UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form
(Mark One)
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number:
(Exact name of registrant as specified in its charter)
|
||
(State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer Identification No.) |
(Address of principal executive offices, including zip code)
Registrant’s telephone number, including area code: (
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class |
Trading Symbol(s) |
Name of Each Exchange on Which Registered |
The |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15 of the Act. Yes ☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period than the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer |
☐ |
|
Accelerated filer |
☐ |
☒ |
|
Smaller reporting company |
||
Emerging growth company |
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes
The aggregate market value of the voting stock held by non-affiliates of the registrant was approximately $
There were
DOCUMENTS INCORPORATED BY REFERENCE
Part III incorporates information by reference from the definitive Proxy Statement for the 2022 annual meeting of stockholders, which is expected to be filed not later than 120 days after the Registrant’s fiscal year ended December 31, 2022.
DURECT CORPORATION
ANNUAL REPORT ON FORM 10-K
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2022
TABLE OF CONTENTS
|
|
|
|
Page |
|
|
|
|
|
ITEM 1. |
|
|
1 |
|
|
|
|
|
|
ITEM 1A. |
|
|
30 |
|
|
|
|
|
|
ITEM 1B. |
|
|
59 |
|
|
|
|
|
|
ITEM 2. |
|
|
59 |
|
|
|
|
|
|
ITEM 3. |
|
|
59 |
|
|
|
|
|
|
ITEM 4. |
|
|
59 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ITEM 5. |
|
|
60 |
|
|
|
|
|
|
ITEM 6. |
|
|
60 |
|
|
|
|
|
|
ITEM 7. |
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
|
61 |
|
|
|
|
|
ITEM 7A. |
|
|
76 |
|
|
|
|
|
|
ITEM 8. |
|
|
77 |
|
|
|
|
|
|
ITEM 9. |
|
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
|
109 |
|
|
|
|
|
ITEM 9A. |
|
|
109 |
|
|
|
|
|
|
ITEM 9B. |
|
|
110 |
|
|
|
|
|
|
ITEM 9C. |
|
Disclosure Regarding Foreign Jurisdiction that Prevent Inspections |
|
110 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ITEM 10. |
|
|
111 |
|
|
|
|
|
|
ITEM 11. |
|
|
111 |
|
|
|
|
|
|
ITEM 12. |
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
|
111 |
|
|
|
|
|
ITEM 13. |
|
Certain Relationships and Related Transactions, and Director Independence |
|
111 |
|
|
|
|
|
ITEM 14. |
|
|
111 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ITEM 15. |
|
|
112 |
|
|
|
|
|
|
|
113 |
|||
|
|
|
|
|
ITEM 16. |
|
|
117 |
|
|
|
|
|
|
|
118 |
|||
Item 1. Business.
Overview
We are a biopharmaceutical company advancing novel and potentially lifesaving investigational therapies derived from our Epigenetic Regulator Program. Larsucosterol (also known as “DUR‑928”), a new chemical entity in clinical development, is the lead candidate in our Epigenetic Regulator Program. An endogenous, orally bioavailable small molecule, larsucosterol has been shown in both in vitro and in vivo studies to play an important regulatory role in lipid metabolism, stress and inflammatory responses, and cell death and survival. We are developing larsucosterol for alcohol-associated hepatitis (also called “alcoholic hepatitis” or “AH”), a life-threatening acute liver condition with no approved therapeutics and a 28-Day and 90-Day historical mortality rate of 20%-26% and 29%-31%, respectively. After completing a Phase 2a trial in which 100% of AH patients treated with larsucosterol survived the 28-Day study period, we are now conducting a ~300-patient, double-blind, placebo-controlled Phase 2b clinical trial called "AHFIRM" (trial in AH to evaluate saFety and effIcacy of laRsucosterol treatMent). Through our AHFIRM trial, we are evaluating larsucosterol’s potential to reduce mortality or liver transplantation compared to a placebo with or without steroids at the investigators’ discretion. Currently, we anticipate dosing the last patient in the AHFIRM trial in the second quarter of 2023, which should enable top-line results to be reported in the second half of 2023. If the AHFIRM trial is successful, it may support a New Drug Application (“NDA”) filing and we may decide to develop our own commercial, sales and marketing organization. We have also investigated larsucosterol in patients with non-alcoholic steatohepatitis (“NASH”) with encouraging results in a Phase 1b clinical trial and are considering further development of larsucosterol for this and other indications.
In addition to our Epigenetic Regulator Program, we developed a novel and proprietary post-surgical pain product called POSIMIR that utilizes our innovative SABER platform technology to enable continuous sustained delivery of bupivacaine, a non-opioid local analgesic, over three days in adults. In February 2021, POSIMIR received U.S. FDA approval for post-surgical pain reduction for up to 72 hours following arthroscopic subacromial decompression. In December 2021, we entered into a license agreement (the “Innocoll Agreement”) with Innocoll Pharmaceuticals Limited (“Innocoll”), pursuant to which the Company granted to Innocoll an exclusive, royalty-bearing, sublicensable right and license to develop, manufacture and commercialize POSIMIR in the U.S. In September 2022, Innocoll launched POSIMIR in the U.S.
NOTE: POSIMIR® is a trademark of Innocoll Pharmaceuticals, Ltd. in the U.S. and a trademark of DURECT Corporation outside of the U.S. SABER®, CLOUD™, ORADUR™ and ALZET® are trademarks of DURECT Corporation. Other trademarks referred to belong to their respective owners. Full prescribing information for POSIMIR, including BOXED WARNING and Medication Guide can be found at www.posimir.com. Full prescribing information for PERSERIS, including BOXED WARNING and Medication Guide can be found at www.perseris.com.
As a result of the assignment of certain patent rights, we also receive single digit sales-based earn-out payments from U.S. net sales of Indivior UK Limited (“Indivior”)’s PERSERIS™(risperidone) drug for schizophrenia and single-digit royalties from net sales of Orient Pharma Co., Ltd. (“Orient Pharma”)’s Methydur Sustained Release Capsules ("Methydur") for the treatment of attention deficit hyperactivity disorder ("ADHD") in Taiwan. We also manufacture and sell ALZET® osmotic pumps used in laboratory research.
Epigenetic Regulator Program and New Chemical Entities
Epigenetic regulation influences the expression of genes through the silencing or initiation of gene activity without modifying the DNA sequence. For instance, methylation (the chemical binding of a methyl group) of cytosine nucleotides in promoter regions of DNA, facilitated by DNA methyltransferases (“DNMTs”), will generally result in downregulation of gene expression, while demethylation (removal of a methyl group) generally results in upregulation. DNA methylation/demethylation can thus regulate the expression of relevant genes, especially clusters of master genes that further modulate crucial cellular activities.
1
Our Epigenetic Regulator Program involves a multi-year collaborative effort with the Department of Internal Medicine at Virginia Commonwealth University (“VCU”), the VCU Medical Center and the McGuire VA Medical Center. The knowledge base supporting this program is a result of more than 30 years of lipid research by Shunlin Ren, M.D., Ph.D., Professor of Internal Medicine at the VCU Medical Center. The lead compound from this program, larsucosterol, is an endogenous sulfated oxysterol, which acts as an epigenetic regulator. Under a license with VCU, we hold the exclusive royalty-bearing worldwide right to develop and commercialize larsucosterol and related molecules discovered in the program.
In March 2021, a peer-reviewed research paper regarding the proposed mechanism of action of larsucosterol was published in The Journal of Lipid Research. The research showed that larsucosterol (referred to in the paper as “25HC3S”) bound to and inhibited the activities of DNMTs 1, 3a and 3b, enzymes that add methyl groups to DNA (a process called “DNA methylation”), as well as reduced DNA hypermethylation. DNMTs 1 and 3a have been shown to be over-expressed in the livers of patients with severe AH. As such, by inhibiting DNMTs 1 and 3a activities, larsucosterol may inhibit DNA hypermethylation, thereby modulating the expression of genes and pathways that are involved in crucial cellular activities, including those associated with cell death, stress response, and lipid biosynthesis. These modulations may lead to improved cell survival, reduced lipid accumulation or lipotoxicity, minimized inflammation, and enhanced liver regeneration, as has been observed in various in vivo animal models and in results from our completed clinical trials in AH and NASH patients.
The biological activity of larsucosterol has been demonstrated in over a dozen different animal disease models involving three animal species. Some of these models represent acute organ injuries (e.g., LPS-induced endotoxin shock, drug-induced acute oxidative stress injury, ischemic-reperfusion-induced kidney and brain injury), and some represent chronic metabolic disorders (e.g., NASH).
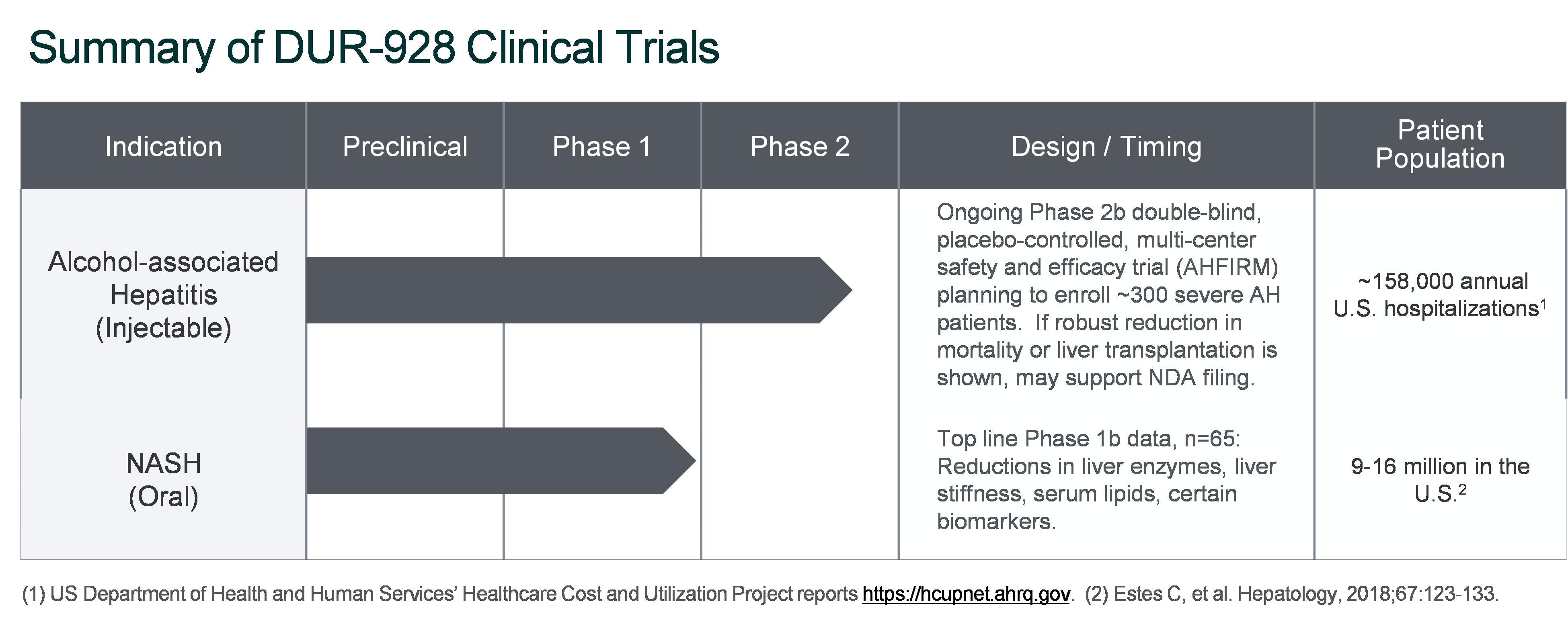
Our major product research and development efforts for larsucosterol are described in the following table:
In pharmacokinetic (“PK”) and toxicology studies conducted in mice, rats, rabbits, dogs, minipigs and monkeys, larsucosterol has been found to be well tolerated and safe by all routes of administration tested to date. These results support the use of larsucosterol in completed and ongoing human safety, PK, proof-of-concept, and efficacy trials. The chronic toxicity of larsucosterol was further assessed in a 6-month oral study in rats and in a 9-month oral study in dogs. These studies support the use of larsucosterol in long duration human trials.
2
Acute Organ Injury Program with Injectable Larsucosterol
Market Opportunity. AH, an acute form of alcohol-associated liver disease, is associated with long-term heavy intake of alcohol and often occurs after a recent period of increased alcohol consumption. AH is typically characterized by recent onset jaundice and hepatic failure. A Model of End-Stage Liver Disease (“MELD”) score is a commonly used scoring system to assess the severity and prognosis of AH patients. AH was associated with approximately 158,000 U.S. hospitalizations in 2020 according to the available data published for that year. A retrospective analysis of 77 studies published between 1971 and 2016, which included data from a total of 8,184 patients, showed the overall mortality from AH was 26% at 28 days, 29% at 90 days and 44% at 180 days. A subsequent global study published in December 2021, which included 85 tertiary centers in 11 countries across 3 continents, prospectively enrolled 2,581 AH patients with a median MELD score of 23.5, reported mortality at 28 and 90 days of approximately 20% and 31%, respectively.
There are no FDA approved therapies for AH and stopping alcohol consumption is necessary, but frequently not sufficient for recovery in many moderate and severe patients. Corticosteroids do not improve survival at 90 days or one year, and have demonstrated an increased risk of infection. In addition, fewer than 50% of AH patients are eligible for corticosteroids. According to a recent study, the healthcare costs associated with treating hospitalized AH patients and their length of hospital stay are significant.
Each hospitalization episode with AH diagnosis for patients who: |
Average length of stay |
Average total charges during hospital stay |
Died during the hospitalization |
9 days |
$147,000 |
Were discharged |
6 days |
$53,000 |
Marlowe, N., Lam, D., Krebs, W., Lin, W. & Liangpunsakul, S. (2022) Prevalence, co-morbidities, and in-hospital mortality of patients hospitalized with alcohol-associated hepatitis in the United States from 2015 to 2019. Alcoholism: Clinical and Experimental Research. |
||
The rate of AH patients undergoing liver transplantation has increased in recent years, although the total number of such transplants is still relatively small. Average charges for a liver transplant exceed $875,000, and patients require lifelong immunosuppressive therapy to prevent organ rejection.
Clinical Program. In 2019, we completed a Phase 2a clinical trial evaluating safety and PK of intravenously (“IV”) infused larsucosterol in patients with moderate and severe AH. Severity of AH was determined by MELD scores with moderate defined as MELD 11-20 and severe as MELD 21-30. This was an open label, dose escalation (30 mg, 90 mg and 150 mg), multi-center U.S. study, designed to be conducted in two sequential parts. Part A included patients with moderate AH and Part B included patients with severe AH.
In this Phase 2a trial, dose escalation was permitted following review of safety and PK results of the prior dose level by a Dose Escalation Committee. The target number of patients for the study was 4 per dose group. Final enrollment included 19 patients with moderate (7 of 19) and severe AH (12 of 19), who received IV larsucosterol at 30 mg, 90 mg, or 150 mg doses. Eight patients (four moderate and four severe) were dosed at 30mg, seven patients (three moderate and four severe) were dosed at 90mg and four patients (all severe) were dosed at 150mg. After being discharged on Day 2, one patient did not return for the scheduled Day 7 and Day 28 follow-up visits; therefore Lille, bilirubin and MELD data reported below are based on 18 patients. The objectives of this study included assessment of safety, PK and pharmacodynamic signals, including liver biochemistry, biomarkers and prognostic scores, including the Lille score, following larsucosterol treatment.
3
In November 2019, the results from this Phase 2a clinical trial of larsucosterol in AH were presented as a late-breaking oral presentation at The Liver Meeting®. The study summary results were also selected for inclusion in the ‘Best of The Liver Meeting’ presentation in the alcohol-related liver disease category.
All 19 patients treated with larsucosterol in this trial survived the 28-day follow-up period and there were no drug-related serious adverse events. Using an alternative measure of AH severity to MELD, Maddrey’s Discrimination Function (“DF”), 15 of the 19 patients had DF scores of 32 or greater, indicating that they had severe AH. Patients treated with larsucosterol had a statistically significant reduction from baseline in bilirubin at Day 7 and Day 28, and MELD at Day 28. Lille scores, described below, were also statistically significantly lower than those from a well-matched group of patients in a contemporary trial as well as several published historical controls. 74% of all larsucosterol treated patients and 67% of those with severe AH were discharged from the hospital within 4 days after receiving a single dose of larsucosterol.
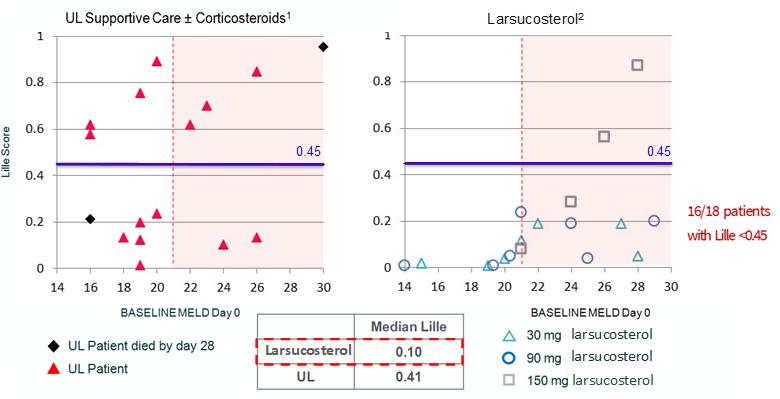
Lille
Lille scores are used in clinical practice to help determine the prognosis and response of AH patients after 7 days of treatment. The lower the Lille score, the better the prognosis. Patients with a Lille score below 0.45, treatment responders, have a six-month survival rate of 85% compared to those with Lille scores above 0.45, who have only a 25% six-month survival rate. The chart below shows the Lille scores for individual AH patients treated with larsucosterol plotted as a function of their baseline MELD scores. In our study, the median Lille score for patients treated with larsucosterol was 0.10. The median Lille score among a cohort of 16 patients treated with standard of care at the University of Louisville (“UL”) was 0.41 (shown as historical control).
4
As shown in the table below, all patients in the 30 mg and 90 mg larsucosterol dosing groups were treatment responders based on their Lille scores, regardless of disease severity by DF, MELD, or baseline serum bilirubin levels. 89% of the overall larsucosterol patient population (16 of 18) were treatment responders based on Lille. Patients with severe AH, as defined by DF ≥32 or MELD 21-30, and baseline serum bilirubin above 8 mg/dL, had similarly high response rates to larsucosterol treatment.
AH Patient Category |
|
n1 |
Responders (Lille<0.45) |
Lille Median (Quartile) |
All Patients2 30 mg or 90 mg larsucosterol 3 |
|
18 14 |
89% 100% |
0.10 (0.04, 0.20) 0.05 (0.04, 0.19) |
DF≥32 (SAH)2, 4 30 mg or 90 mg larsucosterol 3 |
|
15 11 |
87% 100% |
0.19 (0.05, 0.22) 0.12 (0.05, 0.19) |
MELD 21-302 30 mg or 90 mg larsucosterol 3 |
|
12 8 |
83% 100% |
0.19 (0.11, 0.25) 0.19 (0.10, 0.19) |
Baseline bilirubin >8mg/dl2 30 mg or 90 mg larsucosterol 3 |
|
11 8 |
82% 100% |
0.10 (0.05, 0.20) 0.10 (0.05, 0.19) |
The Lille scores of patients treated with larsucosterol in this trial were also significantly lower than several selected published historical studies (Hepatology 2007, 45:1348-1354; Gut 2011, 60:255-260), in which patients had similar baseline bilirubin, albumin, creatinine, prothrombin time and DF scores, and were treated with standard of care with or without corticosteroids. Due to the historical nature of these studies, such comparisons should be interpreted with caution.
5
A sub-group analysis was conducted to compare severe AH patients in the 30 mg and 90 mg dosing groups (n=8) with well-matched severe AH patients (n=13) who received corticosteroids for 28 days in a contemporaneous study at UL. Patients shown in the chart below in the UL steroid group had a mean baseline MELD of 24.46 and mean baseline Maddrey’s DF score of 62.98. The 8 patients in the larsucosterol group had baseline mean MELD of 24.50 and mean baseline Maddrey’s DF score of 61.25. All patients treated with larsucosterol survived the 28-Day follow up period, while 3 of the 13 patients (23%) in the UL steroid group died within the first 28 days.
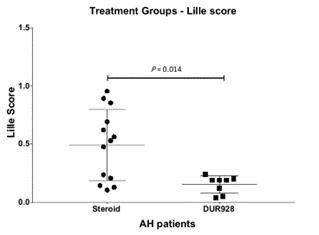
The steroid group in the above graph includes the 7 severe AH patients treated with steroids from the UL group shown in the MELD vs Lille graph above plus an additional 6 severe AH patients subsequently treated in the UL study.
Bilirubin
Bilirubin is formed by the breakdown of red blood cells in the body. The level of total bilirubin in the blood is an indication of how the liver is functioning. Elevated bilirubin levels usually signify liver dysfunction and disease. In this trial (shown in the chart below), patients treated with larsucosterol had a significant early reduction from baseline in bilirubin by Day 7. Patients with more elevated bilirubin at baseline (serum bilirubin >8 mg/dL) had a median reduction from baseline of 25% by Day 7 and 48% by Day 28.
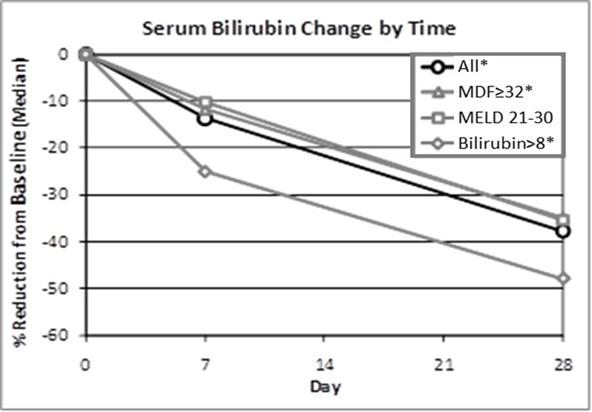
*p<0.05 compared to baseline (Wilcoxon's Signed Rank Test)
6
As discussed above, MELD is another commonly used scoring system used to assess the severity and prognosis of AH patients. Patients with MELD scores of 11-20 are classified as having moderate AH and patients with MELD scores of 21-30 are classified as having severe AH. As with Lille scores, the lower the MELD score, the better the prognosis for the AH patient. In this trial (shown in the chart below), the median reduction from baseline in MELD among all larsucosterol treated patients was >2 points and among those with baseline bilirubin levels >8 mg/dL was 5 points by Day 28.
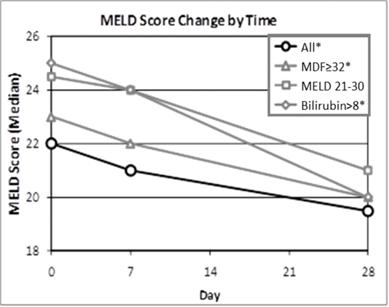
*p<0.05 compared to baseline (Wilcoxon's Signed Rank Test)
MELD is calculated based on (a) bilirubin, (b) serum creatinine (sCr), and (c) International Normalized Ratio ("INR"), which is a measure of prothrombin time.
Safety and Pharmacokinetics
In the Phase 2a study of larsucosterol in AH, larsucosterol was well tolerated at all doses tested. There were no drug-related serious adverse events and only three adverse events designated as possibly or probably related to larsucosterol: one occurrence of moderate generalized pruritus, one mild rash and one grade two alkaline phosphatase elevation. There were no discontinuations, early withdrawals or termination of study drug or study participation due to adverse events. All patients treated with larsucosterol survived through the 28-Day follow-up period. Drug exposures were dose proportional and were not affected by the severity of the disease.
In December 2020, we announced that the FDA had granted larsucosterol Fast Track Designation for the treatment of AH. The FDA grants Fast Track Designation to facilitate development and expedite the review of therapies with the potential to treat a serious condition where there is an unmet medical need. A therapeutic that receives Fast Track Designation may benefit from early and frequent communication with the agency in addition to a rolling submission of the marketing application, with the objective of getting important new therapies to patients more quickly.
In January 2021, we announced the dosing of the first patient in our Phase 2b AHFIRM study of patients with severe AH. AHFIRM is a randomized, double-blind, placebo-controlled, international, multi-center Phase 2b study to evaluate the safety and efficacy of larsucosterol in approximately 300 patients with severe AH. The study is comprised of three arms targeting approximately 100 patients each: (1) Placebo plus supportive care, with or without methylprednisolone capsules at the investigators’ discretion; (2) larsucosterol (30 mg); and (3) larsucosterol (90 mg). Patients in the larsucosterol arms receive the same supportive care without steroids. In order to maintain blinding, patients in the two active arms receive matching placebo capsules if the investigator prescribes steroids. Patients receive an IV dose of larsucosterol or placebo (sterile water) on Day 1 and a second identical IV dose on Day 4 if they are still hospitalized. The primary outcome measure will be the 90-Day incidence of mortality or liver
7
transplantation for patients treated with larsucosterol compared to those treated with placebo. Secondary endpoints include the difference in 90-Day mortality between patients treated with larsucosterol compared to those treated with placebo, the difference in 28-Day mortality or liver transplantation for patients treated with larsucosterol compared to those treated with placebo, and the difference in mortality between patients treated with larsucosterol compared to those treated with placebo. We now have over 60 clinical trial sites across the United States, U.K., E.U. and Australia. We currently expect to enroll the last patient in the AHFIRM trial in the second quarter of 2023, which should enable top-line results to be reported in the second half of 2023.
Phase 1 trials of larsucosterol administered through injection have supported the development of larsucosterol in AH. The initial Phase 1 trial in healthy subjects was a single-site, randomized, double-blinded, placebo-controlled, single-ascending-dose study that evaluated the safety, tolerability and PK of intramuscular ("IM") injected larsucosterol. The 24-subject study (16 healthy volunteers on larsucosterol and 8 on placebo) of four escalating dose levels resulted in dose proportional systemic exposure of larsucosterol. Larsucosterol was well-tolerated at all dose levels, with no serious treatment-related adverse events reported. We also conducted a multiple-dose study involving 10 healthy subjects, in which participants received IM-injected larsucosterol for five consecutive days (8 subjects on larsucosterol, 2 on placebo) using the next to highest dose from the single dose study. No serious treatment related adverse events were reported, no subjects withdrew from the study, no accumulation in plasma concentrations were observed with repeat dosing, and the pain scores and injection site reactions were minimal. We also conducted a single-ascending IV dose infusion study with 16 healthy subjects and observed no treatment-related serious adverse events. The systemic exposure following IV infusion was dose proportional.
A Phase 1 drug-drug interaction study conducted in healthy subjects demonstrated that neither orally administered nor IV injected larsucosterol at doses tested affected the safety and PK of midazolam, a drug metabolized by CYP3A4, which is one of the important enzymes associated with clinically relevant drug-drug interactions.
We also conducted a Phase 1b study with injected larsucosterol in patients with impaired kidney function (stage 3 and 4 chronic kidney disease ("CKD")) and matched control subjects ("MCS"), matched by age, body mass and gender with normal kidney function. This study was a single-site, open-label, single-ascending-dose study in two successive cohorts (first a low dose of 30 mg and then a high dose of 120 mg) evaluating safety and PK of IM injected larsucosterol. The low dose cohort consisted of 6 patients with CKD and 3 MCS; the high dose cohort consisted of 5 CKD patients and 3 MCS. In this trial, larsucosterol was well tolerated among all subjects and the PK parameters between the kidney function impaired patients and the MCS were comparable.
Chronic Liver Disease Program with Orally Administered Larsucosterol
Market Opportunity. NAFLD is the most common form of chronic liver disease in both children and adults. It is estimated that NAFLD affects approximately 30% to 40% of adults and 10% of children in the United States. NASH, a more severe and progressive form of NAFLD, is one of the most common chronic liver diseases worldwide, with an estimated prevalence of 3-5% globally. No drug is currently approved for treatment of NAFLD or NASH. In addition to these liver diseases, there are a number of orphan liver diseases for which we may seek to develop larsucosterol.
Clinical Program. In 2020, we completed a Phase 1b randomized, multi-center, and open-label clinical study in the United States to evaluate safety, PK and signals of biological activity of larsucosterol in NASH patients with stage 1-3 fibrosis. Larsucosterol (at doses of 50 mg QD, 150 mg QD and 300 mg BID) was administered orally for 28 days with 20 patients or more per dose group for a total of 65 patients in the trial. Key endpoints included safety and PK, and clinical chemistry/efficacy signals, such as liver enzymes (e.g., ALT, AST and GGT (each as defined below)), serum lipids (e.g., triglycerides), biomarkers (e.g., CK-18s, inflammatory cytokines), and insulin resistance (i.e., HOMA-IR), as well as liver fat content and liver stiffness by imaging (e.g., MRI-PDFF (as defined below) and FibroScan®).
8
Both the 50 mg and 600 mg dose groups showed a statistically significant median reduction at Day 28 from baseline of serum alanine aminotransferase (“ALT”) levels at -16% and -17%, respectively. The 600 mg dose group also showed statistically significant median reductions at Day 28 from baseline of serum aspartate aminotransferase (“AST”) (-18%) and gamma-glutamyl transferase (“GGT”) (-8%), and the 50 mg dose group had a statistically significant reduction at Day 28 from baseline in liver stiffness as measured by Fibroscan® (-10%).
Patients in the 50 mg or 150 mg dose groups also had statistically significant median reduction at Day 28 from baseline of serum triglycerides (-13% in the 50 mg group) or LDL-C (-11% in the 150 mg group). Patients with elevated baseline triglycerides (≥200 mg/dL; n=16) across all dose groups had a median reduction at Day 28 from baseline of -24% (p <0.01). Furthermore, patients in the 50 mg and 150 mg groups had 22% and 18%, respectively, median reductions (not statistically significant) of HOMA-IR from baseline respectively after 4 weeks of daily oral dosing of larsucosterol. The 600 mg group did not show a change in HOMA-IR.
At Day 28, 43% of patients in all three dose groups showed greater than or equal to 10% liver fat reduction from baseline as measured by magnetic resonance imaging - proton density fat fraction ("MRI-PDFF"). In this subgroup, there was a significant reduction from baseline in median liver fat content (-18%, -19%, and -23%, in the 50 mg, 150 mg and 600 mg groups, respectively). The reduction of liver fat content was accompanied by a significant median reduction from baseline of serum ALT (-21%, -19%, and -32%, in the 50 mg, 150 mg and 600 mg groups, respectively), as well as both CK-18, M30 and M65 (each as defined below) in the 50 mg and 600 mg groups.
Larsucosterol was well tolerated at all three doses evaluated. There were no serious adverse events reported during the study, and no discontinuations, early withdrawals or termination of study drug or study participation due to adverse events. PK parameters after repeat dosing were comparable to those after a single dose (from a prior study), indicating no accumulation of the drug after repeat dosing.
We have completed multiple Phase 1 trials in healthy subjects with orally administered larsucosterol. These include single-ascending-dose and multiple-ascending-dose studies as well as a food effect study. In all of these studies larsucosterol was well-tolerated at all dose levels, with no serious treatment-related adverse events reported. Dose-related increases in plasma concentrations were observed and no accumulation in plasma concentrations or food effects were observed with repeat dosing.
We also conducted a Phase 1b trial in cirrhotic and non-cirrhotic NASH patients and MCS (matched by age, body mass index and gender with normal liver function) utilizing orally administered larsucosterol. This was an open-label, single-ascending-dose safety and PK study conducted in Australia in two successive dose cohorts (first a low dose of 50 mg and then a high dose of 200 mg). Both cohorts consisted of 10 NASH patients and 6 MCS. Data from this study were presented at the International Liver Congress™ 2017 organized by the European Association for the Study of the Liver (EASL) in Amsterdam in April 2017. All patients and MCS in this study tolerated larsucosterol well. One patient (with a prior history of arrhythmia and an ongoing viral infection) in the high dose cohort experienced a serious adverse event (i.e., shortness of breath), which occurred without unusual biochemical changes and resolved without intervention but was considered possibly treatment related by the physician due to its temporal association with dosing. In both low and high dose cohorts, the PK parameters were comparable between the NASH patients and the MCS. In addition, the systemic exposure following the low and high doses of larsucosterol was dose dependent.
While this study was not designed to assess efficacy, we observed statistically significant reductions from baseline levels of several biomarkers after both doses of larsucosterol. A single oral dose of larsucosterol significantly reduced the levels of both full-length (“M65”) and cleaved (“M30”) cytokeratin-18 (“CK-18”), bilirubin, hsCRP, and IL-18 in these subjects. The mean reduction of full-length CK-18 (a generalized cell death marker) at the measured time point of greatest effect (12 hours after dosing) was 33% in the low dose cohort and 41% in the high dose cohort. The mean decrease of cleaved CK-18 (a cell apoptosis marker) at the measured time point of greatest effect (12 hours after dosing) was 37% in the low dose cohort and 47% in the high dose cohort. The mean reduction of total bilirubin (a liver function marker) at the measured time point of greatest effect (12 hours after dosing) was 27% in the low dose cohort and 31% in the high dose cohort. The mean decrease of hsCRP (a marker of inflammation) at the measured time point of greatest effect (24 hours after dosing) was 8% in the low
9
dose cohort and 13% in the high dose cohort. The mean decrease of IL-18 (an inflammatory mediator) at the measured time point of greatest effect (8 hours after dosing) was 4% in the low dose cohort and 8% in the high dose cohort.
We also conducted a Phase 1b open-label, multi-center U.S. study to evaluate the safety, tolerability, and PK of larsucosterol in subjects with moderate (Child-Pugh B scores, n=10) and severe (Child-Pugh C scores, n=7) hepatic function impairment ("HI"), and MCS (n=10) with normal hepatic function. Each subject received a single oral dose of 200 mg larsucosterol. Results from this study were presented at the International Liver Conference 2021 (EASL). Larsucosterol was safe and well-tolerated by all moderate and severe HI subjects with no adverse events and no dose-limiting toxicity reported throughout the study. As expected, clearance of larsucosterol was decreased in HI subjects compared to MCS with normal hepatic function, resulting in a 4-10-fold higher drug exposure (Cmax and AUC) in HI subjects. Additionally, a single oral dose of 200 mg of larsucosterol in subjects with HI resulted in statistically significant median reductions from baseline of the apoptosis biomarker M30 (cCK-18) at 12 hours post-dose.
Collectively, the biological signals observed in NASH and HI patients plus results from our animal models and cell culture studies suggest potential therapeutic activity of larsucosterol for patients with liver diseases. However, additional studies are required to evaluate the safety and efficacy of larsucosterol, and there is no assurance that these biomarker, clinical chemistry and liver imaging effects will be associated with clinically relevant benefits, or that larsucosterol will demonstrate safety or efficacy in treating liver diseases in our ongoing or future trials.
Approved and Commercial Pharmaceutical Products
POSIMIR® (bupivacaine solution)
POSIMIR (bupivacaine solution) for infiltration use is a novel and proprietary product that combines the strength of 660 mg of bupivacaine base with the innovative SABER platform technology, enabling continuous sustained delivery of a non-opioid local analgesic over three days in adults, which we believe coincides with the time period of the greatest need for post-surgical pain control in most patients. POSIMIR contains more bupivacaine than any other approved single-dose sustained-release bupivacaine
10
product. At the end of surgery, POSIMIR is administered into the subacromial space under direct arthroscopic visualization, where it continuously releases bupivacaine for 72 hours or more.
In February 2021, the FDA approved POSIMIR for infiltration use in adults for administration into the subacromial space under direct arthroscopic visualization to produce post-surgical analgesia for up to 72 hours following arthroscopic subacromial decompression.
In December 2021, we entered into the Innocoll Agreement, pursuant to which we granted to Innocoll an exclusive, royalty-bearing, sublicensable right and license to develop, manufacture and commercialize POSIMIR in the U.S. with respect to all uses and applications in humans. The Innocoll Agreement provides for the assignment of our supply agreement with a contract manufacturing organization to Innocoll and also provides Innocoll with the right, within the U.S., to expand the approved indications of POSIMIR. We retain, outside the U.S., all of the global rights to POSIMIR. Innocoll paid us an initial non-refundable, upfront fee of $4.0 million as well as a fee in the amount of $1.3 million primarily to cover the manufacturing supplies and excipients and certain equipment transferred to Innocoll pursuant to the terms of the Innocoll Agreement, and certain recently incurred DURECT expenses the parties negotiated for Innocoll to reimburse. In the fourth quarter of 2021, we recognized $4.1 million as collaborative research and development and other revenue, $1.1 million as product revenue, and a reduction of $0.1 million in net equipment. At December 31, 2021, we included $5.3 million due from Innocoll in accounts receivable on our balance sheet; these funds were received in January 2022. In August 2022, we were issued a new patent by the U.S. Patent and Trademark Office, extending U.S. patent coverage of POSIMIR to at least 2041, resulting in an $8.0 million milestone payment by Innocoll to the Company. In September 2022, Innocoll launched POSIMIR in the U.S., triggering a $2.0 million milestone payment to the Company for the first commercial sale of POSIMIR. As the commercial launch of POSIMIR progresses, we will also earn tiered, low double-digit to mid-teen royalties on net product sales of POSIMIR in the U.S. We may earn additional milestone payments up to $122.0 million in the aggregate, depending on the achievement of certain commercial, regulatory and intellectual property milestones with respect to POSIMIR. Pursuant to the terms of the Innocoll Agreement, except as otherwise expressly provided in the Innocoll Agreement, Innocoll is responsible for expenses relating to the manufacturing, development and commercialization of POSIMIR in the U.S.
PERSERIS™ (risperidone)
In September 2017, we entered into an agreement with Indivior, under which we assigned to Indivior certain patents that may provide further intellectual property protection for PERSERIS, Indivior’s extended-release injectable suspension for the treatment of schizophrenia in adults. In consideration for such assignment, Indivior made non-refundable upfront and milestone payments to DURECT totaling $17.5 million. Additionally, under the terms of the agreement with Indivior, DURECT receives quarterly earn-out payments that are based on a single-digit percentage of U.S. net sales of PERSERIS into 2026. Indivior commercially launched PERSERIS in the U.S. in February 2019.
ORADUR™-ADHD Program
We developed a proprietary drug product for the treatment of ADHD called Methydur in collaboration with Orient Pharma, a diversified multinational pharmaceutical, healthcare and consumer products company with headquarters in Taiwan. We have licensed worldwide Methydur rights to Orient Pharma and they launched Methydur commercially in Taiwan in September 2020. Orient Pharma may seek commercialization partners in other countries throughout the world, including China and the U.S. We receive a single-digit royalty on sales of Methydur by Orient Pharma or its commercialization partners as well as potential milestones and sub-license fees.
Drug Delivery Technologies and Programs
Our drug delivery technologies are designed to deliver the right drug to the right place, in the right amount and at the right time to treat a variety of chronic, acute and episodic diseases and conditions. We aim to improve therapy for a given disease or patient population by controlling the rate and duration of drug administration. In addition, if advantageous for the therapy, our technologies can target the delivery of the drug to its intended site of action.
11
Our technologies are suitable for providing long-term drug therapy because they can often store highly concentrated, stabilized drugs in a small volume and protect the drug from degradation by the body. This, in combination with the ability to continuously deliver desired doses of a drug, can extend the therapeutic value of a wide variety of drugs, including, in some cases, those which would otherwise be ineffective, too unstable, too potent or cause adverse side effects. In some cases, delivering the drug directly to the intended site of action can improve efficacy while minimizing unwanted side effects elsewhere in the body, which may limit the long-term use of many drugs. Our technologies may thus provide better therapy for chronic diseases or conditions, or for certain acute conditions where longer drug dosing is required or advantageous, by replacing multiple injection therapy or oral dosing, improving drug efficacy, reducing side effects and ensuring dosing compliance. Our technology may thereby improve patients’ quality of life by eliminating more repetitive treatments, reducing dependence on caregivers and allowing patients to lead more independent lives.
We currently focus our drug delivery technology efforts around our SABER® and CLOUD™ Bioerodible Injectable Depot Systems. SABER uses a high viscosity base component, such as sucrose acetate isobutyrate (SAIB), to provide controlled release of a drug. When the high viscosity SAIB is formulated with drug, biocompatible excipients and other additives, the resulting formulation is easily injectable with standard syringes and needles. After injection of a SABER formulation, the excipients diffuse away over time, leaving a viscous depot which provides controlled sustained release of drug. CLOUD is a class of bioerodible injectable depot technology which generally does not contain SAIB but includes various other release rate modifying excipients and/or bioerodible polymers to achieve the delivery of drugs for periods of days to months from a single injection.
The SABER technology is the basis of POSIMIR (described above).
Our Strategy
Our objective is to develop multiple pharmaceutical products that address significant unmet medical needs and improve patients’ quality of life. To achieve this objective, our strategy includes the following key elements:
Complete Clinical Development and Seek Regulatory Approval of Larsucosterol for the Treatment of AH. We are currently enrolling our Phase 2b clinical trial (AHFIRM). We expect to complete enrollment in the second quarter of 2023 and to report topline results in the second half of 2023. If the AHFIRM trial is successful, we intend to meet with the FDA and comparable foreign regulatory authorities to discuss registration.
Maximize the Commercial Potential of Larsucosterol in AH. We intend to build a highly specialized commercial organization to support the commercialization of larsucosterol, if approved, in the United States. We believe this market can be effectively addressed with a modest-sized commercial organization, including a hospital-focused sales force focused on hospitals. We may also seek strategic collaborations to commercialize larsucosterol outside the United States.
Apply Our Drug Development Expertise to New Chemical Entities Derived from Our Epigenetic Regulator Program. We have assembled a core team of employees with considerable experience in drug development, and it is our intent to leverage their capabilities by developing product candidates utilizing expertise from our Epigenetic Regulator Program. We may develop these new chemical entities ourselves or with partners.
Expand Development Pipeline by Identifying Potential New Indications for Epigenetic Regulator Programs Focused on Conditions with High Unmet Need. We believe our Epigenetic Regulator Program has the potential to address many of the diseases and disorders that present great challenges to medicine including acute organ injury, metabolic disorders, and other acute and chronic conditions. Our current efforts focus on exploiting our Epigenetic Regulator Program through which we have identified new chemical entities, including larsucosterol, our lead compound, that may have utility in conditions such as acute organ injuries, including AH, and chronic metabolic/lipid disorders, including NASH.
Enable Product Development Through Strategic Agreements. We believe that entering into selective strategic collaborations and other arrangements with respect to our product development programs and technology can enhance the success of our product development and commercialization, leverage and exploit the value of our intellectual property portfolio, mitigate our risk and enable us to
12
better manage our operating costs. Additionally, such collaborations and arrangements enable us to leverage investment by third parties and reduce our net cash burn, while retaining significant economic rights.
Strategic Agreements
We have entered into the following strategic collaboration and other key agreements:
Innocoll Pharmaceuticals Limited. In December 2021, we entered into the Innocoll Agreement, pursuant to which, we granted to Innocoll an exclusive, royalty-bearing, sublicensable right and license to develop, manufacture and commercialize POSIMIR in the U.S. with respect to all uses and applications in humans. The Innocoll Agreement provides for the assignment of our supply agreement with a contract manufacturing organization to Innocoll and also provides Innocoll with the right, within the U.S., to expand the approved indications of POSIMIR. We retain, outside the United States, all of the global rights to POSIMIR. Innocoll paid us an initial non-refundable, upfront fee of $4.0 million as well as a fee in the amount of $1.3 million primarily to cover the manufacturing supplies and excipients and certain equipment transferred to Innocoll pursuant to the terms of the Innocoll Agreement, and certain recently incurred DURECT expenses the parties negotiated for Innocoll to reimburse. In the fourth quarter of 2021, we recognized $4.1 million as collaborative research and development and other revenue, $1.1 million as product revenue, and a reduction of $0.1 million in net equipment. At December 31, 2021, we included $5.3 million due from Innocoll in accounts receivable on our balance sheet; these funds were received in January 2022. In August 2022, we were issued a new patent by the U.S. Patent and Trademark Office, extending U.S. patent coverage of POSIMIR to at least 2041, resulting in an $8.0 million milestone payment by Innocoll to the Company. In September 2022, Innocoll launched POSIMIR in the U.S., triggering a $2.0 million milestone payment to the Company for the first commercial sale of POSIMIR. As the commercial launch of POSIMIR progresses, we will receive tiered, low double-digit to mid-teen royalties on net product sales of POSIMIR in the U.S. We may earn additional milestone payments up to $122.0 million in the aggregate, depending on the achievement of certain commercial, regulatory and intellectual property milestones with respect to POSIMIR. Thus, we recognized $10.0 million of milestone revenue under the agreement with Innocoll during the twelve months ended December 31, 2022.
Pursuant to the terms of the Innocoll Agreement, except as otherwise expressly provided in the Innocoll Agreement, Innocoll is responsible for expenses relating to the manufacturing, development and commercialization of POSIMIR in the United States. The Innocoll Agreement includes customary representations and warranties on behalf of us and Innocoll, including representations as to the licensed intellectual property, regulatory matters and compliance with applicable laws. The Innocoll Agreement also provides for certain mutual indemnities for breaches of representations, warranties and covenants.
Virginia Commonwealth University Intellectual Property Foundation. In December 2012, we entered into an exclusive in-license and research and development agreement with the Virginia Commonwealth University Intellectual Property Foundation regarding certain new chemical entities under development through our Epigenetic Regulator Program, including larsucosterol. Under this licensing arrangement, we agreed to undertake certain efforts to bring licensed products to market, pay for prosecution of related patents and report on progress to VCU. In addition, we are obligated to pay low single-digit percentage patent royalties on net sales of licensed products, subject to annual minimum payments and additional milestone payments. This license includes rights to ten patent families. We may terminate this agreement at any time by written notice, and VCU may terminate this agreement by written notice if there is an uncured material breach.
13
Indivior UK Ltd. In September 2017, we entered into an agreement with Indivior, under which we assigned to Indivior certain patents that may provide further intellectual property protection for PERSERIS, Indivior’s extended-release injectable suspension for the treatment of schizophrenia in adults. In consideration for such assignment, Indivior made non-refundable upfront and milestone payments to DURECT totaling $17.5 million. Additionally, under the terms of the agreement with Indivior, DURECT receives quarterly earn-out payments that are based on a single digit percentage of U.S. net sales of PERSERIS into 2026. Indivior commercially launched PERSERIS in the U.S. in February 2019. We also receive a non-exclusive right under the assigned patents to develop and commercialize certain risperidone-containing products and products that do not contain risperidone or buprenorphine. The agreement contains customary representations, warranties and indemnities of the parties.
Commercial Product Lines
ALZET
The ALZET product line consists of miniature, implantable osmotic pumps and accessories used for research in mice, rats and other laboratory animals. These pumps are neither approved nor intended for human use. ALZET pumps continuously deliver drugs, hormones and other test agents at controlled rates from one day to six weeks without the need for external connections, frequent handling or repeated dosing. In laboratory research, these infusion pumps can be used for systemic administration when implanted under the skin or in the body. They can be attached to a catheter for IV, intracerebral, or intra-arterial infusion or for targeted delivery, where the effects of a drug or test agent are localized in a particular tissue or organ. The wide use and applications of the ALZET product line is evidenced by the more than 21,000 scientific references that now exist.
LACTEL® Absorbable Polymers
On December 31, 2020, we signed an Asset Purchase Agreement with Evonik Corporation (Evonik), pursuant to which Evonik purchased certain assets related to our LACTEL Absorbable Polymers product line. Under the terms of the Asset Purchase Agreement, Evonik paid us approximately $15.1 million and also agreed to assume certain liabilities with respect to the transferred assets and assembled workforce.
Marketing and Sales
Historically, we have established strategic distribution and marketing alliances for our product candidates to leverage the established sales organizations that certain pharmaceutical companies have in markets we are targeting. In the future, we may elect to build our own commercial, sales and marketing capability in order to capture more of the economic value of certain products that we may develop. If we choose to enter into third-party collaborations to commercialize our pharmaceutical product candidates, we may in the future enter into these alliances under circumstances that allow us to participate in the sales and marketing of these products. We will continue to pursue strategic alliances and collaborators from time to time consistent with our strategy to leverage the established sales organizations of third-party collaborators.
We market and sell our ALZET product line through a direct sales force in the U.S. and through a network of distributors outside of the U.S.
Suppliers
We purchase the larsucosterol drug substance from a third-party manufacturer and larsucosterol clinical trial materials from another third-party manufacturer. As needed, we purchase sucrose acetate isobutyrate, a raw material for our SABER-based pharmaceutical systems, including Methydur, POSIMIR, and ORADUR. We expect that we will continue to be able to obtain sufficient supply of these raw materials to meet our needs for the foreseeable future. We do not have in place long term supply agreements with respect to all of the components of any of our pharmaceutical product candidates, however, and are subject to the risk that we may not be able to procure all required components in adequate quantities with acceptable quality, within acceptable time frames or at reasonable cost.
Customers
Our product revenues principally are derived from sales of the ALZET product line to academic and pharmaceutical industry researchers, and from the sale of certain key excipients that are included in POSIMIR, Methydur Sustained Release Capsules, and other products. Until such time that we are able to
14
bring our pharmaceutical product candidates to market, if at all, we expect these to be our principal sources of product revenue. We also receive revenue from collaborative research and development arrangements with our third-party collaborators and earn out revenue from our patent purchase agreement. In 2022, Innocoll accounted for 52% of our total revenue. In 2021, Innocoll accounted for 37% of our total revenue.
Manufacturing
The process for manufacturing our pharmaceutical product candidates is technically complex, requires special skills, and must be performed in qualified facilities. We have entered into development and commercial manufacturing agreements with third parties for the manufacture of larsucosterol and POSIMIR (now assigned to Innocoll). In addition, we have a small multi-discipline manufacturing facility in California that we have used to manufacture research and clinical supplies of several of our pharmaceutical product candidates under good manufacturing practice (“GMP”), including larsucosterol dosage forms and POSIMIR. In the future, we may develop additional manufacturing capabilities for our pharmaceutical product candidates and components to meet our demands and those of our third-party collaborators by contracting with third party manufacturers and by potentially constructing additional manufacturing space at our current facilities in California. We manufacture our ALZET product line and certain key components for POSIMIR and Methydur at one of our California facilities.
Patents and Proprietary Rights
Our success depends in part on our ability to obtain patents, to protect trade secrets, to operate without infringing upon the proprietary rights of others and to prevent others from infringing on our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, filing U.S. and foreign patent applications related to our proprietary molecules and technology, inventions and improvements that are important to the development of our business. As of March 3, 2023, we owned or exclusively in-licensed over 25 unexpired issued U.S. patents and over 170 unexpired issued foreign patents (which include granted European patent rights that have been validated in various EU member states). In addition, we have over 30 pending U.S. patent applications and over 190 foreign applications pending in Europe, Australia, Japan, Canada and other countries.
The patent status of our most advanced drug candidates is as follows:
Our Epigenetic Regulator Program includes ten in-licensed patent families and ten patent families solely owned by us. Seven patent families each include at least one granted patent that could provide protection until at least 2026, 2032, 2033, 2034, 2035, 2037 and 2037, respectively. The other patent families include pending patent applications, which if granted, could result in patents providing protection until at least 2037 to 2043. Patent terms are potentially subject to terminal disclaimers as well as patent term adjustments and extensions. Of the twenty patent families covering larsucosterol and/or other molecules in the Epigenetic Regulator Program, two were only filed in the United States, and the other eighteen have been filed or likely will be filed both in the U.S. and internationally. Since larsucosterol is an endogenous molecule, patent claims directed to larsucosterol compositions of matter may be more difficult to maintain or enforce in the United States under Myriad Genetics and other recent court decisions. One of the U.S. patents issued before Myriad Genetics, and nine of the larsucosterol U.S. patents issued after Myriad Genetics. The granted claims in the U.S. include both composition of matter and method of treatment claims. There can be no assurance that the pending patent applications will be granted. Further, there can be no assurance that VCU will not attempt to terminate their license to us, which termination could result in the loss of our rights to these patent families.
In the United States, POSIMIR is covered by four patent families. Three patent families include granted patents that could provide protection until 2025, 2026 and 2041, respectively. The other patent family includes a pending patent application, which if granted, could result in a patent expiring in 2042. In Europe, POSIMIR is covered by two granted patents with one that could expire in 2025 and one that could expire in 2026. The families that could provide protection until at least 2041 and 2042, respectively, have either been filed or will likely be filed in Europe.
15
Proprietary rights relating to our planned and potential products will be protected from unauthorized use by third parties only to the extent that they are covered by valid and enforceable patents or are effectively maintained as trade secrets. Patents owned by or licensed to us may not afford protection against competitors, and our pending patent applications now or hereafter filed by or licensed to us may not result in patents being issued. In addition, the laws of certain foreign countries may not protect our intellectual property rights to the same extent as do the laws of the U.S.
The patent positions of biopharmaceutical companies involve complex legal and factual questions and, therefore, their enforceability cannot be predicted with certainty. Our patents or patent applications, or those licensed to us, if issued, may be challenged, invalidated or circumvented, and the rights granted thereunder may not provide proprietary protection or competitive advantages to us against competitors with similar technology. Furthermore, our competitors may independently develop similar technologies or duplicate any technology developed by us. Because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that, before any of our products can be commercialized, any related patent may expire or remain in existence for only a short period following commercialization, thus reducing any advantage of the patent, which could adversely affect our ability to protect future product development and, consequently, our operating results and financial position.
Because patent applications in the U.S. are typically maintained in secrecy for at least 18 months after filing and since publication of discoveries in the scientific or patent literature often lag behind actual discoveries, we cannot be certain that we were the first to make inventions or file for protection of inventions set forth in our patents or patent applications.
Our planned or potential products may be covered by third-party patents or other intellectual property rights, in which case we would need to obtain a license to continue developing or marketing these products. Any required licenses may not be available to us on acceptable terms, if at all. If we do not obtain any required licenses, we could encounter delays in product introductions while we attempt to design around these patents, or could find that the development, manufacture or sale of products requiring such licenses is foreclosed. Litigation may be necessary to defend against or assert such claims of infringement, to enforce patents issued to us, to protect trade secrets or know-how owned by us, or to determine the scope and validity of the proprietary rights of others. In addition, interference, derivation, post-grant oppositions, and similar proceedings may be necessary to determine rights to inventions in our patents and patent applications. Litigation or similar proceedings could result in substantial costs to and diversion of effort by us and could have a material adverse effect on our business, financial condition and results of operations. These efforts by us may not be successful.
We may rely, in certain circumstances, on trade secrets to protect our technology. However, trade secrets are difficult to protect. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements with our employees and certain contractors. There can be no assurance that these agreements will not be breached, that we will have adequate remedies for any breach, or that our trade secrets will not otherwise become known or be independently discovered by competitors. To the extent that our employees, consultants or contractors use intellectual property owned by others in their work for us, disputes may also arise as to the rights in related or resulting know-how and inventions.
Government Regulation
The FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries impose substantial requirements upon the development, manufacture and marketing of pharmaceutical products. These agencies and other federal, state and local entities regulate research and development activities and, among other things, the testing, manufacture, quality control, safety, effectiveness, labeling, storage, distribution, record keeping, approval, advertising and promotion of our products. We believe that our products in development will be regulated as drugs by the FDA rather than as biologics or devices.
16
U.S. Drug Development Process
The standard process required by the FDA under the new drug provisions of the Federal Food, Drug and Cosmetics Act (the “FDCA”) before our products in development may be marketed in the U.S. generally involves the following:
Section 505 of the FDCA describes three types of NDAs: (1) an application that contains full reports of investigations of safety and effectiveness (section 505(b)(1)); (2) an application that contains full reports of investigations of safety and effectiveness but where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference (section 505(b)(2)); and (3) an application that contains information to show that the proposed pharmaceutical product candidate is identical in active ingredient, dosage form, strength, route of administration, labeling, quality, performance characteristics and intended use, among other things, to a previously approved product (section 505(j)). We expect that our drug candidates deriving from our Epigenetic Regulator Program will be evaluated for approval after submission of an NDA under section 505(b)(1).
The testing and approval process require substantial time, effort, and financial resources, and we cannot be certain that any approval will be granted on a timely basis, if at all. Preclinical development of a drug candidate can take several years to complete, with no guarantee that an IND based on those studies will become effective to even permit clinical testing to begin. Even though several of our pharmaceutical product candidates utilize active drug ingredients that are commercially marketed in the United States in other dosage forms, we need to establish safety and effectiveness of those active ingredients in the formulation and dosage forms that we are developing.
Preclinical tests include laboratory evaluation of the product candidate, its chemistry, formulation and stability, as well as animal studies to assess the potential safety and efficacy of the pharmaceutical product candidate. We then submit the results of the preclinical tests, together with manufacturing information and analytical data, to the FDA as part of an IND, which must become effective before we may begin human clinical trials. Each subsequent new clinical protocol must also be submitted to the FDA under the IND. An IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-Day time period, raises concerns or questions about the conduct of the trials as
17
outlined in the IND and imposes a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin. Our submission of an IND may not result in FDA authorization to commence clinical trials. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development. Further, an independent IRB at each medical center proposing to conduct the clinical trials must review and approve any clinical study as well as the related informed consent forms and authorization forms that permit us to use individually identifiable health information of study participants.
Human clinical trials are typically conducted in three sequential phases, which may overlap:
In the case of product candidates for severe diseases, such as chronic pain, or life-threatening diseases such as cancer, the initial human testing is often conducted in patients with the target diseases or conditions rather than in healthy volunteers. Since these patients already have the target disease or condition, these studies may provide initial evidence of efficacy traditionally obtained in Phase 2 trials, and thus these trials are frequently referred to as Phase 1/2 clinical trials or Phase 1b trials. Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. We cannot be certain that we will successfully complete Phase 1, Phase 2 or Phase 3 clinical trials of our pharmaceutical product candidates in development within any specific time period, if at all. Furthermore, the FDA or the IRB or the sponsor may suspend clinical trials at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk. During the clinical development of product candidates, sponsors frequently meet and consult with the FDA to ensure that the design of their studies will likely provide data both sufficient and relevant for later regulatory review; however, no assurance of approvability can be given by the FDA.
NDA Review and Approval Processes
Assuming successful completion of all testing in accordance with all applicable regulatory requirements, the results of product development, preclinical studies and clinical studies are submitted to the FDA as part of an NDA for approval of the marketing and commercial shipment of the product. Submission of an NDA may require the payment of a substantial user fee to the FDA, and although the agency has defined user fee goals for the time in which to respond to sponsor applications, there can be no assurance that the FDA will act in any particular timeframe. The FDA may deny approval of an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical trials be conducted. Even if such data is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. An NDA may be approved with significant restrictions on its labeling, marketing and distribution under a Risk Evaluation and Mitigation Strategy or otherwise that could restrict the commercial applications of a product or impose costly procedures in connection with the commercialization or use of the product. Once issued, the FDA may withdraw product approval if ongoing regulatory standards are not maintained or if safety problems occur after the product reaches the market. Requirements for additional Phase 4 studies (post approval marketing studies) to confirm safety and effectiveness in a broader commercial use population may be imposed as a condition of marketing approval. In addition, the FDA may require testing and surveillance programs to monitor the effects of approved products which have been commercialized, and the FDA has the power to require changes in
18
labeling or to prevent further marketing of a product based on the results of these post-marketing programs. Any comparative claims comparing a product to other dosage forms or competitive products typically need to be supported by two adequate and well-controlled head-to-head clinical trials.
Satisfaction of FDA requirements or similar requirements of state, local and foreign regulatory agencies typically takes several years and the actual time required may vary substantially, based upon the type, complexity and novelty of the pharmaceutical product and of the disease or condition. Government regulation may delay or prevent marketing of potential products for a considerable period of time and impose costly procedures upon our activities. We cannot be certain that the FDA or any other regulatory agency will grant approval for any of our pharmaceutical products under development on a timely basis, if at all. Success in preclinical or early-stage clinical trials does not assure success in later stage clinical trials. Targets and pathways identified in vitro may be determined to be less relevant in clinical studies and results in animal model studies may not be predictive of human clinical results. Furthermore, data obtained from clinical activities is not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. Evolving safety concerns can result in the imposition of new requirements for expensive and time-consuming tests, such as for QT interval cardiotoxicity testing. Even if a product receives regulatory approval, the approval may be significantly limited to specific indications. Further, even after regulatory approval is obtained, any problems associated with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Any pharmaceutical products that we may develop and obtain approval for would also be subject to adverse findings of the active drug ingredients being marketed in different dosage forms and formulations. Delays in obtaining, or failures to obtain regulatory approvals would have a material adverse effect on our business. Marketing our pharmaceutical products abroad will require similar regulatory approvals and is subject to similar risks. In addition, we cannot predict what adverse governmental regulations may arise from future U.S. or foreign governmental action.
Expedited Development and Review Programs
There are several FDA programs intended to help facilitate the development of new drugs that meet certain criteria. Specifically, new drugs are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new drug may request the FDA to designate the drug as a Fast Track product at any time during the clinical development of the product. Under a Fast Track designation, the FDA may consider for review sections of the marketing application on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the application, the FDA agrees to accept sections of the application and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the application.
A product candidate is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biological product designated for priority review to facilitate the review. Under priority review, the FDA’s goal is to review an application in six months once it is filed, compared to ten months for a standard review. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Additionally, a product candidate that is being studied for safety and effectiveness in treating serious or life-threatening illnesses and provides meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that it may be approved on the basis of adequate and well-controlled clinical trials establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on an intermediate clinical endpoint other than survival or irreversible morbidity, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials with due diligence and, under the Food and Drug
19
Omnibus Reform Act of 2022 (“FDORA”), the FDA is now permitted to require, as appropriate, that such trials be underway prior to approval or within a specific time period after the date of approval for a product granted accelerated approval. In addition, for products being considered for accelerated approval, unless otherwise informed by the FDA, the FDA generally requires that all advertising and promotional materials intended for dissemination or publication within 120 days following marketing approval be submitted to the agency for review during the pre-approval review period, and that after 120 days following marketing approval, all advertising and promotional materials must be submitted at least 30 days prior to the intended time of initial dissemination or publication. Under FDORA, the FDA has increased authority for expedited procedures to withdraw approval of a drug or indication approved under accelerated approval if, for example, the confirmatory trial fails to verify the predicted clinical benefit of the product.
A product may also be eligible for receipt of a Breakthrough Therapy designation under the provisions of the Food and Drug Administration Safety and Innovation Act (“FDASIA”). The Breakthrough Therapy designation is intended to expedite the FDA’s review of a potential new drug for serious or life-threatening diseases where “preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.” The designation of a drug as a Breakthrough Therapy provides the same benefits as are available under the Fast Track program, as well as intensive FDA guidance on the product’s development program. If a drug is designated as a breakthrough therapy, the FDA will provide more intensive guidance on the drug development program and expedite its review. Fast Track designation, priority review, accelerated approval and Breakthrough Therapy designation do not change the standards for approval, but they may expedite the development or approval process.
Orphan Drug Designation and Exclusivity
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or 200,000 or more individuals in the United States and for which there is no reasonable expectation that the cost of developing and making the product available in the United States for this type of disease or condition will be recovered from sales of the product. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process. If a product that has orphan drug designation subsequently receives FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indication, except in limited circumstances, for seven years. These circumstances are an inability to supply the drug in sufficient quantities or a situation in which a new formulation of the orphan drug has shown superior safety or efficacy or a major contribution to patient care. Competitors, however, may receive approval of either a different product for the same indication or the same product for a different indication but that could be used off-label in the orphan indication. Orphan drug exclusivity could also block the approval of one of our products for seven years if a competitor obtains earlier approval of the same product, as defined by the FDA, for the same indication we are seeking approval, or if our product is determined to be contained within the scope of the competitor’s product for the same indication or disease. If we pursue marketing approval for an indication broader than the orphan drug designation we have received, we may not be entitled to orphan drug exclusivity. Orphan drug status in the European Union (“EU”) has similar, but not identical, requirements and benefits.
Post-Approval Regulation
Any pharmaceutical products manufactured or distributed by us pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including record-keeping requirements and reporting of adverse experiences with the product or the active pharmaceutical ingredient or other components of the product. The FDA may also require post-approval clinical or non-clinical trials. Drug
20
manufacturers and their subcontractors are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and state agencies for compliance with good manufacturing practices, which impose procedural and documentation requirements upon us and our third-party manufacturers. We cannot be certain that we or our present or future suppliers will be able to comply with the GMP regulations and other FDA regulatory requirements.
The FDCA strictly regulates drug product marketing, prohibits manufacturers from marketing drug products for off-label use and regulates the distribution of drug samples. The FDA has actively enforced regulations prohibiting the marketing of products for unapproved uses, and federal and state authorities are also actively litigating against sponsors who promote their drugs for unapproved uses under various fraud and abuse and false claims act statutes. We and our products are also subject to a variety of state laws and regulations in those states or localities where our products are or will be marketed. Any applicable state or local regulations may hinder our ability to market our products in those states or localities. We are also subject to numerous federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control, and disposal of hazardous or potentially hazardous substances. We may incur significant costs to comply with such laws and regulations now or in the future.
The FDA’s policies may change and additional government regulations may be enacted which could prevent or delay regulatory approval of our product candidates. Moreover, increased attention to the containment of health care costs in the U.S. and in foreign markets could result in new government regulations that could have a material adverse effect on our business. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or abroad.
The Drug Enforcement Administration ("DEA") regulates chemical compounds as Schedule I, II, III, IV or V substances, with Schedule I substances considered to present the highest risk of substance abuse and Schedule V substances the lowest risk. Certain active ingredients in ORADUR-Methylphenidate are listed by the DEA as Schedule II under the Controlled Substances Act of 1970. Consequently, their manufacture, research, shipment, storage, sale and use are subject to a high degree of oversight and regulation. For example, all Schedule II drug prescriptions must be signed by a physician, physically presented to a pharmacist and may not be refilled without a new prescription. Furthermore, the amount of Schedule II substances we can obtain for clinical trials and commercial distribution is limited by the DEA and our quota may not be sufficient to complete clinical trials or meet commercial demand. There is a risk that DEA regulations may interfere with the supply of the drugs used in our clinical trials, and, in the future, our ability to produce and distribute our products in the volume needed to meet commercial demand, which could negatively impact us and our collaborators.
Other Healthcare Laws
In addition to FDA and DEA restrictions on the marketing of pharmaceutical products, other foreign, federal and state healthcare regulatory laws restrict business practices in the pharmaceutical industry. These laws include, but are not limited to, federal and state anti‑kickback, false claims, data privacy and security, and physician payment and drug pricing transparency laws.
The U.S. federal Anti‑Kickback Statute prohibits, among other things, any person or entity from knowingly and willfully offering, paying, soliciting, receiving or providing any remuneration, directly or indirectly, overtly or covertly, to induce or in return for purchasing, leasing, ordering, or arranging for or recommending the purchase, lease, or order of any good, facility, item or service reimbursable, in whole or in part, under Medicare, Medicaid or other federal healthcare programs. The term “remuneration” has been broadly interpreted to include anything of value. The Anti‑Kickback Statute has been interpreted to apply to arrangements between pharmaceutical and medical device manufacturers on the one hand and prescribers, purchasers, formulary managers and beneficiaries on the other hand. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases, or recommendations may be subject to scrutiny if they do not meet the requirements of a statutory or regulatory exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor
21
does not make the conduct per se illegal under the U.S. federal Anti‑Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case‑by‑case basis based on a cumulative review of all its facts and circumstances. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated. In addition, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. Moreover, a claim including items or services resulting from a violation of the U.S. federal Anti‑Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. The majority of states also have anti‑kickback laws, which establish similar prohibitions and, in some cases, may apply to items or services reimbursed by any third‑party payor, including commercial insurers.
The federal false claims laws, including the civil False Claims Act, prohibit, among other things, any person or entity from knowingly presenting, or causing to be presented, a false, fictitious or fraudulent claim for payment to, or approval by, the federal government, knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government, or knowingly making a false statement to avoid, decrease, or conceal an obligation to pay money to the U.S. federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. Actions under the civil False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Moreover, a claim including items or services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act.
The federal Physician Payments Sunshine Act requires certain manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services (“CMMS”), information related to payments or other “transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists, and chiropractors), certain non-physician providers (physician assistants, nurse practitioners, clinical nurse specialists, certified registered nurse anesthetists and anesthesiologist assistants, and certified-nurse midwives), and teaching hospitals, and applicable manufacturers and applicable group purchasing organizations to report annually to CMMS ownership and investment interests held by physicians and their immediate family members.
There are federal price reporting laws, which require manufacturers to calculate and report complex pricing metrics to government programs, and such reported prices may be used in the calculation of reimbursement and/or discounts on approved products.
Similar state and local laws and regulations may also restrict business practices in the pharmaceutical industry, such as state anti-kickback and false claims laws, which may apply to business practices, including but not limited to, research, distribution, sales, and marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers, or by patients themselves; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws and regulations that require drug manufacturers to file reports relating to pricing information and marketing expenditures or which require tracking gifts, compensation and other remuneration and items of value provided to physicians, other healthcare professionals and entities ; and state and local laws that require the registration of pharmaceutical sales representatives. Finally, the Physician Self-Referral (“Stark”) Law prohibits physicians from referring Medicare or Medicaid patients to providers of “designated health services” with whom the physician or a member of the physician’s immediate family has an ownership interest or compensation arrangement, unless a statutory or regulatory exception applies.
Violations of any of these laws and other applicable healthcare fraud and abuse laws may be punishable by criminal and civil sanctions, including fines and civil monetary penalties, the possibility of exclusion from federal healthcare programs (including Medicare and Medicaid), disgorgement and corporate integrity agreements, which impose, among other things, rigorous operational and monitoring requirements on companies. Similar sanctions and penalties, as well as imprisonment, also can be imposed upon executive officers and employees of such companies.
22
Coverage and Reimbursement
Sales of any pharmaceutical product depend, in part, on the extent to which such product will be covered by third-party payors, such as federal, state and foreign government healthcare programs, commercial insurance and managed healthcare organizations, and the level of reimbursement for such product by third-party payors. In the United States, no uniform policy exists for coverage and reimbursement for pharmaceutical products among third-party payors. Therefore, decisions regarding the extent of coverage and amount of reimbursement to be provided are made on a plan-by-plan basis. The process for determining whether a third-party payor will provide coverage for a product typically is separate from the process for setting the price of such product or for establishing the reimbursement rate that the payor will pay for the product once coverage is approved.
Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the FDA-approved products for a particular indication, or place products at certain formulary levels that result in lower reimbursement levels and higher cost-sharing obligation imposed on patients. One third-party payor’s decision to cover a particular medical product or service does not ensure that other payors will also provide coverage for the medical product or service, and the level of coverage and reimbursement can differ significantly from payor to payor. As a result, the coverage determination process will often require us to provide scientific and clinical support for the use of our products to each payor separately, which can be a time-consuming process, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. Additionally, a third-party payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved.
Moreover, as a condition of participating in, and having products covered under, certain federal healthcare programs, such as Medicare and Medicaid, we may become subject to federal laws and regulations that require pharmaceutical manufacturers to calculate and report certain price reporting metrics to the government, such as Medicaid Average Manufacturer Price (“AMP”), and Best Price, Medicare Average Sales Price, the 340B Ceiling Price and Non-Federal AMP reported to the Department of Veteran Affairs, and with respect to Medicaid, pay statutory rebates on utilization of manufacturers’ products by Medicaid beneficiaries. Compliance with such laws and regulations require significant resources and any findings of non-compliance may have a material adverse effect on our revenues.
Healthcare Reform
In the United States and certain foreign jurisdictions, there have been, and we expect there will continue to be, a number of legislative and regulatory changes to the healthcare system. In the United States, by way of example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care Education and Reconciliation Act (collectively, the “Affordable Care Act”), was enacted in the United States. Among the provisions of the Affordable Care Act of importance to our potential product candidates, the Affordable Care Act: established an annual, nondeductible fee on any entity that manufactures or imports certain specified branded prescription drugs and biologic agents; expanded eligibility criteria for Medicaid programs; increased the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program; created a Medicare Part D coverage gap discount program; established a Patient-Centered Outcomes Research Institute to oversee, identify priorities in and conduct comparative clinical effectiveness research, along with funding for such research; and established a Center for Medicare & Medicaid Innovation at Centers for Medicare & Medicaid Services to test innovative payment and service delivery models to lower Medicare and Medicaid spending.
23
There have been executive, judicial and Congressional challenges to certain aspects of the Affordable Care Act. For example, the Tax Cuts and Jobs Act of 2017 included a provision that repealed, effective January 1, 2019, the tax-based shared responsibility payment imposed by the Affordable Care Act on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” On June 17, 2021, the U.S. Supreme Court dismissed a challenge on procedural grounds that argued the Affordable Care Act is unconstitutional in its entirety because the individual mandate was repealed by Congress. Thus, the Affordable Care Act will remain in effect in its current form. However, it is possible that the Affordable Care Act will be subject to additional judicial or Congressional challenges in the future.
Further, the Affordable Care Act has been subject to various health reform measures. For example, prior to the U.S. Supreme Court ruling, on January 28, 2021, the Biden administration issued an executive order that initiated a special enrollment period for purposes of obtaining health insurance coverage through the Affordable Care Act marketplace. The executive order also instructed certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the Affordable Care Act. In addition, on August 16, 2022, President Biden signed the Inflation Reduction Act of 2022 (the “IRA”) into law, which among other things, extends enhanced subsidies for individuals purchasing health insurance coverage in Affordable Care Act marketplaces through plan year 2025 and includes several measures intended to lower the cost of prescription drugs and related healthcare reforms. Specifically, the IRA authorizes and directs the Department of Health and Human Services to set drug price caps for certain high-cost Medicare Part B and Part D qualified drugs, with the initial list of drugs to be selected by September 1, 2023, and the first year of maximum price applicability to begin in 2026. The IRA further authorizes the Department of Health and Human Services to penalize pharmaceutical manufacturers that increase the price of certain Medicare Part B and Part D drugs faster than the rate of inflation. Finally, the IRA creates significant changes to the Medicare Part D benefit design by capping Part D beneficiaries’ annual out-of-pocket spending at $2,000 and eliminates the “donut hole” under the Medicare Part D program, both beginning in 2025, by significantly lowering the beneficiary maximum out-of-pocket cost through a newly established manufacturer discount program. The implementation of government imposed cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our product candidates if approved. It is unclear how any such challenges and other litigation, and the healthcare reform measures of the current U.S. presidential administration will impact the Affordable Care Act and our business.
Other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. These changes include reductions to Medicare payments to providers of up to 2% per fiscal year that will remain in effect through 2031 unless additional Congressional action is taken, except for a temporary suspension from May 1, 2020 through March 31, 2022 and limited reductions to 1% from April 1, 2022 through June 30, 2022 due to the COVID-19 pandemic with the 2% payment reduction having resumed on July 1, 2022. Following the resumption of the sequester, under current legislation, the actual reduction in Medicare payments will vary from 1% in 2022 to up to 4% in the final fiscal year of this sequester. Further, the American Taxpayer Relief Act of 2012, which, among other things, further reduced Medicare payments to several types of providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Additionally, on March 11, 2021, President Biden signed the American Rescue Plan Act of 2021 into law, which eliminates the statutory Medicaid drug rebate cap, currently set at 100% of a drug’s average manufacturer price, for single source and innovator multiple source drugs, beginning January 1, 2024.
Further, there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several presidential executive orders, Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for pharmaceutical products. For example, the marketing, pricing and sale of the Company’s products are subject to regulation, investigations and legal actions including under the Medicaid Drug Rebate Program under the Affordable Care Act, which has increased the statutory minimum rebates a manufacturer must pay under the program as well as a new methodology by which rebates are owed for drugs that are
24
inhaled, infused, instilled, implanted or injected. We are also subject to federal and state false claims acts, as well as federal and state antitrust and consumer protection laws. Increased scrutiny of health care industry business practices in recent years by government agencies and state attorneys general in the U.S., and any resulting investigations and prosecutions, carry risk of significant civil and criminal penalties including, but not limited to, debarment from participation in such government healthcare programs.
Individual states in the United States have also become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical product and medical device pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm our business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and medical devices to purchase and which suppliers will be included in their prescription drug and other healthcare programs. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm our business, results of operations, financial condition and prospects.
We expect that the Affordable Care Act, as well as other healthcare reform measures that have been adopted and may be adopted in the future, may result in more rigorous coverage criteria, new payment methodologies and in additional downward pressure on the price that we receive for any approved or cleared product and product candidates, if approved, and could seriously harm our future revenues. Any reduction in reimbursement from Medicare, Medicaid, or other government programs may result in a similar reduction in payments from private payors. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we or our partners are slow or unable to adapt to new requirements or policies, or if we or our partners are not able to maintain regulatory compliance, our products and product candidates may lose any regulatory approval that may have been obtained, which would reduce the likelihood that we may achieve or sustain profitability, or be able to enter attractive collaboration agreements, which would adversely affect our business.
Foreign Regulation
Outside the United States, our ability to market a product is contingent upon receiving a marketing authorization from the appropriate regulatory authorities. The requirements governing the conduct of clinical trials, marketing authorization, pricing and reimbursement vary widely from country to country. At present, foreign marketing authorizations are applied for at a national level, although within the EU regional registration procedures are available to companies wishing to market a product in more than one EU member state. If the competent regulatory authority is satisfied that adequate evidence of safety, quality and efficacy has been presented, a marketing authorization may be granted. This foreign regulatory approval process involves all of the risks associated with FDA approval discussed above and may also include additional risks.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in non-US countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a process that requires the submission of a clinical trial application (“CTA”) much like an IND prior to the commencement of human clinical trials. In the EU, a CTA must be submitted for each trial to the competent health authority and to independent ethics committees by national procedure for a single country trial or by EMA submission portal Clinical Trials Information System for a multinational study. Once the CTA is approved in accordance with the requirements in the concerned countries, clinical trial development may proceed in those countries and are conducted in accordance with GCP and other applicable regulatory requirements.
To obtain regulatory approval of an investigational drug under EU regulatory systems, we must submit a marketing authorization application (“MAA”). This application is similar to the NDA in the United States, with the exception of, among other things, regional and/or country-specific document requirements. Drugs can be authorized in the EU by using the centralized, mutual recognition, decentralized or national authorization procedures described below.
25
The EMA implemented the centralized procedure for the approval of human drugs to facilitate marketing authorizations that are valid throughout the EU. This procedure results in a single marketing authorization granted by the European Commission that is valid across the EU. Under the centralized procedure, the maximum timeframe for the evaluation of an MAA by the EMA is 210 days (excluding clock stoppages for requests by the Committee for Medicinal Products for Human Use (“CHMP”) for additional written or oral information to be provided by the applicant). A positive opinion on the MAA by the CHMP then needs to be endorsed by the European Commission within approximately 67 days. Accelerated assessment might be granted by the CHMP in exceptional cases, in which case the EMA ensures that the evaluation for the opinion of the CHMP is completed within 150 days (excluding clock stops) and the opinion issued thereafter.
The mutual recognition procedure (“MRP”) for the approval of human drugs is an alternative approach to facilitate individual national marketing authorizations within the EU. The MRP may be applied for all human drugs for which the centralized procedure is not obligatory. The MRP is based on the principle of the mutual recognition by EU member states of their respective national marketing authorizations. Based on a marketing authorization in the reference member state, the applicant may apply for marketing authorizations in other member states. In such case, the reference member state shall update its existing assessment report about the drug. After the assessment is completed, copies of the report are sent to all member states, together with the approved summary of product characteristics, labeling and package leaflet. The concerned member states then recognize the decision of the reference member state and the summary of product characteristics, labeling and package leaflet. National marketing authorizations shall be granted within 30 days after acknowledgement of the agreement.
Should any member state refuse to recognize the marketing authorization by the reference member state, the member states shall make all efforts to reach a consensus. If this fails, the procedure is submitted to an EMA scientific committee for arbitration. The opinion of this EMA Committee is then forwarded to the Commission, for the start of the decision making process. As in the centralized procedure, this process entails consulting various European Commission Directorates General and the Standing Committee on Human Medicinal Products or Veterinary Medicinal Products, as appropriate.
Legislation similar to the Orphan Drug Act has been enacted in other countries outside of the United States, including the EU. The orphan legislation in the EU is available for therapies addressing conditions that affect five or fewer out of 10,000 persons, are life-threatening or chronically debilitating conditions and for which no satisfactory treatment is authorized. The market exclusivity period is for ten years, although that period can be reduced to six years if, at the end of the fifth year, available evidence establishes that the product does not justify maintenance of market exclusivity.
For other countries outside of the EU, such as non-EU countries in Eastern Europe, Middle-East, Latin America, Japan or other countries in Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary. In all cases, again, the clinical trials are conducted in accordance with GCP and the other applicable regulatory requirements.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension of clinical trials, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Competition
We may face competition from other companies in numerous industries including pharmaceuticals, medical devices and drug delivery.
Competition for larsucosterol, if approved, will depend on the specific indication(s) for which larsucosterol is approved. Akaza Bioscience Ltd., Aldeyra Therapeutics, Inc., Boehringer Ingelheim International GmbH, Immuron Ltd., Intercept Pharmaceuticals, Inc., Mallinckrodt plc, Novartis Pharma AG, PharmaKing Co. Ltd, Surrozen, Inc., and others have development plans for products to treat AH. Our current and potential competitors may succeed in obtaining patent protection or commercializing products before us. Many of these entities have significantly greater research and development capabilities than we do, as well as substantially more marketing, manufacturing, financial and managerial resources. These entities represent significant competition for us. Acquisitions of, or investments in, competing pharmaceutical or biotechnology companies by large corporations could increase such competitors’ financial, marketing, manufacturing and other resources.
26
Competition for our ALZET product line primarily consists of customers choosing to utilize delivery methods for their research projects other than an osmotic pump. We also face competition for our ALZET product line from other companies including low-cost foreign competitors.
Any pharmaceutical products we develop will compete in highly competitive markets. Many of our potential competitors in these markets have greater development, financial, manufacturing, marketing, and sales resources than we do and we cannot be certain that they will not succeed in developing products or technologies which will render our technologies and products obsolete or noncompetitive. In addition, many of those potential competitors have significantly greater experience than we do in their respective fields.
Corporate History, Headquarters and Website Information
We were incorporated in Delaware in February 1998. Our principal executive offices are located at 10260 Bubb Road, Cupertino, California 95014. Our telephone number is (408) 777-1417, and our website address is www.durect.com. Information contained on our website is not a part of this Annual Report on Form 10-K and the inclusion of our website address in this Annual Report on Form 10-K is an inactive textual reference only. We make our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to these reports available free of charge on our website as soon as reasonably practicable after we file these reports with the Securities and Exchange Commission ("SEC"). The SEC maintains an internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The SEC’s website to access all of this information is www.sec.gov. Our Code of Ethics can be found on our website.
We also use our website, including the investor relations section of our website to announce important information about us and other matters in order to achieve broad, non-exclusionary distribution of information to the public. We encourage investors and others to review the information we make public in these locations, as such information could be deemed to be material information. Information contained on or accessible through our website is not a part of this report, and all website addresses in this report are intended to be inactive textual references only.
Human Capital
Our approach to human capital resource management starts with our mission to advance novel and potentially lifesaving investigational therapies derived from our Epigenetic Regulator Program. Our industry exists in a complex regulatory environment. The unique demands of our industry, together with the challenges of running an enterprise focused on the discovery, development, manufacture and commercialization of innovative medicines, require talent that is highly educated and/or has significant industry experience. Additionally, for certain key functions, we require specific scientific expertise to oversee and conduct R&D activities and the complex manufacturing requirements for biopharmaceutical products.
The biopharmaceutical industry is highly competitive and recruiting and retaining employees is critical to the continued success of our business. We are an equal opportunity employer and we are fundamentally committed to creating and maintaining a work environment in which employees are treated with respect and dignity. All human resources policies, practices and actions related to hiring, promotion, compensation, benefits and termination are administered in accordance with the principal of equal employment opportunity, meaning that they are made on the basis of individual skills, knowledge, abilities, job performance and other legitimate criteria and without regard to race, color, religion, sex, sexual orientation, gender expression or identity, ethnicity, national origin, ancestry, age, mental or physical disability, genetic information, any veteran status, any military status or application for military service, or membership in any other category protected under applicable law. By focusing on employee retention and engagement, we also improve our ability to support our clinical trials, our pipeline, our platform technologies, business and operations, and also protect the long-term interests of our stockholders. Our success also depends on our ability to attract, engage and retain a diverse group of employees.
27
Our base pay program aims to compensate management and staff members relative to the value of the contributions of their role, which takes into account the skills, knowledge and abilities required to perform each position, as well as the experience brought to the job. We also provide annual incentive programs to reward our management team and staff members in alignment with achievement of Company-wide goals that are established annually and designed to drive aspects of our strategic priorities that support and advance our strategy across our Company. Our management team and staff members are eligible for the grant of equity awards under our long-term incentive program that are designed to align the experience of these staff with that of our stockholders. All management team and staff members also participate in a regular performance measurement process that aligns pay to performance and through which they receive performance and development feedback.
Our benefit programs are also generally broad-based, promote health and overall well-being and emphasize saving for retirement. All management team and regular staff members are eligible to participate in the same core health and welfare and retirement savings plans. Other employee benefits include employee stock purchase plan, medical plans, dental plans, vacation and sick-pay plans, employee assistance programs, flexible spending accounts, life and accident insurance and short and long-term disability benefits.
Our Compensation Committee provides oversight of our compensation plans, policies and programs.
As of March 3, 2023, we had 79 employees, including 43 in research and development, 10 in manufacturing and 26 in selling, general and administrative. At least 37 of our employees have advanced degrees of some sort (e.g., MD, PhD, DVM, JD, MBA). The Company strives for gender diversity and diversity beyond gender throughout the organization. Of our employees, 35% are male and 65% are female. From time to time, we also employ independent contractors to support our research, development and administrative organizations. None of our employees are represented by a collective bargaining unit, and we have never experienced a work stoppage. We consider our relations with our employees to be good.
Executive Officers of the Registrant
Our executive officers and their ages as of March 3, 2023 are as follows:
Name |
|
Age |
|
Position |
James E. Brown, D.V.M. |
|
66 |
|
President, Chief Executive Officer and Director |
Timothy M. Papp, M.B.A. |
|
47 |
|
Chief Financial Officer |
Norman L. Sussman, M.D. |
|
70 |
|
Chief Medical Officer |
Judy R. Joice |
|
66 |
|
Senior Vice President, Operations and Corporate Quality Assurance |
James E. Brown, D.V.M. co-founded DURECT in February 1998 and has served as our President, Chief Executive Officer and a Director since June 1998. He previously worked at ALZA Corporation as Vice President of Biopharmaceutical and Implant Research and Development from June 1995 to June 1998. Prior to that, Dr. Brown held various positions at Syntex Corporation, a pharmaceutical company, including Director of Business Development from May 1994 to May 1995, Director of Joint Ventures for Discovery Research from April 1992 to May 1995, and held a number of positions including Program Director for Syntex Research and Development from October 1985 to March 1992. Dr. Brown holds a B.A. from San Jose State University and a D.V.M. (Doctor of Veterinary Medicine) from the University of California, Davis where he also conducted post-graduate work in pharmacology and toxicology.
Timothy M. Papp, MBA, joined DURECT in July 2022 as Chief Financial Officer and brings over 25 years of corporate finance experience to DURECT, including 15 years in the Biopharma sector. Prior to joining DURECT, he was a Managing Director of Healthcare Investment Banking at RBC Capital Markets, LLC from 2020 to 2021. Previously he was a Managing Director of Healthcare Investment Banking at Stifel, Nicolaus & Company, Inc. (“Stifel”), where he worked from 2010 to 2020. Prior to Stifel, he was a Vice President of Healthcare Investment Banking at Cowen and Company, LLC (“Cowen”), where he worked from 2007 to 2010. Mr. Papp also held positions at KeyBanc Capital
28
Markets Inc. and Rodman & Renshaw LLC prior to joining Cowen. Mr. Papp graduated from Duke University with a Bachelor of Science in economics and earned a Master of Business Administration from The Wharton School with a concentration in finance.
Norman L. Sussman, M.D., FAASLD, joined DURECT as Chief Medical Officer in November 2020. He has extensive clinical experience and expertise in the field of liver disease and brings over 30 years of clinical research and development experience in academia and industry. Prior to joining DURECT he was an Associate Professor of Medicine and Surgery at Baylor College of Medicine and a faculty member of Baylor College of Medicine intermittently since 1985. During that time, he served as a Principal Investigator for research focused on the assessment and management of acute liver failure and artificial liver support. Dr. Sussman gained leadership experience in industry as the founder and Vice President of both Amphioxus Cell Technologies from 1995 to 2003 and Hepatix, Inc from 1993 to 1995. Most recently, he has also served in senior leadership roles as a member of the Baylor Faculty Senate and as Director of the telehealth program, Project ECHO®, at Baylor St. Luke’s Medical Center. Dr. Sussman received his MBBCh from the University of the Witwatersrand in Johannesburg, South Africa. He then completed his residency at St. Louis University Hospital and his post-doctoral fellowship at Washington University. He is Board Certified in Internal Medicine, Gastroenterology, and Transplant Hepatology. Dr. Sussman is also a Fellow of the American Association of the Study of Liver Disease, which is a designation that recognizes his superior level of professional achievement in the field of hepatology.
Judy R. Joice has served as our Senior Vice President, Operations and Corporate Quality Assurance since March 2014 and as our Vice President, Operations and Corporate Quality Assurance since April 2011. Previously, Ms. Joice served as our Vice President, Corporate Quality Assurance since July 2008 and as our Executive Director, Quality Assurance from July 2007 to July 2008. She has over 30 years’ experience in the pharmaceutical industry with such companies as Nektar Therapeutics, Oread, Roche Pharmaceuticals, and Syntex Research. During her career, Ms. Joice has gained broad experience in CMC development activities including novel excipients, new chemical entities, devices, and combination products. She has developed, implemented and managed all aspects of company-wide quality systems and compliance functions, ranging from drug development through commercial manufacturing. Ms. Joice has a B.S. in Chemistry from California State University, Hayward.
29
Item 1A. Risk Factors.
In addition to the other information in this Annual Report on Form 10-K, a number of factors may affect our business and prospects. These factors include but are not limited to the following, which you should consider carefully in evaluating our business and prospects. If any of the following risks actually occur, our business, financial condition, results of operations and growth prospects may be materially and adversely affected.
Summary
30
31
Risks Related To Our Business
We are dependent on the success of larsucosterol and the path to regulatory approval is uncertain; we cannot be certain that it will receive regulatory approval or be commercialized
Our business depends substantially on the successful development of larsucosterol, which has completed multiple clinical trials, including a Phase 1b clinical trial in NASH and a Phase 2a clinical trial in AH, and is continuing to enroll patients for a Phase 2b clinical trial (AHFIRM) in patients with severe AH and anticipate enrolling the last patient in the AHFIRM trial in the second quarter of 2023 with top-line results to be reported in the second half of 2023. In AH and NASH, there are no currently approved drugs. Ongoing and future clinical trials will need to establish clinically and statistically significant proof of efficacy, and sufficient evidence of safety to support filing for regulatory approval and/or additional clinical trials and ultimately regulatory approval. Larsucosterol will require additional development, including completion of the ongoing AHFIRM clinical trial and potentially additional clinical trials as well as potentially further preclinical studies, and other non-clinical parameters, to obtain regulatory clearances before it can be commercialized. We will have to interact with the FDA and other regulatory agencies regarding important aspects of the clinical development program, potentially including the size and design of clinical trials, the specific primary and secondary endpoints for the clinical trials, inclusion and exclusion criteria, stopping rules, duration of follow up, size of the safety databases, statistical analysis plans and other matters. Positive results obtained during early development do not necessarily mean later development will succeed or that regulatory clearances or approvals will be obtained. Changes to any of these aspects of our clinical trial design could result in the requirement for additional trials or delay development and approval of larsucosterol. Our drug development efforts may not lead to commercial drugs for several reasons, such as if larsucosterol fails to be shown to be safe and effective or if we do not have adequate financial or other resources to advance larsucosterol through clinical development and the approval processes. We consider larsucosterol to be our lead and most important asset. If larsucosterol fails to demonstrate safety or efficacy at any time or during any phase of development, we would experience potentially significant delays in, or be required to abandon development of larsucosterol, which would materially harm our business. Even if the Phase 2b AHFIRM trial successfully demonstrates a reduction in mortality or liver transplantation over placebo plus standard of care, (1) additional clinical trial(s) may be required to support an NDA filing and ultimately to support approval by FDA and/or other regulatory bodies; and (2) accelerated regulatory pathways (such as an FDA priority review designation) may not be available.
We do not anticipate that larsucosterol will be eligible to receive regulatory approval from the FDA or comparable foreign authorities and begin commercialization for a number of years, if ever. This uncertainty may make it difficult to predict the timing or expense required to obtain regulatory approval for larsucosterol. We also may need to revise our clinical development plans after trials have commenced or have been completed, which could add to the time and expense associated with the clinical development of larsucosterol. If we are unable to reach an agreement with the FDA or other regulatory agencies regarding clinical development plans for larsucosterol, we may curtail or limit our development activities for this product candidate. Even if we ultimately receive regulatory approval for larsucosterol, we or our potential future partners, if any, may be unable to commercialize it successfully for a variety of reasons. These include, for example, the availability of alternative, potentially superior or less expensive treatments, lack of cost-effectiveness, the lack of favorable access and/or commercial pricing, the cost or technical challenges of manufacturing the product on a commercial scale and competition with other treatments. The success of larsucosterol may also be limited by the prevalence and severity of any adverse side effects, including mortality. If we fail to obtain regulatory approval and successfully commercialize larsucosterol, we may be unable to raise sufficient capital or generate sufficient revenues to attain or maintain profitability, and our financial condition and stock price may decline.
We will require and may have difficulty or be unsuccessful in raising needed capital in the future to continue to operate as a going concern
Our business currently does not generate sufficient revenues to meet our capital requirements and we do not expect that it will do so in the near future. We have expended and will continue to expend substantial funds to conduct the research, development, manufacturing and clinical testing of our product candidates, funding and establishing additional clinical- and commercial-scale manufacturing
32
arrangements and facilities, and to provide for the pre-commercial and commercial activities associated with the marketing, sales and distribution of our products and product candidates.
Presently, we do not have sufficient cash resources to meet our plans for the next twelve months from the issuance of these financial statements. Our recurring losses from operations, negative cash flows and need for additional capital raise substantial doubt about our ability to continue as a going concern. As a result, our independent registered public accounting firm included an explanatory paragraph in its report on our financial statements as of, and for the year ended, December 31, 2022. We will require additional financing to fund our operations or we will have to significantly curtail our operations to conserve our capital resources. Additional funds may not be available on acceptable terms, if at all, and such availability will depend on a number of factors, some of which are outside of our control, including general capital markets conditions and investors’ view of our prospects and valuation. Further, investors’ perception of our ability to continue as a going concern may make it more difficult for us to obtain financing, or necessitate that we obtain financing on terms that are more favorable to investors, and could result in the loss of confidence by investors, suppliers and employees. Our continued operations are contingent on our ability to enter into new collaborative agreements, raise additional capital and obtain financing and success in future operations. If we do not acquire sufficient additional funding or alternative sources of capital to meet our working capital needs, we will have to substantially curtail our operations and business plan resulting in delays in generating future revenue.
Our actual capital requirements will depend on many factors, including:
We may consume available resources more rapidly than currently anticipated, resulting in the need for additional funding. We may seek to raise additional funds through equity or debt financings, convertible debt financings, collaborative arrangements with corporate collaborators or other sources, which, in each case, may be dilutive to existing stockholders and may cause the price of our common
33
stock to decline. In addition, in the event that additional funds are obtained through arrangements with collaborators or other sources, we may have to relinquish rights to some of our technologies, products or product candidates that we would otherwise seek to develop or commercialize ourselves.
The FDA’s Fast Track Designation of larsucosterol may not lead to a faster development or regulatory review or approval
The FDA grants Fast Track Designation to therapies that are considered capable of addressing unmet medical needs and possess the potential to treat serious or life-threatening disease conditions in order to facilitate its development and expedite the review procedure. Even though larsucosterol has received Fast Track Designation for the treatment of AH, we may not experience a faster development process, review or approval compared to conventional FDA procedures, or receive FDA approval at all, in that indication or any other. A Fast Track Designation does not change the standards for approval. The FDA may also withdraw Fast Track Designation if it believes that the designation is no longer supported by data from our clinical development program.
In addition, the statutes and regulations that define the timelines and criteria for approval of drugs and biologics are subject to change by the U.S. Congress and the responsible administrative agencies. For example, the Prescription Drug User Fee Act (“PDUFA”) authorizes the FDA to collect fees and use them for the review of human drug applications and defines the review time targets for such applications. The current legislative authority for PDUFA will expire in September 2027. New legislation will then be required for the FDA to continue collecting prescription drug user fees in future fiscal years and for manufacturers to have clarity regarding the time the FDA will spend in review before granting regulatory approval. If PDUFA reauthorization is not completed in the future, the review and approval times for new drugs like larsucosterol could be significantly longer than currently expected, which could delay potential marketing approval and launch.
Safety data and indications of activity from completed Phase 1 and 2 clinical trials of larsucosterol may not predict safety, activity or therapeutic efficacy in future trials
Safety data and indications of activity from completed Phase 1 and 2 clinical trials of larsucosterol may ultimately not be correlated with treatment or improvement in the associated disease, and there is a risk that larsucosterol may not demonstrate therapeutic efficacy in larger placebo-controlled trials such as AHFIRM. The failure of larsucosterol to show efficacy in one indication may negatively affect its perceived value in other indications, and the emergence of safety signals in ongoing or future clinical trials would significantly harm our business.
Open-label trials of larsucosterol in NASH and AH have inherent limitations
The most recently completed NASH and AH trials of larsucosterol were open-label trials with no control groups. Open label trials have inherent risk of bias given that the patients and physicians know that the patients received active study drug, which can lead to placebo effects. Trials without control groups have an inherent risk in that the comparisons used to determine the study drug’s effect and side effect profile are based on comparisons with baseline (pre-treatment) levels (for blood chemistry and biomarker endpoints) and/or with historical controls, which may not have been conducted under similar enough conditions to make accurate comparisons and/or draw accurate conclusions from those comparisons. Additionally, larger placebo-controlled clinical trials are required to evaluate the safety and efficacy of larsucosterol to treat any indication, including AH and NASH. There can be no assurance that ongoing or future studies will demonstrate the safety or efficacy of larsucosterol in a statistically significant or clinically meaningful manner.
Ongoing and future clinical trials for larsucosterol may be delayed and may not demonstrate efficacy or safety
The Phase 2b AHFIRM trial of larsucosterol in patients with AH is subject to potential delays. For example, the uncertainty of COVID-19 has impacted and may in the future impact our clinical trial sites in the U.S., U.K., E.U. and Australia, which affects the predictability and timing of availability of top-line data from this trial. There can be no assurance that the trial will complete enrollment as anticipated if at all, and delays in enrollment could add to the costs and expenses of this trial and harm our business. There can also be no assurance that biological activity demonstrated in previous animal disease models or earlier clinical trials of larsucosterol will also be seen in ongoing trials or future clinical trials, or that any
34
clinically relevant biological activity will be observed, or that enrollment rates will be favorable or that these additional trials will not identify safety issues. Failure of the AHFIRM trial to achieve desired results in its anticipated timeframe would negatively impact our business and ability to raise additional capital.
We may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates
We may experience delays in completing our preclinical studies and initiating or completing clinical trials, and we may experience numerous unfavorable events during, or as a result of, any future clinical trials that we may conduct that could delay or prevent our ability to receive marketing approval or commercialize our product candidates, including:
We could encounter delays if a clinical trial is suspended or terminated by us, by the IRBs of the institutions at which such trials are being conducted, by the Data Safety Monitoring Board for such trial or by the FDA or other regulatory authorities. Such authorities may impose such a suspension or termination or clinical hold due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities, changes in clinical trial design, safety
35
issues or adverse side effects, failure to demonstrate a benefit from using a product, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. Many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates. Further, the FDA may disagree with our clinical trial design and our interpretation of data from clinical trials, or may change the requirements for approval even after it has reviewed and commented on the design for our clinical trials.
Our product development costs will also increase if we experience delays in testing or regulatory approvals. We do not know whether any of our future clinical trials will begin as planned, or whether any of our current or future clinical trials will need to be restructured or will be completed on schedule, if at all. Significant preclinical study or clinical trial delays, also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our product candidates and may harm our business and results of operations. Any delays in our ongoing or future preclinical or clinical development programs may harm our business, financial condition and prospects significantly.
Key components of larsucosterol are provided by a limited number of suppliers, and supply shortages or loss of these suppliers could result in delays or interruptions in supply or increased costs
While we have entered into contract manufacturing agreements with multiple vendors for larsucosterol, we currently have a third-party sole supplier for GMP supplies of larsucosterol. This third party is our sole source for the drug product required for development and commercialization of this drug candidate.
The reliance on a sole or limited number of suppliers could result in:
There can be no assurance that we will receive sufficient quantities of larsucosterol to commence and conduct the non-clinical trials, clinical trials and CMC activities we are planning, and delays in supply could delay development of larsucosterol. In addition, certain of our third-party manufacturers and suppliers may be experiencing delays as a result of the COVID-19 pandemic or have otherwise encountered delays in providing their goods and services. As a result, we may not be able to manufacture our product candidates for our clinical trials and conduct other research and development operations and maintain current clinical and pre-clinical timelines. In addition, if additional third parties in our supply chain are adversely impacted by restrictions resulting from the pandemic, including staffing shortages, raw material shortages, production slowdowns and/or disruptions in delivery systems, our supply chain may be disrupted in other ways, further limiting our ability to manufacture our product candidates for our clinical trials and conduct our research and development operations.
We have supply agreements in place for certain components of our products and product candidates, but do not have in place long term supply agreements with respect to all of the components of any of our products or product candidates. Therefore, the supply of a particular component could be terminated without DURECT’s consent at any time without penalty to the supplier. In addition, we may not be able to procure required components or drugs from third-party suppliers at a commercially reasonable quantity, quality, cost and timing. In addition, certain of our suppliers may be experiencing delays as a result of the COVID-19 pandemic or have otherwise encountered delays in providing their services. Any interruption in the supply of single source components (including active pharmaceutical ingredients, excipients, or components like vials, stoppers, filters and the like), products or product candidates, could cause us to seek alternative sources of supply or attempt to manufacture these items internally if feasible. Furthermore, in some cases, we are relying on our third-party collaborators to procure supply of necessary components. If the supply of any components for our products or product candidates is interrupted,
36
components from alternative suppliers may not be available in sufficient volumes or at acceptable quality levels within required timeframes, if at all, to meet our needs or those of our third-party collaborators. This could delay our ability to obtain commercial product supplies or complete development and obtain approval for commercialization and marketing of our product candidates, causing us to lose sales, incur additional costs, delay new product introductions and could harm our reputation and make access to capital more difficult, expensive or impossible. Supply chain disruptions have affected and are likely to continue to affect the manufacturing and shipment of goods globally. Any delay in production or delivery of the components and drug substances used in our products or product candidates for any reason, including due to an extended closure of our suppliers’ plants as a result of efforts to limit the spread of COVID-19, could adversely impact our business and hinder our growth.
Macroeconomic uncertainties have in the past impacted and may continue to adversely impact our business, including posing challenges to conducting clinical trials
Global economic and business activities continue to face widespread macroeconomic uncertainties, including labor shortages and supply chain disruptions, inflation and monetary supply shifts, as well as recession risks, which may continue for an extended period, which may have an adverse impact on the economies and financial markets of many countries, resulting in a severe and prolonged global economic downturn that could continue to affect demand for our ALZET product line and could have an adverse impact on our business operations and financial condition. Further, such macroeconomic uncertainties may also adversely impact our ability to raise additional capital to provide sufficient funding to continue our product development efforts, including clinical trials, which would make it more difficult for companies such as ours to access capital. For example, the global COVID-19 pandemic delayed the initiation of our AHFIRM Phase 2b clinical trial to evaluate the safety and efficacy of larsucosterol in severe AH patients and it has delayed, and may in the future delay, the pace of enrollment in this trial and other clinical trials.
Additionally, inflation has the potential to adversely affect our liquidity, business, financial condition and results of operations by increasing our overall cost structure. The existence of inflation in the economy has resulted in, and may continue to result in, higher interest rates and capital costs, supply shortages, increased costs of labor, increased manufacturing costs and clinical trial costs, weakening exchange rates and other similar effects. As a result of inflation, we have experienced and may continue to experience cost increases.
The extent to which such macroeconomic uncertainties impact our operations will depend on future developments, which are highly uncertain and cannot be predicted with confidence. As a result, there have been and may continue to be longer lead times required for acquiring components and supplies used in manufacturing of larsucosterol, and there have been periods of reduced demand for our ALZET products, which are used in scientific and pre-clinical research. We may continue to experience disruptions that could severely impact our business, preclinical studies and clinical trials including:
37
Delays or difficulties in the enrollment of subjects in clinical trials may increase our overall development expenses and delay clinical trial data and receipt of necessary regulatory approvals
Successful and timely completion of clinical trials will require that we enroll a sufficient number of subjects and/or patients within a reasonable period of time. Enrollment, a significant factor in the timing of clinical trials, is affected by many factors including the size and nature of the patient population, our ability to recruit clinical sites and the ability of clinical sites to successfully recruit subjects to participate in clinical trials. Initiation of and enrollment in many clinical trials has been and may in the future be adversely affected by COVID-19, which has at times caused some institutions to stop enrolling patients, has created a large number of clinical trial proposals for potential clinical trial sites to review and consider, and has caused many individuals to avoid contact with hospitals or other healthcare providers. Additionally, some of the patients in our clinical trials, including AH patients, are hospitalized and concerns about exposure to COVID-19 limit clinical trial staff’s access to patients, the frequency of interactions between patients and staff, the ability to obtain blood draws and other biological sample collection, and may limit the ability to ship samples to outside laboratories for analysis. In areas heavily impacted by COVID-19, there may be limited hospital staff available for clinical trial activities due to staff becoming infected or due to de-prioritization of clinical trial activities. Trials may be subject to delays as a result of patient enrollment taking longer than anticipated or patient withdrawal. We may not be able to initiate or continue clinical trials for larsucosterol if we are unable to sign and maintain sufficient clinical sites, locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA or other regulatory authorities or if we are unable to collect and analyze biological samples required for trial endpoints. It is possible that the inclusion and exclusion criteria for patients to be enrolled in these trials or COVID-19-related issues may make the trials more difficult to conduct or may significantly extend the time required for enrollment and the cost of these trials.
We cannot predict how successful we will be at enrolling patients in our clinical trials. Enrollment is affected by many factors including:
38
Our inability to sign up and maintain sufficient clinical trial sites and/or enroll a sufficient number of patients for clinical trials would result in significant delays and could require us to abandon one or more clinical trials altogether. Enrollment delays in these clinical trials may result in increased development costs for our drug candidates or delays in regulatory filings and approvals, which would cause the value of our Company to decline and limit our ability to obtain additional financing.
The FDA or other regulatory agencies may require more information or clinical studies for our product candidates, and our product candidates may never be approved
The failure to adequately demonstrate the safety and effectiveness of a pharmaceutical product candidate under development to the satisfaction of FDA and other regulatory agencies will result in delays to the regulatory approval or non-approvability of our product candidates and could materially harm our business. Clinical trials may not demonstrate the sufficient levels of safety and efficacy necessary to obtain the requisite regulatory approvals for our product candidates or may require such significant numbers of patients or additional costs to make it impractical to satisfy the regulatory agency’s requirements, and thus our product candidates may not be approved for marketing. During the review process, the FDA or other regulatory agencies may request additional information regarding the efficacy or safety of our product candidates and providing such additional information could require significant additional work and expense, and take a significant amount of time, resulting in a material delay of approval or the failure to obtain approval or lead our Company to abandon the development of that product candidate. During the review process, the FDA, or other regulatory agencies, may also request more information regarding the chemistry, manufacturing or controls related to our product candidates, and answering such questions could require significant additional work and expense, and take a significant amount of time, resulting in a material delay of approval or the failure to obtain approval or abandonment of the product candidate. Additionally, even if our product candidates receive FDA or other regulatory agency approval, the regulatory agency may require that we conduct additional clinical or non-clinical studies after such approval, place limitations on the use of our products in applicable labels, require marketing under a Risk Evaluation and Mitigation Strategy program, include commercially unattractive language in the approved product label, delay approval to market our products or limit the indicated use of our products, which may harm our business and results of operations.
We have a significant amount of debt. Compliance with repayment obligations and other covenants may be difficult, and failure to fulfill our obligations under the applicable loan agreements may cause our repayment obligations to accelerate
In July 2016, we entered into a Loan and Security Agreement (as amended, the “Loan Agreement”) with Oxford Finance LLC ("Oxford Finance"), pursuant to which Oxford Finance provided a $20 million secured single-draw term loan to us with an initial maturity date of August 1, 2020. The term loan was fully drawn at close and the proceeds may be used for working capital and general business requirements. The term loan repayment schedule provided initially for interest only payments for the first 18 months, followed by consecutive monthly payments of principal and interest in arrears starting on March 1, 2018 and continuing through the maturity date of August 1, 2020. Following five amendments, we make interest only payments under the amended Loan Agreement until June 1, 2023 and the final maturity date of the loan is September 1, 2025. The Loan Agreement provides for a floating interest rate (7.95% initially and 11.45% as of December 31, 2022) based on an index rate plus a spread and an additional payment equal to 10% of the principal amount of the term loan, which is due when the term loan becomes due or upon the prepayment of the facility. Any increases in prevailing interest rates could increase our
39
expenses under the Loan Agreement. If we elect to prepay the loan, there is also a prepayment fee between 0.75% and 2.5% of the principal amount of the term loan depending on the timing of prepayment. Our debt repayment obligations under the Loan Agreement, as amended, may prove a burden to our Company as they become due, particularly following the expiration of the interest-only period. Increased payment requirements that are scheduled to start after June 2023 will increase our cash expenditures and require us to raise additional capital or renegotiate or refinance the Loan Agreement. There can be no assurance that additional capital will be available on acceptable terms, if at all, or that we would be able to successfully renegotiate or refinance the Loan Agreement on acceptable terms, if at all. If our debt repayments increase, we may be required to scale back development programs or other operations, which could have an adverse effect on our business.
The Loan Agreement contains customary events of default, including, among other things, our failure to fulfill certain of our obligations under the Loan Agreement and the occurrence of a material adverse change in our business, operations or condition (financial or otherwise), a material impairment of the prospect of repayment of any portion of the loan, the failure to deliver an unqualified audit report and board approved financial projections within time periods set forth in the Loan Agreement, or a material impairment in the perfection or priority of lender’s lien in the collateral or in the value of such collateral. In the event of default by us under the Loan Agreement, the lender would be entitled to exercise its remedies thereunder, including the right to accelerate the debt, upon which we may be required to repay all amounts then outstanding under the Loan Agreement, which could harm our business, operations and financial condition.
In addition, the term loan is secured by substantially all of our assets, except that the collateral does not include any equity interests in our Company, any intellectual property (including all licensing, collaboration and similar agreements relating thereto), and certain other excluded assets. The Loan Agreement contains customary representations, warranties and covenants by us, which covenants limit our ability to convey, sell, lease, transfer, assign or otherwise dispose of certain of our assets; engage in any business other than the businesses currently engaged in by us or reasonably related thereto; liquidate or dissolve; make certain management changes; undergo certain change of control events; create, incur, assume, or be liable with respect to certain indebtedness; grant certain liens; pay dividends and make certain other restricted payments; make certain investments; make payments on any subordinated debt; and enter into transactions with any of our affiliates outside of the ordinary course of business or permit our subsidiaries to do the same. Complying with these covenants may make it more difficult for us to successfully execute our business strategy.
We do not control the commercialization of POSIMIR, PERSERIS or Methydur
We rely on Innocoll for the commercialization of POSIMIR. The current approved labeling for POSIMIR is limited, and Innocoll is responsible for completing post-marketing non-clinical studies and any additional studies required by FDA, and negative results from these studies could adversely affect commercialization of POSIMIR. Innocoll is also responsible for manufacturing POSIMIR. If Innocoll does not successfully grow POSIMIR sales, the royalty payments we receive under our agreement with them will be limited and we may not receive additional milestone payments from them. We rely on Indivior for the commercialization of PERSERIS. There can be no assurance that PERSERIS sales will maintain current levels or grow materially. If Indivior does not successfully grow PERSERIS sales, future earn-out payments we receive under our agreement with them will be limited. Both POSIMIR and PERSERIS are subject to boxed warnings that may make them more difficult to commercialize. We rely on Orient Pharma for the commercialization of Methydur. If Orient Pharma does not successfully grow Methydur sales, the royalty payments we receive under our agreement with them will be limited. Further, the sales of each of these products may be negatively impacted by challenging macroeconomic conditions.
For certain of our product candidates, we depend to a large extent on third-party collaborators, and we have limited or no control over their development, sales, distribution and disclosure for those product candidates
Our performance for certain of our product candidates depends to a large extent on the ability of our third-party collaborators to successfully develop and obtain regulatory approvals. We have entered into
40
agreements with Innocoll, Indivior and Orient Pharma under which we granted such third parties the right to develop, apply for regulatory approval for, market, promote or distribute certain products or product candidates, subject to payments to us in the form of product royalties, earn-out and other payments. We have limited or no control over the expertise or resources that any collaborator may devote to the development, clinical trial strategy, regulatory approval, marketing or sale of these product candidates, or the timing of their activities. Any of our present or future collaborators may not perform their obligations as expected. These collaborators may breach or terminate their agreement with us or otherwise fail to conduct their collaborative activities successfully and in a timely manner. Enforcing any of these agreements in the event of a breach by the other party could require the expenditure of significant resources and consume a significant amount of management time and attention. Our collaborators may also conduct their activities in a manner that is different from the manner we would recommend or would have chosen had we been developing such product candidates ourselves. Further, our collaborators may elect not to develop or commercialize product candidates arising out of our collaborative arrangements or not devote sufficient resources to the development, clinical trials, regulatory approval, manufacture, marketing or sale of these product candidates. If any of these events occur, we may not recognize revenue from the commercialization of our product candidates based on such collaborations. In addition, these third parties may have similar or competitive products to the ones which are the subject of their collaborations with us, or relationships with our competitors, which may reduce their interest in developing or selling our products or product candidates. We may not be able to control public disclosures made by some of our third-party collaborators, which could negatively impact our stock price.
Cancellation of third-party collaborations may adversely affect potential economic benefits
Third-party collaboration agreements typically allow the third party to terminate the agreement (or a specific program within an agreement) at will by providing notice. Termination can result from failure of the collaboration to achieve anticipated milestones, from changes in strategy of the other party or for other reasons. In these cases, the product rights revert to us or certain rights of the partner to use our proprietary technology are terminated. If there have been payments under such agreements that are being recognized over time, termination of such agreements (or programs) can lead to a near-term increase in our reported revenues resulting from the immediate recognition of the balance of such payments. Termination deprives us of potential future economic benefits under such agreements, and may make it more difficult, unattractive or impossible to enter into agreements with other third parties for use of the assets and/or technologies that were subject to the terminated agreement. For example, termination of our agreements with Innocoll or Orient Pharma could have negative effects on our Company.
If we do not enter into new collaboration agreements, our revenues and/or cash flows will be reduced relative to prior periods
Our revenues have been based to a significant extent on collaborative arrangements with third parties, pursuant to which we receive payments based on our performance of research and development activities set forth in these agreements. For example, approximately 58% of our total revenues in 2019 were derived from our collaboration agreement with Gilead. In June 2020, Gilead notified us that they were terminating this collaboration, resulting in accelerated recognition of $22.7 million in deferred revenue related to a nonrefundable upfront license fee and a milestone payment totaling $35.0 million that had previously been received. In addition, we have seen periodic fluctuations in revenues associated with our other collaboration agreements, which reflect the current development stage of the product candidates subject to those agreements, and our collaborator’s needs for our services. Long-term growth of our collaboration revenues requires us to enter into new collaboration agreements, and there can be no assurance that we will do so. Even if we enter into new collaboration agreements, we may not be able to fulfill our obligations or attain milestones set forth in any specific agreement, which could cause our revenues and/or cash flows to fluctuate or be less than anticipated and may expose us to liability for contractual breach. In addition, these agreements may require us to devote significant time and resources to communicating with and managing our relationships with such collaborators and resolving possible issues of contractual interpretation which may detract from time our management would otherwise devote to managing our operations. Such agreements are generally complex and contain provisions that could give rise to legal disputes, including potential disputes concerning ownership of intellectual property. Such disputes can delay or prevent the development of potential new product candidates, or can lead to
41
lengthy, expensive litigation or arbitration. In general, our collaboration agreements, may be terminated by the other party at will or upon specified conditions including, for example, if we fail to satisfy specified performance milestones or if we breach the terms of the agreement. Acquisitions of our collaborators or strategic changes or re-organizations or re-prioritizations of our collaborators can lead to turnover of program staff, a review of development programs and strategies by the acquirer, and other events that can disrupt a program, resulting in program delays or discontinuations.
If we do not enter into new collaboration agreements, our anticipated revenues and/or cash flows will be reduced relative to periods of increased collaborative research and development revenues, such as occurred in 2020.
Our cash flows are likely to differ from our reported revenues and earnings
Our revenues and earnings may differ from our cash flows from revenue-generating activities. Upfront payments received upon execution of collaborative agreements may be recorded as deferred revenue, in which case they would be recognized over the period of performance for the related performance obligations with the third-party collaborator pursuant to the applicable agreement. The period of performance obligations may also be revised on a prospective basis. As of December 31, 2022, we recognized $812,000 of revenue that was previously classified as deferred revenue, which resulted in our reported revenue being greater than cash flows from our ongoing revenue-generating activities. Assumptions related to revenue recognition for performance obligations provided over time are reviewed in each accounting period and changes are recorded in the current period. In certain circumstances, changes in assumptions related to the measure of progress for a performance obligation performed over time could result in negative revenue or the acceleration of revenue for an accounting period.
Our business strategy includes relying on third parties to support development, clinical testing, manufacturing and commercialization of our products and product candidates.
Our current business strategy includes reliance on third-party CROs, consultants, service providers and suppliers to provide critical services to support development, clinical testing, and manufacturing of our products and product candidates, including, but not limited to larsucosterol and others. For example, we currently depend on third-party vendors to manage and monitor most of our clinical trials. We rely on third parties to manufacture or perform manufacturing steps relating to our products, product candidates and components. We anticipate that we will continue to rely on these and other third-party contractors to support development, clinical testing, and manufacturing of our products and product candidates. Third parties may not execute their responsibilities and tasks competently in compliance with their contractual obligations to us, applicable laws and regulations or in a timely or cost-effective fashion. Failure of these contractors to provide the required services in a competent or timely manner or on reasonable commercial terms could materially delay the development and approval of our product candidates or commercialization of our products, increase our expenses and materially harm our business, financial condition, results of operations and access to capital.
Failure to comply with governmental regulations could materially harm our business
Developing, manufacturing, marketing or promoting a drug is subject to very strict regulations and controls. Furthermore, clearance or approval may entail ongoing requirements for post-marketing studies or surveillance. The manufacture and marketing of drugs are subject to continuing FDA and foreign regulatory review and requirements that we update our regulatory filings. Later discovery of previously unknown problems with a product, manufacturer or facility, or our failure to update regulatory files, may result in restrictions, including withdrawal of the product from the market. Any of the following or other similar events, if they were to occur, could delay or preclude us from further developing, marketing or realizing full commercial value of our products or product candidates, which in turn would materially harm our business, financial condition and results of operations:
42
Manufacturers of drugs must comply with the applicable FDA GMP regulations, which include production design controls, testing, quality control and quality assurance requirements as well as the corresponding maintenance of records and documentation. Compliance with current GMP regulations is difficult and costly. Manufacturing facilities are subject to ongoing periodic inspection by the FDA and corresponding state and in some cases, foreign agencies, including unannounced inspections, and must be licensed before they can be used for the commercial manufacture of our products and product candidates. We and/or our present or future suppliers and distributors may be unable to comply with the applicable GMP regulations and other FDA and/or foreign regulatory requirements. If we, our third-party collaborators or our respective suppliers do not achieve compliance for our products or product candidates we or they manufacture, the FDA or foreign equivalents may refuse or withdraw marketing clearance or approvals, put our or our partner’s clinical trial on hold, withdraw or reject an investigational NDA or require product recall, which may cause interruptions or delays in the development, manufacture and sale of our products and product candidates.
We have a history of operating losses, expect to continue to have losses and may never achieve or maintain profitability and we may not successfully manage our Company through varying business cycles
We have incurred significant operating losses since our inception in 1998 and, as of December 31, 2022, had an accumulated deficit of approximately $561.4 million. We expect to continue to incur significant operating losses over the next several years as we continue to incur significant costs for research and development, clinical trials, manufacturing, sales, and general and administrative functions. Our ability to achieve profitability depends upon our ability, alone or with others, to successfully complete the development of our proposed product candidates, obtain the required regulatory clearances, manufacture and market our proposed product candidates and successfully commercialize our approved products. Development of pharmaceutical product candidates is costly and requires significant investment. In addition, we may choose to license from third parties either rights to particular drugs or other appropriate technology and/or intellectual property rights for use in our products and product candidates. The license fees as well as the operating costs of using or developing these technologies or rights would increase the costs of our products and product candidates as well as our operating costs generally.
Our current revenues are from milestones and royalties from Innocoll related to POSIMIR, the ALZET product line, from certain excipient sales, from earn-out payments from Indivior related to sales of PERSERIS, from royalty payments from Orient Pharma related to sales of Methydur in Taiwan, and from payments under collaborative research and development agreements with third parties. We expect our revenues to decrease in the near future, and we do not expect that our revenues will exceed our operating expenses in the near future. We do not anticipate meaningful revenues to derive from the commercialization and marketing of our products and product candidates in the near future, do not expect to receive additional milestone payments in the near term or meaningful royalties from POSIMIR until the product achieves meaningful sales (if ever) and therefore do not expect to generate sufficient revenues to cover expenses or achieve profitability in the near future.
43
Our success will depend on properly sizing our Company through growth and contraction cycles caused in part by changing business conditions, which places a significant strain on our management and on our administrative, operational and financial resources. For example, in connection with the COVID-19 pandemic, we required most of our personnel, including all of our administrative employees, to work remotely, restricted on-site staff to only mandatory personnel, implemented social distancing on-site, and closed certain of our offices temporarily. Our increased reliance on personnel working remotely makes us more susceptible to reduced productivity, disruptions, delays, and other adverse impacts on our business. In addition, this model could increase our cyber security risk, create data accessibility concerns, and make us more susceptible to communication disruptions, any of which could adversely impact our business operations or delay necessary interactions with the FDA, manufacturing sites, research or clinical trial sites. To mitigate future cycles, we may expand or contract our facilities, our operational, financial and management systems and our personnel. If we are unable to manage growth and contractions effectively, our business would be harmed.
Changes in tax law could adversely affect our business and financial condition
The rules dealing with U.S. federal, state, and local income taxation are constantly under review by persons involved in the legislative process and by the IRS and the U.S. Treasury Department. Changes to tax laws (which changes may have retroactive application) could adversely affect us or holders of our common stock. In recent years, many such changes have been made and changes are likely to continue to occur in the future. For example, on March 11, 2021, President Biden signed into law the “American Rescue Plan Act”, which included extenders to the refundable employee retention credit under the Coronavirus Aid, Relief, and Economic Security ("CARES") Act and limitations to executive compensation effective for tax years beginning after 2026. Future changes in tax laws could have a material adverse effect on our business, cash flow, financial condition or results of operations.
We may develop our own sales force and commercial group to market future products but we have limited sales and marketing experience and may not be able to do so effectively
We have a small sales and marketing group focused on our ALZET product line. We may choose to develop our own sales force and commercial group to market larsucosterol, if approved, or other products that we may develop in the future. Developing a sales force and commercial group would require substantial expenditures and the hiring of qualified personnel. We have limited sales and marketing experience, and may not be able to effectively recruit, train or retain sales and marketing personnel. If we are not able to put in place an appropriate sales force and commercial group for our products in development and provide that commercial team with sufficient financial and other resources, we may not be able to effectively launch or commercialize these or any other products. We may not be able to effectively sell our products and product candidates, if approved, and our failure to do so could limit or materially harm our business.
We and our third-party collaborators may not sell our product candidates effectively
We and our third-party collaborators (including Innocoll, Indivior and Orient Pharma) compete with many other companies that currently have extensive and well-funded marketing and sales operations. Our marketing and sales efforts and those of our third-party collaborators may be unable to compete successfully against these other companies. We and our third-party collaborators, where applicable, may be unable to establish a sufficient sales and marketing organization on a timely basis, if at all. We and our third-party collaborators, where applicable, may be unable to engage qualified distributors. Even if engaged, these collaborators and distributors may:
44
The failure of us or our third-party collaborators to effectively develop, gain regulatory approval for, sell, manufacture and market our products and product candidates will hurt our business, prospects, financial results and may impact our access to capital.
Write-offs related to impairment of goodwill, long-lived assets, inventories and other non-cash charges may adversely impact profitability and cause cash flows to differ from reported earnings
We may incur significant non-cash charges related to impairment write-downs of our long-lived assets, including goodwill. We are required to perform periodic impairment reviews of our goodwill at least annually. The carrying value of goodwill on our balance sheet was $6.2 million at December 31, 2022. To the extent these reviews conclude that the expected future cash flows generated from our business activities are not sufficient to recover the cost of our long-lived assets, we will be required to measure and record an impairment charge to write-down these assets to their realizable values. We completed our last review during the fourth quarter of 2022 and determined that goodwill was not impaired as of December 31, 2022. However, there can be no assurance that upon completion of subsequent reviews a material impairment charge will not be recorded. If future periodic reviews determine that our assets are impaired and a write-down is required, it will adversely impact or delay our profitability.
Inventories, in part, include certain excipients that are sold to customers and included in products and product candidates in development. These inventories are capitalized based on management’s judgment of probable sale prior to their expiration date which in turn is primarily based on management’s internal estimates. The valuation of inventory requires us to estimate the value of inventory that may become expired prior to use. We may be required to expense previously capitalized inventory costs upon a change in our judgment, due to, among other potential factors, a denial or delay of approval of a product by the necessary regulatory bodies, changes in product development timelines, or other information that suggests that the inventory will not be saleable.
Global credit and financial market conditions could negatively impact the value of our investments
Our cash and cash equivalents are maintained in highly liquid investments with remaining maturities of 90 days or less at the time of purchase. Our short-term investments consist primarily of readily marketable debt securities with original maturities of greater than 90 days from the date of purchase but remaining maturities of less than one year from the balance sheet date. Our long-term investments consist primarily of readily marketable debt securities with maturities of one year or beyond from the balance sheet date. No assurance can be given that deterioration in conditions of the global credit and financial markets would not negatively impact our current portfolio of cash equivalents, short-term investments or long-term investments or our ability to meet our financing objectives.
We depend upon key personnel who may terminate their employment with us at any time, and we may not be able to attract and retain sufficient qualified personnel on a timely basis, if at all
Our success will depend to a significant degree upon the continued services of key management, technical and scientific personnel. In addition, our success will depend on our ability to attract and retain other highly skilled personnel, particularly as we develop and expand our Epigenetic Regulator Program. Competition for qualified personnel is intense, and the process of hiring and integrating such qualified personnel is often lengthy. The market for qualified personnel in the San Francisco Bay Area is very competitive and we may be unable to recruit such personnel on a timely basis, if at all. Our management and other employees may voluntarily terminate their employment with us at any time. Further, in 2021 and 2022, the labor market in the U.S. experienced a significant increase in workers leaving their positions (often referred to as the “Great Resignation”), which made the market to replace these individuals competitive and resulted in significant wage inflation in response to labor shortages. During the Great Resignation we faced and may continue to face increased challenges of employee attraction and
45
retention. The loss of the services of key personnel, or the inability to attract and retain additional qualified personnel, could result in delays to product development or approval, loss of sales and diversion of management resources as well as difficulties or inability to raise sufficient capital to fund our Company’s operations.
Cyber-attacks or other failures in telecommunications or information technology systems could result in information theft, data corruption and significant disruption of our business operations
We utilize information technology, systems and networks to process, transmit and store electronic information in connection with our business activities. As use of digital technologies has increased, cyber incidents, including deliberate attacks and attempts to gain unauthorized access to computer systems and networks, have increased in frequency and sophistication. These threats pose a risk to the security of our systems and networks and the confidentiality, availability and integrity of our data, and may cause a disruption in our operations, harm our reputation, cause us to pay to retrieve our data if it becomes infected or otherwise subject to ransomware, and increase our stock trading risk. There can be no assurance that we will be successful in preventing cyber-attacks or successfully mitigating their effects. Similarly, there can be no assurance that our third-party collaborators, distributors and other contractors and consultants will be successful in protecting our clinical and other data that is stored on their systems. Any cyber-attack or destruction or loss of data could have a material adverse effect on our business and prospects. In addition, we may suffer reputational harm or face litigation or adverse regulatory action as a result of cyber-attacks or other data security breaches and may incur significant additional expense to implement further data protection measures.
Our corporate headquarters, certain manufacturing facilities and personnel are located in a seismically active area near wildfire zones
Our corporate headquarters, certain manufacturing facilities and personnel are located in a geographical area that is known to be seismically active and prone to earthquakes, as well as wildfires and related power outages or power shortages. Should such a natural disaster or power outage or power shortage occur, our ability to conduct our business could be severely restricted, and our business and assets, including the results of our research, development and manufacturing efforts, could be harmed or destroyed.
Our business involves environmental risks and risks related to handling regulated substances
In connection with our research and development activities and our manufacture of materials, products and product candidates, we are subject to federal, state and local laws, rules, regulations and policies governing the use, generation, manufacture, storage, air emission, effluent discharge, handling and disposal of certain materials, biological specimens and wastes. Although we believe that we have complied with the applicable laws, regulations and policies in all material respects and have not been required to correct any material noncompliance, we may be required to incur significant costs to comply with environmental and health and safety regulations in the future. Our research and development involves the use, generation and disposal of hazardous materials, including but not limited to certain hazardous chemicals, solvents, agents and biohazardous materials. Although we believe that our safety procedures for storing, handling and disposing of such materials comply with the standards prescribed by state and federal regulations, we cannot completely eliminate the risk of accidental contamination or injury from these materials. We currently contract with third parties to dispose of these substances generated by us, and we rely on these third parties to properly dispose of these substances in compliance with applicable laws and regulations. If these third parties do not properly dispose of these substances in compliance with applicable laws and regulations, we may be subject to legal action by governmental agencies or private parties for improper disposal of these substances. The costs of defending such actions and the potential liability resulting from such actions are often very large. In the event we are subject to such legal action or we otherwise fail to comply with applicable laws and regulations governing the use, generation and disposal of hazardous materials and chemicals, we could be held liable for any damages that result, and any such liability could exceed our resources.
46
As a non-accelerated filer, we are not required to comply with the auditor attestation requirements of the Sarbanes-Oxley Act and, consequently, some investors may find our common stock less attractive
We are a non-accelerated filer as defined by Rule 12b-2 of the Exchange Act, and as such, are not required to provide an auditor attestation of management’s assessment of internal control over financial reporting, which is generally required for SEC reporting companies under Section 404(b) of the Sarbanes-Oxley Act. Therefore, our internal controls over financial reporting will not receive the level of review provided by the process relating to the auditor attestation included in annual reports of issuers that are subject to the auditor attestation requirements. Because we are not required to have our auditors provide an attestation of our management’s assessment of internal control over financial reporting, a material weakness in internal control may remain undetected for a longer period. In addition, investors may find our common stock less attractive because we are not required to comply with the auditor attestation requirements. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and the trading price for our common stock as well as our ability to raise capital may be negatively affected.
If we seek approval to commercialize our current or future drug candidates outside of the United States, a variety of risks associated with international operations could harm our business
If we seek approval of our current or future drug candidates outside of the United States, we expect that we will be subject to additional risks including:
We have no prior experience in these areas. In addition, there are complex regulatory, tax, labor and other legal requirements imposed by many of the individual countries in and outside of Europe with which we will need to comply. Many biopharmaceutical companies have found the process of marketing their own products in foreign countries to be very challenging.
47
Risks Related to Our Intellectual Property
If we are unable to protect, maintain or enforce our intellectual property rights or secure rights to third-party intellectual property, we may lose valuable assets, lose market share or incur costly litigation or our third-party collaborators may choose to terminate their agreements with us
Our ability to commercially exploit our products will depend significantly on our ability to obtain and maintain patents, maintain trade secret protection and operate without infringing the proprietary rights of others.
As of March 3, 2023, we owned or exclusively in-licensed over 25 unexpired issued U.S. patents and over 170 unexpired issued foreign patents (which include granted European patent rights that have been validated in various EU member states). In addition, we have over 30 pending U.S. patent applications and over 190 foreign applications pending in Europe, Australia, Japan, Canada and other countries.
There can be no assurance that the pending patent applications will be granted. Further, there can be no assurance that VCU will not attempt to terminate their license to us, which termination could result in the loss of our rights to certain of these patent families.
The patent positions of pharmaceutical companies, including ours, are uncertain and involve complex legal and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued. Consequently, our patent applications or those that are licensed to us may not issue into patents, and any issued patents may not provide protection against competitive technologies or may be held invalid if challenged. Our competitors may also independently develop products similar to ours or design around or otherwise circumvent patents issued to us or licensed by us. In addition, the laws of some foreign countries may not protect our proprietary rights to the same extent as U.S. law, if at all.
We also rely upon trade secrets, technical know-how and continuing technological innovation to develop and maintain our competitive position. We require our employees, consultants, advisors and collaborators to execute confidentiality and assignment-of-inventions agreements with us. These agreements typically provide that all materials and confidential information developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances, and that all inventions arising out of the individual’s relationship with us will be our exclusive property. These agreements may be breached, and in some instances, we may not have an appropriate remedy available for breach of the agreements. Furthermore, our competitors may independently develop substantially equivalent proprietary information and techniques, reverse engineer our information and techniques, or otherwise gain access to our proprietary technology.
We may be unable to meaningfully protect our rights in trade secrets, technical know-how and other non-patented technology. We may have to resort to litigation or arbitration to protect our intellectual property rights, or to determine their scope, validity or enforceability. In addition, interference, derivation, post-grant oppositions, and similar proceedings may be necessary to determine rights to inventions in our patents and patent applications. Enforcing or defending our proprietary rights is expensive, could cause diversion of our resources and may be unsuccessful. Any failure to enforce or protect our rights could cause us to lose the ability to exclude others from using our technology to develop or sell competing products. In addition, in some circumstances our collaborators have the first right to enforce our patents against third party infringers, and such collaborators may not enforce such claims adequately or successfully or in the manner that we would do ourselves.
48
Our collaboration agreements may depend on our intellectual property
We are party to collaborative agreements with Innocoll and Orient Pharma, among others. Our third-party collaborators have entered into these agreements based on the exclusivity that our intellectual property rights confer on the products being developed. The loss or diminution of our intellectual property rights could result in a decision by our third-party collaborators to terminate their agreements with us. In addition, these agreements are generally complex and contain provisions that could give rise to legal disputes, including potential disputes concerning ownership of intellectual property and data under collaborations. Such disputes can lead to lengthy, expensive litigation or arbitration requiring us to devote management time and resources to such disputes which we would otherwise spend on our business.
We may be sued by third parties claiming that our products or product candidates infringe on their intellectual property rights, particularly because there is substantial uncertainty about the validity and breadth of biopharmaceutical patents
We or our collaborators may be exposed to future litigation by third parties based on claims that our products, product candidates or activities infringe the intellectual property rights of others. We may also be subject to claims asserting that we, our collaborators, our employees, consultants or advisors have wrongfully used or disclosed alleged trade secrets of their current or former employers or claims asserting ownership of what we regard as our own intellectual property. These risks are exacerbated by the fact that the validity and breadth of claims covered in medical technology, pharmaceutical and biotechnology patents and the breadth and scope of trade secret protection involve complex legal and factual questions for which important legal principles are unresolved. Any litigation or claims against us or our collaborators, whether or not valid, could result in substantial costs, could place a significant strain on our financial resources and could harm our reputation and business prospects. We also may not have sufficient funds to litigate, particularly against parties with substantially greater resources. In addition, pursuant to our collaborative agreements, we have provided our collaborators with the right, under specified circumstances, to defend against any claims of infringement of the third-party intellectual property rights, and such collaborators may not defend against such claims adequately or successfully or in the manner that we would do ourselves. Intellectual property litigation or claims could force us or our collaborators to do one or more of the following, any of which could harm our business or financial results:
Risks Related To Our Industry
The markets for our pharmaceutical products, product candidates and for our ALZET product line are rapidly changing and competitive, and new products or technologies developed by others could impair our ability to establish, maintain or grow our business and remain competitive
The pharmaceutical industry is subject to rapid and substantial technological change. Developments by others may render our products, product candidates under development or technologies noncompetitive or obsolete, or we may be unable to keep pace with technological developments or other market factors. Technological competition in the industry from pharmaceutical and biotechnology companies, universities, governmental entities and others diversifying into the field is intense and is expected to increase.
49
We may face competition from other companies in numerous industries including pharmaceuticals, biotechnology, medical devices and drug delivery. Competition for larsucosterol, if approved, will depend on the specific indication(s) for which larsucosterol is approved. Akaza Bioscience Ltd., Aldeyra Therapeutics, Inc., Boehringer Ingelheim International GmbH, Immuron Ltd., Intercept Pharmaceuticals, Inc., Mallinckrodt plc, Novartis Pharma AG, PharmaKing Co. Ltd, Surrozen, Inc., and others have development plans for products to treat AH.
Competition for our ALZET product line primarily consists of customers choosing to utilize delivery methods for their research projects other than an osmotic pump. We also face competition for our ALZET product line from other companies including low cost foreign competitors.
We are engaged in the development of novel therapeutic technologies. Our resources are limited and we may experience technical challenges inherent in such novel technologies. Competitors have developed or are in the process of developing technologies that are, or in the future may be, the basis for competitive products. Some of these products may have an entirely different approach or means of accomplishing similar therapeutic effects than our products and product candidates. Our competitors may develop products that are safer, more effective or less costly than our products and product candidates and, therefore, present a serious competitive threat to our product candidates and product offerings.
The widespread acceptance of therapies that are alternatives to ours may limit market acceptance of our products and product candidates if commercialized. For example, post-operative pain is currently being treated by oral medication, transdermal drug delivery systems, such as drug patches, long-acting and short-acting injectable products and implantable drug delivery devices which are competitive with our products and product candidates. Many of these treatments are widely accepted in the medical community and have a long history of use. The established use of these competitive products may limit the potential for our products and product candidates to receive widespread acceptance if and when commercialized.
Our relationships with physicians, patients and third-party payers are subject to anti-kickback, fraud and abuse, privacy and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings
Healthcare providers, physicians and third-party payers will play a primary role in the recommendation and prescription of POSIMIR and any additional product candidates for which we obtain marketing approval. Our future arrangements with third-party payers and customers may expose us and our partners to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we and our partners may market, sell and distribute our products. As a pharmaceutical company, even though we do not and may not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payers, federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. These regulations include:
50
Efforts to ensure that our business arrangements with third parties comply with applicable healthcare and privacy laws and regulations do and will in the future involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare or privacy laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any physicians or other healthcare providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
51
Healthcare reform measures could hinder or prevent our product candidates’ commercial success
In the United States and some foreign jurisdictions, there have been, and we expect there will continue to be, a number of legislative and regulatory changes to the healthcare system, including cost-containment measures that may reduce or limit coverage and reimbursement for newly approved drugs, that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities, affect our ability to profitably sell any product or product candidates for which we obtain marketing approval and otherwise affect our future revenue and profitability and the future revenue and profitability of our collaborators or potential collaborators. In particular, there have been and continue to be a number of initiatives at the U.S. federal and state levels that seek to reduce healthcare costs and improve the quality of healthcare. For examples of healthcare reform measures, see “Part I, Item 1. Business—Government Regulation—Healthcare Reform” above.
We could be exposed to significant product liability claims which could be time consuming and costly to defend, divert management attention and adversely impact our ability to obtain and maintain insurance coverage
The testing, clinical development, manufacture, marketing and sale of our products and product candidates involve an inherent risk that product liability claims will be asserted against us. Our present product liability insurance may be inadequate and may not fully cover the costs of any claim(s) or any ultimate damages we might be required to pay. Product liability claims or other claims related to our products and product candidates, regardless of their outcome, could require us to spend significant time and money in litigation or to pay significant damages. Any successful product liability claim may prevent us from obtaining adequate product liability insurance in the future on commercially desirable or reasonable terms. In addition, product liability coverage may cease to be available in sufficient amounts or at an acceptable cost. An inability to obtain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or inhibit the commercialization of our products or product candidates if and when approved. A product liability claim could also significantly harm our reputation and delay or prevent market acceptance of our products and product candidates.
Market acceptance of, and market opportunity for, our products or product candidates is uncertain, and failure to achieve market acceptance will delay our ability to generate or grow revenues
Our future financial performance will depend upon the successful introduction and customer acceptance of our products or products we have licensed to others, including larsucosterol, if approved, and Innocoll’s POSIMIR, Indivior’s PERSERIS and Orient Pharma’s Methydur. Even if approved for marketing, these products and product candidates may not achieve market acceptance or the market opportunities for our current and potential future product candidates may be smaller than we predicted, which could adversely affect our future product revenues and could cause our business to suffer. The degree of market acceptance will depend upon a number of factors, including:
Physicians, patients, payers or the medical community in general may be unwilling to accept, utilize or recommend any of the products we have developed. If these products do not achieve widespread market acceptance, we will not achieve meaningful revenues.
52
If users of our products are unable to obtain adequate reimbursement from third-party payers, obtain access to our product(s), or if new restrictive legislation is adopted, market acceptance of our products may be limited and we may not achieve meaningful revenues or profitability
The continuing efforts of government and insurance companies, health maintenance organizations and other payers of healthcare costs to contain or reduce costs of health care may affect our future revenues and profitability, and the future revenues and profitability of our potential customers, suppliers and third-party collaborators and the availability of capital. For example, in certain foreign markets, pricing, access and/or profitability of prescription pharmaceuticals is subject to government control. In the United States, recent federal and state government initiatives have been directed at lowering the total cost of health care, and the U.S. Congress and state legislatures will likely continue to focus on health care reform, the cost of prescription pharmaceuticals and on the reform of the Medicare and Medicaid systems. While we cannot predict whether any such legislative or regulatory proposals will be adopted, the announcement or adoption of such proposals could materially harm our business, financial condition and results of operations.
The successful commercialization of our current and future products will depend in part on the extent to which appropriate reimbursement levels for the cost of our products and related treatment are obtained from governmental authorities, private health insurers and other organizations, such as health maintenance organizations (“HMOs”). Third-party payers often limit access, payments and/or reimbursement for medical products and services. Also, the trend toward managed health care in the United States and the concurrent growth of organizations such as HMOs, which could control or significantly influence the purchase of health care services and products, as well as legislative proposals to reform health care or reduce government insurance programs, may limit access, reimbursement or payment for our products. The cost containment measures that health care payers and providers are instituting and the effect of any health care reform could materially harm our ability to operate profitably and access capital.
If we or our third-party collaborators are unable to train physicians to use our products and product candidates to treat patients’ diseases or medical conditions, we may not achieve market acceptance of our products
Broad use of certain of our products or out-licensed products, such as POSIMIR, will require extensive training of numerous physicians on their proper and safe use. The time required to train physicians could delay adoption of our products and adversely affect market acceptance of our products. We or third parties selling our products may be unable to rapidly train physicians in numbers sufficient to generate adequate demand for our products. Any delay in training would materially delay the demand for our products and harm our business and financial results. In addition, we or our partners may expend significant funds towards such training before any orders are placed for our products, which would increase our expenses and harm our financial results.
Risks related to actions on trade by the U.S. and foreign governments could adversely affect our Company's results of operations and financial condition
The U.S. government under the previous administration indicated its intent to adopt a new approach to trade policy and in some cases to renegotiate, or potentially terminate, certain existing bilateral or multilateral trade agreements. It also initiated the imposition of tariffs on certain foreign products. Changes in U.S. trade policy have resulted in, and could continue to result in, one or more U.S. trading partners adopting responsive trade policy making it more difficult or costly for us to export our products to those countries. These measures could also result in increased costs for goods imported into the United States. This in turn could require us to increase prices to our customers which may reduce demand, or, if we are unable to increase prices, result in lowering our margin on products sold.
53
There is also a concern that the imposition of additional tariffs by the United States could result in the adoption of additional tariffs by other countries. A potential resulting trade war could have a significant adverse effect on world trade and the world economy. We cannot predict future trade policy or the terms of any renegotiated trade agreements and their impact on our business. The adoption and expansion of trade restrictions, the occurrence of a trade war, or other governmental action related to tariffs or trade agreements or policies has the potential to adversely impact demand for our products, our costs, our customers, our suppliers, and the U.S. economy, which in turn could adversely impact our business, financial condition, access to capital and results of operations.
Risks Related To Our Common Stock
Our stock price has in the past and may in the future not meet the minimum bid price for continued listing on Nasdaq. Our ability to continue operations or to publicly or privately sell equity securities and the liquidity of our common stock could be adversely affected if we are delisted from Nasdaq
In several instances in the past, including as recently as February 9, 2022, we received written notifications from Nasdaq informing us that because the closing bid price of our common stock was below $1.00 for 30 consecutive trading days (the “Minimum Closing Bid Price Requirement”), our shares no longer complied with the Minimum Closing Bid Price Requirement for continued listing on Nasdaq under Nasdaq Marketplace Rules. Each time, we were given a period of 180 days from the date of the notification and in one case an extra 180-day period to regain compliance with Nasdaq’s listing requirements by having the closing bid price of our common stock listed on Nasdaq be at least $1.00 for at least 10 consecutive trading days.
While we have regained compliance within the applicable time periods in the past, if our shares again no longer comply with the Minimum Closing Bid Price Requirement for continued listing on the Nasdaq Capital Market under Nasdaq Marketplace Rule 5550(a)(2) and we do not regain compliance within the applicable 180-day time period, Nasdaq will notify us that our securities will be subject to delisting. One strategy to regain compliance in such circumstances would be to implement a reverse stock split. For example, we implemented such a strategy to regain compliance with the Minimum Closing Bid Price Requirement when we completed a 1-for-10 reverse stock split on December 5, 2022 (the “Stock Split”). We could also appeal Nasdaq’s determination to delist our securities to a Hearings Panel. During any appeal process, shares of our common stock would continue to trade on the Nasdaq Capital Market.
There can be no assurance that we will maintain compliance with the requirements for listing our common stock on the Nasdaq Capital Market. Delisting from Nasdaq would constitute an event of default under our loan facility with Oxford, entitling Oxford to accelerate our obligations under such facility, among other actions. Under such circumstances, we could be required to renegotiate the repayment terms of our loan facility, on terms which would not be as favorable to our Company as our current terms, or we could be required to take other actions, such as discontinuing some or all of our operations, selling assets, or other action. Delisting could also adversely affect our ability to raise additional capital through the public or private sale of equity securities, would significantly affect the ability of investors to trade our securities and would negatively affect the value and liquidity of our common stock. Delisting could also have other negative results, including the potential loss of confidence by employees, the loss of institutional investor interest and fewer business development opportunities.
Additionally, there can be no assurance that the Stock Split will result in a per-share market price that will maintain compliance with the Minimum Closing Bid Price Requirement, that will attract institutional investors or investment funds or that such share price will satisfy investing guidelines of institutional investors or investment funds. As a result, the trading liquidity of our common stock may not improve. Further, if the market price of our common stock declines, the percentage decline may be greater than would have occurred in the absence of a reverse stock split.
Our operating history makes evaluating our stock difficult
Our quarterly and annual results of operations have historically fluctuated and we expect will continue to fluctuate for the foreseeable future. We believe that period-to-period comparisons of our operating results should not be relied upon as predictive of future performance. Our prospects must be considered in light of the risks, expenses and difficulties encountered by companies with a limited number
54
of approved pharmaceutical products, particularly companies in new and rapidly evolving markets such as pharmaceuticals and biotechnology. To address these risks, we must, among other things, obtain regulatory approval for and commercialize our product candidates, which may not occur. We may not be successful in addressing these risks and difficulties. We expect to require additional funds to complete the development of larsucosterol or our other product candidates, and to fund operating losses to be incurred in the next several years.
Investors may experience substantial dilution of their investment
In order to raise capital and for other purposes, we may in the future offer and issue additional shares of our common stock or other securities convertible into or exchangeable for our common stock, and the price per share at which we sell additional shares of our common stock or other securities convertible into or exchangeable for our common stock in future transactions may be higher or lower than the price per share at which investors in our common stock bought their shares. In July 2021, we filed the 2021 Registration Statement to sell up to $250 million of securities from time to time in one or more public offerings, including up to $75.0 million of shares of common stock through the 2021 Sales Agreement. Any sales in the public market of our common stock, under the 2021 Sales Agreement, in offerings under our shelf registration statement or otherwise, could adversely affect prevailing market prices for our common stock. Through several financings between 2019 and 2022, and through our 2015 Sales Agreement, 2018 Sales Agreement and 2021 Sales Agreement with Cantor Fitzgerald during this period, we have raised an aggregate of $79.5 million. Further, on February 3, 2023, we consummated a registered direct offering financing pursuant to which we sold an aggregate of 1,700,000 shares of our common stock, pre-funded warrants to purchase up to 300,000 shares of our common stock and common warrants to purchase up to 2,000,000 shares of our common stock. Each share of common stock and accompanying common warrant and each pre-funded warrant and accompanying common warrant were sold together at a combined offering price of $5.00 per share and accompanying warrant or, in the case of pre-funded warrants, $4.99999 per pre-funded warrant and accompanying common warrant. You could experience substantial dilution of your investment as a result of subsequent exercises of the outstanding warrants. As of March 3, 2023, we had up to $240.0 million of our securities available for sale under the 2021 Registration Statement, of which $75.0 million of our common stock are available pursuant to the 2021 Sales Agreement.
In addition, as of December 31, 2022, 2,843,416 shares of our common stock were issuable upon exercise of stock options outstanding under our stock option plans at a weighted average exercise price of $12.97 per share, 2,171,128 additional shares of common stock were reserved for potential future issuance under our stock option plan, and an aggregate of 25,455 shares of common stock were reserved for potential future issuance under our 2000 Employee Stock Purchase Plan. At December 31, 2022, we had 150,000,000 authorized shares of common stock and, as such, we have the ability to issue significantly more shares and options in the future, which would result in substantial dilution to our stockholders, including investors in this offering.
Our ability to use net operating losses and certain other tax attributes is uncertain and may be limited
Our ability to use our federal and state net operating losses to offset potential future taxable income and related income taxes that would otherwise be due is dependent upon our generation of future taxable income before the expiration dates of the net operating losses, and we cannot predict with certainty when, or whether, we will generate sufficient taxable income to use any or all of our net operating losses. In addition, utilization of net operating losses to offset potential future taxable income and related income taxes that would otherwise be due is subject to annual limitations under the “ownership change” provisions of Sections 382 and 383 of the Internal Revenue Code of 1986, as amended (the "Internal Revenue Code") and similar state provisions, which may result in the expiration of net operating losses before future utilization. In general, under the Code, if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period, the corporation’s ability to use its pre-change net operating losses and other pre-change tax attributes (such as research and development credit carryforwards) to offset its post-change taxable income or taxes may be limited. Our equity offerings and other changes in our stock ownership, some of which are outside of our control, may have resulted or could in the future result in an ownership change.
55
If an ownership change limitation were to apply, utilization of our net operating losses and tax credit carryforwards could be limited in future periods and a portion of the carryforwards could expire before being available to reduce future income tax liabilities.
Because our Company is a “smaller reporting company,” we may take advantage of certain scaled disclosures available to us, resulting in holders of our securities receiving less Company information than they would receive from a public company that is not a smaller reporting company
We are a “smaller reporting company” as defined in the Exchange Act. As a smaller reporting company, we may take advantage of certain of the scaled disclosures available to smaller reporting companies and will be able to take advantage of these scaled disclosures for so long as (i) our voting and non-voting common stock held by non-affiliates is less than $250 million measured on the last business day of our second fiscal quarter, or (ii) our annual revenue is less than $100 million during the most recently completed fiscal year and our voting and non-voting common stock held by non-affiliates is less than $700 million measured on the last business day of our second fiscal quarter. To the extent we take advantage of any reduced disclosure obligations, it may make it harder for investors to analyze our Company’s results of operations and financial prospectus in comparison with other public companies.
The price of our common stock may be volatile
The stock markets in general, and the markets for pharmaceutical stocks in particular, have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. These broad market fluctuations may adversely affect the trading price of our common stock.
Price declines in our common stock could result from general market and economic conditions and a variety of other factors, including:
56
The market price of our common stock may fluctuate significantly in response to factors which are beyond our control. The stock market in general has periodically experienced extreme price and volume fluctuations. For example, the COVID-19 pandemic, pronouncements by the Federal Reserve, inflation, outbreaks of war such as between Russia and Ukraine, oil price volatility and other factors have caused broad stock market and industry fluctuations. In addition, the market prices of securities of technology and pharmaceutical companies have also been extremely volatile, and have experienced fluctuations that often have been unrelated or disproportionate to the operating performance of these companies. These broad market fluctuations could result in extreme fluctuations in the price of our common stock, which could cause a decline in the value of our common stock.
In the past, following periods of volatility in the market price of a particular company’s securities, litigation has often been brought against that company. If litigation of this type is brought against us, it could be extremely expensive, particularly if we were to lose the lawsuit and have to pay damages, and divert management’s attention and our Company’s resources.
We have broad discretion over the use of our cash and investments, and their investment may not always yield a favorable return
Our management has broad discretion over how our cash and investments are made and used. We may from time to time invest in ways with which our stockholders may not agree and that do not yield favorable returns.
Our certificate of incorporation, our bylaws and Delaware law contain provisions that could discourage another company from acquiring us
Provisions of Delaware law, our certificate of incorporation and bylaws may discourage, delay or prevent a merger or acquisition that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions include:
57
Our bylaws provide that the Court of Chancery of the State of Delaware is the exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees
Our bylaws provide that the Court of Chancery of the State of Delaware is the exclusive forum for any derivative action or proceeding brought on behalf of our Company, any action asserting a claim of breach of a fiduciary duty owed by any director, officer or other employee of our Company, any action asserting a claim arising pursuant to any provision of the General Corporation Law of Delaware or our Certificate of Incorporation or bylaws or any action asserting a claim governed by the internal affairs doctrine. The choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees.
Alternatively, if a court were to find the choice of forum provision contained in our certificate of incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could adversely affect our business and financial condition.
58
Item 1B. Unresolved Staff Comments.
None.
Item 2. Properties.
The following chart indicates the facilities that we lease, the location and size of each such facility and their designated use.
Location |
|
Approximate Square Feet |
|
Operation |
|
Expiration |
Cupertino, CA |
|
30,149 sq. ft. |
|
Office, Laboratory and Manufacturing |
|
Lease expires 2024 (with an option to renew for an additional five years) |
|
|
|
|
|
|
|
Cupertino, CA |
|
20,100 sq. ft. |
|
Office and Laboratory |
|
Lease expires 2024 (with an option to renew for an additional five years) |
|
|
|
|
|
|
|
Vacaville, CA |
|
24,634 sq. ft. |
|
Manufacturing |
|
Lease expires 2023 (with an option to renew for an additional five years) |
|
|
|
|
|
|
|
We believe that our existing facilities are adequate to meet our current and foreseeable requirements or that suitable additional or substitute space will be available as needed.
Item 3. Legal Proceedings.
We are not a party to any material legal proceedings.
Item 4. Mine Safety Disclosures.
Not applicable.
59
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information
Our common stock is traded on the Nasdaq Capital Market under the symbol “DRRX”.
Holders
As of March 3, 2023, there were approximately 79 holders of record of the common stock. This does not include the number of persons whose stock is in nominee or “street name” accounts through brokers.
Dividend Policy
We have never paid cash dividends on our common stock. We currently intend to retain any future earnings to fund the development and growth of our business. Therefore, we do not currently anticipate paying any cash dividends in the foreseeable future.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
None.
Item 6. [Reserved]
60
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
This Management’s Discussion and Analysis of Financial Condition and Results of Operations as of December 31, 2022 and 2021 should be read in conjunction with our Financial Statements, including the Notes thereto, and “Risk Factors” section included elsewhere in this Annual Report on Form 10-K. References to the “Company,” “DURECT,” “we,” “us” and “our” refer to DURECT Corporation.
Special Note Regarding Forward-Looking Statements
This Form 10-K contains forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended, and Section 27A of the Securities Act of 1933, as amended. When used in this annual report or elsewhere by management from time to time, the words “believe,” “anticipate,” “intend,” “plan,” “estimate,” “expect,” “may,” “will,” “could,” “potentially,” “possibility,” and similar expressions are forward-looking statements. Such forward-looking statements contained herein are based on current expectations and beliefs. Actual events or results may differ materially from those discussed in the forward-looking statements as a result of various factors.
Forward-looking statements made in this report include, but are not limited to, statements about:
61
We caution you that the foregoing list may not contain all of the forward-looking statements made in this Annual Report on Form 10-K. Forward-looking statements are not guarantees of future performance and involve risks and uncertainties. Actual events or results may differ materially from those discussed in the forward-looking statements as a result of various factors. For a more detailed discussion of such forward looking statements and the potential risks and uncertainties that may impact upon their accuracy, see the “Risk Factors” section and “Overview” section of this Management’s Discussion and Analysis of Financial Condition and Results of Operations. These forward-looking statements reflect our view only as of the date of this report. We undertake no obligations to update any forward-looking statements. You should also carefully consider the factors set forth in other reports or documents that we file from time to time with the Securities and Exchange Commission (“SEC”).
This discussion and analysis generally addresses 2022 and 2021 items and year-over-year comparisons between 2022 and 2021. Discussions of 2020 items and year-over-year comparisons between 2021 and 2020 that are not included in this Annual Report on Form 10-K can be found in “Item
62
7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our Annual Report on Form 10-K for the fiscal year ended December 31, 2021, filed with the SEC on March 8, 2022, which is available free of charge on the SEC’s website at www.sec.gov and the investor relations section of our website at www.durect.com/investors/sec-filings/. These website addresses are intended to be inactive, textual references only. None of the materials on, or accessible through, these websites are part of this report or are incorporated by reference herein. Throughout this section, references to number of shares, stock price and exercise price have generally been conformed to reflect the effects of the Company’s 1-for-10 reverse stock split, effective December 5, 2022, unless otherwise specified herein.
Overview
We are a biopharmaceutical company advancing novel and potentially lifesaving investigational therapies derived from our Epigenetic Regulator Program. Larsucosterol (also known as “DUR‑928”), a new chemical entity in clinical development, is the lead candidate in our Epigenetic Regulator Program. An endogenous, orally bioavailable small molecule, larsucosterol has been shown in both in vitro and in vivo studies to play an important regulatory role in lipid metabolism, stress and inflammatory responses, and cell death and survival. We are developing larsucosterol for alcohol-associated hepatitis (“AH”), a life-threatening acute liver condition with no approved therapeutics and a 28-Day and 90-Day historical mortality rate of 20%-26% and 29%-31%, respectively. After completing a Phase 2a trial in which 100% of AH patients treated with larsucosterol survived the 28-Day study period, we are now conducting a ~300-patient, double-blind, placebo-controlled Phase 2b clinical trial called AHFIRM (trial in AH to evaluate saFety and effIcacy of laRsucosterol treatMent). Through our AHFIRM trial, we are evaluating larsucosterol’s potential to reduce mortality or liver transplantation compared to a placebo with or without steroids at the investigators’ discretion. Currently we anticipate dosing the last patient in the AHFIRM trial in the second quarter of 2023. If the AHFIRM trial is successful, it may support a New Drug Application (“NDA”) filing and we may decide to develop our own commercial, sales and marketing organization. We have also investigated larsucosterol in patients with NASH with encouraging results in a Phase 1b clinical trial and are considering further development of larsucosterol for this and other indications.
In addition to our Epigenetic Regulator Program, we developed a novel and proprietary post-surgical pain product called POSIMIR that utilizes our innovative SABER® platform technology to enable continuous sustained delivery of bupivacaine, a non-opioid local analgesic, over three days in adults. In February 2021, POSIMIR received U.S. FDA approval for post-surgical pain reduction for up to 72 hours following arthroscopic subacromial decompression. In December 2021, we entered into a license agreement (the “Innocoll Agreement”) with Innocoll Pharmaceuticals Limited (“Innocoll”), pursuant to which the Company granted to Innocoll an exclusive, royalty-bearing, sublicensable right and license to develop, manufacture and commercialize POSIMIR in the United States. In September 2022, Innocoll launched POSIMIR in the U.S.
As a result of the assignment of certain patent rights, we also receive single digit sales-based earn-out payments from U.S. net sales of Indivior UK Limited’s (“Indivior”) PERSERIS™ (risperidone) drug for schizophrenia and single-digit royalties from net sales of Orient Pharma Co., Ltd.’s (“Orient Pharma”) Methydur Sustained Release Capsules (“Methydur”) for the treatment of attention deficit hyperactivity disorder (“ADHD”) in Taiwan. We also manufacture and sell ALZET® osmotic pumps used in laboratory research.
NOTE: POSIMIR® is a trademark of Innocoll Pharmaceuticals, Ltd. in the U.S. and a trademark of DURECT Corporation outside of the U.S. SABER®, CLOUD™, ORADUR™ and ALZET® are trademarks of DURECT Corporation. Other trademarks referred to belong to their respective owners. Full prescribing information for POSIMIR, including BOXED WARNING and Medication Guide can be found at www.posimir.com. Full prescribing information for PERSERIS, including BOXED WARNING and Medication Guide can be found at www.perseris.com.
63
Collaborative Research and Development and Other Revenue
Collaborative research and development and other revenue consists of three broad categories: (a) the recognition of upfront license payments over the period of our continuing involvement with the third party, (b) the reimbursement of qualified research expenses by third parties, (c) milestone payments in connection with our collaborative agreements and (d) royalties and earn out payments from our agreements with third parties. During the last two years, we generated collaborative research and development revenues from collaborative agreements with Innocoll and others.
Product Revenue
We also currently generate product revenue from the sale of two product lines:
Because we consider our core business to be developing and commercializing pharmaceuticals, we do not intend to significantly increase our investments in or efforts to sell or market any of our existing product lines. However, we expect that we will continue to make efforts to increase our revenues related to collaborative research and development by extending and expanding our current collaborations, and by entering into new collaborations.
Operating Results
Since our inception in 1998, we have generally had a history of operating losses. At December 31, 2022, we had an accumulated deficit of $561.4 million. Our net losses were $35.3 million and $36.3 million for the years ended December 31, 2022 and 2021, respectively. These losses have resulted primarily from costs incurred to research and develop our product candidates and, to a lesser extent, from selling, general and administrative costs associated with our operations and product sales. We expect our research and development expenses to decrease in 2023 compared to 2022 as we expect to complete the AHFIRM trial and to incur lower contract manufacturing costs for larsucosterol in 2023. We expect our selling, general and administrative expenses to increase in 2023 compared to 2022 due to higher market research expenses and employee expenses in 2023. We expect to incur continuing losses and negative cash flows from operations for the foreseeable future. As disclosed in the “Liquidity and Capital Resources” section, we have concluded that substantial doubt exists about our ability to continue as a going concern for a period of at least 12 months from the date of issuance of these financial statements.
Recent Developments
Reverse Stock Split
On December 5, 2022, we effected a 1-for-10 reverse stock split of our outstanding common stock. The reverse stock split also affected our outstanding stock options, purchase rights and equity incentive plans and resulted in the shares underlying such instruments being reduced and the exercise price being increased proportionately.
Registered Direct Offering
On February 3, 2023, we entered into a securities purchase agreement (the “Purchase Agreement”), with two institutional healthcare investors (the “Purchasers”), relating to the purchase and sale of an aggregate of (i) 1,700,000 shares (the “Shares”) of our common stock, par value $0.0001 per share (“Common Stock”), (ii) pre-funded warrants to purchase 300,000 shares of Common Stock, and (iii) accompanying common warrants, to purchase an aggregate of 2,000,000 shares of Common Stock in a registered direct offering (the “Offering”). The aggregate gross proceeds to us from the Offering were $10.0 million before deducting placement agent fees and other estimated offering expenses payable by DURECT and excluding the proceeds, if any, from the exercise of the pre-funded warrants and common warrants issued in the Offering. The aggregate net proceeds to us from the Offering were approximately $8.8 million after deducting placement agent fees and other estimated offering expenses payable by us.
64
The pre-funded warrants are exercisable immediately following the closing date of the Offering and have an unlimited term and an initial exercise price of $0.00001 per share. The common warrants will be immediately exercisable and have a five year term and an initial exercise price of $5.00 per share. The combined offering price is $5.00 per Share and accompanying common warrant, or in the case of pre-funded warrants, $4.99999 per pre-funded warrant and accompanying common warrant. A holder (together with its affiliates) may not exercise any portion of a pre-funded warrant or common warrant to the extent that the holder would own more than 4.99% (or, at the election of the holder 9.99%) of our outstanding Common Stock immediately after exercise.
Critical Accounting Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the dates of the financial statements and the reported amounts of revenues and expenses during the reporting periods. We believe that the most significant accounting estimates and assumptions relate to revenue recognition, prepaid and accrued clinical costs, prepaid and accrued manufacturing costs, and stock-based compensation. We base our estimates on historical experience, current circumstances and various other assumptions that our management believes to be reasonable under the circumstances. In many instances, we could reasonably use different accounting estimates, and in some instances changes in the accounting estimates are reasonably likely to occur from period to period. Accordingly, actual results could differ significantly from the estimates made by our management. To the extent that there are differences between our estimates and actual results, our future financial statement presentation, financial condition, results of operations and cash flows will be affected. We believe that the critical accounting estimates discussed below are critical to understanding our historical and future performance, as these estimates involve a significant level of estimation uncertainty and have had or are reasonably likely to have a material impact on the financial condition or results of operations of the registrant.
Revenue Recognition
Product Revenue, Net
We manufacture and sell ALZET osmotic pumps used in laboratory research, and manufacture and sell certain excipients used by pharmaceutical companies as raw materials in certain of their products, including POSIMIR, an animal health product and Methydur.
Revenue from product sales is recognized when the customer obtains control of our product, which occurs at a point in time, typically upon shipment to the customer. We expense incremental costs of obtaining a contract as and when incurred if the expected amortization period of the asset that we would have recognized is one year or less.
Trade Discounts and Allowances: We provide certain customers with discounts that are explicitly stated in our contracts and are recorded as a reduction of revenues in the period the related product revenue is recognized.
Product Returns: Consistent with industry practice, we generally offer customers a limited right of return for products that have been purchased from us. We estimate the amount of our product sales that may be returned by our customers and record this estimate as a reduction of revenue in the period the related product revenue is recognized. We currently estimate product return liabilities primarily using our own historical sales information. We expect product returns to be minimal.
Collaborative Research and Development and Other Revenue
We enter into license agreements under which we license certain rights to our product candidates or products to third parties. The terms of these arrangements typically include payment to us of one or more of the following: non-refundable, up-front license fees; reimbursement of development costs incurred by us under approved work plans; development, regulatory, intellectual property and commercial milestone payments; payments for manufacturing supply services we provide ourselves or through our contract manufacturers; and royalties on net sales of licensed products. Each of these payments results in
65
collaborative research and development revenues, except for revenues from royalties on net sales of licensed products and earn-out revenues, which are classified as other revenues.
In determining the appropriate amount of revenue to be recognized as we fulfill our obligations under each of our agreements, we perform the following steps: (i) identification of the promised goods or services in the contract; (ii) determination of whether the promised goods or services are performance obligations, including whether they are distinct in the context of the contract; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue when (or as) we satisfy each performance obligation. For arrangements that are determined to include multiple performance obligations, we must develop assumptions that require judgment to determine the estimated stand-alone selling price for each performance obligation identified. These assumptions may include: forecasted revenues, development timelines, reimbursement rates for personnel costs, discount rates and probabilities of technical and regulatory success. We expect to recognize revenue for the variable consideration currently being constrained when it is probable that a significant revenue reversal will not occur.
Licenses of Intellectual Property: If the license to our intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, we recognize revenues from the transaction price allocated to the license when the license is transferred to the customer and the customer is able to use and benefit from the license. For performance obligations comprised of licenses that are bundled with other promises, we utilize judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time and, if over time, we apply an appropriate method of measuring progress for purposes of recognizing related revenue from the allocated transaction price. For performance obligations recognized over time, we evaluate the measure of progress each reporting period and recognize revenue on a cumulative catch-up basis as collaborative research and development revenues.
Milestone Payments: At the inception of each arrangement that includes development milestone payments, we evaluate whether the milestones are considered probable of being reached and estimate the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price. Milestone payments that are not within the control of us or the licensee, such as regulatory approvals, are not considered probable of being achieved until those approvals are received. The transaction price is then allocated to each performance obligation on a relative stand-alone selling price basis, for which we recognize revenue as or when the performance obligations under the contract are satisfied. At the end of each subsequent reporting period, we re-evaluate the probability of achievement of such development milestones and any related constraint, and if necessary, adjust our estimate of the overall transaction price.
Manufacturing Supply Services: Arrangements that include a promise for future supply of raw materials or drug product for either clinical development or commercial supply at the customer’s discretion are generally considered as options. We assess if these options provide a material right to the customer and, if so, they are accounted for as separate performance obligations and allocate a portion of the transaction price based on the estimated standalone selling price of the material right. If we are entitled to additional payments when the customer exercises these options, the deferred transaction price and any additional payments are recorded in collaborative research and development revenue when the customer obtains control of the goods.
Royalties and Earn-outs: For arrangements that include sales-based royalties or earn-outs, including milestone payments based on first commercial sale or the level of sales, and the license is deemed to be the predominant item to which the royalties relate, we recognize revenue at the later of (i) when the related sales occur, or (ii) when the performance obligation to which some or all of the royalty or earn-out has been allocated has been satisfied (or partially satisfied). To date, we have not recognized
66
material royalty revenue resulting from our collaborative arrangements or material earn-out revenues from any of our agreements.
Research and development services: Revenue from research and development services that are determined to represent a distinct performance obligation related to services performed under the collaborative arrangements with our third-party collaborators is recognized over time as the related research and development services are performed using an appropriate method of measuring progress. We evaluate the measure of progress each reporting period and recognize revenue on a cumulative catch-up basis, as collaborative research and development revenue. Research and development expenses under the collaborative research and development agreements generally approximate or exceed the revenue recognized under such agreements over the term of the respective agreements. Deferred revenue may result when we do not expend the required level of effort during a specific period in comparison to funds received under the respective agreement.
We receive payments from our customers based on development cost schedules established in each contract. Up-front payments are recorded as deferred revenue upon receipt or when due, and may require deferral of revenue recognition to a future period until we performs our obligations under these arrangements. Amounts are recorded as accounts receivable when our right to consideration is unconditional. We do not assess whether a contract has a significant financing component if the expectation at contract inception is such that the period between payment by the customer and the transfer of the promised goods or services to the customer will be one year or less.
Prepaid and Accrued Clinical Costs
We incur significant costs associated with third party consultants and organizations for pre-clinical studies, clinical trials, contract research, regulatory advice and other research and development-related services. We are required to estimate periodically the cost of services rendered but unbilled based on management’s estimates. Estimates are determined each reporting period by reviewing the terms and conditions of the underlying contracts, reviewing open purchase orders and by having detailed discussions with internal clinical personnel and third-party service providers as to the nature and status of the services performed in relation to amounts billed. The costs for unbilled services are estimated by applying the rates and fees applicable in the underlying contracts. If these good faith estimates are inaccurate, actual expenses incurred could materially differ from our estimates.
Prepaid and Accrued Manufacturing Costs
We incur significant costs associated with third party consultants and organizations for manufacturing, validation, testing and other research and development-related services. We are required to estimate periodically the cost of services rendered but unbilled based on management’s estimates. Estimates are determined each reporting period by reviewing the terms and conditions of the underlying contracts, reviewing open purchase orders and by having detailed discussions with internal personnel and third-party service providers as to the nature and status of the services performed in relation to amounts billed. The costs for unbilled services are estimated by applying the rates and fees applicable in the underlying contracts. If these good faith estimates are inaccurate, actual expenses incurred could materially differ from these estimates.
Stock-Based Compensation
Employee stock-based compensation is estimated at the date of grant based on the employee stock award’s fair value using the Black-Scholes option-pricing model and is recognized as expense ratably over the requisite period.
We estimate the volatility of our common stock at the date of grant based on the historical volatility of our common stock. We base the risk-free rate that we use in the Black-Scholes option valuation model on the implied yield in effect at the time of option grant on U.S. Treasury zero-coupon issues with equivalent remaining terms. We have never paid any cash dividends on our common stock and we do not anticipate paying any cash dividends in the foreseeable future. Consequently, we use an expected dividend yield of zero in the Black-Scholes option valuation model. We determine the expected life using historical options experience. Upon the adoption of ASU 2016-09, we account for forfeitures as they occur and record stock-based compensation expense only for those awards that vest. We amortize the fair value of options granted on a straight-line basis. All options are amortized over the requisite service
67
periods of the awards, which are generally the vesting periods. We may elect to use different assumptions under the Black-Scholes option valuation model in the future, which could materially affect our net income or loss and net income or loss per share. See Note 9 “Stockholders’ Equity” to our financial statements for further information regarding stock-based compensation.
Results of Operations
Comparison of years ended December 31, 2022 and 2021
Revenue
Collaborative research and development and other revenue
We recognize revenue from collaborative research and development activities and service contracts. Collaborative research and development and other revenue primarily represents reimbursement of qualified expenses related to collaborative agreements with various third parties to research, develop and commercialize potential products using our drug delivery technologies, and revenue from the recognition of upfront fees and milestone payments in connection with our collaborative or license agreements.
We expect our collaborative research and development and other revenue to fluctuate in future periods pending our efforts to enter into potential new collaborations, our existing third-party collaborators’ commitment to and progress in the research and development programs, and any royalty or earn-out revenue recognized from collaborators or counterparties. The collaborative research and development and other revenue associated with our major collaborators or counterparties are as follows (in thousands):
|
|
Year ended December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Collaborator/Counterparty |
|
|
|
|
|
|
||
Innocoll (1) |
|
$ |
10,015 |
|
|
$ |
4,100 |
|
Other (2) |
|
|
3,189 |
|
|
|
2,231 |
|
Total collaborative research and development and other revenue |
|
$ |
13,204 |
|
|
$ |
6,331 |
|
The increase in collaborative research and development and other revenue in 2022 compared with 2021 was primarily due to higher revenue recognized from our agreements with Innocoll, Indivior and feasibility agreements with other companies.
Innocoll paid us a $4.0 million upfront fee and a payment of $1.3 million primarily for the sale of manufacturing supplies and excipients in connection with the license agreement signed in December 2021. Through December 31, 2021, $5.2 million had been recognized as revenue as there were no remaining substantive performance obligations to be provided to Innocoll by the Company. During the twelve months ended December 31, 2022, we recognized $8.0 million of patent milestone revenue and $2.0 million of first commercial sale milestone revenue under the license agreement with Innocoll.
68
As of December 31, 2022, we had potential milestones of up to $122.0 million that we may receive in the future under our collaborative arrangements, of which $10.0 million are development-based milestones, $2.0 million are patent-based milestones and $110.0 million are sales-based milestones. Within the category of development-based milestones, $10.0 million are related to regulatory approvals. In January 2023, we received a $2.0 million sales-based milestone payment that was achieved in September 2022 for the first commercial sale of POSIMIR by Innocoll.
Product revenue, net
A portion of our revenues is derived from product sales, which include our ALZET osmotic pump product line, and certain excipients that are included in POSIMIR, Methydur and in a marketed animal health product. Net product revenues were $6.1 million and $7.6 million in 2022 and 2021, respectively.
The decrease in product revenues in 2022 was primarily attributable to lower revenue from our ALZET osmotic pump product line as a result of lower units sold and lower product revenue related to the sale of manufacturing supplies and excipients that are included in POSIMIR, Methydur and a marketed animal health product compared to 2021.
Operating Expenses
Cost of product revenues
Cost of product revenues includes the cost of product revenue from our ALZET product line, and certain excipients that are included in POSIMIR, Methydur and a marketed animal health product. Cost of product revenues was $1.6 million and $2.0 million in 2022 and 2021, respectively.
The decrease in cost of product revenues in 2022 was primarily attributable to lower cost of goods sold related to our ALZET product line arising from fewer units sold and lower cost of goods sold related to certain excipients and materials that are included in POSIMIR, Methydur and a marketed animal health product compared with 2021.
Stock-based compensation related to cost of product revenues was $20,000 and $19,000 in 2022 and 2021, respectively.
As of December 31, 2022 and 2021, we had 10 manufacturing employees.
Research and development
Research and development expenses are primarily comprised of salaries, benefits, stock-based compensation and other compensation costs associated with research and development personnel, overhead and facility costs, preclinical and non-clinical development costs, clinical trial and related clinical manufacturing costs, contract services, and other outside costs. Research and development expenses were $36.9 million and $31.8 million in 2022 and 2021, respectively. Stock-based compensation recognized related to research and development personnel was $1.2 million in each of 2022 and 2021.
Research and development expenses increased by $5.0 million in 2022 compared to 2021. The increase in 2022 was primarily attributable to higher research and development costs associated with larsucosterol and the depot injectable programs, partially offset by lower research and development costs associated with POSIMIR and other research programs compared to 2021, as more fully discussed below. We expect our research and development expenses to decrease in 2023 compared to 2022 as we expect to complete the AHFIRM trial and to incur lower contract manufacturing costs for larsucosterol in 2023.
Research and development expenses associated with our major development programs were as follows (in thousands):
|
|
Year Ended December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Larsucosterol |
|
$ |
34,048 |
|
|
$ |
25,074 |
|
Depot injectable programs |
|
|
1,588 |
|
|
|
1,388 |
|
POSIMIR |
|
|
524 |
|
|
|
4,249 |
|
Other |
|
|
702 |
|
|
|
1,135 |
|
Total research and development expenses |
|
$ |
36,862 |
|
|
$ |
31,846 |
|
69
Larsucosterol
Our research and development expenses for larsucosterol increased to $34.0 million in 2022 from $25.1 million in 2021, primarily due to higher clinical trial related expenses, higher contract manufacturing expenses and higher employee-related costs for this drug candidate compared with 2021.
We continue to assess the impact of macroeconomic uncertainties, including the COVID-19 pandemic, on our business, including our larsucosterol Phase 2b trial in alcohol-associated hepatitis; COVID-19 may affect our ability to complete recruitment and data analysis for our clinical trials, including larsucosterol trials, in our planned timeframe. For additional information regarding these risks and uncertainties, see “Risk Factors” above.
Depot injectable programs
Our research and development expenses for depot injectable programs increased to $1.6 million in 2022 from $1.4 million in 2021 primarily due to higher employee-related costs, partially offset by lower outside expenses for these programs.
POSIMIR
Our research and development expenses for POSIMIR decreased to $524,000 in 2022 from $4.2 million in 2021, primarily due to lower employee-related costs and lower consulting expenses for POSIMIR.
Other DURECT research programs
Our research and development expenses for all other research activities decreased to $702,000 in 2022 from $1.1 million in 2021, primarily due to lower employee-related costs incurred for these programs.
As of December 31, 2022 and 2021, we had 42 and 45 research and development employees, respectively.
Our research and development programs may span as many as ten years or more, and estimation of completion dates or costs to complete are highly speculative and subjective due to numerous risks and uncertainties associated with developing pharmaceutical products, including significant and changing government regulation, uncertainties of future preclinical and clinical study results, uncertainties related to the COVID-19 pandemic, uncertainties with our collaborators’ commitment to and progress in the programs and uncertainties associated with process development and manufacturing as well as sales and marketing. In addition, with respect to our development programs subject to third-party collaborations, the timing and expenditures to complete the programs are subject to the control of our collaborators. Therefore, we cannot reasonably estimate the timing and costs of the efforts necessary to complete the research and development programs. For additional information regarding these risks and uncertainties, see “Risk Factors” above.
Selling, general and administrative
Selling, general and administrative expenses are primarily comprised of salaries, benefits and stock-based compensation associated with finance, legal, business development, sales and marketing (including sales and marketing expenses for our ALZET product line) and other administrative personnel, overhead and facility costs, and other general and administrative costs. Selling, general and administrative expenses were $15.9 million and $14.4 million in 2022 and 2021, respectively. Selling, general and administrative expenses increased by $1.5 million in 2022 compared to 2021, primarily due to higher patent expenses as well as higher employee expenses in 2022 compared with 2021. Stock-based compensation recognized related to selling, general and administrative personnel was $1.2 million and $1.4 million in 2022 and 2021, respectively. We expect our selling, general and administrative expenses to increase in 2023 compared to 2022 due to higher market research and employee expenses in 2023.
As of December 31, 2022 and 2021, we had 26 and 24 selling, general and administrative personnel, respectively.
70
Other Income (Expense)
Interest and other income
Interest and other income were $2.1 million and $156,000 in 2022 and 2021, respectively. The increase in interest and other income in 2022 compared to 2021 was primarily due to a $1.25 million settlement payment received from a former collaborator as well as higher interest income generated as a result of higher interest rates associated with our cash and investments in 2022 compared with 2021.
Interest expense
Interest expense was $2.4 million and $2.1 million in 2022 and 2021, respectively. The increase in interest expense in 2022 compared to 2021 was primarily due to higher interest rates associated with the term loan with Oxford Finance in 2022 compared to 2021.
Income Taxes
As of December 31, 2022, we had net operating loss ("NOL") carryforwards for federal income tax purposes of approximately $317.7 million, of which approximately $245.2 million will expire in the years 2023 through 2037, and approximately $72.5 million will not expire under current tax laws. As of December 31, 2022, we had federal research and development tax credits of approximately $18.0 million, which expire at various dates beginning in 2023 through 2042, if not utilized. As of December 31, 2022, we had NOL carryforwards for state income tax purposes of approximately $243.8 million, which expire in the years 2023 through 2042, and state research and development tax credits of approximately $17.8 million, which do not expire under current tax laws. Utilization of the NOLs may be subject to a substantial annual limitation due to federal and state ownership change limitations. The annual limitation may result in the expiration of NOLs and credits before utilization.
As of December 31, 2022 and 2021, we had net deferred tax assets of $117.6 million and $116.4 million, respectively. Deferred tax assets reflect the net tax effects of NOLs and credit carryforwards and the temporary differences between the carrying amounts of assets and liabilities for financial reporting and the amounts used for income tax purposes. Realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Accordingly, the net deferred tax assets have been fully offset by a valuation allowance.
Because realization of such tax benefits is uncertain, we provided a 100% valuation allowance as of December 31, 2022 and 2021. Utilization of the NOL and R&D credits carryforwards may be subject to a substantial annual limitation due to ownership change limitations that have occurred previously or that could occur in the future provided by Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, as well as similar state and foreign provisions. These ownership changes may limit the amount of NOL and R&D credits carryforwards that can be utilized annually to offset future taxable income and tax, respectively. In general, an ownership change is defined as a greater than 50% change (by value) in its equity ownership over a three-year period, the corporation’s ability to use its pre-change NOLs and other pre-change tax attributes (such as research and development credit carryforwards) to offset its post-change taxable income or taxes may be limited. Since our formation, we have raised capital through the issuance of capital stock on several occasions which, combined with the purchasing shareholders’ subsequent disposition of those shares, may have resulted in a change of control, as defined by Section 382, or could result in a change of control in the future upon subsequent disposition. We issued $60.0 million of convertible notes in 2003 and subsequently all of these notes had been converted as of December 31, 2008 into approximately 1.9 million shares of our common stock. We also issued approximately 440,000 shares of our common stock to an institutional investor in connection with an equity financing in September 2009. In December 2012, November 2013, April 2016, June 2019 and February 2021, we completed underwritten public offerings in which we sold an aggregate of approximately 1.4 million, 820,000, 1.4 million, 2.9 million and 2.0 million shares, respectively, of our common stock pursuant to effective registration statements. In 2016, 2017, 2018, 2019, 2020 and 2021, we issued approximately 520,000, 890,000, 960,000, 230,000, 530,000 and 95,000 shares, respectively, of our common stock in the open market through Controlled Equity Offering sales agreements with Cantor Fitzgerald pursuant to effective registration statements. These transactions may also have resulted in a change of control as defined by Section 382 or could result in a change of control in the future upon the subsequent disposition of the shares.
71
We have not currently completed a study to assess whether a change in control has occurred or whether there have been multiple changes of control since our formation due to the significant complexity and cost associated with such a study and the fact that there could be additional changes in the future. If we have experienced a change of control at any time since our formation, utilization of our NOL or R&D credits carryforwards would be subject to an annual limitation under Sections 382 and 383 which is determined by first multiplying the value of our stock at the time of the ownership change by the applicable long-term tax-exempt rate, and then could be subject to additional adjustments, as required. Any limitation may result in expiration of a portion of our NOL or R&D credits carryforwards before utilization. Tax years 2000 to 2022 remain subject to future examination by the major tax jurisdictions in which we are subject to tax.
Liquidity and Capital Resources
We had cash, cash equivalents and investments totaling $43.6 million at December 31, 2022, which includes $150,000 of interest-bearing marketable securities classified as restricted investments on our balance sheet as of December 31, 2022 as compared to cash, cash equivalents, cash held in escrow, investments and restricted investments totaling $70.0 million at December 31, 2021.
Our cash and investments policy emphasizes liquidity and preservation of principal over other portfolio considerations. We select investments that maximize interest income to the extent possible given these two constraints. We satisfy liquidity requirements by investing excess cash in securities with different maturities to match projected cash needs and limit concentration of credit risk by diversifying our investments among a variety of high credit-quality issuers.
As discussed below, we do not have sufficient cash resources to fund our planned operations, existing debt and contractual commitments and planned capital expenditures. Our auditors have issued a going concern opinion. Unless we secure additional equity or debt financing, of which there can be no assurance, we may not be able to continue operations.
Cash Flows
We used $26.3 million and $37.3 million of cash in operating activities in the years ended December 31, 2022 and 2021, respectively. The cash provided by or used for operations was primarily to fund operations as well as working capital requirements. Our cash provided by or used in operating activities differs from our net loss in part due to the timing and recognition of up-front payments under collaborative agreements. Depending on the nature of the upfront payments received upon execution of collaborative agreements, which can either be recognized as revenue upfront in full or primarily recorded as deferred revenue and generally recognized over the period using a basis that best reflects the satisfaction of our performance obligations with the third-party collaborator pursuant to the applicable agreement. The decrease in cash used in operating activities in 2022 compared to 2021 was primarily due to changes in accounts payable, accounts receivable, prepaid expenses and other assets, and accrued liabilities as well as $13.3 million received under the agreement with Innocoll in 2022.
We generated $19.8 million and $15.3 million of cash from investing activities in the years ended December 31, 2022 and 2021, respectively. The increase in cash received from investing activities in 2022 was due to a decrease in net purchases of available-for-sale securities, partially offset by a decrease in proceeds from maturities of available-for-sale securities in 2022 compared with 2021. In addition, we received $15 million of cash in 2021 from the sale of the LACTEL product line. We anticipate incurring capital expenditures of approximately $100,000 over the next 12 months. The amount and timing of these capital expenditures will depend on, among other things, our research and development activities and needs, and the need for equipment replacements.
We generated $83,000 and $50.5 million of cash from financing activities in the years ended December 31, 2022 and 2021, respectively. The decrease in cash received from financing activities was primarily due to lower net proceeds from shares sold under our shelf registration statement as well as lower proceeds from the exercise of stock options in 2022 compared to 2021. In 2021, we completed an underwritten public offering of 2,036,458 shares of our common stock at a price of $22.39 per share pursuant to an underwriting agreement with Cantor Fitzgerald, raising net proceeds of approximately $45.4 million. In 2021, we also raised net proceeds of approximately $2.4 million from the sale of 95,000
72
shares of our common stock in the open market at a weighted average price of $26.00 per share pursuant to the 2018 Registration Statement (as defined below) and 2015 Sales Agreement (as defined below).
Shelf Registration Statement
In July 2021, we filed a shelf registration statement on Form S-3 with the SEC (the “2021 Registration Statement”) (File No. 333-258333), which upon being declared effective in August 2021, terminated our registration statement filed in August 2018 (File No. 333-226518) and allowed us to offer up to $250.0 million of securities from time to time in one or more public offerings, inclusive of up to $75.0 million of shares of our common stock which we may sell, subject to certain limitations, pursuant to a sales agreement dated July 30, 2021 with Cantor Fitzgerald (the “2021 Sales Agreement”). The 2021 Sales Agreement replaced a prior 2015 Sales Agreement.
Following the closing of the Offering described above under “Recent Developments”, as of March 3, 2023, we had up to $240.0 million of our securities available for sale under the 2021 Registration Statement, of which $75.0 million of our common stock are available pursuant to the 2021 Sales Agreement.
Any material sales in the public market of our common stock, under the 2021 Sales Agreement or otherwise under the 2021 Registration Statement, could adversely affect prevailing market prices for our common stock.
Term Loan
In July 2016, we entered into a Loan and Security Agreement (as amended, the "Loan Agreement") with Oxford Finance LLC ("Oxford Finance"), pursuant to which Oxford Finance provided a $20.0 million secured single-draw term loan to us with an initial maturity date of August 1, 2020. The term loan was fully drawn at close and the proceeds may be used for working capital and general business requirements. The term loan repayment schedule provided initially for interest only payments for the first 18 months, followed by consecutive monthly payments of principal and interest in arrears starting on March 1, 2018 and continuing through the maturity date of August 1, 2020. Following five amendments, we make interest only payments under the amended Loan Agreement until June 1, 2023 and the final maturity date of the loan is September 1, 2025. The Loan Agreement provides for a floating interest rate (7.95% initially and 11.45% as of December 31, 2022) based on an index rate plus a spread and an additional payment equal to 10% of the principal amount of the term loan, which is due when the term loan becomes due or upon the prepayment of the facility. If we elect to prepay the loan, there is also a prepayment fee between 0.75% and 2.5% of the principal amount of the term loan depending on the timing of prepayment. Our debt repayment obligations under the Loan Agreement, as amended, may prove a burden to the Company as they become due, particularly following the expiration of the interest-only period.
The term loan is secured by substantially all of our assets, except that the collateral does not include any intellectual property (including licensing, collaboration and similar agreements relating thereto), and certain other excluded assets. The Loan Agreement contains customary representations, warranties and covenants by us, which covenants limit our ability to convey, sell, lease, transfer, assign or otherwise dispose of certain assets; engage in any business other than the businesses currently engaged in by us or reasonably related thereto; liquidate or dissolve; make certain management changes; undergo certain change of control events; create, incur, assume, or be liable with respect to certain indebtedness; grant certain liens; pay dividends and make certain other restricted payments; make certain investments; and make payments on any subordinated debt.
73
The Loan Agreement also contains customary indemnification obligations and customary events of default, including, among other things, our failure to fulfill certain obligations under the 2016 Loan Agreement and the occurrence of a material adverse change which is defined as a material adverse change in our business, operations, or condition (financial or otherwise), a material impairment of the prospect of repayment of any portion of the loan, or a material impairment in the perfection or priority of lender’s lien in the collateral or in the value of such collateral. In the event of default by us under the 2016 Loan Agreement, the lender would be entitled to exercise its remedies thereunder, including the right to accelerate the debt, upon which we may be required to repay all amounts then outstanding under the Loan Agreement. As a result, the term loan was reclassified to current liabilities from non-current liabilities on our balance sheet as of December 31, 2022 due to recurring losses, liquidity concerns and a subjective acceleration clause in the Loan Agreement.
Going Concern
As of December 31, 2022, we had approximately $43.6 million in cash, cash equivalents and investments. In February 2023, we received $8.8 million of net proceeds from a registered direct offering after deducting placement agent fees and other estimated offering expenses payable by us. In accordance with ASU No. 2014-15 Presentation of Financial Statements – Going Concern (subtopic 205-40), our management evaluates whether there are conditions or events, considered in the aggregate, that raise substantial doubt about our ability to continue as a going concern within one year after the date that the financial statements are issued. Based on our evaluation, substantial doubt exists regarding our ability to continue as a going concern for a period of one year from the issuance of our financial statements.
Cash used in our operating activities is heavily influenced by the timing and structure of new corporate collaborations. While one feature of our business strategy is seeking new corporate collaborations, assuming no new collaborations and no milestone payments, we anticipate that cash used in operating activities will increase in the near term. In 2022, there were no significant changes in our commercial commitments and contractual obligations. In aggregate, we are required to make future payments pursuant to our existing contractual obligations as follows (in thousands):
Contractual Obligations |
|
2023 |
|
|
2024 |
|
|
2025 |
|
|
Total |
|
||||
Term loan (1) |
|
$ |
5,714 |
|
|
$ |
8,571 |
|
|
$ |
7,715 |
|
|
$ |
22,000 |
|
Operating lease obligations |
|
|
1,970 |
|
|
|
275 |
|
|
|
— |
|
|
|
2,245 |
|
Total contractual cash obligations |
|
$ |
7,684 |
|
|
$ |
8,846 |
|
|
$ |
7,715 |
|
|
$ |
24,245 |
|
(1) Includes principal, interest and final payments and assumes no acceleration of obligations.
Presently, we do not have sufficient cash resources to fund our planned operations, existing debt and contractual commitments and planned capital expenditures through at least the next 12 months from issuance of these financial statements. We may consume available resources more rapidly than currently anticipated, resulting in the need for additional funding. We expect to incur continuing losses and negative cash flows from operations for the foreseeable future.
Depending on whether we enter into additional collaborative agreements in the near term and the extent to which we earn revenues from our collaborative agreements, we may decide to raise additional capital through a variety of sources in the short-term and in the long-term, including:
There can be no assurance that we will enter into additional collaborative agreements or maintain existing collaborative agreements, will earn collaborative revenues or that additional capital will be available on favorable terms, if at all. If adequate funds are not available, we may be required to significantly reduce or re-focus our operations or to obtain funds through arrangements that may require us to relinquish rights to certain of our products, technologies or potential markets, either of which could
74
have a material adverse effect on our business, financial condition and results of operations. To the extent that additional capital is raised through the sale of equity or convertible debt securities, the issuance of such securities would result in ownership dilution to our existing stockholders (assuming convertible debt securities were converted into shares). These factors raise substantial doubt regarding our ability to continue as a going concern. Our inability to obtain required funding in the near future or our inability to obtain funding on favorable terms will have a material adverse effect on our operations and strategic development plan for future growth. If we cannot successfully raise additional capital and implement our strategic development plan, our liquidity, financial condition and business prospects will be materially and adversely affected, and we may have to cease operations.
As a result of our recurring losses from operations, negative cash flows from operating activities and need to raise additional capital, our independent registered public accounting firm included an explanatory paragraph in its report on our audited financial statements for the year ended December 31, 2022 expressing substantial doubt as to our ability to continue as a going concern.
Recent Accounting Pronouncements
See Note 1 “Summary of Significant Accounting Policies” – “Recent Accounting Pronouncements”, to our financial statements for a full description of recent accounting pronouncements, including the expected dates of adoption and estimated effects on financial condition and results of operations, which is incorporated herein by reference.
75
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Interest Rate Risk
Our exposure to market risk for changes in interest rates relates primarily to our investment portfolio and to our term loan. Fixed rate securities and borrowings may have their fair market value adversely impacted due to fluctuations in interest rates, while floating rate securities may produce less income than expected if interest rates fall and floating rate borrowings may lead to additional interest expense if interest rates increase. Due in part to these factors, our future investment income may fall short of expectations due to changes in interest rates or we may suffer losses in principal if forced to sell securities which have declined in market value due to changes in interest rates. Our interest expense on the term loan may rise if the interest rates increase.
Our primary investment objective is to preserve principal while at the same time maximizing yields without significantly increasing risk. Our portfolio includes money markets funds, certificates of deposit, commercial paper, corporate debt, and U.S. government agencies. The diversity of our portfolio helps us to achieve our investment objectives. As of December 31, 2022, 100% of our investment portfolio was composed of investments maturing less than 90 days from the date of purchase.
The following table presents the amounts of our cash equivalents and investments that may be subject to interest rate risk and the average interest rates as of December 31, 2022 by year of maturity (dollars in thousands):
|
|
2023 |
|
|
Cash equivalents: |
|
|
|
|
Fixed rate |
|
$ |
633 |
|
Average fixed rate |
|
|
4.01 |
% |
Variable rate |
|
$ |
40,465 |
|
Average variable rate |
|
|
4.72 |
% |
Short-term investments: |
|
|
|
|
Fixed rate |
|
$ |
— |
|
Average fixed rate |
|
|
— |
|
Restricted investments: |
|
|
|
|
Fixed rate |
|
$ |
150 |
|
Average fixed rate |
|
|
0.11 |
% |
Total investment securities |
|
$ |
41,248 |
|
Average rate |
|
|
4.48 |
% |
As of December 31, 2022, the fair value of our term loan was estimated to be $21.2 million. The Loan Agreement provides for interest only payments through June 1, 2023, followed by consecutive monthly payments of principal and interest in arrears starting on June 1, 2023 and continuing through the maturity date of the term loan of September 1, 2025. The Loan Agreement provides for a floating interest rate (7.95% initially and 11.45% as of December 31, 2022) based on an index rate plus a spread. In addition, a payment equal to 10% of the principal amount of the term loan is due when the term loan becomes due or upon the prepayment of the facility. If the Company elects to prepay the loan, there is also a prepayment fee of between 0.75% and 2.5% of the principal amount of the term loan depending on the timing of prepayment. The obligation under the term loan is subject to interest rate risk because the interest rates under the obligation may exceed current interest rates.
76
Item 8. Financial Statements and Supplementary Data.
DURECT CORPORATION
INDEX TO FINANCIAL STATEMENTS
|
|
Page No. |
Report of Independent Registered Public Accounting Firm (PCAOB ID: |
|
78 |
|
|
|
|
80 |
|
|
|
|
|
81 |
|
|
|
|
|
82 |
|
|
|
|
|
83 |
|
|
|
|
|
84 |
77
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of DURECT Corporation
Opinion on the Financial Statements
We have audited the accompanying balance sheets of DURECT Corporation (the Company) as of December 31, 2022 and 2021, the related statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2022, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2022 and 2021, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2022, in conformity with U.S. generally accepted accounting principles.
The Company’s Ability to Continue as a Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has suffered recurring losses from operations and has stated that substantial doubt exists about the Company’s ability to continue as a going concern. Management's evaluation of the events and conditions and management’s plans regarding these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical audit matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of the critical audit matter
78
does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Accrued clinical costs |
|
Description of the Matter |
At December 31, 2022, the Company had $1,965 thousand in accrued clinical costs. As described in Note 1 of the financial statements, the Company incurs significant costs associated with third-party service providers for its clinical trials. The Company is required to estimate periodically the cost of services rendered but unbilled based on management’s estimates. Estimates are determined each reporting period by reviewing the terms and conditions of the underlying contracts, reviewing open purchase orders and by having detailed discussions with internal clinical personnel and third-party service providers as to the nature and status of the services performed in relation to amounts billed. The costs for unbilled services are estimated by applying the rates and fees applicable in the underlying contracts.
Auditing management’s accounting for accrued clinical costs was especially challenging as evaluating the nature and status of the services performed under the Company’s clinical agreements in relation to amounts billed was dependent upon the accumulation of a high volume of information from internal clinical personnel and third-party service providers.
|
How We Addressed the Matter in Our Audit |
Our audit procedures included, among others, obtaining an understanding of accrued clinical costs by performing a fluctuation analysis over the accrued clinical costs, gaining an understanding of key movements against our expectations, reviewing significant agreements in place with the Company’s third-party clinical service providers, performing inquiries with management to verify the nature and status of the services performed, confirming applicable information directly with third-party clinical service providers, including: total amounts invoiced during the year, estimated unbilled amounts, unpaid amounts, patient enrollment status for clinical sites at December 31, 2022 and the terms and conditions of the underlying contracts. Our procedures also included vouching significant accrued clinical costs at December 31, 2022 to subsequent cash disbursements and tracing a sample of subsequent cash disbursements to related invoices received by the Company to verify whether they were appropriately included or excluded from the accrued clinical costs balance. |
We have served as the Company’s auditor since 1998.
/s/
March 8, 2023
79
DURECT CORPORATION
BALANCE SHEETS
(in thousands, except per share amounts)
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
A S S E T S |
|
|
|
|
|
|
||
Current assets: |
|
|
|
|
|
|
||
Cash and cash equivalents |
|
$ |
|
|
$ |
|
||
Short-term investments |
|
|
— |
|
|
|
|
|
Accounts receivable (net of allowances of $ |
|
|
|
|
|
|
||
Inventories, net |
|
|
|
|
|
|
||
Prepaid expenses and other current assets |
|
|
|
|
|
|
||
Total current assets |
|
|
|
|
|
|
||
Property and equipment, net |
|
|
|
|
|
|
||
Operating lease right-of-use assets |
|
|
|
|
|
|
||
Goodwill |
|
|
|
|
|
|
||
Long-term restricted investments |
|
|
|
|
|
|
||
Other long-term assets |
|
|
|
|
|
|
||
Total assets |
|
$ |
|
|
$ |
|
||
L I A B I L I T I E S A N D S T O C K H O L D E R S’ E Q U I T Y |
|
|
|
|
|
|
||
Current liabilities: |
|
|
|
|
|
|
||
Accounts payable |
|
$ |
|
|
$ |
|
||
Accrued liabilities |
|
|
|
|
|
|
||
Deferred revenue, current portion |
|
|
— |
|
|
|
|
|
Term loan, current portion, net |
|
|
|
|
|
— |
|
|
Operating lease liabilities, current portion |
|
|
|
|
|
|
||
Total current liabilities |
|
|
|
|
|
|
||
Deferred revenue, non-current portion |
|
|
— |
|
|
|
|
|
Operating lease liabilities, non-current portion |
|
|
|
|
|
|
||
Term loan, non-current portion, net |
|
|
— |
|
|
|
|
|
Other long-term liabilities |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
Stockholders’ equity: |
|
|
|
|
|
|
||
Preferred stock, $ |
|
|
|
|
|
|
||
Common stock, $ |
|
|
|
|
|
|
||
Additional paid-in capital |
|
|
|
|
|
|
||
Accumulated other comprehensive loss |
|
|
( |
) |
|
|
( |
) |
Accumulated deficit |
|
|
( |
) |
|
|
( |
) |
Stockholders’ equity |
|
|
|
|
|
|
||
Total liabilities and stockholders’ equity |
|
$ |
|
|
$ |
|
||
The accompanying notes are an integral part of these financial statements.
80
DURECT CORPORATION
STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(in thousands, except per share amounts)
|
|
Year ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
Collaborative research and development and other revenue |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Product revenue, net |
|
|
|
|
|
|
|
|
|
|||
Total revenues |
|
|
|
|
|
|
|
|
|
|||
Operating expenses: |
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
||||
Research and development |
|
|
|
|
|
|
|
|
|
|||
Selling, general and administrative |
|
|
|
|
|
|
|
|
|
|||
Total operating expenses |
|
|
|
|
|
|
|
|
|
|||
Loss from operations |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Other income (expense): |
|
|
|
|
|
|
|
|
|
|||
Interest and other income |
|
|
|
|
|
|
|
|
|
|||
Interest expense |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Net other expense |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Loss from continuing operations |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Income from discontinued operations (Note 11) |
|
|
— |
|
|
|
— |
|
|
|
|
|
Net loss |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Net change in unrealized loss on available-for-sale |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Total comprehensive loss |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Net loss per share |
|
|
|
|
|
|
|
|
|
|||
Basic and diluted |
|
|
|
|
|
|
|
|
|
|||
Loss from continuing operations |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Income from discontinued operations |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
Net loss per common share, basic and diluted |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
Weighted-average shares used in computing net loss per share, basic and diluted |
|
|
|
|
|
|
|
|
|
|||
The accompanying notes are an integral part of these financial statements.
81
DURECT CORPORATION
STATEMENTS OF STOCKHOLDERS’ EQUITY
(in thousands)
|
|
Common Stock |
|
|
Additional |
|
|
Accumulated |
|
|
Accumulated |
|
|
Total |
|
|||||||||
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Income (loss) |
|
|
Deficit |
|
|
Equity |
|
||||||
Balance at December 31, 2019 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
Issuance of common stock upon exercise of stock options and purchases of ESPP shares |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
Issuance of common stock upon equity financings, net of issuance costs of $ |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Stock-based compensation expense from stock options and ESPP shares |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Change in unrealized loss on available-for-sale securities, net of tax |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
Balance at December 31, 2020 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
Issuance of common stock upon exercise of stock options and purchases of ESPP shares |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Issuance of common stock upon equity financings, net of issuance costs of $ |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Stock-based compensation expense from stock options and ESPP shares |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Change in unrealized loss on available-for-sale securities, net of tax |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
Balance at December 31, 2021 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
Issuance of common stock upon exercise of stock options and purchases of ESPP shares |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
Issuance of common stock upon equity financings, net of issuance costs of $ |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
Stock-based compensation expense from stock options and ESPP shares |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Change in unrealized loss on available-for-sale securities, net of tax |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
Balance at December 31, 2022 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
The accompanying notes are an integral part of these financial statements.
82
DURECT CORPORATION
STATEMENTS OF CASH FLOWS
(in thousands)
|
|
Year ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
Cash flows from operating activities |
|
|
|
|
|
|
|
|
|
|||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Adjustments to reconcile net loss to net cash used in operating |
|
|
|
|
|
|
|
|
|
|||
Gain on sale of equipment |
|
|
— |
|
|
|
( |
) |
|
|
— |
|
Depreciation and accretion |
|
|
|
|
|
|
|
|
|
|||
Stock-based compensation |
|
|
|
|
|
|
|
|
|
|||
Inventory write-down |
|
|
— |
|
|
|
— |
|
|
|
|
|
Amortization of debt issuance cost |
|
|
|
|
|
|
|
|
|
|||
Net accretion/amortization on investments |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
Changes in operating lease liabilities |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Gain on sale of LACTEL product line |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
Changes in assets and liabilities: |
|
|
|
|
|
|
|
|
|
|||
Accounts receivable |
|
|
|
|
|
( |
) |
|
|
|
||
Inventories |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Prepaid expenses and other assets |
|
|
|
|
|
|
|
|
( |
) |
||
Accounts payable |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
Accrued liabilities |
|
|
|
|
|
|
|
|
( |
) |
||
Deferred revenue |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
Total adjustments |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
Net cash used in operating activities |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Cash flows from investing activities |
|
|
|
|
|
|
|
|
|
|||
Purchases of property and equipment |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Purchases of available-for-sale securities |
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Proceeds from maturities of available-for-sale securities |
|
|
|
|
|
|
|
|
|
|||
Proceeds from sales of available-for-sale securities |
|
|
— |
|
|
|
|
|
|
— |
|
|
Net proceeds from sale of LACTEL product line |
|
|
— |
|
|
|
|
|
|
— |
|
|
Net cash provided by investing activities |
|
|
|
|
|
|
|
|
|
|||
Cash flows from financing activities |
|
|
|
|
|
|
|
|
|
|||
Payments on equipment financing obligations |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Net proceeds from issuances of common stock upon exercise of |
|
|
|
|
|
|
|
|
|
|||
Net proceeds from issuances of common stock in connection with |
|
|
|
|
|
|
|
|
|
|||
Term loan amendment cost |
|
|
— |
|
|
|
( |
) |
|
|
— |
|
Net cash provided by financing activities |
|
|
|
|
|
|
|
|
|
|||
Net (decrease) increase in cash and cash equivalents |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
Cash, cash equivalents, and restricted cash, beginning of the |
|
|
|
|
|
|
|
|
|
|||
Cash, cash equivalents, and restricted cash, end of the period (1) |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Supplemental disclosure of cash flow information |
|
|
|
|
|
|
|
|
|
|||
Cash paid for interest |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Supplementary disclosure of non-cash investing and financing information |
|
|
|
|
|
|
|
|
|
|||
Cash held in escrow |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
|
(1)
The accompanying notes are an integral part of these financial statements.
83
DURECT CORPORATION
NOTES TO FINANCIAL STATEMENTS
Nature of Operations
DURECT Corporation (the “Company”) was incorporated in the state of Delaware on
Basis of Presentation and Use of Estimates
The Company’s financial statements have been prepared in accordance with U.S. generally accepted accounting principles (U.S. GAAP). The preparation of the accompanying Financial Statements conforms to accounting principles generally accepted in the U.S. which requires management to make judgments, estimates and assumptions that affect the reported amounts of assets, liabilities, equity, revenues and expenses, and related disclosures. On an ongoing basis, management evaluates its estimates including, but not limited to, those related to revenue recognition, the period of performance, identification of performance obligations and evaluation of milestones with respect to our collaborations, the amounts of revenues, recoverability of inventory, certain accrued liabilities including accrued clinical costs, asset retirement obligations, and stock-based compensation. The Company bases its estimates on historical experience and on various other market-specific and other relevant assumptions that the Company believes to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results could differ materially from those estimates.
Reverse Stock Split
On December 5, 2022, the Company effected a
For all financial statement periods presented, references to number of shares, net loss per share, stock price and exercise price have been conformed to reflect the effects of the Company’s
Discontinued Operations
On December 31, 2020, the Company sold its LACTEL Absorbable Polymer (LACTEL) product line to Evonik. The accompanying financial statements have been recast to reflect the assets, liabilities, revenue and expenses related to the Company’s LACTEL product line as discontinued operations for the years ended December 31, 2020 (see Note 11). The Company believes this format provides comparability with its previously filed financial statements.
84
Liquidity and Need to Raise Additional Capital
As of December 31, 2022, the Company has an accumulated deficit of $
There are no assurances that such additional funding will be obtained and that the Company will succeed in its future operations. If the Company cannot successfully raise additional capital and implement its strategic development plan, its liquidity, financial condition and business prospects will be materially and adversely affected, and the Company may have to cease operations. As further described in Note 8, the Company reclassified the remaining balance of its term loan to current liabilities from non-current liabilities on the Company’s balance sheet as of December 31, 2022 due to recurring losses, liquidity concerns and a subjective acceleration clause in the Company’s Loan Agreement. These financial statements have been prepared on a going concern basis and do not include any adjustments to the amounts and classification of assets and liabilities that may be necessary in the event the Company can no longer continue as a going concern.
Cash, Cash Equivalents and Investments
The Company considers all highly liquid investments with maturities of
The Company invests in debt instruments of government agencies, corporations, and money market funds with high credit ratings. The Company has established guidelines regarding diversification of its investments and their maturities with the objectives of maintaining safety and liquidity, while maximizing yield.
Total cash held in escrow (related to the sale of the LACTEL product line) was $
Concentrations of Credit Risk
Financial instruments that potentially subject the Company to credit risk consist principally of interest-bearing investments and trade receivables. The Company maintains cash, cash equivalents and investments with various major financial institutions. The Company performs periodic evaluations of the relative credit standing of these financial institutions. In addition, the Company performs periodic evaluations of the relative credit quality of its investments.
85
Pharmaceutical companies and academic institutions account for a substantial portion of the Company’s trade receivables. The Company provides credit in the normal course of business to its customers and collateral for these receivables is generally not required. The risk associated with this concentration is limited to a certain extent due to the large number of accounts and their geographic dispersion. The Company monitors the creditworthiness of its customers to which it grants credit terms in the normal course of business. The Company maintains reserves for estimated credit losses and, to date, such losses have been immaterial in all periods presented.
Customer and Product Line Concentrations
Revenue from the sale of products from the ALZET product line accounted for
Total revenue by geographic region for the years 2022, 2021 and 2020 are as follows (in thousands):
|
|
Year ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
United States |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Europe |
|
|
|
|
|
|
|
|
|
|||
Japan |
|
|
|
|
|
|
|
|
|
|||
Others |
|
|
|
|
|
|
|
|
|
|||
Total |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Revenue by geography is determined by the location of the customer.
Allowance for Doubtful Account
Allowance for doubtful accounts as of December 31, 2022, 2021 and 2020 were comprised as follows (in thousands):
|
|
Balance at |
|
|
Additions (Reductions) to allowances |
|
|
Deductions |
|
|
Balance at |
|
||||
Allowance for doubtful accounts |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Year ended December 31, 2022 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
— |
|
|
$ |
( |
) |
Year ended December 31, 2021 |
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
||
Year ended December 31, 2020 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
Inventories
Inventories are stated at the lower of cost or net realizable value, with cost determined on a first-in, first-out basis. The Company capitalizes inventories produced in preparation for product launches after receiving regulatory approval on a product. The Company may be required to expense previously capitalized inventory costs upon a change in management’s judgment due to new information that suggests that the inventory will not be saleable. If the Company is able to subsequently sell products made with raw materials that were previously written down, the Company will report an unusually high gross profit as there will be no or little associated cost of goods for these materials.
86
The Company’s inventories consisted of the following (in thousands):
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Raw materials |
|
$ |
|
|
$ |
|
||
Work in-process |
|
|
|
|
|
|
||
Finished goods |
|
|
|
|
|
|
||
Total inventories |
|
$ |
|
|
$ |
|
||
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation, which is computed using the straight-line method over the estimated useful lives of the assets, which range from to
Goodwill
Goodwill is periodically assessed and evaluated for impairment. The Company operates in
The Company evaluates goodwill for impairment at least annually. To date, the Company has not recorded any impairment charge related to goodwill.
Impairment of Long-Lived Assets
The Company reviews long-lived assets, including property and equipment, intangible assets, and other long-term assets, for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable.
An impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition is less than its carrying amount. Impairment, if any, is calculated as the amount by which an asset’s carrying value exceeds its fair value, typically using discounted cash flows to determine fair value. Through December 31, 2022, there have been
Leases
ASC 842 requires the Company to recognize an operating lease right-of-use asset and corresponding operating lease liability for the Company’s leased properties. The Company’s operating lease right-of-use assets and liabilities are recognized under ASC 842 based on the present value of lease payments over the remaining lease term at the lease commencement date. In determining the net present value of lease
87
payments, we estimate the incremental borrowing rate based on the information available, including remaining lease term. As of December 31, 2022, the weighted-average remaining lease term was 0.99 years for the Company’s leased properties.
Stock-Based Compensation
The Company accounts for share-based payments using a fair-value based method for costs related to all share-based payments, including stock options and stock issued under the Company’s employee stock purchase plan (ESPP). The Company estimates the fair value of share-based payment awards on the date of grant using an option-pricing model. The Company recognizes compensation costs on a straight-line basis over the requisite service period and accounts for forfeitures as they occur. See Note 9 for further information regarding stock-based compensation.
Revenue Recognition
Product Revenue, Net
The Company manufacture and sell ALZET osmotic pumps used in laboratory research, and manufacture and sell certain excipients used by pharmaceutical companies as raw materials in certain of their products, including POSIMIR, an animal health product and Methydur.
Revenues from product sales are recognized when the customer obtains control of the Company’s product, which occurs at a point in time, typically upon shipment to the customer. The Company expenses incremental costs of obtaining a contract as and when incurred if the expected amortization period of the asset that the Company would have recognized is one year or less.
Trade Discounts and Allowances: The Company provides certain customers with discounts that are explicitly stated in the Company’s contracts and are recorded as a reduction of revenue in the period the related product revenue is recognized.
Product Returns: The Company generally offers customers a limited right of return for products that have been purchased. The Company estimates the amount of its product sales that are probable of being returned by its customers and records this estimate as a reduction of revenue in the period the related product revenue is recognized. The Company currently estimates product return liabilities primarily using its historical sales information. The Company expects product returns to be minimal.
Collaborative Research and Development and Other Revenue
The Company enters into license agreements, under which it licenses certain rights to its product candidates or products to third parties. The terms of these arrangements typically include payment to the Company of one or more of the following: non-refundable, up-front license fees; reimbursement of development costs incurred by the Company under approved work plans; development, regulatory, intellectual property and commercial milestone payments; payments for manufacturing supply services the Company provides itself or through its contract manufacturers; and royalties on net sales of licensed products. Each of these payments results in collaborative research and development revenues, except for revenues from royalties on net sales of licensed products and earn-out revenues, which are classified as other revenues.
88
In determining the appropriate amount of revenue to be recognized as it fulfills its obligations under each of its agreements, the Company performs the following steps: (i) identification of the promised goods or services in the contract; (ii) determination of whether the promised goods or services are performance obligations including whether they are distinct in the context of the contract; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue when (or as) the Company satisfies each performance obligation. For arrangements that are determined to include multiple performance obligations, the Company must develop assumptions that require judgment to determine the estimated stand-alone selling price for each performance obligation identified. These assumptions may include: forecasted revenues, development timelines, reimbursement rates for personnel costs, discount rates and probabilities of technical and regulatory success. The Company expects to recognize revenue for the variable consideration currently being constrained when it is probable that a significant revenue reversal will not occur.
Licenses of intellectual property: If the license to the Company’s intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, the Company recognizes revenues from the transaction price allocated to the license when the license is transferred to the customer and the customer is able to use and benefit from the license. For performance obligations comprised of licenses that are bundled with other promises, the Company utilizes its judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time and, if over time, the Company applies an appropriate method of measuring progress for purposes of recognizing related revenues from the allocated transaction price. For performance obligations recognized over time, the Company evaluates the measure of progress each reporting period and recognizes revenues on a cumulative catch-up basis as collaborative research and development revenues.
Milestone Payments: At the inception of each arrangement that includes development milestone payments, the Company evaluates whether the milestones are considered probable of being reached and estimates the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price. Milestone payments that are not within the control of the Company or the licensee, such as regulatory approvals, are not considered probable of being achieved until those approvals are received. The transaction price is then allocated to each performance obligation on a relative stand-alone selling price basis, for which the Company recognizes revenue as or when the performance obligations under the contract are satisfied. At the end of each subsequent reporting period, the Company re-evaluates the probability of achievement of such development milestones and any related constraint, and if necessary, adjusts its estimate of the overall transaction price.
Manufacturing Supply Services: Arrangements that include a promise for future supply of raw materials or drug product for either clinical development or commercial supply at the customer’s discretion are generally considered as options. The Company assesses if these options provide a material right to the customer and if so, they are accounted for as separate performance obligations and allocated a portion of the transaction price based on the estimated standalone selling price of the material right. If the Company is entitled to additional payments when the customer exercises these options, the deferred transaction price and any additional payments are recorded in collaborative research and development revenue when the customer obtains control of the goods.
89
Royalties and Earn-outs: For arrangements that include sales-based royalties or earn-outs, including milestone payments based on first commercial sale or the level of sales, and the license is deemed to be the predominant item to which the royalties relate, the Company recognizes revenue at the later of (i) when the related sales occur, or (ii) when the performance obligation to which some or all of the royalty has been allocated has been satisfied (or partially satisfied). To date, the Company has not recognized material royalty revenue resulting from the Company’s collaborative arrangements or material earn-out revenues from any of the Company’s agreements.
Research and development services: Revenue from research and development services that are determined to represent a distinct performance obligation with the Company’s third-party collaborators is recognized over time as the related research and development services are performed using an appropriate method of measuring progress. The Company evaluates the measure of progress each reporting period and recognizes revenue on a cumulative catch-up basis, as collaborative research and development revenue. Research and development expenses under the collaborative research and development agreements generally approximate or exceed the revenue recognized under such agreements over the term of the respective agreements. Deferred revenue may result when the Company does not expend the required level of effort during a specific period in comparison to funds received under the respective agreement.
The Company receives payments from its customers based on development cost schedules established in each contract. Up-front payments are recorded as deferred revenue upon receipt or when due and may require deferral of revenue recognition to a future period until the Company performs its obligations under these arrangements. Amounts are recorded as accounts receivable when the Company’s right to consideration is unconditional. The Company does not assess whether a contract has a significant financing component if the expectation at contract inception is such that the period between payment by the customer and the transfer of the promised goods or services to the customer will be one year or less.
Prepaid and Accrued Clinical Costs
The Company incurs significant costs associated with third party consultants and organizations for pre-clinical studies, clinical trials, contract research, regulatory advice and other research and development-related services. The Company is required to estimate periodically the cost of services rendered but unbilled based on management’s estimates. Estimates are determined each reporting period by reviewing the terms and conditions of the underlying contracts, reviewing open purchase orders and by having detailed discussions with internal clinical personnel and third-party service providers as to the nature and status of the services performed in relation to amounts billed. The costs for unbilled services are estimated by applying the rates and fees applicable in the underlying contracts. If these good faith estimates are inaccurate, actual expenses incurred could materially differ from these estimates.
Prepaid and Accrued Manufacturing Costs
The Company incurs significant costs associated with third party consultants and organizations for manufacturing, validation, testing and other research and development-related services. The Company is required to estimate periodically the cost of services rendered but unbilled based on management’s estimates. Estimates are determined each reporting period by reviewing the terms and conditions of the underlying contracts, reviewing open purchase orders and by having detailed discussions with internal personnel and third-party service providers as to the nature and status of the services performed in relation to amounts billed. The costs for unbilled services are estimated by applying the rates and fees applicable in the underlying contracts. If these good faith estimates are inaccurate, actual expenses incurred could materially differ from these estimates.
90
Research and Development Expenses
Research and development expenses are primarily comprised of salaries and benefits associated with research and development personnel, overhead and facility costs, preclinical and non-clinical development costs, clinical trial and related clinical manufacturing costs, contract services, and other outside costs. Research and development costs are expensed as incurred. Research and development costs paid to third parties under sponsored research agreements are recognized as the related services are performed. In addition, research and development expenses incurred that are reimbursed by the Company’s partners are recorded as collaborative research and development revenue.
Comprehensive Loss
Components of other comprehensive loss are comprised entirely of unrealized gains and losses on the Company’s available-for-sale securities for all periods presented. Total comprehensive loss has been disclosed in the Company’s Statements of Operations and Comprehensive Loss.
Segment Reporting
The Company operates in
Net Loss Per Share
Basic net loss per share is calculated by dividing the net loss by the weighted-average number of common shares outstanding. Diluted net loss per share is computed using the weighted-average number of common shares outstanding and common stock equivalents (i.e., options to purchase common stock) outstanding during the period, if dilutive, using the treasury stock method for options.
The numerators and denominators in the calculation of basic and diluted net loss per share were as follows (in thousands except per share amounts):
|
|
Year Ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
Numerators: |
|
|
|
|
|
|
|
|
|
|||
Net loss |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Denominators: |
|
|
|
|
|
|
|
|
|
|||
Weighted average shares used to compute basic |
|
|
|
|
|
|
|
|
|
|||
Effect of dilutive securities: |
|
|
|
|
|
|
|
|
|
|||
Dilution from stock options |
|
|
— |
|
|
|
— |
|
|
|
— |
|
Dilution from ESPP |
|
|
— |
|
|
|
— |
|
|
|
— |
|
Dilutive common shares |
|
|
— |
|
|
|
— |
|
|
|
— |
|
Weighted average shares used to compute diluted |
|
|
|
|
|
|
|
|
|
|||
Net loss per share: |
|
|
|
|
|
|
|
|
|
|||
Basic |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Diluted |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
The computation of diluted net loss per share for 2022, 2021 and 2020 excludes the impact of options to purchase
91
Shipping and Handling
Costs related to shipping and handling are included in cost of revenues for all periods presented.
Recent Accounting Pronouncements
In June 2016, the FASB issued Accounting Standards Update No. 2016-13 (ASU 2016-13) “Financial Instruments - Credit Losses: Measurement of Credit Losses on Financial Instruments.” ASU 2016-13 requires measurement and recognition of expected credit losses for financial assets. This standard is effective for fiscal years beginning after December 15, 2022 for small reporting companies, including interim reporting periods within those years and must be adopted using a modified retrospective approach, with certain exceptions. Early adoption is permitted. The Company adopted the standard on January 1, 2023 and the adoption did not have a material effect on the financial statements.
In March 2020, the FASB issued ASU 2020-04, Reference Rate Reform (Topic 848): Facilitation of the Effects of Reference Rate Reform on Financial Reporting. In response to concerns about structural risks of the cessation of London Interbank Offered Rate (LIBOR), the amendments in this ASU provide optional guidance for a limited time to ease the potential burden in accounting for (or recognizing the effects of) reference rate reform on financial reporting. The amendments in this ASU provide optional expedients and exceptions for applying GAAP to contracts, hedging relationships and other transactions affected by reference rate reform if certain criteria are met. The amendments in this ASU apply only to contracts and hedging relationships that reference LIBOR or another reference rate expected to be discontinued due to reference rate reform. The expedients and exceptions provided by the amendments do not apply to contract modifications made and hedging relationships entered into or evaluated after December 31, 2022. The amendments in this ASU are elective and are effective for all entities as of March 12, 2020 through December 31, 2022. The adoption of the standard did not have a material effect on the financial statements.
The collaborative research and development and other revenue associated with the Company’s major third-party collaborators are as follows (in thousands):
|
|
Year ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
Collaborator/Counterparty |
|
|
|
|
|
|
|
|
|
|||
Innocoll (1) |
|
$ |
|
|
$ |
|
|
$ |
— |
|
||
Gilead (2) |
|
|
— |
|
|
|
— |
|
|
|
|
|
Other (3) |
|
|
|
|
|
|
|
|
|
|||
Total collaborative research and development and other revenue |
|
$ |
|
|
$ |
|
|
$ |
|
|||
92
As of December 31, 2022, the Company had potential milestones of up to $
Agreement with Innocoll
On December 21, 2021, the Company entered into a license agreement (the “Innocoll Agreement”) with Innocoll Pharmaceuticals Limited (“Innocoll”). Pursuant to the Innocoll Agreement, the Company has granted Innocoll an exclusive, royalty-bearing, sublicensable right and license to develop, manufacture and commercialize in the United States, POSIMIR®, the Company’s FDA-approved post-surgical pain product, with respect to all uses and applications in humans. The Innocoll Agreement provides for the assignment of the Company’s supply agreement with its contract manufacturing organization to Innocoll and also provides Innocoll with the right, within the United States, to expand the approved indications of POSIMIR. The Company retains, outside the United States, all of the global rights to POSIMIR.
Upon execution of the Innocoll Agreement, Innocoll paid the Company an initial nonrefundable, upfront fee of $
The Company also evaluated Innocoll’s future purchases of an excipient from the Company and concluded that these purchases are option rights, and are at market rates, and do not constitute a material right performance obligation. As such, these future purchases have been excluded from the allocation of transaction price and the Company will account for them as separate contracts when and if Innocoll elects to issue purchase orders for the excipient.
During December 2021, the upfront fee of $
93
In August 2022, the Company was issued a new patent by the U.S. Patent and Trademark Office, extending U.S. patent coverage of POSIMIR to at least 2041, resulting in an $
Agreement with Gilead Sciences, Inc.
On July 19, 2019, the Company entered into a license agreement (the “Gilead Agreement”) with Gilead Sciences, Inc. (“Gilead”). Pursuant to the Gilead Agreement, the Company granted Gilead the exclusive worldwide rights to develop and commercialize a long-acting injectable HIV product utilizing DURECT’s SABER® technology. Gilead also received exclusive access to the SABER platform for HIV and Hepatitis B Virus (HBV) and the exclusive option to license additional SABER-based products directed to HIV and HBV.
Under the terms of the Gilead Agreement, Gilead made a non-refundable upfront payment to DURECT of $
During the twelve months ended December 31, 2019, the upfront and milestone consideration of $
During the twelve months ended December 31, 2019, the Company recognized $
In June 2020, Gilead terminated the Gilead Agreement and a related R&D agreement between Gilead and the Company. As a result, the Company recognized $
Patent Purchase Agreement with Indivior
In September 2017, we entered into an agreement with Indivior (the “Indivior Agreement”), under which we assigned to Indivior certain patents that may provide further intellectual property protection for PERSERIS, Indivior’s extended-release injectable suspension for the treatment of schizophrenia in adults. In consideration for such assignment, Indivior made non-refundable upfront and milestone payments to DURECT totaling $
94
receives quarterly earn-out payments into 2026 that are based on a single digit percentage of U.S. net sales of PERSERIS. Indivior commercially launched PERSERIS in the U.S. in February 2019. The Indivior Agreement contains customary representations, warranties and indemnities of the parties. Amounts recognized during the twelve months ended December 31, 2022, 2021 and 2020 related to earn-out revenues from PERSERIS have been immaterial and are included in collaborative research and development and other revenue.
Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. The Company’s valuation techniques used to measure fair value maximize the use of observable inputs and minimize the use of unobservable inputs. The Company follows a fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value. These levels of inputs are the following:
The Company’s financial instruments are valued using quoted prices in active markets or based upon other observable inputs.
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Total |
|
||||
Money market funds |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
Certificates of deposit |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Commercial paper |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Total |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
The following table sets forth the fair value of our financial assets that were measured at fair value on a recurring basis as of December 31, 2021 (in thousands):
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Total |
|
||||
Money market funds |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
Certificates of deposit |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Commercial paper |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
U.S. Government agencies |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Total |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
|
|||
The Company’s financial instruments are valued using quoted prices in active markets or based upon other observable inputs. Money market funds are classified as Level 1 financial assets. Certificates of deposit, commercial paper and U.S. Government agency securities are classified as Level 2 financial assets. The fair value of the Level 2 assets is estimated using pricing models using current observable market information for similar securities. The Company’s Level 2 investments include U.S. government-backed securities and corporate securities that are valued based upon observable inputs that may include benchmark yields, reported trades, broker/dealer quotes, issuer spreads, two-sided markets, benchmark securities, bids, offers and reference data including market research publications. The fair value of
95
commercial paper is based upon the time to maturity and discounted using the three-month treasury bill rate. The average remaining maturity of the Company’s Level 2 investments as of December 31, 2022 is less than twelve months and these investments are rated by S&P and Moody’s at AAA or AA- for securities and A1 or P1 for commercial paper.
The following is a summary of available-for-sale securities as of December 31, 2022 and 2021 (in thousands):
|
|
December 31, 2022 |
|
|||||||||||||
|
|
Amortized |
|
|
Unrealized |
|
|
Unrealized |
|
|
Estimated |
|
||||
Money market funds |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
Certificates of deposit |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Commercial paper |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
|
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
|
||
Reported as: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Cash and cash equivalents |
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
|
||
Long-term restricted investments |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
|
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
|
||
|
|
December 31, 2021 |
|
|||||||||||||
|
|
Amortized |
|
|
Unrealized |
|
|
Unrealized |
|
|
Estimated |
|
||||
Money market funds |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
Certificates of deposit |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Commercial paper |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
Corporate debt |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
|
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Reported as: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Cash and cash equivalents |
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
|
||
Short-term investments |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
Long-term restricted investments |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
|
|
$ |
|
|
$ |
— |
|
|
$ |
( |
) |
|
$ |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
The following is a summary of the cost and estimated fair value of available-for-sale securities at December 31, 2022, by contractual maturity (in thousands):
|
|
December 31, 2022 |
|
|||||
|
|
Amortized |
|
|
Estimated |
|
||
Mature in one year or less |
|
$ |
|
|
$ |
|
||
Mature after one year through five years |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
There were
As of December 31, 2022, unrealized losses on available-for-sale investments are not attributed to credit risk and are considered to be temporary. The Company believes that it is more-likely-than-not that investments in an unrealized loss position will be held until maturity or the recovery of the cost basis of
96
the investment. To date, the Company has not recorded any impairment charges on marketable securities related to other-than-temporary declines in market value.
Property and equipment consist of the following (in thousands):
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Equipment |
|
$ |
|
|
$ |
|
||
Leasehold improvements |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Less accumulated depreciation and amortization |
|
|
( |
) |
|
|
( |
) |
Property and equipment, net |
|
$ |
|
|
$ |
|
||
Depreciation expense was $
As of December 31, 2022 and 2021, the Company recorded $
As of December 31, 2022 and 2021, the Company had $
Operating Leases
The Company has lease arrangements for its facilities in California as follows.
Location |
|
Approximate Square Feet |
|
Operation |
|
Expiration |
Cupertino, CA |
|
|
Office, Laboratory and Manufacturing |
|
||
|
|
|
|
|
|
|
Cupertino, CA |
|
|
Office and Laboratory |
|
||
|
|
|
|
|
|
|
Vacaville, CA |
|
|
Manufacturing |
|
||
|
|
|
|
|
|
|
Under these leases, the Company is required to pay certain maintenance expenses in addition to monthly rent. Rent expense is recognized on a straight-line basis over the lease term for leases that have scheduled rental payment increases. Rent expense under all operating leases was $
97
Future minimum payments under these noncancelable leases are as follows (in thousands):
Year ending December 31, |
|
Operating |
|
|
2023 |
|
|
|
|
2024 |
|
|
|
|
|
|
|
|
|
Less present value adjustment |
|
|
( |
) |
Operating lease liabilities recognized |
|
$ |
|
|
Accrued liabilities as of December 31, 2022 and 2021 were comprised as follows (in thousands):
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Accrued compensation and benefits |
|
$ |
|
|
$ |
|
||
Accrued clinical costs |
|
|
|
|
|
|
||
Accrued contract research and manufacturing cost |
|
|
|
|
|
|
||
Others |
|
|
|
|
|
|
||
Total |
|
$ |
|
|
$ |
|
||
In July 2016, the Company entered into a $
The term loan is secured by substantially all of the assets of the Company, except that the collateral does not include any intellectual property (including licensing, collaboration and similar agreements relating thereto), and certain other excluded assets. The 2016 Loan Agreement contains customary representations, warranties and covenants by the Company, which covenants limit the Company’s ability to convey, sell, lease, transfer, assign or otherwise dispose of certain assets of the Company; engage in any business other than the businesses currently engaged in by the Company or reasonably related thereto; liquidate or dissolve; make certain management changes; undergo certain change of control events; create, incur, assume, or be liable with respect to certain indebtedness; grant certain liens; pay dividends and make certain other restricted payments; make certain investments; and make payments on any subordinated debt.
98
The Loan Agreement also contains customary indemnification obligations and customary events of default, including, among other things, the Company’s failure to fulfill certain obligations of the Company under the Loan Agreement and the occurrence of a material adverse change which is defined as a material adverse change in the Company’s business, operations, or condition (financial or otherwise), a material impairment of the prospect of repayment of any portion of the loan, or a material impairment in the perfection or priority of lender’s lien in the collateral or in the value of such collateral. In the event of default by the Company under the Loan Agreement, the lender would be entitled to exercise its remedies thereunder, including the right to accelerate the debt, upon which the Company may be required to repay all amounts then outstanding under the Loan Agreement, which could harm the Company’s financial condition. The conditionally exercisable call option related to the event of default is considered to be an embedded derivative which is required to be bifurcated and accounted for as a separate financial instrument. In the periods presented, the value of the embedded derivative is not material, but could become material in future periods if an event of default became more probable than is currently estimated.
As of December 31, 2022, the Company was in compliance with all material covenants under the Loan Agreement and there had been no material adverse change. In accordance with ASC 470-10-45-2, the term loan was reclassified to current liabilities from non-current liabilities on the Company’s balance sheet as of December 31, 2022 due to recurring losses, liquidity concerns and a subjective acceleration clause in the Company’s Loan Agreement.
The fair value of the term loan approximates the carrying value. Future maturities due under the term loan as of December 31, 2022, are as follows (in thousands):
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
Total minimum payments |
|
|
|
|
Less unamortized debt discount and accrued final payment |
|
|
( |
) |
Carrying value of term loan, net |
|
|
|
Common Stock
In August 2018, the Company filed a new shelf registration statement on Form S-3 with the SEC, which upon being declared effective in October 2018, terminated an earlier registration statement and allowed the Company to offer up to $
On June 20, 2019, the Company entered into a transaction to sell
In February 2021, the Company completed an underwritten public offering of
99
financing were approximately $
In 2019, the Company raised net proceeds of approximately $
In July 2021, the Company filed a shelf registration statement on Form S-3 with the SEC (the “2021 Registration Statement”) (File No. 333-258333), which upon being declared effective in August 2021, terminated the 2018 Registration Statement and allows the Company to offer up to $
On December 5, 2022, the Company effected a
Description of Stock-Based Compensation Plans
2000 Stock Plan (Incentive Stock Plan)
In January 2000, the Company’s Board of Directors and stockholders adopted the DURECT Corporation 2000 Stock Plan, under which incentive stock options and non-statutory stock options and stock purchase rights may be granted to employees, consultants and non-employee directors. The 2000 Stock Plan was amended by written consent of the Board of Directors in March 2000 and written consent of the stockholders in August 2000.
In April 2005, the Board of Directors approved certain amendments to the 2000 Stock Plan. At the Company’s annual stockholders meeting in June 2005, the stockholders approved the amendments of the 2000 Stock Plan to: (i) expand the types of awards that the Company may grant to eligible service providers under the Stock Plan to include restricted stock units, stock appreciation rights and other similar types of awards (including other awards under which recipients are not required to pay any purchase or exercise price) as well as cash awards; and (ii) include certain performance criteria that may be applied to awards granted under the Stock Plan.
At the Company’s annual stockholders meeting in June 2010, the stockholders approved amendments of the 2000 Stock Plan to: (i) provide that the number of shares that remain available for issuance will be reduced by two shares for each share issued pursuant to an award (other than an option or stock appreciation right) granted on or after the date of the 2010 Annual Meeting; (ii) expand the types of transactions that might be considered repricings and option exchanges for which stockholder approval is required; (iii) provide that shares tendered or withheld in payment of the exercise price of an option or withheld to satisfy a withholding obligation, and all shares with respect to which a stock appreciation right is exercised, will not again be available for issuance under the Stock Plan; (iv) require that options and stock appreciation rights have an exercise price or base appreciation amount that is at least fair market value on the grant date, except in connection with certain corporate transactions, and that stock appreciation rights may not have longer than a
100
cover mergers in which the consideration payable to stockholders is not solely securities of the successor corporation.
At the Company’s annual stockholders meeting in June 2011, June 2014, June 2016 and June 2018, the stockholders approved amendments of the 2000 Stock Plan to increase the number of shares of the Company’s common stock available for issuance by
At the Company’s annual stockholders meeting in June 2019, the stockholders approved an amendment of the 2000 Stock Plan to extend the term of the 2000 Stock Plan to the date that is ten (
In April 2013, the Board of Directors approved certain amendments to the 2000 Stock Plan to: (i) increase the number of stock options granted to a non-employee director on the date which such person first becomes a director from
Options granted under the 2000 Stock Plan expire no later than ten years from the date of grant. Options may be granted with different vesting terms from time to time not to exceed
At the Company’s annual stockholders meeting in June 2022, the stockholders approved an amendment of the 2000 Stock Plan to increase the number of shares of the Company’s common stock available for issuance by
A total of
As of December 31, 2022,
2000 Employee Stock Purchase Plan
In August 2000, the Company adopted the 2000 Employee Stock Purchase Plan. This purchase plan is implemented by a series of overlapping offering periods of
101
In April 2010, the Board of Directors approved certain amendments to the 2000 Employee Stock Purchase Plan. At the Company’s annual stockholders meeting in June 2010, the stockholders approved the amendment of the 2000 Employee Stock Purchase Plan to: (i) increase the number of shares of our common stock authorized for issuance under the ESPP by
In March 2015, the Board of Directors approved certain amendments to the 2000 Employee Stock Purchase Plan. At the Company’s annual stockholders meeting in June 2015, the stockholders approved the amendments of the 2000 Employee Stock Purchase Plan to: (i) increase the number of shares of our common stock authorized for issuance under the ESPP by
The plan expires in June 2030. A total of
As of December 31, 2022, shares of common stock reserved for future issuance consisted of the following:
|
|
December 31, |
|
|
Stock options outstanding |
|
|
|
|
Stock options available for grant |
|
|
|
|
Employee Stock Purchase Plan |
|
|
|
|
|
|
|
|
|
A summary of stock option activity under all stock-based compensation plans is as follows:
|
|
Number of |
|
|
Weighted |
|
|
Weighted |
|
|
Aggregate |
|
||||
Outstanding at December 31, 2021 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
Options granted |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
Options exercised |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
Options forfeited |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
Options expired |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
Outstanding at December 31, 2022 |
|
|
|
|
$ |
|
|
|
|
|
$ |
— |
|
|||
Exercisable at December 31, 2022 |
|
|
|
|
$ |
|
|
|
|
|
$ |
— |
|
|||
Vested and expected to vest at |
|
|
|
|
$ |
|
|
|
|
|
$ |
— |
|
|||
The weighted-average grant date fair value of options granted during the years ended December 31, 2022, 2021 and 2020 was $
102
in the table above represents the total intrinsic value (i.e., the difference between the Company’s closing stock price on the last trading day of 2022 and the exercise price, multiplied by the number of in-the-money options) that would have been received by the option holders had all option holders exercised their in-the-money options on December 31, 2022. This amount changes based on the fair market value of the Company’s common stock. The total value of options exercised was $
Expenses for non-employee stock options are recorded over the vesting period of the options, which closely approximates the non-employee’s performance period, with the value determined by the Black-Scholes option valuation method and remeasured over the vesting term.
As of December 31, 2022, the Company had three stock-based equity compensation plans, which are described above. The employee stock-based compensation cost that has been included in the statements of operations and comprehensive loss is shown as below (in thousands):
|
|
Year ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
Cost of product revenues |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Research and development |
|
|
|
|
|
|
|
|
|
|||
Selling, general and administrative |
|
|
|
|
|
|
|
|
|
|||
|
|
$ |
|
|
$ |
|
|
$ |
|
|||
Because the Company had a net operating loss carryforward as of December 31, 2022,
Determining Fair Value
Valuation and Expense Recognition. The Company estimates the fair value of stock options granted using the Black-Scholes option valuation model. The Company recognizes the expense on a straight-line basis. The expense for options is recognized over the requisite service periods of the awards, which is generally the vesting period.
Expected Term. The expected term of options granted represents the period of time that the options are expected to be outstanding. The Company determines the expected life using historical options experience. This develops the expected life by taking the weighted average of the actual life of options exercised and cancelled and assumes that outstanding options are exercised uniformly from the current holding period through the end of the contractual life.
Expected Volatility. The Company estimates the volatility of its common stock at the date of grant based on the historical volatility of the Company’s common stock.
Risk-Free Rate. The Company bases the risk-free rate that it uses in the Black-Scholes option valuation model on the implied yield in effect at the time of option grant on U.S. Treasury zero-coupon issues with substantially equivalent remaining terms.
Dividends. The Company has never paid any cash dividends on its common stock and the Company does not anticipate paying any cash dividends in the foreseeable future. Consequently, the Company uses an expected dividend yield of
103
The Company used the following assumptions to estimate the fair value of options granted and shares purchased under its stock plans and employee stock purchase plan for the years ended December 31, 2022, 2021 and 2020:
|
|
Year ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
Stock Options |
|
|
|
|
|
|
|
|
|
|||
Risk-free rate |
|
|
|
|
|
|
||||||
Expected dividend yield |
|
|
— |
|
|
|
— |
|
|
|
— |
|
Expected term (in years) |
|
|
|
|
|
|
||||||
Volatility |
|
|
|
|
|
|
||||||
Forfeiture rate (1) |
|
|
% |
|
|
% |
|
|
% |
|||
|
|
Year ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
Employee Stock Purchase Plan |
|
|
|
|
|
|
|
|
|
|||
Risk-free rate |
|
|
|
|
|
|
||||||
Expected dividend yield |
|
|
— |
|
|
|
— |
|
|
|
— |
|
Expected term (in years) |
|
|
|
|
|
|
|
|
|
|||
Volatility |
|
|
|
|
|
|
||||||
There were
As of December 31, 2022, $
The following table summarizes information about stock options outstanding at December 31, 2022:
|
|
Options Outstanding |
|
|
Options Exercisable |
|
||||||||||||||
Range of |
|
Number of |
|
|
Weighted- |
|
|
Weighted- |
|
|
Number of |
|
|
Weighted- |
|
|||||
$ |
|
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
$ |
|
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
$ |
|
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
$ |
|
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
$ |
|
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
$ |
|
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
$ |
|
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
$ |
|
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
$ |
|
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
$ |
|
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
$ |
|
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
The Company received $
104
The Company accounts for income taxes using the liability method under ASC 740, Income Taxes. Under this method, deferred tax assets and liabilities are determined based on temporary differences resulting from the different treatment of items for tax and financial reporting purposes. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to reverse. Additionally, the Company must assess the likelihood that deferred tax assets will be recovered as deductions from future taxable income. The Company has provided a full valuation allowance on the Company’s deferred tax assets because the Company believes it is more likely than not that its deferred tax assets will not be realized. The Company evaluates the realizability of its deferred tax assets on a quarterly basis. The Company recorded a deferred tax liability of $
The reconciliation of income tax expenses (benefit), at the statutory federal income tax rate of
|
|
Year Ended December 31, |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
U.S. federal taxes benefit at statutory rate |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
Change in valuation allowance |
|
|
|
|
|
|
|
|
( |
) |
||
Stock-based compensation |
|
|
|
|
|
|
|
|
|
|||
Research and development tax credits |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
Expiring net operating losses |
|
|
|
|
|
|
|
|
|
|||
Other |
|
|
|
|
|
|
|
|
|
|||
Total income tax (benefit) provision |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
In 2022, 2021 and 2020, total income tax provision (benefit) expense was
Significant components of the Company’s deferred tax assets and liabilities are as follows (in thousands):
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Deferred tax assets: |
|
|
|
|
|
|
||
Net operating loss carryforwards |
|
|
|
|
|
|
||
Research and other credits |
|
|
|
|
|
|
||
Section 174 R&D capitalization |
|
|
|
|
|
— |
|
|
Deferred revenue |
|
|
— |
|
|
|
|
|
Stock-based compensation |
|
|
|
|
|
|
||
Other |
|
|
|
|
|
|
||
Total deferred tax assets |
|
|
|
|
|
|
||
Valuation allowance for deferred tax assets |
|
|
( |
) |
|
|
( |
) |
Deferred tax liabilities - right of use asset |
|
|
( |
) |
|
|
( |
) |
Net deferred tax assets and liabilities |
|
$ |
( |
) |
|
$ |
( |
) |
The Company recognizes deferred tax assets to the extent that the Company believes that these assets are more likely than not to be realized. In making such a determination, all available positive and negative evidence is considered, including future reversals of existing taxable temporary differences,
105
projected future taxable income, tax-planning strategies, and results of recent operations. If it is determined that the Company would be able to realize deferred tax assets in the future in excess of their net recorded amount, the Company would make an adjustment to the deferred tax asset valuation allowance, which would cause a provision benefit to be recognized. The recognition and measurement of tax benefits requires significant judgment. Judgments concerning the recognition and measurement of tax benefit might change as new information becomes available. Given the Company’s history of operating losses, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance increased by $
As of December 31, 2022, the Company had net operating loss carryforwards for federal income tax purposes of approximately $
As of December 31, 2022, the Company had net operating loss carryforwards for state income tax purposes of approximately $
Utilization of the net operating losses may be subject to a substantial annual limitation due to federal and state ownership change limitations. The annual limitation may result in the expiration of net operating losses before utilization.
At December 31, 2022 and December 31, 2021, the Company had unrecognized tax benefits of approximately $
A reconciliation of the beginning and ending amount of unrecognized tax benefits is as follows (in thousands):
|
|
December 31, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Balance at beginning of the year |
|
$ |
|
|
$ |
|
||
Increase related to prior year tax positions |
|
|
( |
) |
|
|
|
|
Increase related to current year tax positions |
|
|
|
|
|
|
||
Balance at end of the year |
|
$ |
|
|
$ |
|
||
Interest and penalty costs related to unrecognized tax benefits, if any, are classified as a component of interest and other income, net in the Statements of Operations and Comprehensive Loss. The Company did
The Company files income tax returns in the U.S. federal jurisdiction and various state jurisdictions. The Company is subject to U.S. federal and state income tax examination for calendar tax years ending 2002 through 2022 due to unutilized net operating losses and research credits.
California Assembly Bill 85 (AB 85) was signed into law in June 2021. The legislation suspended the use of California Net Operating Loss deductions for 2020, 2021, and 2022 for certain taxpayers and imposed a limitation on the use of certain California Tax Credits for 2020, 2021, and 2022. The carryover periods for Net Operating Loss deductions disallowed by this provision were extended. California enacted Senate Bill 113 on February 14, 2022, which removed the net operating loss suspension and limited use of business tax credits for 2022. Senate Bill 113 had no income tax impact as the Company continues to record full valuation allowance against the deferred tax assets due to the cumulative tax losses.
106
Beginning with 2022, the Tax Cuts and Jobs Act eliminated the option to deduct research and development expenditures when incurred under Section 174 and requires taxpayers to capitalize and amortize domestic expenditures over five years and foreign expenditures over fifteen years. Given the lack of current legislative guidance available, the Company has provided for an estimate of their expenditures subject to the Section 174 requirements and established a deferred tax asset, offset by the Company’s
On December 31, 2020, the Company completed the sale of its LACTEL Absorbable Polymers product line to Evonik. Under the terms of the Asset Purchase Agreement, Evonik paid DURECT approximately $
As a result of the sale of the LACTEL product line, the operating results from our LACTEL product line have been excluded from continuing operations and presented as discontinued operations in the accompanying Statements of Operations and Comprehensive Loss for all periods presented. During the twelve months ended December 31, 2020, we recorded a gain on sale of the LACTEL product line of $
The components of income from discontinued operations as reported in the Company’s statement of operations were as follows (in thousands):
|
|
Year ended December 31, |
|
|
|
|
2020 |
|
|
Total revenues |
|
|
|
|
Operating expenses: |
|
|
|
|
Cost of product revenues |
|
|
|
|
Research and development |
|
|
|
|
Selling, general and administrative |
|
|
|
|
Total costs and expenses |
|
|
|
|
Income from discontinued operations |
|
|
|
|
Other income: |
|
|
|
|
Gain on sale of the LACTEL product line |
|
|
|
|
Net income from discontinued operations |
|
$ |
|
|
Net income per share |
|
|
|
|
Basic and diluted |
|
$ |
|
|
Weighted-average shares used in computing net income per share basic and diluted |
|
|
|
|
Basic and diluted |
|
|
|
|
|
|
|
|
|
107
The following table presents certain non-cash items related to discontinued operations, which are included in the Company’s statement of cash flows (in thousands):
|
Years ended December 31, |
|
|
|
2020 |
|
|
Depreciation |
$ |
|
|
Stock-based compensation expense |
|
|
|
Goodwill |
|
|
|
Loss on disposal of property and equipment |
|
|
|
|
$ |
|
|
Gain on sale of the LACTEL product line |
$ |
|
|
Non-cash items, net |
$ |
( |
) |
The pre-funded warrants are exercisable immediately following the closing date of the Offering and have an unlimited term and an initial exercise price of $
108
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
Not applicable.
Item 9A. Controls and Procedures.
Disclosure Controls and Procedures
As required by paragraph (b) of Exchange Act Rules 13a-15 or 15d-15, DURECT’s management, including our Chief Executive Officer and Chief Financial Officer, conducted an evaluation as of the end of the period covered by this report, of the effectiveness of DURECT’s disclosure controls and procedures as defined in Exchange Act Rule 13a-15(e) and 15d-15(e). Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that DURECT’s disclosure controls and procedures were effective as of the end of the period covered by this report.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Under the supervision of our Chief Executive Officer and Chief Financial Officer and with the participation of our management, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2022 based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 Framework). Based on that evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2022.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting identified in connection with the evaluation required by paragraph (d) of Exchange Act Rules 13a-15 or 15d-15 that occurred during our last fiscal quarter that have materially affected or are reasonably likely to materially affect, our internal control over financial reporting.
109
Item 9B. Other Information.
Adjustment of certain items in our Unaudited Condensed Balance Sheet as of December 31, 2022 included within our Fourth Quarter and Full Year 2022 Financial Results earnings release.
Subsequent to the issuance of the Company's earnings announcement on March 7, 2023, the Company recorded an adjustment in its Unaudited Condensed Balance Sheet as of December 31, 2022 to reclassify the $15,895,000 noncurrent portion of its term loan to current liabilities. The adjustment had no effect on the previously reported total assets and total liabilities and stockholders’ equity as of December 31, 2022. The effects of this adjustment also did not change its previously reported Unaudited Condensed Statements of Operations and Comprehensive Loss for the year ended December 31, 2022.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
110
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The names of the executive officers of the Company and their ages, titles and biographies as of the date hereof are incorporated by reference from Part I, Item 1, above.
Information required by this item will be contained in our definitive proxy statement to be filed with the Securities and Exchange Commission on Schedule 14A in connection with our 2023 Annual Meeting of Stockholders (the Proxy Statement), which is expected to be filed not later than 120 days after the end of our fiscal year ended December 31, 2022, under the headings “Election of Directors,” “The Board, Board Committees and Meetings,” “Code of Ethics,” and “Section 16(a) Beneficial Ownership Reporting Compliance,” and is incorporated herein by reference.
Item 11. Executive Compensation
Information required by this item will be contained in the Proxy Statement under the headings “Executive Compensation,” “Director Compensation,” and “Compensation Committee Report” and is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Information required by this item will be contained in the Proxy Statement under the headings “Common Stock Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information,” and is incorporated herein by reference.
Information required by this item will be contained in the Proxy Statement under the headings “Certain Relationships,” “Other Transactions,” and “The Board, Board Committees and Meetings,” and is incorporated herein by reference.
Item 14. Principal Accountant Fees and Services
Information required by this item will be contained in the Proxy Statement under the heading “Fees Billed for Services Rendered by Principal Accountant,” and is incorporated herein by reference.
111
PART IV
Item 15. Exhibits and Financial Statement Schedules.
The Index to financial statements in Item 8 of this report is incorporated herein by reference as the list of the financial statements required as part of this report.
Schedules not listed above have been omitted because the information required to be set forth therein is not applicable or is not present in amounts sufficient to require submission of the schedule, or because the information required is included in the financial statements or notes thereto.
112
Exhibit Index
Exhibit Number |
|
Description |
|
|
|
|
|
|
3.1 |
|
|
|
|
|
3.2 |
|
|
|
|
|
3.3 |
|
|
|
|
|
3.4
|
|
|
|
|
|
3.5 |
|
|
|
|
|
3.6 |
|
|
|
|
|
3.7 |
|
|
|
|
|
3.8* |
|
|
|
|
|
3.9 |
|
|
|
|
|
4.1* |
|
|
|
|
|
10.1+ |
|
|
|
|
|
113
Exhibit Number |
|
Description |
10.2+ |
|
|
|
|
|
10.3+ |
|
|
|
|
|
10.4 |
|
|
|
|
|
10.5 |
|
|
|
|
|
10.6 |
|
|
|
|
|
10.7 |
|
|
|
|
|
10.8 |
|
|
|
|
|
10.9 |
|
|
|
|
|
10.10 |
|
|
|
|
|
10.11 |
|
|
|
|
|
10.12** |
|
114
Exhibit Number |
|
Description |
|
|
|
10.13 |
|
|
|
|
|
10.14 |
|
|
|
|
|
10.15 |
|
|
|
|
|
10.16 |
|
|
|
|
|
10.17 |
|
|
|
|
|
10.18 |
|
|
|
|
|
10.19 |
|
|
|
|
|
10.20 |
|
|
|
|
|
10.21 |
|
|
10.22+ |
|
|
10.23 |
|
115
Exhibit Number |
|
Description |
|
|
|
10.24
|
|
|
|
|
|
10.25++ |
|
|
|
|
|
10.26 |
|
|
|
|
|
10.27+ |
|
|
|
|
|
10.28+ |
|
|
|
|
|
10.29+* |
|
Offer letter between Judy Joice and DURECT Corporation dated May 9, 2007. |
|
|
|
10.30+* |
|
Amendment to Offer Letter between Judy Joice and DURECT Corporation dated March 31, 2014. |
|
|
|
10.31+ |
|
|
|
|
|
23.1* |
|
|
|
|
|
31.1* |
|
|
|
|
|
31.2* |
|
|
|
|
|
32.1*** |
|
|
|
|
|
32.2*** |
|
|
|
|
|
101 |
|
The following financial statements from the Company's Annual Report on Form 10-K for the year ended December 31, 2022, formatted in Inline XBRL: (i) Consolidated Balance Sheets as of December 31, 2022 and 2021, (ii) Consolidated Statements of Operations and Comprehensive Loss for the years ended December 31, 2022, 2021 and 2020, (iii) |
116
Exhibit Number |
|
Description |
|
|
Condensed Statements of Stockholders' Equity for the years ended December 31, 2022, 2021 and 2020, (iv) Condensed Statements of Cash Flows for the years ended December 31, 2022, 2021 and 2020 and (v) Notes to Condensed Financial Statements, tagged as blocks of text and including detailed tags. |
|
|
|
104 |
|
Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101). |
* Filed herewith.
** Confidential treatment granted with respect to certain portions of this Exhibit.
*** Furnished, not filed.
+ Indicates a management contract or compensatory plan or arrangement.
++ Certain portions of this exhibit (indicated by “[***]”) have been omitted in accordance with Item 601(b)(10) of Regulation S-K.
Item 16. Form 10-K Summary.
The Company has elected not to include summary information.
117
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
DURECT CORPORATION |
|
|
|
By: |
/s/ james e. brown |
|
James E. Brown President and Chief Executive Officer |
Date: March 8, 2023
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature |
|
Title |
|
Date |
|
|
|
|
|
/s/ james e. brown |
|
President, Chief Executive Officer and Director (Principal Executive Officer) |
|
March 8, 2023 |
James E. Brown |
|
|
|
|
|
|
|
|
|
/s/ TIMOTHY M. PAPP |
|
Chief Financial Officer (Principal Accounting Officer) |
|
March 8, 2023 |
Timothy M. Papp |
|
|
|
|
|
|
|
|
|
/s/ MOHAMMAD AZAB |
|
Director |
|
March 8, 2023 |
Mohammad Azab |
|
|
|
|
|
|
|
|
|
/s/ terrence f. blaschke |
|
Director |
|
March 8, 2023 |
Terrence F. Blaschke |
|
|
|
|
|
|
|
|
|
/s/ Gail M. farfel |
|
Director |
|
March 8, 2023 |
Gail M. Farfel |
|
|
|
|
|
|
|
|
|
/s/ PETER S. GARCIA |
|
Director |
|
March 8, 2023 |
Peter S. Garcia |
|
|
|
|
|
|
|
|
|
/s/ david r. hoffmann |
|
Director, Chairman of the Board |
|
March 8, 2023 |
David R. Hoffmann |
|
|
|
|
|
|
|
|
|
/s/ GAIL J. MADERIS |
|
Director |
|
March 8, 2023 |
Gail J. Maderis |
|
|
|
|
|
|
|
|
|
/s/ judith j. Robertson |
|
Director |
|
March 8, 2023 |
Judith J. Robertson |
|
|
|
|
118