August 15 (or the next business day if the date is not a business day). Beginning in 2020, the Company changed the timing of its annual equity award grants to the last trading day of February and on the same date in August or, if that date is not a trading day, the next trading day. The February NSO grants vest 1/8 upon completion of 6 months of service and 1/48 per month thereafter. The August NSO grants vest 7/48 at the end of one month and 1/48 per month thereafter through a 3.5-year vesting period. NSOs granted to new hires generally vest 1/4 upon completion of one year of service and 1/48 per month thereafter. NSOs granted to existing non-employee directors vest one year from the date of grant or at the next Annual Shareholders Meeting, whichever comes first. New non-employee directors receive pro-rated NSO grants that vest on the same term as the annual NSO grants. Option vesting terms are determined by the Board and, in the future, may vary from past practices.
Performance Stock Units. Beginning in 2022, the Company granted PSUs to officers and other key employees, which are designed to provide further incentives for the achievement of the Company’s long-term objectives. The PSUs are subject to three-year cliff vesting and pre-established, quantitative goals. Whether any PSUs vest, and the amount that does vest, is tied to completion of service over three years and the achievement of three equally-weighted, quantitative metrics that directly align with or help drive the Company’s strategy and long-term total shareholder return.
2000 Non-Employee Directors’ Stock Option Plan. In March 2000, the Board of Directors adopted the 2000 Non-Employee Directors’ Stock Option Plan (the “Directors’ Plan”). In October 2009, the automatic evergreen increase provisions were eliminated so that no further automatic increases would be made to the number of shares reserved for issuance under the Directors’ Plan. In addition, the common stock authorized for issuance under the Directors’ Plan was reduced to 1,350,000. Options are granted at an exercise price not less than the fair market value of the stock on the date of grant and have a term not to exceed 10 years. Prior to 2016, initial stock option grants to new non-employee directors vested over a three-year period with 1/3 of the shares vesting after one year from the date of grant and 1/36 of the shares vesting monthly thereafter. Annual stock option grants vested one year from the date of the grant. Since 2016, new non-employee directors received pro-rated stock option grants that vest on the same term as the annual stock option grants. The Directors’ Plan was terminated in November 2020 and, therefore, there are no shares reserved for future grants under the Directors’ Plan. However, options granted prior to the plan’s termination continue to remain outstanding until their original expiration date.
2000 Employee Stock Purchase Plan. In March 2000, the Board adopted the ESPP. Employees are generally eligible to participate in the ESPP if they are customarily employed by the Company for more than 20 hours per week and more than 5 months in a calendar year and are not 5% stockholders of the Company. Under the ESPP, eligible employees may select a rate of payroll deduction up to 15% of their eligible compensation subject to certain maximum purchase limitations. The duration for each offering period is 24 months and is divided into four purchase periods of approximately six months in length. Offerings are concurrent. The purchase price of the shares under the offering is the lesser of 85% of the fair market value of the shares on the offering date or 85% of the fair market value of the shares on the purchase date. A two-year look-back feature in the ESPP causes the offering period to reset if the fair value of the Company’s common stock on the first or last day of the purchase period is less than that on the original offering date. ESPP purchases by employees are settled with newly-issued common stock from the ESPP’s previously authorized and available pool of shares. In April 2017, the Company’s stockholders approved an amended and restated ESPP to provide for an increase in the number of shares of common stock reserved for issuance
from 18,270,945 to 22,770,945. As of December 31, 2022, there were approximately 2.4 million shares reserved for future issuance under the ESPP.
Restricted Stock Units
RSU activity for the year ended December 31, 2022, was as follows (in millions, except per share amounts):
| | | | | | | | | | | |
| |
Shares | | Weighted-Average
Grant Date Fair Value Per Share |
Unvested balance as of December 31, 2021 | 4.8 | | | $ | 207.37 | |
| Granted | 2.0 | | | $ | 272.97 | |
| Vested | (1.9) | | | $ | 191.19 | |
| Forfeited | (0.3) | | | $ | 235.38 | |
Unvested balance as of December 31, 2022 | 4.6 | | | $ | 241.47 | |
As of December 31, 2022, 4.2 million shares of RSUs were expected to vest with an aggregate intrinsic value of $1.12 billion. The aggregate vesting date fair value of RSUs vested was $536 million, $578 million, and $478 million during the years ended December 31, 2022, 2021, and 2020, respectively.
Stock Options
NSO activity during 2022 under all of the stock plans was as follows (in millions, except per share amounts):
| | | | | | | | | | | |
| | Stock Options Outstanding |
| | Number
Outstanding | | Weighted-Average Exercise Price Per Share |
Balance as of December 31, 2021 | 11.7 | | | $ | 125.07 | |
| Options granted | 1.2 | | | $ | 249.62 | |
| Options exercised | (1.9) | | | $ | 77.59 | |
| Options forfeited/expired | (0.2) | | | $ | 240.02 | |
Balance as of December 31, 2022 | 10.8 | | | $ | 144.86 | |
The aggregate intrinsic value of stock options exercised under the Company’s stock plans determined as of the date of option exercise was $315 million, $613 million, and $598 million during the years ended December 31, 2022, 2021, and 2020, respectively. Cash received from option exercises and employee stock purchase plans for the years ended December 31, 2022, 2021, and 2020, was $234 million, $276 million, and $309 million, respectively. The income tax benefit from stock options exercised was $70 million for the year ended December 31, 2022.
The following table summarizes significant ranges of outstanding and exercisable options as of December 31, 2022 (number of shares and aggregate intrinsic value in millions):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Options Outstanding | | Options Exercisable |
Range of
Exercise Prices | | Number
of Shares | | Weighted-Average
Remaining
Contractual Life | | Weighted-Average
Exercise Price
Per Share | | Aggregate
Intrinsic
Value (1) | | Number
of Shares | | Weighted-Average
Remaining
Contractual Life | | Weighted-Average
Exercise Price
Per Share | | Aggregate
Intrinsic
Value (1) |
$29.91-$49.34 | | 1.1 | | | 0.9 | | $ | 45.33 | | | | | 1.1 | | | | | $ | 45.33 | | | |
$51.02-$59.23 | | 1.5 | | | 2.1 | | $ | 55.61 | | | | | 1.5 | | | | | $ | 55.61 | | | |
$59.46-$77.00 | | 1.4 | | | 2.6 | | $ | 67.23 | | | | | 1.4 | | | | | $ | 67.23 | | | |
$77.04-$139.52 | | 1.4 | | | 4.6 | | $ | 108.37 | | | | | 1.4 | | | | | $ | 108.37 | | | |
$143.49-$174.26 | | 1.3 | | | 6.1 | | $ | 168.86 | | | | | 1.2 | | | | | $ | 168.99 | | | |
$175.53-$182.83 | | 1.2 | | | 6.6 | | $ | 179.92 | | | | | 1.0 | | | | | $ | 180.26 | | | |
$182.90-$242.34 | | 1.3 | | | 7.1 | | $ | 219.80 | | | | | 0.6 | | | | | $ | 224.29 | | | |
$243.26-$290.33 | | 1.1 | | | 7.2 | | $ | 268.20 | | | | | 0.4 | | | | | $ | 261.46 | | | |
$292.49-$341.16 | | — | | | 6.8 | | $ | 311.17 | | | | | — | | | | | $ | 330.70 | | | |
$347.42-$347.42 | | 0.5 | | | 8.6 | | $ | 347.42 | | | | | 0.2 | | | | | $ | 347.42 | | | |
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | |
| Total | | 10.8 | | | 4.7 | | $ | 144.87 | | | $ | 1,354 | | | 8.8 | | | 4.2 | | $ | 121.60 | | | $ | 1,292 | |
(1)The aggregate intrinsic value represents the total pre-tax intrinsic value, based on the Company’s closing stock price of $265.35 as of December 31, 2022, which would have been received by the option holders had all in-the-money option holders exercised their options as of that date.
As of December 31, 2022, a total of 10.6 million shares of stock options vested and expected to vest had a weighted-average remaining contractual life of 4.7 years, an aggregate intrinsic value of $1.35 billion, and a weighted-average exercise price of $143.06.
Performance Stock Units
The 2022 PSU grant metrics are focused on relative total shareholder return (“TSR”), year-over-year da Vinci procedure growth for 2023, and two-year compound annual da Vinci procedure growth for 2024. TSR is considered a market condition, and the expense is determined at the grant date. The two procedure growth goals are considered performance conditions, and the expense is recorded based on the forecasted performance, which is reassessed each reporting period based on the probability of achieving the two performance conditions. The number of shares earned at the end of the three-year period will vary, based on actual performance, from 0% to 125% of the target number of PSUs granted. PSUs are subject to forfeiture if employment terminates prior to the vesting date. PSUs are not considered issued or outstanding shares of the Company.
The Company calculates the fair value for each component of the PSUs individually. The fair value for the component with the TSR metric was determined using Monte Carlo simulation. The fair value per share for the components with the procedure growth metrics is equal to the closing stock price on the grant date.
PSU activity during 2022 was as follows (in millions, except per share amounts):
| | | | | | | | | | | |
| |
Shares | | Weighted-Average
Grant Date Fair Value Per Share |
Unvested balance as of December 31, 2021 | — | | | $ | — | |
| Granted | 0.1 | | | $ | 299.32 | |
| Vested | — | | | $ | — | |
| Performance change | — | | | $ | — | |
| Forfeited | — | | | $ | — | |
Unvested balance as of December 31, 2022 | 0.1 | | | $ | 299.32 | |
As of December 31, 2022, 0.1 million shares of PSUs were expected to vest with an aggregate intrinsic value of $18 million.
Employee Stock Purchase Plan
Under the ESPP, employees purchased approximately 0.4 million, 0.5 million, and 0.5 million shares, representing approximately $87.9 million, $75.9 million, and $71.2 million in employee contributions for the years ended December 31, 2022, 2021, and 2020, respectively.
Share-Based Compensation Expense
The following table summarizes share-based compensation expense (in millions):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2022 | | 2021 | | 2020 |
| Cost of revenue—product | $ | 67.6 | | | $ | 68.9 | | | $ | 58.9 | |
| Cost of revenue—service | 23.6 | | | 22.2 | | | 24.0 | |
| Total cost of revenue | 91.2 | | | 91.1 | | | 82.9 | |
| Selling, general and administrative | 261.1 | | | 231.6 | | | 202.2 | |
| Research and development | 164.2 | | | 134.1 | | | 113.6 | |
| Share-based compensation expense before income taxes | 516.5 | | | 456.8 | | | 398.7 | |
| Income tax benefit | 101.7 | | | 93.7 | | | 81.4 | |
| Share-based compensation expense after income taxes | $ | 414.8 | | | $ | 363.1 | | | $ | 317.3 | |
The Black-Scholes-Merton option pricing model is used to estimate the fair value of stock options granted under the Company’s share-based compensation plans and rights to acquire stock granted under the ESPP. The weighted-average estimated fair values of stock options, the rights to acquire stock under the ESPP, and RSUs, as well as the weighted-average assumptions used in calculating the fair values of stock options and rights to acquire stock under the ESPP that were granted during the years ended December 31, 2022, 2021, and 2020, were as follows:
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| 2022 | | 2021 | | 2020 |
| RSUs | | | | | |
| Fair value at grant date | $272.97 | | $256.52 | | $181.89 |
| STOCK OPTIONS | | | | | |
| Risk-free interest rate | 2.6% | | 0.8% | | 0.6% |
| Expected term (in years) | 3.2 | | 4.1 | | 4.1 |
| Expected volatility | 38% | | 32% | | 32% |
| Fair value at grant date | $73.65 | | $78.23 | | $54.34 |
| PSUs | | | | | |
| Fair value at grant date | $299.32 | | $— | | $— |
| ESPP | | | | | |
| Risk-free interest rate | 2.1% | | 0.1% | | 0.9% |
| Expected term (in years) | 1.2 | | 1.2 | | 1.2 |
| Expected volatility | 39% | | 29% | | 30% |
| Fair value at grant date | $80.61 | | $89.98 | | $57.29 |
As share-based compensation expense recognized in the Consolidated Statements of Income during the years ended December 31, 2022, 2021, and 2020, is based on awards ultimately expected to vest, it has been reduced for estimated forfeitures.
As of December 31, 2022, there was $700 million, $121 million, $15 million, and $65 million of total unrecognized compensation expense related to unvested restricted stock units, unvested stock options, unvested performance stock units, and rights granted to acquire common stock under the ESPP, respectively. The unrecognized compensation expense is expected to be recognized over a weighted-average period of 2.3 years for unvested restricted stock units, 2.4 years for unvested stock options, 2.2 years for unvested performance stock units, and 1.5 years for rights granted to acquire common stock under the ESPP.
NOTE 11. INCOME TAXES
Income before provision for income taxes for the years ended December 31, 2022, 2021, and 2020, consisted of the following (in millions):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2022 | | 2021 | | 2020 |
| U.S. | $ | 956.7 | | | $ | 1,298.7 | | | $ | 926.8 | |
| Foreign | 650.1 | | | 591.6 | | | 280.2 | |
| Total income before provision for income taxes | $ | 1,606.8 | | | $ | 1,890.3 | | | $ | 1,207.0 | |
The provision for income taxes for the years ended December 31, 2022, 2021, and 2020, consisted of the following (in millions):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2022 | | 2021 | | 2020 |
| Current | | | | | |
| Federal | $ | 350.4 | | | $ | 158.8 | | | $ | 34.2 | |
| State | 49.2 | | | 17.3 | | | 21.5 | |
| Foreign | 48.1 | | | 50.1 | | | 26.9 | |
| 447.7 | | | 226.2 | | | 82.6 | |
| Deferred | | | | | |
| Federal | (188.8) | | | (21.4) | | | 23.8 | |
| State | (16.4) | | | 0.5 | | | 1.6 | |
| Foreign | 19.9 | | | (43.1) | | | 32.2 | |
| (185.3) | | | (64.0) | | | 57.6 | |
| Total income tax expense | $ | 262.4 | | | $ | 162.2 | | | $ | 140.2 | |
The provision for income taxes for the year ended December 31, 2022, reflected the impact of a change in U.S. tax law effective January 1, 2022, which requires the capitalization and amortization of research and development expenditures incurred after December 31, 2021.
On August 16, 2022, the Inflation Reduction Act of 2022 (the “IRA”) was enacted in the United States. The IRA introduces a 15% alternative minimum tax based on the financial statement income of certain large corporations, effective for tax years beginning after December 31, 2022. The IRA also includes a 1% excise tax on the net fair market value of stock repurchases made after December 31, 2022. The Company considered the applicable tax law changes, and there is no impact to the Company’s tax provision for the twelve months ended December 31, 2022. The Company will continue to evaluate the impact of these tax law changes on future periods.
The Company’s 2021 income tax expense included a one-time benefit of $66.4 million from re-measurement of its Swiss deferred tax assets resulting from the extension of the economic useful life of certain intangible assets.
Income tax expense differs from amounts computed by applying the statutory federal income rate of 21% for the years ended December 31, 2022, 2021, and 2020, as a result of the following (in millions):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2022 | | 2021 | | 2020 |
| Federal tax at statutory rate | $ | 337.4 | | | $ | 397.0 | | | $ | 253.5 | |
| Increase (reduction) in tax resulting from: | | | | | |
| State taxes, net of federal benefits | 34.9 | | | 33.1 | | | 23.1 | |
| Foreign rate differential | (64.2) | | | (54.3) | | | (19.3) | |
| U.S. tax on foreign earnings | 75.4 | | | 40.1 | | | 29.3 | |
Research and development credit | (41.7) | | | (30.7) | | | (37.1) | |
| Share-based compensation not benefited | 24.1 | | | 17.8 | | | 14.3 | |
| Unrecognized tax benefit related to share-based compensation | 3.3 | | | 13.6 | | | 39.3 | |
| Reversal of unrecognized tax benefits | (11.1) | | | (3.0) | | | (4.0) | |
| Excess tax benefits related to share-based compensation | (98.7) | | | (185.8) | | | (166.2) | |
| Deferred tax re-measurement | — | | | (66.4) | | | — | |
| Other | 3.0 | | | 0.8 | | | 7.3 | |
| Total income tax expense | $ | 262.4 | | | $ | 162.2 | | | $ | 140.2 | |
Deferred income taxes reflect tax carryforwards and the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets and liabilities are as follows (in millions):
| | | | | | | | | | | |
| | December 31, |
| | 2022 | | 2021 |
| Deferred tax assets: | | | |
| Intangible assets | $ | 342.8 | | | $ | 369.1 | |
Capitalized research and development expenditures | 172.5 | | | 4.8 | |
| Research and development credits | 156.7 | | | 98.5 | |
| Share-based compensation expense | 121.3 | | | 110.9 | |
| Expenses deducted in later years for tax purposes | 57.5 | | | 38.4 | |
| Lease liabilities | 16.6 | | | 15.2 | |
| Net operating losses | 6.4 | | | 9.7 | |
| Net unrealized losses on available-for-sale securities and other | 45.5 | | | 5.3 | |
| Gross deferred tax assets | 919.3 | | | 651.9 | |
| Valuation allowance | (168.6) | | | (104.6) | |
| Deferred tax assets | 750.7 | | | 547.3 | |
| Deferred tax liabilities: | | | |
| Property, plant, and equipment | (64.1) | | | (79.4) | |
| Right-of-use assets | (11.8) | | | (12.3) | |
| Intangible assets | (9.3) | | | (9.7) | |
| Other | (1.0) | | | (5.1) | |
| Deferred tax liabilities | (86.2) | | | (106.5) | |
| Net deferred tax assets | $ | 664.5 | | | $ | 440.8 | |
As of December 31, 2022, the Company had $27.4 million of U.S. and foreign federal net operating loss carryforwards, certain of which will expire starting in 2028 if not utilized. Utilization of these net operating loss carryforwards may be subject to certain limitations. The Company does not expect the limitations to result in any permanent loss of these tax benefits. As of December 31, 2022, the Company had $221.9 million in California research and development credit carryforwards, which do not expire.
As of December 31, 2022, and 2021, the Company had valuation allowances of $168.6 million and $104.6 million, respectively, primarily related to California deferred tax assets, for which the Company does not believe a tax benefit is more likely than not to be realized.
The Company intends to repatriate earnings from its Swiss subsidiary and joint venture in Hong Kong, as needed, and the U.S. and foreign tax implications of such repatriations are not expected to be significant. The Company will continue to indefinitely reinvest earnings from the rest of its foreign subsidiaries, which are not significant.
A reconciliation of the beginning and ending amounts of gross unrecognized income tax benefits for the years ended December 31, 2022, 2021, and 2020, are as follows (in millions):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2022 | | 2021 | | 2020 |
| Beginning balance | $ | 222.5 | | | $ | 176.3 | | | $ | 96.7 | |
| Increases related to tax positions taken during the current year | 49.5 | | | 40.6 | | | 40.1 | |
| Increases related to tax positions taken during a prior year | 4.9 | | | 11.2 | | | 46.1 | |
| Decreases related to tax positions taken during a prior year | (16.5) | | | (1.3) | | | — | |
| Decreases related to settlements with tax authorities | (1.2) | | | (0.2) | | | (0.5) | |
| Decreases related to expiration of statute of limitations | (6.6) | | | (4.1) | | | (6.1) | |
| Ending balance | $ | 252.6 | | | $ | 222.5 | | | $ | 176.3 | |
As of December 31, 2022, 2021, and 2020, gross interest related to unrecognized tax benefits accrued was $21.0 million, $14.9 million, and $11.0 million, respectively. The Company’s net unrecognized tax benefits and related interest are presented in other long-term liabilities and long-term deferred tax assets on the Consolidated Balance Sheets.
Total gross unrecognized tax benefits as of December 31, 2022, were $252.6 million, of which $187.4 million, if recognized, would have an impact on the Company’s effective tax rate.
The Company recorded an increase in the income tax provision of $39.3 million during the year ended December 31, 2020, as a result of a Ninth Circuit Court of Appeals opinion involving an independent third party related to charging foreign subsidiaries for share-based compensation. An additional charge of $13.6 million related to this matter was recorded to income tax expense in 2021, after additional IRS guidance was issued in July 2021. The Company will continue to monitor future IRS actions or other developments regarding this matter and will assess the impact of any such developments on its income tax provision in the quarter that they occur.
The Company files federal, state, and foreign income tax returns in many U.S. and OUS jurisdictions. Years before 2016 are closed for the significant jurisdictions. Certain of the Company’s unrecognized tax benefits could change due to activities of various tax authorities, including potential assessment of additional tax, possible settlement of audits, or through normal expiration of various statutes of limitations, which could affect the Company’s effective tax rate in the period in which they change. Due to the uncertainty related to the timing and potential outcome of audits, the Company cannot estimate the range of reasonably possible changes in unrecognized tax benefits that may occur in the next 12 months.
The Company is subject to the examination of its income tax returns by the Internal Revenue Service and other tax authorities. The outcome of these audits cannot be predicted with certainty. The Company’s management regularly assesses the likelihood of adverse outcomes resulting from these examinations to determine the adequacy of the Company’s provision for income taxes. If any issues addressed in the Company’s tax audits are resolved in a manner not consistent with management’s expectations, the Company could be required to adjust its provision for income taxes in the period such resolution occurs.
NOTE 12. NET INCOME PER SHARE
The following table presents the computation of basic and diluted net income per share attributable to Intuitive Surgical, Inc. (in millions, except per share amounts):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2022 | | 2021 | | 2020 |
| Numerator: | | | | | |
| Net income attributable to Intuitive Surgical, Inc. | $ | 1,322.3 | | | $ | 1,704.6 | | | $ | 1,060.6 | |
| Denominator: | | | | | |
| Weighted-average shares outstanding used in basic calculation | 355.7 | | | 356.1 | | | 351.1 | |
| Add: dilutive effect of potential common shares | 6.3 | | | 9.7 | | | 9.9 | |
| Weighted-average shares outstanding used in diluted calculation | 362.0 | | | 365.8 | | | 361.0 | |
| Net income per share attributable to Intuitive Surgical, Inc.: | | | | | |
| Basic | $ | 3.72 | | | $ | 4.79 | | | $ | 3.02 | |
| Diluted | $ | 3.65 | | | $ | 4.66 | | | $ | 2.94 | |
Share-based compensation awards of approximately 3.4 million, 0.8 million, and 1.9 million shares for the years ended December 31, 2022, 2021, and 2020, respectively, were outstanding but were not included in the computation of diluted net income per share attributable to Intuitive Surgical, Inc. common stockholders, because the effect of including such shares would have been anti-dilutive in the periods presented.
NOTE 13. EMPLOYEE BENEFIT PLANS
The Company sponsors various retirement plans for its eligible U.S. and non-U.S. employees. For employees in the U.S., the Company maintains the Intuitive Surgical, Inc. 401(k) Plan (the “Plan”). As allowed under Section 401(k) of the Internal Revenue Code, the Plan provides tax-deferred salary contributions for eligible U.S. employees. The Plan allows employees to contribute up to 100% of their annual compensation to the Plan on a pre-tax and/or after-tax basis. Employee contributions are limited to a maximum annual amount as set periodically by the Internal Revenue Code. The Company matches 200% of employee contributions up to $1,500 per calendar year per person. All matching employer contributions vest immediately.
SCHEDULE II
VALUATION AND QUALIFYING ACCOUNTS
(IN MILLIONS)
| | | | | | | | | | | | | | | | | | | | | | | |
| Balance at
Beginning of
Year | | Additions | | Deductions (1) | | Balance at
End of Year |
| Sales returns and allowances | | | | | | | |
Year ended December 31, 2022 | $ | 13.1 | | | $ | 44.4 | | | $ | (43.0) | | | $ | 14.5 | |
Year ended December 31, 2021 | $ | 15.5 | | | $ | 41.7 | | | $ | (44.1) | | | $ | 13.1 | |
Year ended December 31, 2020 | $ | 11.7 | | | $ | 39.7 | | | $ | (35.9) | | | $ | 15.5 | |
(1)Primarily represents products returned.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURES
None.
ITEM 9A. CONTROLS AND PROCEDURES
Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow for timely decisions regarding required disclosure.
As required by SEC Rule 13a-15(b), we carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Based on the foregoing, our principal executive officer and principal financial officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level.
Inherent Limitations Over Internal Controls
Our internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of the Consolidated Financial Statements for external purposes in accordance with GAAP. Our internal control over financial reporting includes those policies and procedures that:
(i)pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets;
(ii)provide reasonable assurance that transactions are recorded as necessary to permit preparation of the Consolidated Financial Statements in accordance with GAAP and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and
(iii)provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on the Consolidated Financial Statements.
Management, including our principal executive officer and principal financial officer, does not expect that our internal controls will prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of internal controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. Also, any evaluation of the effectiveness of controls in future periods are subject to the risk that those internal controls may become inadequate because of changes in business conditions or that the degree of compliance with the policies or procedures may deteriorate.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in the Exchange Act Rules 13a-15(f). Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an assessment of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on the results of our assessment under the framework in the Internal Control—Integrated Framework (2013), our management concluded that our internal control over financial reporting was effective as of December 31, 2022.
The effectiveness of our internal control over financial reporting as of December 31, 2022, has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report, which is included under “Item 8. Financial Statements and Supplementary Data” of this Annual Report.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the quarter ended December 31, 2022, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
None.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
PART III
Certain information required by Part III is omitted from this report on Form 10-K and is incorporated herein by reference to our definitive Proxy Statement for our next Annual Meeting of Stockholders (the “Proxy Statement”), which we intend to file pursuant to Regulation 14A of the Securities Exchange Act of 1934, as amended, within 120 days after December 31, 2022.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS, AND CORPORATE GOVERNANCE
The information required by this item concerning our directors and corporate governance is incorporated by reference to the information set forth in the section titled “Directors and Corporate Governance” in our Proxy Statement. Information required by this item concerning our executive officers is incorporated by reference to the information set forth in the section entitled “Executive Officers of the Company” in our Proxy Statement. Information regarding our Section 16 reporting compliance and code of business conduct and ethics is incorporated by reference to the information set forth in the section entitled “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” in our Proxy Statement.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item regarding executive compensation is incorporated by reference to the information set forth in the sections titled “Executive Compensation” and “Compensation for Directors” in our Proxy Statement.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item regarding security ownership of certain beneficial owners and management is incorporated by reference to the information set forth in the section titled “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” in our Proxy Statement for the 2023 Annual Meeting of Stockholders to be filed with the SEC within 120 days of December 31, 2022.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE
The information required by this item regarding certain relationships and related transactions and director independence is incorporated by reference to the information set forth in the sections titled “Certain Relationships and Related Transactions” and “Directors and Corporate Governance” in our Proxy Statement.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this item regarding principal accountant fees and services is incorporated by reference to the information set forth in the section titled “Principal Accountant Fees and Services” in our Proxy Statement.
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULE
(a)The following documents are filed as part of this Annual Report on Form 10-K.
1)Financial Statements—See Index to Consolidated Financial Statements at Item 8 of this report on Form 10-K.
2)The following financial statement schedule of Intuitive Surgical, Inc. for 2022, 2021, and 2020 is filed as part of this report and should be read in conjunction with the Consolidated Financial Statements of Intuitive Surgical, Inc.: All other schedules have been omitted, because they are not applicable, not required under the instructions, or the information requested is set forth in the Consolidated Financial Statements or related notes thereto.
3)Exhibits
The exhibits filed as part of this report are listed under “Exhibits” at subsection (b) of this Item 15.
(b)Exhibits
EXHIBIT INDEX | | | | | | | | |
| 3.1(1) | | |
| | |
| 3.2(2) | | |
| |
| 3.3(3) | | |
| |
| 4.1(4) | | |
| |
| 4.2(5) | | |
| | |
| 10.1(6) | | |
| |
| 10.2(7) | | |
| |
| 10.3(8) | | |
| |
| 10.4(9) | | |
| | |
| 10.5(10) | | |
| |
| 10.6(11) | | |
| |
| 10.7(12) | | |
| |
| 10.8(13) | | |
| |
| 10.9 | | |
| |
| 10.10 | | |
| | |
| 10.11 | | |
| | |
| 21.1 | | |
| |
| 23.1 | | |
| | |
| 31.1 | | |
| |
| 31.2 | | |
| |
| 32.1 | | |
| | |
| 101 | | The following materials from Intuitive Surgical, Inc.’s Annual Report on Form 10-K for the year ended December 31, 2022, formatted in Inline XBRL (Inline Extensible Business Reporting Language): (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Income, (iii) Consolidated Statements of Comprehensive Income, (iv) Consolidated Statements of Stockholders’ Equity, (v) Consolidated Statements of Cash Flows, and (vi) Notes to Consolidated Financial Statements, tagged at Level I through IV. |
| | |
| 104 | | The cover page from Intuitive Surgical, Inc.’s Annual Report on Form 10-K for the year ended December 31, 2022, formatted in Inline XBRL and contained in Exhibit 101. |
1.Incorporated by reference to Exhibit 3.1 filed with the Company’s Quarterly Report on Form 10-Q filed on July 23, 2020 (File No. 000-30713).
2.Incorporated by reference to Exhibit 3.1 filed with the Company’s Quarterly Report on Form 10-Q filed on October 20, 2021 (File No. 000-30713).
3.Incorporated by reference to Exhibit 3.1 filed with the Company’s Current Report on Form 8-K filed on February 1, 2021 (File No. 000-30713).
4.Incorporated by reference to Exhibit 4.2 filed with the Company’s Registration Statement Amendment on Form S-1/A filed on May 2, 2000 (File No. 333-33016).
5.Incorporated by reference to Exhibit 4.2 filed with the Company’s Annual Report on Form 10-K filed on February 3, 2022 (File No. 333-33016).
6.Incorporated by reference to exhibits filed with the Company’s Registration Statement on Form S-1 filed on March 22, 2000 (File No. 333-33016).
7.Incorporated by reference to Exhibit 10.1 filed with the Company’s Current Report on Form 8-K filed on August 3, 2015 (File No. 000-30713).
8.Incorporated by reference to Exhibit 4.2 filed with the Company’s Registration Statement on Form S-8 filed on May 1, 2015 (File No. 333-203793).
9.Incorporated by reference to Exhibit 10.1 filed with the Company’s Current Report on Form 8-K filed on April 26, 2017 (File No. 000-30713).
10.Incorporated by reference to Exhibit 10.1 filed with the Company’s Current Report on Form 8-K filed on May 3, 2022 (File No. 000-30713).
11.Incorporated by reference to Exhibit 10.1 filed with the Company’s Current Report on Form 8-K filed on December 2, 2008 (File No. 000-30713).
12.Incorporated by reference to Exhibit 10.9 filed with the Company’s 2015 Annual Report on Form 10-K filed on February 2, 2016 (File No. 000-30713).
13.Incorporated by reference to Exhibit 10.10 filed with the Company’s 2015 Annual Report on Form 10-K filed on February 2, 2016 (File No. 000-30713).
* Management contract or compensatory plan or arrangement.
ITEM 16. FORM 10-K SUMMARY
None.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| | | | | | | | |
| INTUITIVE SURGICAL, INC. |
| |
| By: | | /S/ GARY S. GUTHART |
| | Gary S. Guthart, Ph.D.
President and Chief Executive Officer |
Date: February 10, 2023
Power of Attorney
Each person whose individual signature appears below hereby authorizes and appoints Gary Guthart, Ph.D., and Jamie Samath, and each of them, with full power of substitution and re-substitution and full power to act without the other, as his or her true and lawful attorney-in-fact and agent to act in his or her name, place, and stead and to execute in the name and on behalf of each person, individually and in each capacity stated below, and to file any and all amendments to this annual report on Form 10‑K and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing, ratifying and confirming all that said attorneys-in-fact and agents or any of them or their or his substitute or substitutes may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| | | | | | | | | | | | | | |
| Signature | | Title | | Date |
| | |
/S/ GARY S. GUTHART | | President, Chief Executive Officer, and Director
(Principal Executive Officer) | | February 10, 2023 |
| Gary S. Guthart, Ph.D. | | |
/S/ JAMIE E. SAMATH | | Senior Vice President and Chief Financial Officer
(Principal Financial Officer) | | February 10, 2023 |
| Jamie E. Samath | | |
/S/ FREDRIK C. WIDMAN | | Vice President, Corporate Controller
(Principal Accounting Officer) | | February 10, 2023 |
| Fredrik C. Widman | | |
/S/ CRAIG H. BARRATT | | Chairman of the Board of Directors | | February 10, 2023 |
| Craig H. Barratt, Ph.D. | | |
/S/ JOSEPH C. BEERY | | Director | | February 10, 2023 |
| Joseph C. Beery | | |
/S/ AMAL M. JOHNSON | | Director | | February 10, 2023 |
| Amal M. Johnson | | |
/S/ DON R. KANIA | | Director | | February 10, 2023 |
| Don R. Kania, Ph.D. | | |
/S/ AMY L. LADD | | Director | | February 10, 2023 |
| Amy L. Ladd, Ph.D. | | |
/S/ KEITH R. LEONARD JR. | | Director | | February 10, 2023 |
| Keith R. Leonard Jr. | | | | |
/S/ ALAN J. LEVY | | Director | | February 10, 2023 |
| Alan J. Levy, Ph.D. | | |
/S/ JAMI DOVER NACHTSHEIM | | Director | | February 10, 2023 |
| Jami Dover Nachtsheim | | |
/S/ MONICA P. REED | | Director | | February 10, 2023 |
| Monica P. Reed | | | | |
/S/ MARK J. RUBASH | | Director | | February 10, 2023 |
| Mark J. Rubash | | | | |
