UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
|
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
OR
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number:
(Exact name of registrant as specified in its charter)
|
|
(IRS Employer |
|
|
|
|
|
|
|
(Address of principal executive offices) |
(Zip Code) |
Registrant’s telephone number, including area code: (
Securities Registered Pursuant to Section 12(b) of the Act:
|
Title of each class |
Trading Symbol |
Name of each exchange on which registered |
||
|
|
|
The |
Securities Registered Pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically, every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
Accelerated filer ☐ |
|
|
Large accelerated filer ☐ |
Smaller reporting company |
|
|
Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes
The aggregate market value of the registrant’s common stock, excluding shares beneficially owned by affiliates, computed by reference to price at which the registrant’s common stock was last sold as of June 30, 2023, was $
As of March 22, 2024, there were
TABLE OF CONTENTS
| Page | ||
| Cautionary Note Regarding Forward-Looking Statements | ii | |
| PART I | ||
| Item 1. | Business | 1 |
| Item 1A. | Risk Factors | 29 |
| Item 1B. | Unresolved Staff Comments | 41 |
| Item 1C. | Cybersecurity | 41 |
| Item 2. | Properties | 41 |
| Item 3. | Legal Proceedings | 42 |
| Item 4. | Mine Safety Disclosures | 42 |
| PART II | ||
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 42 |
| Item 6. | [Reserved] | 42 |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations | 42 |
| Item 7A. | Quantitative and Qualitative Disclosures about Market Risk | 54 |
| Item 8. | Financial Statements and Supplementary Data | 54 |
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | 54 |
| Item 9A. | Controls and Procedures | 54 |
| Item 9B. | Other Information | 55 |
| Item 9C. | Disclosure Regarding Foreign Jurisdictions that Prevent Inspections | 55 |
| PART III | ||
| Item 10. | Directors, Executive Officers and Corporate Governance | 55 |
| Item 11. | Executive Compensation | 58 |
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 62 |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence | 63 |
| Item 14. | Principal Accounting Fees and Services | 64 |
| PART IV | ||
| Item 15. | Exhibits, Financial Statements Schedules | 65 |
| Item 16. | Form 10-K Summary | 68 |
| SIGNATURES | ||
| Report of Independent Registered Public Accounting Firm | ||
| Financial Statements | F-1 | |
i
Cautionary Note Regarding Forward-Looking Statements
This annual report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”).
In some cases, you can identify forward-looking statements by the following words: “anticipate,” “assume,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “ongoing,” “plan,” “potential,” “predict,” “project,” “should,” “will,” “would,” or the negative of these terms or other comparable terminology, although not all forward-looking statements contain these words. Forward-looking statements are not a guarantee of future performance or results and will not necessarily be accurate indications of the times at, or by, which such performance or results will be achieved. Forward-looking statements are based on information available at the time the statements are made and involve known and unknown risks, uncertainties and other factors that may cause our results, levels of activity, performance or achievements to be materially different from the information expressed or implied by the forward-looking statements in this report. These factors include:
|
● |
our lack of diversification and the corresponding risk of an investment in our Company; |
|
● |
potential deterioration of our financial condition and results due to failure to diversify; |
|
● |
our ability to successfully complete acquisitions and integrate operations for new product candidates; |
|
● |
our ability to obtain additional capital, on acceptable terms or at all, required to implement our business plan; |
|
● |
results of our Phase Ia/Ib, and phase II clinical trials; |
|
● |
progress and success of our randomized Phase III clinical trial; |
|
● |
our ability to demonstrate safety and effectiveness of our product candidate; |
|
● |
our ability to obtain regulatory approvals for our product candidate in the United States, the European Union, or other international markets; |
|
● |
the market acceptance and future sales of our product candidate; |
|
● |
the cost and delays in product development that may result from changes in regulatory oversight applicable to our product candidate; |
|
● |
the rate of progress in establishing reimbursement arrangements with third-party payors; |
|
● |
the effect of competing technological and market developments; |
|
● |
the costs involved in filing and prosecuting patent applications and enforcing or defending patent claims; and |
|
● |
other risk factors included under the caption “Risk Factors” starting on page 30 of this report. |
You should read the matters described in “Risk Factors” and the other cautionary statements made in this report as being applicable to all related forward-looking statements wherever they appear in this report. We cannot assure you that the forward-looking statements in this report will prove to be accurate and therefore you are encouraged not to place undue reliance on forward-looking statements. You should read this report completely. Other than as required by law, we undertake no obligation to update or revise these forward-looking statements, even though our situation may change in the future.
We caution readers not to place undue reliance on any forward-looking statement that speaks only as of the date made and to recognize that forward-looking statements are predictions of future results, which may not occur as anticipated. Actual results could differ materially from those anticipated in the forward-looking statements and from historical results due to the risks and uncertainties described in Part I, Item 1A, of this annual report, as well as others that we may consider immaterial or do not anticipate at this time. Although we believe that the expectations reflected in our forward-looking statements are reasonable, we do not know whether our expectations will prove correct. Our expectations reflected in our forward-looking statements can be affected by inaccurate assumptions that we might make or by known or unknown risks and uncertainties, including those described in Part I, Item 1A, of this annual report. The risks and uncertainties described in Part I, Item 1A, of this annual report are not exclusive and further information concerning us and our business, including factors that potentially could materially affect our financial results or condition, may emerge from time to time. We assume no obligation to update forward-looking statements to reflect actual results or changes in factors or assumptions affecting such forward-looking statements. We advise stockholders and investors to consult any further disclosures we may make on related subjects in our subsequent annual reports on Form 10-K, quarterly reports on Form 10-Q, and current reports on Form 8-K that we file with or furnish to the U.S. Securities and Exchange Commission (the “SEC”).
ii
PART I
Item 1. Business
Panbela Therapeutics, Inc. and its wholly owned subsidiaries Panbela Research, Inc., Cancer Prevention Pharma Limited (Ireland) and Cancer Prevention Pharmaceuticals, Inc. (collectively “we,” “us,” “our,” “Panbela” and the “Company”) exist for the primary purpose of developing disruptive therapeutics for the treatment of patients with urgent unmet medical needs. Panbela Therapeutics Pty Ltd is a wholly owned subsidiary of Panbela Research, Inc. Cancer Prevention Pharmaceuticals, LLC., and Cancer Prevention Pharma Limited (UK and Wales) are wholly owned subsidiaries of Cancer Prevention Pharmaceuticals Inc. The original business entity predecessor to our Company was incorporated under the laws of the State of Delaware in 2011. The term “common stock” refers to our common stock, par value $0.001 per share.
Cancer Prevention Pharmaceuticals, Inc. Acquisition
On June 15, 2022, Panbela acquired Cancer Prevention Pharmaceuticals, Inc. (“CPP”), a private clinical stage company developing therapeutics to reduce the risk and recurrence of cancer and rare diseases, via merger for consideration consisting of (a) 304 shares of common stock including shares that were held back until June 15, 2023 subject to a holdback escrow (as defined in the Merger Agreement), (b) replacement options to purchase up to 42 shares of common stock at a weighted average exercise price of $6,743.41 per share, and (d) replacement warrants to purchase up to 4 shares of common stock at a weighted average exercise price of $6,720.00 per share, and post-closing contingent payments up to a maximum of $60 million, subject to satisfaction of milestones.
Holding Company Reorganization
Effective June 15, 2022, Panbela became a successor issuer to Panbela Research, Inc. (formerly known as Panbela Therapeutics, Inc., the “Predecessor”) pursuant to a holding company reorganization in which the Predecessor became a direct, wholly owned subsidiary of Panbela. Panbela became a successor issuer to the Predecessor by operation of Rule 12g-3(a) promulgated under the Securities Exchange Act of 1934, as amended the (“Exchange Act”).
Reverse Stock Splits
On January 18, 2024, we effected a reverse stock split at a ratio of one-for-twenty (1:20) shares of the Company’s common stock. On June 1, 2023, we effected a reverse stock split at a ratio of one-for-thirty (1:30) shares of the Company’s common stock and on January 13, 2023, we effected a reverse stock split at a ratio of one-for-forty (1:40) shares of the Company’s common stock. All share and per share amounts of our common stock presented have been retroactively adjusted to reflect these reverse stock splits.
Business Overview
Panbela is a clinical stage biopharmaceutical company developing disruptive therapeutics for the treatment of patients with urgent unmet medical needs. We are currently enrolling patients in our randomized double-blind placebo controlled clinical trial for the treatment of pancreatic cancer, a Phase III clinical trial funded by the National Cancer Institute (the “NCI”) for the study of colon cancer risk reduction and colon adenoma therapy (“CAT”), a preventative treatment approach for survivors of colorectal cancer or those who have high-risk colon polyps. In addition, we are designing a Phase III registration trial for familial adenomatous polyposis (“FAP”), a rare inherited condition that can cause the growth of thousands of colorectal adenomas (i.e., adenomatous polyps), which are recognized as a key risk factor for colon cancer. We also support several investigator initiated trials and company sponsored preclinical trials including: (1) Phase I and Phase II clinical trials for the treatment of early-onset type 1 diabetes funded by the Juvenile Diabetes Research Foundation; (2) Phase II clinical trial for the treatment of gastric cancer funded by the NCI; (3) Phase I/II clinical trial for the treatment of non-small cell lung cancer (“NSCLC”) possessing the STK11 mutation; (4) Phase II clinical trial for the treatment of metastatic castration-resistant prostate cancer; and (5) preclinical studies that we have sponsored in the orphan disease and cancer fields.
The Company’s lead assets are ivospemin (SBP-101), FlynpoviTM (eflornithine (CPP-1X) and sulindac), and eflornithine (CPP-1X), which provide a multi-targeted approach to reset dysregulated biology present in many types of diseases such as cancer and autoimmunity. Many tumors require greatly elevated levels of polyamines to support their growth and survival. These agents target the polyamine pathway at complementary junctions, which have been shown to be altered in disease. In particular, our lead assets have the potential to suppress and prevent tumor growth, enhance anti-tumor activity of other anti-cancer agents, and modulate the immune system.
Ivospemin is a proprietary polyamine analogue designed to induce polyamine metabolic inhibition. Ivospemin has demonstrated encouraging activity against metastatic disease in a clinical trial of patients with pancreatic cancer. The efficacy and safety results demonstrated in our completed Phase I clinical trial of ivospemin in combination with gemcitabine and nab-paclitaxel in the first line treatment of metastatic pancreatic cancer provide support for the current randomized, double-blind, placebo-controlled study of ivospemin in combination with gemcitabine and nab-paclitaxel in patients previously untreated for metastatic pancreatic cancer. We believe that ivospemin, if successfully developed, may represent a novel approach that effectively treats patients with pancreatic cancer and could become a dominant product in that market. In the past decade, two combination chemotherapy regimens, a quadruplet of fluorouracil, leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX) and a doublet, nab-paclitaxel and gemcitabine have been utilized as first-line standard of care. The first was based on a phase III trial but not Food and Drug Administration (“FDA”) approved and the latter based on a phase III trial which led to FDA approval. Most recently, the FDA approved Onivyde (irinotecan liposome injection) plus oxaliplatin, fluorouracil and leucovorin (NALIRIFOX) as a first-line treatment in adults living with metastatic pancreatic adenocarcinoma (“mPDAC”). This is the first FDA approval in first line mPDAC in over ten years. Ivospemin has received Fast Track status and orphan drug designation status for pancreatic cancer in the United States and we have also received orphan drug designation in Europe.
Our June 2022 acquisition of CPP added the Company’s second lead asset, eflornithine, in multiple forms. First, an investigational new drug product, Flynpovi, is a combination of the polyamine synthesis inhibitor eflornithine and the non-steroidal anti-inflammatory drug sulindac and then secondly, eflornithine as a single agent. Eflornithine is an enzyme-activated, irreversible inhibitor of the enzyme ornithine decarboxylase (“ODC”), the first rate-limiting enzyme in the biosynthesis of polyamines. Sulindac, a non-steroidal anti-inflammatory drug (“NSAID”), facilitates the export and catabolism of polyamines. Flynpovi has a unique dual mechanism of action whereby it suppresses the synthesis of new polyamines and increases the export and catabolism of polyamines from the diet and microbiome. We believe Flynpovi is unique in that it is designed to treat the risk factors (e.g., polyps) that lead to FAP surgeries and colon cancer and therefore may have the ability to prevent various types of colon cancer. In the FAP-310 Phase III trial, the efficacy and safety of Flynpovi (eflornithine (CPP-1X) and sulindac), as compared with either drug alone, in adults with FAP was conducted. While the study missed the primary composite endpoint (Burke et al. 2020), a post-hoc analysis showed that none of the patients in the combination arm progressed to a need for lower gastrointestinal (“LGI”) surgery for up to 48 months compared to 13.2% and 15.7% of patients in the sulindac and eflornithine arms (Balaguer et al. 2022). These data corresponded to risk reductions for the need for LGI surgery approaching 100% between combination and either monotherapy. Given the statistical significance of the LGI group, a new drug application (“NDA”) was filed with the FDA; however, since this was based on the results of an exploratory analysis, a complete response letter was issued. To address this deficiency concern, the Company must submit the results of one or more adequate and well-controlled clinical trial which demonstrates an effect on a clinical endpoint. There are no currently approved pharmaceutical therapies for FAP.
Additional programs are evaluating a single agent tablet eflornithine or high dose powder eflornithine sachets for several indications including prevention of gastric cancer, recent onset Type 1 diabetes, metastatic castration-resistant prostate cancer, and STK-11 mutant NSCLC. Preclinical studies and Phase I or Phase II investigator-initiated trials suggest that eflornithine treatment is well tolerated and has potential activity.
Flynpovi has received Fast Track designation in the United States and orphan drug designation status for FAP in the United States and Europe. In addition, we have received orphan drug designation status for eflornithine as a single agent for neuroblastoma in the United States and Europe and for gastric cancer in the United States.
Clinical Trials
Ivospemin (SBP-101)
In August 2015, the FDA accepted our Investigational New Drug (“IND”) application for our ivospemin product candidate. We have completed an initial clinical trial of ivospemin in patients with previously treated locally advanced or metastatic pancreatic cancer. This was a Phase I, first-in-human, dose-escalation, safety study. From January 2016 through September 2017, we enrolled twenty-nine patients into six cohorts, or groups, in the dose-escalation phase of the Phase I trial. No drug-related bone marrow toxicity or peripheral neuropathy was observed at any dose level. In addition to being evaluated for safety, 23 of the 29 patients were evaluable for preliminary signals of efficacy prior to or at the eight-week conclusion of their first cycle of treatment using the Response Evaluation Criteria in Solid Tumors (“RECIST”), the currently accepted standard for evaluating change in the size of tumors. A summary of both the safety and preliminary signals of efficacy for this completed clinical trial is contained later in this “Business” section under ivospemin (SBP-101) Clinical Development – Pancreatic Cancer, Phase I Clinical Trial Design and Completion (ivospemin Monotherapy).
In 2018, we began enrolling patients in our second clinical trial, a Phase Ia/Ib study of the safety, efficacy, and pharmacokinetics of ivospemin administered in combination with two standard-of-care chemotherapy agents, gemcitabine and nab-paclitaxel. A total of 25 subjects were enrolled in 4 cohorts to evaluate the dosage level and schedule. An additional 25 subjects were enrolled in the expansion phase of the trial. Interim results were presented in January of 2022. Best response in evaluable subjects (cohorts 4 and Ib N=29) was a Complete Response (“CR”) in 1 (3%), Partial Response (“PR”) in 13 (45%), Stable Disease (“SD”) in 10 (34%) and Progressive Disease (“PD”) in 5 (17%). One subject did not have post baseline scans with RECIST tumor assessments. Median Progression Free Survival (“PFS”), now final at 6.5 months, may have been negatively impacted by drug dosing interruptions to evaluate potential toxicity. Median overall survival in Cohort 4 + Phase Ib was 12.0 months when data was presented in January 2022 and is now final at 14.6 months. Two patients from cohort 2 have demonstrated long term survival: one at 30.3 months (final data) and one at 33.0 months and still alive as of March 18, 2022. Seven subjects were still alive at the data cutoff date of March 18, 2022, one from cohort 2 and six from cohort 4 plus Ib. Further details regarding the study design, safety and interim signals of efficacy are contained later in this “Business” section under ivospemin (SBP-101) Clinical Development – Pancreatic Cancer, Phase Ia/Ib Clinical Trial Interim Results (First Line Combination Therapy).
The safety results and tumor growth inhibition demonstrated in our Phase Ia/b study provides support for the randomized study of ivospemin initiated in January of 2022. The trial, referred to as the ASPIRE trial, is a randomized double-blind placebo-controlled trial in combination with gemcitabine and nab-paclitaxel in patients previously untreated for metastatic pancreatic cancer. The trial is being conducted globally at approximately 95 sites in the United States, Europe and Asia-Pacific. The ASPIRE trial commenced in 2022 and all countries are open to enrollment.
The ASPIRE trial will evaluate overall survival as the primary endpoint and will also be examined at the interim analysis. PFS will also be analyzed to provide additional efficacy evidence. This trial design was supported by the final data from the Phase Ia/Ib first line metastatic pancreatic cancer trial which completed enrollment in December of 2020. The ASPIRE study will enroll 600 subjects and is anticipated to take 36 months to complete enrollment with the interim analysis available in mid- 2024. The Independent Data Safety Monitoring Board (“DSMB”) has met twice, the most recent taking place in November 2023. The DSMB members evaluated the safety of 214 patients. The result of both DSMB meetings confirmed no safety concerns and the trial continuing without modification. On January 25, 2024, the Company announced that the ASPIRE trial had surpassed fifty percent enrollment and expects that the trial will be fully enrolled by the first quarter of 2025. Further details regarding the study design and anticipated timing are contained later in this “Business” section under ivospemin (SBP-101) Clinical Development – Pancreatic Cancer, Randomized Clinical Trial design and anticipated timing (ASPIRE trial).
If we successfully complete all FDA recommended clinical studies, we intend to seek marketing authorization from the FDA, the European Medicines Agency (“EMA”) (European Union), and Therapeutic Goods Administration (“TGA”) (Australia). The submission fees in the US and Europe may be waived for ivospemin as it has been designated an orphan drug in these geographic regions.
In early April 2022, the Company announced a poster presentation highlighting the results for ivospemin (also known as SBP-101) as a polyamine metabolism modulator in ovarian cancer at the American Association for Cancer Research Annual Conference which was subsequently published in June 2022 in the International Journal of Molecular Sciences (Holbert et al. 2022). The poster and publication conclude that the ivospemin treatment of C57Bl/6 mice injected with VDID8+ ovarian cancer cells significantly prolonged survival and decreased overall tumor burden. The results suggest that ivospemin may have a role in the clinical management of ovarian cancer, and the Company intends to continue pre-clinical and clinical studies in ovarian cancer. In April 2023, the Company announced a poster presentation highlighting additional preclinical work in ovarian cancer. The poster highlights the efficacy of SBP-101 in combination with standard of care chemotherapy agents used to treat platinum-resistant ovarian cancer. Treatment with gemcitabine, topotecan, and doxorubicin have been shown to significantly increase the in vitro toxicity of SBP-101 in both cisplatin-sensitive and cisplatin-resistant ovarian cancer cell lines. Paclitaxel and docetaxel have been shown to not have any added benefit in vitro to SBP-101 alone. The poster concludes that the treatment of C57Bl/6 mice containing VDID8+ ovarian cancer with SBP-101 in combination with doxorubicin significantly prolonged survival and decreased overall tumor burden.
Additional preclinical work is underway evaluating ivospemin and eflornithine (also known as CPP-1X or DFMO) in multiple myeloma (cell lines). Data published in the November supplemental issue of the Journal Blood investigated the effects of polyamine inhibition by ivospemin and CPP-1X on myeloma cell lines growth and viability in vitro. Results showed that ivospemin and CPP-1X treatment significantly decreased cell proliferation and induced apoptosis in a panel of multiple myeloma cell lines. When ivospemin and CPP-1X were combined an almost complete abolition of cell growth occurred. These results demonstrate the anti-neoplastic potential of ivospemin and CPP-1X and offer a compelling rationale for its clinical development as a potentially promising treatment option for multiple myeloma. The work reflects the company’s on-going collaboration with researchers from The University of Texas MD Anderson Cancer Center for the evaluation of polyamine metabolic inhibitor therapies in combination with CAR-T cell therapies in preclinical models.
Flynpovi
In December 2009, the FDA accepted our IND application for the combination product, Flynpovi. Flynpovi showed promising results in an NCI supported randomized, placebo-controlled Phase IIb/III clinical trial to prevent recurrent colon adenomas, particularly high-risk pre-cancerous polyps in which 375 subjects who had resected sporadic adenoma were treated for 3 years with eflornithine (500 mg once a day) + sulindac (150 mg once a day [N = 191]) or matched placebo/placebo (N = 184). Results demonstrated a marked risk reduction (70%) in developing metachronous adenomas, 92% risk reduction in developing advanced adenomas, and 95% risk reduction in developing multiple adenomas with the active combination regimen compared to placebo (Meyskens et al. 2008). This combination regimen was generally well tolerated.
Given the similar mechanism of disease in sporadic and FAP-associated adenomatous polyposis, and the mechanism of action of Flynpovi in prevention of progressive polyposis in both the general population with sporadic adenomas and in patients with FAP, a Phase III program in FAP and a Phase III program to study colon cancer risk reduction in partnership with the Southwest Oncology Group (“SWOG”) and the NCI were initiated.
In the FAP-310 Phase III study completed in 2019, the efficacy and safety of the combination of eflornithine and sulindac, as compared with either drug alone, in adults with familial adenomatous polyposis was conducted (Burke et al. 2020). The patients were randomly assigned in a 1:1:1 ratio to receive eflornithine, sulindac, or both once daily for up to 48 months. The primary end point, assessed in a time-to-event analysis, was disease progression, defined as a composite of major surgery, endoscopic excision of advanced adenomas, diagnosis of high-grade dysplasia in the rectum or pouch, or progression of duodenal disease. A total of 171 patients underwent randomization. Disease progression occurred in 18 of 56 patients (32%) in the eflornithine-sulindac group, 22 of 58 (38%) in the sulindac group, and 23 of 57 (40%) in the eflornithine group, with a hazard ratio of 0.71 (95% confidence interval [CI], 0.39 to 1.32) for eflornithine-sulindac as compared with sulindac (P = 0.29) and 0.66 (95% CI, 0.36 to 1.23) for eflornithine-sulindac as compared with eflornithine (Burke et al. 2020). Adverse and serious adverse events were similar across the treatment groups. In a post-hoc analysis, none of the patients in the combination arm progressed to a need for LGI surgery for up to 48 months compared with 7 (13.2%) and 8 (15.7%) patients in the sulindac and eflornithine arms (Balaguer et al. 2022). These data corresponded to risk reductions for the need for LGI surgery approaching 100% between combination and either monotherapy with HR = 0.00 (95% CI, 0.00-0.48; p = 0.005) for combination versus sulindac and HR = 0.00 (95% CI, 0.00-0.44; p = 0.003) for combination versus eflornithine. Given the statistical significance of the LGI group, an NDA was filed with the FDA. As the study failed to meet the primary endpoint, and the NDA was based on the results of an exploratory analysis, a complete response letter was issued. To address this deficiency concern, the Company must submit the results of one or more adequate and well-controlled clinical trial which demonstrates an effect on a clinical endpoint.
In collaboration with the NCI, and SWOG, a Phase III clinical trial has been initiated to study the benefits of Flynpovi as a therapeutic treatment for use by colon cancer survivors. The trial is named PACES for “Prevention of Adenomas and Cancer with eflornithine and sulindac.” The PACES trial is funded by the NCI and managed by SWOG. This is an ongoing Phase III double blind placebo-controlled trial of Flynpovi to prevent recurrence of high risk adenomas and second primary colorectal cancers in patients with stage 0-III colon or rectal cancer. The purpose of this study is to assess whether Flynpovi (compared to corresponding placebos) has a reduced rate of cancer or high-risk adenoma recurrence compared to comparator arms after three years of daily dosing. We have exclusive rights to the data that comes from the trial for regulatory and commercial purposes. The Company is evaluating its options for CAT in the European Union and Asia.
In April 2023, the Company announced that it regained the North American rights to develop and commercialize Flynpovi in patients with FAP, as a result of the termination of the licensing agreement between CPP and with One-Two Therapeutics Assets Limited effective July 4, 2023.
Eflornithine (CPP-1X) and eflornithine sachets (CPP-1X-S)
For the single agent eflornithine, there is a trial ongoing evaluating eflornithine sachets (CPP-1X-S) in a Phase I/II trial in STK11 mutation patients with non-small cell lung cancer which began this year and two trials ongoing to evaluate eflornithine tablets (CPP-1X-T), a Phase II trial in Recent Onset Type I diabetes with eflornithine and a Phase II trial in metastatic castration-resistant prostate cancer both of which began last year. Lastly, a Phase II trial evaluating eflornithine for the prevention of gastric cancer was completed in 2021 with data analysis ongoing.
Through March 22, 2024, we had:
|
● |
secured an orphan drug designation for ivospemin from the FDA; |
|
● |
submitted and received acceptance from the FDA for an IND application for ivospemin; |
|
● |
completed a Phase Ia monotherapy safety study of ivospemin in the treatment of patients with metastatic pancreatic ductal adenocarcinoma; |
|
● |
received “Fast Track” designation from the FDA for ivospemin for metastatic pancreatic cancer; |
|
● |
completed enrollment and released interim results in our second trial a Phase Ia /Ib clinical study of ivospemin, a first-line study with ivospemin given in combination with a current standard of care in patients with pancreatic ductal adenocarcinoma who were previously untreated for metastatic disease; a total of 50 subjects were enrolled in this study, 25 in the Phase Ia and 25 in the Phase Ib or expansion phase; |
|
● |
secured a two-year research agreement with Johns Hopkins School of Medicine led by Professor Robert Casero, an internationally recognized researcher in polyamine biology; |
|
● |
completed process improvement measures expected to be scalable for commercial use and received issue notification for a patent covering this new shorter synthesis of ivospemin in several territories; |
|
● |
initiated a randomized, double-blind, placebo-controlled study, referred to as ASPIRE, with ivospemin given in combination with gemcitabine and nab-paclitaxel in patients with pancreatic ductal adenocarcinoma who are previously untreated for metastatic disease; |
|
● |
completed preclinical evaluation of ivospemin for use as neoadjuvant therapy in resectable pancreatic cancer prior to surgery; |
|
● |
obtained early, preclinical, indication of tumor growth inhibition activity in ovarian cancer and presented the results at ASCO-GI and AACR conferences; |
|
● |
received USAN adoption of the nonproprietary name of ivospemin for SBP-101; |
|
● |
acquired and integrated CPP, adding a second lead asset in multiple forms and an expansive clinical development program ranging from pre-clinical to registration level clinical trials; |
|
● |
EMA Committee for Orphan Medicinal Products issued a positive opinion on Panbela’s application for orphan designation of ivospemin in combination with gemcitabine and nab-Paclitaxel in patients with metastatic pancreatic ductal adenocarcinoma; |
|
● |
announced the initiation of Phase II program through Indiana University for early onset Type I diabetes utilizing eflornithine; |
|
● |
ASPIRE is open to enrollment in every planned country within NA, EMEA, and APAC, Completed two Independent DSMB meetings for ASPIRE with no safety concerns or modifications to study design; |
|
● |
announced the initiation of the Phase I/II clinical trial for the treatment of NSCLC possessing the STK11 mutation through Moffitt Cancer Center; |
|
● |
entered into a sponsored research agreement with The University of Texas MD Anderson Cancer Center for the evaluation of polyamine metabolic inhibitor therapies in combination with CAR-T cell therapies in preclinical models; |
|
● |
announced the SWOG Cancer Research Network’s PACES S0820 Phase III trial passed a single planned futility analysis and will continue; |
|
● |
announced the approval of US WorldMeds’ NDA Approval for Eflornithine (DFMO) in Pediatric Neuroblastoma, the first polyamine approval in oncology; and |
|
● |
exceeded 50% enrollment in ASPIRE global clinical trial. |
Pancreatic Cancer
Pancreatic cancer afflicts approximately 151,000 people in Europe (Epidemiology in Europe and Recommendations for Screening in High-Risk Populations, Partyka, et al July 2023), approximately 64,000 people in the United States annually (American Cancer Society. Cancer Facts & Figures 2023. Atlanta, GA: American Cancer Society; 2023 and Overview of Pancreatic Cancer) and 293,000 people worldwide – excluding Europe and United States (GLOBOCAN 2020). It has been identified as the fourth leading cause of death from cancer in Europe (GLOBOCAN 2020) and the third leading cause of death from cancer in the United States (SEER Cancer Statistics Factsheets 2021). On average, Pancreatic Ductal Adenocarcinoma (“PDA”) represents approximately 95% of all pancreatic cancers diagnosed in a given calendar year. Considering that the median overall survival for previously untreated patients with good performance status is between 8.5 months (Von Hoff 2013) and 11.1 months (Conroy 2011) with the two most commonly available treatment regimens, effective treatment for PDA has remained a major unmet medical need.
Pancreatic cancer is generally not diagnosed early because the initial clinical signs and symptoms are vague and non-specific. The most common presenting symptoms include weight loss, epigastric (upper central region of the abdomen) and/or back pain, and jaundice. The back pain is typically dull, constant, and of visceral origin radiating to the back, in contrast to the epigastric pain which is vague and intermittent. Less common symptoms include nausea, vomiting, diarrhea, anorexia, and new onset diabetes (which can be an early signal) or glucose intolerance (Hidalgo 2010).
Surgery remains the only treatment option with curative intent, although only about 20% of patients are candidates for surgical resection at the time of the diagnosis. Patients who undergo radical surgery still have a limited survival rate, averaging 23 months (Macarulla T, et al Clin Transl Oncol 2017).
For the minority of patients who present with resectable disease, surgery is the treatment of choice. Depending on the location of the tumor, the operative procedures may involve cephalic pancreatoduodenectomy, referred to as a “Whipple procedure” distal pancreatectomy or total pancreatectomy. Pancreatic enzyme deficiency and diabetes are frequent complications of both the disease and these surgical procedures. Up to 70% of patients with pancreatic cancer present with biliary obstruction that can be relieved by percutaneous or endoscopic stent placement. However, even if the tumor is fully resected, the outcome in patients with pancreatic cancer has been disappointing (Hidalgo 2010, Seufferlein 2012). Post-operative administration of chemotherapy improved progression-free and overall survival in three large randomized clinical trials (Hidalgo 2010), but median post-surgical survival in patients treated in all three trials was similar, only 20-22 months. Pre-operative (neo-adjuvant) chemotherapy is of increasing interest, with the goal of improved successful resections and long-term outcomes.
For patients who present with unresectable, locally advanced or metastatic disease, which represent a majority of PDA patients, management options range from chemotherapy alone to combined forms of treatment with radiation therapy and chemotherapy. However, due to the increased toxicity of combined treatment, randomized trials of such combined regimens have had low enrollment, precluding a firm conclusion as to any advantage of adding radiation to chemotherapy (Hidalgo 2010).
Gemcitabine was the first chemotherapeutic agent approved for the treatment of patients with PDA in the modern regulatory era, providing a median survival duration of 5.65 months (Burris 1997). Gemcitabine monotherapy was the standard of care for patients with metastatic pancreatic cancer until combination therapy with gemcitabine plus erlotinib (Tarceva®) was shown to increase median survival by two weeks. This modest benefit was tempered by a significant side effect profile and high cost, limiting its adoption as a standard treatment regimen. Subsequently, the multidrug chemotherapy combination FOLFIRINOX was shown to provide a median survival benefit of 4.3 months (Overal Survival (“OS”) = 11.1 months) over gemcitabine alone (6.8 months), but its significant side effect profile limits the regimen to select patients with a good performance status and often requires supplementation with WBC growth factor therapy. Nab-paclitaxel (Abraxane®) received marketing authorization for use in combination with gemcitabine (FDA approved 2013) after showing an increase in overall survival of seven weeks compared to gemcitabine alone (Von Hoff 2013).
On February 13, 2024 Onivyde® (irinotecan liposome injection) plus oxaliplatin, fluorouracil and leucovorin (NALIRIFOX) was approved by the FDA as a first-line treatment in adults living with mPDAC. This is the first drug approved as a first-line treatment of PDA since the approval of Abraxane. Lynparza® was approved in December 2019 for maintenance therapy of patients with deleterious or suspected deleterious germline BRCA-mutated (“gBRCAm”) metastatic pancreatic adenocarcinoma whose disease has not progressed on at least 16 weeks of a first-line platinum-and chemotherapy regimen.
Familial adenomatous polyposis
Familial adenomatous polyposis (“FAP”) is a rare and potentially life threatening genetic condition occurring in approximately one in 10,000 individuals in the United States. FAP is caused primarily by mutations in the adenomatous polyposis coli (“APC”) tumor suppressor gene. APC mutations are usually inherited as autosomal dominant genetic traits, but as many as 25% of those afflicted with FAP with an identical germline mutation have no family history. Only 1 in 10,000 people will develop FAP. Estimated annual prevalence in the U.S. is approximately 30,000 and in Europe approximately 50,000. If untreated, patients will develop hundreds to thousands of polyps throughout the colon and rectum. FAP often develops in the early teens and results in a nearly 100% lifetime risk of colorectal cancer by age forty if untreated. No approved FAP drug is on the market.
Most patients are asymptomatic for years until the adenomas are large and numerous, and cause rectal bleeding or even anemia, or cancer develops. Generally, cancers start to develop a decade after the appearance of the polyps. Nonspecific symptoms may include constipation or diarrhea, abdominal pain, palpable abdominal masses and weight loss.
Cancer prevention and maintaining a good quality of life are the main goals of management of patients with FAP. By the late teens or early twenties, colorectal cancer prophylactic surgery is advocated. Prophylactic surgery often requires total abdominal colectomy with ileal-rectal anastomoses (“IRA”) and subsequent frequent endoscopic surveillance, with polypectomy and cautery/laser ablation as needed. Patients with extensive rectal involvement must undergo total proctocolectomy with ileal pouch-anal reconstruction. Despite this, approximately 50% of patients who have had total proctocolectomy with ileal pouch-anal reconstruction will develop adenomatous polyps in the neo-rectum (ileal pouch). Duodenal cancer and desmoids are the two main causes of mortality after total colectomy; they need to be identified early and treated. Upper endoscopy is necessary for surveillance to reduce the risk of ampullary and duodenal cancer. Patients with progressive tumors and unresectable disease may respond or stabilize with a combination of cytotoxic chemotherapy and surgery (when possible, to perform). Individuals with FAP carry a 100% risk of CRC; however, this risk is reduced significantly when patients enter a screening-treatment program.
A major unmet need in the treatment of patients with FAP is a therapeutic means to defer or obviate the need for major surgical interventions, particularly colectomy with IRA or proctocolectomy with an ileal surgical pouch IPAA. Such interventions often require temporary or permanent ileostomy, and with it, long-term or permanent quality of life (“QoL”) deficits such as frequent bowel movements (average 6 per day), nocturnal fecal incontinence and, in female patients, reduced reproductive potential. It is critical to find non-surgical alternatives that will delay or obviate the need of repeated endoscopic and surgical procedures to maintain patient QoL. For those patients who have an intact colon in particular, pharmacotherapy offers the opportunity to meaningfully control or delay polyposis progression and offer a greater choice over when or if they undergo prophylactic colectomy/proctocolectomy in order to optimize QoL.
This potential benefit is in fact likely the most powerful potential benefit possible since the long-term course of FAP essentially mandates ultimate colectomy for most patients. The value to a younger patient in safely delaying such a radical procedure by years cannot be overstated.
There are currently no approved and marketed pharmacotherapeutic treatments for patients with FAP. While in 1999 celecoxib was conditionally approved by the FDA for the treatment of FAP based on reductions of polyp number observed in a randomized double-blind placebo controlled study conducted in patients with FAP, it was subject to the marketing authorization holder, Pfizer, providing additional data. In 2011, the FDA requested that Pfizer voluntarily withdraw the FAP indication for CELEBREX (celecoxib) Capsules from the market because the post-marketing study intended to verify clinical benefit and required as a condition of approval under subpart H was never completed. In a letter in 2011, Pfizer requested that the FDA withdraw the FAP indication for CELEBREX (celecoxib) Capsules from the market. Effective in 2012, the approval for the FAP indication for CELEBREX Capsules was withdrawn. Celecoxib was also authorized for FAP treatment centrally by the European Commission after the EMA’s scientific review in October 2003 under “exceptional circumstances”. Authorization was granted subject to specific obligations during product life cycle, chiefly to provide further data on its efficacy and safety; however, the applicant/authorization holder could not fulfill this central post-authorization obligation. According to publicly available information, the post- authorization study was initiated in the first quarter of 2004 and the EU Centralized Marketing Authorisation was withdrawn because the holder was unable to provide the data as required.
Ovarian Cancer
Worldwide Ovarian Cancer has annual incidence of approximately 314,000 and annual deaths of approximately 207,000 (Globocan 2020). In the United States, Ovarian represents approximately 1% of all new cancer cases at approximately 22,000 (American Cancer Society. Cancer Facts & Figures 2021. Atlanta, GA: American Cancer Society; 2021) and the five-year survival rate for metastatic disease is approximately 29% (SEER fact sheet Ovarian 2022). According to the American Cancer Society, ovarian cancer is the fifth leading cause of cancer deaths among women, accounting for more deaths than any other cancer of the female reproductive system.
Nearly 70 % of the patients are diagnosed with advanced-stage due to the failure of screening methods for detecting early-stage disease (Giornelli 2016; Partridge et al. 2009; Bast et al. 2007; Gohagan et al. 2000; Chudecka-Głaz 2015). Thus, most patients will relapse within the first 2 years after diagnosis, even after an optimal primary cytoreductive surgery and six cycles of the standard adjuvant chemotherapy with carboplatin/paclitaxel.
The second line chemotherapy depends mainly on the disease-free interval (“DFI”) (time between completion of first line chemotherapy and clinical relapse); or progression-free interval (“PFI”) (time between the last chemotherapy given for relapsed disease and progression). There are three classifications: Platinum-refractory/resistant with relapse during platinum treatment (refractory) or with a DFI/PFI <6 months (resistant), Platinum-sensitive relapse occurring >12 m of last platinum-based chemotherapy, or partially sensitive to platinum with disease-free survival DFS/ PFS between 6 and 12 months from the last platinum-based chemotherapy.
According to Pignata et al. 2017, in platinum-sensitive patients, treatment with platinum-based combinations is associated with a PFS advantage compared with single agents or non-platinum combinations. For patients with partially sensitive relapse (PFI between 6 and 12 months), two options are available: platinum doublets or non-platinum therapy (single agent or combination). Last, patients with resistant or refractory relapse (PFI < 6 months) disease there are few options. For these patients, monotherapy with a non-platinum drug or participation in clinical trials is indicated.
Colorectal Cancer
According to United States Cancer Statistics published by the American Cancer Society, in the United States in 2022, it is estimated that CRC will be the third most commonly occurring cancer among males and females and the third leading cause of cancer-related deaths. High-risk adenomatous polyps are considered the key risk factor for CRC. In 2015, the disease will be responsible for an estimated 52,000 deaths in the United States. An even higher rate of incidence occurs in the European Union, where approximately 255,000 people per year die from CRC according to the Globocan 2020 Fact Sheets.
Globally, there are approximately 1,931,000 new diagnoses each year (approximately 180,000 expected in North America in 2020). Rates of presentation are also becoming significant in Asia (China and Japan). Colorectal adenomas (or “polyps”) are considered the key risk factor for CRC. The general consensus in the medical and scientific communities is that these polyps are the precursors to more than 90% of all colorectal cancers.
Colon cancer represents nearly three-fourths of all colorectal cancers in the U.S. Despite potentially curative treatment with surgery (with or without adjuvant chemotherapy), local stage and locally advanced stage colon cancer patients remain at considerable risk for colorectal adenomas, distant recurrence, secondary colonic tumor formation, and colorectal cancer related mortality. Polypectomy appears to be an effective way to decrease mortality from colon cancer, but widespread adoption of this approach is limited by both cost and patient acceptability (Newcomb et al. 1992; Selby et al. 1992). Certain types of colorectal polyps have increased risk of progression to colorectal cancer. High-risk polyps (polyps with villous histology, size ≥ 1 cm, high grade dysplasia, or multiple adenomas defined as 3 or more) have become the focus of colorectal tumorigenesis research due to the higher rate of malignant potential for these lesion (Lotfi et al. 1986; Spencer et al. 1984; Winawer et al. 1993; Martinez et al. 2009). The current standard of care for resected colon cancer patients (beyond surgery, and adjuvant chemotherapy when indicated) is surveillance monitoring with clinical exams, laboratory analyses, and colonoscopic evaluation. However, data suggest that colonoscopy does not predict death from colorectal cancer uniformly throughout the colon – in fact, right-sided colorectal cancers were not observed to gain any mortality benefit from colonoscopy (Baxter et al. 2009). Other potential problems with colonoscopy include (rarely) perforations, infection, bleeding, and non-adherence with current recommendations. Safe and effective chemo preventive interventions, therefore, offer great potential to complement and improve upon the current colon cancer surveillance paradigm. Unlike other therapies used to treat CAT, Flynpovi is a non-surgical and non-invasive option that has the potential to both improve patient quality of life and reduce higher healthcare system-wide expense burdens.
Proprietary Technology
Function and Characteristics of Polyamines
Polyamines are metabolically distinct entities within human cells that bind to and facilitate DNA replication, RNA transcription and processing, and protein (such as pancreatic enzymes) synthesis. Human cells contain three essential and naturally occurring polyamines – putrescine, spermidine, and spermine. Polyamines perform many functions necessary for cellular proliferation, apoptosis and protein synthesis. The critical balance of polyamines within cells is maintained by several enzymes such as ODC and spermidine/spermine N1 acetyl transferase (“SSAT”). All of these homeostatic enzymes are short-lived, rapidly inducible intracellular proteins that serve to regulate native polyamine pools tightly and continuously. These enzymes constantly maintain polyamines within a very narrow range of concentration inside the cell.
Polyamine metabolism and cancer
Polyamines are required for cell proliferation. It is believed that many cancers, especially oncogene-driven cancers, might be sensitive to interference with polyamine metabolism. The natural polyamines putrescine, spermidine and spermine are intimately involved in growth-related processes, wound healing, and the development of cancer. Under normal conditions, the pool of polyamines is tightly controlled through regulation of synthesis, catabolism, and transport mechanisms (Gerner and Meyskens 2004). The loss of this tight control can result in an excessive accumulation of polyamines, which favors malignant transformation of cells. Consequently, with the loss of growth control in cancer cells, the transformed cells may be more sensitive to polyamine depletion than normal cells. Thus, the polyamine metabolic pathway is a rational target for therapeutic intervention (Casero 2018).
Immune systems require multiple soluble and cellular components, including polyamines, for a normal immune function. As such, polyamines are important modulators of the immune response, particularly in the tumor microenvironment where they are found in high concentrations. High levels of polyamines are present in tumor cells and in autoreactive B- and T-cells in autoimmune diseases. Dysregulation of polyamines can result in tumor immune evasion, elevated cell stress, and increased autoimmunity. By resetting the polyamine pathway through therapeutic interventions, there is the potential to restore normal immune functions.
Pharmacotherapeutic Approaches to Reset the Polyamine Pathway
The Company’s lead assets are ivospemin and eflornithine (including Flynpovi), which provide a multi-targeted approach to reset dysregulated biology present in many types of diseases such as cancer and autoimmunity. For instance, many tumors require greatly elevated levels of polyamines to support their growth and survival. These agents target the polyamine pathway at complementary junctions which have been shown to be altered in disease. In particular, these agents have the potential to suppress and prevent tumor growth, enhance anti-tumor activity of other anti-cancer agents, and modulate the immune system.
Polyamine Analogue–- ivospemin (SBP-101)
Many tumors, including pancreatic cancer, display an increased uptake rate of polyamines. Polyamine analogues such as ivospemin are structurally similar to naturally occurring polyamines and are recognized by the cell’s polyamine uptake system, allowing these compounds to gain ready entrance to the cell. We believe that pancreatic acinar cells, because of their extraordinary protein synthesis capacity, exhibit enhanced uptake of polyamines and polyamine analogues. Because of this preferential uptake by pancreatic acinar cells, polyamine analogues such as ivospemin disrupt the cell’s polyamine balance and biosynthetic network, and induce programmed cell death, or apoptosis, via processes including caspase 3 activation and poly ADP ribose polymerase (PARP) cleavage. Proof of concept has been demonstrated in multiple human pancreatic cancer models, both in vivo and in vitro, that pancreatic ductal adenocarcinoma exhibits sensitivity to ivospemin.
Ivospemin is a proprietary polyamine analogue, which we believe accumulates in the exocrine pancreas acinar cells due to its unique chemical structure. Ivospemin was discovered and extensively studied by Professor Raymond J. Bergeron at the University of Florida College of Pharmacy.
As laboratory studies suggest, the primary mechanism of action for ivospemin has been demonstrated to include the enhanced uptake of the compound in the exocrine pancreas; therefore, pancreatic cancer was logical for the initial development of this compound. Sufficiently high dosing in animal models leads to correspondingly depressed levels of native polyamines, with caspase 3 activation, PARP cleavage and apoptotic destruction (programmed cell death) of the exocrine pancreatic acinar and ductal cells without an inflammatory response. Importantly, pancreatic islet cells, which secrete insulin, are structurally and functionally dissimilar to acinar cells and are not impacted by ivospemin. In animal models at two independent laboratories, ivospemin has demonstrated significant suppression of transplanted human pancreatic cancer cells, including metastatic pancreatic cancer growth.
We believe that ivospemin exploits the natural affinity of the exocrine pancreas, the liver and kidney, and pancreatic ductal adenocarcinoma cells while leaving the insulin-producing islet cells unharmed. Most current cancer therapies, including chemotherapy, radiation, and surgery, are associated with significant side effects that further reduce the patient’s quality of life. However, based on data evaluated from clinical studies to date, we believe that the adverse effects of ivospemin in causing bone marrow suppression or peripheral neuropathy do not overlap with or exacerbate those seen with typical chemotherapy options. The dose-limiting toxicities observed in cohort five of our first Phase I study, as noted below, were not observed at lower doses and are not expected to overlap with the adverse events of bone marrow suppression and peripheral neuropathy commonly associated with standard chemotherapy. The dose and dosing schedule evaluated in the expansion phase of the recently completed Phase Ia/Ib is below the maximum tolerable dose MTD and at this dose level, neither the exocrine nor the endocrine human pancreas is expected to be affected by ivospemin, resulting in no treatment impact on pancreatic enzyme or insulin levels. This dose level and dosing schedule in the new ASPIRE trial will be the same as evaluated in the expansion phase of the Ia/Ib study.
Ornithine Decarboxylase Inhibitor–- eflornithine (CPP-1X)
Ornithine decarboxylase is the first and rate-limiting enzyme in the biosynthesis of polyamines which catalyzes the conversion of ornithine to putrescine and regulates the biosynthesis of polyamines in mammalian as well as many other eukaryotic cells. Eflornithine, also known as α-difluoromethylornithine (DFMO), is an ornithine analogue. Eflornithine irreversibly binds to ODC1 and prevents the natural ODC1 substrate, ornithine, from accessing the active site of the enzyme (Meyskens and Gerner 1999). The administration of eflornithine decreases both ODC activity and polyamine concentrations. In genetic mouse models with an APC gene mutation, the administration of eflornithine reduces intestinal carcinogenesis, decreasing the concentration of polyamines through inhibition of ODC and inhibiting tumor development (Erdman et al 1999).
Treatment of animals with eflornithine results in inhibition of ODC activity, especially in tissues and organs with rapidly dividing cells. Polyamine biosynthesis has been shown to be critical for eukaryotic cellular growth and differentiation, and inhibition of polyamine biosynthesis can stimulate or inhibit cellular differentiation depending on the model studied (Gerner and Meyskens 2004). Accordingly, eflornithine has promoted or inhibited cell differentiation in a variety of models.
Polyamine biosynthesis is also a critical step in experimental chemical-induced carcinogenesis, cell transformation, and tumor cell proliferation, and there is a growing body of evidence that eflornithine's inhibitory effect on cell proliferation and tumorigenesis may involve a complex inter-relationship between oncogenes, polyamine metabolism, and ODC activity. MYC is an oncogene that encodes a transcription factor that is required for the proliferation of normal cells but when overexpressed can lead to aberrant cell growth (Gerner and Meyskens 2004). Additionally, c-Myc is a transcriptional activator of the ODC gene (Pena et al. 1993) (Bello-Fernandez, Packham, and Cleveland 1993). Furthermore, eflornithine has been shown to decrease N-Myc mRNA in neuroblastoma cells and c-Myc mRNA in human colon carcinoma cells (Celano et al. 1988) and spermidine preferentially stimulated transcription and expression of c-Myc, but not c-Fos (Tabib and Bachrach 1999). Taken together, these results suggest that polyamines play a feedback role in the regulation of expression of certain oncogenes at the level of transcription.
Mice with a mutation of the adenomatous polyposis coli (“Apc”) tumor suppressor gene develop intestinal tumors in numbers similar to those found in patients with FAP. Mutations of the Apc gene increases the activity of ODC and leads to increased intestinal polyamine levels. Studies in animal models of FAP indicate that eflornithine alone is effective in reducing the number of intestinal tumors (Erdman et al. 1999) and colonic tumor burden (Yerushalmi et al. 2006). Eflornithine may lower polyamine levels in colorectal mucosa and skin cells (Gerner and Meyskens 2004).
The major clinical evidence for benefit of eflornithine derives from prospective, randomized, placebo-controlled clinical studies of eflornithine monotherapy in patients with elevated risk for developing certain forms of cancer (prostate and basal cell skin cancer). In a randomized, placebo-controlled, clinical study in subjects with a history of resected colon polyps, eflornithine reduced polyamines in rectal mucosal tissue. This marker study is especially relevant to patients with FAP, in whom target tissues include intestinal and colonic mucosa (Meyskens et al. 1998).
Eflornithine has received regulatory approvals as a high dose, intravenously delivered medication for the treatment of a form of African sleeping sickness, as a topical agent for the treatment of hirsutism (excess hair growth on body parts where hair growth is usually absent or minimal) and in 2023 as an oral dosage form to reduce the risk of relapse in adults and children with high-risk neuroblastoma.
Activator of Spermidine/Spermine N-Acetyltransferase (“SSAT1”) – Sulindac
Transport of polyamines is maintained by the peroxisome-proliferator activated receptor-g (“PPARg”). This receptor positively regulates SSAT transcription facilitating polyamine acetylation and transport of polyamines out of the cell. Under normal conditions, the K-Ras molecule has no activity on PPARg. However, mutation of the K-Ras gene produces a product that inhibits PPARg’s effect on SSAT translation resulting in elevated polyamine pools and tumorigenesis (Babbar et al. 2003). NSAIDs, such as sulindac, act through PPARg to enhance transcriptional of SSAT which increases catabolism and export of polyamines.
Sulindac is a member of the arylalkanoic acid class of NSAIDs and is a non-selective inhibitor of cyclooxygenases involved in prostaglandin synthesis. To understand potential mechanisms of action of sulindac, patterns of gene expression resulting from treatment with sulindac sulfone, a sulindac metabolite lacking cyclooxygenase inhibitory activity, were measured in human colon tumor-derived cells (Babbar et al. 2003). Sulindac sulfone inhibited cell growth, and induced apoptosis and the expression of spermidine/spermine N-acetyltransferase (SSAT1), a polyamine catabolic enzyme implicated in polyamine export (Xie, Gillies, and Gerner 1997). Sulindac sulfone induction of SAT1 expression occurs via a cyclooxygenase-independent transcriptional activation of SAT1 at a specific peroxisomal proliferator activated receptor gamma (PPARγ) responsive element PPRE in the SAT1 gene. Treatment of cells with sulindac sulfone induces SAT1 expression and stimulates polyamine export.
Experimental findings in human cell and mouse models indicate that sulindac and other NSAIDS activate polyamine catabolism (Gerner and Meyskens 2009). Thus, NSAIDs complement inhibitors of polyamine synthesis, like eflornithine, to reduce tissue polyamine levels. In cell culture, sulindac metabolites reduce cell survival in vitro in a dose-dependent manner at doses above 150 µM at 24-hour exposure times (Lawson et al. 2000).
Experiments in both mouse and rat models of colon cancer have demonstrated a preventative effect for sulindac (Babbar et al. 2003). Sulindac blocked tumor formation in the multiple intestinal neoplasia (Min) mouse, a murine model of APC mutation-associated intestinal carcinogenesis, mimicking FAP. In the Min mouse, tumor-preventing doses of sulindac inhibited tissue levels of prostaglandin-E2 and COX-2 (Boolbol et al. 1996). In other nonclinical studies, sulindac had an inhibitory effect on bladder, lung, and forestomach tumor formation in rat and mouse models (Kelloff, Boone, et al. 1994, Kelloff, Crowell, et al. 1994).
Dual Targeting–- Flynpovi
The ability to decrease the polyamine pools by a dual mechanism of action, i.e., suppressed synthesis and enhanced catabolism and export, led to the hypothesis that Flynpovi would complement one another in the prevention of tumor development in a patient population where elevated polyamine pools lead to enhanced tumorigenesis. Eflornithine is the irreversible inhibitor of ODC which is responsible for de novo synthesis of polyamines and sulindac regulates SSAT which plays a role in polyamine export and catabolism. Hence the combination, Flynpovi, inhibits the generation of new polyamines and also removes polyamines obtained from the diet and microbiome.
The ability of Flynpovi to reduce polyamines in the GI tract has been demonstrated in both the preclinical and clinical settings. In the study by Igantenko et al, the effect of eflornithine alone and in combination with NSAIDs sulindac or celecoxib on intestinal tumor number and grade and polyamine content was evaluated in ApcMin/+ mice (Ignatenko et al. 2008). Administration of eflornithine in combination with sulindac was superior to each single agent at significantly (P< 0.05) decreasing putrescine, spermidine, and total intestinal polyamine concentrations to below baseline levels in the ApcMin/+ mice. Additionally, in this study with the exception of the 0.5% eflornithine treatment group, all treatment groups developed significantly (P<0.05) fewer tumors/animal than the control group. The combination treatment of 2% eflornithine and sulindac suppressed intestinal tumorigenesis to a level that was not statistically significantly different from that for sulindac alone. Although sulindac alone produced a significant decrease in the number of intestinal tumors in ApcMin/+ mice, it did not reduce the percentage of high‑grade adenomas. However, the combination of eflornithine and sulindac significantly (P<0.05) decreased the number of high-grade adenomas compared to the sulindac alone group.
The ability of the eflornithine and sulindac treatment group to suppress high grade adenomas is a key finding as it is the high-grade adenomas in this model which correlate to the high-grade adenomas seen in FAP patients that are indicators for excisional and surgical events clinically. These data support the rationale for treatment of FAP patients with eflornithine combined with sulindac to reduce intestinal polyamine contents and the incidence of high-grade intestinal adenomas.
More importantly, combination treatment with Flynpovi dramatically reduces the incidence of metachronous colorectal adenomas in patients with prior sporadic adenomas (Meyskens et al. 2008). Meyskens and colleagues performed a Phase IIb/III, double-blind pharmacoprevention of Sporadic Colorectal Adenomas Study (PSCA Study) in which 375 subjects who had resected sporadic adenoma were treated for 3 years with eflornithine (500 mg once a day) + sulindac (150 mg once a day [N = 191]) or matched placebo/placebo (N = 184). Results demonstrated a marked risk reduction (70%) in developing metachronous adenomas, 92% risk reduction in developing advanced adenomas, and 95% risk reduction in developing multiple adenomas with the active combination regimen compared to placebo. This combination regimen was generally well tolerated.
The mechanism of disease in sporadic and FAP-associated adenomatous polyposis, and the mechanism of eflornithine and NSAID action in prevention of progressive polyposis in both the general population with sporadic adenomas and in patients with FAP, led to the development of the FAP-310 trial in patients with FAP associated with APC germline mutations.
The FAP-310 Phase III study that evaluated the efficacy and safety of the combination of eflornithine and sulindac, as compared with either drug alone, in adults with familial adenomatous polyposis was conducted (Burke et al. 2020). The patients were randomly assigned in a 1:1:1 ratio to receive eflornithine, sulindac, or both once daily for up to 48 months. In a post-hoc analysis, none of the patients in the combination arm progressed to a need for LGI surgery for up to 48 months compared with 7 (13.2%) and 8 (15.7%) patients in the sulindac and eflornithine arms (Balaguer et al. 2022). These data corresponded to risk reductions for the need for LGI surgery approaching 100% between combination and either monotherapy with HR = 0.00 (95% CI, 0.00-0.48; P= 0.005) for combination versus sulindac and HR = 0.00 (95% CI, 0.00-0.44; P= 0.003) for combination versus eflornithine.
Development Plan for Ivospemin (SBP-101)
Development of ivospemin for the pancreatic cancer indication has included a pre-clinical and a clinical phase. The pre-clinical phase, which was substantially completed during 2015, consisted of four primary components: chemistry, manufacturing and controls (“CMC”), preclinical (laboratory and animal) pharmacology studies, preclinical toxicology studies, and regulatory submissions in Australia and the United States.
Preparation of the ivospemin IND for pancreatic cancer required collaboration by our manufacturing, preclinical toxicology, pharmacokinetic, and metabolism experts, our regulatory affairs project management, and our in-house clinical expertise. In August 2015, the FDA accepted our application.
In Australia, a Human Research Ethics Committee application was submitted with subsequent Clinical Trial Notification CTN to the TGA.
Our initial clinical trial in previously treated patients with locally advanced or metastatic pancreatic cancer was a Phase I, first-in-human, dose-escalation, safety study conducted at clinical sites in both Australia and the United States. We engaged expert clinicians who treat pancreatic cancer at major cancer treatment centers in Melbourne and Adelaide, Australia as well as the Mayo Clinic Scottsdale and HonorHealth in Scottsdale, Arizona. These Key Opinion Leaders, with proven performance in pancreatic cancer studies, agreed to participate as investigators for our Phase I First-in-Human study.
Enrollment in our initial Phase I safety trial of ivospemin in previously treated pancreatic cancer patients commenced in January 2016 and was completed in September 2017. Results from this trial are discussed in ivospemin (SBP-101) Clinical Development – Pancreatic Cancer, Phase I Clinical Trial Design and Completion (ivospemin Monotherapy) below.
We completed enrollment of patients in our second clinical trial in December 2020. This second clinical trial was a Phase Ia/Ib study of the safety, efficacy and pharmacokinetics of ivospemin administered in combination with two standard-of-care chemotherapy agents, gemcitabine and nab-paclitaxel. A total of 25 subjects were enrolled in four cohorts of Phase Ia and an additional 25 subjects were enrolled in the expansion Phase Ib by December of 2020. Safety and interim efficacy results from this trial are discussed in ivospemin (SBP-101) Clinical Development–- Pancreatic Cancer, Phase Ia/Ib Clinical Trial Interim Results (First Line Combination Therapy) below.
In January of 2022, we initiated our third clinical trial. This new trial is a randomized, double blind, placebo-controlled study of safety and efficacy of ivospemin administered in combination with two standard-of-care chemotherapy agents, gemcitabine and nab-paclitaxel. Trial design and expected timing are discussed in Clinical Development–- Pancreatic Cancer, Randomized Clinical Trial Design and Anticipated Timing (ASPIRE trial).
In addition, we are exploring ivospemin for neoadjuvant treatment in appropriate pancreatic cancer patients. There is also preclinical data to suggest that ivospemin may have potential therapeutic uses for cancers other than pancreatic. In February 2021, we entered into a research agreement with the Johns Hopkins University School of Medicine. The collaboration has focused on the further development of Panbela’s investigative agent ivospemin, including activity in cell lines outside of pancreatic cancer, biomarkers informing diagnostics and potential combination with checkpoint inhibitors. In December 2021, the Company announced positive preclinical data supporting the activity of ivospemin in ovarian cancer cell lines which was presented and published in 2022 (Holbert et al. 2022). Further data resulting from the ongoing relationship with Johns Hopkins University School of Medicine is expected.
Ivospemin (SBP-101) Clinical Development – Pancreatic Cancer
Our clinical development in Pancreatic Cancer thus far includes:
|
● |
a Phase I SBP-101 Monotherapy study completed in 2017; |
|
● |
a Phase Ia/Ib SBP-101 First Line Combination Therapy study, Study enrollment completed in 2020 and data base locked in early 2022; and |
|
● |
ASPIRE, a Randomized, Double-Blind Placebo Controlled First Line Combination Therapy study was initiated in January of 2022. |
Details of these programs follow.
Phase I Clinical Trial Design and Completion (ivospemin Monotherapy)
We have completed an initial clinical trial of ivospemin in patients with previously treated locally advanced or metastatic pancreatic cancer. This was a Phase I, first-in-human, dose-escalation, safety study. From January 2016 through September 2017, we enrolled twenty-nine patients into six cohorts, or groups, in the dose-escalation phase of the Phase I trial. No drug-related bone marrow toxicity or peripheral neuropathy was observed at any dose level. In addition to being evaluated for safety, 23 of the 29 patients were evaluable for preliminary signals of efficacy prior to or at the eight-week conclusion of their first cycle of treatment using the RECIST, the currently accepted standard for evaluating change in the size of tumors.
The absence of adverse events which could potentially overlap with adverse events typically observed in the use of conventional chemotherapeutic agents, supports the case for combination of ivospemin with conventional chemotherapeutic agents, such as gemcitabine, nab-paclitaxel, or even FOLFIRINOX.
Phase Ia/Ib Clinical Trial Interim Results (First Line Combination Therapy)
In 2018, we began enrolling patients in our second clinical trial, a Phase Ia/Ib study of the safety, efficacy and pharmacokinetics of ivospemin administered in combination with two standard-of-care chemotherapy agents, gemcitabine and nab-paclitaxel. A total of 25 subjects were enrolled in 4 cohorts to evaluate the dosage level and schedule. An additional 25 subjects were enrolled in the expansion phase of the trial. Interim results were presented in January of 2022. Best response in evaluable subjects (cohorts 4 and Ib N=29) was a CR in 1 (3%), PR in 13 (45%), SD in 10 (34%) and PD in 5 (17%). One subject did not have post baseline scans with RECIST tumor assessments. Median PFS, now final at 6.5 months, may have been negatively impacted by drug dosing interruptions to evaluate potential toxicity. Median overall survival in Cohort 4 + Phase Ib was 12.0 months when data was presented in January 2022 and is now final at 14.6 months. Two patients from cohort 2 have demonstrated long term survival: one at 30.3 months (final data) and one at 33.0 months and still alive.
Figure 4. Evaluation of SBP 101 Phase Ib First-line combo-therapy Safety Trial -
Best Overall Response
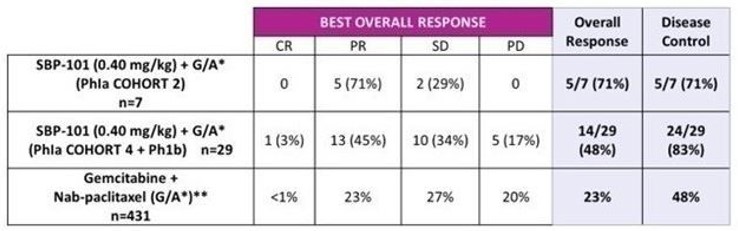
Source: Singhal, N., Poster Presentation, ASCO GI 2022
Randomized Clinical Trial design and anticipated timing (ASPIRE trial)
In January of 2022, the Company announced the initiation of a new clinical trial. Referred to as ASPIRE, the trial is a randomized double-blind placebo-controlled trial in combination with gemcitabine and nab-paclitaxel, a standard pancreatic cancer treatment regimen in patients previously untreated for metastatic pancreatic cancer. The trial will be conducted globally at approximately 88 sites in the United States, Europe and Asia–- Pacific.
While opening of clinical sites in the United States and the rest of the world has been slower than originally anticipated, due in part to resource fatigue in the medical community, all sites were opened in the quarter ending March 31, 2024.
The trial was originally designed as a Phase II/III trial with a smaller sample size (150) to support the events required for interim analysis based on PFS and a primary endpoint of overall survival (“OS”). In response to European and FDA regulatory feedback, the study was amended to include the total trial sample size (600) and the design modified to utilize overall survival as the primary endpoint to be examined at interim analysis. PFS will also be analyzed to provide additional efficacy evidence. This amendment was supported by the final data from the Phase Ia/Ib first line metastatic pancreatic cancer trial which completed enrollment in December of 2020. The study will enroll 600 subjects and is anticipated to take 36 months for complete enrollment.
On January 25, 2024, the Company announced that the trial had exceeded 50% enrollment. The Company projects that full enrollment will be completed by the first quarter of 2025 and that interim data analysis based on overall survival should be available by the middle of 2024.
If we can successfully complete all FDA recommended clinical studies, we intend to seek marketing authorization from the FDA, the EMA, Ministry of Health and Welfare (Japan) and TGA (Australia). The submission fees may be waived in geographies where ivospemin has been designated an orphan drug, as described under “Orphan Drug Status.”
Development Plan for Flynpovi and Eflornithine (CPP-1X)
In December 2009, the FDA accepted CPP’s IND application for the combination product, Flynpovi, product candidate and in November 2009 and August 2018, the FDA accepted IND applications for eflornithine.
The Development plan executed by CPP of Flynpovi for FAP and colon cancer prevention has included both a pre-clinical/non-clinical and a clinical phase. The non-clinical phase consisted of four primary components: CMC, preclinical (laboratory and animal) pharmacology studies, preclinical toxicology studies, and regulatory submissions in the U.S. and Europe. Similarly, the development plan for eflornithine and eflornithine sachets in several different indications included much of the same primary components and regulatory submission in the U.S.
Clinical Development –Flynpovi
Our clinical development of Flynpovi thus far includes:
|
● |
The FAP-310 Phase III |
|
● |
The PACES Phase III trial |
FAP-310 Phase III Trial
In the FAP-310 Phase III study, the efficacy and safety of the combination of Flynpovi (ES combo), as compared with either drug eflornithine or sulindac alone, in adults with FAP was conducted. A total of 171 patients underwent randomization. Disease progression occurred in 18 of 56 patients (32%) in the Flynpovi, 22 of 58 (38%) in the sulindac group, and 23 of 57 (40%) in the eflornithine group, with a hazard ratio of 0.71 (95% CI, 0.39 to 1.32) for Flynpovi as compared with sulindac (P = 0.29) and 0.66 (95% CI, 0.36 to 1.23) for Flynpovi as compared with eflornithine. In a post-hoc analysis, none of the patients in the combination arm progressed to a need for LGI surgery for up to 48 months compared with 7 (13.2%) and 8 (15.7%) patients in the sulindac and eflornithine arms. These data corresponded to risk reductions for the need for LGI surgery approaching 100% between combination and either monotherapy with HR = 0.00 (95% CI, 0.00–0.48; P = 0.005) for combination versus sulindac and HR = 0.00 (95% CI, 0.00–0.44; P = 0.003) for combination versus eflornithine.

Given the statistical significance of the LGI group, an NDA was filed with the FDA. As the study failed to meet the primary endpoint, and the NDA was based on the results of an exploratory analysis, a complete response letter was issued. To address this deficiency concern, the Company must submit the results of one or more adequate and well-controlled clinical trial which demonstrates an effect on a clinical endpoint.
Phase III Clinical Trial in Colon Cancer Survivors
In collaboration with the NCI, and SWOG, a Phase III clinical trial has been initiated to study the benefits of Flynpovi as a therapeutic treatment for use by colon cancer survivors. The trial is named PACES for “Prevention of Adenomas and Cancer with eflornithine and sulindac.” The PACES trial is funded by the NCI and managed by SWOG. This is an ongoing double-blind placebo-controlled trial of Flynpovi to prevent recurrence of high-risk adenomas and second primary colorectal cancers in patients with stage 0-III colon or rectal cancer, Phase III PACES. The purpose of this study is to assess whether the Flynpovi, combination of eflornithine and sulindac, (compared to corresponding placebos) has a reduced rate of cancer or high-risk adenoma recurrence compared to comparator arms after three years of daily dosing. We have exclusive rights to the data that comes from the trial for regulatory and commercial purposes.
Clinical Development –Eflornithine (CPP – 1X)
Our clinical development of eflornithine thus far includes:
|
● |
Phase II Gastric Cancer Prevention Trial |
|
● |
Phase I and Phase II Recent Onset Type 1 Diabetes Trials |
|
● |
Phase I/II STK-11 Mutant NSCLC Trial |
|
● |
Phase II Metastatic Castration-Resistant Prostate Cancer Trial |
Phase II Gastric Cancer Prevention Trial
H. pylori is the most common bacterial infection in humans and causes gastritis in all individuals. Gastritis progresses along the “Correa cascade” from gastritis to the precancerous stages of atrophic gastritis (loss of specialized gastric epithelium) and intestinal metaplasia, to gastric adenocarcinoma (Correa 1992). In response to H. pylori infection the host elicits a robust innate and adaptive immune response, which results in mucosal inflammation but fails to eradicate the organism. Several studies have demonstrated that the failure of the immune response may be related to dysregulated L-arginine metabolism and polyamines including the upregulation of ornithine decarboxylase (“ODC”) by macrophages (Chaturvedi et al. 2010; Chaturvedi, de Sablet, Coburn, et al. 2012) (Chaturvedi, de Sablet, Peek, et al. 2012), (Chaturvedi et al. 2011) (Xu et al. 2004) (Chaturvedi et al. 2014) (Chaturvedi et al. 2004). Levels of polyamines are increased in H. pylori-induced gastritis in mice, and oral DFMO treatment reduces gastric polyamine levels, and severity of both H. pylori colonization and gastritis (Chaturvedi et al. 2010). In the gerbil model of H. pylori-induced gastric cancer, polyamine levels correlate with levels of gastritis, DNA damage, and progression to dysplasia/carcinoma. In this model, eflornithine reduces polyamine levels and DNA damage, and reduces rates of dysplasia and carcinoma by >50% (Chaturvedi et al. 2014).
In collaboration with investigators at Vanderbilt University and funding by the NCI, the investigator-initiated Phase II trial performed in Honduras and Puerto Rico was a randomized, double-blinded study comparing once daily eflornithine versus placebo for an up to 18-month treatment period in patients with gastric premalignant lesions. This trial has completed and is undergoing data analysis. The Company has received orphan drug designation for the use of eflornithine for the treatment of gastric cancer in the United States.
Phase I and II Recent Onset Type 1 Diabetes (“T1D”) Trials
T1D is an organ-specific autoimmune disease characterized by chronic immune-mediated destruction of pancreatic β-cells, leading to partial, or in most cases, absolute insulin deficiency. The majority of cases result from autoimmune mediated pancreatic β-cell destruction, which occurs at a variable rate. Patients become clinically symptomatic when approximately 90% of pancreatic β-cells are destroyed. Therefore, preserving β-cell function is a target for promising treatments (Couper et al. 2014). The activity of ODC is upregulated in early diabetic kidney disease, contributing to renal hypertrophy and hyperfiltration (Pedersen et al. 1992; Deng et al. 2003). In vivo studies in experimental models of recent-onset T1D evaluating eflornithine demonstrate roles in suppressing the development of renal hypertrophy and hyperplasia, decreasing the incidence of diabetes, augmenting the survival and regeneration of β-cell populations, decreasing insulinitis, and maintaining an immune-tolerant balance of T-cell subpopulations.
The Company collaborated with investigators at Indiana University on a JDRF funded Phase I study to evaluate the safety and efficacy of increasing doses of eflornithine in patients with recent onset Type 1 diabetes. The completed Phase I trial demonstrated that a 3-month course of oral eflornithine was well tolerated with a favorable adverse event profile in children and adults with recent-onset T1D (Sims et al 2023). Urinary polyamine data showed that eflornithine treatment inhibited ornithine decarboxylase activity effectively, reflected by a dose dependent reduction in urinary putrescine values. Furthermore, although not powered to detect metabolic efficacy, subjects treated with 750 mg/m2/day and 1000 mg/m2/day of eflornithine exhibited higher C-peptide AUC 6 months after treatment indicative of improved β cell function compared to placebo (Sims et al 2023). These data suggest that eflornithine may improve β cell function alone and in combination regimens to treat or prevent type 1 diabetes that also include immunotherapy. Based on the Phase I trial results, a randomized, double-blind, placebo-controlled Phase II study to evaluate the efficacy and safety of eflornithine treatment to preserve insulin production in Type 1 diabetes initiated in early 2023.This is a JDRF funded trial being led by investigators at Indiana University. The primary objectives are to examine the clinical efficacy of 1000 mg/m2/day of oral DFMO to stabilize or improve loss of β cell function in persons with recent onset T1D based on mixed-meal stimulated C-peptide AUC in the treatment group compared to placebo after 6 months of DFMO treatment and to evaluate the safety and tolerability of 1000 mg/m2/day of oral DFMO in persons with new onset T1D. The secondary objectives are to elucidate the relationship between DFMO treatment and markers of endoplasmic reticulum stress, polyamine concentrations, immunologic and other mechanistic outcomes in persons with new onset T1D and characterize the dietary polyamine intake and urinary polyamine excretion of persons with recent-onset T1D. This study is a multicenter, double-blind, placebo-controlled, 2:1 random assigned targeting a total of 70 patients.
Phase I/II STK-11 Mutant NSCLC Trial
STK11 is the fourth-most frequently mutated gene in lung adenocarcinoma, with loss of function occurring in up to 30% of all cases (Laderian et al. 2020). Patients with LKB1 loss have reduced infiltrates of cytotoxic T-cells and respond poorly to anti PD1 or anti-PDL-1 therapies regardless of the PDL-1 status. CheckMate-057 trial lung tumors harboring co-mutations in KRAS and STK11 had an inferior response to PD-1 axis inhibitors (Skoulidis et al. 2018). These results suggest that STK11-mutated tumors were found to have a cold immune microenvironment regardless of KRAS status.
Bioinformatic analyses using two well-annotated lung adenocarcinoma datasets identified upregulation of ornithine decarboxylase (the target for eflornithine). Additionally, LKB1-loss tumors show a significant up-regulation of several solute transporters (SLC7A2, SLC14A2, and SLC16A4). SLC7A2 is known to be responsible for the membrane transport of cationic amino acids arginine, lysine, and ornithine. Furthermore, LKB1 loses the arginine pathway in which arginine is converted to ornithine and urea (by arginase) and ornithine is converted to putrescine (by ODC1). Together, the results suggested that ODC1 may be a key metabolic driver in LKB1-loss lung cancer.
In other model systems eflornithine treatment has been shown to modulate the tumor microenvironment. A previously studied cohort revealed that ODC1 may be instrumental to immune suppression (Chamaillard et al. 1997). Since eflornithine is an ODC1 inhibitor, it is hypothesized that inhibiting the metabolic enzyme ODC1 using eflornithine will increase the number of tumor-infiltrating lymphocytes in LKB1-loss tumors and restore benefit of PD(L)-1 blockade to these patients.
The Company has initiated a Phase I/II investigator-initiated trial to assess eflornithine in patients with STK-11 mutant NSCLC 2024.
Phase II Metastatic Castration-Resistant Prostate Cancer Trial
Prostate cancer is uniformly lethal once it has escaped the confines of the prostate gland, resulting in the death of over ~30,000 American patients each year (Jemal et al. 2008). Androgen ablation therapy has remained the standard of care for patients with recurrent/metastatic cancer since its discovery by Charles Huggins in the 1940s (Huggins 1941). While androgen ablation therapy provides significant palliative benefit, all patients undergoing androgen ablation eventually relapse and no longer respond to androgen ablation no matter how completely given (Crawford et al. 1989, Laufer et al. 2000). Recent clinical trials from investigators at Johns Hopkins University have demonstrated that rapidly cycling from the polar extremes of high dose to castrate serum levels of androgen, Bipolar Androgen Therapy (“BAT”) (Denmeade et al. 2010), was well tolerated and showed signs of clinical benefit. Preclinical studies have demonstrated that the addition of eflornithine enhanced the anti-tumor effects of BAT therapy.
The Company has initiated a Phase II investigator-initiated trial in to determine if treatment with the combination of difluoromethylornithine (DFMO or eflornithine) and high dose testosterone will improve the prostate-specific antigen (“PSA”) response rate in patients with metastatic castrate-resistant prostate cancer compared with historical controls. This trial opened in October 2023 and is recruiting subjects.
Neuroblastoma Trial
Neuroblastoma, a rare cancer originating from immature nerve cells, contributes to nearly 15% of pediatric cancer deaths. Panbela Therapeutic’s subsidiary, Cancer Prevention Pharmaceuticals, has extensively collaborated with leading neuroblastoma research groups such as the Neuroblastoma Medulloblastoma Translational Research Consortium (“NMTRC”) (now Beat Childhood Cancer), New Advances in Neuroblastoma Therapy (“NANT”), the Children’s Oncology Group (“COG”), and the NCI in the clinical development of eflornithine as a treatment for neuroblastoma.
In July of 2023, the Company announced it had divested certain assets in its eflornithine pediatric neuroblastoma program to US WorldMeds® (“USWM”), a Kentucky-based specialty pharmaceutical company. Under the terms of the agreement, Panbela is entitled to receive up to approximately $9.5 million non-dilutive funding in exchange for the sale of certain assets within its pediatric neuroblastoma program for eflornithine. Panbela will receive payments upon USWM’s successful completion of milestones related to eflornithine's clinical development, regulatory approval, and commercial sales.
In December of 2023, the Company announced the USWM received FDA approval of their NDA for the use of eflornithine as a maintenance therapy for neuroblastoma in remission.
Total Development Costs
The development of ivospemin involves a preclinical and clinical development phase. We completed our initial preclinical development work for pancreatic cancer and two Phase I clinical trials. The Phase II/III trial was initiated in 2022 and is currently ongoing. Additional clinical trials may be required to receive FDA or other foreign jurisdictions approvals. The cost and timing of additional clinical trials is highly dependent on the nature and size of the trials.
The development of Flynpovi also has involved preclinical and clinical development work for FAP and colon cancer prevention. The Company intends to secure consensus from the FDA and the EMA on a global registration trial, before seeking a development or licensing partner.
Orphan Drug Status
The Orphan Drug Act provides special status to drugs which are intended for the safe and effective treatment, diagnosis or prevention of rare diseases that affect fewer than 200,000 people in the United States, or that affect more than 200,000 persons but for which a manufacturer is not expected to recover the costs of developing and marketing such a drug. Orphan drug designation has the advantage of reducing drug development costs by: (i) streamlining the FDA’s approval process, (ii) providing tax breaks for expenses related to the drug development, (iii) allowing the orphan drug manufacturer to receive assistance from the FDA in funding the clinical testing necessary for approval of an orphan drug, and (iv) facilitating drug development efforts. More significantly, the orphan drug manufacturer’s ability to recover its investment in developing the drug is also greatly enhanced by the FDA granting the manufacturer seven years of exclusive U.S. marketing rights upon approval. Designation of a product candidate as an orphan drug therefore may provide its sponsor with the opportunity to adopt a faster and less expensive pathway to commercializing its product.
We obtained U.S. Orphan Drug Status for ivospemin in 2014 and in Europe in early 2023.
We have obtained orphan drug designation status for Flynpovi and eflornithine for FAP in the United States (2013 and 2011 respectively) and Europe (2013 and 2011 respectively). In addition, we have received orphan drug designation status for eflornithine as a single agent for neuroblastoma in the United States (2010) and Europe (2011) and for gastric cancer (2015) in the United States.
Fast Track
In June 2020, we received Fast Track Designation from the FDA for development of ivospemin for the treatment of first-line patients with metastatic PDA when administered in combination with gemcitabine and nab-paclitaxel.
Additionally, in September 2017, we received Fast Track Designation from the FDA for the development of Flynpovi for the treatment of FAP.
With the designation of Fast Track Designation, we, or our North American partners, may engage in more frequent interactions with the FDA, and the FDA may review sections of a NDA before the application is complete. This rolling review is available if we provide, and the FDA approves, a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, the FDA’s application review period does not begin until the last section of the NDA is submitted. Additionally, Fast Track Designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
Intellectual Property
As the result of efforts at our contract manufacturer Syngene International Ltd to refine the synthetic process, a new shorter synthetic process has been developed on which a patent (US 11,098,005 B2) “METHODS FOR PRODUCING (65,155)-3,8,13,18-TETRAAZAICOSANE-6, 15-DIOL” issued on Aug. 24, 2021, and was assigned to Panbela. The patent claims cover a novel process for the production of ivospemin and reduces the number of synthetic steps from nineteen to six. This patent has issued in territories, Australia, US, Europe (Germany, France, Spain, Great Britain and Italy), India, and Japan and patent prosecution is ongoing in several other territories.
Additionally, for ivospemin, a Method of Use Patent for the treatment of cancer in undergoing patent prosecution in several different territories and has the potential for patent coverage through 2041.
For Flynpovi, there is a composition of matter patent for the fixed dose combination of eflornithine and sulindac that is broadly nationalized, providing potential protection through 2037. Additionally, we hold several Method of Use patents for Flynpovi and/or eflornithine for the treatment of Familial Adenomatous Polyposis, neuroblastoma, and the Treatment of Recent Onset Type 1 Diabetes.
We are evaluating other opportunities to provide additional intellectual property.
Human Capital Management
As of March 22, 2024, we had eight employees, seven of which were full time. None of our employees are represented by a labor union or covered by a collective bargaining agreement. We believe our relationship with our employees is good.
Our human capital resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing, and integrating our existing and new employees, advisors, and consultants. The principal purposes of our equity and cash incentive plans are to attract, retain and reward personnel through the granting of stock-based and cash-based compensation awards, to motivate such individuals to perform to the best of their abilities and achieve our objectives and lead to the success of the Company and increase value to our stockholders.
We value diversity of backgrounds and perspectives in our workforce and our policy is that we do not discriminate based on race, religion, creed, color, national origin, ancestry, physical disability, mental disability, medical condition, genetic information, marital status, sex, gender, gender identity, gender expression, age, military and veteran status, sexual orientation or any other protected characteristic as established by federal, state or local laws.
We believe that operational responsibilities can be managed by our current employees, independent consultants and our global CRO. We have historically used the services of independent consultants and contractors to perform various professional services. We believe that this use of third-party service providers enhances our ability to minimize general and administrative expenses. We intend to periodically evaluate our staffing and talent requirements and expect to add employees if that becomes a more appropriate resource alternative.
Competition
The development and commercialization of new products to treat cancer is intensely competitive and subject to rapid and significant technological change. While we believe that our knowledge, experience and scientific resources provide us with competitive advantages, we face substantial competition from major pharmaceutical companies, specialty pharmaceutical companies, and biotechnology companies worldwide. Many of our competitors have significantly greater financial, technical, and human resources. Smaller and early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. As a result, our competitors may discover, develop, license or commercialize products before or more successfully than we do.
We face competition with respect to our current product candidates and will face competition with respect to future product candidates, from segments of the pharmaceutical, biotechnology and other related markets that pursue approaches to targeting molecular alterations and signaling pathways associated with cancer. Our competitors may obtain regulatory approval of their products more rapidly than we do or may obtain patent protection or other intellectual property rights that limit our ability to develop or commercialize our product candidates. Our competitors may also develop drugs that are more effective, more convenient, less costly, or possessing better safety profiles than our products, and these competitors may be more successful than us in manufacturing and marketing their products.
In addition, we may need to develop our product candidates in collaboration with diagnostic companies, and we will face competition from other companies in establishing these collaborations. Our competitors will also compete with us in recruiting and retaining qualified scientific, management and commercial personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Furthermore, we also face competition more broadly across the market for cost-effective and reimbursable cancer treatments. The most common methods of treating patients with cancer are surgery, radiation and drug therapy, including chemotherapy, immunotherapy, hormone therapy and targeted drug therapy or a combination of such methods. There are a variety of available drug therapies marketed for cancer. In many cases, these drugs are administered in combination to enhance efficacy. While our product candidates, if any are approved, may compete with these existing drug and other therapies, to the extent they are ultimately used in combination with or as an adjunct to these therapies, our product candidates may be approved as companion treatments and not be competitive with current therapies. Some of these drugs are branded and subject to patent protection, and others are available on a generic basis. Insurers and other third-party payors may also encourage the use of generic products or specific branded products. We expect that if our product candidates are approved, they will be priced at a premium over competitive generic, including branded generic, products. As a result, obtaining market acceptance of, and gaining a significant share of the market for, any of our product candidates that we successfully introduce to the market will pose challenges. In addition, many companies are developing new therapeutics, and we cannot predict what the standard of care will be as our product candidate progresses through clinical development.
Commercialization
We have not established a sales, marketing or product distribution infrastructure nor have we devoted significant management resources to planning such an infrastructure because our lead product candidate is still in early clinical development. We currently anticipate that we will partner with a larger pharmaceutical organization having the expertise and capacity to perform these functions.
Manufacturing and Suppliers
We do not own or operate, and currently have no plans to establish, any manufacturing facilities. We currently rely, and expect to continue to rely, on third parties for the manufacture of our product candidates for preclinical and clinical testing as well as for commercial manufacture of any products that we may commercialize. If needed, we intend to engage, by entering into a supply agreement or through another arrangement, third-party manufacturers to provide us with additional ivospemin clinical supply. We identified and qualified manufacturers to provide the active pharmaceutical ingredient and fill-and-finish services for our initial product candidate prior to our submission of an NDA to the FDA and expect to continue utilizing this approach for other product candidates.
Material Agreements
The Standard Exclusive License Agreement (“License Agreement”) dated December 22, 2011, between us and UFRF grants us an exclusive license to the proprietary technology covered by issued United States Patents Nos. US 5,962,533, which expired in February 2016, and US 6,160,022 which expired in July 2020 and Know-How as defined by the License Agreement, with reservations by UFRF for academic or government uses. Under this agreement, we had agreed to pay various royalties, expenses and milestone payments to UFRF. The License Agreement was amended in December 2016 (“First Amendment”) and again in October 2019 (“Second Amendment”). Under the Second Amendment all minimum royalty payments and milestone payments defined in the License Agreement were eliminated. In addition, the period for payment of royalties was changed to be shorter of (i) ten (10) years from first commercial sale or (ii) the period of market exclusivity on a country-by-country basis. UFRF may also terminate this license for standard and similar causes such as material breach of the agreement, bankruptcy, failure to pay royalties and other customary conditions. The agreement allows for UFRF to terminate if the first commercial sale is not made by December 31, 2025.
Government Regulation
FDA Approval Process
In the United States, pharmaceutical products are subject to extensive regulation by the FDA. The Federal Food, Drug and Cosmetic Act and other federal and state statutes and regulations govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling and import and export of pharmaceutical products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution.
Pharmaceutical product development for a new product or certain changes to an approved product in the United States typically involves preclinical laboratory and animal tests, the submission to the FDA of an IND which must become effective before clinical testing may commence, and adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug for each indication for which FDA approval is sought. Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease.
Preclinical tests include laboratory evaluation of product chemistry, formulation and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements, including good laboratory practices. The results of preclinical testing are submitted to the FDA as part of an IND along with other information, including the Investigator’s Brochure, information about product chemistry, manufacturing and controls, potential perceived side effects and risks, and a proposed clinical trial protocol. Long-term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has neither commented on nor questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin.
Clinical trials involve the administration of the investigational new drug to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted: (i) in compliance with federal regulations; (ii) in compliance with good clinical practice (“GCP”), an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators and monitors; as well as (iii) under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. patients and certain subsequent protocol amendments must be submitted to the FDA as part of the IND.
The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent information for patients in clinical trials must also be submitted to an institutional review board (“IRB”) for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In Phase I, the initial introduction of the drug into healthy human subjects/patients, the drug is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses, and, if possible, early evidence of effectiveness. Phase II usually involves trials in a limited patient population to determine the drug's effectiveness for a particular indication, dosage tolerance and optimum dosage, and to identify common adverse effects and safety risks. If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase II evaluations, pivotal, or Phase III trials are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the drug. In many cases the FDA requires two adequate and well-controlled Phase III clinical trials to demonstrate the efficacy of the drug. A single Phase III trial with other confirmatory evidence may be sufficient in instances where the study is a large multicenter trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome, and confirmation of the result in a second trial would be practically or ethically impossible. After an NDA is approved, a Phase IV trial may be undertaken to evaluate safety over a long period of time, quality of life or cost effectiveness.
After completion of the required clinical testing, an NDA is prepared and submitted to the FDA. FDA approval of the NDA is required before marketing of the product may begin in the United States. The NDA must include the results of all preclinical, clinical and other testing and a compilation of data relating to the product’s pharmacology, chemistry, toxicology, manufacture, controls and any proposed labeling. The cost of preparing and submitting an NDA is substantial, and the fees are typically increased annually.
The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of NDAs to encourage timeliness. Most applications for standard review drug products are reviewed within twelve months from submission; most applications for priority review drugs are reviewed within eight months from submission. Priority review can be applied to drugs that the FDA determines offer major advances in treatment or provide a treatment where no adequate therapy exists. If priority review is achieved, the FDA’s goal is to act on the application within six months. The review process for both standard and priority review may be extended by the FDA for three additional months to consider certain late-submitted information, or information intended to clarify information already provided in the submission. The FDA may also refer applications for novel drug products, or drug products that present difficult questions of safety or efficacy, to an outside advisory committee—typically a panel that includes clinicians and other experts—for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations.
Before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product unless compliance with current good manufacturing practice (“cGMP”), a quality system regulating manufacturing, is satisfactory and the NDA contains data that provide substantial evidence that the drug is safe and effective in the indication studied.
After the FDA evaluates the NDA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing, or information, in order for the FDA to reconsider the application. If, or when, those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included.
An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require a risk evaluation and mitigation strategy (“REMS”) to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use (“ETASU”). ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained, or problems are identified following initial marketing.
Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing NDAs.
Fast Track Designation and Accelerated Approval
The FDA is required to facilitate the development, and expedite the review, of drugs that are (1) intended for the treatment of a serious or life-threatening disease or (2) condition for which there is no effective treatment, and which demonstrate the potential to address unmet medical needs for the condition. Under the Fast Track program, the sponsor of a new product candidate may request that the FDA designate the product candidate for a specific indication as a Fast Track drug concurrent with, or after, the filing of the IND for the product candidate. The FDA must determine if the product candidate qualifies for Fast Track Designation within 60 days of receipt of the sponsor’s request.
Under the Fast Track program and the FDA’s accelerated approval regulations, the FDA may approve a drug for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments.
In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions, or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A product candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase IV or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, will allow the FDA to withdraw the drug from the market on an expedited basis. All promotional materials for product candidates approved under accelerated regulations are subject to priority review by the FDA.
If a submission is granted Fast Track Designation, the sponsor may engage in more frequent interactions with the FDA, and the FDA may review sections of the NDA before the application is complete. This rolling review is available if the applicant provides, and the FDA approves, a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, the FDA’s time period goal for reviewing an application does not begin until the last section of the NDA is submitted. Additionally, Fast Track Designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
Breakthrough Therapy Designation
The FDA is also required to expedite the development and review of the application for approval of drugs that are intended to treat a serious or life-threatening disease or condition where preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. Under the Breakthrough Therapy program, the sponsor of a new product candidate may request that the FDA designate the product candidate for a specific indication as a breakthrough therapy. The FDA must determine if the product candidate qualifies for Breakthrough Therapy designation within 60 days of receipt of the sponsor’s request.
Orphan Drug Designation and Exclusivity
The Orphan Drug Act provides incentives for the development of products intended to treat rare diseases or conditions. Under the Orphan Drug Act, the FDA may grant orphan designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug available in the United States for this type of disease or condition will be recovered from sales of the product. If a sponsor demonstrates that a drug is intended to treat a rare disease or condition, the FDA will grant orphan designation for that product for the orphan disease indication, assuming that the same drug has not already been approved for the indication for which the sponsor is seeking orphan designation. If the same drug has already been approved for the indication for which the sponsor is seeking orphan designation, the sponsor must present a plausible hypothesis of clinical superiority in order to obtain orphan designation. Orphan designation must be requested before submitting an NDA. After the FDA grants orphan designation, the FDA discloses the identity of the therapeutic agent and its potential orphan use.
Orphan designation may provide manufacturers with benefits such as research grants, tax credits, Prescription Drug User Fee Act (“PDUFA”) application fee waivers and eligibility for orphan drug exclusivity. If a product that has orphan designation subsequently receives the first FDA approval of the active moiety for that disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which for seven years prohibits the FDA from approving another product with the same active ingredient for the same indication, except in limited circumstances. Orphan drug exclusivity will not bar approval of another product under certain circumstances, including if a subsequent product with the same active ingredient for the same indication is shown to be clinically superior to the approved product on the basis of greater efficacy or safety, or providing a major contribution to patient care, or if the company with orphan drug exclusivity is not able to meet market demand. Further, the FDA may approve more than one product for the same orphan indication or disease as long as the products contain different active ingredients. Moreover, competitors may receive approval of different products for the indication for which the orphan drug has exclusivity or obtain approval for the same product but for a different indication for which the orphan drug has exclusivity.
In the European Union, orphan drug designation also entitles a party to financial incentives such as reduction of fees or fee waivers and 10 years of market exclusivity is granted following drug or biological product approval. This period may be reduced to 6 years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
Post-Approval Requirements
Once an NDA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved labeling.
Adverse event reporting and submission of periodic reports are required following FDA approval of an NDA. The FDA also may require post-marketing testing, known as Phase IV testing, risk evaluation and mitigation strategies, or REMS, and surveillance to monitor the effects of an approved product, or the FDA may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control, drug manufacture, packaging and labeling procedures must continue to conform to current good manufacturing practices, or cGMPs, after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies. Registration with the FDA subjects' entities to periodic unannounced inspections by the FDA, during which the Agency inspects manufacturing facilities to assess compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality-control to maintain compliance with cGMPs. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
Additional Regulations and Environmental Matters
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including but not limited to, the Centers for Medicare and Medicaid Services, or CMS, other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice, or DOJ, and individual U.S. Attorney offices within the DOJ, and state and local governments. For example, sales, marketing and scientific/educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the false claims laws, and our activities may implicate the privacy provisions of the Health Insurance Portability and Accountability Act (“HIPAA”) and similar state laws, each as amended.
The federal Anti-Kickback Statute prohibits, among other things, any person or entity, from knowingly and willfully offering, paying, soliciting or receiving any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any item or service reimbursable under Medicare, Medicaid or other federal healthcare programs. The term remuneration has been interpreted broadly to include anything of value. The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers, and formulary managers on the other. There are statutory exceptions and regulatory safe harbors protecting some common activities from prosecution. The exceptions and safe harbors are drawn narrowly and practices that involve remuneration that may be alleged to be intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all its facts and circumstances. While we reasonably believe our practices to be in compliance with the Anti-Kickback Statute, our practices may not in all cases meet all the criteria for protection under a statutory exception or regulatory safe harbor.
Additionally, the intent standard under the Anti-Kickback Statute was amended by the Affordable Care Act (“ACA”) to a stricter standard such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it to have committed a violation. In addition, the ACA codified case law that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act (as further discussed below).
The Civil Monetary Penalties statute authorizes the imposition of severe financial penalties against any person or entity who, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent.
The federal False Claims Act prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false claim for payment to, or approval by, the federal government or knowingly making, using, or causing to be made or used, a false record or statement material to a false or fraudulent claim to the federal government. As a result of a modification made by the Fraud Enforcement and Recovery Act of 2009, a claim includes “any request or demand” for money or property presented to the U.S. government. Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies’ marketing of the product for unapproved, and thus non-reimbursable, uses.
In addition, federal fraud statutes prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud or to obtain, by means of false or fraudulent pretenses, representations or promises, any money or property owned by, or under the control or custody of, any healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up by trick, scheme or device, a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of, or payment for, healthcare benefits, items or services.
Also, many states have similar fraud and abuse statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
We may be subject to data privacy and security regulations by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”) and its implementing regulations, imposes requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to business associates, independent contractors or agents of covered entities that receive or obtain protected health information in connection with providing a service on behalf of a covered entity. HITECH also created four new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in specified circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
Additionally, the federal Physician Payments Sunshine Act within the ACA, and its implementing regulations, require that certain manufacturers of drugs, devices, biological and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) report information related to certain payments or other transfers of value made or distributed to physicians, other specified health care professionals and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, the physicians, other specified health care professionals and teaching hospitals and report annually certain ownership and investment interests held by physicians and other specified health care professionals and their immediate family members. Some states have analogous laws requiring manufacturers to report certain transfers of value to covered individuals and entities. To distribute products commercially, we must comply with state laws that require the registration of manufacturers and wholesale distributors of drug and biological products in a state, including, in certain states, manufacturers and distributors who ship products into the state even if such manufacturers or distributors have no place of business within the state. Some states also impose requirements on manufacturers and distributors to establish the pedigree of product in the chain of distribution, including some states that require manufacturers and others to adopt new technology capable of tracking and tracing product as it moves through the distribution chain. Several states have enacted legislation requiring pharmaceutical and biotechnology companies to establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities, and/or register their sales representatives, as well as to prohibit pharmacies and other healthcare entities from providing certain physician prescribing data to pharmaceutical and biotechnology companies for use in sales and marketing, and to prohibit certain other sales and marketing practices. All our activities are potentially subject to federal and state consumer protection and unfair competition laws.
If our operations are found to be in violation of any of the federal and state healthcare laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including without limitation, civil, criminal and/or administrative penalties, damages, fines, disgorgement, exclusion from participation in government programs, such as Medicare and Medicaid, injunctions, private “qui tam” actions brought by individual whistleblowers in the name of the government, or refusal to allow us to enter into government contracts, contractual damages, reputational harm, administrative burdens, diminished profits and future earnings, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any product candidates for which we obtain regulatory approval. In the United States and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend, in part, on the extent to which third-party payors provide coverage and establish adequate reimbursement levels for such products. In the United States, third-party payors include federal and state healthcare programs, privately managed care providers, health insurers and other organizations. The process for determining whether a third-party payor will provide coverage for a product may be separate from the process for setting the price of a product or for establishing the reimbursement rate that such a payor will pay for the product. Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all the FDA-approved products for a particular indication. Third-party payors are increasingly challenging the price, examining the medical necessity and reviewing the cost-effectiveness of medical products, therapies and services, in addition to questioning their safety and efficacy. We may need to conduct expensive pharmaco-economic studies to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain the FDA approvals. Our product candidates may not be considered medically necessary or cost-effective. A payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage for the product. This is also true of Medicare reimbursement, where different vendors process payments, so that coverage by one vendor does not assure that all other vendors will provide coverage. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. In addition, the United States federal government position on matters related to drug pricing is evolving and uncertain, and any changes could have a material impact on drug pricing generally in the United States, including for our product candidates if approved.
Different pricing and reimbursement schemes exist in other countries. In the EU, governments influence the price of pharmaceutical products through their pricing and reimbursement rules and control of national health care systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines but monitor and control company profits. The National Institute for Health and Care Excellence (“NICE”) in the United Kingdom also requires consideration of cost-benefit analysis. The downward pressure on health care costs has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert a commercial pressure on pricing within a country.
The marketability of any product candidates for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, emphasis on managed care in the United States has increased and we expect will continue to increase the pressure on healthcare pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Available Information
Our website is located at www.Panbela.com. The information contained on or connected to our website is not a part of this prospectus. We have included our website address as a factual reference and do not intend it to be an active link to our website.
We make available, free of charge, through our website materials we file or furnish to the SEC pursuant to Section 13(a) or 15(d) of the Exchange Act, including our annual report on Form 10-K, our quarterly reports on Form 10-Q, our current reports on Form 8-K and amendments to those reports. These materials are posted to our website as soon as reasonably practical after we electronically file them with or furnish them to the SEC.
Members of the public may read and copy any materials we file with the SEC at its Public Reference Room at 450 Fifth Street, NW, Washington, DC 20549. Information on the operation of the Public Reference Room is available by calling the SEC at 1-800-SEC-0330. The SEC maintains a website that contains reports, proxy and information statements and other information about us and other issuers that file electronically at http://www.sec.gov.
|
Item 1A. |
Risk Factors |
You should carefully consider the following information about risks, together with the other information contained in this report before making an investment in our common stock. If any of the circumstances or events described below actually arise or occur, our business, results of operations, cash flows and financial condition could be harmed.
Risks Related to Our Business and Financial Position
We are a pre revenue company with a history of negative operating cash flow.
We have experienced negative cash flows for our operating activities since inception, primarily due to the investments required to commercialize our primary drug candidates. Our financing cash flows historically have been positive due to proceeds from the sale of equity securities and promissory notes issuances. Our net cash used in operating activities was $25.2 million and $15.3 million for the years ended December 31, 2023, and 2022, respectively, and we had negative working capital of $9.3 million on December 31, 2023, and negative working capital of $6.0 million as December 31, 2022. Working capital is defined as current assets less current liabilities.
Our operations are subject to all the risks, difficulties, complications and delays frequently encountered in connection with the development of new products, as well as those risks that are specific to the pharmaceutical and biotechnology industries in which we compete. Investors should evaluate us considering the delays, expenses, problems and uncertainties frequently encountered by companies developing markets for new products, services and technologies. We may never overcome these obstacles.
As a result of our limited financial liquidity, we and our auditors have expressed substantial doubt regarding our ability to continue as a “going concern.”
As a result of our current limited financial liquidity, our auditors’ report for our 2023 financial statements, which is included as part of this report, contains a statement concerning our ability to continue as a “going concern.” Our limited liquidity could make it more difficult for us to secure additional financing or enter into strategic relationships on terms acceptable to us, if at all, and may materially and adversely affect the terms of any financing that we may obtain and our public stock price generally.
Our continuation as a “going concern” is dependent upon, among other things, achieving positive cash flow from operations and, if necessary, augmenting such cash flow using external resources to satisfy our cash needs. Our plans to achieve positive cash flow primarily include engaging in offerings of securities. Additional potential sources of funds include negotiating up-front and milestone payments on our current and potential future product candidates or royalties from sales of our products that secure regulatory approval and any milestone payments associated with such approved products. These cash sources could, potentially, be supplemented by financing or other strategic agreements. However, we may be unable to achieve these goals or obtain required funding on commercially reasonable terms, or at all, and therefore may be unable to continue as a going concern.
We may be unable to obtain the additional capital that is required to execute our business plan, which could restrict our ability to grow.
Our current capital and our other existing resources will be sufficient only to provide a limited amount of working capital and will not be sufficient to fund our expected continuing opportunities. While we project that our current capital resources will fund our operations, including increased clinical trial costs, into the second quarter of 2024, we will require additional capital to continue to operate our business and complete our clinical development plans.
Future research and development, including clinical trial cost, capital expenditures and possible acquisitions, and our administrative requirements, such as salaries, insurance expenses and general overhead expenses, as well as legal compliance costs and accounting expenses, will require a substantial amount of additional capital and cash flow. There is no guarantee that we will be able to raise the additional capital required to fund our ongoing business on commercially reasonable terms or at all.
We intend to pursue sources of additional capital through various financing transactions or arrangements, including collaboration arrangements, debt financing, equity financing or other means. We may not be successful in locating suitable financing transactions on commercially reasonable terms, in the time period required or at all, and we may not obtain the capital we require by other means. If we do not succeed in raising additional capital, our resources will not be sufficient to fund our operations going forward.
Any additional capital raised through the sale of equity may dilute the ownership percentage of our stockholders. This could also result in a decrease in the fair market value of our equity securities because our assets would be owned by a larger pool of outstanding equity. The terms of securities we issue in future capital transactions may be more favorable to our new investors, and may include preferences, superior voting rights and the issuance of warrants or other derivative securities which may have a further dilutive effect.
Our ability to obtain needed financing may be impaired by such factors as the capital markets, both generally and in the pharmaceutical and other drug development industries in particular, the limited diversity of our activities and/or the loss of key personnel. If the amount of capital we are able to raise from financing activities is not sufficient to satisfy our capital needs, even to the extent that we reduce our operations, we may be required to cease our operations.
We may incur substantial costs in pursuing future capital financing, including investment banking fees, legal fees, accounting fees, securities law compliance fees, printing and distribution expenses and other costs, which may adversely impact our financial condition.
The markets for our product candidates are highly competitive and are subject to rapid scientific change, which could have a material adverse effect on our business, results of operations and financial condition.
The pharmaceutical and biotechnology industries in which we compete are highly competitive and characterized by rapid and significant technological change. We face intense competition from organizations such as pharmaceutical and biotechnology companies, as well as academic and research institutions and government agencies. Some of these organizations are pursuing products based on technologies similar to our technology. Other of these organizations have developed and are marketing products or are pursuing other technological approaches designed to produce products that are competitive with our product candidates in the therapeutic effect these competitive products have on the diseases targeted by our product candidates. Our competitors may discover, develop or commercialize products or other novel technologies that are more effective, safer or less costly than any that we may develop. Our competitors may also obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for our product candidates.
Many of our competitors are substantially larger than we are and have greater capital resources, research and development staff and facilities than we have. In addition, many of our competitors are more experienced in drug discovery, development and commercialization, obtaining regulatory approvals and drug manufacturing and marketing.
We anticipate that the competition with our product candidates and technology will be based on a number of factors including product efficacy, safety, availability and price. The timing of market introduction of our planned future product candidates and competitive products will also affect competition among products. We expect the relative speed with which we can develop our product candidates, complete the required clinical trials, establish strategic partners and supply appropriate quantities of the product candidate for late-stage trials, if required, to be important competitive factors. Our competitive position will also depend upon our ability to attract and retain qualified personnel, to obtain patent protection in non-U.S. markets, which we currently do not have, or otherwise develop proprietary products or processes and to secure sufficient capital resources for the period between technological conception and commercial sales or out-license to pharmaceutical partners. If we fail to develop and deploy a proposed product candidate in a successful and timely manner, we will in all likelihood not be competitive.
Our lack of diversification increases the risk of an investment in our Company and our financial condition and results of operations may deteriorate if we fail to diversify.
Our Board of Directors has centered our attention on our drug development activities, which are currently focused on a limited number of product candidates. Our ability to diversify our investments will depend on our access to additional capital and financing sources and the availability and identification of suitable opportunities.
Larger companies have the ability to manage their risk by diversification. However, we lack and expect to continue to lack diversification, in terms of both the nature and geographic scope of our business. As a result, we will likely be impacted more acutely by factors affecting pharmaceutical and biotechnology industries in which we compete than we would if our business were more diversified, enhancing our risk profile. If we cannot diversify our operations, our financial condition and results of operations could deteriorate.
Our business may suffer if we do not attract and retain talented personnel.
Our success will depend in large measure on the abilities, expertise, judgment, discretion, integrity and good faith of our management and other personnel in conducting our business. We have a small management team, and the loss of a key individual or inability to attract suitably qualified staff could materially adversely impact on our business.
Our success depends on the ability of our management, employees, consultants and strategic partners, if any, to interpret market data correctly and to interpret and respond to economic market and other conditions in order to locate and adopt appropriate investment opportunities, monitor such investments, and ultimately, if required, to successfully divest such investments. Further, no assurance can be given that our key personnel will continue their association or employment with us or that replacement personnel with comparable skills can be found. We will seek to ensure that management and any key employees are appropriately compensated; however, their services cannot be guaranteed. If we are unable to attract and retain key personnel, our business may be adversely affected.
We may be required to defend lawsuits or pay damages for product liability claims.
Product liability is a major risk in testing and marketing biotechnology and pharmaceutical products. We may face substantial product liability exposure in human clinical trials and in the sale of products after regulatory approval. Product liability claims, regardless of their merits, could exceed policy limits, divert management’s attention and adversely affect our reputation and the demand for our product. In any such event, your investment in our securities could be materially and adversely affected.
Risks Related to the Development and Approval of New Drugs
Clinical trials required for our product candidates are expensive and time-consuming, and their outcome is highly uncertain. If any of our drug trials are delayed or yield unfavorable results, we will have to delay or may be unable to obtain regulatory approval for our product candidate.
We must conduct extensive testing of each product candidate before we can obtain regulatory approval to market and sell it. We need to conduct both preclinical animal testing and human clinical trials. Conducting these trials is a lengthy, time-consuming and expensive process. These tests and trials may not achieve favorable results for many reasons, including, among others, failure of the product candidate to demonstrate safety or efficacy, the development of serious or life-threatening adverse events, or side effects, caused by or connected with exposure to the product candidate, difficulty in enrolling and maintaining subjects in the clinical trial, lack of sufficient supplies of the product candidate or comparator drug, and the failure of clinical investigators, trial monitors, contractors, consultants, or trial subjects to comply with the trial protocol. A clinical trial may fail because it did not include a sufficient number of patients to detect the endpoint being measured or reach statistical significance. A clinical trial may also fail because the dose(s) of the investigational drug included in the trial were either too low or too high to determine the optimal effect of the investigational drug in the disease setting. Many clinical trials are conducted under the oversight of Independent Data Monitoring Committees (“IDMCs”) also known as DSMB’s. These independent oversight bodies are made up of external experts who review the progress of ongoing clinical trials, including available safety and efficacy data, and make recommendations concerning a trial’s continuation, modification, or termination based on interim, unblinded data. Any of our ongoing clinical trials may be discontinued or amended in response to recommendations made by responsible IDMCs based on their review of such interim trial results.
We will need to reevaluate our product candidates if they do not test favorably and either conduct new trials, which are expensive and time consuming, or abandon our drug development program. Even if we obtain positive results from preclinical or clinical trials, we may not achieve the same success in future trials. Many companies in the biopharmaceutical industry have suffered significant setbacks in clinical trials, even after promising results have been obtained in earlier trials. The failure of clinical trials to demonstrate safety and effectiveness for the desired indication could harm the development of our product candidate and our business, financial condition and results of operations may be materially harmed.
We face significant risks in our product candidate development efforts.
Our business depends on the successful development and commercialization of our product candidates. We are currently focused on developing our initial product candidate, SBP-101, for the treatment of PDA and are not permitted to market it in the United States until we receive approval of an NDA from the FDA, or in any foreign jurisdiction until we receive the requisite approvals from such jurisdiction. The process of developing new drugs and/or therapeutic products is inherently complex, unpredictable, time-consuming, expensive and uncertain. We must make long-term investments and commit significant resources before knowing whether our development programs will result in drugs that will receive regulatory approval and achieve market acceptance. A product candidate that appears to be promising at all stages of development may not reach the market for a number of reasons that may not be predictable based on results and data from the clinical program. A product candidate may be found ineffective or may cause harmful side effects during clinical trials, may take longer to progress through clinical trials than had been anticipated, may not be able to achieve the pre-defined clinical endpoints even though clinical benefit may have been achieved, may fail to receive necessary regulatory approvals, may prove impracticable to manufacture in commercial quantities at reasonable cost and with acceptable quality, or may fail to achieve market acceptance.
We cannot predict whether or when we will obtain regulatory approval to commercialize our product candidates and we cannot, therefore, predict the timing of any future revenues from this or other product candidates, if any. The FDA has substantial discretion in the drug approval process, including the ability to delay, limit or deny approval of a product candidate for many reasons. For example, the FDA:
|
● |
could determine that the information provided by us was inadequate, contained clinical deficiencies or otherwise failed to demonstrate the safety and effectiveness of any of our product candidates for any indication; |
|
● |
may not find the data from clinical trials sufficient to support the submission of an NDA or to obtain marketing approval in the United States, including any findings that the clinical and other benefits of our product candidates outweigh their safety risks; |
|
● |
may disagree with our trial design or our interpretation of data from preclinical studies or clinical trials or may change the requirements for approval even after it has reviewed and commented on the design for our trials; |
|
● |
may identify deficiencies in the manufacturing processes or facilities of third-party manufacturers with which we enter into agreements for the manufacturing of our product candidates; |
|
● |
may approve our product candidates for fewer or more limited indications than we request or may grant approval contingent on the performance of costly post-approval clinical trials; |
|
● |
may change its approval policies or adopt new regulations; or |
|
● |
may not approve the labeling claims that we believe are necessary or desirable for the successful commercialization of our product candidates. |
Any failure to obtain regulatory approval of our initial product candidate or future product candidates we develop, if any, would significantly limit our ability to generate revenues, and any failure to obtain such approval for all of the indications and labeling claims we deem desirable could reduce our potential revenues.
Our product candidates are based on a new formulation of an existing technology which has never been approved for the treatment of any cancer and, consequently, is inherently risky. Concerns about the safety and efficacy of our product candidate could limit our future success.
We are subject to the risks of failure inherent in the development of product candidates based on new technologies. These risks include the possibility that any product candidates we create will not be effective, that our current product candidate will be unsafe, ineffective or otherwise fail to receive the necessary regulatory approvals or that our product candidate will be hard to manufacture on a large scale or will be uneconomical to market.
Many pharmaceutical products cause multiple potential complications and side effects, not all of which can be predicted with accuracy and many of which may vary from patient to patient. Long-term follow-up data may reveal additional complications associated with our product candidate. The responses of potential physicians and others to information about complications could materially affect the market acceptance of our product candidate, which in turn would materially harm our business.
Due to our reliance on third parties to conduct our clinical trials, we are unable to directly control the timing, conduct, expense and quality of our clinical trials, which could adversely affect our clinical data and results and related regulatory approvals.
We extensively outsource our clinical trial activities and expect to directly perform only a small portion of the preparatory stages for planned trials. We rely on independent third-party CROs to perform most of our clinical trials, including document preparation, site identification, screening and preparation, pre-study visits, training, program management and bio-analytical analysis. Many important aspects of the services performed for us by the CROs are out of our direct control. If there is any dispute or disruption in our relationship with our CROs, our clinical trials may be delayed. Moreover, in our regulatory submissions, we rely on the quality and validity of the clinical work performed by third-party CROs. If a CRO’s processes, methodologies or results are determined to be invalid or inadequate, our own clinical data and results and related regulatory approvals could be adversely affected or invalidated.
We rely on third-party suppliers and other third parties for production of our product candidate and our dependence on these third parties may impair the advancement of our research and development programs and the development of our product candidates.
We rely on, and expect to continue to rely on, third parties for the supply of raw materials and manufacture of drug supplies necessary to conduct our preclinical studies and clinical trials. During 2021 the Company, in collaboration with our manufacturing partner confirmed a new shorter and less expensive synthesis of the active drug substance. However, delays in production by third parties could delay our clinical trials or have an adverse impact on any commercial activities. In addition, the fact that we are dependent on third parties for the manufacture of and formulation of our product candidates means that we are subject to the risk that the products may have manufacturing defects that we have limited ability to prevent or control. Although we oversee these activities to ensure compliance with our quality standards, budgets and timelines, we have had and will continue to have less control over the manufacturing of our product candidates than potentially would be the case if we were to manufacture our product candidates. Further, the third parties we deal with could have staffing difficulties, might undergo changes in priorities or may become financially distressed, which would adversely affect the manufacturing and production of our product candidates.
The results of pre-clinical studies and completed clinical trials are not necessarily predictive of future results, and our current product candidates may not have favorable results in later studies or trials.
Pre-clinical studies and Phase I clinical trials are not primarily designed to test the efficacy of a product candidate in the general population, but rather to test initial safety, to study pharmacokinetics and pharmacodynamics, to study limited efficacy in a small number of study patients in a selected disease population, and to identify and attempt to understand the product candidate’s side effects at various doses and dosing schedules. Success in pre-clinical studies or completed clinical trials does not ensure that later studies or trials, including continuing pre-clinical studies and large-scale clinical trials, will be successful nor does it necessarily predict future results. Favorable results in early studies or trials may not be repeated in later studies or trials, and product candidates in later stage trials may fail to show acceptable safety and efficacy despite having progressed through earlier trials.
Risks Related to the Regulation of our Business
Federal and state pharmaceutical marketing compliance and reporting requirements may expose us to regulatory and legal action by state governments or other government authorities.
The Food and Drug Administration Modernization Act (the “FDMA”) established a public registry of open clinical trials involving drugs intended to treat serious or life-threatening diseases or conditions in order to promote public awareness of and access to these clinical trials. Under the FDMA, pharmaceutical manufacturers and other trial sponsors are required to post the general purpose of these trials, as well as the eligibility criteria, location and contact information of the trials. Failure to comply with any clinical trial posting requirements could expose us to negative publicity, fines and other penalties, all of which could materially harm our business.
In recent years, several states, including California, Vermont, Maine, Minnesota, New Mexico and West Virginia, have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs and file periodic reports on sales, marketing, pricing and other activities. Similar legislation is being considered in other states. Many of these requirements are new and uncertain, and available guidance is limited. Unless we are in full compliance with these laws, we could face enforcement actions and fines and other penalties and could receive adverse publicity, all of which could harm our business.
If the product candidate we develop becomes subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, our ability to successfully commercialize our product candidate may be impaired.
Our future revenues, profitability and access to capital will be affected by the continuing efforts of governmental and private third-party payors to contain or reduce the costs of health care through various means. We expect several federal, state and foreign proposals to control the cost of drugs through government regulation. We are unsure of the impact recent health care reform legislation may have on our business or what actions federal, state, foreign and private payors may take in response to the recent reforms. Therefore, it is difficult to predict the effect of any implemented reform on our business. Our ability to commercialize our product candidate successfully will depend, in part, on the extent to which reimbursement for the cost of such product candidate and related treatments will be available from government health administration authorities, such as Medicare and Medicaid in the United States, private health insurers and other organizations. Significant uncertainty exists as to the reimbursement status of newly approved health care products, particularly for indications for which there is no current effective treatment or for which medical care typically is not sought. Adequate third-party coverage may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product research and development. If adequate coverage and reimbursement levels are not provided by government and third-party payors for the use of our product candidates, our product candidates may fail to achieve market acceptance and our results of operations will be harmed.
Healthcare legislative reform measures may have a material adverse effect on our business and results of operations.
Legislative and regulatory actions affecting government prescription drug procurement and reimbursement programs occur relatively frequently. In the United States, the ACA was enacted in 2010 to expand healthcare coverage. Since then, numerous efforts have been made to repeal, amend or administratively limit the ACA in whole or in part. For example, the Tax Cuts and Jobs Act, signed into law by President Trump in 2017, repealed the individual health insurance mandate, which is considered a key component of the ACA. In December 2018, a Texas federal district court struck down the ACA on the grounds that the individual health insurance mandate is unconstitutional, although this ruling has been stayed pending appeal. The ongoing challenges to the ACA and new legislative proposals have resulted in uncertainty regarding the ACA's future viability and destabilization of the health insurance market. The resulting impact on our business is uncertain and could be material.
Efforts to control prescription drug prices could also have a material adverse effect on our business. For example, in 2018, President Trump and the Secretary of the U.S. Department of Health and Human Services (“HHS”) released the "American Patients First Blueprint" and have begun implementing certain portions. The initiative includes proposals to increase generic drug and biosimilar competition, enable the Medicare program to negotiate drug prices more directly and improve transparency regarding drug prices and ways to lower consumers' out-of-pocket costs. The Trump administration also proposed to establish an “international pricing index” that would be used as a benchmark to determine the costs and potentially limit the reimbursement of drugs under Medicare Part B. Among other pharmaceutical manufacturer industry-related proposals, Congress has proposed bills to change the Medicare Part D benefit to impose an inflation-based rebate in Medicare Part D and to alter the benefit structure to increase manufacturer contributions in the catastrophic phase. The volume of drug pricing-related bills has dramatically increased under the current Congress, and the resulting impact on our business is uncertain and could be material.
In addition, many states have proposed or enacted legislation that seeks to regulate pharmaceutical drug pricing indirectly or directly, such as by requiring biopharmaceutical manufacturers to publicly report proprietary pricing information or to place a maximum price ceiling on pharmaceutical products purchased by state agencies. For example, in 2017, California’s governor signed a prescription drug price transparency state bill into law, requiring prescription drug manufacturers to provide advance notice and explanation for price increases of certain drugs that exceed a specified threshold. Both Congress and state legislatures are considering various bills that would reform drug purchasing and price negotiations, allow greater use of utilization management tools to limit Medicare Part D coverage, facilitate the import of lower-priced drugs from outside the United States and encourage the use of generic drugs. Such initiatives and legislation may cause added pricing pressures on our products.
Changes to the Medicaid program at the federal or state level could also have a material adverse effect on our business. Proposals that could impact coverage and reimbursement of our products, including giving states more flexibility to manage drugs covered under the Medicaid program and permitting the re-importation of prescription medications from Canada or other countries, could have a material adverse effect by limiting our products’ use and coverage. Furthermore, state Medicaid programs could request additional supplemental rebates on our products as a result of an increase in the federal base Medicaid rebate. To the extent that private insurers or managed care programs follow Medicaid coverage and payment developments, they could use the enactment of these increased rebates to exert pricing pressure on our products, and the adverse effects may be magnified by their adoption of lower payment schedules.
Other proposed regulatory actions affecting manufacturers could have a material adverse effect on our business. It is difficult to predict the impact, if any, of any such proposed legislative and regulatory actions or resulting state actions on the use and reimbursement of our products in the United States, but our results of operations may be adversely affected.
Risks Related to our Intellectual Property
If we are unable to obtain, maintain and enforce our proprietary rights, we may not be able to compete effectively or operate profitably.
For ivospemin, we are party to a license agreement with the University of Florida Research Foundation (“UFRF”) and for Flynpovi, we are party to a license agreement with the Arizona Board of Regents of the University of Arizona. The patents underlying the licensed intellectual property and those of other biopharmaceutical companies are generally uncertain and involve complex legal, scientific and factual questions.
Our ability to develop and commercialize drugs depends in significant part on our ability to: (i) obtain and/or develop broad, protectable intellectual property; (ii) obtain additional licenses, if required, to the proprietary rights of others on commercially reasonable terms; (iii) operate without infringing upon the proprietary rights of others; (iv) prevent others from infringing on our proprietary rights; and (v) protect our corporate know-how and trade secrets.
Patents that we may acquire, and those that might be issued in the future, may be challenged, invalidated or circumvented, and the rights granted thereunder may not provide us with proprietary protection or competitive advantages against competitors with similar technology. Furthermore, our competitors may independently develop similar technologies or duplicate any technology we develop. Because of the extensive time required for development, testing and regulatory review of potential product candidates, it is possible that, before any of our product candidates can be commercialized, any related patent may expire or remain in force for only a brief period following commercialization, thus reducing any advantage of the patent.
Because patent applications in the U.S. and many foreign jurisdictions are typically not published until at least 12 months after filing, or in some cases not at all, and because publications of discoveries in the scientific literature often lag behind actual discoveries, neither we nor our licensors can be certain that either we or our licensors were the first to make the inventions claimed in issued patents or pending patent applications, or that we were the first to file for protection of the inventions set forth in these patent applications.
Additionally, UFRF previously elected to seek protection for certain elements of the licensed technology only in the United States, and the time to file for international patent protection has expired. This limits the strength of the Company’s intellectual property position in certain markets and could affect the overall value of the Company to a potential corporate partner.
Obtaining and maintaining our patent protection depends upon compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The United States Patent and Trademark Office and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case.
We may be exposed to infringement or misappropriation claims by third parties, which, if determined adversely to us, could cause us to pay significant damage awards.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical and biotechnology industries. We may become a party to various types of patent litigation or other proceedings regarding intellectual property rights from time to time even under circumstances where we are not using and do not intend to use any of the intellectual property involved in the proceedings.
The cost of any patent litigation or other proceeding, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the cost of such litigation or proceedings more effectively than we will be able to because our competitors may have substantially greater financial resources. If any patent litigation or other proceeding is resolved against us, we or our collaborators may be enjoined from developing, manufacturing, selling or importing our drugs without a license from the other party and we may be held liable for significant damages. We may not be able to obtain any required license(s) on commercially acceptable terms or at all.
Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Patent litigation and other proceedings may also absorb significant management time.
Confidentiality agreements with employees and others may not adequately prevent disclosure of our know-how, trade secrets and other proprietary information and may not adequately protect our intellectual property, which could impede our ability to compete.
Because we operate in the highly technical field of medical technology development, we rely in part on trade secret protection in order to protect our proprietary trade secrets and unpatented know-how. However, trade secrets are difficult to protect, and we cannot be certain that others will not develop the same or similar technologies on their own. We have taken steps, including entering into confidentiality agreements with all of our employees, consultants and corporate partners, to protect our trade secrets and unpatented know-how. These agreements generally require that the other party keep confidential and not disclose to third parties any confidential information developed by the party or made known to the party by us during the course of the party’s relationship with us. We also typically obtain agreements from these parties which provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us. Enforcing a claim that a party illegally obtained and is using our trade secrets or know-how is difficult, expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets or know-how. The failure to obtain or maintain trade secret protection could adversely affect our competitive position.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology industry, we employ individuals who were previously employed at other biotechnology companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Risks Associated with Our Common Stock
Failure to maintain the listing of our common stock on a national securities exchange could seriously harm the liquidity of our stock and our ability to raise capital.
On March 5, 2024, Nasdaq notified us that the Nasdaq Hearings Panel had determined to delist our common stock and trading of our common stock was suspended on March 7, 2024. Nasdaq will complete the delisting by filing a Form 24 Notification of Delisting with the SEC after applicable appeal periods have lapsed. The panel reached its decision because our Company was in violation of the minimum $2.5 million stockholders equity requirement in Listing Rule 5550(b)(1) and unable to comply with any of the alternative requirements in Listing Rule 5550(b) (collectively, the “Minimum Stockholders’ Equity Requirement”). The period during which we could appeal the decision to the Nasdaq Listing and Hearing Review Council has lapsed, but the Council may, on its own motion, determine to review the panel’s decision within 45 calendar days after the Company was notified of the decision. Although we are seeking all possible opportunities to regain compliance with the Minimum Stockholders’ Equity Requirement or to obtain an alternative listing on a national securities exchange, we believe that even if we were able to regain compliance with all applicable Nasdaq continued listing requirements, it is likely that Nasdaq will proceed with delisting our common stock.
As previously disclosed, in the past we have received notices from Nasdaq’s Listing Qualifications Department indicating that, for 30 consecutive business days, our common stock had not maintained a minimum closing bid price of $1.00 per share as required by Nasdaq Listing Rule 5550(a)(2) (“Minimum Bid Price Requirement”); (ii) the Minimum Stockholders’ Equity Requirement; and the minimum requirement of 500,000 publicly held shares as required by Nasdaq Listing Rule 5550(a)(4) (the “Minimum Float Requirement”). In February 2024, we received a letter from Nasdaq confirming that we had cured the most recently identified deficiencies under the Minimum Bid Price Requirement and Minimum Float Requirement.
While we have regained compliance in the past, if, for any reason, Nasdaq were to delist our securities from trading on The Nasdaq Capital Market and we were unable to obtain listing on another reputable national securities exchange, a reduction in some or all of the following may occur, each of which could materially adversely affect our stockholders:
|
● |
the liquidity and marketability of our common stock; |
|
● |
the market price of our common stock; |
|
● |
our ability to obtain financing for the continuation of our operations; |
|
● |
the number of institutional and general investors that will consider investing in our common stock; |
|
● |
the number of market makers in our common stock; |
|
● |
the availability of information concerning the trading prices and volume of our common stock; and |
|
● |
the number of broker-dealers willing to execute trades in shares of our common stock. |
In addition, if we cease to be listed on The Nasdaq Capital Market, we may have to pursue trading on a less recognized or accepted market, such as the over the counter markets, our stock may be traded as a “penny stock”, which would make transactions in our stock more difficult and cumbersome, and we may be unable to access capital on favorable terms or at all, as companies trading on alternative markets may be viewed as less attractive investments with higher associated risks, such that existing or prospective institutional investors may be less interested in, or prohibited from, investing in our common stock. This may also cause the market price of our common stock to further decline.
Raising additional capital may cause dilution to our stockholders or restrict our operations.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, stockholders’ ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect their rights as stockholders. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring additional debt, making capital expenditures or declaring dividends. Any of these events could adversely affect our ability to achieve our product development and commercialization goals and harm our business. We do not anticipate any adverse effects stemming from the lack of available credit facilities at this time.
Issuance of common stock in offerings or pursuant to the exercise of rights to purchase shares may cause the price of our common stock to decline and cause investors to lose a significant portion of their investment.
If our stockholders sell substantial amounts of our common stock in the public market or upon the expiration of any statutory holding period under Rule 144, or upon expiration of lock-up periods applicable to outstanding shares or issued upon the exercise of outstanding options or warrants, it could create a circumstance commonly referred to as an “overhang” and in anticipation of which the market price of our common stock could fall. The existence of an overhang, whether sales have occurred or are occurring, also could make our ability to raise additional financing through the sale of equity or equity-related securities in the future at a time and price that we deem reasonable or appropriate more difficult. As of March 22, 2024, we had outstanding options to purchase 607 shares of our common stock at a weighted-average exercise price of $14,410.38 per share with a remaining contractual life of 8.50 years and outstanding warrants to purchase 9,095,943 of common stock at a weighted-average exercise price of $3.13 per share and an average remaining exercise period of 5.23 years.
Securities analysts may not initiate coverage or continue to cover our common stock, and this may have a negative impact on the market price of our common stock.
Common stock prices are often significantly influenced by the research and reports that securities analysts publish about companies and their business. We do not have any control over these analysts. There is no guarantee that securities analysts will cover, or continue to cover, our common stock. If securities analysts do not cover our common stock, the lack of research coverage may adversely affect the market price of our common stock. If our common stock is covered by securities analysts and our stock is downgraded, our stock price will likely decline. If one or more of these analysts ceases to cover us or fails to publish regular reports on us, we can lose visibility in the financial markets, which can cause our stock price or trading volume to decline.
Our charter documents and Delaware law may inhibit a takeover that stockholders consider favorable.
The provisions of our certificate of incorporation and bylaws and applicable provisions of Delaware law may make it more difficult for or prevent a third party from acquiring control of us without the approval of our board of directors. These provisions:
|
● |
set limitations on the removal of directors; |
|
● |
limit who may call a special meeting of stockholders; |
|
● |
establish advance notice requirements for nominations for election to our board of directors or for proposing matters that can be acted upon at stockholder meetings; |
|
● |
do not permit cumulative voting in the election of our directors, which would otherwise permit less than a majority of stockholders to elect directors; |
|
● |
establish a classified board of directors limiting the number of directors that are elected each year; and |
|
● |
provide our board of directors with the ability to designate the terms of and issue preferred stock without stockholder approval. |
In addition, Section 203 of the Delaware General Corporation Law generally limits our ability to engage in any business combination with certain persons who own 15% or more of our outstanding voting stock or any of our associates or affiliates who at any time in the past three years have owned 15% or more of our outstanding voting stock unless our board of directors has pre-approved the acquisitions that lead to such ownership. These provisions may have the effect of entrenching our management team and may deprive stockholders of the opportunity to sell their shares to potential acquirers at a premium over prevailing prices. This potential inability to obtain a control premium could reduce the price of our common stock.
We cannot assure that a reverse stock split will increase our stock price for the required time period or maintain our listing on a national securities exchange.
Our recent reverse stock splits were each accompanied by an initial increase in the market price of our common stock; however, the ongoing effect of reverse stock splits on the market price of our common stock cannot be predicted with any certainty, and the history of reverse stock splits for other companies is varied. Some investors may view one or more reverse stock splits negatively.
While the rules of national securities exchanges do not impose specific limits on the number of times a listed company may effect a reverse stock split, certain exchanges, such as Nasdaq, have stated that a series of reverse stock splits may undermine investor confidence, especially where the reverse stock splits follow dilutive transactions. Accordingly, a national securities exchange may determine that it is not in the public interest to maintain or accept our listing, even if we comply with all applicable initial or continued listing standards.
Furthermore, the reverse stock split may not result in a per share price that attracts investors who do not trade in lower priced stocks. Although we believe the reverse stock split may enhance the marketability of our common stock to certain potential investors, we cannot assure you that our common stock will be more attractive to investors following the reverse stock split. Even though we have implemented a reverse stock split, the market price of our common stock may decrease due to factors unrelated to the reverse stock split, including our future performance or general market trends. If the trading price of the common stock declines, the percentage declines as an absolute number and as a percentage of our overall market capitalization may be greater than would occur in the absence of a reverse stock split.
Recent and future reverse stock splits may decrease the liquidity of our common stock and result in higher transaction costs.
The liquidity of our common stock may be negatively impacted by a reverse stock split, given the reduced number of shares that are outstanding after such a reverse stock split, particularly if the stock price does not increase as a result of the reverse stock split. Additionally, if the reverse stock split is implemented, it will increase the number of our stockholders who own “odd lots” of fewer than 100 shares of common stock. Brokerage commissions and other costs of transactions in odd lots are generally higher than the costs of transactions of more than 100 shares of common stock. Accordingly, reverse stock splits may not achieve the desired results of increasing marketability of our common stock as described above.
Recent reverse stock splits have not been accompanied by decreases in our authorized shares.
Although the reverse stock splits have not had have any directly dilutive effects on our stockholders, the reduction in outstanding shares that resulted from the reverse stock split reduced the proportion of shares owned by our stockholders relative to the number of shares authorized for issuance, giving the Board of Directors an effective increase in the relative number of authorized shares available for issuance, in its discretion. The Board of Directors from time to time may deem it to be in the best interests of the Company and its stockholders to enter into transactions and other ventures that may include the issuance of shares of our common stock. If the Board of Directors authorizes the issuance of additional shares of common stock subsequent to a reverse stock split, the dilution to the ownership interest of our existing stockholders may be greater than would occur had such reverse stock split not been effected.
If we issue preferred stock, the rights of the holders of our common stock and the value of such common stock could be adversely affected.
Our Board of Directors is authorized to issue classes or series of preferred stock, without any action on the part of the stockholders. The Board of Directors also has the power, without stockholder approval, to set the terms of any such classes or series of preferred stock, including voting rights, dividend rights and preferences over the common stock with respect to dividends or upon the liquidation, dissolution or winding-up of our business and other terms. If we issue preferred stock in the future that has a preference over the common stock with respect to the payment of dividends or upon liquidation, dissolution or winding-up, or if we issue preferred stock with voting rights that dilute the voting power of the common stock, the rights of holders of the common stock or the value of the common stock would be adversely affected.
If we fail to maintain effective internal control over financial reporting, the price of our common stock may be adversely affected.
We are required to establish and maintain appropriate internal controls over financial reporting. Failure to establish those controls, or any failure of those controls once established, could adversely impact our public disclosures regarding our business, financial condition or results of operations. Any failure of these controls could also prevent us from maintaining accurate accounting records and discovering accounting errors and financial fraud.
Management’s assessment of internal controls over financial reporting may identify weaknesses that need to be addressed or other potential matters that may raise concerns for investors. Any actual or perceived weaknesses that need to be addressed in our internal control over financial reporting or disclosure of management’s assessment of our internal controls over financial reporting may have an adverse impact on the price of our common stock.
We will need to raise additional capital in the future to finance our operations, which may not be available on acceptable terms, or at all. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate our product development efforts or other operations.
We have had recurring losses from operations, negative operating cash flow and have an accumulated deficit. We must raise additional funds in order to continue financing our operations. If additional capital is not available to us when needed or on acceptable terms, we may not be able to continue to operate our business pursuant to our business plan or we may have to discontinue our operations entirely. Any additional capital raised through the sale of equity or equity-backed securities may dilute our stockholders’ ownership percentages and could also result in a decrease in the market value of our equity securities. The terms of any securities issued by us in future capital transactions may be more favorable to new investors, and may include preferences, superior voting rights and the issuance of warrants or other derivative securities, which may have a further dilutive effect on the holders of any of our securities then outstanding.
If we are unable to secure additional funds when needed or on acceptable terms, we may be required to defer, reduce or eliminate significant planned expenditures, restructure, curtail or eliminate some or all of our operations, dispose of technology or assets, pursue an acquisition of our company by a third party at a price that may result in a loss on investment for our stockholders, file for bankruptcy or cease operations altogether. Any of these events could have a material adverse effect on our business, financial condition and results of operations. Moreover, if we are unable to obtain additional funds on a timely basis, there will be substantial doubt about our ability to continue as a going concern and increased risk of insolvency and up to a total loss of investment by our stockholders.
|
Item 1B. |
Unresolved Staff Comments |
None.
|
Item 1C. |
Cybersecurity |
We operate in the biotechnology sector, which is subject to various cybersecurity risks that could adversely affect our business, financial condition, and results of operations, including intellectual property theft; fraud; extortion; harm to employees or customers; violation of privacy laws and other litigation and legal risk; and reputational risk. We have implemented a risk-based approach to identify and assess the cybersecurity threats that could affect our business and information systems. We use various tools and methodologies to manage cybersecurity risk that are tested on a regular cadence. We also monitor and evaluate our cybersecurity posture and performance on an ongoing basis through regular vulnerability scans, penetration tests and threat intelligence feeds. We require third-party service providers with access to personal, confidential or proprietary information to implement and maintain comprehensive cybersecurity practices consistent with applicable legal standards and industry best practices. Our business depends on the availability, reliability, and security of our information systems, networks, data, and intellectual property. Any disruption, compromise, or breach of our systems or data due to a cybersecurity threat or incident could adversely affect our operations, customer service, product development, and competitive position. They may also result in a breach of our contractual obligations or legal duties to protect the privacy and confidentiality of our stakeholders. Such a breach could expose us to business interruption, lost revenue, ransom payments, remediation costs, liabilities to affected parties, cybersecurity protection costs, lost assets, litigation, regulatory scrutiny and actions, reputational harm, customer dissatisfaction, harm to our vendor relationships, or loss of market share. The Company is currently in the process of implementing a more formalized cybersecurity program.
|
Item 2. |
Properties |
Our primary business functions are conducted by our employees and independent contractors on a distributed basis. Accordingly, we do not lease or own any real property and all employees currently work from their homes. We maintain our principal mailing address at Suite 305 at 712 Vista Boulevard in Waconia, Minnesota.
|
Item 3. |
Legal Proceedings |
We are not currently party to any material legal proceedings. From time to time, we may be named as a defendant in legal actions arising from our normal business activities. We believe that we have obtained adequate insurance coverage or rights to indemnification in connection with potential legal proceedings that may arise.
|
Item 4. |
Mine Safety Disclosures |
None.
PART II
|
Item 5. |
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information
Our common stock is listed on the Nasdaq Capital Market under the symbol “PBLA.” On March 5, 2024, Nasdaq notified us that the Nasdaq Hearings Panel has determined to delist our common stock and trading of our common stock on Nasdaq was suspended on March 7, 2024. Nasdaq will complete the delisting by filing a Form 25 Notification of Delisting with the SEC after applicable appeal periods have lapsed. In the interim, notwithstanding the suspension of trading on Nasdaq, we expect that our common stock will remain eligible for quotation on the OTC Pink Market under our existing symbol, “PBLA.” We are evaluating all available opportunities to regain compliance with Nasdaq’s applicable continued listing standards or to obtain a listing on another national securities exchange. In the interim, we may seek eligibility for quotation on the OTCQB Market.
As of March 22, 2024, there were 55 holders of record of our common stock.
Dividends
We have never paid cash dividends on any of our securities. We currently intend to retain any earnings for use in operations and do not anticipate paying cash dividends in the foreseeable future.
Recent Sales of Unregistered Equity Securities
None.
Purchases of Equity Securities by the Company
None.
|
Item 6. |
[Reserved] |
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following discussion of our financial condition and results of operations should be read in conjunction with our financial statements and the notes to those financial statements included elsewhere in this annual report. This discussion contains forward-looking statements, which are based on our assumptions about the future of our business. Our actual results will likely differ materially from those contained in the forward-looking statements. Please read “Cautionary Note Regarding Forward-Looking Statements” included at the beginning of this annual report for additional information.
Overview
Panbela Therapeutics, Inc. (“Panbela” and together with its direct and indirect subsidiaries, “we,” “us,” “our,” and the “Company”) is a clinical stage biopharmaceutical company developing disruptive therapeutics for the treatment of patients with urgent unmet medical needs.
Our lead candidates are ivospemin (SBP-101) for which we have exclusively licensed the worldwide rights from the University of Florida Research Foundation, Inc. and Flynpovi (eflornithine (CPP-1X) and Sulindac). Flynpovi is delivered in an oral form. The Company has an exclusive worldwide license to commercialize Flynpovi from the Arizona Board of Regents of the University of Arizona.
As Panbela is focused on utilizing a polyamine platform to develop disruptive therapeutics for the treatment of patients with urgent unmet medical needs, we are engaged in two sponsored research agreements to evaluate the polyamines individually and combined for various diseases. At present, the collaboration with Johns Hopkins University School of Medicine has been focused on mechanism of action and solid tumors while the MD Anderson Cancer Center has been focused on the hematologic malignancies. An abstract about ivospemin and CPP-1X (also known as DFMO or Eflornithine) research in multiple myeloma (cell lines), has been accepted for an online publication on the American Society of Hematology (ASH) meeting site in the November 2023 supplemental issue of the journal Blood and an abstract about ivospemin in combination with doxorubicin in models of ovarian cancer has been accepted for presentation on April 10, 2024 at the annual AACR conference.
Ivospemin (SBP-101)
In 2015, the Food and Drug Administration (“FDA”) accepted our Investigational New Drug (“IND”) application for our ivospemin product candidate. In May of 2022 we were notified that the United States Adopted Names (“USAN”) had adopted ivospemin as a USAN for SBP-101. The USAN information on ivospemin was posted on the USAN Web site (www.ama-assn.org/go/usan).
We have completed an initial clinical trial of ivospemin in patients with previously treated locally advanced or metastatic pancreatic cancer. This was a Phase I, first-in-human, dose-escalation, safety study. From January 2016 through September 2017, we enrolled twenty-nine patients into six cohorts, or groups, in the dose-escalation phase of the Phase I trial. No drug-related bone marrow toxicity or peripheral neuropathy was observed at any dose level. In addition to being evaluated for safety, 23 of the 29 patients were evaluable for preliminary signals of efficacy prior to or at the eight-week conclusion of their first cycle of treatment using the Response Evaluation Criteria in Solid Tumors (“RECIST”), the currently accepted standard for evaluating change in the size of tumors.
In 2018, we began enrolling patients in our second clinical trial, a Phase Ia/Ib study of the safety, efficacy and pharmacokinetics of ivospemin administered in combination with two standard-of-care chemotherapy agents, gemcitabine and nab-paclitaxel. A total of 25 subjects were enrolled in four cohorts to evaluate the dosage level and schedule. An additional 25 subjects were enrolled in the expansion phase of the trial. Interim results were presented in January of 2022. Best response in evaluable subjects (cohorts 4 and Ib N=29) was a CR in 1 (3%), PR in 13 (45%), SD in 10 (34%) and PD in 5 (17%). One subject did not have post baseline scans with RECIST tumor assessments. Median PFS, now final at 6.5 months, may have been negatively impacted by drug dosing interruptions to evaluate potential toxicity. Median overall survival in cohort 4 + Phase Ib was 12.0 months when data was presented in January 2022 and is now final at 14.6 months. Two patients from cohort 2 have demonstrated long term survival. One at 30.3 months (final data) and one at 33.0 months and still alive at database lock on March 18, 2022. Seven subjects are still alive at database lock, one from cohort 2 and six from cohort 4 plus Phase Ib.
In January of 2022, the Company announced the initiation of a new clinical trial. Referred to as ASPIRE, the trial is a randomized double-blind placebo-controlled trial in combination with gemcitabine and nab-paclitaxel, a standard pancreatic cancer treatment regimen in patients previously untreated for metastatic pancreatic cancer. The trial will be conducted globally at approximately 94 sites in the United States, Europe and Asia – Pacific. The Company announced the first patient enrolled in the trial in Australia in August of 2022. In September 2022, the company announced that they had obtained regulatory approval to open sites in Spain, France and Italy. On December 31, 2023, there were 85 sites open in 10 countries.
While opening of clinical sites in the United States and the rest of the world has been slower than originally anticipated, due in part to resource fatigue in the medical community, the Company expects all sites to be open by first quarter-2024.
The trial was originally designed as a Phase II/III with a smaller initial sample size. In response to European and FDA regulatory feedback, the study was amended to include the total trial sample size (600) and the design modified to utilize overall survival (the primary endpoint) to be examined at interim analysis as well. All Countries are open, and the full complement of sites is expected to be open by the end of the first quarter of 2024. The independent Data Safety Monitoring Board (DSMB) met for a prespecified safety analysis and recommended the trial continue without modification. The study is anticipated to take 36 months for complete enrollment of 600 subjects with the interim analysis available in mid-2024. On January 25, 2024, the Company announced that the trial had exceeded 50% enrollment. The Company projects that full enrollment will be completed by the first quarter of 2025 and that interim data analysis based on overall survival should be available by the middle of 2024.
In early April 2023, the Company announced a poster presentation highlighting the results for ivospemin as a polyamine metabolism modulator in ovarian cancer at the American Association for Cancer Research Annual Conference. The poster concludes that the ivospemin chemotherapy treatment of C57Bl/6 mice injected with VDID8+ ovarian cancer cells significantly prolonged survival and decreased overall tumor burden. The results suggest that ivospemin in combination with standard of care chemotherapy may have a role in the clinical management of ovarian cancer, and the Company intends to continue pre-clinical and clinical studies in ovarian cancer.
Additional clinical trials may be required for FDA or other country approvals. The cost and timing of additional clinical trials are highly dependent on the nature and size of the trials.
Flynpovi (eflornithine (CPP-1X) and sulindac)
In 2009, the FDA accepted our IND application for the combination product, Flynpovi, product candidate.
In a Phase III study, the efficacy and safety of the combination of eflornithine and sulindac known as Flynpovi, as compared with either drug eflornithine or sulindac alone, in adults with familial adenomatous polyposis (“FAP”) was conducted. A total of 171 patients underwent randomization. Disease progression occurred in 18 of 56 patients (32%) in the Flynpovi group, 22 of 58 (38%) in the sulindac group, and 23 of 57 (40%) in the eflornithine group, with a hazard ratio of 0.71 (95% confidence interval [CI], 0.39 to 1.32) for Flynpovi as compared with sulindac (p = 0.29) and 0.66 (95% CI, 0.36 to 1.23) for Flynpovi as compared with eflornithine. In a post-hoc analysis, none of the patients in the Flynpovi arm progressed to a need for lower gastrointestinal (“LGI”) surgery for up to 48 months compared with 7 (13.2%) and 8 (15.7%) patients in the sulindac and eflornithine (CPP-1X) arms. These data corresponded to risk reductions for the need for LGI surgery approaching 100% between Flynpovi and either monotherapy with HR = 0.00 (95% CI, 0.00–0.48; p = 0.005) for Flynpovi versus sulindac and HR = 0.00 (95% CI, 0.00–0.44; p = 0.003) for Flynpovi versus eflornithine. Given the statistical significance of the LGI group, a new drug application (“NDA”) was filed with the FDA. As the study failed to meet the primary endpoint, and the NDA was based on the results of an exploratory analysis, a complete response letter was issued. To address this deficiency concern, the Company must submit the results of one or more adequate and well-controlled clinical trials which demonstrate an effect on a clinical endpoint.
In April of 2023 the Company regained the North American rights to develop and commercialize Flynpovi in patients with FAP, as a result of the termination of the licensing agreement between CPP and One-Two Therapeutics Assets Limited.
We also have an ongoing double-blind placebo-controlled trial of Flynpovi to prevent recurrence of high-risk adenomas and second primary colorectal cancers in patients with stage 0-III colon or rectal cancer, Phase III – Preventing Adenomas of the Colon with Eflornithine and Sulindac (“PACES”). The purpose of this study is to assess whether the combination of eflornithine and sulindac (compared to corresponding placebos) has efficacy against colorectal lesions with respect to high-grade dysplasia, adenomas with villous features, adenomas one cm or greater, multiple adenomas, any adenomas >/= 0.3 cm, total advanced colorectal events, or total colorectal events. The PACES trial is funded by the National Cancer Institute (“NCI”) in collaboration with Southwest Oncology Group (“SWOG”). The Company announced on June 28, 2023, that the PACES trial passed a pre-planned futility analysis.
Eflornithine (CPP-1X)/eflornithine sachets (CPP-1X-S)
In 2009 and 2018, the FDA accepted our IND applications for eflornithine.
There is a trial evaluating eflornithine sachets in STK11 mutation patients with non-small cell lung cancer scheduled to begin this year. For eflornithine tablets, a Phase II trial in early onset Type I diabetes was opened on January 11, 2023, in collaboration with Indiana University and the Juvenile Diabetes Research Foundation (“JDRF”). Two poster presentations were given discussing the Phase I T1D results, one at the Endocrine Society meeting and the other at the Immunology of Diabetes Society Meeting in June 2023and published in Cell Reports Medicine in November 2023. Additionally, eflornithine is being evaluated with high dose testosterone and enzalutamide in metastatic castration-resistant prostate cancer in a Phase II trial.
On July 17, 2023, the Company divested certain rights, titles and interests in its eflornithine pediatric neuroblastoma program. Included in these assets is an ongoing trial evaluating eflornithine sachets in relapsed refractory neuroblastoma supported by the Children’s Oncology Group (“COG”) /NCI Under the terms of the agreement with US World Meds®, the Company is entitled to receive up to approximately $9.5 million in non-dilutive funding in exchange for the sale of these assets. An initial payment of $400,000 was received by the Company at the time of closing, remaining payments will be receivable if the acquiring company successfully completes certain milestones related to clinical development, regulatory approval and commercial sales.
Financial Overview
On January 18, 2024 we effected a reverse stock split at a ratio of one-for-twenty (1:20) shares of the Company’s common stock. On June 1, 2023, we effected a reverse stock split at a ratio of one-for-forty (1:30) shares of the Company’s common stock and on January 13, 2023, we effected a reverse stock split at a ratio of one-for-forty (1:40) shares of the Company’s common stock. All share and per share amounts of our common stock presented have been retroactively adjusted to reflect these reverse stock splits.
We have incurred losses of $125.5 million since 2011. For the year ended December 31, 2023, we incurred a net loss of $25.3 million. Not reflected in the net loss for the year, but recorded as an increase to accumulated loss was approximately $9.1 million of a noncash adjustment for the value offered to induce warrant exercise. We also incurred negative cash flows from operating activities of approximately $25.2 million for this period. We expect to continue to incur substantial losses, which will generate negative net cash flows from operating activities, as we continue to pursue research and development activities and commercialize.
Our cash was approximately $2.6 million and $1.3 million as of December 31, 2023, and December 31, 2022, respectively. An increase in cash of approximately $1.3 for the year ended December 31, 2023, was due to approximately $25.2 million negative cash flow from operations offset by approximately $26.1 million generated from net financing activities. Due to drug shortages of Abraxane, which is utilized in addition to ivospemin for the current randomized clinical trial, the Company has become responsible for procuring this standard of care component to the clinical trial and in the year ended December 31, 2023 approximately $3.7 million was charged to research and development when the drug was made available to our clinical sites. The Company continues to explore all avenues to procure supply and prepayments may be required in the future. These prepayments are required well in advance of delivery and will be held on the balance sheet as a prepaid expense and reflected in the periods cash used in operations. Net financing activities included a registered public offering of common stock, pre-funded warrants, and warrants with net proceeds of approximately $21.5 million. The Company also sold common stock through its at-the market sales arrangement, with net proceeds of approximately $1.6 million. In the same period, the Company also recorded $1.6 million in loan repayments. The Company also had voluntary and induced warrant exercises in the fourth quarter totaling approximately $4.7 million in net proceeds.
On January 31, 2024, the Company completed a best efforts offering of common stock and warrants. The net proceeds from this offering totaled approximately $8.2 million.
The Company paid its required payment of principal and interest totaling approximately $1.26 million on March 7, 2024, which was paid within an applicable grace period, as extended by the lender.
We need to raise additional capital to continue our operations and execute our business plan past the first quarter of 2024 including completing required future trials and pursuing regulatory approvals in the United States, the European Union, and other international markets. Historically we have financed our operations principally from the sale of equity securities and debt. While we have been successful in the past in obtaining the necessary capital to support our operations and we are likely to seek additional financing through similar means, there is no assurance that we will be able to obtain additional financing under commercially reasonable terms and conditions, or at all. This risk would increase if our clinical data were not positive or if economic or market conditions deteriorate. The accompanying consolidated financial statements have been prepared assuming that we will continue as a going concern which contemplates the realization of assets and liquidation of liabilities in the normal course of business.
If we are unable to obtain additional financing when needed, we would need to scale back our operations, taking actions which may include, among other things, reducing use of outside professional service providers, reducing staff or staff compensation, significantly modifying, or delaying the development of our product candidates, licensing to third parties the rights to commercialize our product candidates, or ceasing operations.
The Company did not experience any significant disruptions to our operations as a result of the COVID-19 pandemic.
Key Components of Our Results of Operations
General and Administrative Expenses
Our selling, general and administrative expenses consist primarily of salaries, benefits and other costs, including stock-based compensation, for our executive and administrative personnel; legal and other professional fees; travel, insurance and other corporate costs.
Research and Development Expenses
Research and development costs include expenses incurred in the conduct of our human clinical trials, for third-party service providers performing various testing and accumulating data related to our preclinical studies; sponsored research agreements; developing and scaling the manufacturing process necessary to produce sufficient amounts of the SBP-101, Flynpovi and CPP-1X compounds for use in our pre-clinical studies and human clinical trials; consulting resources with specialized expertise related to execution of our development plan for our product candidates; personnel costs, including salaries, benefits and share-based compensation; and costs to license and maintain our licensed intellectual property.
Completion of clinical trials may take several years or more, and the length of time generally varies according to the type, complexity, novelty and intended use of a product candidate. The cost of clinical trials may vary significantly over the life of a project as a result of differences arising during clinical development, including, among others:
|
● |
per patient trial costs; |
|
● |
the number of trials required for approval; |
|
● |
the number of sites included in the trials; |
|
● |
the length of time required to enroll suitable patients; |
|
● |
the number of doses that patients receive; |
|
● |
the number of patients that participate in the trials; |
|
● |
the drop-out or discontinuation rates of patients; |
|
● |
the duration of patient follow-up; |
|
● |
potential additional safety monitoring or other studies requested by regulatory agencies; |
|
● |
the number and complexity of analyses and tests performed during the trial; |
|
● |
the phase of development of the product candidate; and |
|
● |
the efficacy and safety profile of the product candidate. |
We charge research and development costs, including clinical trial costs, to expense when incurred. Our human clinical trials are performed at clinical trial sites and are administered jointly by us with assistance from CROs. Costs of setting up clinical trial sites are accrued upon execution of the study agreement. Expenses related to the performance of clinical trials generally are accrued based on contracted amounts and the achievement of agreed upon milestones, such as site openings, patient enrollment, patient follow-up, etc. We monitor levels of performance under each significant contract, including the extent of patient enrollment and other activities through communications with the clinical trial sites and CROs, and adjust the estimates, if required, on a quarterly basis so that clinical expenses reflect the actual effort expended at each clinical trial site and by each CRO.
Research and development costs also include IPR&D. This asset was acquired from the security holders of CPP and written off to research and development expense immediately subsequent to the asset acquisition on June 15, 2022.
We expense costs associated with obtaining licenses for patented technologies when it is determined there is no alternative future use of the intellectual property subject to the license.
Other Income (Expense)
Other income (expense) consists of interest income, cash and non-cash interest expense and transaction gains and losses resulting from transactions denominated in other than our functional currency.
Critical Accounting Estimates
Our management’s discussion and analysis of financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”). The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses. On an ongoing basis, we evaluate these estimates and judgments, including those described below. We base our estimates on our historical experience and on various other assumptions that we believe to be reasonable under the circumstances. These estimates and assumptions form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results and experiences may differ materially from these estimates.
While our significant accounting policies are more fully described in Note 4 to our Consolidated Financial Statements starting on page F-1, we believe that the following description of critical accounting estimates are the most critical to aid you in fully understanding and evaluating our reported financial results and affect the more significant judgments and estimates that we use in the preparation of our financial statements.
Stock-based Compensation
In accounting for share-based incentive awards we measure and recognize the cost of employee and non-employee services received in exchange for awards of equity instruments based on the grant date fair value of those awards. Calculating share-based compensation expense requires the input of highly subjective assumptions, which represent our best estimates and involve inherent uncertainties and the application of management’s judgment. Compensation cost is recognized ratably using the straight-line attribution method over the vesting period, which is considered to be the requisite service period.
The fair values of share-based awards are estimated at the date of grant using the Black-Scholes option pricing model. The determination of the fair value of share-based awards is affected by our stock price, as well as assumptions regarding a number of complex and subjective variables. Risk free interest rates are based upon U.S. Treasury rates appropriate for the expected term of each award. Expected volatility rates are based on the Company’s own volatility rates. The assumed dividend yield is zero, as we do not expect to declare any dividends in the foreseeable future. The expected term of options granted is determined using the “simplified” method. Under this approach, the expected term is presumed to be the mid-point between the average vesting date and the end of the contractual term.
We grant options to employees and non-employees, including our directors. Grants made to new employees are awarded on a case-by-case basis. Option grants to employees generally vest annually over three years from the date of the grant. Options granted to our non-employee directors generally vest over the year following the date of the grant. Options granted to other non-employees generally vest over three years. Options issued to employees and non-employees generally have a maximum term of ten years.
Option grants to non-employees have been made in conjunction with their service as advisors to us. Certain of these advisors have also purchased shares of stock in private placements, but none are known to beneficially own 5% or more of our outstanding common stock.
Accounting for Mergers and Acquisition
ASC 805, Business Combinations, provides a model for determining whether an acquisition represents a business combination. In order to be a business, the integrated set of activities of the acquired entity needs to have an input and a substantive process that together significantly contribute to the ability to create outputs. The acquired entity must also pass the “Screen Test” which involves determining whether the acquisition represents an in-substance asset acquisition based on whether the fair value of the gross assets acquired is “substantially all” concentrated in a single asset or group of similar assets. This evaluation excludes certain acquired assets such as cash, deferred taxes, and goodwill associated with deferred taxes, but includes all other gross assets, including any consideration transferred in excess of the identified assets.
The Company accounted for the acquisition of CPP as an asset acquisition after determining that the transaction did not meet the criteria of a business combination. The Company determined that substantially all of the fair value of the gross assets acquired was concentrated in a single identifiable asset or group of similar identifiable assets, the most significant of which is the IPR&D asset associated with CPP’s product candidates. The net assets were recorded at fair value at the date of acquisition and the IPR&D was recorded as research and development expense in the Company’s consolidated statement of operations.
Results of Operations
Comparison of the Results of Operations (in thousands) for the Years Ended December 31, 2023, and 2022
| Year Ended December 31, | ||||||||||||
|
2023 |
2022 |
Percent Change |
||||||||||
|
Operating Expenses |
||||||||||||
|
General and administrative |
$ | 5,033 | $ | 6,044 | -16.7 | % | ||||||
|
Research and development |
20,614 | 28,049 | -26.5 | % | ||||||||
|
Total operating expenses |
25,647 | 34,093 | -24.8 | % | ||||||||
|
Other (expense) income, net |
198 | (956 | ) | -120.7 | % | |||||||
|
Income tax benefit |
186 | 116 | 60.3 | % | ||||||||
|
Net Loss |
$ | (25,263 | ) | $ | (34,933 | ) | -27.7 | % | ||||
General and administrative (“G&A”) and research and development (“R&D”) expenses include non-cash stock-based compensation expense as a result of our issuance of stock options. The terms and vesting schedules for stock-based awards vary by type of grant and the employment status of the grantee. The awards granted through December 31, 2023, vest based upon time-based and performance conditions. We expect to record additional non-cash compensation expense in the future, which may be significant. The following table summarizes the stock-based compensation expense in our Consolidated Statements of Operations and Comprehensive Loss for the years ended December 31, 2023, and 2022 (in thousands):
| Year ended December 31, | ||||||||
|
2023 |
2022 |
|||||||
|
General and administative |
$ | 643 | $ | 889 | ||||
|
Research and development |
180 | 199 | ||||||
|
Total stock based compensation |
$ | 823 | $ | 1,088 | ||||
General and administrative expense
G&A expenses decreased 16.7% to $5.0 million in 2023, down from $6.0 million in 2022. The decrease in G&A expenses is primarily the result of decreased legal and financial advisory expenses.
Research and product development expense
Our R&D expenses decreased 26.5% to $20.6 million in 2023, down from $28.0 million in 2022. After removing the non-cash write off of approximately $17.7 million of IPR&D in the year ended December 31,2022, R&D expense for 2023 increased by $10.3 million. This increase is due primarily to increased clinical trial costs related to our ivospemin randomized trial. As we expand our clinical studies it is expected that R&D expenses will continue to increase. The write-off of IPR&D was a one-time occurrence related to the accounting for the acquisition of CPP.
Other income (expense), net
Other income, net, was $0.2 million for the year ended December 31, 2023, versus other net expense of $1.0 million for the year ended December 31, 2022. The income in 2023 was primarily associated with the gain on sale of intellectual property and the expense in 2022 was primarily related to a foreign currency transaction loss.
Income tax benefit
Income tax benefit increased to $186,000 in 2023, up from $116,000 in 2022. Our income tax benefit is derived primarily from refundable tax incentives associated with our R&D activities conducted in Australia which have increased in 2023 as the ASPIRE trial expanded in 2023 to several sites in Australia.
Liquidity and Capital Resources
The following table summarizes our liquidity and capital resources as of December 31, 2023, and 2022 and for each of the fiscal years ended December 31, 2023, and 2022, and is intended to supplement the more detailed discussion that follows (in thousands):
|
Liquidity and Capital Resources |
December 31, |
|||||||
|
2023 |
2022 |
|||||||
|
Cash and cash equivalents |
$ | 2,578 | $ | 1,285 | ||||
|
Working capital deficit |
$ | (9,258 | ) | $ | (6,056 | ) | ||
|
Cash Flow Data |
Year Ended December 31, |
|||||||
|
2023 |
2022 |
|||||||
|
Cash Provided by (used in): |
||||||||
|
Operating Activities |
$ | (25,249 | ) | $ | (15,276 | ) | ||
|
Investment Activities |
400 | (656 | ) | |||||
|
Financing Activities |
26,142 | 5,354 | ||||||
|
Effect of exchange rate changes on cash |
- | (4 | ) | |||||
|
Net change in cash and cash equivalents |
$ | 1,293 | $ | (10,582 | ) | |||
Working Capital
Our total cash and cash equivalents was $2.6 million as of December 31, 2023, compared to $1.3 million as of December 31, 2022. As of December 31, 2023, we had $12.3 million in current liabilities and negative working capital of $9.3 million. As of December 31, 2022, we had $7.8 million in current liabilities and negative working capital of $6.0 million. Working capital is calculated as current assets less current liabilities.
Cash Flows
Net Cash Used in Operating Activities
Net cash used in operating activities was $25.2 million during 2023, compared to $15.3 million during 2022. The net cash used in each of these periods primarily reflects the net loss for these periods and is partially offset by the effects of changes in operating assets and liabilities. For the year ended December 31, 2022 the non-cash write-off of IPR&D of approximately $17.7 million had a positive impact on our cash used in Operations.
Net Cash Used in Investing Activities
Cash provided from investing activities in the year ended December 31, 2023 was the result of proceeds from the sale of intellectual property. Net cash used in investing activities was $0.7 million during the year ended December 31, 2022. The net cash used in 2022 is primarily cash expended for legal and financial advisor fees incurred at the end of the process to acquire CPP.
Net Cash Provided by Financing Activities
Net cash provided by financing activities was $26.1 million for 2023 which primarily represents the net proceeds from several public offerings during the year as well as the exercise for cash of certain warrants to purchase common stock. This capital raised was offset by the payment of $1.65 million in principal due on outstanding notes payable. Net cash provided from financing activities for 2022 was approximately $5.4 million which represents the net proceeds of a registered offering in October of 2022.
Net Cash Provided by Financing Activities subsequent to December 31, 2023
On January 31, 2024, the Company completed a registered public offering of common stock and warrants. The securities were issued for a combined offering price of $2.06 per share of common stock and 2 warrants, or $2.059 per pre-funded warrant and 2 warrants. Net proceeds from the offering totaled approximately $8.2 million.
Capital Requirements
As we continue to pursue our operations and execute our business plan, including the completion of clinical development plan for our initial product candidate, ivospemin, in pancreatic cancer, and pursuing regulatory approvals in the United States, the European Union and other international markets, we expect to continue to incur substantial and increasing losses, which will continue to generate negative net cash flows from operating activities.
Our future capital uses and requirements depend on numerous current and future factors. These factors include, but are not limited to, the following:
|
● |
the progress of clinical trials required to support our applications for regulatory approvals, including the completion the ASPIRE trial, our global, randomized Phase II/III trial initiated in January of 2022; |
|
● |
our ability to negotiated payment terms with critical vendors |
|
● |
the cost to implement development efforts for SBP-101 in ovarian cancer and expand development efforts for assets acquired as the result of the acquisition of CPP; |
|
● |
the cost, if any, to develop our product candidate, Flynpovi, in geographies outside of North America; |
|
● |
the cost to develop eflornithine in various indications if early clinical trials underway now, and funded through third party collaborations are successful; |
|
● |
our ability to demonstrate the safety and effectiveness of our product candidates; |
|
● |
our ability to obtain regulatory approval of our product candidates in the United States, the European Union or other international markets; |
|
● |
the cost and delays in product development that may result from changes in regulatory oversight applicable to our product candidates; |
|
● |
the market acceptance and level of future sales of our product candidates; |
|
● |
the rate of progress in establishing reimbursement arrangements with third-party payors; |
|
● |
the effect of competing technological and market developments; and |
|
● |
the costs involved in filing and prosecuting patent applications and enforcing or defending patent claims. |
As of December 31, 2023, we did not have any existing credit facilities under which we could borrow funds. We historically have financed our operations principally from the sale of equity securities and debt. While we have been successful in the past in obtaining the necessary capital to support our operations and we are likely to seek additional financing through similar means, there is no assurance that we will be able to obtain additional financing under commercially reasonable terms and conditions, or at all.
Indebtedness
CPP issued to Sucampo GmbH (“Lender”) an Amended and Restated Promissory Note (the “Note”) on June 15, 2022, for the principal sum of approximately $6.2 million (the “Principal”). The note bears simple interest on any outstanding Principal at a rate of 5% per annum. On March 8, 2024 the company paid the second installment on the balance due of $1.0 plus accrued and unpaid interest of approximately $259,000. This payment was made prior to the expiration of a grace period provided by the lender. All remaining unpaid principal, together with any then unpaid and accrued interest is payable as follows: (i) $1.0 million, plus all interest accrued but unpaid on or before each of January 31, 2025 and January 31, 2026; and (ii) all remaining Principal plus accrued but unpaid interest on or before January 31, 2027. The Company made the scheduled January 31, 2023, payment of $1.0 million plus accrued interest. The outstanding principal balance on December 31, 2023, was approximately $5.2 million. Accrued and unpaid interest as of December 31, 2023, totaled approximately $238,000. See Footnote 7.
Panbela has provided a Guarantee of payment in favor of the Lender for the full amount of the Note issued to the Lender.
Issuances of common stock and warrants after December 31, 2023
On January 31, 2024, the Company completed a registered public offering and issued an aggregate of 794,000 shares of its common stock, pre-funded warrants to purchase up to an aggregate of 3,581,000 shares of common stock at an exercise price of $0.001 per shares and warrants to purchase up to an aggregate of 8,750,000 shares of its common stock. The initial exercise price of the warrants is $2.06 per underlying share. The securities were issued for a combined offering price of $2.06 per share of common stock and warrants to purchase up to two additional shares of common stock, or $2.059 per pre-funded warrant and warrants. Net proceeds from the offering totaled approximately $8.2 million. As of March 7, 2024 no pre-funded warrants remained outstanding. The securities were offered pursuant to an effective registration statement on Form S-1.
Issuances of Common Stock and warrants during 2023 and 2022
On December 21, 2023, we entered into warrant exercise inducement offer letters with the holders of our existing Class C warrants to purchase shares of our common stock, pursuant to which the holders agreed to exercise for cash their Class C warrant to purchase an aggregate of 127,800 shares of our common stock, in the aggregate, at the existing exercise price of $15.60 per share, in exchange for our agreement to issue new warrants on substantially the same terms as the Class C warrants, to purchase up to 255,600 shares of our common stock. We received aggregate gross proceeds of approximately $2.0 million from the exercise of the Class C warrants by the holders. Net proceeds from the offering totaled approximately $1.9 million. The shares of common stock issuable upon exercise of the Class C warrants were registered for resale by the holders pursuant to an existing registration statement on Form S-1.
On November 2, 2023, we entered into warrant exercise inducement offer letters with certain holders of our existing warrants to purchase shares of common stock, pursuant to which the holders agreed to exercise for cash their existing warrants to purchase 106,500 shares of our common stock, in the aggregate, at a reduced exercise price of $15.60 per share, in exchange for our agreement to issue new warrants on substantially the same terms as the existing warrants, to purchase up to 213,000 shares of common stock and a cash payment of $0.125 per existing warrant share which was paid in full upon the exercise of the existing warrants. We received aggregate gross proceeds of approximately $1.9 million from the exercise of the existing warrants by the holders and the sale of the inducement warrants. Net proceeds from the offering totaled approximately $1.8 million. The shares of the Company’s common stock issuable upon exercise of the Existing Warrants were registered pursuant to an existing registration statement on Form S-1.
On June 21, 2023, the Company completed a registered public offering and issued an aggregate of 29,300 shares of its common stock, pre-funded warrants to purchase up to an aggregate of 84,200 shares of common stock at an exercise price of $0.001 per share and warrants to purchase up to an aggregate of 227,000 shares of its common stock at an original exercise price of $75.00 per share. The securities were issued for a combined offering price of $75.00 per share of common stock and warrants to purchase two shares, or $74.999 per pre-funded warrant and warrants to purchase two shares. Net proceeds from the offering totaled approximately $7.7 million. All Prefunded warrants were exercised by December 31, 2023. The securities were offered pursuant to an effective registration statement on Form S-1.
On January 31, 2023, the Company completed a registered public offering and issued an aggregate of 8,070 shares of its common stock, pre-funded warrants to purchase up to an aggregate of 3,054 shares of common stock at an exercise price of $0.001 per shares and warrants to purchase up to an aggregate of 22,250 shares of its common stock. The exercise price on these warrants after being repriced for applicable dilutive public offerings is now at 1.0992 per share. The securities were issued for a combined offering price of $1,350.00 per share of common stock and 2 warrants, or $1,349.999 per pre-funded warrant and 2 warrants. Net proceeds from the offering totaled approximately $13.7 million. All pre-funded warrants were exercised by December 31, 2022. The securities were offered pursuant to an effective registration statement on Form S-1.
On October 4, 2022, the Company completed a registered public offering and issued an aggregate of 295 shares of its common stock, pre-funded warrants to purchase up to an aggregate of 542 shares of common stock at an exercise price of $0.001 per shares and warrants to purchase up to an aggregate of 1,256 shares of its common stock. The exercise price on these warrants after being repriced for a dilutive public offering and subsequently for reverse stock splits is $12.386 per share. The securities were issued for a combined offering price of $7,200 per share of common stock and 1.5 warrants, or $7,199.999 per pre-funded warrant and 1.5 warrants. Net proceeds from the offering totaled approximately $5.3 million. All pre-funded warrants were exercised by December 31, 2022. The securities were offered pursuant to an effective registration statement on Form S-1.
On July 19, 2022, Panbela Therapeutics, Inc. (the “Company”), entered into a Sales Agreement with Roth Capital Partners, LLC (the “Agent”) to sell shares of the Company’s common stock having an aggregate gross sales price of up to $8,400,000, from time to time, through an “at-the-market” equity offering program (the “ATM Program”). During the last month of the year ended December 31, 2022, the Company sold 47 shares of common stock under the ATM Offering and generated approximately $93,000 in gross proceeds. The Company incurred financing costs of approximately $44,000, which were charged to additional paid in capital in December 2022 when the Company began selling shares under the ATM Offering. Under the ATM Program, the Company pays the Agent a commission equal to 3.0% of the aggregate gross proceeds of any sales of common stock under the ATM Program. Net proceeds for the year ended December 31, 2022, was approximately $46,000. During the year ended December 31, 2023, the Company sold 915 shares of common stock under the ATM Program for approximately $1.6 million in gross proceeds. Net proceeds for sales during the year ended December 31, 2023, were approximately $1.6 million.
Future Capital Requirements
We require additional funds to continue our operations and execute our business plan, including completion of required future trials and pursuing regulatory approvals in the United States, the European Union and other international markets. We historically have financed our operations principally from the sale of equity securities and debt. While we have been successful in the past in obtaining the necessary capital to support our operations and we are likely to seek additional financing through similar means, there is no assurance that we will be able to obtain additional financing under commercially reasonable terms and conditions, or at all. We believe that our existing cash and cash raised through public offerings in January 2024 will be sufficient to fund our operating expenses into the second quarter of 2024.
If we are unable to obtain additional financing when needed, we would need to scale back our operations, taking actions which may include, among other things, reducing use of outside professional service providers, reducing staff or staff compensation, significantly modifying or delaying the development of our product candidates, licensing to third parties the rights to commercialize our product candidate for pancreatic cancer or other applications that we would otherwise seek to pursue, or suspending operations.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, the interests of our current stockholders would be diluted, and the terms may include liquidation or other preferences that adversely affect the rights of our current stockholders. If we issue preferred stock, it could affect the rights of our stockholders or reduce the value of our common stock. In particular, specific rights granted to future holders of preferred stock may include voting rights, preferences as to dividends and liquidation, conversion and redemption rights, sinking fund provisions, and restrictions on our ability to merge with or sell our assets to a third party. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. Any of these events could adversely affect our ability to achieve our regulatory approvals and commercialization goals and harm our business.
Our future success is dependent upon our ability to obtain additional financing, Phase II/III clinical trials and required future trials, and our ability to obtain marketing approval for our product candidates in the United States, the European Union and other international markets. If we are unable to obtain additional financing when needed, if our Phase II/III clinical trial is not successful, if we do not receive regulatory approval required for future trials or if once these studies are concluded, we do not receive marketing approval for our ivospemin product candidate, we would not be able to continue as a going concern and would be forced to cease operations. The financial statements included in this report have been prepared assuming that we will continue as a going concern and do not include any adjustments relating to the recoverability or classification of assets or the amounts of liabilities that might result from the outcome of these uncertainties.
License Agreement
Pursuant to our exclusive license agreement with UFRF, which was last amended on October 4, 2019, we are required to pay royalties ranging from 2.5% to 5% of net sales of licensed products developed from the licensed technology for the shorter of: ten (10) years from the first commercial sale of a licensed product or the period of market exclusivity on a country-by-country basis. The latest amendment eliminated all future milestone payments. The Company remains committed to paying an annual license maintenance fee of $10,000.
|
Item 7A. |
Quantitative and Qualitative Disclosures about Market Risk |
As a smaller reporting company, we are not required to provide disclosure pursuant to this item.
|
Item 8. |
Financial Statements and Supplementary Data |
The financial statements and notes thereto required pursuant to this Item begin on page F-1 of this annual report on Form 10-K.
|
Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
|
Item 9A. |
Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, has evaluated our disclosure controls and procedures. Based on such evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that, as of December 31, 2023, our disclosure controls and procedures were effective in ensuring that information relating to the Company required to be disclosed in the reports that we file or submit under the Exchange Act, is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, including ensuring that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
Changes in Internal Control Over Financial Reporting
We have not identified any change in our internal control over financial reporting during our most recently completed fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Internal control over financial reporting refers to the processes designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer, and effected by our Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
|
(1) |
Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; |
|
(2) |
Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorization of our management and directors; and |
|
(3) |
Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting cannot provide absolute assurance of preventing and detecting misstatements on a timely basis. It is possible to design into the process safeguards to reduce, though not eliminate, the risk that misstatements are not prevented or detected on a timely basis.
Our management conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework set forth in the report entitled Internal Control—Integrated Framework published by the Committee of Sponsoring Organizations of the Treadway Commission, known as COSO (2013 Framework). Based on this assessment, management has concluded that, as of December 31, 2023, our internal control over financial reporting was effective.
This report does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our independent registered public accounting firm pursuant to Section 989G of the Dodd-Frank Wall Street Reform and Consumer Protection Act, which exempts smaller reporting companies from the auditor attestation requirement.
|
Item 9B. |
Other Information |
.
|
Item 9C. |
Disclosure Regarding Foreign Jurisdictions that Prevent Inspections |
None.
PART III
|
Item 10. |
Directors, Executive Officers and Corporate Governance |
Information about our Executive Officers
Jennifer K. Simpson, Ph.D., MSN, CRNP, age 55, has served as President and Chief Executive Officer and as a director of our Company since July 2020. Prior to joining the Company, Dr. Simpson served as President and Chief Executive Officer and as a member of the board of directors of Delcath Systems, Inc. (Nasdaq: DCTH) from 2015 to June 2020. She had previously held various other leadership roles at Delcath since 2012. From 2011 to 2012, Dr. Simpson served as Vice President, Global Marketing, Oncology Brand Lead at ImClone Systems, Inc. (a wholly owned subsidiary of Eli Lilly and Company), where she was responsible for all product commercialization activities and launch preparation for one of the late-stage assets. From 2009 to 2011, Dr. Simpson served as Vice President, Product Champion and from 2008 to 2009 as the Associate Vice President, Product Champion for ImClone’s product Ramucirumab. From 2006 to 2008, Dr. Simpson served as Product Director, Oncology Therapeutics Marketing at Ortho Biotech (now Janssen Biotech), a Pennsylvania-based biotech company that focuses on innovative solutions in immunology, oncology and nephrology. Earlier in her career, Dr. Simpson spent over a decade as a hematology/oncology nurse practitioner and educator. Dr. Simpson has served on the board of directors and nominating and corporate governance committee of Eagle Pharmaceuticals, Inc. since August 2019 and on the board of Directors of CytRx Corporation since July 2021. Dr. Simpson earned a Ph.D. in Epidemiology from the University of Pittsburgh, an M.S. in Nursing from the University of Rochester, and a B.S. in Nursing from the State University of New York at Buffalo.
Susan Horvath, age 64, has served as our Vice President and Chief Financial Officer since April 2018. Ms. Horvath has held both finance and operating positions within pharmaceutical, healthcare and consumer organizations. In addition to her position with the Company, Ms. Horvath sits on the board of directors and provides financial consulting services for Photonic Pharma, LLC, a privately held company focused on efficiency in early-stage drug discovery. Prior to joining the Panbela team, Ms. Horvath served as Chief Financial Officer of Eyebobs, LLC, a private company focused on eyewear for corrective vision, from 2016 to January 2018; Vice President and Chief Financial Officer of Tenacious Holdings, Inc. (d/b/a ergodyne), a privately held, safety products company, from 2014 to 2016; Chief Financial Officer and Vice President of Human Resources at Healthsense, Inc., a next generation technology (SaaS) and remote monitoring company focused on providing safety and improving quality of life while reducing overall costs of healthcare for seniors and fragile adults, from 2011 to 2014; Chief Financial Officer, Vice President of Operations & Human Resources of Hemosphere, Inc., an early commercialization stage medical device company, from 2008 to 2010; and Vice President & Team Leader International of CNS, Inc, a publicly traded consumer health care products company focused on the development and marketing of strong consumer brands, from 2004 to 2007. Ms. Horvath holds a Bachelor of Science degree in Accounting from the University of Illinois, Champaign, and is a Certified Management Accountant and Certified Public Accountant, inactive.
Information about our Board of Directors
Our business is overseen by a Board of Directors divided into three classes as nearly equal in number as possible, and directors typically are elected to a designated class for a term of three years. The following sets forth certain information regarding the current members of our Board of Directors:
Class II Directors–- Term Expiring in 2024
Michael T. Cullen, M.D., M.B.A., age 78, has served as Chairman of the Board and a non-employee director of our Company since his retirement as an employee of the Company in May 2021. Dr. Cullen had served as Executive Chairman and as a director of our Company since its co-founding in November 2011. Dr. Cullen brings 33 years of pharmaceutical experience to our Company, including expertise in working with development-stage companies in planning, designing and advancing drug candidates from preclinical through clinical development. Dr. Cullen served as our President and Chief Executive Officer between October 2018 and July 2020. He previously served as our Chief Medical Officer and President from November 2011 to June 2015. Dr. Cullen provided due diligence consulting to the pharmaceutical industry from 2009 to 2011, after one year in transition consulting to Eisai Pharmaceuticals. He developed several oncology drugs as Chief Medical Officer for MGI Pharma Inc. from 2000 to 2008, and previously at G.D. Searle, SunPharm Corporation, and as Vice President for Clinical Consulting at IBAH Inc., the world’s fifth largest contract research organization, where he provided consulting services on business strategy, creating development plans, regulatory matters and designing clinical trials for several development stage companies in the pharmaceutical industry. Dr. Cullen was also a co-founder and Chief Executive Officer of IDD Medical, a pharmaceutical start-up company. Dr. Cullen joined 3M Pharmaceuticals in 1988 and contributed to the development of cardiovascular, pulmonary, rheumatology and immune-response modification drugs. Over the course of his career Dr. Cullen has been instrumental in obtaining the approval of ten drugs, including three since 2004: Aloxi®, Dacogen® and Lusedra®. Board-certified in Internal Medicine, Dr. Cullen practiced from 1977 to 1988 at Owatonna Clinic, Owatonna, MN, where he served as president. Dr. Cullen earned his MD and BS degrees from the University of Minnesota and his M.B.A. from the University of St. Thomas and completed his residency and Board certification in Internal Medicine through the University of North Carolina in Chapel Hill and Wilmington, NC.
D. Robert Schemel, age 68, has served as a director since September 2015. Mr. Schemel had previously served as a director of Sun BioPharma Research, Inc. since March 2012. Mr. Schemel has over 39 years’ experience in the agriculture industry. From 1973-2005, Mr. Schemel owned and operated a farming operation in Kandiyohi County, Minnesota, building a 5,000-acre operation producing corn, soybeans and sugar beets. Mr. Schemel has extensive experience in serving on boards of directors. From 1992-1996 he served as a board member for ValAdCo and then from 1996-2003 he served as the Chairman of the Board for Phenix Biocomposites.
Class III Directors -Terms Expiring in 2025
Arthur J. Fratamico, age 58, has served as a director of our Company since December of 2019. He is a registered pharmacist with over 30 years of experience in the pharmaceutical industry and has been the Chief Executive Officer of Radiant Biotherapeutics, which is advancing a novel antibody platform that is focused on the development of Multabodies, which are multi-valent and multi-specific antibodies since May 2021. Prior to Radiant, Mr. Fratamico served as Chief Business Officer at Galera Therapeutics, Inc., a biopharmaceutical company dedicated to discovering and developing novel dismutase mimetics with the goal of transforming cancer radiotherapy, since January 2017. Prior to joining Galera, Mr. Fratamico served as Chief Business Officer of Vitae Pharmaceuticals, Inc., a Nasdaq-listed clinical-stage biotechnology company, from May 2014 until its sale to Allergan in December 2016. Prior to Vitae Pharmaceuticals, he held similar executive roles with a number of biotechnology companies leading their business development efforts, including facilitating the sales of Gemin X Pharmaceuticals, Inc. and MGI Pharma, Inc. In addition to being responsible for numerous licensing transactions and acquisitions, he also directed corporate strategy and managed external corporate communications. He also served in several senior marketing, product planning and new product development positions. Mr. Fratamico earned a bachelor’s degree in pharmacy from the Philadelphia College of Pharmacy and Science and an M.B.A. from Drexel University.
Jeffrey S. Mathiesen, age 63, has served as a director of our Company since September 2015. Mr. Mathiesen also serves as a director and audit committee chairman of NeuroOne Medical Technologies Corporation, a publicly traded medical device company. Since June 2021, Mr. Mathiesen has served as Chief Financial Officer, Treasurer and Secretary of Helius Medical Technologies, Inc. (Nasdaq: HSDT), a publicly traded medical device company, developing noninvasive platform technologies focused on neurological wellness, and he served as director since May 2022 and previously served as director and Audit Committee Chair from June 2020 through June 2021. Additionally, Mr. Mathiesen previously served as a director and Audit Committee Chair of Healthcare Triangle, Inc. (Nasdaq: HCTI), a publicly traded provider of cloud and data transformation platform and solutions for healthcare and life sciences, from March 2022 to December 2022 and as a director and audit committee chairman of eNeura, Inc., a privately held medical technology company providing therapy for both acute treatment and prevention of migraine, from July 2018 to February 2020. Mr. Mathiesen has served as Advisor to the CEO of Teewinot Life Sciences, a privately held biopharmaceutical company focused on the biosynthetic production of pure pharmaceutical grade cannabinoids from October 2019 to December 2019, and as Chief Financial Officer from March 2019 to October 2019. In August 2020, Teewinot Life Sciences filed a voluntary petition under Chapter 11 of the United States Bankruptcy Code. Previously he served as Chief Financial Officer of Gemphire Therapeutics Inc., a publicly traded biopharmaceutical company from September 2015 to September 2018. From August 2015 to September 2015, he was a consultant to Gemphire. He served as Chief Financial Officer of Sunshine Heart, Inc., a publicly traded medical device company, from March 2011 to January 2015. Mr. Mathiesen has held executive positions with publicly traded companies dating back to 1993, including vice president and chief financial officer positions. Mr. Mathiesen holds a B.S. in Accounting from the University of South Dakota and is also a Certified Public Accountant.
Class I Directors -Terms Expiring in 2026
Daniel J. Donovan, age 59, has served as a director since June 2022. He had served as a director and Chief Business Officer, a non-employee position, of CPP from 2011 until immediately before the completion of its acquisition by Panbela in June 2022. He has served as chief executive officer of rareLife Solutions, Inc., a private company since he founded it in 2014. He served on the Board of Directors at Intensity Therapeutics since January of 2023 and is a member of the audit committee. Before rareLife, Mr. Donovan founded Envision Pharma in 2001, serving as managing director then president until 2011. Envision Pharma was acquired by United BioSource Corporation in 2008, where Mr. Donovan served as Senior Vice President Strategy and Market Development and was a member of the leadership team. Mr. Donovan began his career at Pfizer serving in a variety of positions of increasing responsibility, ranging from sales to market research and marketing in the U.S. and internationally, culminating in his position as Director and European Team Leader. During his time at Pfizer, he played a pivotal role in the commercialization of some of the pharmaceutical industry’s most successful product launches.
Jeffrey E. Jacob, age 62, has served as a director since June 2022. He served as Chief Executive Officer of CPP from 2009 until immediately before the completion of its acquisition by Panbela in June 2022. He is also the principal of Tucson Pharma Ventures LLC, an Arizona-based biopharmaceutical consulting and investment firm, a role he’s held since 2004. In 2004, Mr. Jacob founded Systems Medicine Inc., a startup company applying systems biology, predictive pharmacogenomics, and clinical trial design innovations to the development of new cancer drugs and served as its chief executive officer until its sale in 2007, after which he served as a divisional chief executive officer until late 2008. Between 1987 and 2004, Mr. Jacob was employed by Research Corporation Technologies, most recently as Senior Vice President. During that time, he led the transformation of Research Corporation Technologies from a patent development and licensing organization to an early stage-technology incubation and venture deployment firm. He has served as a member of the board of directors of Research Corporation Technologies and currently serves as its chair. He is also a founding board member and previously served as the chief program officer of Critical Path Institute. Mr. Jacob holds a master’s degree in engineering and a master’s degree in technology and policy from the Massachusetts Institute of Technology and a bachelor’s degree in engineering from the University of Arizona.
Jennifer K. Simpson Ph.D., MSN, CRNP, has served as our President and Chief Executive Officer and as a director of our Company since July 2020. See “Information about our Executive Officers” above for further information regarding Dr. Simpson’s background and experience.
Delinquent Section 16(a) Reports
Section 16(a) of the Securities and Exchange Act of 1934 requires that our directors, executive officers and beneficial owners of more than 10% of our common stock file initial reports of ownership and reports of changes in ownership with the SEC. Directors, executive officers and beneficial owners of greater than 10% of our common stock are required to furnish us with copies of all Section 16(a) forms they file. Based solely on a review of the copies of such forms furnished to us and written representations from our directors and executive officers, all Section 16(a) filing requirements were met for the fiscal year ended December 31, 2023.
Code of Ethics and Business Conduct
We have adopted a code of ethics and business conduct (the “Code”) that applies to our principal executive officer, principal financial officer, principal accounting officer or controller or persons performing similar functions, as well as other employees and our directors. The Code is posted to the Investor Relations-Corporate Governance section of our website at www.panbela.com. We intend to include on our website, within the time period required by Form 8-K, an amendment to, or waiver from, any provision of our Code that applies to our principal executive officer, principal financial officer, principal accounting officer, controller, or persons performing similar functions, and that relates to any element of the Code of Ethics definition enumerated in Item 406(b) of SEC Regulation S-K.
Certain Information Regarding Board Committees
Audit Committee
The Audit Committee is composed of three members, Mr. Mathiesen, its chair, and Messrs. Donovan and Schemel. The committee’s primary functions, among others, are to: (a) assist the Board of Directors in discharging its statutory and fiduciary responsibilities with regard to audits of the books and records of our Company and the monitoring of its accounting and financial reporting practices; (b) carry on appropriate oversight to determine that our Company and its subsidiaries have adequate administrative and internal accounting controls and that they are operating in accordance with prescribed procedures and codes of conduct; and (c) independently review our Company’s financial information that is distributed to stockholders and the general public.
All of the members of the Audit Committee meet the requirements for financial literacy under the applicable rules and regulations of the Securities and Exchange Commission (the “SEC”). Our Board of Directors has determined that Jeffrey S. Mathiesen is qualified to serve as an audit committee financial expert, as that term is defined under the applicable rules of the SEC. Each member of the Audit Committee satisfies the independence requirements of Rule 10A-3(b)(1) of the Securities Exchange Act.
|
Item 11. |
Executive Compensation |
Compensation of Named Executive Officers
The following disclosure focuses on our named executive officers. For fiscal 2023, our “named executive officers” consisted of our only executive officers, Dr. Simpson and Ms. Horvath.
Base salaries for each of our named executive officers were initially established based on arm’s-length negotiations with the applicable executive. The Compensation Committee of our Board of Directors reviews our executive officers’ salaries annually. When negotiating or reviewing base salaries, the Compensation Committee considers market competitiveness based on the experience of its members, the executive’s expected future contribution to our success and the relative salaries and responsibilities of our other executives.
Summary Compensation Table
The following table provides information regarding the compensation earned by our named executive officers for fiscal 2023 (collectively referred to as the “Executives”):
|
Name and Principal Positions |
Year |
Salary |
Option Awards (a) |
Nonequity Incentive Plan Compensation (b) |
Total |
|||||||||||||
|
Jennifer K. Simpson |
2023 |
527,000 | 44,911 | - | 571,911 | |||||||||||||
| President and Chief Executive Officer |
2022 |
506,000 | - | 188,324 | 694,324 | |||||||||||||
|
Susan Horvath |
2023 |
333,000 | 15,526 | 119,394 | 467,920 | |||||||||||||
| Chief Financial Officer and Vice President of Finance |
2022 |
330,200 | - | 103,459 | 423,459 | |||||||||||||
|
(a) |
The values of option awards in this table represent the fair value of such awards granted during the fiscal year, as computed in accordance with Financial Accounting Standards Board Accounting Standards Codification Topic 718. The assumptions used to determine the valuation of the awards are discussed in Note 9 to the consolidated financial statements. No equity awards were granted during fiscal 2022. |
|
(b) |
Represents payments to be made in 2024 under Panbela’s 2023 Cash Incentive Program and payments made in 2023 under Panbela’s 2022 Cash Incentive Programs as described further below. On February 8, 2024 the Compensation Committee certified actual performance for purposes of determining the cash incentive payments earned under the 2023 Cash Incentive Program to be paid in 2024. Dr. Simpson has waived her payment which would have been paid in 2024. |
Outstanding Equity Awards as of December 31, 2023
|
Option Awards |
||||||||||||||||
|
Name |
Grant Date |
Number of securities underlying unexercised options (#) exercisable |
Number of securities underlying unexercised options (#) un- exercisable |
Option exercise price |
Option expiration Date |
|||||||||||
|
Jennifer K. Simpson |
7/17/2020 |
8 | – | 239,760 |
7/17/2030 |
|||||||||||
|
3/30/2021 |
5 |
2(a) |
98,160 |
3/30/2031 |
||||||||||||
|
3/27/2023 |
– |
165(b) |
300 |
3/27/2033 |
||||||||||||
|
Susan Horvath |
4/17/2018 |
1 | – | 138,000 |
4/17/2028 |
|||||||||||
|
5/21/2019 |
2 | – | 70,800 |
5/21/2029 |
||||||||||||
|
9/24/2019 |
1 | – | 120,000 |
9/24/2029 |
||||||||||||
|
6/24/2020 |
1 | – | 119,520 |
6/24/2030 |
||||||||||||
|
3/30/2021 |
1 | – | 98,160 |
3/30/2031 |
||||||||||||
|
3/27/2023 |
– |
57(b) |
300 |
3/27/2033 |
||||||||||||
|
(a) |
Scheduled to vest with respect to all remaining shares on March 30, 2024. |
|
(b) |
Scheduled to vest with respect to all remaining shares on March 27, 2024. |
Cash Incentive Compensation
For 2023 and 2022, the Compensation Committee established performance objectives for each of the Executives based on clinical development and financial milestones. Each Executive’s potential payment upon satisfaction of the objectives was equal to the target set forth in the Executive’s employment agreement as described further below. In the first quarter of 2024, the Compensation Committee determined that Dr. Simpson’s bonus for 2023 was approved for payment at 86.18% of target and Ms. Horvath’s bonus was approved for payment at 89.64% of target. Dr. Simpson has waived her 2023 cash incentive compensation. Ms. Horvath’s 2023 bonus is expected to be paid in the first quarter of 2024. The 2022 cash incentive which was approved by the Compensation committee and was paid in the first quarter of 2023.
Employment Agreements
We are party to employment agreements with each of the Executives. In addition to the specific terms summarized below, each Executive is eligible to participate in the other compensation and benefit programs generally available to our employees, including our other executive officers, if any. Each such employment agreement also includes customary non-competition and non-solicitation covenants and a requirement that the Executive carry out a supplemental agreement regarding confidentiality and assignment of intellectual property.
In accordance with the employment agreements, the base salary of each Executive is reviewed annually by the Compensation Committee of our Board of Directors. Pursuant to the employment agreements, the committee may authorize an increase for the applicable year but may not reduce an Executive’s base salary below its then-current level other than with the Executive’s consent or pursuant to a general wage reduction in respect of substantially all of our executive officers. As discussed above, the Compensation Committee established performance criteria for 2023 and, based upon achievement of those objectives, cash payments were approved. Dr Simpson has waived payment of her cash payment and Ms. Horvath’s cash payment was paid in the first quarter of 2024.
President and Chief Executive Officer
Under her employment agreement, Dr. Simpson is eligible to receive an annual performance-based cash bonus with a target amount equal to no less than 50% of her base salary. Payment of the bonus amount is subject to achievement of metrics to be established by our Board of Directors and her continued employment with the Company through the end of the applicable cash bonus period.
Vice President of Finance and Chief Financial Officer
Under her employment agreement, Ms. Horvath is eligible to receive an annual performance-based cash bonus with a target amount equal to no less than 40% of her base salary. Payment of the bonus amount is subject to achievement of metrics to be established by our Board of Directors and her continued employment with the Company through the end of the applicable cash bonus period.
Potential Payments Upon Termination or Change-in-Control
Under their respective employment agreements, if any of the Executive’s employment is terminated by us for any reason other than for “cause” (as defined in the applicable employment agreement) or by him or her for “good reason” (as defined in the applicable employment agreement), then he or she will be eligible to receive an amount equal to their respective annualized salary plus an amount equal to a prorated portion of their cash bonus target, if any, for the year in which the termination occurred, in addition to other amounts accrued on or before the date of termination. If any such termination occurs within six months prior to or two years after a “change of control” (as defined in the applicable employment agreement), then the Executive would instead receive an amount equal to his or her respective annualized salary, plus an amount equal to his or her full cash bonus target for the year in which the termination occurred.
Director Compensation
The following table sets forth certain information regarding compensation of the persons who served as non-employee directors during the most recently completed fiscal year.
|
Name |
Fees Earned or |
Option Awards ($) |
Total |
|||||||||
|
Michael T. Cullen (a) |
72,500 | 3,631 | 76,131 | |||||||||
|
Arthur J. Fratamico (b) |
49,000 | 3,631 | 52,631 | |||||||||
|
Jeffrey S. Mathiesen (c) |
81,500 | 3,631 | 85,131 | |||||||||
|
D. Robert Schemel (d) |
57,500 | 3,631 | 61,131 | |||||||||
|
Daniel J. Donovan (e) |
52,500 | 3,631 | 56,131 | |||||||||
|
Jeffrey J. Jacob (f) |
40,000 | 3,631 | 43,631 | |||||||||
|
(a) |
Dr. Cullen held options to purchase an aggregate of 30 shares as of December 31, 2023. |
|
(b) |
Mr. Fratamico held options to purchase an aggregate of 14 shares as of December 31, 2023. |
|
(c) |
Mr. Mathiesen held options to purchase an aggregate of 14 shares as of December 31, 2023. |
|
(d) |
Mr. Schemel held options to purchase an aggregate of 14 shares as of December 31, 2023. |
|
(e) |
Mr. Donovan held options to purchase an aggregate of 14 shares as of December 31, 2023. |
|
(f) |
Mr. Jacob held options to purchase an aggregate of 24 shares as of December 31, 2023. |
During 2023, our Company reimbursed non-employee directors for out-of-pocket expenses incurred in connection with attending meetings of our Board of Directors and its committees.
In February 2022, the Compensation Committee approved an update to the cash compensation for non-employee directors. The revised annual amounts described below were effective January 1, 2022, and have not been changed for the year beginning January 1, 2024. These annual amounts will be paid out monthly.
|
Annual Retainer |
General |
Audit Committee |
Nominating & Governance Committee |
Compensation Committee |
||||||||||||
|
Nonemployee director |
40,000 |
|
‐ |
|
‐ |
|
‐ | |||||||||
|
Chairman |
32,500 | (a) | ||||||||||||||
|
Lead independent director |
|
22,500 | (a) |
|
‐ |
|
‐ |
|
‐ | |||||||
|
Committee chair |
|
‐ | 15,000 | 7,500 | 10,000 | |||||||||||
|
Committee member |
|
‐ | 7,500 | 4,000 | 5,000 | |||||||||||
|
(a) |
Paid in addition to nonemployee director retainer. |
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
Equity Compensation Plan Information
The following table presents the number of shares of common stock authorized for issuance under the Company’s equity compensation plans as of December 31, 2023:
|
Plan Category |
Number of Securities to Be Issued Upon Exercise of Outstanding Options, Warrants and Rights |
Weighted-Average Exercise Price of Outstanding Options, Warrants and Rights |
Number of Securities Remaining Available for Future Issuance under Equity Compensation Plans |
|||||||||
|
Equity compensation plans approved by security holders |
607 | (a) | $ | 14,410.38 | – | (b) | ||||||
|
Equity compensation plans not approved by security holders |
– | – | – | |||||||||
|
Total |
607 | - | ||||||||||
|
(a) |
Includes 561 shares underlying common stock options under the 2016 Plan, 5 shares underlying common stock options under the 2011 plan and 41 shares underlying the CPP option plan. We ceased issuing awards under the 2011 Plan upon stockholder approval of the 2016 plan in 2016. |
| (b) |
The 2016 Plan provides that the number of shares of common stock available for issuance under the plan will increase on January 1 of each year beginning in 2021 and ending on January 1, 2025 in an amount equal to the lesser of (i) the number of shares necessary to increase the total option pool to 20% of the total number of fully diluted shares (as defined in the Amended 2016 Plan) as of December 31 of the immediately preceding calendar year and (ii) such lesser number of shares as may be determined by the Board of Directors or its Compensation Committee prior to January 1st of any calendar year. |
Security Ownership of Certain Beneficial Owners and Management
The following table sets forth certain information with respect to the beneficial ownership of our outstanding common stock as of March 22, 2024 by (i) each of our named executive officers identified in the Summary Compensation Table below; (ii) each of our directors; (iii) all of our executive officers, directors and director nominees as a group; and (iv) each other beneficial owner of 5% or more of our outstanding common stock. Ownership percentages are based on 4,854,861 shares of common stock outstanding as of the close of business on the same date. Beneficial ownership is determined in accordance with the rules of the SEC. To our knowledge and subject to applicable community property laws, each of the holders of stock listed below has sole voting and investment power as to the stock owned unless otherwise noted. The table below includes the number of shares underlying rights to acquire common stock that are exercisable within 60 days from March 22, 2024. Except as otherwise noted below, the address for each director or officer listed in the table is c/o Panbela Therapeutics, Inc., 712 Vista Blvd #305, Waconia, Minnesota 55387.
|
Name |
Amount and Nature of Beneficial Ownership |
Percentage of Outstanding Shares |
|||||
|
Executive Officers and Directors |
|||||||
|
Jennifer K. Simpson |
201 |
(a) | * | ||||
|
Susan Horvath |
84 |
(b) | * | ||||
|
Michael T. Cullen |
63 |
(c) | * | ||||
|
Daniel Donovan |
25 |
(d) | * | ||||
|
Arthur J. Fratamico |
14 |
(e) | * | ||||
|
Jeffrey Jacob |
27 |
(f) | * | ||||
|
Jeffrey S. Mathiesen |
14 |
(g) | * | ||||
|
D. Robert Schemel |
48 |
(h) | * | ||||
|
All directors and current executive officers as a group (8 persons) |
476 |
(i) | * | ||||
|
(a) |
Includes 180 shares subject to stock options and 14 shares subject to warrants. |
|
(b) |
Includes 63 shares subject to stock options and 14 shares subject to warrants. |
|
* |
Less than 1% |
|
(c) |
Includes 14 shares held by the Cullen Living Trust. 30 shares subject to stock options and 11 shares subject to warrants. |
|
(d) |
Includes 14 shares subject to stock options. Also includes 3 shares held by Westport Boys, LLC (“Westport”) and 6 shares held by GDB Investments, LLP (“GDB”). Mr. Donovan is a managing member of Westport and a designated member of GDB. Mr. Donovan disclaims beneficial ownership of the securities owned by Westport and GDB except to the extent of his pecuniary interest therein. |
|
(e) |
Includes 14 shares subject to stock options. |
|
(f) |
Includes 24 shares subject to options and 2 shares subject to warrants. |
|
(g) |
Includes 14 shares subject to options. |
|
(h) |
Includes 19 shares held by spouse, 14 shares subject to stock options and 12 shares subject to warrants. |
|
(i) |
Includes 353 shares subject to stock options and 53 shares subject to warrants. |
|
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
Certain Relationships and Related Party Transactions
The following is a summary of transactions since January 1, 2022 to which our Company has been a party and in which the amount involved exceeded $86,000, which is approximately 1% of the average of our total assets as of the ends of our last two completed fiscal years, and in which any of our directors, executive officers, or beneficial owners of more than 10% of our capital stock had or will have a direct or indirect material interest, other than the compensation arrangements that are described under the heading “Executive Compensation: Employment Agreements” in Item 11.
On June 15, 2022, CPP entered into a Separation and Release Agreement (the “Separation Agreement”) with Mr. Jacob, now a director of Panbela, whereby he resigned as its Chief Executive Officer, employee, and all other capacities, immediately prior to the closing of our acquisition of CPP. In consideration for Mr. Jacob’s acknowledgements, representations, warranties, covenants, releases and agreements set forth in the Separation Agreement, CPP has agreed to pay to Mr. Jacob a total of $350,000, representing one times his base salary at the time of his resignation. Such payment will become due upon the earlier of (i) CPP or its parent completing a material financing and (ii) the two-year anniversary of the Closing Date, or June 15, 2024. As of December 31, 2023, the remaining installment of $25,000 remained payable to Mr. Jacob. This amount was paid on January 31, 2024. As further consideration, CPP has also agreed to reimburse Mr. Jacob for the employer’s portion of the premium payments for him to continue his current medical insurance coverage for 12 months through the Consolidated Omnibus Budget Reconciliation Act (COBRA).
Limitation of Liability of Directors and Officers and Indemnification
Our certificate of incorporation limits the liability of the directors to the fullest extent permitted by Delaware law.
Our bylaws provide that we will indemnify and advance expenses to the directors and officers to the fullest extent permitted by law or, if applicable, pursuant to indemnification agreements. They further provide that we may choose to indemnify other employees or agents of our Company from time to time. The Delaware General Corporation Law and the bylaws also permit us to secure insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in connection with their services to our Company, regardless of whether the bylaws permit indemnification. We maintain a directors’ and officers’ liability insurance policy.
At present there is no pending litigation or proceeding involving any of the current or former directors or officers as to which indemnification is required or permitted, and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the SEC this indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Related Person Transaction Approval Policy
Our Board of Directors has adopted a written policy regarding transactions with related persons, which we refer to as our related party transaction approval policy. Our related party transaction approval policy requires that any executive officer proposing to enter into a transaction with a “related party” generally must promptly disclose to our Audit Committee the proposed transaction and all material facts with respect thereto. In reviewing a transaction, our Audit Committee will consider all relevant facts and circumstances, including (1) the commercial reasonableness of the terms, (2) the benefit and perceived benefits, or lack thereof, to us, (3) the opportunity costs of alternate transactions and (4) the materiality and character of the related party’s interest, and the actual or apparent conflict of interest of the related party.
Our Audit Committee will not approve or ratify a related party transaction unless it determines that, upon consideration of all relevant information, the transaction is beneficial to our Company and stockholders, and the terms of the transaction are fair to our Company. No related party transaction will be consummated without the approval or ratification of our Audit Committee. It will be our policy that a director will recuse him- or herself from any vote relating to a proposed or actual related party transaction in which they have an interest. Under our related party transaction approval policy, a “related party” includes any of our directors, director nominees, executive officers, any beneficial owner of more than 5% of our common stock and any immediate family member of any of the foregoing. Related party transactions exempt from our policy include transactions available to all of our employees and stockholders on the same terms and transactions between us and the related party that, when aggregated with the amount of all other transactions between us and the related party or its affiliates, involved less than one percent of the average of our Company’s total assets at yearend for the last two completed fiscal years.
Director Independence
The continued listing rules of The Nasdaq Stock Market, LLC (the “Nasdaq Rules”) require that a majority of our Board of Directors be “independent directors” as that term is defined in the Nasdaq Rules. Our Board has determined that each of Messrs. Donovan, Fratamico, Mathiesen, and Schemel are “independent directors.”
|
Item 14. |
Principal Accounting Fees and Services |
Audit Fees
Cherry Bekaert LLP (PCAOB ID 00677) served as our independent registered public accounting firm for the years ended December 31, 2023, and 2022. The following table presents the aggregate fees for professional services provided by Cherry Bekaert LLP related to those fiscal years:
|
Year Ended |
||||||||
|
December 31, 2023 |
December 31, 2022 |
|||||||
|
Audit Fees (a) |
$ | 141,750 | $ | 124,800 | ||||
|
Audit-Related Fees (b) |
16,327 | 5,575 | ||||||
|
Total |
$ | 158,077 | $ | 130,375 | ||||
|
(a) |
Audit Fees consisted of fees for the audit of our annual consolidated financial statements, including audited consolidated financial statements presented in our annual report on Form 10-K, review of the consolidated financial statements presented in our quarterly reports on Form 10-Q and services that are normally provided by the independent registered public accountants in connection with statutory and regulatory filings or engagements for those fiscal years. This category also includes advice on audit and accounting matters that arose during, or as a result of, the audit or the review of interim financial statements and statutory audits required by non-U.S. jurisdiction. |
|
(b) |
Audit Related Fees consisted of fees for assurances and related services that are reasonably related to the performance of the audit of the consolidated financial statements and are not reported under “Audit Fees”. |
Pre-approval Policy
The Audit Committee has established a policy governing our use of the services of our independent registered public accountants. Under the policy, the Audit Committee is required to pre-approve all audit and permitted non-audit services performed by our independent registered public accountants in order to ensure that the provision of such services does not impair the public accountants’ independence. In 2023 and 2022, all fees identified above under the captions “Audit Fees” that were billed by Cherry Bekaert LLP were approved by the Audit Committee in accordance with SEC requirements.
PART IV
|
Item 15. |
Exhibits, Financial Statements Schedules |
|
(a) |
Financial Statements, Financial Statement Schedules, and Exhibits. |
|
(1) |
Financial Statements |
The following financial statements are filed as part of this report:
| Report of Independent Registered Public Accounting Firm (PCAOB ID 00 |
F-1 |
| Consolidated Balance Sheets | F-2 |
| Consolidated Statements of Operations and Comprehensive Loss | F-3 |
| Consolidated Statements of Stockholders’ Deficit | F-4 |
| Consolidated Statements of Cash Flows | F-5 |
| Notes to Consolidated Financial Statements | F-6 |
|
(2) |
Financial Statement Schedules |
Schedules not listed above have been omitted because they are not applicable or not required or the information required to be set forth therein is included in the Consolidated Financial Statements and notes thereto identified above.
|
(3) |
Exhibits |
|
Exhibit No. |
Description |
|
|
3.1 |
||
|
3.2 |
||
|
3.3 |
||
|
4.1 |
||
|
4.2 |
||
|
4.3 |
||
|
4.4 |
||
|
4.5 |
||
|
4.6 |
Form of Common Stock Purchase Warrant (included in Exhibit 4.5) |
|
Exhibit No. |
Description | |
|
4.7 |
||
|
4.8 |
||
|
4.9 |
||
|
4.9 |
||
|
4.10 |
||
|
4.12 |
||
|
4.13 |
||
|
4.14 |
||
|
4.15 |
||
|
4.16 |
||
|
4.17 |
||
|
4.18 |
||
|
4.19 |
||
|
10.1 |
||
|
10.2* |
||
|
10.3* |
||
|
10.4* |
||
|
10.5* |
||
|
10.6* |
||
|
10.7** |
|
Exhibit No. |
Description | |
|
10.8 |
||
|
10.9 |
||
|
10.10* |
||
|
10.11* |
||
|
10.12 |
||
|
10.13 |
||
|
10.14* |
||
|
10.15* |
||
|
10.16 |
||
|
10.17 |
||
|
10.18 |
||
|
10.19 |
||
|
10.20 |
||
|
10.21 |
||
|
10.22 |
||
|
10.23 |
||
|
10.24 |
||
|
10.25 |
||
|
10.26 |
||
|
10.27 |
|
Exhibit No. |
Description | |
|
10.28 |
||
|
21.1 |
||
|
23.1† |
||
|
24.1† |
||
|
31.1† |
||
|
31.2† |
||
|
32.1 |
||
|
32.2‡ |
||
|
97† |
||
|
101† |
Financial statements from the annual report on Form 10-K of the Company for the year ended December 31, 2023, formatted in inline XBRL: (i) the Consolidated Balance Sheets, (ii) Consolidated Statements of Comprehensive Loss, (iii) the Consolidated Statements of Stockholders’ Equity, (iv) the Consolidated Statements of Cash Flows, and (v) the Notes to Consolidated Financial Statements |
|
|
104† |
Cover Page Data File (formatted as inline XBRL and contained in Exhibit 101) |
|
* |
Management compensatory plan or arrangement required to be filed as an exhibit to this report. |
|
† |
Filed herewith |
|
‡ |
Furnished herewith |
|
** |
Portions of exhibit omitted pursuant to order granting confidential treatment issued by the Securities and Exchange Commission. |
|
Item 16. |
Form 10-K Summary |
None.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized on March 26, 2024.
|
PANBELA THERAPEUTICS, INC. |
||
|
By: |
/s/ JENNIFER K. SIMPSON |
|
|
Jennifer K. Simpson |
||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities indicated on March 26, 2024.
|
/s/ JENNIFER K. SIMPSON |
/s/ SUSAN HORVATH |
|
|
Jennifer K. Simpson, |
Susan Horvath, |
|
|
* |
* |
|
|
Michael T. Cullen, Chairman and Director |
Arthur J. Fratamico, Director |
|
|
* |
* |
|
|
Daniel J. Donovan, Director |
Jeffrey S. Mathiesen, Director |
|
|
* |
* |
|
|
Jeffrey E. Jacob, Director |
D. Robert Schemel, Director |
|
* |
Jennifer K. Simpson, by signing her name hereto, does hereby sign this document on behalf of each of the above-named directors of the registrant pursuant to powers of attorney duly executed by such persons. |
|
By: |
/s/ JENNIFER K. SIMPSON |
|
|
Jennifer K. Simpson, |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders
Panbela Therapeutics, Inc.
Waconia, Minnesota
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Panbela Therapeutics, Inc. and Subsidiaries (the “Company”) as of December 31, 2023 and 2022, and the related consolidated statements of operations and comprehensive loss, stockholders’ deficit, and cash flows for each of the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the years then ended in conformity with accounting principles generally accepted in the United States of America.
Substantial Doubt about the Company's Ability to Continue as a Going Concern
The accompanying financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has recurring losses and negative cash flows from operations that raise substantial doubt about its ability to continue as a going concern. Management’s evaluations of the events and conditions and management’s plans regarding those matters are described in Note 3. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter – Equity-linked transactions
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the Company’s Audit Committee and that: (i) relates to accounts or disclosures that are material to the financial statements and (ii) involved especially challenging, subjective, or complex judgements. The communication of critical audit matters does not alter, in any way, our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Critical Audit Matter Description
As disclosed in Notes 4, 9, and 10 to the consolidated financial statements, the Company has various equity-linked transactions, including stock-based compensation and warrant inducements, which require management to make fair value estimates. These fair value estimates include the use of the Black-Scholes option pricing model and require management to make several assumptions regarding the underlying inputs.
Auditing the Company’s accounting for equity-linked transactions required auditor judgment due to the subjectivity of significant assumptions used in the option pricing models and the accounting for the transactions.
How the Critical Audit Matter Was Addressed in the Audit
To test the Company’s accounting for the Transaction, we performed the following audit procedures:
|
● |
We obtained an understanding over management’s estimation process, including the process of estimating the fair value of these equity-linked instruments. |
|
● |
We obtained a listing of all equity transactions, including stock-based compensation and warrant inducements, and management’s accounting analysis supporting these transactions. We evaluated the conclusions reached to ensure these were recorded in accordance with the relevant accounting guidance. |
|
● |
We assessed the accuracy and completeness of the equity-linked transactions during the year by reading the relevant Board of Directors minutes and agreements. |
|
● |
We evaluated the significant assumptions used by management to calculate the fair value of equity transactions. Such evaluation included independent calculation of the critical inputs into the Black-Scholes option pricing model. |
|
● |
We evaluated the disclosures surrounding equity-linked transactions, including stock-based compensation and warrant inducements, to ensure these were disclosed in accordance with the relevant accounting guidance. |
/s/
We have served as the Company’s auditors since 2014.
March 26, 2024
Panbela Therapeutics, Inc.
Consolidated Balance Sheets
(In thousands, except share amounts)
|
December 31, 2023 |
December 31, 2022 |
|||||||
|
ASSETS |
||||||||
|
Current assets: |
||||||||
|
Cash and cash equivalents |
$ | $ | ||||||
|
Prepaid expenses and other current assets |
||||||||
|
Income tax receivable |
||||||||
|
Total current assets |
||||||||
|
Other noncurrent assets |
||||||||
|
Total assets |
$ | $ | ||||||
| LIABILITIES AND STOCKHOLDERS' DEFICIT | ||||||||
|
Current liabilities: |
||||||||
|
Accounts payable |
$ | $ | ||||||
|
Accrued expenses |
||||||||
|
Accrued interest payable |
||||||||
|
Note payable |
||||||||
|
Debt, current portion |
||||||||
|
Total current liabilities |
||||||||
|
Debt, net of current portion |
||||||||
|
Total non current liabilities |
||||||||
|
Total liabilities |
||||||||
| Stockholders' deficit: | ||||||||
|
Preferred stock, $ |
||||||||
|
Common stock, $ |
||||||||
|
Treasury Stock at cost; |
( |
) | ||||||
|
Additional paid-in capital |
||||||||
|
Accumulated deficit |
( |
) | ( |
) | ||||
|
Accumulated comprehensive income |
||||||||
|
Total stockholders' deficit |
( |
) | ( |
) | ||||
|
Total liabilities and stockholders' deficit |
$ | $ | ||||||
Share and per share data have been adjusted for all periods presented to reflect the one-for-forty reverse stock split effective January 13, 2023, the one-for-thirty reverse stock split effective June 1, 2023, and the one-for-twenty reverse stock split effective January 18, 2024.
The accompanying notes to the consolidated financial statements are an integral part of these statements.
Panbela Therapeutics, Inc.
Consolidated Statements of Operations and Comprehensive Loss
(In thousands, except share and per share amounts)
|
Year Ended December 31, |
||||||||
|
2023 |
2022 |
|||||||
|
Operating expenses: |
||||||||
|
General and administrative |
$ | $ | ||||||
|
Research and development |
||||||||
|
Operating loss |
( |
) | ( |
) | ||||
|
Other income (expense): |
||||||||
|
Interest income |
||||||||
|
Gain on sale of intellectual property |
||||||||
|
Interest expense |
( |
) | ( |
) | ||||
|
Other expense |
( |
) | ( |
) | ||||
|
Total other income (expense) |
( |
) | ||||||
|
Loss before income tax benefit |
( |
) | ( |
) | ||||
|
Income tax benefit |
||||||||
|
Net loss |
( |
) | ( |
) | ||||
|
Foreign currency translation adjustment |
( |
) | ||||||
|
Comprehensive loss |
$ | ( |
) | $ | ( |
) | ||
|
Basic and diluted net loss per share |
$ | ( |
) | $ | ( |
) | ||
|
Weighted average shares outstanding - basic and diluted |
||||||||
Share and per share data have been adjusted for all periods presented to reflect the one-for-forty reverse stock split effective January 13, 2023, the one-for-thirty reverse stock split effective June 1, 2023, and the one-for-twenty reverse stock split effective January 18, 2024.
The accompanying notes to the consolidated financial statements are an integral part of these statements.
Panbela Therapeutics, Inc.
Consolidated Statements of Stockholders’ (Deficit)
(In thousands, except share amounts)
|
For the Years ended December 31, 2023 and 2022 |
||||||||||||||||||||||||||||||||
|
Common Stock |
Treasury Stock |
Additional Paid in |
Accumulated |
Accumulated Comprehensive |
Total Stockholders' |
|||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital |
Deficit |
Income |
Deficit |
|||||||||||||||||||||||||
|
Balance at January 1, 2022 |
$ | $ | $ | $ | ( |
) | $ | $ | ||||||||||||||||||||||||
|
Issuance of common stock, options and warrants - CPP acquistion |
||||||||||||||||||||||||||||||||
|
Public offering - issuance of common stock and warrants |
||||||||||||||||||||||||||||||||
|
Sale of common stock |
||||||||||||||||||||||||||||||||
|
Exercise of warrants for cash |
||||||||||||||||||||||||||||||||
|
Stock-based compensation |
- | - | ||||||||||||||||||||||||||||||
|
Net Loss |
- | - | ( |
) | ( |
) | ||||||||||||||||||||||||||
|
Adjustment for fractional shares not issued, stock split |
||||||||||||||||||||||||||||||||
|
Foreign Currency translation adjustment |
- | - | ||||||||||||||||||||||||||||||
|
Balance at December 31, 2022 |
( |
) | ( |
) | ||||||||||||||||||||||||||||
|
Public offering - issuance of common stock and warrants |
||||||||||||||||||||||||||||||||
|
Sale of common stock |
||||||||||||||||||||||||||||||||
|
Exercise of warrants, cashless |
||||||||||||||||||||||||||||||||
|
Exercise of warrants for cash |
||||||||||||||||||||||||||||||||
|
Treasury stock |
( |
) | ( |
) | ||||||||||||||||||||||||||||
|
Stock-based compensation |
- | - | ||||||||||||||||||||||||||||||
|
Net Loss |
- | - | ( |
) | ( |
) | ||||||||||||||||||||||||||
|
Adjustment for the value offered to induce warrant exercise |
- | - | ( |
) | ||||||||||||||||||||||||||||
|
Adjustment for fractional shares not issued, stock split |
( |
) | ( |
) | ( |
) | ||||||||||||||||||||||||||
|
Foreign Currency translation adjustment |
- | - | ( |
) | ( |
) | ||||||||||||||||||||||||||
|
Balance at December 31, 2023 |
$ | $ | ( |
) | $ | $ | ( |
) | $ | $ | ( |
) | ||||||||||||||||||||
Share and per share data have been adjusted for all periods presented to reflect the one-for-forty reverse stock split effective January 13, 2023, the one-for-thirty reverse stock split effective June 1, 2023, and the one-for-twenty reverse stock split effective January 18, 2024.
The accompanying notes to the consolidated financial statements are an integral part of these statements.
Panbela Therapeutics, Inc.
Consolidated Statements of Cash Flows
(In thousands)
|
Year Ended December 31, |
||||||||
|
2023 |
2022 |
|||||||
|
Cash flows from operating activities: |
||||||||
|
Net loss |
$ | ( |
) | $ | ( |
) | ||
|
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
|
Write off of in process research and development (IPR&D) |
||||||||
| Gain on sale of intellectual property | ( |
) | ||||||
|
Stock-based compensation |
||||||||
|
Non-cash interest expense |
||||||||
|
Changes in operating assets and liabilities: |
||||||||
|
Income tax receivable |
( |
) | ||||||
|
Prepaid expenses and other current assets |
( |
) | ||||||
|
Deposits held for clinical trial costs |
( |
) | ( |
) | ||||
|
Accounts payable |
||||||||
|
Accrued liabilities |
( |
) | ||||||
|
Net cash used in operating activities |
( |
) | ( |
) | ||||
|
Cash flows from investing activities: |
||||||||
| Proceeds from sale of intellectual property | ||||||||
|
Investment in IPR&D |
( |
) | ||||||
|
Cash acquired in merger |
||||||||
|
Net cash used in investing activities |
( |
) | ||||||
|
Cash flows from financing activities: |
||||||||
|
Proceeds from sale of common stock, net of fees and offering costs, $ |
||||||||
|
Proceeds from public offering of common stock and warrants net of underwriters discount and offering costs of $ |
||||||||
|
Proceeds from induced exercise of warrants, net of costs of $ |
||||||||
|
Proceeds from voluntary exercise of warrants |
||||||||
|
Payments made on notes payable |
( |
) | ||||||
|
Cash paid for fractional shares |
( |
) | ||||||
|
Net cash provided by financing activities |
||||||||
|
Effect of exchange rate changes on cash |
( |
) | ||||||
| Net change in cash | ( |
) | ||||||
|
Cash and cash equivalents at beginning of year |
||||||||
| Cash and cash equivalents at end of year | $ | $ | ||||||
|
Supplemental disclosure of cash flow information: |
||||||||
|
Cash paid during period for interest |
$ | $ | ||||||
|
Supplemental disclosure of non-cash transactions: |
||||||||
|
Fair value of common stock, stock options and stock warrants issued as consideration for asset acquisition |
$ | $ | ||||||
|
Adjustment for the value offered to induce warrant exercise |
$ | $ | ||||||
The accompanying notes to the consolidated financial statements are an integral part of these statements.
Panbela Therapeutics, Inc.
Notes to Consolidated Financial Statements
|
1. |
Business |
Panbela Therapeutics, Inc. (“Panbela”) and its direct wholly owned subsidiaries: Cancer Prevention Pharmaceuticals, Inc. (“CPP”) and Panbela Research, Inc. (“Panbela Research”) and Cancer Prevention Pharmaceuticals, LTD (Ireland) and their respective subsidiaries, all of which are wholly owned, exist for the primary purpose of developing disruptive therapeutics for the treatment of patients with urgent unmet medical needs. Panbela Therapeutics Pty Ltd is a wholly owned subsidiary of Panbela Research organized under the laws of Australia. CPP has two wholly owned dormant subsidiaries: Cancer Prevention Pharma Limited, a United Kingdom entity, and Cancer Prevention Pharmaceuticals, LLC, an Arizona limited liability company. Panbela, together with its direct and indirect subsidiaries is referred to as “we,” “us,” “our,” and the “Company.”
The primary objective of our pipeline is the utilization of pharmacotherapies to reduce or normalize increased disease-associated polyamines using complementary pharmacotherapies. Our lead candidates are ivospemin (SBP-101) for which we have exclusively licensed the worldwide rights to from the University of Florida Research Foundation, Inc. and Flynpovi™ a combination of eflornithine (CPP-1X) and sulindac. We have exclusively licensed rights from the Arizona Board of Regents of the University of Arizona to commercialize Flynpovi.
Acquisition of CPP
On June 15, 2022, we completed the previously announced strategic business reorganization and acquisition of CPP pursuant to the agreement and plan of merger, dated as of February 21, 2022 (the “Merger Agreement”), by and among Panbela, CPP, Panbela Research, Canary Merger Subsidiary I, Inc. (“Merger Sub I”), and Canary Merger Subsidiary II, Inc. (“Merger Sub II”). Pursuant to the terms of the Merger Agreement, (i) Merger Sub I, then a wholly owned subsidiary of Panbela, which was itself a wholly owned subsidiary of Panbela Research, merged with and into Panbela Research (the “First Merger”), with Panbela Research surviving the First Merger, and (ii) Merger Sub II, then a wholly owned subsidiary of Panbela, merged with and into CPP (the “Second Merger” and, together with the First Merger, the “Mergers”), with CPP surviving the Second Merger. As a result of the Mergers, each of Panbela Research and CPP became a wholly owned subsidiary of Panbela. In addition, in connection with the consummation of the Mergers, then “Panbela Therapeutics, Inc.” was renamed to “Panbela Research, Inc.” and then “Canary Merger Holdings, Inc.” was renamed to “Panbela Therapeutics, Inc.” See Note 6, “Asset Acquisition,” for additional information.
Reverse stock splits
Effective January 13, 2023, June 1, 2023, and January 18, 2024, the Company's Board of Directors approved one-for-, one-for-thirty, and one-for-twenty reverse stock splits of its common stock, respectively. The par value and authorized shares of the Company's common stock were not affected by the reverse stock splits. Unless specifically provided otherwise herein, all share and per share amounts of our common stock presented have been retroactively adjusted to reflect all reverse stock splits. See Note 9 for more information.
|
2. |
Risks and Uncertainties |
The Company operates in a highly regulated and competitive environment. The development, manufacturing and marketing of pharmaceutical products require approval from, and are subject to ongoing oversight by, the Food and Drug Administration in the United States, the Therapeutic Goods Administration in Australia, the European Medicines Agency in the European Union, and comparable agencies in other countries. Obtaining approval for a new pharmaceutical product is never certain, may take many years, and is normally expected to involve substantial expenditure.
We have incurred losses of $
The accompanying Consolidated Financial Statements have been prepared assuming that we will continue as a going concern which contemplates the realization of assets and liquidation of liabilities in the normal course of business and do not include any adjustments relating to the recoverability or classification of assets or the amounts of liabilities that might result from the outcome of these uncertainties. Our ability to continue as a going concern, realize the carrying value of our assets and discharge our liabilities in the ordinary course of business is dependent upon a number of factors, including our ability to obtain additional financing, the success of our development efforts, our ability to obtain marketing approval for our ivospemin (SBP-101), eflornithine (CPP-1X) and eflornithine sachets (CPP-1X-S) product candidates in the United States, Australia, the European Union or other markets, and Flynpovi outside of North America and ultimately our ability to market and sell our product candidates. These factors, among others, raise substantial doubt about our ability to continue operations as a going concern. See Note 3 titled “Liquidity and Management Plans”.
|
3. |
Liquidity and Management Plans |
We will need to seek additional sources of funds to support our current business plans. We may seek to raise additional funds through various sources, such as equity and debt financing, or through strategic collaborations and license agreements. We can give no assurances that we will be able to secure additional sources of funds to support our operations, or if such funds are available to us, that such additional financing will be sufficient to meet our needs or on terms acceptable to us. This risk would increase if our clinical data were not positive or economic and market conditions deteriorate.
If we are unable to obtain additional financing when needed, we would need to scale back our operations taking actions that may include, among other things, reducing use of outside professional service providers, reducing staff or staff compensation, significantly modify or delay the development of our product candidates, license to third parties the rights to commercialize our product candidates as therapies in cancer or auto-immune diseases or other applications that we would otherwise seek to pursue, or cease operations.
Subsequent to December 31, 2023, the Company completed a registered public offering of common stock and warrants to purchase shares of common stock for gross proceeds of approximately $
During the year ended December 31, 2023 the Company completed two registered offerings of common stock and warrants to purchase shares of common stock. For June 21, 2023 offering gross proceeds were approximately $
The Company provided inducement warrants to certain shareholders to exercise their warrants. On November 2, 2023 gross proceeds were approximately $
During 2023 and 2022 the company also sold shares of common stock via an At the Market (ATM) facility. During the year ended December 31, 2023 net proceeds were approximately $
On October 4, 2022, the Company completed a registered public offering offerings of common stock and warrants to purchase shares of common stock for gross proceeds of approximately $
Much of the development efforts under way regarding the asset acquired in the CPP acquisition are funded by a licensing partner, see Note 8, “License Agreement for the Development and Commercialization of Flynpovi”, or collaborations with outside organizations including National Cancer Institute (“NCI”) and the Juvenile Diabetes Research Foundation (“JDRF”).
Based on the proceeds from the January 2024 offering it is expected that our cash will last into the second quarter of 2024.
Our future success is dependent upon our ability to obtain additional financing, the success of our development efforts, our ability to obtain marketing approval for ivospemin (SBP-101), Flynpovi, eflornithine (CPP-1X) and eflornithine sachets (CPP-1X-S) in the United States or other markets and ultimately our ability to market and sell our product candidates. If we are unable to obtain additional financing when needed, if our clinical trials are not successful or if we are unable to obtain marketing approval, we would not be able to continue as a going concern and would be forced to cease operations and liquidate our company.
There can be no assurances that we will be able to obtain additional financing on commercially reasonable terms, or at all. The sale of additional equity securities or convertible debt would likely result in dilution to our current stockholders.
|
4. |
Summary of Significant Accounting Policies |
Basis of presentation
We have prepared the accompanying Consolidated Financial Statements in conformity with accounting principles generally accepted in the United States of America (“GAAP”). Our fiscal year ends on December 31.
Principles of consolidation
The accompanying Consolidated Financial Statements include the assets, liabilities and expenses of Panbela Therapeutics, Inc. and our direct and indirect subsidiaries. All significant intercompany transactions and balances have been eliminated in consolidation.
Use of estimates
The preparation of Consolidated Financial Statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the Consolidated Financial Statements and the reported amount of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Business Combinations and Asset Acquisition
We account for acquired businesses using the acquisition method of accounting, which requires that the assets acquired, and liabilities assumed be recorded at the date of acquisition at their respective fair values if the acquisition meets the definition of a business combination. If the acquisition does not meet the definition of a business combination, then it is accounted for as an asset acquisition and the purchase consideration is allocated to the acquired assets.
ASC 805, Business Combinations, provides a model for determining whether an acquisition represents a business combination. In order to be a business, the integrated set of activities of the acquired entity needs to have an input and a substantive process that together significantly contribute to the ability to create outputs. The acquired entity must also pass the “Screen Test” which involves determining whether the acquisition represents an in-substance asset acquisition based on whether the fair value of the gross assets acquired is “substantially all” concentrated in a single asset or group of similar assets. This evaluation excludes certain acquired assets such as cash, deferred taxes, and goodwill associated with deferred taxes, but includes all other gross assets, including any consideration transferred in excess of the identified assets.
Concentration of credit risk
Financial instruments that potentially subject the Company to significant concentrations of credit risk consist primarily of cash. Cash is deposited in demand accounts at commercial banks. At times, such deposits may be in excess of insured limits. As of December 31, 2023, $
Cash and cash equivalents
Cash equivalents include short-term, highly liquid investments with maturities of three months or less.
Other noncurrent assets
Other noncurrent assets are comprised primarily of long-term deposits with contract research organizations (“CROs”). These amounts are recognized as operating expenses or research and development expense as the trial is completed.
Research and development costs
Research and development costs include expenses incurred in the conduct of our human clinical trials, for third-party service providers performing various testing and accumulating data related to our preclinical studies; sponsored research agreements; developing and scaling the manufacturing process necessary to produce sufficient amounts of the SBP-101, Flynpovi and CPP-1X compounds for use in our pre-clinical studies and human clinical trials; consulting resources with specialized expertise related to execution of our development plan for our product candidates; personnel costs, including salaries, benefits and share-based compensation; and costs to license and maintain our licensed intellectual property.
We charge research and development costs, including clinical trial costs, to expense when incurred. Our human clinical trials are performed at clinical trial sites and are administered jointly by us with assistance from CROs. Costs of setting up clinical trial sites are accrued upon execution of the study agreement. Expenses related to the performance of clinical trials generally are accrued based on contracted amounts and the achievement of agreed upon milestones, such as site openings, patient enrollment, patient follow-up, etc. We monitor levels of performance under each significant contract, including the extent of patient enrollment and other activities through communications with the clinical trial sites and CROs, and adjust the estimates, if required, on a quarterly basis so that clinical expenses reflect the actual effort expended at each clinical trial site and by each CRO.
Research and development costs also include IPR&D. This asset was acquired from the security holders of CPP and written off to research and development expense immediately subsequent to the asset acquisition.
We expense costs associated with obtaining licenses for patented technologies when it is determined there is no alternative future use of the intellectual property subject to the license.
Clinical Trial Accruals
Costs for preclinical studies and clinical trial activities are recognized based on an evaluation of the vendors’ progress towards completion of specific tasks, using data such as clinical site activations, patient enrollment or information provided to the Company by its vendors regarding their actual costs incurred. Payments for these activities are based on the terms of individual contracts and payment timing may differ significantly from the period in which the services are performed. The Company determines accrual estimates through reports from and discussions with applicable personnel and outside service providers as to the progress or state of completion, or the services completed. The Company’s estimates of accrued expenses as of each balance sheet date are based on the facts and circumstances known at the time.
Stock-based compensation
In accounting for stock-based incentive awards, we measure and recognize the cost of employee and non-employee services received in exchange for awards of equity instruments based on the grant date fair value of those awards. Compensation cost is recognized ratably using the straight-line attribution method over the vesting period, which is considered to be the requisite service period. We record forfeitures in the periods in which they occur. The compensation expense for performance-based stock option awards is recognized when “performance” has occurred or is probable of occurring.
The fair value of stock-based awards is estimated at the date of grant using the Black-Scholes option pricing model. The determination of the fair value of stock-based awards is affected by our stock price, as well as assumptions regarding a number of complex and subjective variables. Risk free interest rates are based upon U.S. Treasury rates appropriate for the expected term of each award. Expected volatility rates are based primarily on the volatility rates of the Company’s common stock. The assumed dividend yield is , as we do not expect to declare any dividends in the foreseeable future. The expected term of options granted is determined using the “simplified” method. Under this approach, the expected term is presumed to be the mid-point between the average vesting date and the end of the contractual term.
The fair value of restricted stock units is calculated as the fair value of the underlying common stock as of the date of grant.
Income taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the Consolidated Financial Statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted rates, for each of the jurisdictions in which the Company operates, expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in operations in the period that includes the enactment date. Valuation allowances are established when necessary to reduce deferred tax assets to the amount that is more likely than not to be realized. The Company has provided a full valuation allowance against the gross deferred tax assets as of December 31, 2023 and 2022. The Company’s policy is to classify interest and penalties related to income taxes as income tax expense in the Consolidated Statements of Operations and Comprehensive Loss.
Foreign currency translation
The functional currency of Panbela Therapeutics Pty Ltd is the Australian Dollar (“AUD”). Accordingly, assets and liabilities, and equity transactions of Panbela Therapeutics Pty Ltd are translated into U.S. dollars at period-end exchange rates. Expenses are translated at the average exchange rate in effect for the period. The resulting translation gains and losses are recorded as a component of accumulated comprehensive gain (loss) in the Consolidated Statements of Operations and Comprehensive Loss. During the years ended December 31, 2023 and 2022, any reclassification adjustments from accumulated other comprehensive gain to operations were inconsequential.
The Company records transactions denominated in foreign currencies at the exchange rate in effect on that date. Fluctuations between the transaction date and settlement date are recognized as transaction gain/loss.
Net loss per share
We compute net loss per share by dividing our net loss (the numerator) by the weighted-average number of common shares outstanding (the denominator) during the period. Shares issued during the period and shares reacquired during the period, if any, are weighted for the portion of the period that they were outstanding. The computation of diluted earnings per share, or EPS, is similar to the computation of basic EPS except that the denominator is increased to include the number of additional common shares that would have been outstanding if the dilutive potential common shares had been issued. Our diluted EPS is the same as basic EPS due to common equivalent shares being excluded from the calculation, as their effect is anti-dilutive.
For the year ended December 31, 2023 the numerator was adjusted by the cost of the warrant inducement of approximately $
The following outstanding potential common shares were not included in the diluted net loss per share calculations as their effects were not dilutive:
|
December 31, |
||||||||
|
2023 |
2022 |
|||||||
|
Employee and non-employee stock options |
||||||||
|
Common stock issuable under common stock purchase warrants |
||||||||
Reclassifications
Certain prior year amounts have been reclassified for consistency with the current year presentation. These reclassifications had no effect on the reported results of operations. Specifically, we reclassified $
Recently Issued Accounting Pronouncements
In November 2023, the FASB issued ASU 2023-07: Segment Reporting (Topic 280), Improvements to Reportable Segment Disclosures. This update will improve reportable segment disclosure requirements by enhancing disclosures around significant segment expenses and disclosures around the CODM, pertaining to what measures are used to evaluate profit or loss and such measures are used in assessing segment performance. These amendments will improve financial reporting by requiring disclosure of incremental segment information on an annual and interim basis for all public entities. ASU 2023-07 is effective for public entities' fiscal years beginning after December 15, 2023 and interim periods within fiscal years beginning after December 15, 2024. The Company only has one reportable segment and thus, the adoption of this ASU will not have a material impact on the financial statements and related disclosures.
Recently Adopted Accounting Pronouncements Not Yet Adopted
Fair Value Measurement of Equity Securities Subject to Contractual Sale Restrictions
In June 2022, the FASB issued ASU 2022-03, ASC Subtopic 820 “Fair Value Measurement of Equity Securities Subject to Contractual Sale Restrictions”. The FASB is issuing this Update (1) to clarify the guidance in Topic 820,
Fair Value Measurement, when measuring the fair value of an equity security subject to contractual restrictions that prohibit the sale of an equity security, (2) to amend a related illustrative example, and (3) to introduce new disclosure requirements for equity securities subject to contractual sale restrictions that are measured at fair value in accordance with Topic 820. For public business entities, the amendments in this Update are effective for fiscal years beginning after December 15, 2023, and interim periods within those fiscal years. Early adoption is permitted for both interim and annual financial statements that have not yet been issued or made available for issuance. The Company does not expect that the adoption of this guidance will have a material impact on its consolidated financial statements.
Improvements to Income Tax Disclosures
In December 2023, the FASB issued ASU No. 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures (ASU 2023-09), which improves the transparency of income tax disclosures by requiring consistent categories and greater disaggregation of information in the effective tax rate reconciliation and income taxes paid disaggregated by jurisdiction. It also includes certain other amendments to improve the effectiveness of income tax
disclosures. This guidance will be effective for the annual periods beginning the year ended December 31, 2025. Early adoption is permitted. Upon adoption, the guidance can be applied prospectively or retrospectively. The Company does not expect the adoption of this guidance to have a material impact on its consolidated financial statements.
|
5. |
Accrued Expenses |
Accrued expenses consisted of the following (in thousands):
|
December 31, |
||||||||
|
2023 |
2022 |
|||||||
|
Clinical trial and related expenses |
$ | $ | ||||||
|
Incentive compensation |
||||||||
|
Severance pay and other payroll related |
||||||||
|
Professional services |
||||||||
|
Other |
||||||||
|
Total accrued liabilities |
$ | $ | ||||||
|
6. |
Asset Acquisition |
On June 15, 2022, the Company completed the previously announced strategic business reorganization and acquisition of CPP through the Mergers. Under the terms of the Merger Agreement, the holders of CPP’s outstanding capital stock immediately prior to the Merger received shares of common stock of Panbela upon closing of the Merger. The stockholders of Panbela Research retained a majority of the outstanding shares of Panbela, the post-merger holding company. CPP stockholders will be eligible to receive contingent payments totaling a maximum of $
We performed the “screen test,” to determine if substantially all of the fair value of the gross assets acquired in the Mergers is concentrated in a single identifiable asset or group of similar identifiable assets. CPP’s lead asset, eflornithine in three forms, including Flynpovi (eflornithine (CPP-1X) and sulindac), eflornithine (CPP-1X), and eflornithine sachets (CPP-1X-S), were identified as the single identifiable asset consisting of IPR&D. Accordingly, our acquisition of CPP has been recorded as an asset acquisition.
The contract consideration for the assets acquired includes certain contingent consideration which at the acquisition date is neither probable of occurring nor reasonably estimable. As such, the value of this contingent consideration has been excluded from the allocation of the purchase price below. Acquisition-related transaction costs incurred have been recorded as additional investment in IPR&D.
The following is a summary of the purchase consideration and the allocation of that purchase consideration in connection with the CPP asset acquisition:
|
Shares |
Value (in thousands) |
|||||||
|
Common stock issued to CPP shareholders |
$ | |||||||
|
Common stock underlying options continued |
||||||||
|
Common stock underlying warrants replaced |
||||||||
|
Total non cash consideration |
$ | |||||||
|
Transaction costs incurred |
$ | |||||||
|
Total Consideration |
$ | |||||||
|
In process research and development * |
$ | |||
|
Cash |
||||
|
Other current assets |
||||
|
Accounts payable and accrued expenses |
( |
) | ||
|
Accrued interest and notes payable |
( |
) | ||
| $ |
*
|
7. |
Notes Payable |
Sucampo Promissory Note
As of December 31, 2023 and December 31, 2022, CPP had a balance outstanding of approximately $
Tillotts Promissory Note
As of December 31, 2022, CPP had a balance outstanding of approximately $
|
8. |
Commitments and Contingencies |
License agreement with the University of Florida Research Foundation
On December 22, 2011, we entered into an exclusive license agreement with the University of Florida Research Foundation (“UFRF”). This license agreement was amended on December 12, 2016 (“First Amendment”) and again on October 3, 2019 (“Second Amendment”). The license agreement requires the Company to pay royalties to UFRF ranging from
The amended license agreement remains subject to customary and usual termination provisions. The Company must also pay an annual license maintenance fee of $
License Agreement with the University of Arizona
CPP is party to a license agreement with the Arizona Board of Regents of the University of Arizona (the “University”). Pursuant to an Inter-institutional Agreement, the Regents of the University of California on behalf of the University of California, Irvine, has agreed to license certain patents, provisional patents, clinical trial data and other intellectual property related to the chemoprevention of cancer, the prevention of polyps and other technologies to CPP. The University has the right to administer the joint patent rights held between the University and the University of California, Irvine. The license agreement gives CPP exclusive rights to commercialize products based on intellectual property. In exchange for the intellectual property, CPP paid the University certain fees and reimbursements of patent costs and granted the university a warrant to acquire shares of CPP. As a result of the Mergers, the warrant was replaced with a warrant to purchase
CPP also agreed to pay the University additional milestone payments totaling up to $
Sponsored Research Agreement with the Johns Hopkins University
On April 1, 2023, the Company entered a sponsored research agreement with Johns Hopkins University. The Company committed to a total of $
|
9. |
Stockholders’ Equity |
Warrant Exercise Inducements & Private Placement of Class D Warrants
On December 21, 2023, we entered into warrant exercise inducement offer letters with certain holders of existing Class C warrants to purchase our common stock, pursuant to which the holders agreed to exercise for cash their existing warrants to purchase
Warrant Exercise Inducements & Private Placement of Class C Warrants
On November 2, 2023, we entered into warrant exercise inducement offer letters with certain holders of existing Class A and Class B warrants to purchase our common stock, pursuant to which the holders agreed to exercise for cash their existing Class A and Class B warrants to purchase
Public offering of common stock and warrants June 2023
On June 21, 2023, the Company completed a registered public offering and issued an aggregate of
Of the remaining warrants, warrants to purchase
As of December 31, 2023, warrants to purchase
Public offering of common stock and warrants January 2023
On January 30, 2023, the Company completed a registered public offering and issued an aggregate of
All of the pre-funded warrants were exercised by February 3, 2023. The remaining warrants have an alternative cashless exercise provision pursuant to which the holder may provide notice and receive an aggregate number of shares equal to the product of (x) the aggregate number of shares of common stock that would be issuable upon a cash exercise and (y)
Public offering of common stock and warrants
On October 4, 2022, the Company completed a registered public offering and issued an aggregate of
At-the-Market Program
On July 19, 2022, Panbela Therapeutics, Inc. (the “Company”), entered into a Sales Agreement with Roth Capital Partners, LLC (the “Agent”) to sell shares of the Company’s common stock having an aggregate gross sales price of up to $
During the years ended December 31, 2023 and December 31, 2022, the Company sold
Reverse Stock Splits
On December 19, 2023, the Company held a special meeting of its stockholders at which the stockholders approved a proposal to effect an amendment to the Company's certificate of incorporation, as amended, to implement a reverse stock split at a ratio ranging from any whole number between one-for-eight (1:
The Company effected a reverse stock split of one-for-thirty (1:
The Company effected a reverse stock split of one-for-forty (1:
Shares reserved
|
Shares of common stock reserved for future issuance were as follows as of December 31, 2023: |
||||
|
Stock options outstanding |
||||
|
Shares available for grant under equity incentive plan |
||||
|
Common shares issuable under outstanding common stock purchase warrants |
||||
|
10. |
Stock-Based Compensation |
2016 Omnibus Incentive Plan
The 2016 Omnibus Incentive Plan, as last amended effective April 9, 2020 (the “2016 Plan”), has been approved by our Board of Directors and ratified by our stockholders. The 2016 Plan permits the granting of incentive and non-statutory stock options, restricted stock, stock appreciation rights, performance units, performance shares and other stock awards to eligible employees, directors and consultants. We grant options to purchase shares of common stock under the 2016 Plan with an exercise price no less than the fair market value of the underlying common stock as of the date of grant. Options granted under the 2016 Plan have a maximum term of years. As of December 31, 2023, options to purchase
2011 Stock Option Plan
Prior to approval of the 2016 Plan, stock-based awards were granted under the 2011 Stock Option Plan (the “2011 Plan”). In conjunction with stockholder approval of the 2016 Plan, the Board terminated the 2011 Plan, although awards outstanding under the 2011 Plan will remain outstanding in accordance with and pursuant to the terms thereof. Options granted under the 2011 Plan have a maximum term of years and generally vest over to years for employees. As of December 31, 2023, options to purchase
CPP’s 2010 Equity Incentive Plan
As a result of the Mergers, the Company has assumed all remaining rights and obligations with respect to CPP’s 2010 Equity Incentive Plan (the “CPP Plan”) through the issuance of replacement options. As of December 31, 2023, options to purchase
We recognize stock-based compensation based on the fair value of each award as estimated using the Black-Scholes option valuation model. Ultimately, the actual expense recognized over the vesting period will only be for those shares that actually vest.
A summary of option activity is as follows:
|
Shares Underlying Options |
Weighted Average Exercise Price Per Share |
Aggregate Intrinsic Value |
||||||||||
|
Balance at January 1, 2022 |
$ | $ | ||||||||||
|
Options continued from CPP Plan |
||||||||||||
|
Exercised |
||||||||||||
|
Cancelled |
||||||||||||
|
Forfeitures |
( |
) | ||||||||||
|
Balance at December 31, 2022 |
$ | $ | ||||||||||
|
Granted |
||||||||||||
|
Exercised |
||||||||||||
|
Cancelled |
||||||||||||
|
Forfeitures |
||||||||||||
|
Balance at December 31, 2023 |
$ | $ | ||||||||||
Stock-based compensation expense for each of the periods presented is as follows (in thousands):
|
Year ended December 31, |
||||||||
|
2023 |
2022 |
|||||||
|
General and administative |
$ | $ | ||||||
|
Research and development |
||||||||
|
Total stock based compensation |
$ | $ | ||||||
A summary of the status of our unvested shares during the two years ended and as of December 31, 2023 is as follows:
|
Shares Under Option |
Weighted Average Grant Date Fair Value |
|||||||
|
Unvested at January 1, 2022 |
$ | |||||||
|
Granted |
||||||||
|
Vested |
( |
) | ||||||
|
Forfeitures |
( |
) | ||||||
|
Unvested at December 31, 2022 |
$ | |||||||
|
Granted |
||||||||
|
Vested |
( |
) | ||||||
|
Forfeitures |
||||||||
|
Unvested at December 31, 2023 |
$ | |||||||
Information about stock options outstanding, vested and expected to vest as of December 31, 2023, is as follows:
| Outstanding, Vested and Expected to Vest |
Options Vested and Exercisable |
|||||||||||||||||||||||
|
Per Share Exercise Price |
Shares |
Weighted Average Remaining Contractual Life (Years) |
Weighted Average Exercise Price |
Options Exercisable |
Weighted Average Remaining Contractual Life (Years) |
|||||||||||||||||||
| $ |
$ | |||||||||||||||||||||||
| $ |
- | $ |
$ | |||||||||||||||||||||
| $ |
- | $ |
$ | |||||||||||||||||||||
| $ |
- | $ |
$ | |||||||||||||||||||||
| Totals | $ | |||||||||||||||||||||||
As of December 31, 2023, total compensation expense related to unvested employee stock options not yet recognized was $
Options to purchase
The following table reflects the key assumptions used for calculating fair market value for options granted during the year ended December 31, 2023:
|
2023 |
|||||
|
Common stock fair value |
$ |
||||
|
Risk-free interest rate |
|
to | |||
|
Expected dividend yield |
$ |
||||
|
Expected Option life (in years) |
|
to | |||
|
Expected stock price volatility |
|
- | |||
Non-employee stock-based compensation
We account for stock options granted to nonemployees in accordance with Accounting Standards Update (“ASU”) 2019-07, “Compensation – Stock Compensation (Topic 718). In connection with stock options granted to nonemployees, we recorded approximately $
|
11. |
Income Taxes |
We have incurred net operating losses since our inception. We have not reflected the benefit of net operating loss carryforwards in the accompanying financial statements and have established a full valuation allowance against our deferred tax assets.
On December 31, 2023 and 2022, the Company had an income tax receivable of approximately $
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes as well as operating losses and tax credit carryforwards.
The significant components of our deferred tax assets and liabilities are as follows (in thousands):
|
December 31, |
||||||||
|
Deferred tax assets (liabilities) |
2023 |
2022 |
||||||
|
Net operating loss carryforwards |
$ | $ | ||||||
|
Research credit carryforwards |
||||||||
|
Stock-based compensation |
||||||||
|
Section 174 amortization |
||||||||
|
Other |
||||||||
|
Deferred tax assets |
||||||||
|
Valuation allowance |
( |
) | ( |
) | ||||
|
Net deferred tax asset |
$ | $ | ||||||
Realization of the future tax benefits is dependent on our ability to generate sufficient taxable income within the carry-forward period. Because of our history of operating losses, management believes that the deferred tax assets arising from the above-mentioned future tax benefits are currently not likely to be realized and, accordingly, we have provided a full valuation allowance.
A reconciliation of the statutory tax rates and the effective tax rates is as follows:
|
Year Ended December 31, |
||||||||
|
2023 |
2022 |
|||||||
|
Statutory rate |
% | % | ||||||
|
Permanent differences |
( |
) | ||||||
|
Change in effective tax rate |
||||||||
|
States |
||||||||
|
Valuation allowance |
( |
) | ( |
) | ||||
|
Foreign research incentives |
||||||||
|
Other |
||||||||
|
Effective rate |
% | % | ||||||
| 2023 |
2022 |
|||||||
|
Tax Expense: |
||||||||
|
Current |
( |
) | ( |
) | ||||
|
Deferred |
||||||||
|
Noncurrent |
||||||||
| ( |
) | ( |
) | |||||
Net operating losses and tax credit carryforwards as of December 31, 2023, are as follows:
|
(In Thousands) |
Amount |
Expiration Years |
|||
|
Net operating losses--federal |
|
||||
|
2018 to 2022 net operating losses -- federal |
Never expires |
||||
|
2023 - federal |
Limited to 80% usage |
||||
|
Tax credits--federal |
|
||||
Utilization of the net operating loss carryforwards and credits may be subject to a substantial annual limitation due to the ownership change limitations provided by the IRC, and similar state provisions. We have not performed a detailed analysis to determine whether an ownership change under Section 382 of the IRC has occurred. The effect of an ownership change would be the imposition of an annual limitation on the use of net operating loss carryforwards attributable to periods before the change.
The Company is subject to taxation in the United States and Australia. Tax returns for the year ended December 31, 2018 and thereafter are subject to examinations by federal and state tax authorities. Tax returns of Panbela Therapeutics Pty Ltd for the year ended December 31, 2016 and thereafter are subject to examination by the Australian tax authorities.
|
12. |
Subsequent Events |
Issuance of common stock and warrants in January 2024
On January 31, 2024, the Company completed a registered public offering and issued an aggregate of